
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembraneless ethanol fuel cell Pt–Sn–Re nano active catalyst on a mesoporous carbon support
M. Priya a and
B. Muthukumaran*b
a and
B. Muthukumaran*b
aDepartment of Chemistry, School of Basic Sciences, Vels Institute of Science, Technology & Advanced Studies, Chennai, Tamilnadu, India. E-mail: priyamanokhar@gmail.com
bDepartment of Chemistry, Presidency College (Autonomous), Chennai 600 005, India. E-mail: dr.muthukumaran@yahoo.com
First published on 22nd March 2024
Abstract
Herein, we report, for the first-time, mesoporous carbon-supported binary and ternary catalysts with different atomic ratios of Pt/MC (100), Pt–Sn/MC (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:115:05) prepared using a co-impregnation reduction method as anode components for membraneless ethanol fuel cells (MLEFLs). Mechanistic and structural insights into binary Pt–Sn/MC, Pt–Re/MC and ternary Pt–Sn–Re/MC catalysts were obtained using scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD) and energy-dispersive X-ray spectroscopy (EDX) methods. In particular, chemical characterization via cyclic voltammetry, CO stripping voltammetry and chronoamperometry indicated that Pt–Sn–Re/MC (80:15:05) had better dynamics toward ethanol oxidation than Pt–Sn–Re/MC (80:10:10), Pt–Sn/MC (50:50) and Pt–Re/MC (50:50) catalysts. In terms of the single cell performance of the prepared catalysts, Pt–Sn–Re/MC (80:15:05) (31.5 mW cm−2) showed a higher power density and current density than Pt–Sn–Re/MC(80:10:10), Pt–Re/MC (50:50) and Pt–Sn/MC (50:50) at room temperature. The addition of Re into the binary Pt–Sn catalyst improved its electrical performance for ethanol oxidation in a membraneless ethanol fuel cell. As a result, the ternary-based Pt–Sn–Re/MC (80:15:05) catalyst demonstrated enhanced performance compared to monometallic and bimetallic catalysts in the ethanol oxidation reaction in a membraneless fuel cell.
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:115:05) prepared using a co-impregnation reduction method as anode components for membraneless ethanol fuel cells (MLEFLs). Mechanistic and structural insights into binary Pt–Sn/MC, Pt–Re/MC and ternary Pt–Sn–Re/MC catalysts were obtained using scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD) and energy-dispersive X-ray spectroscopy (EDX) methods. In particular, chemical characterization via cyclic voltammetry, CO stripping voltammetry and chronoamperometry indicated that Pt–Sn–Re/MC (80:15:05) had better dynamics toward ethanol oxidation than Pt–Sn–Re/MC (80:10:10), Pt–Sn/MC (50:50) and Pt–Re/MC (50:50) catalysts. In terms of the single cell performance of the prepared catalysts, Pt–Sn–Re/MC (80:15:05) (31.5 mW cm−2) showed a higher power density and current density than Pt–Sn–Re/MC(80:10:10), Pt–Re/MC (50:50) and Pt–Sn/MC (50:50) at room temperature. The addition of Re into the binary Pt–Sn catalyst improved its electrical performance for ethanol oxidation in a membraneless ethanol fuel cell. As a result, the ternary-based Pt–Sn–Re/MC (80:15:05) catalyst demonstrated enhanced performance compared to monometallic and bimetallic catalysts in the ethanol oxidation reaction in a membraneless fuel cell.
1. Introduction
Portable electronic telecommunication and computer devices, including mobile phones, laptops and personal digital assistants, have recently seen a sharp increase in demand.1 The development of electrochemical energy-conversion technologies has received much attention during the past two decades in order to overcome the global energy crisis.2 To date, numerous studies have been developed focusing on miniature power supplies, such as fuel cells, supercapacitors, batteries and other micro energy harvesters.3 Among these choices, membraneless fuel cells are considered one of the most promising miniaturized power generators.4 Fuel cells represent alternative power sources that can convert chemical energy into electrical energy during an electrochemical reaction.5 This power source has the ability to meet the standards for sustainable power sources and is a candidate to replace the existing batteries.6 Alcohols have been used extensively as a fuel under ambient conditions because they have a higher volumetric energy density than H2 fuel, are less poisonous, and are simple to handle, and some alcohols are renewable.7In particular, among proton-exchange membrane fuel cells, direct ethanol fuel cells (DEFCs) have attracted considerable attention owing to their high energy density, abundant production from biomass and non-toxicity.8 Compared to methanol, DEFCs have the potential to achieve electrical activity with apparently a less significant fuel crossover to a cathode.9 Compared to methanol, ethanol is less hazardous, and its full oxidation results in higher energy densities.10 However, numerous drawbacks in the utilization of such systems prevented their commercialization since the kinetics of the ethanol oxidation reaction (EOR) is slow and complicated, with many adsorption intermediates and reaction products.11 At ambient temperature, it is still exceedingly difficult to split the C–C bond, which is necessary for the complete oxidation of ethanol to CO2, which requires 12 electrons per molecule.12 In addition, Pt-based catalysts are costly and ineffective as anode catalysts for ethanol electro-oxidation because they can be easily poisoned by highly adsorbed intermediates.12
A number of co-catalysts are added to the Pt surface in order to circumvent these issues and make it easier to oxidize poisoning intermediates at lower overpotentials. These co-catalysts include gold (Au),13 silver (Ag),14 copper (Cu),15 cobalt,16 tin,17 ruthenium,18 manganese,19 and bismuth.20 To date, Pt–Sn bimetallic catalysts have received the most attention among the candidates and have been discovered to be the most effective for use in ethanol fuel cells. Utilizing oxophilic atoms can accelerate the oxidation of water by creating surface hydroxides, which can more easily oxidize the intermediates of chlorhexidine (CHx) and carbon monoxide (CO) (functional mechanism).21–23 However, it is still desired to start ethanol oxidation with some significant enhancements for improving the catalytic activities.
To overcome these problems, the addition of a third metal is one of the apparent choices for improving the performance of the electrode and lowering the usage of noble metals. In Pt–Sn–M catalysts, the third metal is an oxophilic element, such as tungsten (W), osmium (Os), nickel (Ni), iridium (Ir), rhodium (Rh), or tin (Sn). The role of the third element is to promote CO oxidation and prevent CO from adhering to the Pt.24
Furthermore, a recent study focused on changing the supporting materials to increase the electrochemical reaction. Recently, mesoporous carbon-supported materials have played an essential role in the electrochemical reactions in fuel cells due to their high surface area, good crystalline nature, low electrical resistance, high pore size, and good interaction between the prepared catalysts.25,26 On this basis, the present study describes mesoporous carbon-supported Pt–Sn–Re prepared by a co-impregnation reduction method.27 The chemistry, morphological structure and the electrochemical activity of the ethanol oxidation reaction (EOR) were investigated with sodium perborate as an oxidant, and sulfuric acid as an electrolyte. The oxidation of ethanol was electrochemically studied in H2SO4 solutions and tests were also carried out in membraneless ethanol fuel cells (MLEFLs).
2. Materials and methods
2.1 Materials
The precursors used for the preparation of the catalysts were H2PtCl6·6H2O, SnCl2·2H2O ReCl3 and mesoporous carbon support. Graphite plates (3 cm long and 0.1 cm wide, obtained from E-TEK) were used as substrates for the catalysts to prepare the electrodes. Deionized water (DI) was used as a solvent in all the experiments. Nafion® (DE 521, DuPont USA) dispersion was used to make the catalyst slurry.30 Ethanol (Merck) was used as a fuel, oxidant and electrolyte for the electrochemical analysis. Sodium perborate (Riedel) and H2SO4 (Merck) were used as a fuel, oxidant and electrolyte for the electrochemical analysis, respectively. All the chemicals were of analytical grade. Pt/MC (40 wt%) was used as cathode catalyst.292.2 Preparation of the mesoporous carbon (MC)-supported Pt–Sn–Re catalysts
Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:115:05) was prepared by a co-impregnation reduction method. The precursors were first suspended in propanol and then ultrasonicated for 3 h. Mesoporous carbon with a large surface area was heated to 110 °C, suspended in propanol separately, and then ultrasonically processed for 3 h. Then, the mesoporous carbon suspension was gradually mixed with the precursor suspension.
The weight ratio of Pt–M/MC (M drop-wise = Sn, Re, and Sn–Re) was controlled according to the targeted metal loading. To ensure the proper impregnation on the mesoporous carbon support, the precursor and mesoporous carbon suspension were ultrasonically blended for 3 h.
The suspension was then kept at 70 °C for 12 h till all the propanol had evaporated. Using the impregnation process, the precursor mixtures were reduced in hydrogen atmospheres at various temperatures to produce the binary Pt–Sn/MC, Pt–Re/MC, and ternary catalysts Pt–Sn–Re/MC.
2.3 Physical characterization
The crystal structures of prepared catalysts were determined by powder X-ray diffraction (PXRD) investigations utilizing a Rigaku multiflex diffractometer (model RU-200 B) with Cu Kα1 radiation sources (λKα1 = 1.5406 Å) operating temperature. The tube current was 40 mA with a tube voltage 40 kV. The 2θ angular regions between 20° and 90° were recorded at a scan rate of 5° min−1. The compositional ratios and the particle sizes were determined using an energy-dispersive X-ray (EDAX) system integrated with a transmission electron microscopy (TEM) unit.2.4 Chemical measurements
The thin porous technique was used to conduct the electrochemical measurements. For the working electrode, about 20 mg of the prepared catalyst was added to 50 ml of water containing three drops of 60% polytetrafluoroethylene (PTEF) suspension. Next, the above mixture was ultrasonically treated for 10 min and then transferred to the cavity of the working electrode. The current values (I) in the cyclic voltammetry (CV) and chronoamperometry (CA) studies were represented in amperes and normalized per gram of platinum. The amount of platinum was determined by the electrocatalyst present in the working electrode multiplied by its percentage of platinum. CV and CA measurements were carried out at 25 °C using a solution of 1 M ethanol in 0.5 M H2SO4 that was saturated with N2 gas purged for at least 30 min with high-purity nitrogen gas to assure oxygen-free measurement. The working electrode was a catalyst-coated glass carbon electrode (GCE, 3 mm diameter and 0.071 cm2 electrode area, from CHI, USA). The reference electrode was made of Ag/AgCl in saturated KCl (RHE).Ethanol is a clean and safe biofuel in the liquid phase, which is much easier and safer to store and transport than pure hydrogen. Compared to the technology to extract hydrogen from ethanol and then convert hydrogen to electricity, our technology can directly convert ethanol into electricity by a facile electro-oxidation on a Pt catalyst. It is also renewable, and its complete oxidation to CO2 and H2O produces a high yield of 12 electrons per molecule, as shown below:
| Anode: C2H5OH + 3H2O → 2CO2 + 12H+ + 12e− | (1) |
| Cathode: 6H2O2 + 12H+ + 12e− → 12H2O | (2) |
| Overall: C2H5OH + 6H2O2 → 2CO2 + 9H2O | (3) |
Ethanol has the possibility to achieve an electrochemical reaction analogous to the performance of methanol through a lesser amount of important fuel crossover to the cathode (Table 1). Compared to methanol oxidation, it gives intermediate CO and has a single carbon bond; nevertheless, the ethanol oxidation reaction (EOR) mechanism is slow, and follows complex pathways with numerous adsorbed intermediates and by-products, such as CH3COOH, CH3CHO, CHx, CO and CO2. The process of absolute ethanol oxidation to form CO2 adsorbs 12 electrons per molecule and necessitates cleavage of the C–C bond, which is still extremely difficult to attain at room temperature.
| Fuel | Standard potential E° (V) | Theoretical specific energy Ws (kW h kg−1) | Theoretical density energy We (kW h m−3) | Pure compounds capacity (eA h kg−1) | Energy efficiency η (%) |
|---|---|---|---|---|---|
| Methanol | 1.21 | 6.07 | 4.82 | 5019 | 97 |
| Ethylene glycol | 1.25 | 5.27 | 5.87 | 4318 | 97 |
| Glycol | 1.25 | 5.09 | 6.42 | 4071 | — |
| Ethanol | 1.51 | 8.03 | 6.28 | 6981 | 97 |
3. Results and discussion
3.1 Physical characterization of the Pt–Sn–Re/MC
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:115:05) were prepared by a co-impregnation reduction method as anode components for membraneless ethanol fuel cells (MLEFLs). The X-ray diffraction patterns of the prepared catalysts, including Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:15:05), are shown in Fig. 1. For all the catalysts, the first peak at about 25–35° was related to the diffraction of the (022) plane of mesoporous carbon. The diffraction peaks at around 38.5°, 46.5°, 67.8°, 81.4° and 86° corresponded to the (1 1 1), (2 0 0), (2 2 0), and (3 1 1) planes of Pt, respectively, indicating that Pt existed in the face-centred cubic (fcc) plane. To measure the particle size and lattice parameter of Pt while avoiding disruption of the carbon interaction, the (2 2 0) plane of Pt was used. In the case of Pt–Sn/MC alloy catalysts, the addition of Sn increased the lattice parameter of the Pt crystal structure, as indicated by the shift of the diffraction peaks to lower 2θ values. These shifts may be attributed to the alloy formation between Pt and Sn.
 | ||
| Fig. 1 XRD patterns of the as-prepared catalysts: Pt/MC (100) (blue), Pt–Sn/MC (50:50) (orange), Pt–Re/MC (50:50) (red), Pt–Sn Re/MC(80:15:05) (green) and Pt–Sn–Re/MC(80:15:05) (violet). | ||
The mean particle sizes and lattice parameters related to Pt (220) for the prepared catalysts, including Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10), and Pt–Sn–Re/MC (80:15:05), are given in Table 2.
| Catalysts | Nominal atomic ratio | EDX atomic ratio | Lattice parameter (nm) | Crystallite size (nm) | Particle size obtained from TEM (nm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Pt | Sn | Re | Pt | Sn | Re | ||||
| Pt/MC | 100 | — | — | — | — | — | 0.3926 | 7.4 | 7.1 |
| Pt–Sn/MC | 50 | 50 | — | 49 | 51 | — | 0.3993 | 6.9 | 6.7 |
| Pt–Re/MC | 50 | — | 50 | 47 | 53 | 0.3922 | 7.9 | 7.5 | |
| Pt–Sn–Re/MC | 80 | 10 | 10 | 78 | 9 | 13 | 0.3988 | 9.6 | 9.2 |
| Pt–Sn–Re/MC | 80 | 15 | 05 | 78 | 18 | 4 | 0.3978 | 6.7 | 6.3 |
The average particle size was between 6 and 9 nm. The addition of Sn to Pt did not alter the lattice of the Pt crystal structure and there was no shift of the 2θ values in the diffraction peaks. This may be attributed to their various atomic sizes in comparison to the Pt element (Pt = 1.39, Re = 1.37, and Sn = 1.61). This theoretical value shows that the Sn metal had a bigger size compared to the other two metals, and hence, adding Sn led to expansion of the Pt lattice. Along with the Pt and Re peaks, there were new peaks observed for Pt–Sn combination catalysts that were attributed to Pt–Sn (102), Pt–Sn (110), and Pt–Sn (202), respectively, at around 41.5°, 44.5°, and 63°. According to the joint committee of powder diffraction values (JCPDS-250614), the presence of Re in Pt–Sn shifted the diffraction peak to a lower 2θ value. This shift is an indication of the lattice parameter.27–35
:50), Pt–Re/MC (50:50) and Pt–Sn–Re/MC (80:15:05) catalysts are shown in Fig. 2 and compared. In Fig. 2, the white hue proved the presence of Sn due to the acquired stimulated electron beam. These images clearly show that the prepared catalysts had porous structure and were evenly distributed on the mesoporous carbon support.
 | ||
| Fig. 2 SEM images of (a) Pt–Sn–Re/MC (80:15:05), (b) Pt–Re/Mc (50:50) and (C) Pt–Sn/MC (50:50) catalysts. | ||
:15:05) and Pt–Re/MC (50:50) are shown in Fig. 3. The EDX spectra of Pt–Re/MC (50:50) and Pt–Sn–Re/MC (80:15:05) confirmed the presence of Pt, Sn, and Re. The composition of the prepared catalysts is shown in Table 3 and it could be concluded that the theoretical and experiment percentages were well-matched. While, it is confirmed that reduced metal precursors to metal particles represent the spectrum.
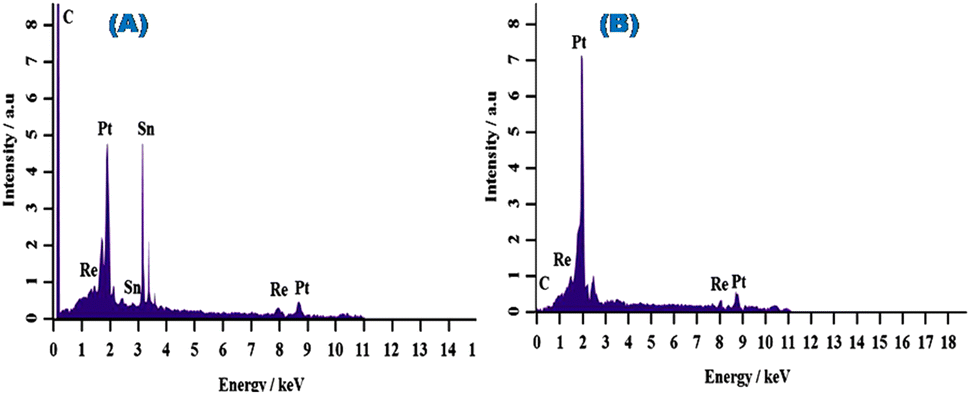 | ||
| Fig. 3 EDX spectra of (A) Pt–Sn–Re/MC (80:15:05) and (B) Pt–Re/MC (50:50). | ||
| Catalyst | QH/μc | QCO/μc | Electrode real surface area (cm2) | SEAS/H (m2 gPt−1)a | SEAS/CO (m2 gPt−1)a | Roughness |
|---|---|---|---|---|---|---|
| a Length of one side. | ||||||
| Pt/MC (100) | 404 | 1776 | 2.8 | 25 | 28 | 78.4 |
| Pt–Sn/MC (50:50) |
243 | 735 | 1.7 | 30 | 35 | 47.6 |
| Pt–Re/MC (50:50) |
239 | 780 | 1.9 | 32 | 40 | 50.3 |
| Pt–Sn–Re/MC (8:10:10) |
454 | 1362 | 3.3 | 43 | 46 | 89.4 |
| Pt–Sn–Re/MC (80:15:05) |
510 | 1431 | 3.3 | 45 | 48 | 96.3 |
:15:05) varied from 1 to 9 nm, with a particle size distribution from 1 to 13 nm. For Pt–Sn–Re/MC (80:10:10), the mean particle size was 9.2 nm, while the size distribution ranged from 1 to 13 nm. This confirmed that the particle size obtained from the TEM image (Fig. 4) and XRD analysis (Fig. 1) were almost similar for the prepared catalyst.
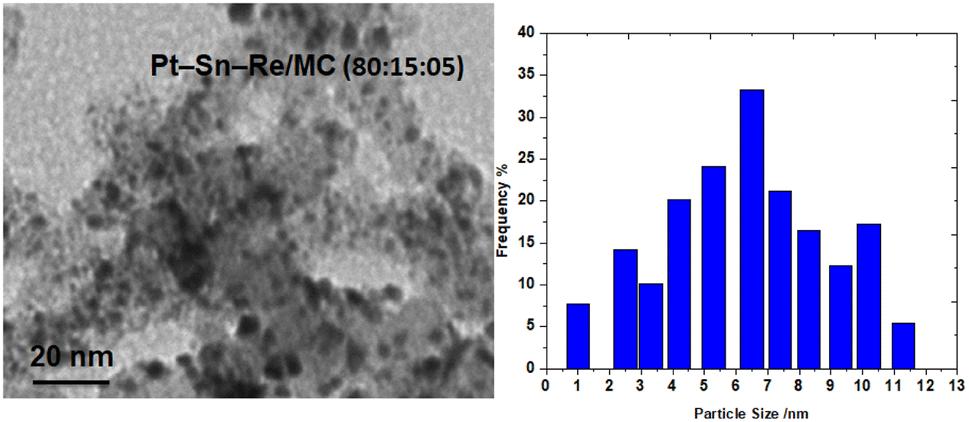 | ||
| Fig. 4 TEM image and particle size distribution of the Pt–Sn–Re/MC (80:15:05) catalyst. | ||
3.2 Chemical characterization of the mesoporous carbon-supported catalysts
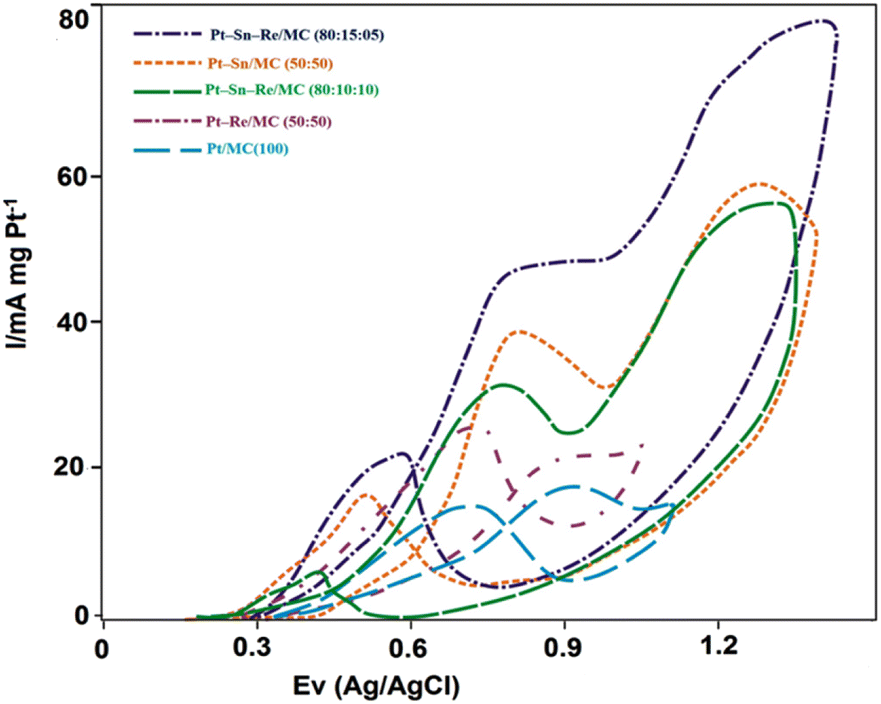
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:15:05) catalysts. In Fig. 5, a single oxidation peak with a central potential of around 795 mV could be seen emerging from the stripping peak. This peak then broadened, and the peak potential for COad stripping changed to a somewhat lower potential (640 mV). Therefore, the low degree of alloying of Sn with Pt can explain the slight shift of the COad stripping peak potential for the binary Pt–Sn/MC (50:50) sample. The stripping curves of the Pt/MC and Pt–Sn/MC (50:50) catalysts showed various shapes and onset potentials that were employed for COad oxidation, in addition to the differentiation in the peak potentials for COad oxidation. Evaluating the sharp single stripping peak in the Pt/MC catalyst, the Pt–Sn/MC (50:50) catalyst had a broad oxidation area of 410 to 640 mV.
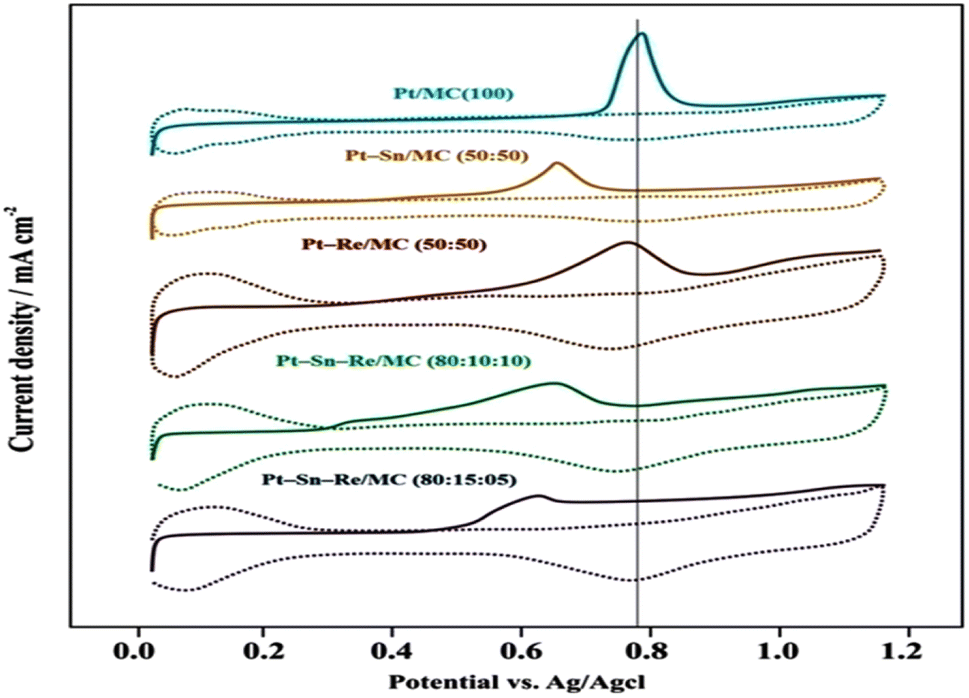 | ||
| Fig. 5 Pre-adsorbed CO stripping voltammograms, with the solid line showing the scan direction and the dotted line indicating the Pt/MC, Pt/MC (100) (blue), Pt–Sn/MC (50:50) (orange), Pt–Re/MC (50:50) (brown), Pt–Sn–Re/MC (80:10:10) (green) and Pt–Sn–Re/MC (80:15:05) (violet) catalysts. | ||
Although the sharp oxidation peak at a higher potential was due to COad oxidation on the pure Pt catalyst surface, the broad oxidation on the pure Pt catalyst surface could be ascribed to COad oxidation on the surface of Pt metal particles that were in close contact with the tin oxide phase.28 Even in the case of Sn not alloyed with Pt entirely, a close contact with Sn or SnO2 was also a consideration and was capable of lowering the potential for COad oxidation on Pt under a bifunctional mechanism.29 For the binary Pt–Re/MC catalyst, the onset potential for COad oxidation on the catalysts involved a value 0.05 V higher than that on Pt–Sn/MC, which occurred at 410 mV, while retaining the same peak potentials at around 750 mV (Fig. 5).
For the Pt–Sn–Re/MC ternary catalyst, the onset potential for COad oxidation on both the Pt–Sn–Re/MC (80:10:10 and 80:15:05) catalysts involved a value 0.05 V lower than that on Pt–Sn/MC, which occurred at 410 mV, while retaining the same peak potentials at around 640 mV (Fig. 5). This observation that the COad oxidation efficiency was improved when Re was added to the Pt–Sn/MC catalyst suggested that the inferior performance of Pt–Sn–Re/MC for COad oxidation may not only be due to the electronic modification of Pt on alloying with Re, but also because of the limited supply of oxygen-containing species to adsorb CO to complete the oxidation reaction.
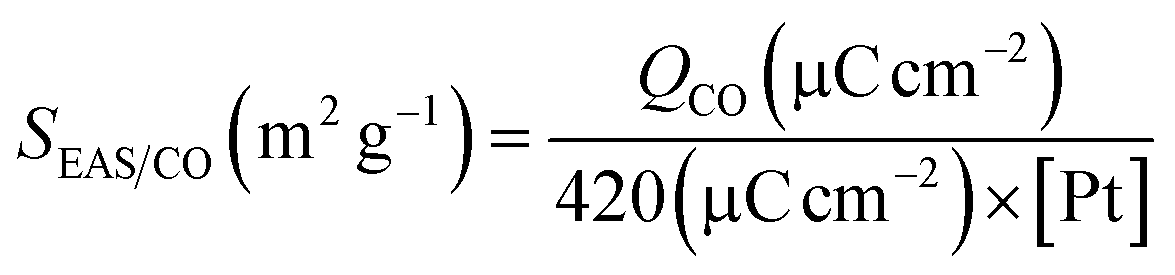
where QCO is the charge corresponding to CO on the Pt surface, [Pt] (mg cm−2) is the Pt loading on the electrode surface and 420 μC per real cm2 is the charge required to oxidize a monolayer of CO on the Pt surface. The roughness of each electrode was calculated by dividing the SEAS obtained with the apparent surface area. The electrochemical surface area and roughness of the Pt-based electrocatalysts are given in Table 3.
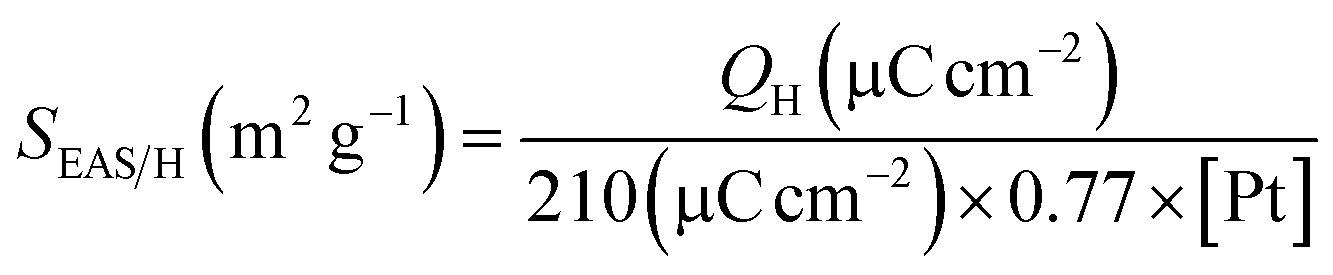
On the basis of these values, the electrochemical surface area increased with the addition of a third metal to the Pt–Sn surface. Stimulatingly, this finding is identical to the lattice parameter. The electrochemical surface area and lattice parameter of crystalline Pt in binary Pt–Sn and ternary Pt–Sn–Re catalysts increase, and the electrocatalytic activity is enhanced. This suggests that the Re-changed Pt–Sn/MC surfaces had actively different sites for adsorption and oxidative desorption, which could be selectively inhabited by CO species, either by straight adsorption of the CO dissolved in the electrolyte or by the adsorption and decomposition reaction of ethanol.
Fig. 6 shows the cyclic voltammograms (CVs) of the Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10), and Pt–Sn–Re/MC (80:15:05) catalysts for CO oxidation in a solution of 0.5 M H2SO4.
 | ||
| Fig. 6 Cyclic voltammograms of Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:15:05) in 0.5 M H2SO4 electrolyte solution at normal temperature. | ||
On account of the strong adsorption of CO on the Pt surface, the hydrogen adsorption–desorption of Pt was completely blocked in the hydrogen region; indicating the presence of a saturated COad layer.
The CV curves were tested in a half-cell between 0.05 and 1.2 V (vs. Ag/AgCl) in the absence of ethanol. The characteristic features of polycrystalline Pt, which is, the hydrogen adsorption/desorption peaks in the potentially low region, the oxide formation/stripping wave/peak in the high potential region, and a flat double layer in between, were observed for all the synthesized catalysts.
Multiple hydrogen adsorption/desorption peaks with different intensities were obtained in the region of 0.05 and 0.35 V for the catalysts. Substantial increases in the double-layer regions at 0.05 and 0.35 V were observed for the catalysts. A substantial increase in the voltammetric charge of the ternary Pt–Sn–Re/MC (80:15:05) catalyst was observed in the double-layer region, showing that adding Re into binary Pt–Sn/MC led to a superior activity in the oxidation reactions.30
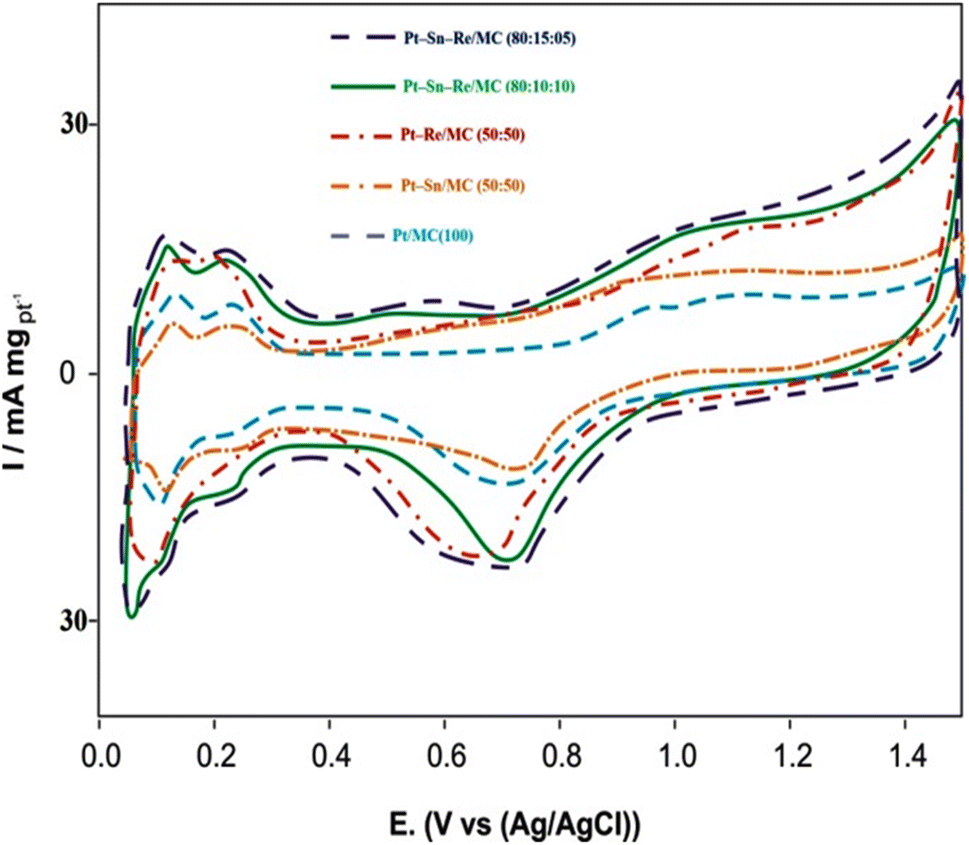
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10), and Pt–Sn–Re/MC (80:15:05), starting at 0.620, 0.515, 0.450, and 0.230 V. The potentials at which the peak currents occur for ethanol oxidation among the catalysts were 0.770, 0.765, 0.726, 0.738 and 0.793 V versus Ag/AgCl.
 | ||
| Fig. 7 Cyclic voltammograms of Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:15:05) in 0.5 M H2SO4 + 1.0 M CH3CH2OH solution at normal temperature and a scan rate of 50 mV s−1. | ||
The main results for the CV tests of the mesoporous carbon-supported Pt (100), Pt–Sn (50:50), Pt–Re (50:50), Pt–Sn–Re (80:10:10), and Pt–Sn–Re (80:15:05) catalysts are listed in Table 4, including the positive peak potentials and corresponding peak current densities for ethanol electro-oxidation. The CV results in Table 4 show that the ternary Pt–Sn–Re/MC (80:15:05) catalyst had a higher positive potential and higher peak current density compared to all the other prepared catalysts. This was due to the addition of Sn and Re, which enhanced the activity of the Pt catalysts for ethanol oxidation. The role of Sn in the electro-oxidation of ethanol was recognized and favoured the cleavage of H2O to OHads, resulting in the oxidation of CO and the CH3CO intermediates to CO2 and CH3COOH. As illustrated by the XRD results, the addition of Sn consider the expansion of the Pt–Pt lattice spaces.35 The extended Pt–Pt lattice space might assist the dissociative adsorption of much bigger ethanol molecules in the lower potential area, and therefore increase the ethanol electro-oxidation. However, the addition of Re into Pt/MC and Pt–Sn/MC led to breaking the C–C bond.
| Catalyst | Scan rate 50 mV s−1 | |
|---|---|---|
| Positive peak potential (mV vs. Ag/AgCl) | Peak current density (mA cm−2) | |
| Pt/MC (100) | 770 | 9.5 |
| Pt–Sn/MC (50:50) |
765 | 44.5 |
| Pt–Re/MC (50:50) |
726 | 18.6 |
| Pt–Sn–Re/MC (80:10:10) |
738 | 35.2 |
| Pt–Sn–Re/MC (80:15:05) |
793 | 63.2 |
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10), and Pt–Sn–Re/MC (80:15:05), to evaluate their catalytic efficacy and stability at electrode potentials of technical relevance over an extended period of time. Here, the ternary Pt–Sn–Re/MC (80:15:05) and Pt–Sn–Re/MC (80:10:10) catalysts demonstrated better performances than the binary Pt–Sn/MC (50:50) and Pt–Re/MC (50:50) catalysts. This kind of activity confirmed various promotion effects of the prepared catalyst for the ethanol oxidation reaction. This long-term analysis showed the slow degradation with the current at 500 mV even after 12 hours. In the first few seconds, the fraction of current used to charge the regional interfacial to the designated overpotential was quickly reduced; following, one observed a slow decrease over longer potential was quickly reduced; following which, there was a slow decrease over longer periods.
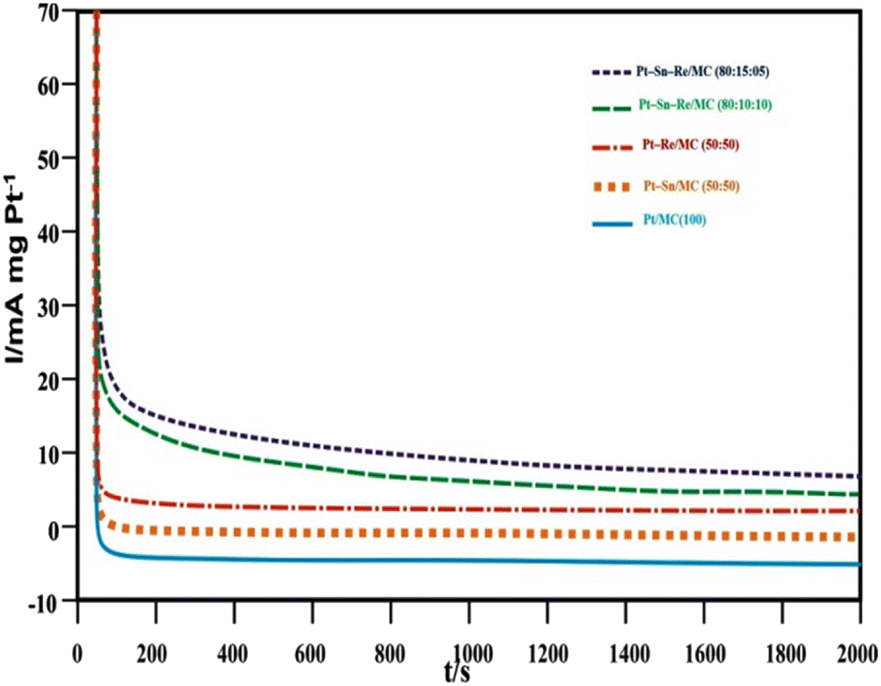 | ||
| Fig. 8 Chronoamperometry tests of Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Sn–Re/MC (80:15:05) catalysts in 0.5 M H2SO4 + 1.0 M CH3CH2OH solution at normal temperature. | ||
This behaviour could be explained by assuming that initially the active sites were free from the adsorbed ethanol molecules, but a new adsorption of ethanol molecules occurred as a function of the liberation of the active sites by ethanol oxidation and/or of the intermediate species (CO, CHx, CH3CHO and CH3COOH) formed during the first few minutes, which were responsible for poisoning the catalytic sites.32
:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10), and Pt–Sn–Re/MC (80:15:05) catalysts prepared by the co-impregnation method are shown in Fig. 10. In that figure, it could be observed that the Pt/MC and Pt–Re/MC anode catalysts had poor electrical performance, whereby the open-circuit voltages (OCV) were 0.56 and 0.58 V and the maximum power densities reached only 4.7 and 11.4 mW cm−2. The addition of Sn to the Pt anode greatly enhanced the electrical performance of the MLEFC by increasing the OCV to 0.73 V and power density to 23 mW cm−2, indicating an improvement in the electrical performance, even superior to that of Pt/MC and Pt–Re/MC. The addition of Re into the Pt anode also greatly enhanced the electrical performance of the MLEFC by increasing the power density to 31.5 mW cm−2, even higher than that of Pt/MC, Pt–Sn/MC and Pt–Re/MC.
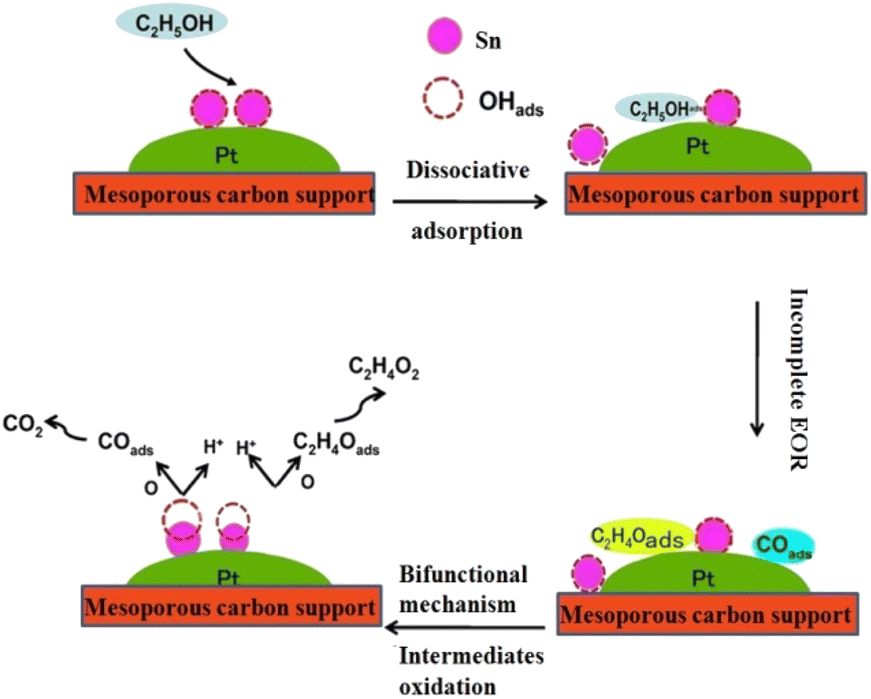 | ||
| Fig. 9 Schematic diagrams of the mechanism of action of Pt-based alloy catalysts in the ethanol oxidation reaction (EOR). | ||
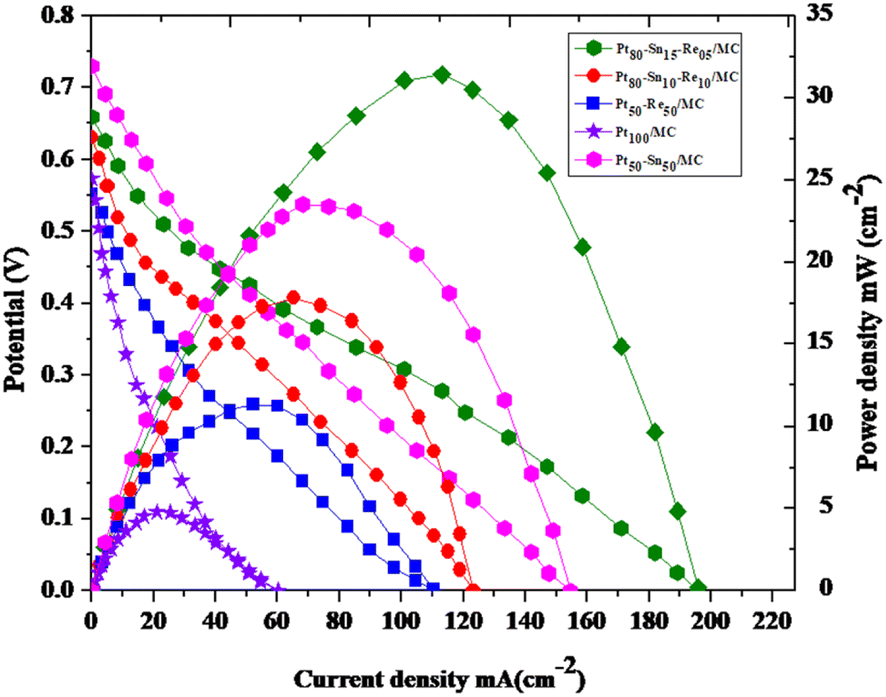 | ||
| Fig. 10 Polarization and power density curves of Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Re–Sn/MC (80:15:05) catalysts at 2 mg cm−2 loading on the anode and Pt/MC (100) cathode at room temperature. | ||
Increased percentage of Re into the Pt anode decreased the power density to 18.6 mA cm−2, which means the electrical performance was higher than that obtained with Pt/MC, Pt–Sn/MC and Pt–Re/MC. Increased percentage of Re into the Pt anode decreases the power density to 18.6 mA cm−2, which means the electrical performances are lower than those obtained with Pt–Sn/MC (Fig. 10).
The bifunctional mechanism was first postulated by Watanabe and Motoo(1973)36 and later added to by Watanable et al. (1975). The mechanism involves facile water activation at the second metal (Sn) sites, thereby promoting the oxidation of COads to CO2 by supplying oxygen-containing species at lower potentials. According to the bifunctional mechanism, ethanol is dissociatively adsorbed on the Pt sites giving adsorbed COads and/or formyl-like species-CHOads; the tin in Pt–Sn catalyst dissociates water into Sn–OHads and H on the surface of the catalyst and, then, the species adsorbed onto Pt and Sn sites combine to form CO2 (Watanabe et al., (1975);37 Swathirajan et al., (1991);38 Markovic et al., (2001);39 Gojkovic et al., (2003)40), as shown below in Fig. 9.
This bifunctional mechanism successfully explains several aspects of ethanol and CO oxidation reactions on Pt–Sn. Furthermore, the activity of Pt–Sn anode catalysts also needs further development for enhancing the ethanol oxidation reaction, and for increasing the stability of the Pt catalyst. One of the best methods to improve cell performance is by adding a third metal into the Pt surface. The addition of Re metal accelerates the second metal by bifunctional and electronic (ligand) mechanisms, giving a higher performance for the ethanol oxidation reaction. This is because the addition of rhenium into the Pt–Sn surface enhances the ethanol molecule breaking of the C–C and C–H bonds, even though too high a percentage rhenium content blocks the ethanol oxidation reaction. Table 5 shows the open-circuit voltage, maximum power density, and maximum current density for the prepared Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Re–Sn/MC (80:15:05) catalysts.
| Anode catalysts | Open-circuit voltage (V) | Maximum power density (mW cm−2) | Current density at maximum power density (mA cm−2) |
|---|---|---|---|
| Pt/MC (100) | 0.57 | 4.7 | 61.2 |
| Pt–Sn/MC (50:50) |
0.73 | 23.4 | 154.01 |
| Pt–Re/MC (50:50) |
0.55 | 11.4 | 110.38 |
| Pt–Sn–Re/MC (80:10:10) |
0.63 | 18.6 | 123.24 |
| Pt–Sn–Re/MC (80:15:05) |
0.66 | 31.5 | 196.45 |
These single cell performances clearly demonstrate the benefits obtained by alloying Pt with Sn and Re. The increase in the open-circuit voltage (OCV) indicates that the bi- and tri-metallic catalysts were less poisoned by the adsorbed intermediates from the ethanol than the Pt/MC catalyst (Fig. 11).33
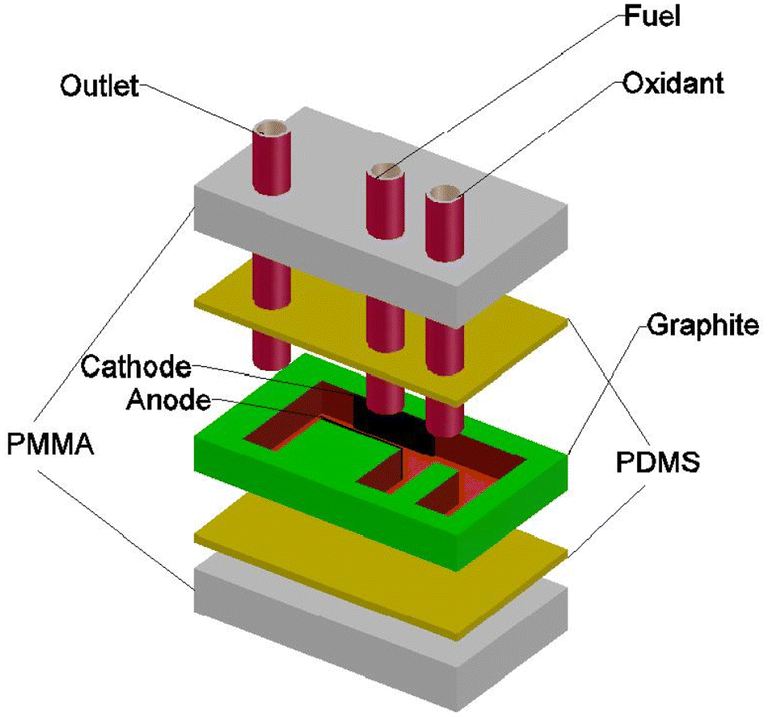 | ||
| Fig. 11 Schematic of an E-shaped membraneless laminar flow-based fuel cell with graphite plates moulded with poly(dimethylsiloxane) PDMS and sealed with poly(methylmethacrylate) PMMA. | ||
The addition of Re and Sn enhanced the activity of the Pt catalysts towards the electro-oxidation of ethanol. In the literature, the role of Sn in the electro-oxidation of ethanol is documented. Sn helps to break H2O to OHads, resulting in the oxidation of CO and CH3CO intermediates to CO2 and CH3COOH. As displayed by the XRD results, the addition of Sn could include inducing extension of the Pt–Pt lattice distance. The extended Pt–Pt lattice distance could then facilitate the dissociative adsorption of much bigger ethanol molecules in the lower potential region, thus enhancing the ethanol electro-oxidation. However, the addition of Re to Pt/MC or Pt–Sn/MC helps break the C–C bond.41–43 Also, the ECSAH of ternary catalysts were found to be larger than those of Pt and binary ones, suggesting better Pt utilization from the former. However, when comparing the calculated ECSACO with ECSAH, larger differences were noted for the same catalysts. In this observation, ternary Pt–Sn–Re catalysts gave a higher current density and power density.44
4. Conclusion
In this present study, for the first-time, mesoporous carbon-supported binary Pt/MC (100), Pt–Sn/MC (50:50), and ternary Pt–Re/MC (50:50), Pt–Sn–Re/MC (80:10:10) and Pt–Re–Sn/MC (80:15:05) catalysts were prepared by a co-impregnation reduction method. These anode catalysts were successfully tested in a single membraneless fuel cell, using 1.0 M ethanol as the fuel and 0.1 M sodium perborate as the oxidant, in the presence of 0.5 M H2SO4 as the electrolyte. A significant enhancement in ethanol oxidation was observed, attributed to the high dispersion of the ternary catalyst and to Re acting as a promotion agent. X-Ray diffraction analysis confirmed the formation of Pt–Sn/MC, Pt–Re/MC, and Pt–Sn–Re/MC metal catalysts and that they had typical Pt crystalline structures, as well as the formation of the Pt–Sn alloy. TEM morphology analysis illustrated that the particle sizes of the entire prepared catalysts varied from 6–9 nm. Energy-dispersive X-ray analysis confirmed that the theoretical value had good agreement with the experimental composition of the prepared catalyst. Cyclic voltammetry tests clearly indicated that Pt–Sn–Re/MC (80:15:05) was more active towards ethanol electro-oxidation than Pt/MC (100), Pt–Sn/MC (50:50), Pt–Re/MC (50:50) and Pt–Sn–Re/MC (80:10:10). This was due to the activation that took place at the electrode surface due to the ligand effect. In the chronoamperometry tests, all the catalysts, at high and low potentials, showed that Pt–Sn–Re/MC (80:15:05) gave high stability at a steady condition. The peak power density of Pt–Sn–Re/MC (80:15:05) (31.5 mW cm−2) was higher than that of Pt/MC (100) (4.7 mW cm−2), Pt–Sn/MC (50:50) (23.4 mW cm−2), Pt–Re/MC (50:50) (11.4 mW cm−2), and Pt–Sn–Re/MC (80:10:10) (18.6 mW cm−2). It was found that the addition of Re on the anode catalyst enhanced the cell performance by helping to break the C–C bond. The platinum electronic density was altered, and the current density and peak potential increased, as a result of the electronic effect linked to the alloy formation between Pt, Sn, and Re. Thus, the two mechanisms mentioned above work in concert to raise the ternary Pt–Sn–Re/MC catalyst's peak potential and current density, demonstrating the best possible performance for the ethanol oxidation reaction. Membraneless ethanol fuel cells offer cleaner emission than fossil fuels and no charging times compared to electric vehicle batteries. The strong mesoporous carbon support of Pt–Sn–Re strengthens the electronic effect, which enhances both the stability and performance of the catalyst. The electronic effect at the interface of Pt–Sn–Re also enhances the electron transfer towards the direct C1-12 e− EOR pathway for MLEFC. From this result, it could be concluded that Pt–Sn–Re/MC (80:15:05) was the best catalyst for the complete electro-oxidation of ethanol among the catalysts tested in membraneless ethanol fuel cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by University grants Commission (UGC), New Delhi, India through a major research project (42-325/20134) (SR) is great fully acknowledged.References
- E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431–3450 CrossRef CAS.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2021, 31(9), 2009070 CrossRef CAS.
- Y. Wang, H. Wang, X. Jin and D. Y. C. Leung, Biosens. Bioelectron., 2020, 166, 112410 CrossRef CAS PubMed.
- K. Jiao, X. Jin, Q. Du, Z. Bao, B. Xie, B. Wang and Y. Zhao, Nature, 2021, 595(7867), 361–369 CrossRef CAS PubMed.
- S. Basu, Fuel cell science and Technology, Springer, New York, 2007 Search PubMed.
- Y. Liang, C. -Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. -Q. Huang and D. Yu, InfoMat, 2019, 1(1), 6–32 CrossRef CAS.
- Z. Zakaria, S. K. Kamarudin and S. N. Timmiati, Appl. Energy, 2016, 163, 334–342 CrossRef CAS.
- N. Shaari, S. K. Kamarudin, R. Bahru, S. H. Osman and N. A. I. Md Ishak, Int. J. Energy Res., 2021, 45(5), 6644–6688 CrossRef CAS.
- Z. Zakaria and S. Kartom Kamarudin, Int. J. Energy Res., 2020, 44(8), 6223–6239 CrossRef CAS.
- C.-C. Kung, P.-Y. Lin, Y. Xue, R. Akolkar, L. Dai, Y. Xiong and C.-C. Liu, J. Power Sources, 2014, 256, 329–335 CrossRef CAS.
- M. A. F. Akhairi and S. K. Kamarudin, Int. J. Hydrogen Energy, 2016, 7, 4214–4228 CrossRef.
- M. A. Rakan and P. G. Pickup, J. Power Sources, 2017, 366, 27–32 CrossRef.
- D. Van Dao, G. Adilbish, T. D. Le, T. T. D. Nguyen, I.-H. Lee and Y. Yeon-Tae, J. Catal., 2019, 377, 589–599 CrossRef.
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2012, 5(8), 1619–1624 CrossRef CAS PubMed.
- A. El Attar, L. Oularbi, S. Chemchoub and M. El Rhazi, Int. J. Hydrogen Energy, 2020, 45(15), 8887–8898 CrossRef CAS.
- A. L. M. Reddy, R. Natarajan and S. Ramaprabhu, Carbon, 2008, 46(1), 2–11 CrossRef CAS.
- R. P. R. Anjos, A. O. Santos, R. M. Antoniassi, O. C. Alves, E. A. Ponzio and J. C. M. Silva, ACS Appl. Energy Mater., 2021, 4(6), 6253–6260 CrossRef CAS.
- K. Tarantseva, N. Politaeva, K. Tarantsev, M. Yackhkind and A. K. Mishra, J. Chem. Technol. Biotechnol., 2022, 97(1), 101–110 CrossRef CAS.
- Q. Liu, K. Jiang, J. Fan, Y. Lin, Y. Min, Q. Xu and W.-B. Cai, Electrochim. Acta, 2016, 203, 91–98 CrossRef CAS.
- F. S. Lima, E. H. Fontes, J. Nandenha, R. F. B. de Souza and A. O. Neto, J. Fuel Chem. Technol., 2021, 49(10), 1540–1548 CrossRef CAS.
- J. Kim, H. Jung, S.-M. Jung, J. Hwang, D. Y. Kim, N. Lee and K.-S. Kim, J. Am. Chem. Soc., 2020, 143(3), 1399–1408 CrossRef PubMed.
- Z. Xie, W. Wang, D. Ding, Y. Zou, Y. Cui, L. Xu and J. Jiang, J. Mater. Chem. A, 2020, 8(24), 12169–12176 RSC.
- L. Su, D. Gong, Y. Jin, D. Wu and W. Luo, J. Energy Chem., 2022, 66, 107–122 CrossRef CAS.
- L. Su, D. Gong, Y. Jin, D. Wu and W. Luo, J. Energy Chem., 2022, 66, 107–122 CrossRef CAS.
- T. S. Zhao, Y. S. Li and S. Y. Shen, Energy Power Eng. China, 2010, 4, 443–458 CrossRef.
- J. Lilloja, E. Kibena-Põldsepp, A. Sarapuu, M. Käärik, J. Kozlova, P. Paiste and A. Kikas, et al., Appl. Catal., B, 2022, 306, 121113 CrossRef CAS.
- L. S. Carvalho, C. L. Pieck, M. C. Rangel, N. S. Fıgoli, J. M. Grau, P. Reyes and J. Miguel Parera, Appl. Catal., A, 2004, 269(1–2), 91–103 CrossRef CAS.
- J. Mann, N. Yao and B. B. Andrew, Langmuir, 2006, 22(25), 10432–10436 CrossRef CAS PubMed.
- D. N. Saritha, M. Reddy and V. Gubbala Ramesh, Mater. Today: Proc., 2022, 585–594 Search PubMed.
- X. Wang, F. Zhu, Y. He, M. Wang, Z. Zhang, Z. Ma and R. Li, J. Colloid Interface Sci., 2016, 468, 200–210 CrossRef CAS PubMed.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10(19), 11011–11045 CrossRef CAS.
- L. Yaqoob, T. Noor and N. Iqbal, RSC Adv., 2021, 11(27), 16768–16804 RSC.
- V. Erduran, M. Bekmezci, M. Akin, R. Bayat, I. Isik, and F. Şen, Nano materials for Direct Alcohol fuel cell, 2021, pp. 209–249 Search PubMed.
- M. Priya, S. Kiruthika and B. Muthukumaran, Ionics, 2017, 23, 1209–1218 CrossRef CAS.
- M. Priya, M. Arun, S. Elumalai, B. Kiruthika and B. Muthukumaran, Adv. Phys. Chem., 2014, 2014, 862691 Search PubMed.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267–273 CrossRef CAS.
- M. Watanabe, J. Electroanal. Chem., 1987, 229, 395 CrossRef CAS.
- S. Swathirajan, Y. M. Mikhail and G. P. Meisner, J. Electrochem. Soc., 1991, 138, 2631 CrossRef CAS.
- N. M. Markovic, T. J. Schmidt, V. Stamenkovic and P. N. Ross, Oxygen reduction reaction on Pt and Pt bimetallic surfaces: A selective review, Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720, US, 2001 Search PubMed.
- S. L. Gojkovic, T. R. Vidakovic and D. R. Đurovic, Electrochim.Acta, 2003, 48(24), 3607–3614 CrossRef CAS.
- C. F. Vigier, A. Coutanceau, E. M. Perrard, C. Belgsir and C. Lamy, J. Appl. Electrochem., 2004, 34, 439–446 CrossRef.
- M. Bonarowska, A. Malinowski and Z. Karpinski, Appl. Catal., A, 1999, 188, 145–154 CrossRef CAS.
- A. K. Aboul-Gheit, M. F. Menoufy and A. K. El-Morsi, Appl. Catal., 1990, 61, 283–292 CrossRef CAS.
- J. Chang, G. Wang, X. C. Z. Yang, H. Wang, B. Li, W. Zhang, L. kovarik, Y. du, N. Orlovskaya, B. Xu, G. Wwang and Y. yang, Nat. Commun., 2023, 14, 1346 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |