
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent perspectives, challenges, and future directions in the electrochemical detection of microplastics
Ayman H. Kamel
 *ab,
A. Hefnawyac,
Layla J. Hazeemd,
Suad A. Rashdana and
Hisham S. M. Abd-Rabbohe
*ab,
A. Hefnawyac,
Layla J. Hazeemd,
Suad A. Rashdana and
Hisham S. M. Abd-Rabbohe
aDepartment, College of Science, University of Bahrain, Zallaq 32038, Kingdom of Bahrain. E-mail: ahkamel76@sci.asu.edu.eg; ahmohamed@uob.edu.bh
bDepartment of Chemistry, Faculty of Science, Ain Shams University, Cairo 11566, Egypt
cDepartment of Materials Science, Institute of Graduate Studies and Research, Alexandria University, El-Shatby, Alexandria 21526, Egypt
dDepartment of Biology, College of Science, University of Bahrain, Zallaq 32038, Bahrain
eChemistry Department, Faculty of Science, King Khalid University, Abha 62529, Saudi Arabia
First published on 10th January 2024
Abstract
Microplastics (5 μm) are a developing threat that contaminate every environmental compartment. The detection of these contaminants is undoubtedly an important topic of study because of their high potential to cause harm to ecosystems. For many years, scientists have been assiduously striving to surmount the obstacle of detection restrictions and minimize the likelihood of receiving results that are either false positives or false negatives. This study covers the current state of electrochemical sensing technology as well as its application as a low-cost analytical platform for the detection and characterization of novel contaminants. Examples of detection mechanisms, electrode modification procedures, device configuration, and performance are given to show how successful these approaches are for monitoring microplastics in the environment. Additionally included are the recent developments in nanoimpact techniques. Compared to electrochemical methods for microplastic remediation, the use of electrochemical sensors for microplastic detection has received very little attention. With an overview of microplastic electrochemical sensors, this review emphasizes the promise of existing electrochemical remediation platforms toward sensor design and development. In order to enhance the monitoring of these substances, a critical assessment of the requirements for future research, challenges associated with detection, and opportunities is provided. In addition to—or instead of—the now-in-use laboratory-based analytical equipment, these technologies can be utilized to support extensive research and manage issues pertaining to microplastics in the environment and other matrices.
 Ayman H. Kamel | He received his PhD degree from Ain Shams University, Cairo, Egypt, in 2005. He has been a Professor of Analytical Chemistry at Ain Shams University since January 2016. He has authored or co-authored over 124 papers in peer-reviewed journals. His research interests include the development of the rational design of solid-state potentiometric sensors for environmental monitoring, development of man-tailored mimic polymers as a novel stationary phase in chromatographic separation, and solid-phase extraction. |
1. Microplastics: sources and occurrence
Owing to numerous transport phenomena like wind and ocean currents, microplastics (MPs) can be found in coastal areas and aquatic ecosystems worldwide in different size fractions. Household sewage discharge, polymeric particles from cosmetic and cleaning goods, feedstocks used to make plastic products, and plastic pellets or powders used for air blasting are the primary sources of these contaminants.1 The secondary source of MPs is the progressive fragmentation of larger plastic items under atmospheric conditions, such as mechanical deterioration and UV-light exposure, which helps to introduce significant amounts of MPs into the environment.2 This increases the consumption of plastic waste by a wide range of creatures and the emergence of new environmental threats.3Another significant source of MP discharge is wastewater treatment facilities.4,5 Large plastic particles are effectively removed during wastewater treatment, but MPs frequently evade the units and enter the water.6 It is noteworthy that many water treatment facilities are situated close to the ocean and seawater, creating a high source of MP release. For instance, in Mainland China, about 1873 wastewater plants (56%), out of 3340, with 78 × 106 m3 day−1 of treatment capacity are located in coastal regions where their effluents can be directly or indirectly discharged into aquatic ecosystems.7 Various studies have addressed the fate, incidence, detection, and removal of MPs in water treatment plants.8,9
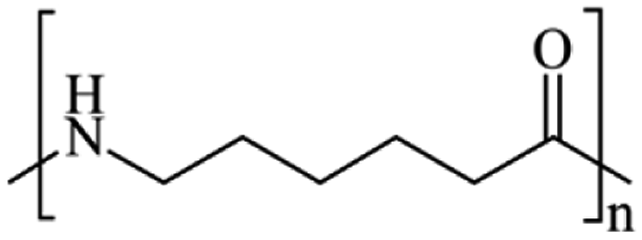
Microbeads (<1 μm) used in cleaning products, cosmetics, and laundry soaps are additional sources of MPs. Polyethylene (PE), polypropylene (PP), polystyrene (PS), polyethylene terephthalate (PET), polyvinyl chloride (PVC), and polyesters are the major components of (Table 1) these particles, which are eventually carried into the ocean (Fig. 1).
| Type of polymer | Density (g cm−3) | Biodegradation | Recycle ID code | Molecular structure |
|---|---|---|---|---|
| High density polyethylene (HDPE) | 0.917–0.965 | Not reported |  |
 |
| Low density polyethylene (LDPE) | Bacteria, fungi, waxworms, mealworms |  |
||
| Linear low density polyethylene (LLDPE) | ||||
| Polypropylene (PP) | 0.90–0.91 | Not reported | 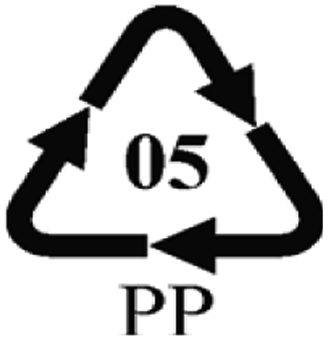 |
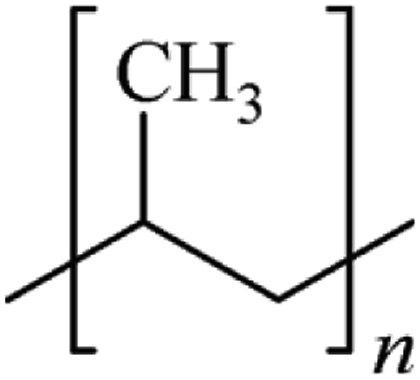 |
| Polystyrene (PS) | 1.04–1.1 | Bacteria, mealworms |  |
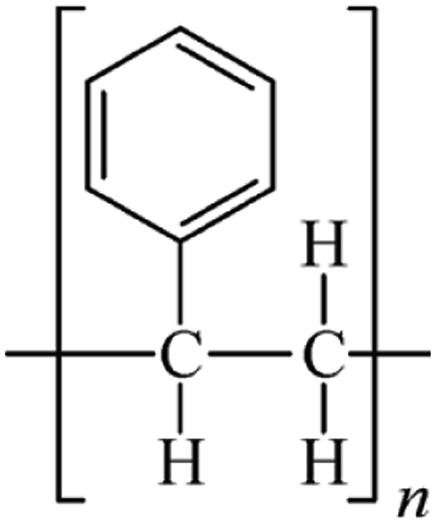 |
| Polyvinylchloride (PVC) | 1.16–1.58 | Fungi |  |
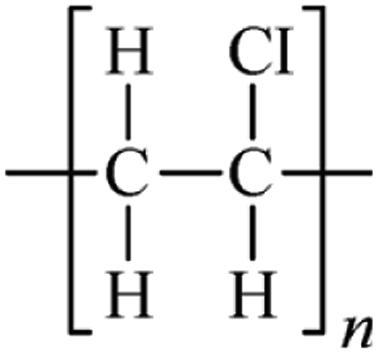 |
| Polymethylacrylate (PMA) | 1.17–1.2 | Cyanobacteria | 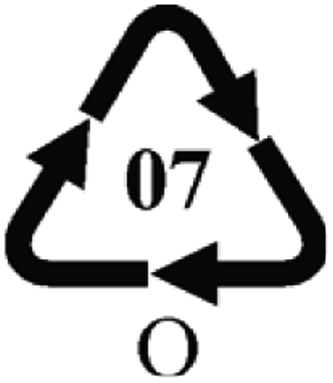 |
 |
| Polyethylene terephthalate (PET) | 1.37–1.45 | Bacteria |  |
 |
| Polyurethane (PU) | 1.20 | Fungi |  |
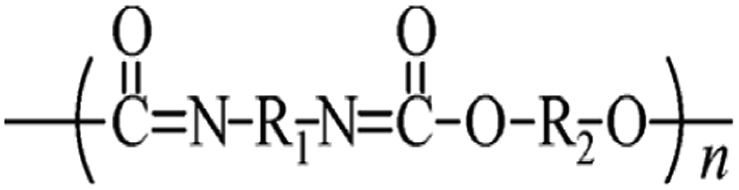 |
| Polyesters (nylon, acrylic, etc.) | 1.24–2.3 | Bacteria |  |
 |
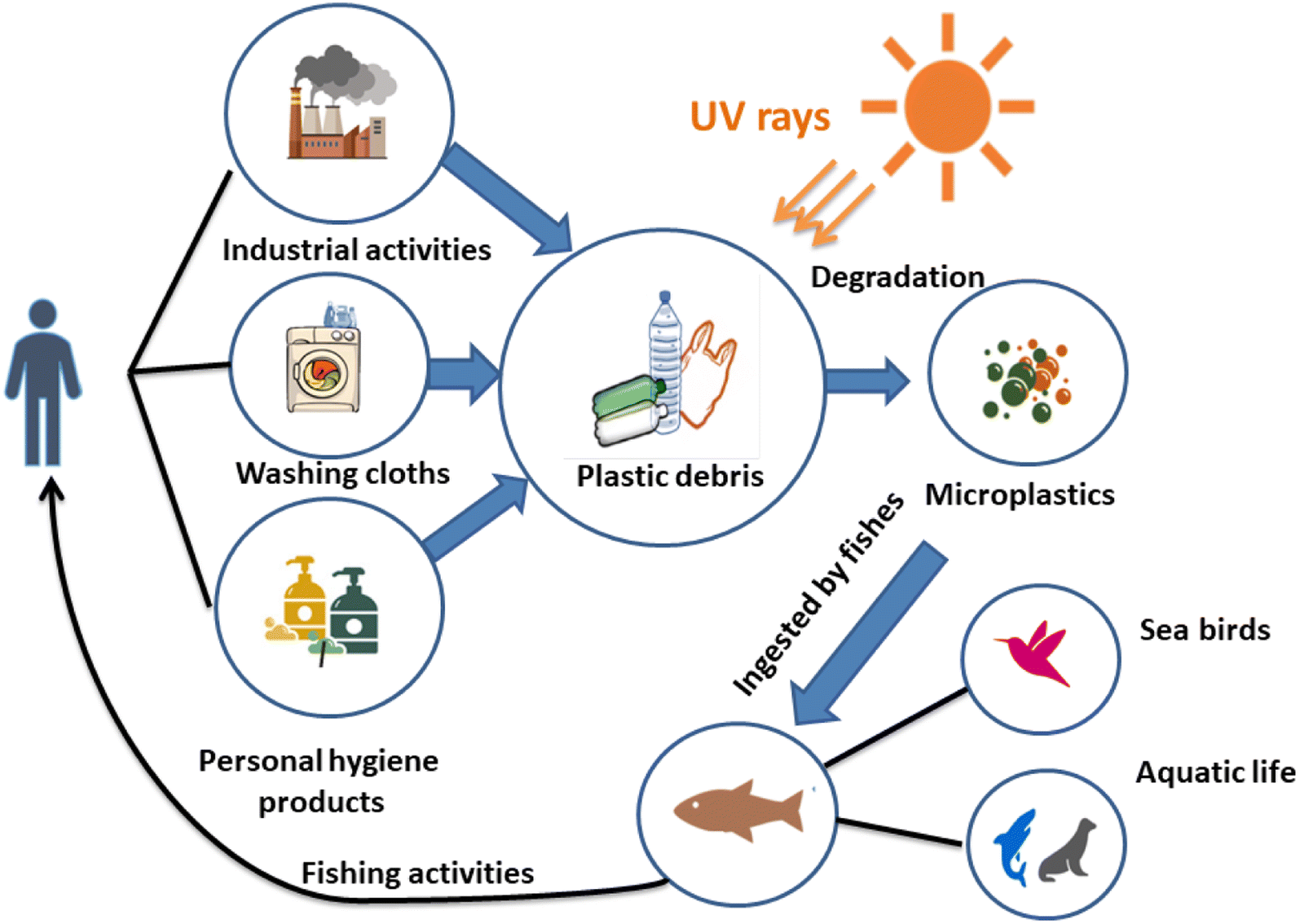 | ||
| Fig. 1 MPs pathways. | ||
2. Microplastics and human health
The ingestion or inhalation of MPs into the body of a person has the potential of negative health impacts (Fig. 2).10 A comparison to particulate air pollution can be made: small particles (2.5 mm), such as those found in diesel exhaust, have been associated with an increased risk of dying from lung cancer, cardiovascular disease, and inflammatory illnesses because they can cross the cell membranes and cause oxidative stress and inflammation.11 This correlation offers strong motivation to learn more about the potential dangers of MP particles.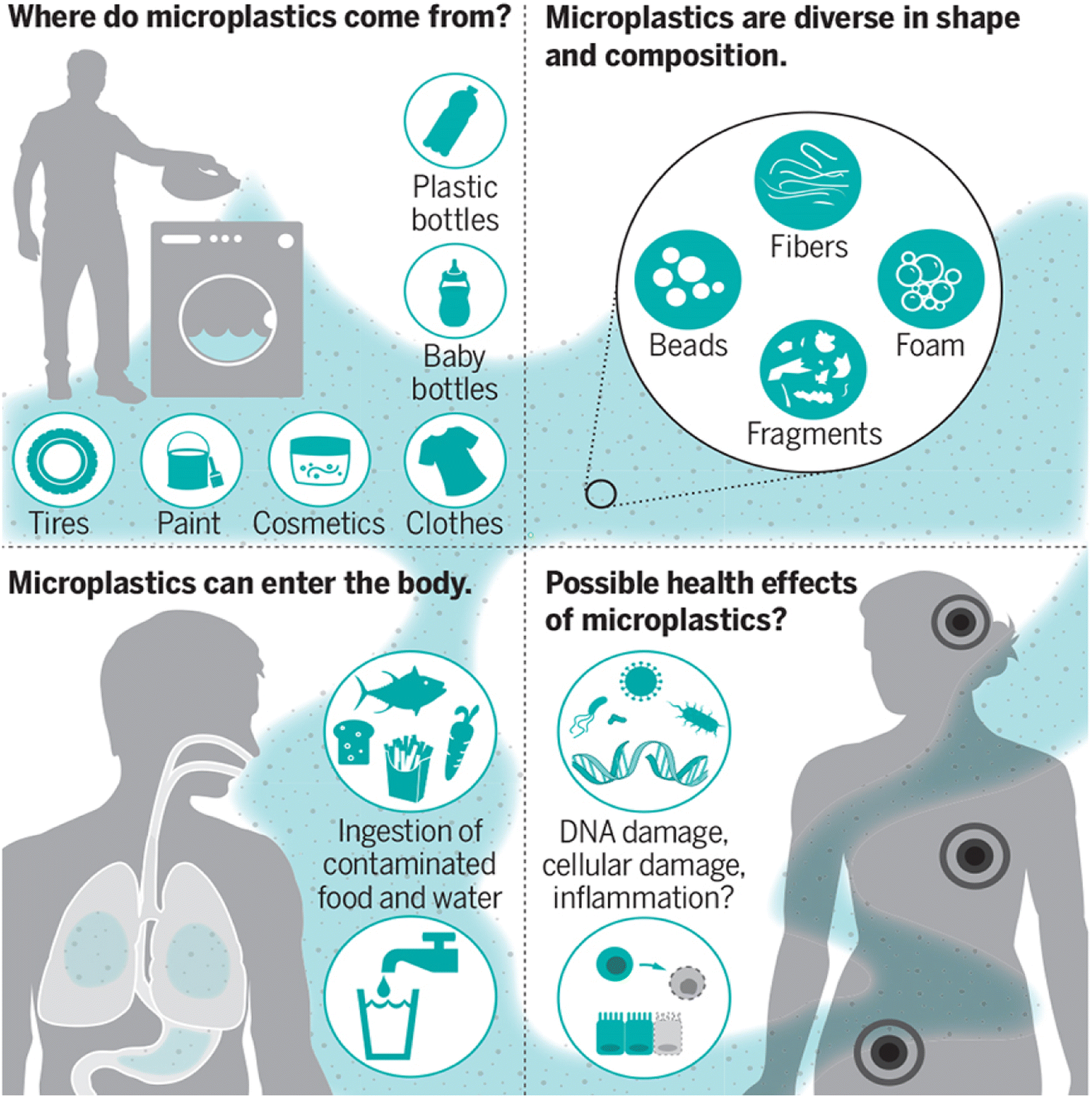 | ||
| Fig. 2 MPs and their effect on human health.10 | ||
The reported concentrations of MPs in tap and bottled water range from 0 to 104 particles per liter, with smaller MPs often having higher particle counts.12 According to the initial air observations of larger, mostly fibrous MPs, the deposition rates in central London, for instance, ranged from 575 to 1008 MPs per square meter per day,13 indicating that plastic particles are a major component of fine dust. Increased exposure from contaminated indoor air, ingestion of household dust or dust that settles on food,14 and direct exposure to food particles discharged from plastic bottles or containers (such as PP baby-feeding bottles15), are of particular concern.
Larger MPs are probably eliminated through feces or mucociliary clearance into the gut after depositing in the respiratory system or lungs.12,13 The fraction of smaller-sized particles (10 μm), which are probably toxic, is typically not included in existing assessments due to methodological limitations and measurement preference toward larger particles, which probably underestimate human external exposure.16,17 Remarkably, the assessments of internal exposure to plastic particles in biological fluids and tissues are still in their early stages.
Reducing the ambiguity in the human risk assessment of MPs requires a deeper comprehension of their capacity to pass through the epithelial barriers of the gastrointestinal system, skin, and airways. A limited amount of in vitro and in vivo data indicates that only a tiny portion of administered MPs can pass through the epithelial barriers of the lungs and intestines.
These MPs have distinct uptake characteristics and generally increase in absorption efficiency as the particle size decreases.18 Because of the potential for accumulation in tissues and organs as well as lifetime exposure, this modest percentage of particle absorption is not insignificant. MPs smaller than 10 μm have been found to move from the gut cavity to the lymph and circulatory systems, inducing systemic exposure and accumulation in tissues such as the brain, kidneys, and liver.17 These findings have also been observed in rodents and aquatic species. Even though the tiniest particles (less than 0.1 μm) have the potential to penetrate all the organs and even the placenta, brain, and cell membranes,17,19,20 there are still significant gaps in our understanding of absorption, distribution, metabolism, and excretion (ADME). It is also unknown if MPs have dose-dependent impacts on humans.
MPs could cause physical, chemical, and microbial toxicity, as well as long-term effects, if they touch the epithelial lining of the intestines or lungs or if they are consumed. Numerous studies on human cell cultures in the lab and on rodents in the wild suggest that breathing or consuming MPs may have a wide range of biological effects, such as physical (particle) toxicity, which can lead to oxidative stress, cytokine secretion, cellular damage, immune and inflammatory responses, DNA damage, and neurotoxic and metabolic effects.17 In these studies, only a small number of pure, commercially available particle types are used. These particles are not the same as those found in the environment, and the effects found are often caused by high doses of MPs. Moreover, it is not always possible to rule out the chemical contamination of these test particles. Epidemiological studies have documented lung damage, including inflammation, fibrosis, and allergies, among employees in the plastic and textile sectors who are exposed to high levels of plastic fiber dust.18 These findings are consistent with the effects seen in research on ambient particle exposure.
Every day, humans are exposed to a variety of natural and synthetic particles; among them, particulate air pollution is acknowledged as one of the primary environmental risk factors for diseases worldwide. It is imperative to assess the possible contribution of MPs in overall ambient particle exposure. In comparison to other ambient particles, MPs may have unique particle properties and a wider and more diverse toxicity profile because of their persistence, wide size range, and complex structure. Important health concerns related to MPs remain largely unexplored to this day. These include internal exposure; ADME processes (including the effect of the eco- or bio-corona); interaction with the immune system; and the possible effects of nanosized plastics on the placenta, fetus, and brain. These problems are essential to innovation, evidence-based policymaking, and risk management strategies. They are starting to be resolved by innovative interdisciplinary research programs like Microplastics & Health in the Netherlands and the European Union Horizon 2020 research programme. To address this potential health risk, multidisciplinary research projects including experts from the medical, environmental, and polymer domains are required. Although comprehensive risk assessment is still way off, it is important to fill in the major research gaps today to enable prompt decision-making about health policy and mitigation techniques.
3. Analytical techniques for the detection of microplastics
3.1. Visual inspection methods
Selecting and classifying MPs as well as sighting the color and size of the examined objects under a microscope or with the naked eye can be undertaken using visual inspection approaches such as the direct visual method, optical microscope observation, and electronic microscope observation21 (Fig. 3). The accuracy of these methods is poor, and they are time consuming.22 MPs are tiny plastic particles with a diameter of less than 1 mm that are easy to overlook or underestimate. Furthermore, the results showed that the error rate is inversely related to the particle size when employing this technique.23,24 Consequently, more accurate and efficient technologies for MP identification must be developed. The advantages and limitations of such methods are summarized in Table 2. | ||
| Fig. 3 Classification of visual inspection methods. | ||
| Method | Methodology | Particle size | Advantages | Limitations |
|---|---|---|---|---|
| Counting microscopy | Direct particle counting is done | It can identify particles with sizes in the micrometer (μm) range | Samples containing a considerable number of large MPs are easily identified, providing a fast and reasonably priced overview of MP abundance | Since the nature of the materials cannot be ascertained, a combination of identification techniques is required |
3.2. Techniques for thermal analysis
Fig. 4 shows the categorization of thermal analysis.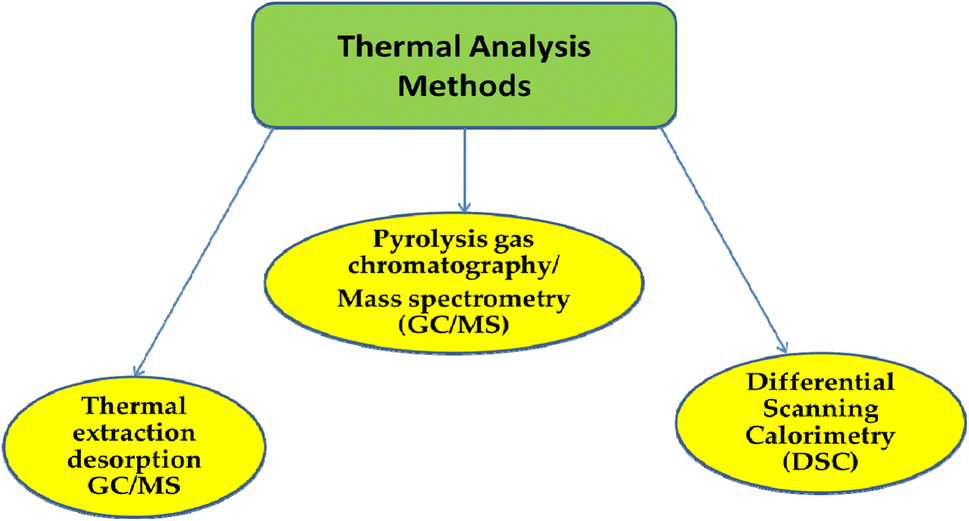 | ||
| Fig. 4 Classification of thermal analysis methods. | ||
The production variables that affect the transition temperature are impurities, additives, and a part of the polymerization chains. Because MP particle size has a significant impact on the DSC values, samples need to be pretreated before testing. This is yet another DSC disadvantage. Furthermore, when the melting point of the polymer mixture is the same or when the sensitivity of the DSC sensors is low, some polymers have limited thermal conductivity. Its limitations hinder its rapid application in MP detection. It was found that thermogravimetric analysis-differential scanning calorimetry (TGA-DSC) could accurately separate PE and PP in environmental samples. The phase-change signals of other types of MP cannot be recognized because of the significant overlap in the phase-change signals. The larger sample size causes the peak area to increase but the resolution to decrease, leading to inaccurate measurement results.
Table 3 provides an overview of the benefits and drawbacks, as well as some significant characteristics of thermal analysis techniques. Table 4 provides an overview of the applications of thermal analytical techniques in MPs.
| Method | Advantages | Disadvantages | Sample mass | Detection limit | Ref. |
|---|---|---|---|---|---|
| Pyr-GC-MS | • Various polymer kinds can be detected | • Lengthy process | 0.5 mg | 0.007 mg g−1 | 27 and 33 |
| • Accurate findings | • Sample deterioration | ||||
| • High sensitivity | • High reaction temperatures | ||||
| • No sample preparation | |||||
| TED-GC-MS | • Large sample size | • High reaction temperatures | 100 mg | — | 29, 34 and 35 |
| • No sample preprocessing | • Sample deterioration. Excessive processing times | ||||
| • No blocking reaction cube | |||||
| • High sample mass | |||||
| DSC | • Reliable outcomes | • Prolonged processing durations | 3–15 mg | — | 31 and 32 |
| • Extensively used methodology | • Sample degradation | ||||
| • Affordable and uncomplicated analysis | • Clear substrate influence | ||||
| • Sample preparation |
| Method | Source of the sample | Type of sample | Limit of detection (LOD) | Sample abundance | Ref. |
|---|---|---|---|---|---|
| a NR: not reported. | |||||
| Pressurized liquid extraction and Pyr-GC-MS | Soil and sediments | PE, PP | NR | PE (3.3 ± 0.3 mg g−1) and PP (0.08 ± 0.02 mg g−1) | 36 |
| Pyr-GC-MS | Soils and sediments of freshwater | PE (fractionation ratio 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), PE (fractionation ratio 17:2), PE (fractionation ratio 18:2), PP, PS (pyrolysis product Sty), and PS (pyrolysis product aMeSty) :2), PE (fractionation ratio 17:2), PE (fractionation ratio 18:2), PP, PS (pyrolysis product Sty), and PS (pyrolysis product aMeSty) |
PE (fractionation ratio 15:2, 4800 μg L−1), PE (fractionation ratio 17:2, 2500 μg L−1), PE (fractionation ratio 18:2, 11300 μg L−1), PP (43200 μg L−1), PS (pyrolysis product Sty, 500 μg L−1) and PS (pyrolysis product aMeSty, 1600 μg L−1) |
NR | 37 |
| Pyr-GC-MS | Stomachs of marine fishes | PVC, PET, nylon, silica gel, and epoxy resin | NR | NR | 38 |
| Pyr-GC-MS | Surface water and wastewater | PS and PE | PS (30 μg L−1) and PE (1000 μg L−1) | NR | 39 |
| Pyr-GC-MS and Nile red dye | Lagoon sludge | NR | NR | Fresh sludge (40.5 ± 11.9 × 103 particles per kg) and dehydrated sludge (36 ± 9.7 × 103 particles per kg) | 40 |
| Pyr-GC-MS and FTIR | Surface water | PVC, PP, and PE | NR | 0–110000 particles per km2 |
38 |
| TED-GC-MS | Freshwater | PE, PS, PET, and PP | PE (20.0 μg mg−1), PP (5.7 μg mg−1), PS (2.2 μg mg−1) and PET (18.0 μg mg−1) | NR | 41 |
| TGA, DSC | Wastewater | PE, PP, PET, PA, PES, PVC, and PU | NR | NR | 42 |
| DSC | Wastewater | PE, PP, PA, and PET | NR | NR | 43 |
| ATR-FTIR and DSC | Dutch beaches | NR | NR | NR | 44 |
3.3. Spectral analysis technique
The information obtained from the spectrum analytical method is more accurate than that obtained via visual recognition.45 At present, spectroscopic methods can identify MPs and confirm their composition. The spectral signal needs to be analyzed to reflect the different characteristic peaks that each type of MP creates. Fourier-transform infrared (FTIR) spectroscopy and Raman spectroscopy have been used to identify the polymer types of MP particles with a minimum particle size of 10 and 1 μm, respectively.Due to its excellent signal-to-noise ratio and lack of fluorescence interference, the nonlinear Raman spectroscopy approach is becoming increasingly popular in the field of MP detection.50,51 Surface-enhanced Raman spectroscopy, on the other hand, is gradually expanding its ability to detect MPs because of its unique chemical specificity and high sensitivity. Future quick in situ MP detection in water may be possible owing to the Raman spectroscopy approach, which has the potential to solve the challenge of target identification in liquids.
| Method | Methodology | Particle size by different analytical methods | Merits | Drawbacks |
|---|---|---|---|---|
| FTIR | • The excitable vibrations produced by a substance rely on its molecular structure and composition | • Particles >500 μm in size can be analyzed using ATR-FTIR | • Non-invasive methods | • Samples must be IR reactive, due to insufficient absorbance |
| • The IR spectra of plastic polymers have very distinctive band structures | • Particles <20 μm in size can be analyzed using microscope coupled FTIR | • Reliable, efficient, and dependable | • Samples <20 μm may not yield spectra that may be understood. It is difficult to evaluate non-transparent particles with this technology | |
| • The newly developed automatic FTIR imaging techniques, such as FPA, which allow thousands of spectra to be quickly captured inside an area with a single measurement, greatly reduces the analysis time | • To operate and process the data from the expensive, highly specialized instruments, competent employees are required | |||
| • The detection is affected by the ambient matrix (biofilm formation on polymer, for example), which complicates the interpretation of the results | ||||
| • The sample needs to be treated to remove the IR-active water | ||||
| Raman spectroscopy | • The interaction between the irradiated laser light and the molecules and atoms of the sample causes the back-scattered light to have a different frequency than that of the irradiating laser | • The microscopy-coupled Raman spectroscopy (RS) technique is suitable for particles larger than 1 μm | • The study of minute particles, ranging from 1 to 20 μm, with good spatial resolution and comparatively low water sensitivity is made possible by microscopy-linked RS | • The fluorescence resulting from inorganic, organic, and biological impurities significantly hinder MP identification |
| • The Raman shift can be used to create Raman spectra that are unique to a given molecule | • As far as particles go, this is the only practical method for sizes between 1 and 20 μm | • In addition to being able to quickly execute chemical mapping and analyze opaque and dark particles, the RS approach enables automatic and speedy data collection and processing | • The sample needs to be cleaned before analysis can begin | |
| • Three important Raman collection parameters are wavelength, laser intensity, and photo-bleaching. Automated mapping using micro-RS is still being developed | ||||
| • RS's analysis takes a long time |
3.4. Other analytical methods
With its low detection limit and good sensitivity, high-performance liquid chromatography (HPLC) is an often-used terminal detection method. It is also more appropriate for finding large, polar, and thermally unstable MPs.58,59 Scanning electron microscopy-energy-dispersive spectroscopy (SEM-EDS) is an analytical technique that combines scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS). Microstructures can be detected using scanning electron microscopy (SEM), which is a technique that falls between transmission electron microscopy and optical microscopy. Depending on the composition of the surface material, SEM can be directly used for microscopic imaging. The ability to capture MP surface features in clear, high-magnification pictures facilitates the ability to distinguish MPs from other organic-matter particles.60 Since elemental analysis can only provide an image of the material's surface morphology, it is critical to integrate the surface properties of plastic particles with elemental analysis in order to more accurately identify MPs. Nowadays, the elemental composition data of MPs are commonly examined using EDS to characterize their surface structure and discover more about their chemical composition. On the basis of the surface emission radiation of MPs, diffraction and reflection can be used to indicate the elemental composition of polymers, and SEM-EDS can be used to look into the surface morphology of MPs with a sample capacity of 5–10 mg.61 Despite the fact that this approach is currently often employed to characterize nanoplastics, it has the drawback of providing no chemical information. Table 6 provides an overview of the advantages and disadvantages of using HPLC and SEM techniques for MP identification.| Method | Methodology | Particle size | Advantages | Limitations |
|---|---|---|---|---|
| SEM | Sample pictures are produced when the sample is exposed to an electron beam, which quantifies the secondary ions in the sample | For particles as small as a micron in diameter, the analysis is feasible | A high-resolution image of the samples can be produced | A high vacuum is required to cover the samples, and there is no precise identification data available |
| HPLC | Samples are dissolved in certain solvents. To evaluate the various molar mass distributions, size exclusion chromatography is employed, and HPLC analysis is performed to quantify the findings | A sample size of several milligrams is required for the chemical extraction | Certain polymers showed higher recoveries | Its applications are limited to environmental samples because of restrictions on polymer types and the difficulties of establishing physical properties such as size. Only a small number of samples can be assessed per run. This process can only be used to analyze specific polymers, such as PS and PET |
3.5. Parameters used in sampling of microplastics
Following sample preparation, several techniques can be used to separate, identify, and quantify MPs from environmental samples. It is feasible to compile the frequently reported techniques in the scientific literature into grouped sampling approaches, including analytical techniques already used and established for other analytes, even though a universally accepted protocol for sampling and quantifying MPs in the environment is not available. Table 7 lists a few parameters for sampling MPs in various matrices, along with the corresponding analytical methods for finding and measuring MP particles.| Matrix | Sampling | Analytical technique | Ref. | ||
|---|---|---|---|---|---|
| Time elapsed (quantity collected) | Equipment | Extraction | |||
| a ATR-FTIR: attenuated total reflectance-Fourier-transform infrared spectroscopy; FESEM-EDX: field-emission scanning electron microscopy with energy-dispersive X-ray spectroscopy; FTIR: Fourier-transform infrared spectroscopy; ICP-MS: inductively coupled plasma-mass spectrometry; Py-GC-MS: pyrolysis-gas chromatography-mass spectrometry; SEM: scanning electron microscopy; EDS: energy-dispersive X-ray spectroscopy; TED-GC-MS: thermal extraction desorption-gas chromatography-mass spectrometry. | |||||
| Water | 2 h (between 2.3 and 310 million L day−1) | Stacked Tyler sieves (0.355 mm and 0.125 mm stainless steel mesh) | — | Stereo microscope | 63 |
| 30 min | Manta trawl (rectangular opening 16 cm high by 61 cm wide, 3 m long, 333 μm mesh) | — | |||
| 15–20 min (total of 2 L) | Neuston nets (0.52 × 0.36 m) of 333 μm mesh | — | Py-GC-MS | 64 | |
| <10 L | Telescopic sampling pole | — | Micro-FTIR spectroscopy | 65 | |
| 30 min | Plankton net (153 μm) | 32 μm steel-wire sieve and saturated NaCl solution | Stereo microscope and ATR-FTIR | 66 | |
| Water, biological tissue | 5 min (water, flow rates between 0.11 and 5.04 m s−1), 30 min (biological tissue) | 80 μm mesh conical net/seine nets, gillnets, conventional tackle, and minnow traps | 10% NaClO, HNO3:NaClO (1:10 v/v) |
Stereo microscope | 67 |
| Water, sediment | — | 1, 2, 4 and 10 mm mesh size | — | TED-GC-MS | 33 |
| Sediment | — | 0.25 × 0.25 m2, 5 mm sieve | Top layer of sediment (3–6 cm) | ATR-FTIR | 68 |
| — | Metal spoon | Directly from the sediment to a depth of 2 cm | Stereo microscope, SEM, Py-GC-MS | 26 | |
| — | Stainless steel shovel and 20 × 20 cm wooden frame | Surface layer (depth of 20 m) | Stereo microscope, micro-FTIR spectroscopy, SEMEDS, ICP-MS | 69 | |
| 4–6 L | Sediment cores (diameter of 10 cm to a depth of 5 cm) | Munich plastic sediment separator. Centrifugation tubes with sieves (750 mm mesh) | Micro-Raman spectroscopy | 70 | |
| — | Stainless steel scoop (10 cm depth) in order to fill a 1 L glass Kilner jar | Concentrated ZnCl2 solution (1.7–1.8 kg L−1) | Raman spectroscopy | 71 | |
| 3 kg | Ekman dredge | Saturated NaCl solution and 30% H2O2 | Stereo microscope and ATR-FTIR | 66 | |
| Biological tissue | Whenever 3 clams of 40–45 mm were retained | 0.5 × 0.5 m2 | 69–71% HNO3 | Stereo microscope | 72 |
| Until approximately 50 mussels were collected | Tweezers | 30% H2O2 | Stereo microscope, micro-FTIR spectroscopy, SEMEDS | 73 | |
| Overnight/4 weeks | Gillnets (mesh of 50 mm) per cages | Trituration of dried samples, 15% H2O2 | Stereo microscope, ATRFTIR | 74 | |
| — | Baka 44/60, 40/60 and GOC 73 trawl gears | Dried samples, NaOH 1 M | Inverted microscope and stereo microscope | 75 | |
| — | 90 mm GF/A 1.6 mm glass fibre filters and magnetic hot plate stirrer | Digestion solution (KOH 10% solution, 60 °C, 24 h) | Py-GC-MS, Raman spectroscopy | 76 | |
| — | Gillnet, demersal trawl | SDS, protease, chitinase and H2O2 treatment; vacuum dried samples, petroleum ether (60/80) | Py-GC-MS | 77 | |
| — | — | Dried fish and excised organs or eviscerated flesh | Micro-Raman spectroscopy, FESEM-EDX | 78 | |
4. Electrochemical detection of microplastics: a solution for low-cost monitoring
4.1. Overview
For the purpose of finding MPs in intricate matrices, extremely sensitive spectroscopic techniques are utilized. When it comes to simultaneously determining the MP concentration, these procedures are fairly adaptable. Additionally, these methods have femtomolar-range detection limits that are extremely low. Nevertheless, the production of several samples requiring challenging analytical methods and the need for highly skilled individuals to operate the sophisticated equipment are the drawbacks of these costly spectroscopic approaches. Additionally, these methods must be combined with other chromatographic methods in order to complete sample speciation, and they are only appropriate for quantitative analysis. This may increase the chance that the sample will alter during handling and storage. Furthermore, these optical techniques necessitate high-precision and high-power operations, making them unsuitable for in-field applications once more. They include expensive and sophisticated equipment with lasers, photodetectors, and so on. Thus, research is still being done to create quick, inexpensive, easy, and dependable methods that are appropriate for in situ and timely measurements of MPs.Conversely, electrochemical methods are more affordable, approachable, dependable, and appropriate for field use. These electrochemical methods enable straightforward processes and are ideal for creating tiny circuits as transportable devices for in situ contamination sample monitoring. In addition, these methods require less time for analysis than conventional spectroscopic methods, which enable real-time water sample monitoring. To increase their performance in MP detection, these electrochemical approaches need to be improved in terms of design. Compared to other spectroscopic and optical techniques, these techniques have lower sensitivity and limits of detection (LODs). Different biosensing electrodes are used with different electrochemical techniques to change the electrode material, thereby increasing the sensitivity and limitations of detection. This study looks at the different electrochemical methods used to find MPs in water samples, as well as the newest progress in making different interface materials that can be used to change the electrodes that are used in these methods.
Low-cost sensors and electrochemical techniques are interesting analytical tools for MP field detection.79,80 Chemical sensors are generally designed by combining an electrochemical transducer with embedded identification elements and a sensing substance.81,82 A chemical or physical change happens when a target molecule interacts with the sensor surface because of the analyte's covalent or non-covalent binding. The target can be either qualitatively or quantitatively detected, owing to the translation of this binding into a readable signal that can be electrochemically tested. Subsequently, while fabricating electrochemical sensors, care must be taken to select and understand the materials that will be exploited to build the sensor as well as the chemical or biological processes that take place at the contact.83 Recognition elements that are designed and selected at the electrode surface are chosen on the basis of their affinity and compatibility with the target analyte.
The physical characteristics of the sensing material, such as light absorbance, electrical conductivity, permittivity, and work function, are altered during the identification process between these components and the analyte. These alterations are converted into signals that can be detected, such as changes in color, voltage, current, resistance, or impedance.84 The work function indicates the amount of thermodynamic effort required to extract one electron from the material's Fermi level, whereas the permittivity indicates the material's ease of electromagnetic radiation transmission.
Electrochemical sensors for MP detection need transducers assembled within a carefully designed sensing interface; ideally, these sensors can be fabricated into a portable unit. These sensors require sensor materials with specific binding sites and recognition abilities to selectively identify the target analyte.85,86 Attaining the requisite sensitivity and selectivity requires the capacity to design specialized recognition sites. A typical architecture of the sensor design for MP and source of exposure detection is shown in Fig. 5. A successful MP-monitoring sensor should operate on par with or better than conventional chromatographic methods. These sensors ought to be lightweight, affordable, and simple to operate. They should deliver accurate, real-time data with little sample preparation.
 | ||
| Fig. 5 Sources of MPs and illustration of sensor design for their electrochemical detection. | ||
4.2. Electrochemical identification of MPs
Distinctions between MPs and other particles in an electronic field in terms of their electronic characteristics provide the basis for electrochemistry-driven MP identification. By qualitatively and quantitatively assessing the electrochemical responses (such as current and impedance) of mixed particles, it is possible to achieve simultaneous material identification and sizing. Table 8 provides a summary of the properties of the three sample systems for the electrochemical sensing of MPs, which are addressed in this section.| Method | Plastic type | Operational conditions | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| Resistive pulse sensor | Poly styrene (PS) | • Concentration range: up to 6.52 × 107 particles per L | • High concentration sensing range | • Restricted size range | 87 |
| • Sizing range: 2–30 μm | • Easy to use | • Sophisticated sensor manufacturing procedure | |||
| • Flow rate: up to 1 mL min−1 | • Suitable for most plastics | ||||
| Impedance spectroscopy | Poly ethylene (PE) | • The concentration range is 6.52 × 107 particles per L | • Simple to carry out | • Sophisticated data processing | 88 |
| • The size range is 300–1000 μm | • Quite accurate | • Comparatively poor MP recovery rate | |||
| • The flow rate is up to about 100 mL min−1 | • Works with the majority of plastics | ||||
| • Wide range of sensors | |||||
| Single micro-particle electrode | Poly ethylene (PE) | • Concentration range: up to 1.4 × 1011 particles per L | • High size precision (<2 μm) | • Restricted size range | 89 |
| • Sizing range: 1–10 μm | • A wide concentration sensing range | • Intricate operation |
5. Electrochemical sensors for MP determination
Although electrochemical sensors have been extensively used to detect environmental contaminants, including leachates from MPs,95 their applications for MP detection are fairly rare and have only recently come to light. Because of the low cost, low response time, easy operation, and mobility of electrochemical devices, the development of electrochemical sensors for MPs is especially interesting. In contrast to other traditional techniques, electrochemical approaches can be readily expanded to the in-field assessment of many sample kinds, as they do not require any preliminary MP isolation or purification. Amperometry, voltammetry, and label-free electrochemical impedance spectroscopy (EIS) have been used to achieve MP monitoring and sensing up to this point.Using an EIS-based sensor in combination with flow cytometry, PE MPs were detected.88 The detector consisted of a flow cell for EIS detection with Au-plated circuit boards that held all the Au-plated electrodes and a flow cytometer for particle detection (Fig. 6). The idea behind this approach was that the real fraction of impedance should change according to the particle volume at low frequencies. Therefore, at low frequencies, the change in impedance is proportional to the MP particle volume and is caused by the MP particles flowing over the electrodes (Fig. 6). The plastic and biological particles may be differentiated with this method at any observed frequency. It is interesting to note that although MPs caused a positive change in impedance, biological particles caused a negative change, making this platform suitable for MP detection in complex media.
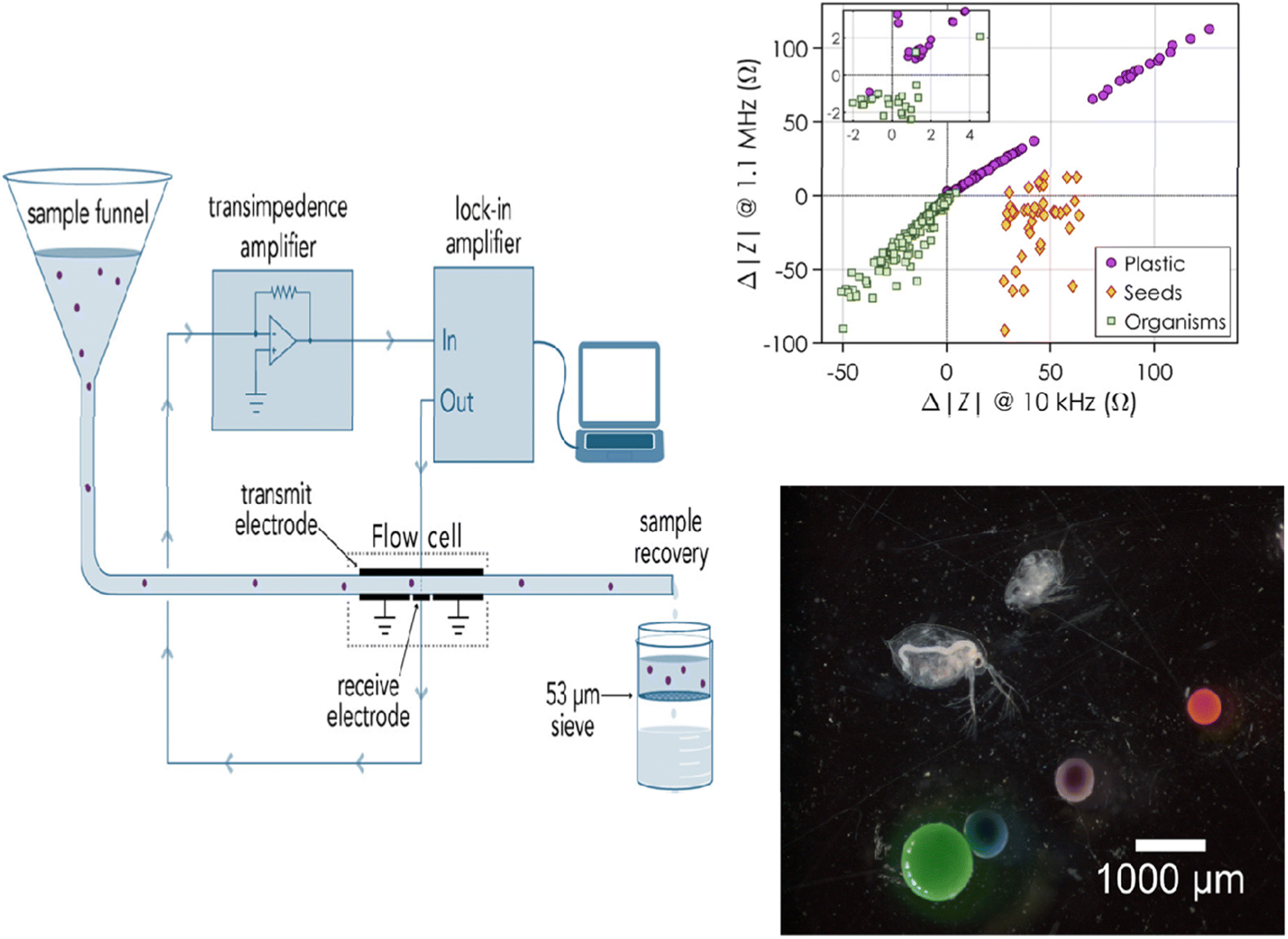 | ||
| Fig. 6 Diagram showing how MPs are measured for impedance while going through a flow cell. Variation in impedance according to the type of particle: plastic, seeds, or organisms.88 | ||
The sensitivity of the sensor and its capacity to detect and distinguish between different MP sizes were assessed in real time by testing the clean water flow against water that had been spiked with known MPs. The impedance would alter, producing peaks and enabling the measurement of particle size if MPs were present in the fluid under test. Using the linear relationship between the particle diameter and the cubic root of the actual impedance change, the impedance change and particle size were related. As a result, this sensor could measure and size both PE MPs (212–1000 μm) and MP beads (containing biological components in the 210–1200 μm size range). According to Collson and Michel,88 the sensor had a recovery rate of over 90% for MPs in the 100–300 μm size range and a false-positive rate of 1% when it came to identifying biological materials as MPs. Although MPs were effectively detected using EIS—a label-free approach—the next generation of sensors should focus on detecting MPs in the field as well as differentiating between various types of MP particles besides PE.
Additionally, spherical PE MPs (1–22 μm) have been detected using particle impact electrochemistry.89 A popular technique for studying particles suspended in solution is the particle–electrode impact method. Measurements using chronoamperometry revealed a fast current response due to particle interaction with the carbon fiber microwire electrode. The undivided three-electrode setup used in the electrochemical analysis setup was maintained at a particular voltage in order to observe the desired reaction.
A particle–electrode collision caused a signal change that was monitored. Fig. 7 shows the transient current response, or spike, that was produced when the particles collided with the electrode and was examined to identify the MP. The current spike in the chronoamperogram was caused by a collision between PE MP particles and the working electrode. This happened because the amount of oxygen in PE MP particles declines. With an R2 value of 0.96, an excellent connection was found using this method between the MP particle concentrations and the frequency of spikes.89 Comparing this detection method to other approaches, the MP measurement was shown to be more accurate and consistent overall. Although the technique was used to find electrically insulating MPs, conductive particles might also be found.
 | ||
| Fig. 7 SEM images for the carbon fiber electrode with and without MPs (a). An enhanced view of a transient current–time signal resulting from a collision event between MPs and electrode can be seen in the inset of the chronoamperogram (b), in which the blue line represents the PE MPs and the black line represents the MP-free state. Histogram showing the distribution of integrated charges extracted from spikes resulting from chronoamperometric measurements (c). | ||
The serial faradaic ion concentration polarization technique is an alternate method for MP detection.96 Based on their electrophoretic mobilities, the MPs are sorted by the faradaic ion concentration polarization. Their electrophoretic mobilities have an impact on the interaction between the MPs and the electric-field gradients. Elevated electrophoretic mobility particles concentrate at a low-electric-field site. On the other hand, lower electrophoretic mobilities are concentrated in greater electric fields. MPs were sorted into distinct chambers using a trifurcated trunk, with the bipolar electrodes positioned close to the trunk. The ion depletion zones and electric-field gradients are positioned across the opening of the split channel by the BPE. As a result, the MPs are sorted, with the higher-electrophoretic-mobility particles going into the lower channel and the lower ones flowing into the upper channel. The flow of PS MPs in a trifurcated microchannel was managed by the faradaic ion concentration polarization. MPs may be continuously focused on, sorted, and separated using this strategy. Because MPs interact with electric-field gradients and can be seen with optical and fluorescence microscopy, they could be put into groups based on how fast they move through an electric field.
MPs were sorted according to their size and electrophoretic mobilities in the trifurcated microchannels, which were oriented toward the MPs' trajectory by the electric-field gradients as they approached the cathodic end of the bipolar electrodes (Fig. 8). In addition to isolating or pre-concentrating different MPs on demand without the need for membrane-based separation, this strategy makes the technology even simpler by allowing MPs in water systems to be continuously monitored and in real time. Crucially, the ability of the faradaic ion concentration polarization to interface with different methods would provide sensor developers and designers with more creative freedom; yet, MP detection has not taken advantage of this potential.
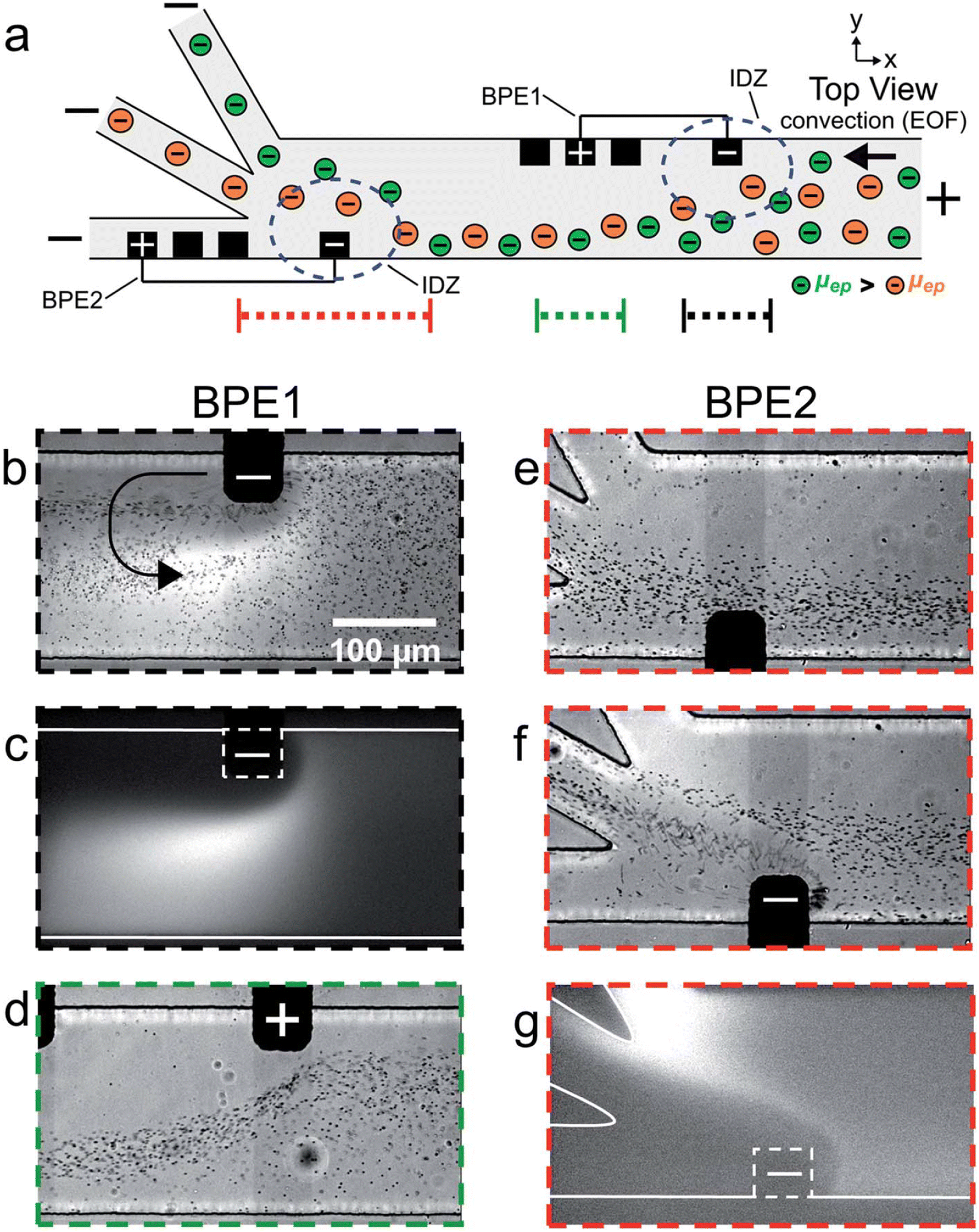 | ||
| Fig. 8 (a) Schematic of the microfluidic configuration used for serial faradaic ion concentration polarization experiments. For frames (b–e), only BPE1 was active. For frames (f) and (g), both BPE1 and BPE2 were active. (b–g) Series of optical and fluorescence micrographs showing the location of mP1 and BODIPY2_ during serial faradaic ion concentration polarization. With reference to the three dotted lines at the bottom of (a), the micrographs were captured along the portion of the channel length indicated by (b and c) the dotted black line; (d) the dotted green line; and (e–g) the dotted red line. The curved black arrow in (b) indicates the location and rotation direction (counterclockwise) of the vortex downstream of the cathodic pole of BPE1.96 | ||
More recently, the multiuse RPS served as the foundation for the development of an inexpensive, high-throughput technique.87 This microfluidic sensor, which was constructed on a silver wire, used an analyte's (MP) translocation via a tight constriction to detect changes in current. The gadget was utilized to track MPs in tea bags and identify algae to show the value of this technique for counting and tracking MPs in the presence of biological particles, which is still a difficult task.
Images of the base unit, lid, sensing zone, and electrodes are displayed in Fig. 9a–d. The lids can have various designs. The first is a flat surface that resembles a Gen 1 acetate film. In the second design, a ridge is positioned to extend into the channel and protrudes 1 mm from the surface. The third, shown in Fig. 9e, features several ridges that can alter the channel's and sensing region's shape. The gadget features printed screw threads to attach pumps because it was made to be integrated into a flow system. The integration of the electrodes' screw threads is a second change from Gen 1, in addition to the lid design (Fig. 1d). The device can quickly screen liquid volumes of 1 mL min−1 for the presence of algae and MPs.
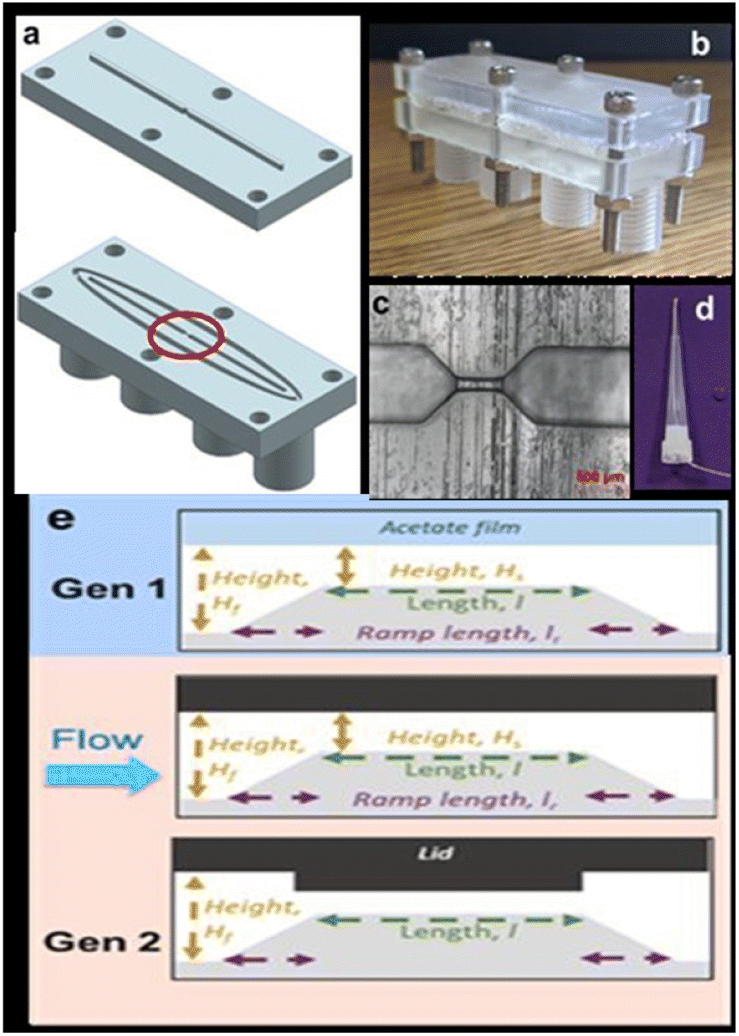 | ||
| Fig. 9 (a) Design of the device, (b) image of the assembled device, (c) an optical microscope image of the sensing zones circled in part (a), (d) silver wire embedded within a pipette tip, and (e) schematic of the sensing zone when using an acetate film, a flat lid, and a ridge lid.87 | ||
There have also been reports of other dual-mode sensor instances, in addition to impedimetric and amperometric sensors for MPs. A sensor for exoelectrogenic-biofilm-based PE MP detection has been created, employing voltammetry and impedance.97 The use of electroactive bacterium film in microbial electrochemical systems has shown a lot of promise as an energy-efficient method for MP detection in wastewater. The biofilms were subjected to MPs to find out how they responded as well as to examine the electrochemical characteristics, shape, EPS, and microbiological organization of the biofilms. A carbon fiber brush as the anode served as the working electrode in this three-electrode arrangement, which also included titanium woven wire mesh serving as the cathode counter electrode and Ag/AgCl as the reference electrode. PE–MP binding to the biofilm was the reason of the elevated internal resistance.97 Using Nyquist plots, EIS was used to quantify the impedance of the MFCs and MECs both before and after binding. The presence of PE–MP boosted the cells' resilience. On the basis of the equivalent circuit model, the charge transfer resistance (Rct) was responsible for most of the resistance. Furthermore, a notable rise in Rct may be caused by an increase in the number of dead cells induced by the toxic effects of PE–MPs. When PE–MPs were present, the current density in the microbial fuel cells remained constant. On the other hand, as MP concentrations grew in the microbial electrolysis cell, the current signal decreased; this tendency persisted for more than 42 days (Fig. 10). The decrease in signal was caused by an increase in internal resistance as a result of PE–MP binding to the biofilm.97 Therefore, the microbial electrolysis cells would need to be used to measure the concentration of the MP present to sense PE–MPs using exoelectrogenic biofilms.
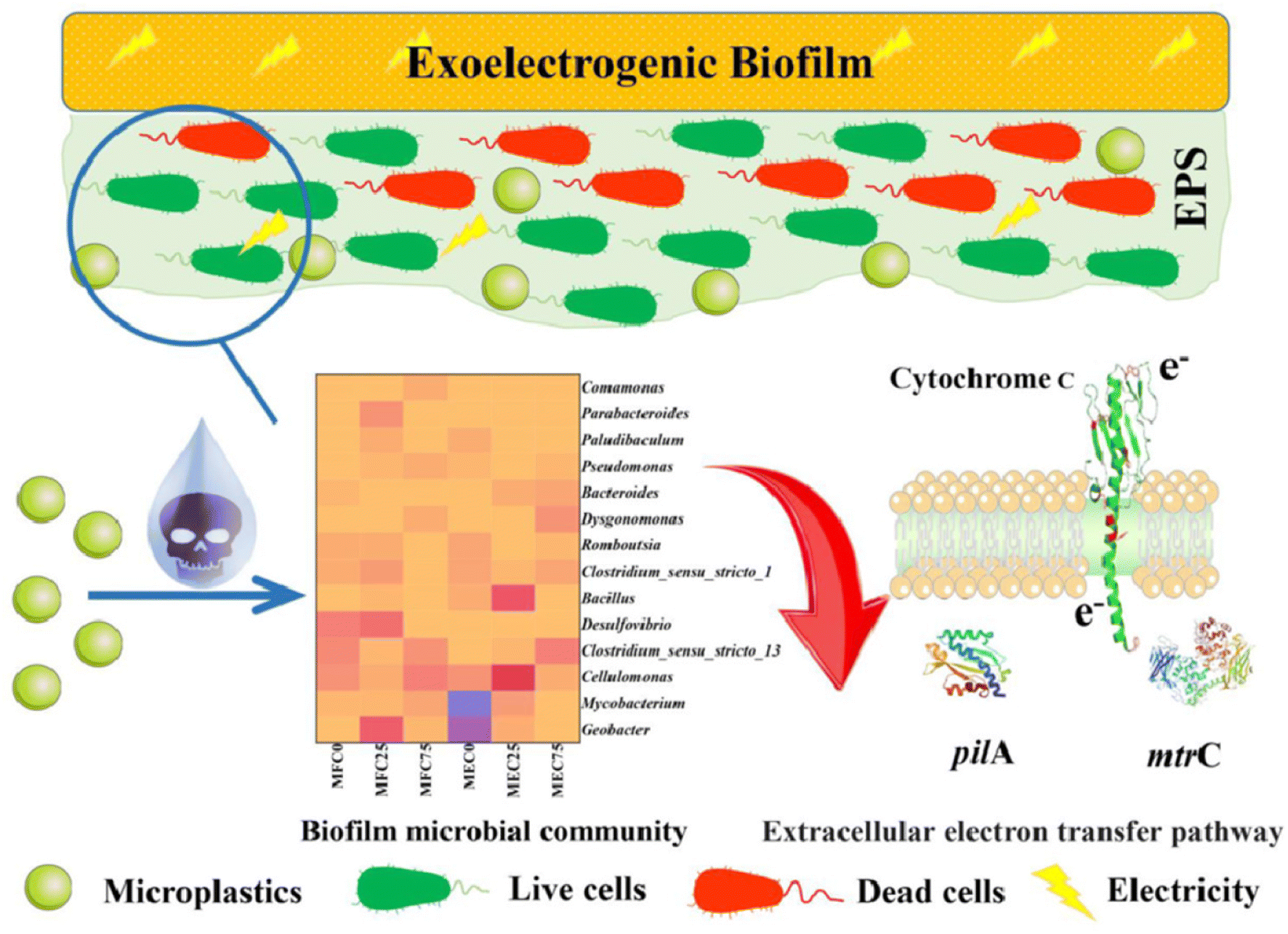 | ||
| Fig. 10 Schematic of exoelectrogenic biofilm on an electrode surface and its interactions with MPs.97 | ||
Furthermore, to expand its application into a real-world environment, a microbial electrolysis cell could be utilized in the future to differentiate between MP types and sizes. The same technique might be applied to MP remediation in microbial electrochemical wastewater treatment systems. Although MP cleanup has used electrochemical techniques, MP sensors are still not well understood. In order to effectively monitor MPs, further research and development of sensors is required, despite the existence of successful electrochemical sensors.
There are numerous MP cleanup procedures that give excellent chances for creating new testing instruments. By applying anodic oxidation or reduction of MPs, for instance, MP remediation can be indirectly employed for sensing applications such as MP coagulation detection, MP byproduct sensing, MP interactions with biofilms or other biologicals, and so on. MP identification and differentiation; standardized methods for MP isolation, characterization, and environmental monitoring and tracking; and MP detection in a range of sample types (soil, soil runoffs, various watersheds and pools, wastewaters and industrial effluents, agricultural watersheds, and atmosphere) are the current challenges facing the advancement of electrochemical sensors for MPs. The electrochemical gadgets may also offer a way to continuously track MPs' movements and destiny in their surroundings and in real time. The breakdown of MPs into smaller NPs is a crucial component of MP chemistry; thus, it is also necessary to identify and track plastic particles downstream from the MPs. Since MPs readily interact with and absorb other biomolecules, understanding their basic interactions through electrochemical techniques may initiate new possibilities for MP cleanup and monitoring.
6. Important elements in MPs' electrochemical identification
Electrochemical systems are susceptible to the following factors: applied current density or potential, analyte property, electrode activity, and electrolyte property. The influence of experimental conditions should be examined in order to direct future research for the electrochemical identification and removal of MPs in water matrixes. The properties of the MPs, the applied current density or potential, and the electrolyte are thoroughly covered in this section.6.1. Plastics' property
The size, shape, density, and type of plastics are significant characteristics that influence their electrochemical behaviors. Because PVC has a far lower electrical resistance than PE, it is more susceptible to electro-precipitation and electro-flotation. Furthermore, electro-flotation makes it simpler to remove MPs with densities lower than that of water (like PE) from water.98 The ideal removal parameters for different-sized MPs vary due to the unique electrical characteristics of polymers of varying sizes.89 The electrocoagulation of small MPs (20–60 μm) requires a current density of 2.88 mA cm−2 and a pH value of 4, whereas large MPs (350–1500 μm) require a current density of 2.88 mA cm−2 and a pH value of 7, possibly due to the larger bubbles generated at neutral pH.99 Furthermore, the shape of MPs and NPs affects the effectiveness of electrochemical removal. Fiber MPs (CA and PP) have a greater electrocoagulation removal efficiency than granular MPs (PE and PMMA).100 As of right now, only plastic beads have been investigated for electrochemical sensing; other MP forms have received less attention. It is important to look into the electrochemical identification of MPs with various shapes (e.g., sheet, fiber, and film) in light of the shape-dependent electrical characteristics of plastics (Shimizu et al., 2017).896.2. Density of current
The applied voltage or current density can directly affect the reaction efficiency. Higher current densities (voltage intensities) generally encourage the elimination of MPs through processes like electrocoagulation. In the electrocoagulation/electro-flotation process, the higher current density helps flocs form (like Al(OH)3) and speeds up the release of the coagulating species from the Al anode. The cathode also releases more hydrogen bubbles when the current density is high. These bubbles help get rid of MPs and provide electro-flotation by sticking to agglomerates.99 In the insulator-based di-electrophoresis process, on the other hand, a high voltage intensity would make particles stick together, which could make the device less effective at separating things by blocking its tiny gaps.101 In addition to electrochemical performance, one should consider the high energy consumption brought about by the high current density.6.3. Electrolyte
Many studies have been conducted on the effects of pH, carrying interfering compounds, and electrolyte concentration on electrochemical studies involving MPs. Better conductivity from a larger electrolyte concentration benefits the electrochemical process, too.102 The electrochemical signal in the RPS-based sensing technique is clearly influenced by the ionic strength of the electrolyte (KCl), with conductive pulses and reversed pulse directions being observed at lower ionic strengths. An increase in electrolyte concentration during the electrocoagulation process might increase the MPs.100 Additionally, Na2SO4-electrolyte-derived reactive species ( SO 4 ˙ ) electro-oxidize PS MPs during the electrooxidation – H2O2 process, and a concentration increase of Na2SO4 from 0.007 to 0.03 M results in noticeably better electro-degradation kinetics. Nevertheless, an additional concentration increase to 0.06 M has no beneficial effect. This could be because there is not enough H2O2 generated in situ to react with S2O82− to form SO4˙− species.103
( SO 4 ˙ ) electro-oxidize PS MPs during the electrooxidation – H2O2 process, and a concentration increase of Na2SO4 from 0.007 to 0.03 M results in noticeably better electro-degradation kinetics. Nevertheless, an additional concentration increase to 0.06 M has no beneficial effect. This could be because there is not enough H2O2 generated in situ to react with S2O82− to form SO4˙− species.103
The pH of the electrolyte controls the characteristics of surface charge and the chemical structure, as well as the forms of many substances such as coagulants, analytes, and electrode materials. A near-neutral solution is advised to help create big flocs in the electrocoagulation of MPs.98–100,102 Comparable outcomes are shown when bisphenol A (BPA) is electrochemically detected. The acid–base dissociation of BPA is influenced by the pH of the electrolyte, which leads to a pH-dependent oxidation current and potential. When the pH goes up, the –COOH groups on multiwalled carbon nanotubes (MWCNTs) become more deprotonated. This makes them act like acidic anions and would reject anionic BPA species if they were used as a BPA sensor in an alkaline solution (MWCNTs-βCD). Accordingly, BPA may be detected at a pH level of 7.4, which is almost neutral.104
Metal ions (such as Pb, Ni, Cu, and Cr) can interact with naturally aged MPs by surface complexation and electrostatic adsorption. It is interesting to note that the presence of MPs and metal ions together promotes the removal of metal ions without altering the MPs' ability to electro-coagulate.105 An increase in Cl− ion concentration has no appreciable impact on the removal performance of PE MPs during electrocoagulation.102 As of right now, research on the function of dissolved organic materials in the electrochemical removal of MPs is lacking. Usually, metal ions, anions, and organics are added to the electrolyte to test how selective the electrodes are for finding contaminants that come from MPs. Some small molecules (like uric acid and ascorbic acid) and inorganics (like KBr, NaNO2, KCl, and NaNO3) do not have much of an effect. However, organics (like tetrabromobisphenol A (TBBPA)) that share a chemical structure with the target pollutants (like BPA) may make identification harder.106
7. Fabrication of electrochemical sensing devices
The construction of electrochemical sensing devices requires the thoughtful design and deliberate selection of electrode materials and device components that are appropriate for the intended use. For the device to consistently function in its intended environment, all of its parts must be mechanically strong and chemically stable. The next stage is to determine the best technique for immobilizing the active material on the surface of the working electrodes after selecting the electrode materials. The approach for altering the electrode and the types of materials utilized can influence the electrode's thickness, adhesion, mass loading, distribution, and homogeneity on the surface. A range of electrodes, including metal foam, carbon-based conductive paper, glassy carbon, and ITO/FTO glass, are accessible and can be utilized as platforms for electrochemical devices involved in MP monitoring. An overview of the many techniques for depositing materials at electrode surfaces is given in this section. These techniques address issues of reproducibility, compatibility, and large-scale processing—all of which are crucial elements in the creation of sensing devices.7.1. Electrophoretic deposition
A tried-and-true technique for directly growing thin films on electrode surfaces is electrophoretic deposition (EPD), which is renowned for its capacity to yield homogeneous coatings with adjustable thickness. Applying a DC electric field to a target substrate containing a suspension of the substance to be deposited is how EPD is carried out (Fig. 11). The electric field can create intricate 2D and 3D patterns on electrode surfaces by forcing the charged particles in suspension in an orderly manner toward the oppositely charged electrode. | ||
| Fig. 11 Schematic of the EPD process. | ||
Biological materials can also be deposited using this technique. Particle size, solvent type, electrophoretic mobility, and electric-field intensity can be changed to create films with diverse properties, such as thickness and homogeneity.107 By providing 0.2–1.6 V, this technique has been used to deposit gold nanoparticles of various sizes onto ITO glass electrodes. Under ideal circumstances, nanoparticles of metal oxides and sulfides, including copper,108,109 iron,110 cobalt,111 zinc,112 and titanium,113 have been deposited. Graphene oxide,114–118 graphene quantum dots,119 carbon nanotubes,120 and their composites were among the carbon-based materials that were deposited using applied voltages ranging from 2 to 100 V. These materials were chosen on the basis of their charge and mobility. Graphene composites were deposited on silicon wafers using higher voltages (300 V).121,122
Additionally, metal–organic frameworks (MOFs) were deposited using EPD to produce homogeneous films with the required shape and thickness. Because MOF particles have defect sites from missing linkers, they are partially charged. When a strong enough electric field is applied, these charges push the MOFs in the direction of the electrode that is oppositely charged. By using EPD to spread MOFs in toluene for different lengths of time, thin films of NU-1000, HKUST-1, UiO-66, and Al-MIL-53 have been micropatterned. These films stick well to the glass electrode (FTO) and maintain their crystal structure.123 To detect explosives and Cr3+ ions, an Ln-BTC MOF thin film was made by dispersing MOF in a CH2Cl2 solution.124 Electrically conductive MOFs have recently attracted a lot of attention for thin-film manufacturing in order to be deposited as electrode materials for supercapacitors and batteries.125,126 To give an example, putting 0.5 V between the electrodes for 10 min made 2D MOF nickel-2,3,6,7,10,11 hexaaminotriphenylene (Ni3(HITP)2) nanosheets stick to nickel foam.127
7.2. Layer-by-layer assembly
Another straightforward and reliable technique for creating multilayer films on solid substrates is layer-by-layer assembly (LBL). The electrode is alternately exposed to positively and negatively charged species in this “bottom-up” method. It is possible to create electrodes that are precisely suited by adjusting the distribution, thickness, and composition of the layers.128 LBL does not need a conductive substrate, in contrast to EPD. Furthermore, the film can be deposited on substrates with various topologies, which makes it easier for 3D microstructured films to form.129 Roll-to-roll,130 spraying,131 atomization,132 magnetic assembly,133 electrocoupling,134 creaming,135 and other processes are examples of LBL technology. The needs of the application and the substrate's ability to hold materials in place determine the choice of the substrate. This technique offers a broadly applicable way to embed active materials on electrode surfaces for target analyte binding or conversion. It is suitable for sequestering various sorts of materials in between the layers, such as metal nanoparticles. Surface-supported MOF thin films (SURMOFs) were made using LBL by first coating the support with a self-assembled monolayer (SAM) and then immersing it several times in solutions containing organic ligands and metal precursors, cleaning it after each immersion (Fig. 12a).136,137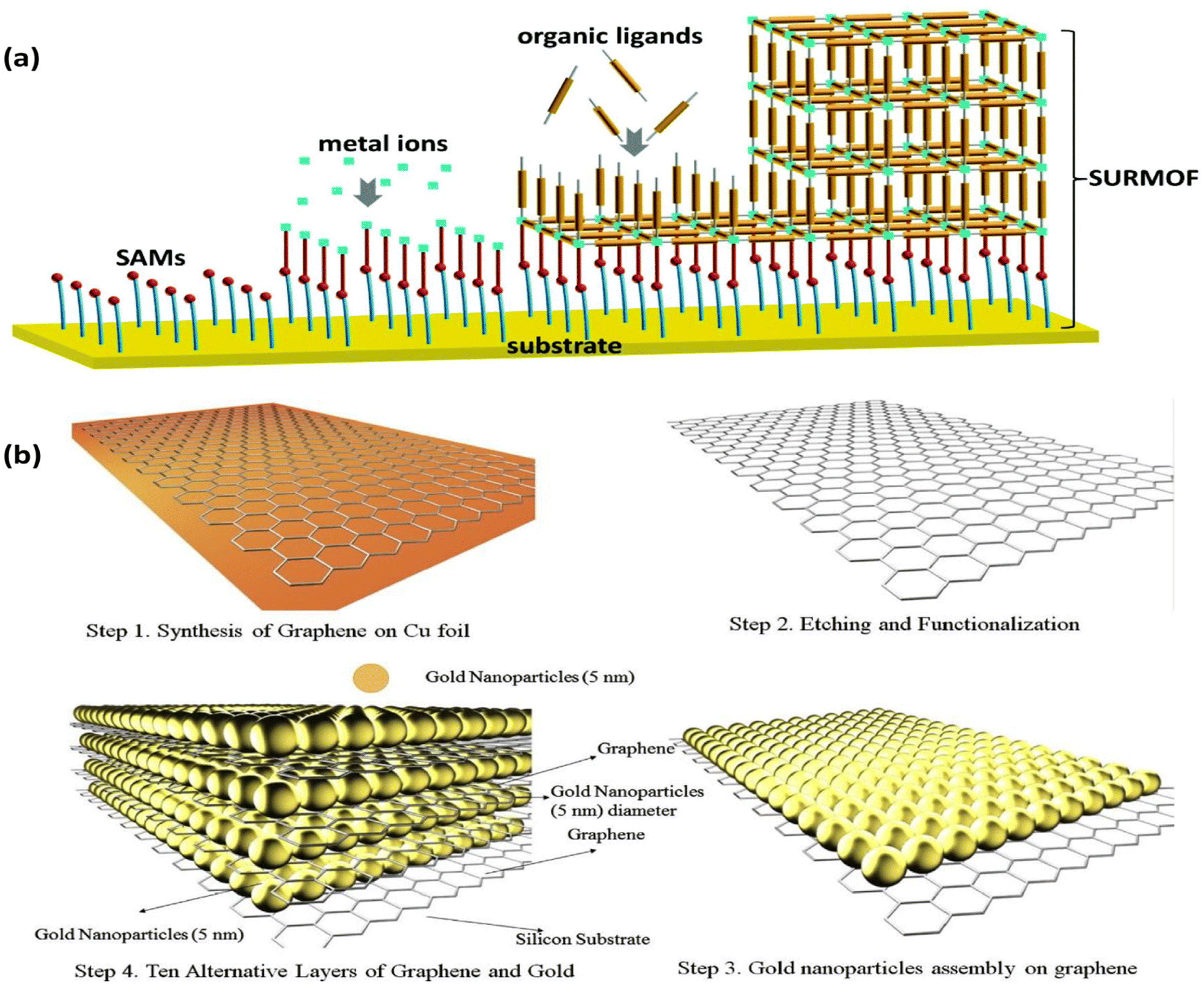 | ||
| Fig. 12 (a) Illustration of SURMOF film formation via an LBL assembly.139 (b) Schematic of the LBL assembly of Au NPs/graphene electrode. Reproduced with permission from ref. 141. | ||
This process encourages the controlled development of highly aligned and uniform MOF sheets for gas sensing138 and electrochemical catalysis.139 Additionally, LBL enables the deposition of MNPs on solid substrates, giving good control over the density and distribution of NPs.140 Electrostatic interaction was used to deposit Au NPs on graphene, which makes it easier to capture the injected electrons from the analyte (Fig. 12b).141 LBL assembly can be utilized to build substrates with tailored surface-enhanced Raman scattering (SERS), regulated Au NP size, and layer thickness for use in sensing applications.142 Shorter fabrication times can be achieved by automating the assembly process with this method. A thorough analysis released in 2016 covered various LBL assembly methods in great depth.143
7.3. Direct 3D and inkjet printing
Large-scale production and quick processing times were made possible by the development of automated procedures, which were spurred by the rapidly growing interest in modern manufacturing. With the use of 3D printing technology, electrodes with a variety of shapes may be consistently produced, lowering the overall production costs and facilitating manufacturability.144 The main obstacle in the development of 3D printable electrodes, electronics, and structures is the formulation of inks and materials that are printable. Metal oxides, carbon-based materials, ceramics, polymers, biological or biomimetic materials, and polymers are some examples of these materials. The direct printing of electrode materials enables high structural design freedom and hastens the compositional optimization of materials for sensing applications. There are several ways to print electrode materials, including binder jetting, fused deposition modeling (FDM), stereolithography (SLA), and direct ink writing (DIW), as shown in Fig. 13A–D.145 | ||
| Fig. 13 Top image shows a schematic of the common printing techniques: (A) direct ink writing (DIW), (B) fused deposition modeling (FDM), (C) stereolithography (SLA), and (D) binder jetting.145 | ||
The most popular 3D printing method, DIW, uses droplet-based and continuous filament techniques to create structures with a sub-50 μm resolution. This method depends on regulating the ink's viscosity, rheological characteristics, and shear-thinning behavior to promote smooth extrusion, enable printability, and preserve the printed shape. The difficulty with electrochemical devices is creating conductive inks and processes that can generate huge-volume shapes with high resolution.146 SLA prints high-resolution structures by using photocurable polymers, which solidify during printing and allow for simultaneous laser scanning. Usually, this process entails printing non-conductive polymers onto conductive substrates and then covering the printed structures with a conductive layer.147 Using carbon filaments that have been heated and extruded, FDM is a popular printing method for creating electrode materials. Nevertheless, commercially available filaments with weak conductivity, such as graphene or PLA, may need to be treated after printing.148 Binder jetting works by putting binder droplets on a bed of powder, which is hardened to create 3D objects. Large structures with intricate geometries may be created using this method, which makes it suitable for use in industrial settings.
Electrodes can be printed by using conductive materials for the powder bed.149 The quick output, repeatability of the electrode geometry, and ease of production have made printing sensor materials appealing. For example, cellulose paper and plastic substrates were inkjet printed with chemiresistor films and CNT designs.150 At room temperature, the printed electrodes responded to NO2 and Cl2 gases in a reversible and selective manner. Charge-transfer p-type doping of CNTs serves as the foundation for the sensing process. As evident in Fig. 14a,b,151 nozzle-jet technology was used to print Ag–rGO composites onto a flexible substrate. These were then used as sensors to directly detect phosphate ions in eutrophic water. The printed sensor showed a low LOD of 0.2 μM and a linear range of 0.005–6 mM.
 | ||
| Fig. 14 (a) Schematic of Ag/rGO hybrid ink formulation, (b) nozzle-jet-printed Ag/rGO-based FET sensor, and (c) optical image of printed Ag/rGO-based FET sensors on a PET substrate.151 | ||
Screen printing is a simple, roll-to-roll method for creating inexpensive electrochemical sensors. It involves depositing layers after layers via open mesh screens that are specifically made to create a thick film. The conductive tracks can be built using a range of commercially available conductive pastes. The last layer necessitates the creation of a printable formulation and is usually tailored to incorporate the sensing material with or without an embedded enzyme. It is hard to put biocomponents like enzymes in the printed layer for biological sensors because they might not work during the serigraphy process or the curing step. The printed electrode can be interfaced with portable wireless potentiostats as a further step in the development of portable electrochemical sensing platforms. Additionally, methods for mass-producing these sensors should be designed. Gravure-printed electrodes made of biocompatible inks with electrode morphologies that offer mechanical stability and produce 150 m of flexible rolls are a recent example of the roll-to-roll technique (Fig. 15).152 The low-cost electrochemical detection of heavy metals and tiny compounds has proven to be applicable. We used tyrosinase carbon enzymatic black ink to show that the technology can be used to print enzyme-based parts for making several electrochemical biosensors.153 Printing offers benefits like low production costs, low repeatability, and manufacturability when it comes to creating sensors.
 | ||
| Fig. 15 Example of roll-to-roll gravure-printed electrodes on flexible PET substrates (a) with optical images showing an array of 3 mm-diameter electrodes consisting of carbon (working electrode), silver (reference electrode) and carbon (counter electrodes), and insulation layer (b) with SEM images showing the cross section of hierarchical carbon ink over silver ink (c), carbon electrode surfaces, and silver electrode (d) with nanostructured ink components (e).152,154 | ||
8. Future challenges
Even though MPs can be electrochemically detected in a fast, precise, selective, and reliable manner, there are still a few areas that require improvement in order to enhance electrochemical identification methods.• Only plastic microbeads with known sizes and concentrations have been investigated for MP sensing; other MP shapes have received less attention. Determining the electrochemical reactions of various MPs and NPs in the sensing process is vital, given the size- and shape-dependent electrical characteristics of plastics.87,89
• Because lab-made waters are used in current research on the electrochemical sensing of MPs, the effect of impurities found in real water, like metal ions, organisms, organic contaminants, and suspended particles, is not taken into account. For instance, metal ions would cause MPs to aggregate, which would have an impact on the sizing procedure. In light of this, more research is advised to determine the function of various elements in MPs' electrochemical sensing process.
• The electrochemical device and MP identification efficiency are closely related. Small sensing zones typically improve the sensing accuracy; however, when large MPs or aggregates are present, obstructions may happen. It would be interesting to create sensing devices with numerous channels in order to address this problem. Advanced additive manufacturing methods can also expedite the device design process.
• The majority of sensing materials require time-consuming, complex, and expensive processing in order to identify contaminants originating from MPs. Therefore, creating high-performing electrodes with simple procedures is crucial to develop electrochemical sensors that are commercially viable.
9. Future outlooks
Benefits from electrochemical approaches include great sensitivity, cheap operating costs, and ease of use. Numerous disposable, single-use electrodes and self-powered devices are readily accessible and offer great platforms for straightforward, low-cost, and onsite contamination monitoring in decentralized laboratories. Developments in advanced material development, microfabrication, and additive manufacturing techniques, such as 3D printing and inkjet printing, have resulted in improved selectivity, stability, and shelf life of developed sensors, as well as increased detection capabilities. Nevertheless, despite the substantial advancements made, a number of enhancements are still required to transfer this technology from the lab to the real world. First off, the majority of research is still conducted using standard solutions that ignore the sample composition and matrix effect. It is still necessary to ascertain issues like interferences, the existence of naturally occurring organic compounds, and electrode passivation. Second, it must be proven that the necessary selectivity and sensitivity can be attained and that the procedure may be carried out in situ with or without a minimal sample pretreatment step. Even though the use of 2D, 3D, and layered materials, as well as advanced manufacturing techniques, has resulted in significant advancements in improving the detection sensitivity and portability, many electrochemical sensors are still in the “proof-of-concept stage,” and their functionality in the field and in real environmental scenarios needs to be established. Last, in order to show competitive advantages, electrochemical sensors must be validated.Several possible paths might be imagined to enhance the potential of electrochemical approaches for MP field detection:
(1) Rationally designed 2D and 3D nanomaterials combined with biological molecules as sensing elements to increase the sensitivity and selectivity.
(2) Large-scale, scalable manufacturing techniques like 2D and 3D printing to fabricate electrodes and improve reproducibility.
(3) High-throughput multi array platforms for simultaneous MP detection, possibly in conjunction with self-reference electrodes to minimize interferences and improve selectivity.
(4) Integration of electrochemical measurements with artificial intelligence and pattern recognition.
(5) Developments in nanoimpact electrochemistry to enable ultrasensitive detection. Ultimately, a sample unit and six electrochemical sensors might be interfaced to produce a field-portable device that combines detection, recognition, and separation functions into one unit. In conclusion, electrochemical instruments may prove to be inexpensive, user-friendly platforms for tracking these newly discovered pollutants in the environment.
10. Conclusions
MPs are a part of everyday life everywhere on Earth, from the Arctic to the ocean floor. There has been a great deal of research done on the dangers that MPs present to the environment and to people. This paper describes the present status of electrochemical approaches for MP detection. To show the different domains in which electrochemistry can be used as an alternative technique for MP field detection, examples of applications have been given. The detection of MPs and their concentrations in samples could be sped up with affordable, field-proven electrochemical techniques. These methods make it possible for extensive research to determine MP concentrations and to track and control the dangers associated with these pollutants' existence in the environment. They can also be employed as an adjunct to existing laboratory apparatus. The advantages of electrochemical methods include high sensitivity, low operating costs, and simplicity of usage. There are several self-powered instruments and disposable, one-time electrodes available on the market that provide excellent platforms for efficient, affordable, and onsite contamination detection in decentralized labs. The development of new materials, microfabrication, and additive manufacturing methods—like 3D printing and inkjet printing—has improved the selectivity, stability, and shelf life of the sensors, as well as their capacity for detection. Even with all of these advancements, a few more improvements are needed to take this technology from the lab to the real world. The advent of electrochemical technologies has facilitated the creation of instruments for cleaning, sensors for monitoring MPs, and basic knowledge about MPs. Thus, electrochemical methods are useful instruments that have the potential to revolutionize our comprehension of MPs in the environment and streamline the identification and elimination of MPs.Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Large Groups Project under grant number (RGP.2/335/44).References
- J.-Q. Jiang, Sustain. Prod. Consum., 2018, 13, 16–23 CrossRef.
- M. Eriksen, L. C. M. Lebreton, H. S. Carson, M. Thiel, C. J. Moore, J. C. Borerro, F. Galgani, P. G. Ryan and J. Reisser, PLoS One, 2014, 9, e111913 CrossRef PubMed.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- M. A. Browne, P. Crump, S. J. Niven, E. Teuten, A. Tonkin, T. Galloway and R. Thompson, Environ. Sci. Technol., 2011, 45, 9175–9179 CrossRef CAS PubMed.
- Z. Long, Z. Pan, W. Wang, J. Ren, X. Yu, L. Lin, H. Lin, H. Chen and X. Jin, Water Res., 2019, 155, 255–265 CrossRef CAS PubMed.
- F. Murphy, C. Ewins, F. Carbonnier and B. Quinn, Environ. Sci. Technol., 2016, 50, 5800–5808 CrossRef CAS PubMed.
- L. Jin, G. Zhang and H. Tian, Water Res., 2014, 66, 85–98 CrossRef CAS PubMed.
- S. A. Carr, J. Liu and A. G. Tesoro, Water Res., 2016, 91, 174–182 CrossRef CAS PubMed.
- J. Sun, X. Dai, Q. Wang, M. C. M. van Loosdrecht and B.-J. Ni, Water Res., 2019, 152, 21–37 CrossRef CAS PubMed.
- A. D. Vethaak and J. Legler, Science, 2021, 371, 672–674 CrossRef CAS PubMed.
- F. J. Kelly and J. C. Fussell, Philos. Trans. R. Soc., A, 2020, 378, 20190322 CrossRef CAS PubMed.
- A. A. Koelmans, N. H. M. Nor, E. Hermsen, M. Kooi, S. M. Mintenig and J. De France, Water Res., 2019, 155, 410–422 CrossRef CAS PubMed.
- S. L. Wright, J. Ulke, A. Font, K. L. A. Chan and F. J. Kelly, Environ. Int., 2020, 136, 105411 CrossRef CAS PubMed.
- A. I. Catarino, V. Macchia, W. G. Sanderson, R. C. Thompson and T. B. Henry, Environ. Pollut., 2018, 237, 675–684 CrossRef CAS PubMed.
- D. Li, Y. Shi, L. Yang, L. Xiao, D. K. Kehoe, Y. K. Gun’ko, J. J. Boland and J. J. Wang, Nat. Food, 2020, 1, 746–754 CrossRef CAS PubMed.
- B. Koelmans, S. Pahl, T. Backhaus, F. Bessa, G. van Calster, N. Contzen, R. Cronin, T. Galloway, A. Hart and L. Henderson, A Scientific Perspective on Microplastics in Nature and Society, SAPEA, 2019 Search PubMed.
- C. Q. Y. Yong, S. Valiyaveettil and B. L. Tang, Int. J. Environ. Res. Public Health, 2020, 17, 1509 CrossRef CAS PubMed.
- S. L. Wright and F. J. Kelly, Environ. Sci. Technol., 2017, 51, 6634–6647 CrossRef CAS PubMed.
- M. M. Gruber, B. Hirschmugl, N. Berger, M. Holter, S. Radulović, G. Leitinger, L. Liesinger, A. Berghold, E. Roblegg and R. Birner-Gruenberger, J. Nanobiotechnol., 2020, 18, 1–14 CrossRef PubMed.
- M. Prüst, J. Meijer and R. H. S. Westerink, Part. Fibre Toxicol., 2020, 17, 1–16 CrossRef PubMed.
- N. L. Fahrenfeld, G. Arbuckle-Keil, N. N. Beni, S. L. Bartelt-Hunt, V. Hidalgo-Ruz, L. Gutow, R. C. Thompson and M. Thiel, TrAC, Trends Anal. Chem., 2012, 46, 248–254 Search PubMed.
- V. Hidalgo-Ruz, L. Gutow, R. C. Thompson and M. Thiel, Environ. Sci. Technol., 2012, 46, 3060–3075 CrossRef CAS PubMed.
- M. Filella, Environ. Chem., 2015, 12, 527–538 CrossRef CAS.
- J. S. Hanvey, P. J. Lewis, J. L. Lavers, N. D. Crosbie, K. Pozo and B. O. Clarke, Anal. Methods, 2017, 9, 1369–1383 RSC.
- J.-L. Xu, K. V Thomas, Z. Luo and A. A. Gowen, TrAC, Trends Anal. Chem., 2019, 119, 115629 CrossRef CAS.
- E. Fries, J. H. Dekiff, J. Willmeyer, M.-T. Nuelle, M. Ebert and D. Remy, Environ. Sci.: Processes Impacts, 2013, 15, 1949–1956 RSC.
- S. Tianniam, T. Bamba and E. Fukusaki, J. Biosci. Bioeng., 2010, 109, 89–93 CrossRef CAS PubMed.
- E. Dümichen, A.-K. Barthel, U. Braun, C. G. Bannick, K. Brand, M. Jekel and R. Senz, Water Res., 2015, 85, 451–457 CrossRef PubMed.
- A. M. Elert, R. Becker, E. Duemichen, P. Eisentraut, J. Falkenhagen, H. Sturm and U. Braun, Environ. Pollut., 2017, 231, 1256–1264 CrossRef CAS PubMed.
- E. Dümichen, P. Eisentraut, M. Celina and U. Braun, J. Chromatogr. A, 2019, 1592, 133–142 CrossRef PubMed.
- M. Rodríguez Chialanza, I. Sierra, A. Pérez Parada and L. Fornaro, Environ. Sci. Pollut. Res., 2018, 25, 16767–16775 CrossRef PubMed.
- S. Huppertsberg and T. P. Knepper, Anal. Bioanal. Chem., 2018, 410, 6343–6352 CrossRef CAS PubMed.
- E. Dümichen, P. Eisentraut, C. G. Bannick, A.-K. Barthel, R. Senz and U. Braun, Chemosphere, 2017, 174, 572–584 CrossRef PubMed.
- Z. Huang, B. Hu and H. Wang, Environ. Chem. Lett., 2023, 21, 383–401 CrossRef CAS PubMed.
- Y. L. Shishkin, Thermochim. Acta, 2006, 444, 26–34 CrossRef CAS.
- G. Dierkes, T. Lauschke, S. Becher, H. Schumacher, C. Földi and T. Ternes, Anal. Bioanal. Chem., 2019, 411, 6959–6968 CrossRef CAS PubMed.
- Z. Steinmetz, A. Kintzi, K. Muñoz and G. E. Schaumann, J. Anal. Appl. Pyrolysis, 2020, 147, 104803 CrossRef CAS.
- C. A. Peters, E. Hendrickson, E. C. Minor, K. Schreiner, J. Halbur and S. P. Bratton, Mar. Pollut. Bull., 2018, 137, 91–95 CrossRef CAS PubMed.
- M. Funck, A. Yildirim, C. Nickel, J. Schram, T. C. Schmidt and J. Tuerk, MethodsX, 2020, 7, 100778 CrossRef CAS PubMed.
- B. El Hayany, L. El Fels, K. Quénéa, M.-F. Dignac, C. Rumpel, V. K. Gupta and M. Hafidi, J. Environ. Manage., 2020, 275, 111249 CrossRef CAS PubMed.
- R. Becker, K. Altmann, T. Sommerfeld and U. Braun, J. Anal. Appl. Pyrolysis, 2020, 148, 104829 CrossRef CAS.
- M. Majewsky, H. Bitter, E. Eiche and H. Horn, Sci. Total Environ., 2016, 568, 507–511 CrossRef CAS PubMed.
- H. Bitter and S. Lackner, Chemosphere, 2020, 258, 127388 CrossRef CAS PubMed.
- S. Kühn, A. Van Oyen, A. M. Booth, A. Meijboom and J. A. Van Franeker, Chemosphere, 2018, 213, 103–113 CrossRef PubMed.
- Y. K. Song, S. H. Hong, M. Jang, G. M. Han, M. Rani, J. Lee and W. J. Shim, Mar. Pollut. Bull., 2015, 93, 202–209 CrossRef CAS PubMed.
- D. Schymanski, C. Goldbeck, H.-U. Humpf and P. Fürst, Water Res., 2018, 129, 154–162 CrossRef CAS PubMed.
- R. Lenz, K. Enders, C. A. Stedmon, D. M. A. Mackenzie and T. G. Nielsen, Mar. Pollut. Bull., 2015, 100, 82–91 CrossRef CAS PubMed.
- A. Dehaut, L. Hermabessiere and G. Duflos, TrAC, Trends Anal. Chem., 2019, 116, 346–359 CrossRef CAS.
- C. Zarfl, Anal. Bioanal. Chem., 2019, 411, 3743–3756 CrossRef CAS PubMed.
- T. S. Galloway, Y. Dogra, N. Garrett, D. Rowe, C. R. Tyler, J. Moger, E. Lammer, R. Landsiedel, U. G. Sauer and G. Scherer, Environ. Sci.: Nano, 2017, 4, 1981–1997 RSC.
- S. A. Borman, Anal. Chem., 1982, 54, 1021A–1026A CrossRef CAS.
- L. Mai, L.-J. Bao, L. Shi, C. S. Wong and E. Y. Zeng, Environ. Sci. Pollut. Res., 2018, 25, 11319–11332 CrossRef CAS PubMed.
- J. Lee and K.-J. Chae, J. Hazard. Mater., 2021, 403, 124049 CrossRef CAS PubMed.
- H. K. Imhof, J. Schmid, R. Niessner, N. P. Ivleva and C. Laforsch, Limnol. Oceanogr.: Methods, 2012, 10, 524–537 CrossRef CAS.
- C. F. Araujo, M. M. Nolasco, A. M. P. Ribeiro and P. J. A. Ribeiro-Claro, Water Res., 2018, 142, 426–440 CrossRef CAS PubMed.
- M. G. J. Löder, M. Kuczera, S. Mintenig, C. Lorenz and G. Gerdts, Environ. Chem., 2015, 12, 563–581 CrossRef.
- Z. Sobhani, X. Zhang, C. Gibson, R. Naidu, M. Megharaj and C. Fang, Water Res., 2020, 174, 115658 CrossRef CAS PubMed.
- W. Fu, J. Min, W. Jiang, Y. Li and W. Zhang, Sci. Total Environ., 2020, 721, 137561 CrossRef CAS PubMed.
- J. Deng, P. Guo, X. Zhang, H. Su, Y. Zhang, Y. Wu and Y. Li, Mar. Pollut. Bull., 2020, 159, 111482 CrossRef CAS PubMed.
- A. M. Mahon, B. O'Connell, M. G. Healy, I. O'Connor, R. Officer, R. Nash and L. Morrison, Environ. Sci. Technol., 2017, 51, 810–818 CrossRef CAS PubMed.
- A. Vianello, A. Boldrin, P. Guerriero, V. Moschino, R. Rella, A. Sturaro and L. Da Ros, Estuarine, Coastal Shelf Sci., 2013, 130, 54–61 CrossRef CAS.
- A. B. Silva, A. S. Bastos, C. I. L. Justino, J. P. da Costa, A. C. Duarte and T. A. P. Rocha-Santos, Anal. Chim. Acta, 2018, 1017, 1–19 CrossRef CAS PubMed.
- R. Sutton, S. A. Mason, S. K. Stanek, E. Willis-Norton, I. F. Wren and C. Box, Mar. Pollut. Bull., 2016, 109, 230–235 CrossRef CAS PubMed.
- A. R. McCormick, T. J. Hoellein, M. G. London, J. Hittie, J. W. Scott and J. J. Kelly, Ecosphere, 2016, 7, e01556 CrossRef.
- A. S. Tagg, M. Sapp, J. P. Harrison and J. J. Ojeda, Anal. Chem., 2015, 87, 6032–6040 CrossRef CAS PubMed.
- Y. Y. Tsang, C. W. Mak, C. Liebich, S. W. Lam, E. T. P. Sze and K. M. Chan, Mar. Pollut. Bull., 2017, 115, 20–28 CrossRef CAS PubMed.
- S. H. Campbell, P. R. Williamson and B. D. Hall, Facets, 2017, 2, 395–409 CrossRef.
- C. C. Wessel, G. R. Lockridge, D. Battiste and J. Cebrian, Mar. Pollut. Bull., 2016, 109, 178–183 CrossRef CAS PubMed.
- A. Käppler, D. Fischer, S. Oberbeckmann, G. Schernewski, M. Labrenz, K.-J. Eichhorn and B. Voit, Anal. Bioanal. Chem., 2016, 408, 8377–8391 CrossRef PubMed.
- H. K. Imhof, C. Laforsch, A. C. Wiesheu, J. Schmid, P. M. Anger, R. Niessner and N. P. Ivleva, Water Res., 2016, 98, 64–74 CrossRef CAS PubMed.
- A. A. Horton, C. Svendsen, R. J. Williams, D. J. Spurgeon and E. Lahive, Mar. Pollut. Bull., 2017, 114, 218–226 CrossRef CAS PubMed.
- K. Davidson and S. E. Dudas, Arch. Environ. Contam. Toxicol., 2016, 71, 147–156 CrossRef CAS PubMed.
- J. Li, X. Qu, L. Su, W. Zhang, D. Yang, P. Kolandhasamy, D. Li and H. Shi, Environ. Pollut., 2016, 214, 177–184 CrossRef CAS PubMed.
- C. G. Avio, L. R. Cardelli, S. Gorbi, D. Pellegrini and F. Regoli, Environ. Pollut., 2017, 227, 207–214 CrossRef CAS PubMed.
- J. Bellas, J. Martínez-Armental, A. Martínez-Cámara, V. Besada and C. Martínez-Gómez, Mar. Pollut. Bull., 2016, 109, 55–60 CrossRef CAS PubMed.
- A. Dehaut, A.-L. Cassone, L. Frère, L. Hermabessiere, C. Himber, E. Rinnert, G. Rivière, C. Lambert, P. Soudant and A. Huvet, Environ. Pollut., 2016, 215, 223–233 CrossRef CAS PubMed.
- M. Fischer and B. M. Scholz-Böttcher, Environ. Sci. Technol., 2017, 51, 5052–5060 CrossRef CAS PubMed.
- A. Karami, A. Golieskardi, Y. Bin Ho, V. Larat and B. Salamatinia, Sci. Rep., 2017, 7, 5473 CrossRef PubMed.
- S. Aralekallu, M. Palanna, S. Hadimani, K. P. CP, V. A. Sajjan, M. O. Thotiyl and L. K. Sannegowda, Dalton Trans., 2020, 49, 15061–15071 RSC.
- M. Palanna, S. Aralekallu, C. P. K. Prabhu and V. A. Sajjan, Electrochim. Acta, 2021, 367, 137519 CrossRef CAS.
- S. Khaire, P. Gaikwad, S. Aralekallu, Z. M. Bhat, A. R. Kottaichamy, M. C. Devendrachari, R. Thimmappa, S. P. Shafi, M. Gautam and M. O. Thotiyl, ChemElectroChem, 2018, 5, 362–366 CrossRef CAS.
- S. Aralekallu, I. Mohammed, N. Manjunatha, M. Palanna and L. K. Sannegowda, Sens. Actuators, B, 2019, 282, 417–425 CrossRef CAS.
- Z. Meng, R. M. Stolz, L. Mendecki and K. A. Mirica, Chem. Rev., 2019, 119, 478–598 CrossRef CAS PubMed.
- G. T. Chandran, X. Li, A. Ogata and R. M. Penner, Anal. Chem., 2017, 89, 249–275 CrossRef CAS PubMed.
- W. R. de Araujo, S. M. Reddy and T. R. L. C. Paixão, Nanomater. Electrochem. Sens., 2017, 1–5 Search PubMed.
- M. A. Al Mamun and M. R. Yuce, Adv. Funct. Mater., 2020, 30, 2005703 CrossRef.
- M. Pollard, E. Hunsicker and M. Platt, ACS Sens., 2020, 5, 2578–2586 CrossRef CAS PubMed.
- B. C. Colson and A. P. M. Michel, ACS Sens., 2021, 6, 238–244 CrossRef CAS PubMed.
- K. Shimizu, S. V. Sokolov, E. Kätelhön, J. Holter, N. P. Young and R. G. Compton, Electroanalysis, 2017, 29, 2200–2207 CrossRef CAS.
- M. Pollard, R. Maugi and M. Platt, Analyst, 2022, 147, 1417–1424 RSC.
- R. Pan, K. Hu, D. Jiang, U. Samuni and M. V Mirkin, J. Am. Chem. Soc., 2019, 141, 19555–19559 CrossRef CAS PubMed.
- M. G. Blevins, H. L. Allen, B. C. Colson, A.-M. Cook, A. Z. Greenbaum, S. S. Hemami, J. Hollmann, E. Kim, A. A. LaRocca and K. A. Markoski, Sensors, 2021, 21, 3532 CrossRef PubMed.
- D. Spencer and H. Morgan, ACS Sens., 2020, 5, 423–430 CrossRef CAS PubMed.
- F. Luo, F. Chen, Y. Xiong, Z. Wu, X. Zhang, W. Wen and S. Wang, Anal. Chem., 2021, 93, 4506–4512 CrossRef CAS PubMed.
- T. M. B. F. Oliveira, F. W. P. Ribeiro, S. Morais, P. de Lima-Neto and A. N. Correia, Curr. Opin. Electrochem., 2022, 31, 100866 CrossRef CAS.
- C. D. Davies and R. M. Crooks, Chem. Sci., 2020, 11, 5547–5558 RSC.
- S. Wang, M. Xu, B. Jin, U. J. Wünsch, Y. Su and Y. Zhang, Water Res., 2022, 211, 118046 CrossRef CAS PubMed.
- C. Akarsu, H. Kumbur and A. E. Kideys, Water Sci. Technol., 2021, 84, 1648–1662 CrossRef CAS PubMed.
- D. Elkhatib, V. Oyanedel-Craver and E. Carissimi, Sep. Purif. Technol., 2021, 276, 118877 CrossRef CAS.
- M. Shen, Y. Zhang, E. Almatrafi, T. Hu, C. Zhou, B. Song, Z. Zeng and G. Zeng, Chem. Eng. J., 2022, 428, 131161 CrossRef CAS.
- L. Weirauch, M. Lorenz, N. Hill, B. H. Lapizco-Encinas, M. Baune, G. R. Pesch and J. Thöming, Biomicrofluidics, 2019, 13, 064112 CrossRef CAS PubMed.
- W. Perren, A. Wojtasik and Q. Cai, ACS Omega, 2018, 3, 3357–3364 CrossRef CAS PubMed.
- M. Kiendrebeogo, M. R. K. Estahbanati, A. K. Mostafazadeh, P. Drogui and R. D. Tyagi, Environ. Pollut., 2021, 269, 116168 CrossRef CAS PubMed.
- M. Y. Ali, A. U. Alam and M. M. R. Howlader, Sens. Actuators, B, 2020, 320, 128319 CrossRef CAS.
- R. Xu, Z. Yang, Y. Niu, D. Xu, J. Wang, J. Han and H. Wang, Sep. Purif. Technol., 2022, 290, 120905 CrossRef CAS.
- Z. Li, J. Hu, Y. Xiao, Q. Zha, L. Zeng and M. Zhu, Anal. Chim. Acta, 2021, 1146, 174–183 CrossRef CAS PubMed.
- A. R. Boccaccini, S. Keim, R. Ma, Y. Li and I. Zhitomirsky, J. R. Soc., Interface, 2010, 7, S581–S613 CrossRef CAS PubMed.
- L. Dörner, P. Schmutz, R. Kägi, M. V Kovalenko and L. P. H. Jeurgens, Langmuir, 2020, 36, 8075–8085 CrossRef PubMed.
- Y. Park, H. Kang, W. Jeong, H. Son and D.-H. Ha, Nanomaterials, 2021, 11, 133 CrossRef CAS PubMed.
- S. C. Mills, C. S. Smith, D. P. Arnold and J. S. Andrew, AIP Adv., 2019, 9, 125028 CrossRef.
- A. Rousta, D. Dorranian and M. Elahi, Surf. Eng., 2020, 36, 919–928 CrossRef CAS.
- M. Verde, M. Peiteado, A. C. Caballero, M. Villegas and B. Ferrari, J. Colloid Interface Sci., 2012, 373, 27–33 CrossRef CAS.
- J. Bandy, Q. Zhang and G. Cao, Mater. Sci. Appl., 2011, 2, 1427–1431 CAS.
- B. P. Singh, S. Nayak, K. K. Nanda, B. K. Jena, S. Bhattacharjee and L. Besra, Carbon, 2013, 61, 47–56 CrossRef CAS.
- Y. J. Kwon, Y. Kim, H. Jeon, S. Cho, W. Lee and J. U. Lee, Composites, Part B, 2017, 122, 23–30 CrossRef CAS.
- Y. Yang, J. Li, D. Chen, T. Fu, D. Sun and J. Zhao, ChemElectroChem, 2016, 3, 757–763 CrossRef CAS.
- J. A. Quezada-Rentería, L. F. Cházaro-Ruiz and J. R. Rangel-Mendez, Carbon, 2017, 122, 266–275 CrossRef.
- C.-Y. Ho, S.-M. Huang, S.-T. Lee and Y.-J. Chang, Appl. Surf. Sci., 2019, 477, 226–231 CrossRef CAS.
- W. Liu, Y. Feng, X. Yan, J. Chen and Q. Xue, Adv. Funct. Mater., 2013, 23, 4111–4122 CrossRef CAS.
- M. Atiq Ur Rehman, Q. Chen, A. Braem, M. S. P. Shaffer and A. R. Boccaccini, Int. Mater. Rev., 2021, 66, 533–562 CrossRef CAS.
- J. Ding, X. Yan and Q. Xue, Mater. Chem. Phys., 2012, 133, 405–409 CrossRef CAS.
- J. Yang, X. Yan, J. Chen, H. Ma, D. Sun and Q. Xue, RSC Adv., 2012, 2, 9665–9670 RSC.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C. Kung, M. J. Katz, M. So, B. Klahr, D. Jin and Y. Chung, Adv. Mater., 2014, 26, 6295–6300 CrossRef CAS PubMed.
- J. Feng, X. Yang, S. Gao, J. Shi and R. Cao, Langmuir, 2017, 33, 14238–14243 CrossRef CAS PubMed.
- M. A. Borysiewicz, J.-H. Dou, I. Stassen and M. Dincă, Faraday Discuss., 2021, 231, 298–304 RSC.
- N. Pan, H. Zhang, B. Yang, H. Qiu, L. Li, L. Song and M. Zhang, Chem. Commun., 2020, 56, 13615–13618 RSC.
- D. K. Nguyen, I. M. Schepisi and F. Z. Amir, Chem. Eng. J., 2019, 378, 122150 CrossRef.
- J. J. Richardson, M. Björnmalm and F. Caruso, science, 2015, 348, aaa2491 CrossRef PubMed.
- A. A. Mamedov, N. A. Kotov, M. Prato, D. M. Guldi, J. P. Wicksted and A. Hirsch, Nat. Mater., 2002, 1, 190–194 CrossRef CAS PubMed.
- K. Fujimoto, S. Fujita, B. Ding and S. Shiratori, Jpn. J. Appl. Phys., 2004, 44, L126 CrossRef.
- J. B. Schlenoff, S. T. Dubas and T. Farhat, Langmuir, 2000, 16, 9968–9969 CrossRef CAS.
- A. Qi, P. Chan, J. Ho, A. Rajapaksa, J. Friend and L. Yeo, ACS Nano, 2011, 5, 9583–9591 CrossRef CAS PubMed.
- X. Hong, J. Li, M. Wang, J. Xu, W. Guo, J. Li, Y. Bai and T. Li, Chem. Mater., 2004, 16, 4022–4027 CrossRef CAS.
- G. Rydzek, J.-S. Thomann, N. Ben Ameur, L. Jierry, P. Mésini, A. Ponche, C. Contal, A. E. El Haitami, J.-C. Voegel and B. Senger, Langmuir, 2010, 26, 2816–2824 CrossRef CAS.
- D. O. Grigoriev, T. Bukreeva, H. Möhwald and D. G. Shchukin, Langmuir, 2008, 24, 999–1004 CrossRef CAS.
- D. Chen, A. E. Sedykh, G. E. Gomez, B. L. Neumeier, J. C. C. Santos, V. Gvilava, R. Maile, C. Feldmann, C. WÖll and C. Janiak, Adv. Mater. Interfaces, 2020, 7, 2000929 CrossRef CAS.
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC.
- S. Okur, Z. Zhang, M. Sarheed, P. Nick, U. Lemmer and L. Heinke, Sens. Actuators, B, 2020, 306, 127502 CrossRef CAS.
- Y.-H. Xiao, Z.-G. Gu and J. Zhang, Nanoscale, 2020, 12, 12712–12730 RSC.
- S. Srivastava and N. A. Kotov, Acc. Chem. Res., 2008, 41, 1831–1841 CrossRef CAS.
- I. Shakir, Z. Ali and D. J. Kang, J. Alloys Compd., 2014, 617, 707–712 CrossRef CAS.
- Z. Liu, L. Bai, G. Zhao and Y. Liu, Beilstein J. Nanotechnol., 2016, 7, 1028–1032 CrossRef CAS PubMed.
- J. J. Richardson, J. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- M. P. Browne, E. Redondo and M. Pumera, Chem. Rev., 2020, 120, 2783–2810 CrossRef CAS PubMed.
- T. Chu, S. Park and K. Fu, Carbon Energy, 2021, 3, 424–439 CrossRef.
- F. Zhang, M. Wei, V. V Viswanathan, B. Swart, Y. Shao, G. Wu and C. Zhou, Nano Energy, 2017, 40, 418–431 CrossRef CAS.
- X. Su, X. Li, C. Y. A. Ong, T. S. Herng, Y. Wang, E. Peng and J. Ding, Advanced Science, 2019, 6, 1801670 CrossRef.
- C. W. Foster, M. P. Down, Y. Zhang, X. Ji, S. J. Rowley-Neale, G. C. Smith, P. J. Kelly and C. E. Banks, Sci. Rep., 2017, 7, 42233 CrossRef PubMed.
- A. Azhari, E. Marzbanrad, D. Yilman, E. Toyserkani and M. A. Pope, Carbon, 2017, 119, 257–266 CrossRef CAS.
- S. Ammu, V. Dua, S. R. Agnihotra, S. P. Surwade, A. Phulgirkar, S. Patel and S. K. Manohar, J. Am. Chem. Soc., 2012, 134, 4553–4556 CrossRef CAS.
- K. S. Bhat, U. T. Nakate, J.-Y. Yoo, Y. Wang, T. Mahmoudi and Y.-B. Hahn, ACS Omega, 2019, 4, 8373–8380 CrossRef CAS PubMed.
- M. Bariya, Z. Shahpar, H. Park, J. Sun, Y. Jung, W. Gao, H. Y. Y. Nyein, T. S. Liaw, L.-C. Tai and Q. P. Ngo, ACS Nano, 2018, 12, 6978–6987 CrossRef CAS PubMed.
- G. R. Cagnani, G. Ibanez-Redin, B. Tirich, D. Gonçalves, D. T. Balogh and O. N. Oliveira Jr, Biosens. Bioelectron., 2020, 165, 112428 CrossRef CAS.
- M. H. Hassan, R. Khan and S. Andreescu, Electrochem. Sci. Adv., 2022, 2, e2100184 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |