DOI:
10.1039/D3RA06812A
(Paper)
RSC Adv., 2024,
14, 1018-1033
Novel pyrazoline linked acyl thiourea pharmacophores as antimicrobial, urease, amylase and α-glucosidase inhibitors: design, synthesis, SAR and molecular docking studies†
Received
9th October 2023
, Accepted 5th December 2023
First published on 2nd January 2024
Abstract
In the present work, a small library of novel pyrazolinyl-acyl thiourea (5a–j) was designed and synthesized through a multistep sequence and the synthesized compounds were screened for their antifungal, antibacterial and antioxidant activities as well as urease, amylase and α-glucosidase inhibitory activities. The synthesized series (5a–o) was characterized using a combination of spectroscopic techniques, including FT-IR, 1H NMR and 13C NMR. All compounds (5a–j) were found to have significant potency against urease, α-glucosidase, α-amylase, and DPPH. The synthesized compounds were also screened for potential antibacterial and anti-fungal inhibition activities. IC50 values for all the prepared compounds for urease, α-glucosidase, amylase, and DPPH inhibition were determined and derivatives 5b and 5g were found to be the most potent urease inhibitors with IC50 values of 54.2 ± 0.32 and 43.6 ± 0.25 μM, respectively. Whilst compound 5b (IC50 = 68.3 ± 0.11 μM) is a potent α-glucosidase inhibitor, compound 5f (90.3 ± 1.08 μM) is a potent amylase inhibitor and compound 5b (103.4 ± 1.15 μM) is a potent antioxidant. The different substitutions on the phenyl ring were the basis for structure–activity relationship (SAR) study. The molecular docking study was performed for the confirmation of binding interactions.
1. Introduction
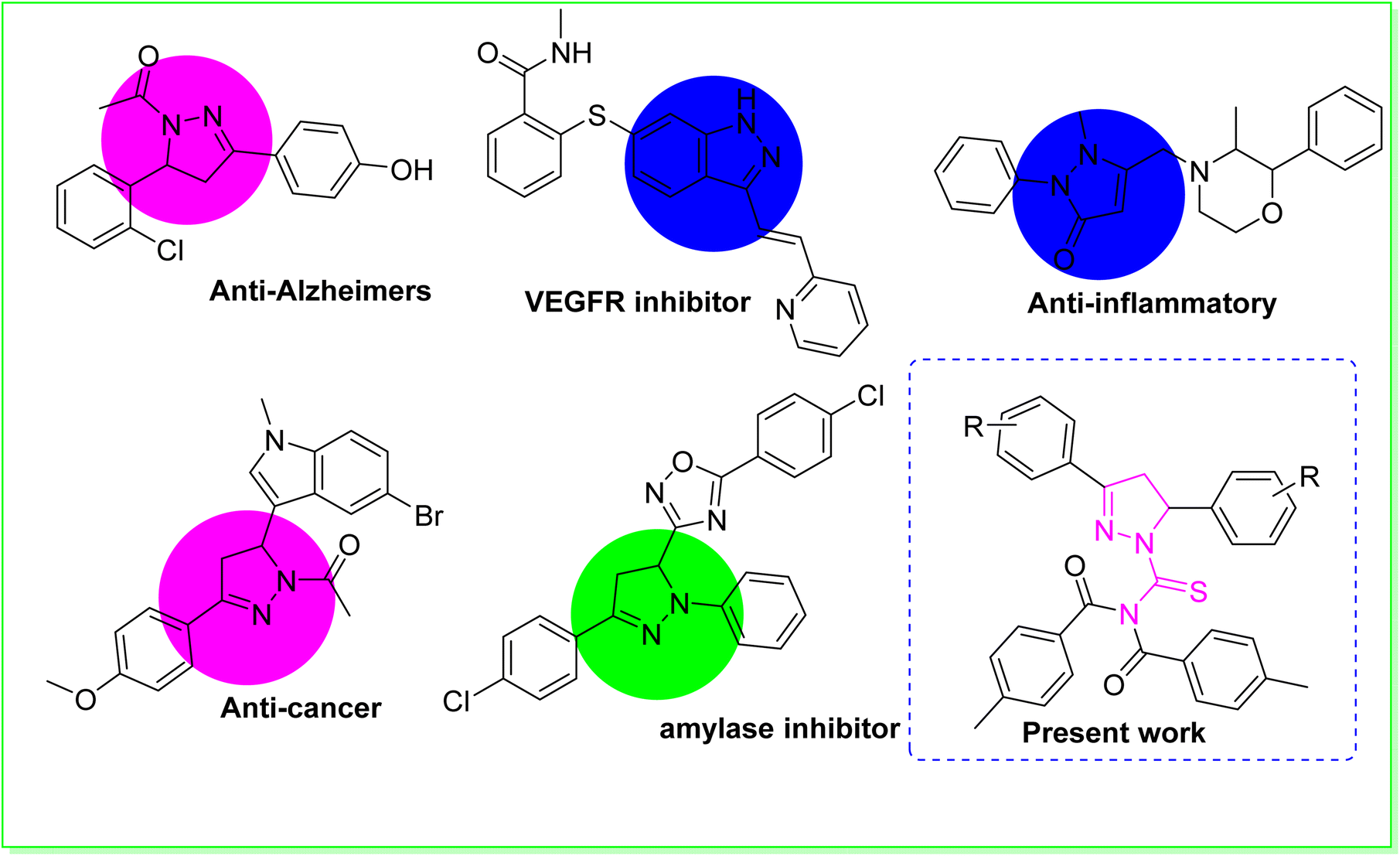
Heterocyclic compounds are the most important category of organic compounds. All biological processes are mainly regulated by organic molecules with a heterocyclic structure.1 In the quest to create novel compounds with vibrant, versatile, and useful properties, researchers discovered that an elegant member of the heterocyclic family known as pyrazoline was intriguing.2 Consequently, a diverse range of molecular hybrids containing the pyrazoline and urea moiety have been successfully conceived and synthesized.3 As a result, pyrazolines are widely incorporated into the structures of various essential medicinal and biological compounds as an appreciated scaffold.4 Structure–activity studies on pyrazoline-based compounds indicated that modifications in the pyrazoline scaffold's substitution pattern have an enormous effect on the biological profile.5 Numerous scientists in the area of medicinal chemistry have performed extensive studies on pyrazoline scaffolds for curing a range of diseases.6 Pyrazolines possess diverse biological activities, including anti-cancer,7 anti-Alzheimer,8 anti-tuberculosis,9 anti-inflammatory,10 antimicrobial,11 anti-EGFR12 and anti-Parkinsonian.13 The pyrazoline ring system is an important structural property of numerous drugs used in clinical trials to treat various disorders.14 Pyrazoline-based pharmaceutical agents, such as phenazone, axitinib, YC-1, ibipinabant, morazone and sulfinpyrazone are utilised as medications.15 One of the most intriguing scaffolds are 1-acetyl-pyrazolines. These scaffolds were found to display anticancer and anti-inflammatory properties. The use of 1-acetyl-pyrazoline derivatives as a monoamine oxidase inhibitor has been reported to be effective in treating Alzheimer's disease (AD),16 which is responsible for the majority of dementia cases diagnosed above the age of 60,17 and also inhibiting kinesin spindle protein (KSP), with potential use in the treatment of cancer.18 In addition to their outstanding antioxidant, anti-inflammatory, antifungal, analgesic, antipyretic, antibacterial, antiangiogenic, antiviral, and antitumoral effects, pyrazolinic scaffolds have an extensive number of uses in pharmacology. Enzymes have become the most important biological targets for therapeutic interventions owing to their regulatory effects in gluconeogenesis and glycogenolysis processes.19 Among many enzymes, α-amylase is a crucial carbohydrate digesting enzyme that is also involved in the hydrolysis of α-(1,4)-glycosidic bonds in starch.20 As a result, α-amylase is regarded as one of the most promising biological targets for the development of type II diabetes treatment drugs.21 Urease (urea amidohydrolase, EC 3.5.1.5) is found across numerous plants, animals, fungi and bacteria.22 Urease is a metallo enzyme that hydrolyses urea to generate carbon dioxide and carbamate subsequently bursting into ammonia gas. The excessive release of ammonia permits Helicobacter pylori to flourish in the stomach. Helicobacter pylori is hazardous to human health because it causes a variety of stomach-related illnesses such as pyelonephritis, hepatic coma, peptic ulcers, and kidney stone production.23 Acyl substituted thiourea substances are efficient reagents used to synthesize a wide range of heterocyclic and organosulfur compounds.24 Acyl thiourea derivatives, on the other hand, exhibit a diverse range of medicinal characteristics, including antifungal,25 anti-tuberculosis, antimicrobial,26 antioxidant, antibacterial,27 anticancer,28 anti-phenoloxidase and rodenticidal activities.29 Many of these applications rely on the structural and conformational features of 1-acyl thiourea. The formation of appropriate hydrogen bonds with specific receptors in particular is a critical factor that plays a role in many fields, including analytical applications of 1-acyl thiourea as a chemo sensor for selective and sensitive naked-eye recognition of anions, as well as chemical biology and drug design.30 The aforesaid biological and synthetic significance of thiourea on one hand and the multifunctional value of the pyrazoline moiety in drug design on the other prompted us to synthesize some new pyrazoline hybrid acyl thiourea conjugates.31
Our research group is currently working on the designing of simple synthetic methods to synthesize low molecular weight compounds due to their significant inhibitory impact on urease, α-amylase, and α-glucosidase enzymes. In the present research work, we discuss the synthesis, identification, and biological evaluation of novel pyrazoline hybrid acyl thiourea pharmacophores as ternary inhibitors of urease, α-amylase, and α-glucosidase enzymes, which were initiated by the biological relevance of pyrazoline structural motifs in medicinal chemistry. The urease, amylase, and glucosidase inhibitory effects of pyrazoline compounds synthesised via different substitution patterns were investigated. Furthermore, molecular docking studies were performed to confirm binding interactions (Fig. 1).
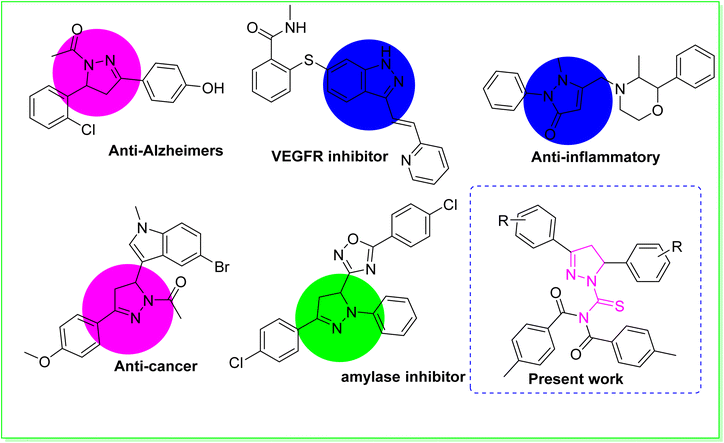 |
| | Fig. 1 Some previously reported biologically potent compounds. | |
2. Results and discussion
2.1. Chemistry
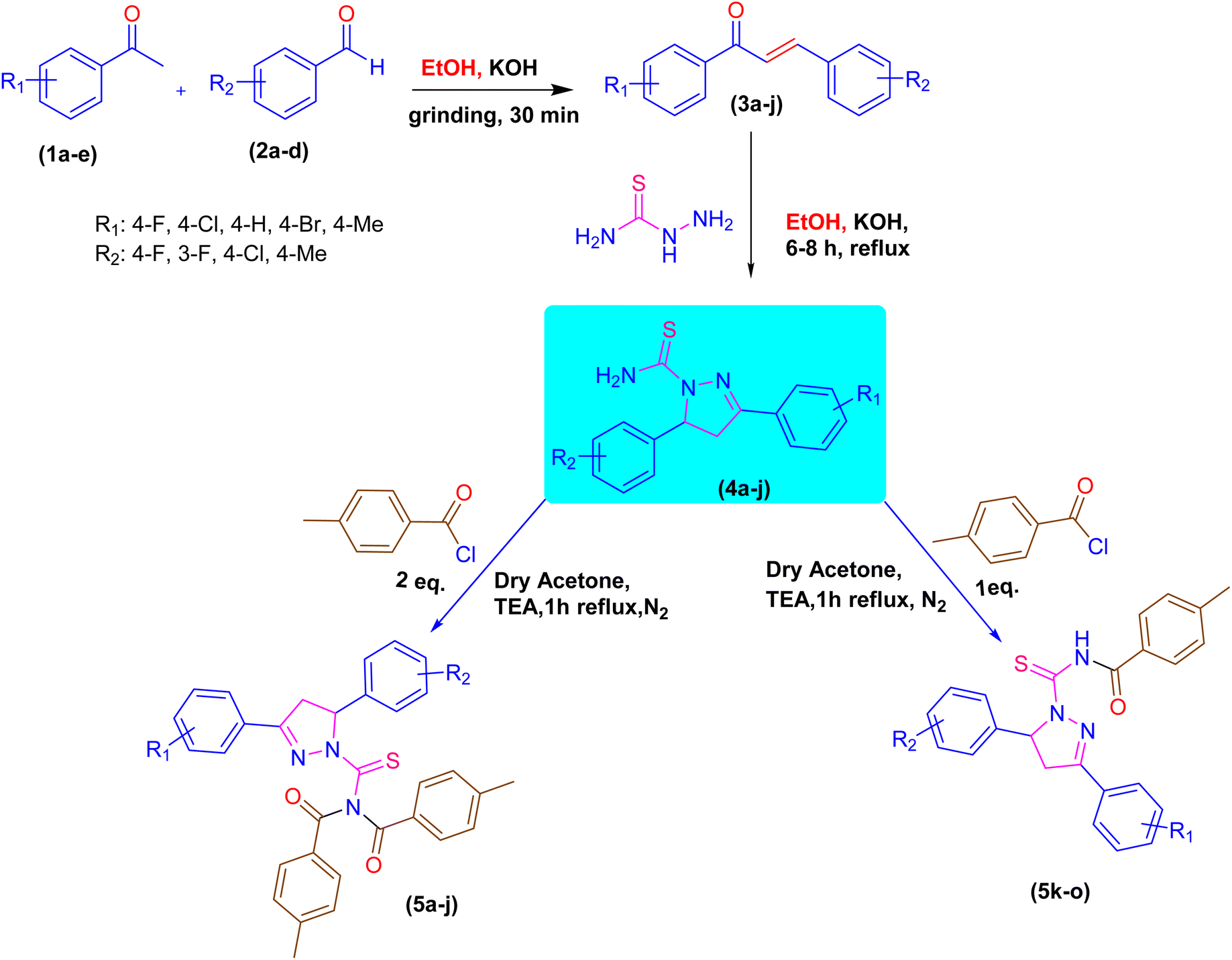
A novel class of pyrazoline hybrid acyl thiourea derivatives (5a–o) were synthesized via three step synthesis. These three steps include condensation, cyclization, and substitution. First, mechanochemical synthesis of chalcone derivatives was carried using aldol condensation reaction between differently substituted aldehydes and acetophenones using a mortar and pestle. Next, chalcones were converted into thiourea based pyrazoline derivatives by cyclization reaction between chalcones and thiosemicarbazide in an excellent yield. Finally, a substitution reaction using p-tolyl chloride was performed, which resulted in the formation of N,N-diacyl pyrazoline thioureas (5a–j) or N-acyl pyrazoline thioureas (5k–o) by using the acid chloride in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 molar ratio, respectively. The derivatives 5b and 5i have a fluoro substituent on the para and meta positions of the phenyl ring and show a moderate yield of 65% when compared to other derivatives. The methyl substituent on the para position of the phenyl ring in derivative 5h was produced with an outstanding yield of 80%. Overall, this is a synthetic method that involves a number of steps, all of which are high yielding and clean. The products showed up as a single spot on the TLC plate under UV and stain, and column chromatography was not required to purify the compounds. The synthetic pathway used to synthesize the target molecules is depicted in Scheme 1.
:1 and 1:1 molar ratio, respectively. The derivatives 5b and 5i have a fluoro substituent on the para and meta positions of the phenyl ring and show a moderate yield of 65% when compared to other derivatives. The methyl substituent on the para position of the phenyl ring in derivative 5h was produced with an outstanding yield of 80%. Overall, this is a synthetic method that involves a number of steps, all of which are high yielding and clean. The products showed up as a single spot on the TLC plate under UV and stain, and column chromatography was not required to purify the compounds. The synthetic pathway used to synthesize the target molecules is depicted in Scheme 1.
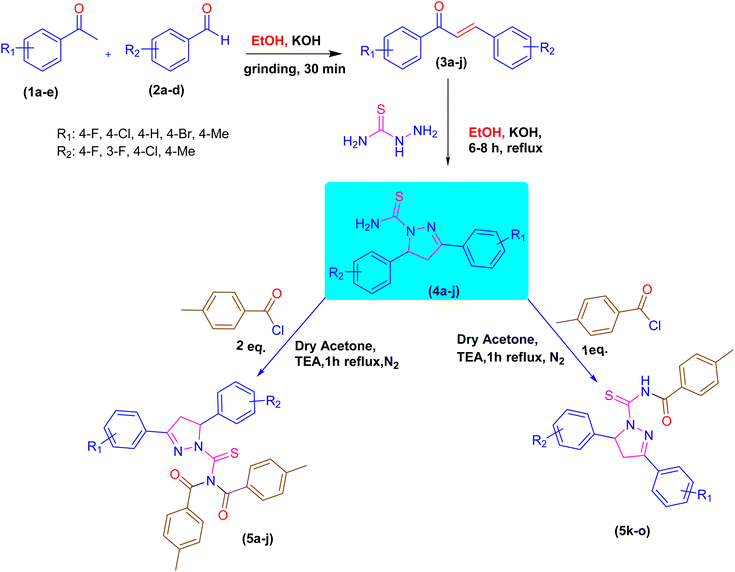 |
| | Scheme 1 Synthetic pathway of pyrazoline acyl thiourea hybrid pharmacophores. | |
The structure of all novel synthesized pyrazoline acyl thiourea-hybrid pharmacophores was investigated via spectroscopic analysis, such as 1H-NMR, 13C-NMR and FTIR. The synthesized compounds were also analysed via FTIR spectroscopy. The two strong peaks at 3050 cm−1 and 2948 cm−1 were assigned to C sp2–H and C sp3–H stretching of aromatic C–H and aliphatic C–H, respectively. The presence of the carbonyl group was confirmed by appearance of an absorption peak around 1694 cm−1. The peak around 1601 cm−1 indicates the presence of an aromatic part in the structure. The peak around 1209 cm−1 was assigned to the thiocarbonyl functional group. In 1H-NMR, three characteristic signals of ABX coupling of three pyrazoline diastereotopic protons (double doublet), proton Hx signal at δ 3.88 and proton Ha and Hb at δ 5.92 and δ 3.20, appeared, respectively. One of the diastereotopic protons Ha was deshielded due to the magnetic anisotropy of the neighbouring double bond, which is in the plane of this double bond, while the other proton lacks this anisotropic effect. The two shielded methyl groups showed a six protons singlet at δ 2.40. The other aromatic protons in the structures appeared in their characteristic regions. The signal of protons attached to sp2 hybridized carbons of the aromatic ring appeared in their respective region ranging from δ 7.60 to 6.95 observed in compound 5a. In 13C-NMR, the signal of the thiocarbonyl group appeared at δ 175.37 and carbonyl group at δ 171.81. Carbon of thiocarbonyl is more deshielded than the carbonyl group since the p orbital overlap between carbon and sulphur is less efficient in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S pi band due to size disparity between C and S as compared to C and O. Hence the resonance contributing structure where carbon carries a positive charge is more prevalent in thiocarbonyl as compared to the carbonyl group. The signals at δ 166.40–115.60 were assigned to sp2 hybridized carbon of the aromatic ring, and other sp3 carbon signals appeared at their respective region δ 43.02–21.77 (Fig. 2).
S pi band due to size disparity between C and S as compared to C and O. Hence the resonance contributing structure where carbon carries a positive charge is more prevalent in thiocarbonyl as compared to the carbonyl group. The signals at δ 166.40–115.60 were assigned to sp2 hybridized carbon of the aromatic ring, and other sp3 carbon signals appeared at their respective region δ 43.02–21.77 (Fig. 2).
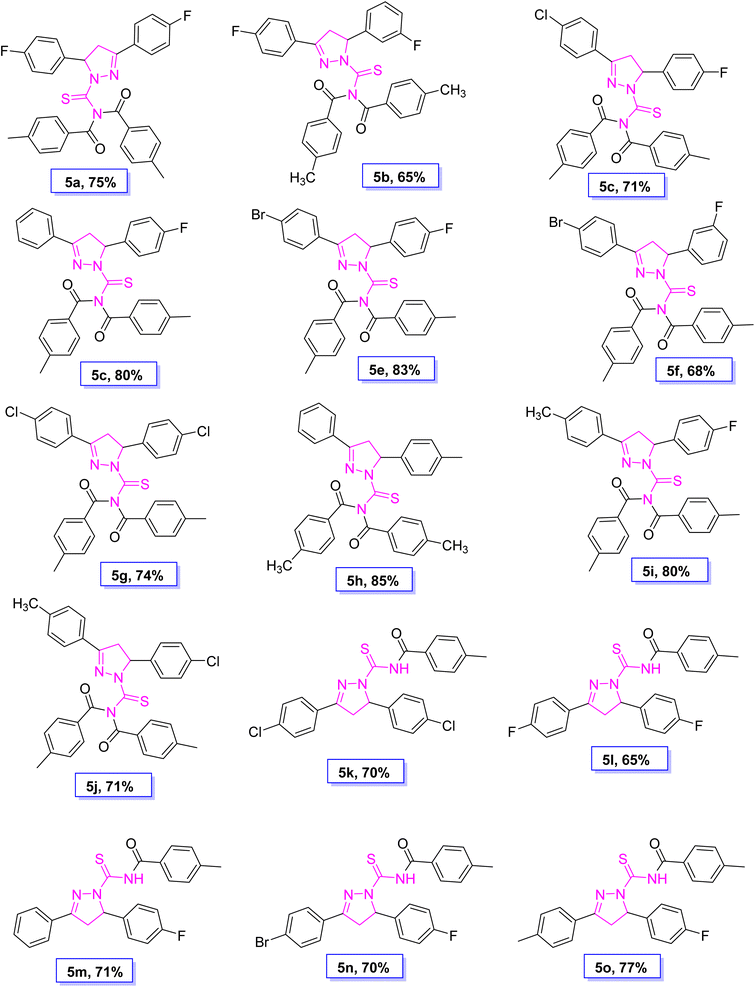 |
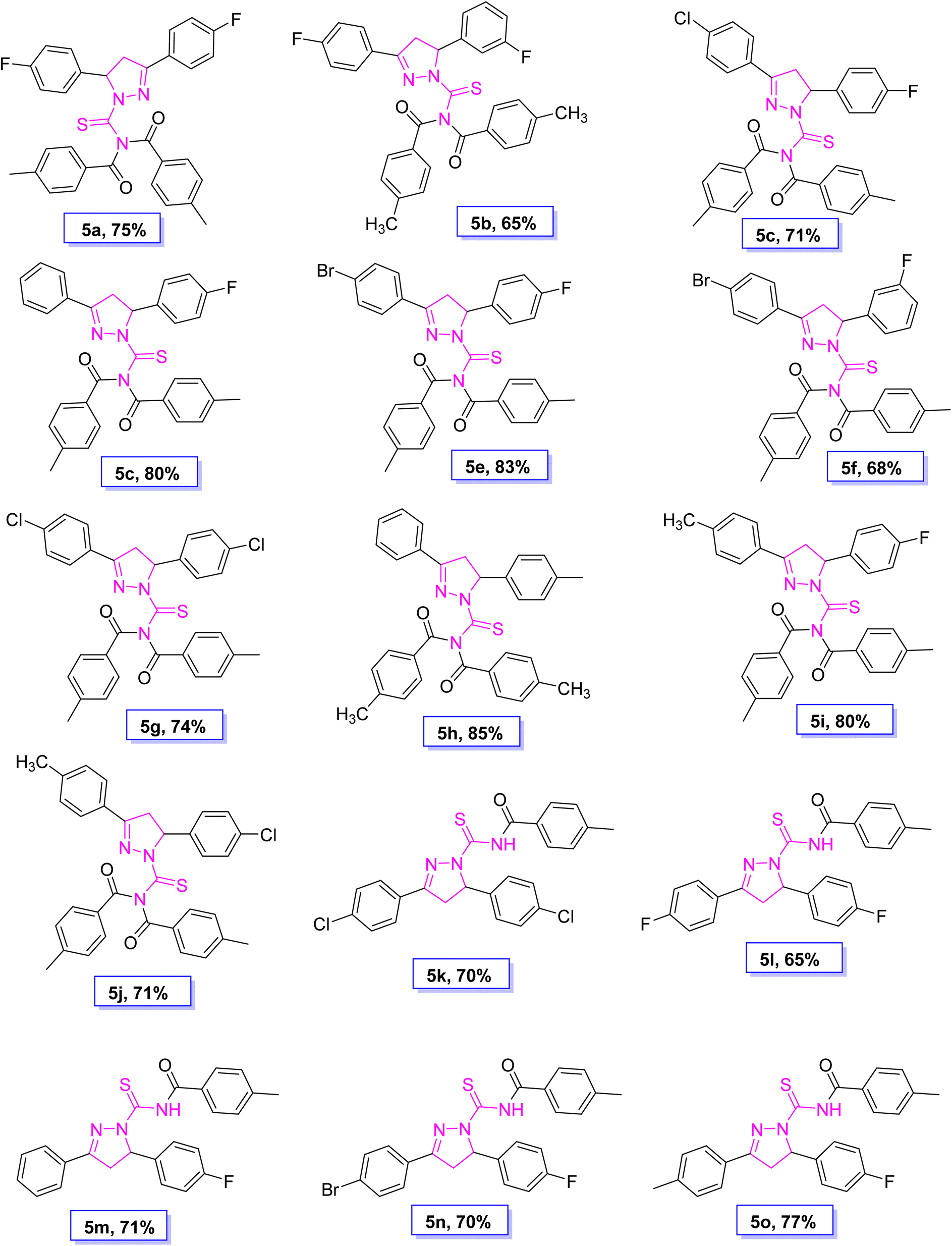
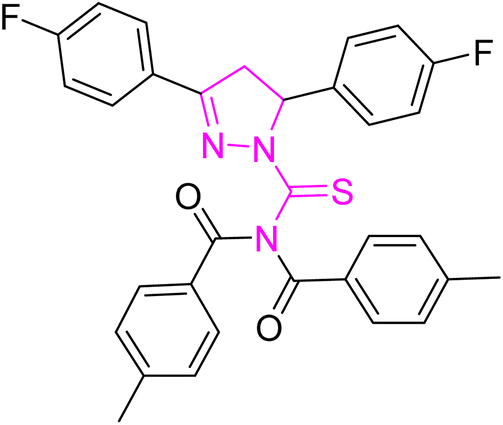
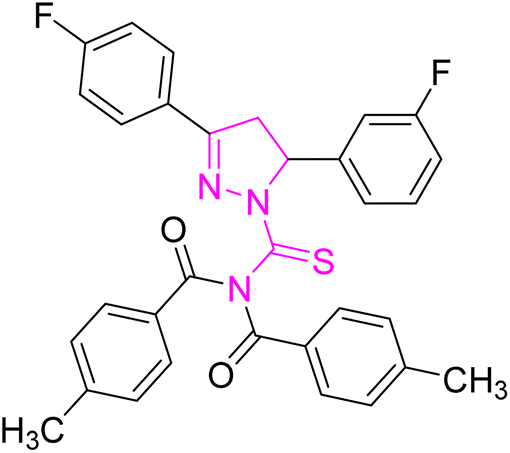
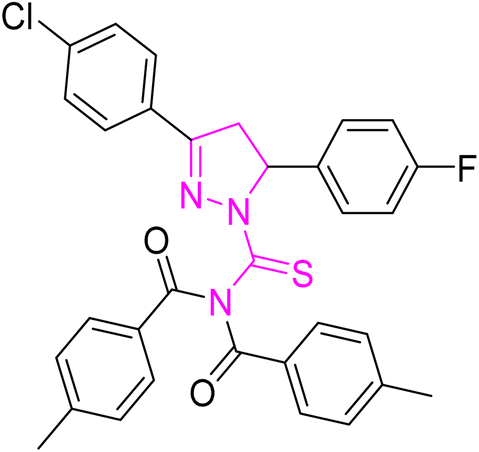
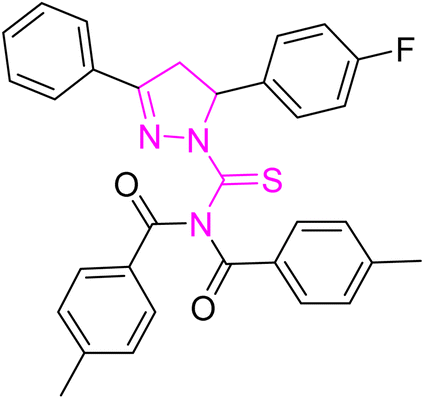
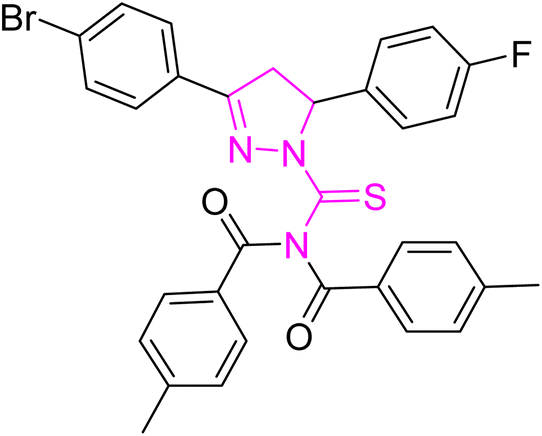
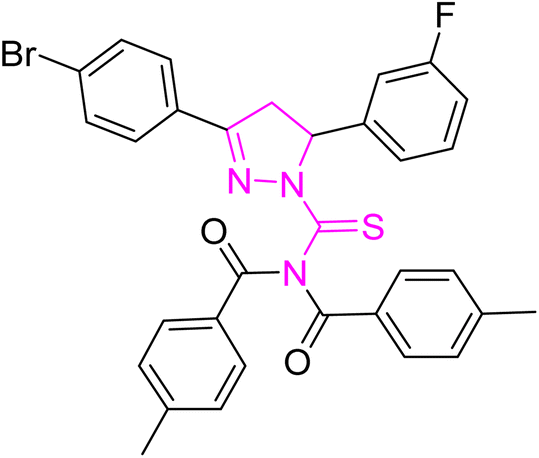
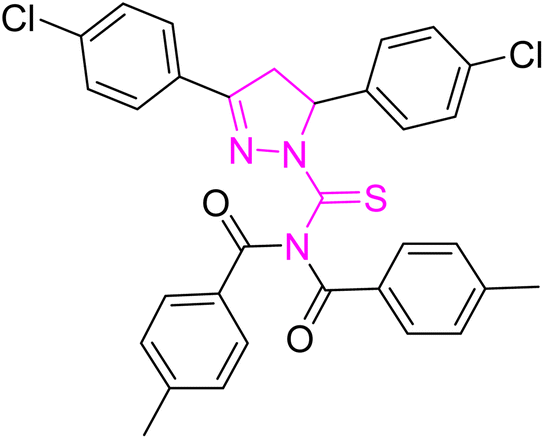
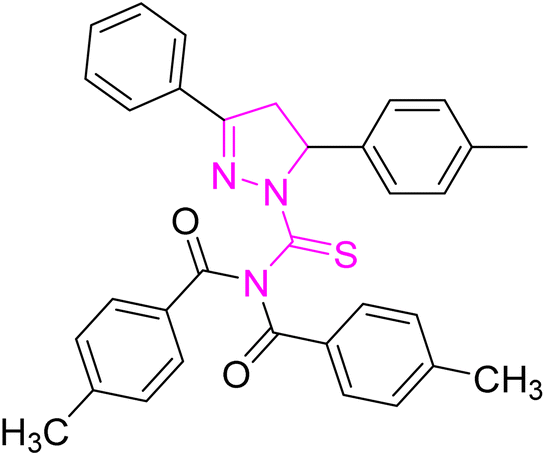
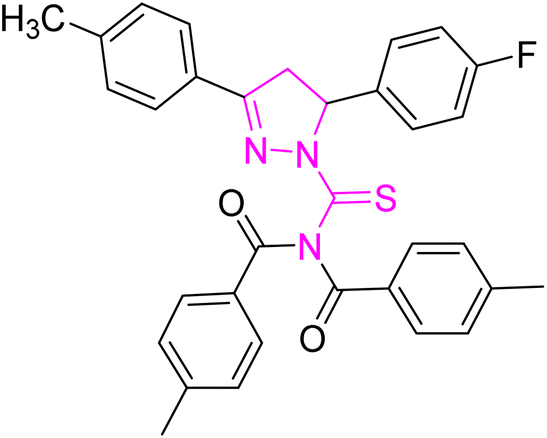
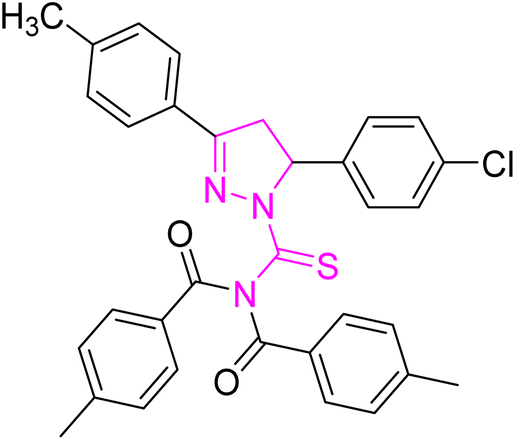
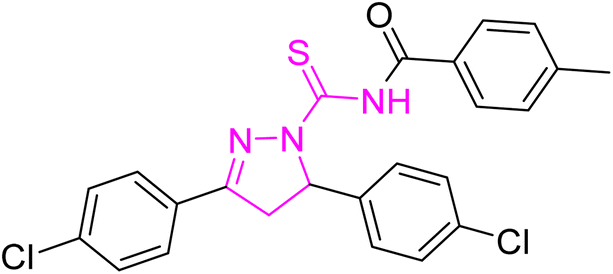
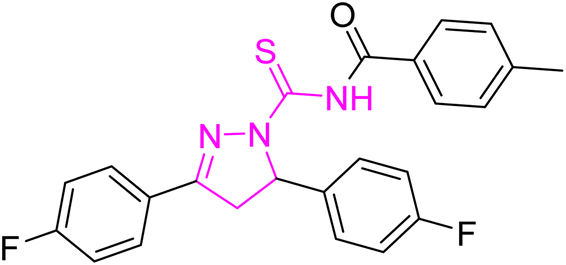
| | Fig. 2 Molecular structure and % yield of synthesized derivatives (5a–o). | |
2.2. In vitro urease inhibitory activity and molecular docking study
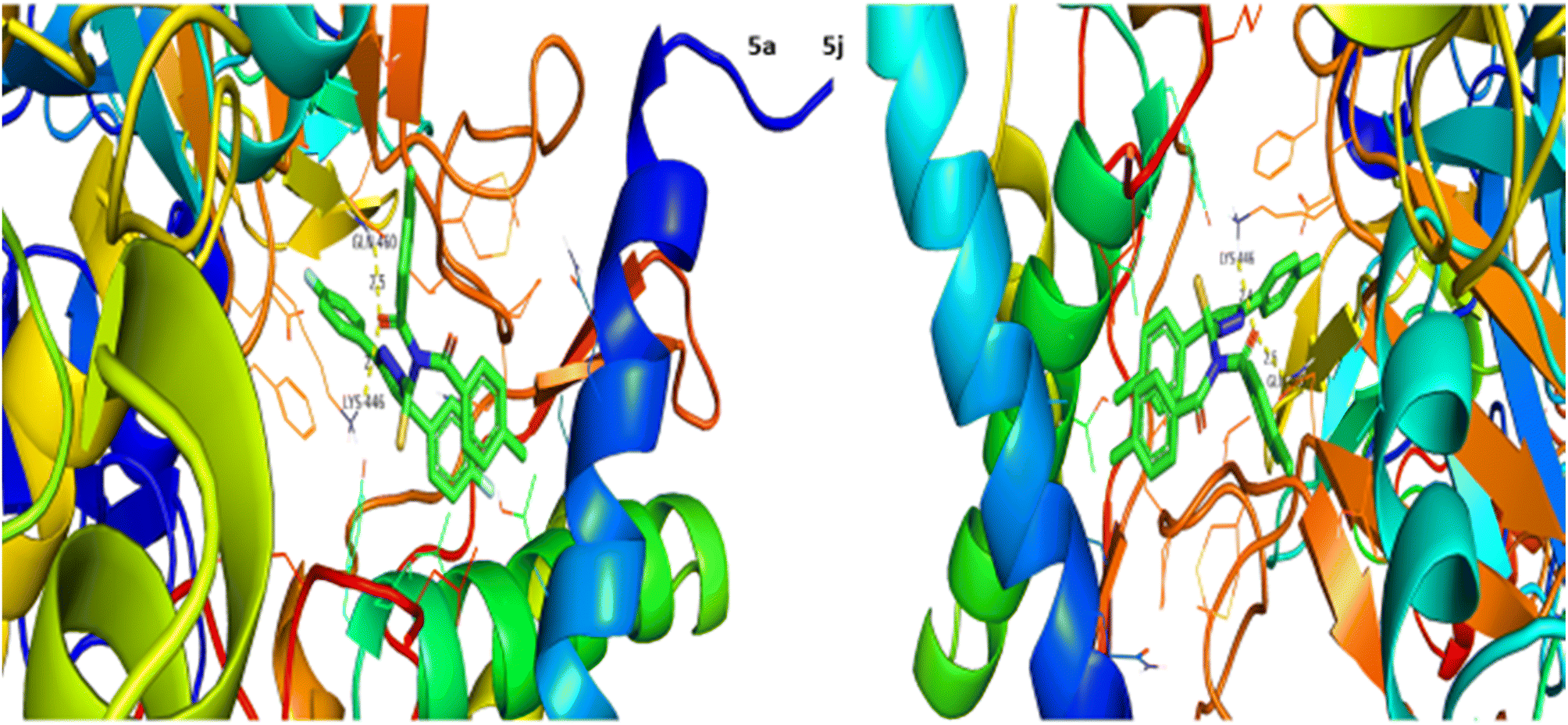
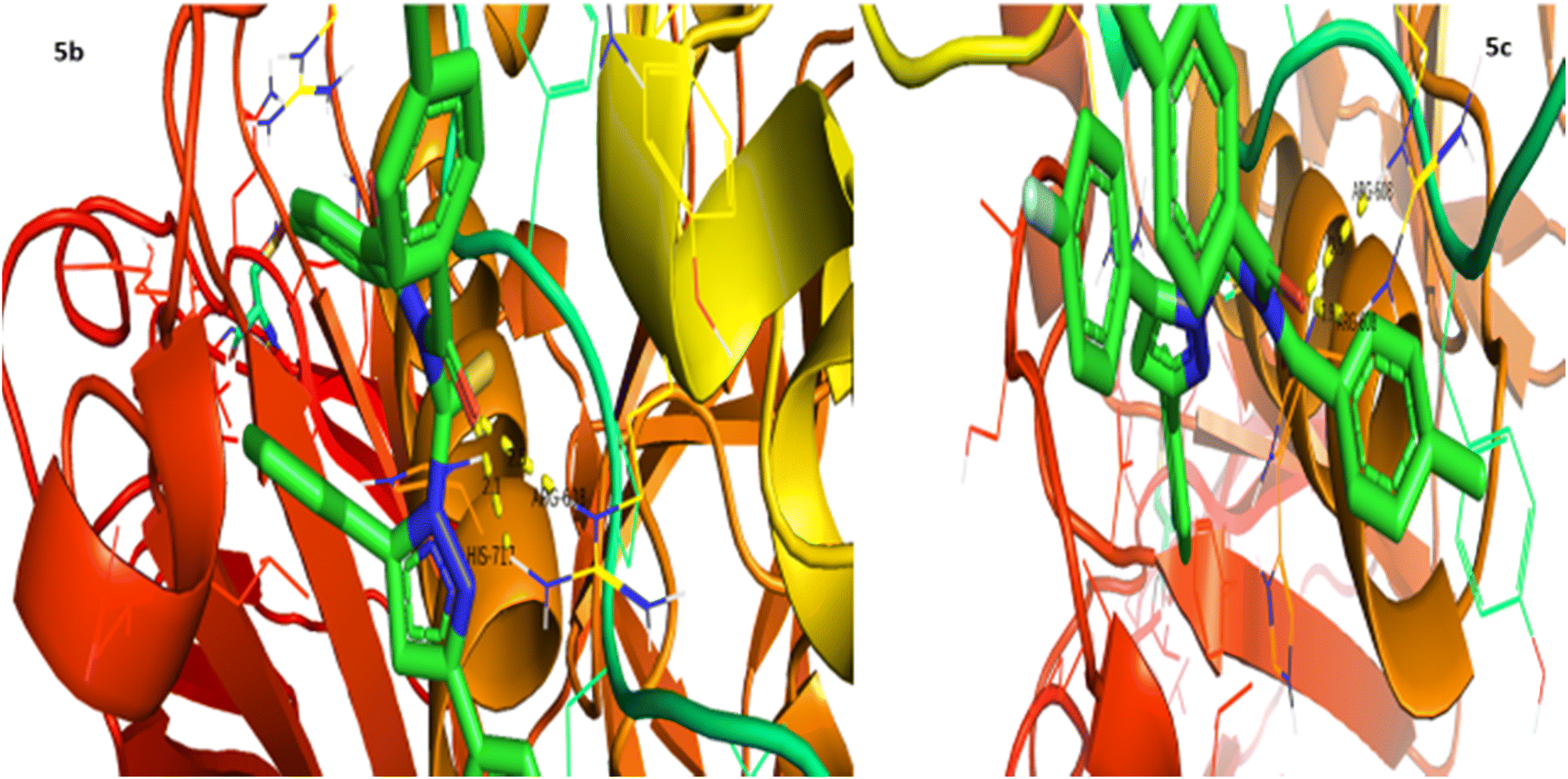
Urease inhibition potential of the (5a–j) series was evaluated using a reported method and the results in the form of IC50 values are given in Table 1. Thiourea was used as a positive control and exhibited prominent enzyme inhibition activity with an IC50 value of 9.8 μM. The results of IC50 represented that the highest activity was shown by the compound 5g with an IC50 value of 43.6 μM, followed by compounds 5b (IC50 54.2 μM) and 5a (IC50 57.9 μM) accordingly. Compounds 5j and 5c showed moderate activity with IC50 values of 72.2 and 80.5 μM, respectively. On the other hand, rest of the compounds presented low activity with IC50 values in the range of 102–137 μM. Overall, the (5a–j) series showed good anti urease activity. After these in vitro evaluations, structures of this series were docked with anti-urease protein receptors and the results are presented in Table 2. Among these ligands 5a and 5j presented the strongest interaction with urease receptors, showing binding energies of −10.9 and −11.2 kcal mol−1 having double interaction at GLN-460 and LYS-446 with bond distances between 2.4 and 2.5 Å (Table 2), respectively. Compound 5f exhibited good interaction with a binding energy of −9.1 kcal mol−1 having interaction at GLN-460 with a bond distance of 2.6 Å, while rest of the compounds showed interaction with urease receptors, showing binding energies in the range of −9.1 to −10.9 kcal mol−1 having interaction at LYS-446 with a bond distance ranging from 2.3 to 2.8 Å accordingly. Overall docking results of this series showed positive correlation with urease inhibition, providing linkage with in vitro activity (Fig. 3).
Table 1 Urease inhibitory activity of compounds (5a–j)a
| Compound |
Urease activity IC50 ± SEM (μM) |
Compound |
Urease activity IC50 ± SEM (μM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. |
| 5a |
57.9 ± 0.15 |
5f |
105.1 ± 0.18 |
| 5b |
54.2 ± 0.32 |
5g |
43.6 ± 0.25 |
| 5c |
80.5 ± 0.12 |
5h |
102.1 ± 0.27 |
| 5d |
118.2 ± 0.17 |
5i |
136.9 ± 1.06 |
| 5e |
124.4 ± 0.49 |
5j |
72.2 ± 0.70 |
| Thiourea |
9.8 ± 0.1 |
|
|
Table 2 Result of docking studies of compounds (5a–j)
| Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
| 5a |
−10.9 |
GLN-460 = 2.5 |
5f |
−9.1 |
GLN-460 = 2.6 |
| LYS-446 = 2.4 |
| 5b |
−9.9 |
LYS-446 = 2.4 |
5g |
−10.9 |
LYS-446 = 2.3 |
| LYS-446 = 2.8 |
| 5c |
−9.8 |
LYS-446 = 2.3 |
5h |
−9.1 |
LYS-446 = 2.4 |
| 5d |
−10.8 |
LYS-446 = 2.3 |
5i |
−9.9 |
LYS-446 = 2.6 |
| 5e |
−9.8 |
LYS-446 = 2.3 |
5j |
−11.2 |
GLN-460 = 2.5 |
| LYS-446 = 2.4 |
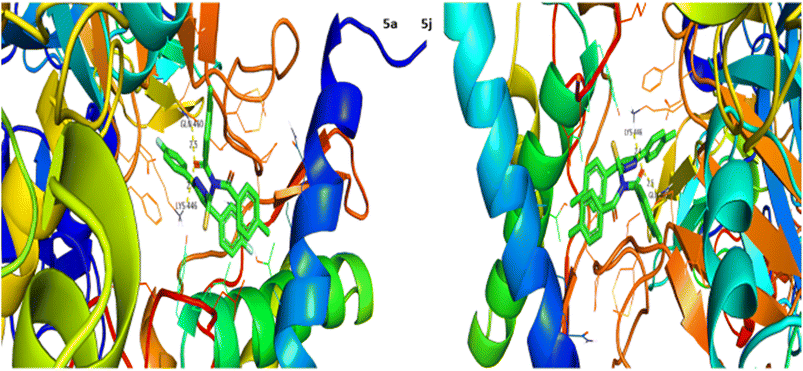 |
| | Fig. 3 Interaction of urease with 5a and 5j. | |
2.3. In vitro α-glucosidase inhibitory activity and molecular docking study
α-Glucosidase inhibition potential of the (5a–j) series was evaluated using a reported method and the results in the form of IC50 values are given in Table 3. Acarbose was used as a positive control and showed prominent enzyme inhibition activity with an IC50 value of 9.5 μM. The results of IC50 represented that the highest activity was shown by the compounds 5a (IC50 69.9 μM) and 5b (IC50 68.3 μM), followed by compound 5j (IC50 73.4 μM). Compounds 5c, 5h and 5f showed moderate activity with IC50 values of 80.6, 81.0 and 95.3 μM, respectively. On the other hand, compounds 5d, 5e and 5i exhibited low activity range IC50 values (113–117 μM). Conclusively, all compounds showed higher/moderate α-glucosidase inhibition potential. After enzyme inhibition activity evaluation, structures of this series were docked with α-glucosidase protein receptors and the results are presented in Table 4. Among all ligands 5c exhibited the strongest interaction with receptors, presenting a binding energy of −10.1 kcal mol−1 having interaction at ARG-608 with a bond distance of 1.9 Å (Table 4). Compounds 5b and 5g showed good interaction with receptors with binding energies of −9.9 and −8.0 kcal mol−1 having interaction at HIS-717 with bond distances of 2.1 and 2.5 Å, respectively. Rest of the compounds also exhibited good interaction with the binding energies in the range of −8.0 to −9.9 kcal mol−1 having interaction at ARG-608 with bond distances ranging from 1.8 to 2.0 Å accordingly. These results represent positive interactions of this series with the enzyme, presenting their inhibitory role (Fig. 4).
Table 3 α-Glucosidase inhibitory activity of compounds (5a–j)a
| Compound |
α-Glucosidase activity IC50 ± SEM (μM) |
Compound |
α-Glucosidase activity IC50 ± SEM (μM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. |
| 5a |
69.9 ± 0.13 |
5f |
95.3 ± 0.57 |
| 5b |
68.3 ± 0.11 |
5g |
128.7 ± 2.2 |
| 5c |
80.6 ± 0.12 |
5h |
81.03 ± 0.31 |
| 5d |
114.8 ± 0.85 |
5i |
113.5 ± 0.17 |
| 5e |
116.6 ± 0.28 |
5j |
73.4 ± 0.19 |
| Acarbose |
9.5 ± 0.2 |
|
|
Table 4 Result of docking studies of compounds (5a–j)
| Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
| 5a |
−9.1 |
ARG-608 = 1.9 |
5f |
−8.8 |
ARG-608 = 1.8 |
| 5b |
−9.9 |
HIS-717 = 2.1 |
5g |
−8.0 |
HIS-717 = 2.5 |
| 5c |
−10.1 |
ARG-608 = 1.9 |
5h |
−8.5 |
ARG-608 = 1.8 |
| 5d |
−9.9 |
ARG-608 = 2.0 |
5i |
−9.8 |
ARG-608 = 1.9 |
| 5e |
−9.6 |
ARG-608 = 1.8 |
5j |
−9.3 |
ARG-608 = 1.9 |
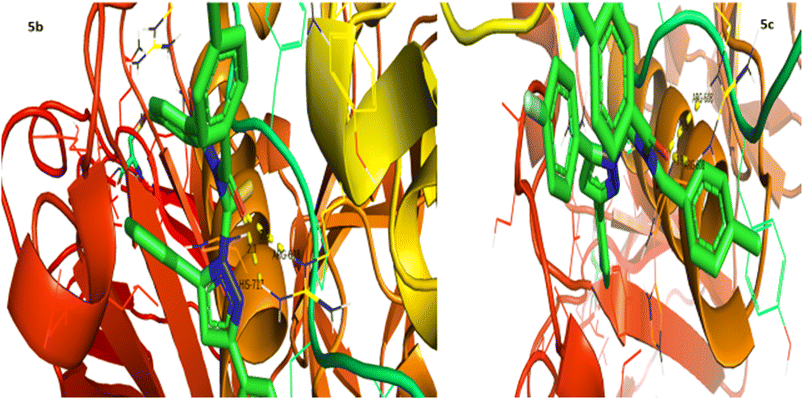 |
| | Fig. 4 Interaction of α-glucosidase with 5b and 5c. | |
2.4. In vitro α-amylase inhibitory activity and molecular docking study
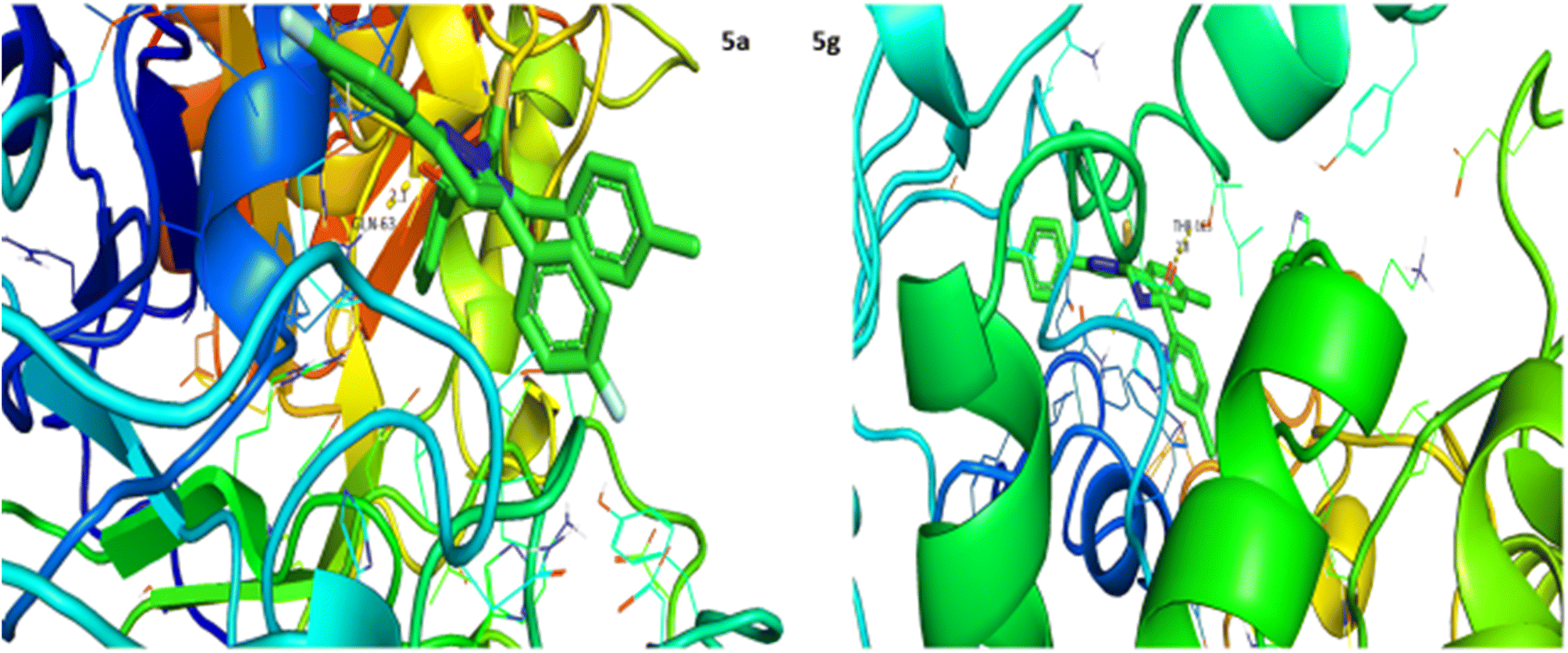
α-Amylase inhibition potential of the (5a–j) series was evaluated using the iodine starch method and the results as IC50 values are expressed in Table 5. Acarbose was used as a positive control and exhibited prominent enzyme inhibition activity with an IC50 value of 10.2 μM. The results of IC50 represented that the compounds 5b (IC50 99.3 μM), 5e (IC50 97.4 μM) and 5f (IC50 90.3 μM) showed comparatively better activity having IC50 values less than 100 μM, while rest of the compounds presented low activity having IC50 values in the range of 103 to 131 μM except 5c which exhibited no activity. Overall, the compounds presented moderate α-amylase inhibition activity. Following the in vitro evaluation, structures of this series were docked with α-amylase protein receptors and the results are presented in Table 6. Among these ligands 5a presented the strongest interaction with enzyme receptors, showing a binding energy of −10.2 kcal mol−1 having interaction at GLN-63 with a bond distance of 2.1 Å (Table 6). Compound 5g exhibited good interaction with a binding energy of −8.9 kcal mol−1 having interaction at THR-163 with a bond distance of 2.8 Å, while no interaction was recorded for compounds 5b and 5f. Rest of the compounds also showed good interaction with the binding energies in the range of −8.6 to −9.6 kcal mol−1 having interaction at GLN-63 with bond distances ranging from 1.9 to 2.8 Å accordingly. These results support the finding of in vitro evaluation, suggesting their potential use as enzyme inhibitors (Fig. 5).
Table 5 α-Amylase inhibitory activity of compounds (5a–j)a
| Compound |
α-Amylase activity IC50 ± SEM (μM) |
Compound |
α-Amylase activity IC50 ± SEM (μM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. |
| 5a |
118.4 ± 0.64 |
5f |
90.3 ± 1.08 |
| 5b |
99.3 ± 0.62 |
5g |
131.3 ± 1.56 |
| 5c |
0 |
5h |
121.3 ± 3.75 |
| 5d |
103.9 ± 3.18 |
5i |
114.6 ± 0.28 |
| 5e |
97.4 ± 1.53 |
5j |
109.9 ± 1.56 |
| Acarbose |
10.2 ± 0.1 |
|
|
Table 6 Result of docking studies of compounds (5a–j)
| Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
Compound |
Binding energy (kcal mol−1) |
Binding residues/bond lengths (Å) |
| 5a |
−10.2 |
GLN-63 = 2.1 |
5f |
Nil |
No interaction |
| 5b |
Nil |
No interaction |
5g |
−8.9 |
THR-163 = 2.8 |
| 5c |
−9.5 |
GLN-63 = 2.1 |
5h |
−8.6 |
GLN-63 = 2.8 |
| 5d |
−9.0 |
GLN-63 = 2.1 |
5i |
−9.4 |
GLN-63 = 1.9 |
| 5e |
−9.6 |
GLN-63 = 2.0 |
5j |
−8.7 |
GLN-63 = 2.1 |
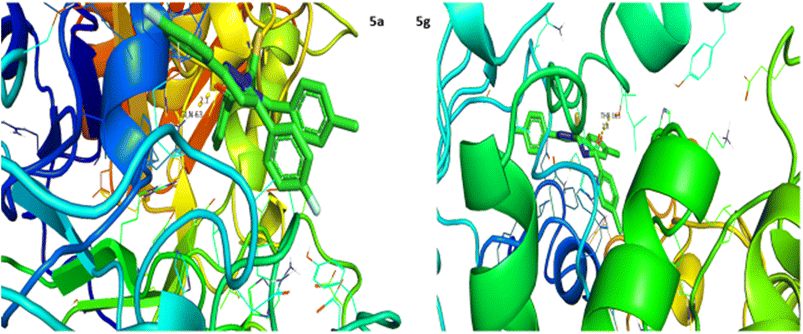 |
| | Fig. 5 Interaction of α-amylase with 5a and 5g. | |
2.5. DPPH assay
Antioxidant potential of the (5a–j) series was determined using DPPH assay and the results in the form of IC50 values are presented in Table 7. Ascorbic acid was used as a positive control and exhibited strong activity with an IC50 value of 9.6 μM. The results of IC50 represented that the complete series have moderate activity with IC50 values greater than 100 μM. The compounds 5a to 5j showed activities in the range of 103–117 μM.
Table 7 DPPH inhibitory activity of compounds (5a–j)a
| Compound |
DPPH activity IC50 ± SEM (μM) |
Compound |
DPPH activity IC50 ± SEM (μM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. |
| 5a |
105.0 ± 0.12 |
5f |
103.6 ± 0.06 |
| 5b |
103.4 ± 1.15 |
5g |
111.9 ± 0.35 |
| 5c |
106.5 ± 1.79 |
5h |
116.5 ± 0.92 |
| 5d |
104.5 ± 0.15 |
5i |
105.4 ± 1.65 |
| 5e |
105.7 ± 0.15 |
5j |
105.0 ± 0.73 |
| Ascorbic acid |
9.6 ± 0.4 |
|
|
2.6. Anti-bacterial assay
Anti-bactericidal activity of the compounds was determined by using the disc diffusion method. The results were measured in terms of inhibition zones and are presented in Table 8. The results showed that 5d has significant antibacterial activity against all tested strains except Pseudomonas aeruginosa. On the other hand, all other compounds showed no activity against the four bacterial strains. The assay was performed in triplicate and kanamycin was used as a positive control.
Table 8 Anti-bacterial activity of compounds (5a–j)
| Compound no. |
Antibacterial zone of inhibition in mm |
| Escherichia coli |
Pseudomonas aeruginosa |
Bacillus subtilis |
Staphylococcus aureus |
| 5a |
— |
— |
— |
— |
| 5b |
— |
— |
— |
— |
| 5c |
— |
— |
|
— |
| 5d |
7 |
— |
9 |
10 |
| 5e |
|
— |
|
|
| 5f |
— |
— |
|
— |
| 5g |
— |
— |
— |
— |
| 5h |
— |
— |
— |
— |
| 5i |
— |
— |
|
— |
| 5j |
— |
— |
— |
— |
| Kanamycin |
16 |
20 |
22 |
18 |
2.7. Anti-fungal assay
Antifungal activity of the compounds was determined by using the disc diffusion method. The results were measured in terms of inhibition zones and are presented in Table 9. The results showed that all compounds have no significant antifungal activity against Aspergillus fumigatus except 5i. On the other hand, no any compound showed activity against Candida albicans. The assay was performed in triplicate and terbinafine was used as a positive control.
Table 9 Anti-fungal activity of compounds (5a–j)
| Compound |
Antifungal zone of inhibition in mm |
| Aspergillus fumigatus |
Candida albicans |
| 5a |
— |
— |
| 5b |
— |
— |
| 5c |
— |
— |
| 5d |
— |
— |
| 5e |
— |
— |
| 5f |
— |
— |
| 5g |
— |
— |
| 5h |
— |
— |
| 5i |
5 |
— |
| 5j |
— |
— |
| Terbinafine |
16 |
20 |
2.8. Brine shrimp cytotoxicity assay
The toxicity of the compounds was determined by brine shrimp cytotoxicity assay and eggs were hatched as reported earlier.32 25 μl of each stock solution (100, 50 and 25 μM) of the test compound was taken in glass vials (25 ml) and solubilized in 2 ml of seawater. Twenty shrimps were transferred to each vial using a Pasteur pipette, and the volume was raised up to 5 ml. Twenty nauplii were added by counting macroscopically in the stem of the pipette against a light background and the experiment was performed in triplicate. The vials were maintained under illumination at room temperature 25 °C to 28 °C after 24 hours of incubation. Survivors were counted with the aid of a 3× magnifying glass and IC50 values were calculated.
2.8.1 Results. This is a rapid, inexpensive, in-house general bioassay that may serve as a pre-screen test for the identification of toxicity levels of the compounds. To test the toxicity of the compounds brine shrimp assay was performed. Shrimps were subjected to different concentrations of compounds. Doxorubicin was used as a positive control drug, which showed an IC50 value of 9.8 ± 5.9 μM, representing toxic effect on the brine shrimps. Compounds 5d, 5e and 5i had IC50 values less than 50 μM, showing significant level of toxicity to the brine shrimps. Rest of the compounds had IC50 values more than 100 μM, showing no toxic response, as summarized in Table 10. Cytotoxic action of a drug is simply provided by disturbing the basic mechanisms concerned with mitotic activity, cell growth, function, and differentiation.33
Table 10 Results of brine shrimp cytotoxicity of compounds (5a–j)
| Compound |
α-Glucosidase activity IC50 ± SEM (μM) |
Compound |
α-Glucosidase activity IC50 ± SEM (μM) |
| 5a |
164.6 ± 17.2 |
5f |
102.9 ± 7.9 |
| 5b |
115.4 ± 9.7 |
5g |
114.1 ± 11.6 |
| 5c |
141.8 ± 8.1 |
5h |
124.2 ± 6.5 |
| 5d |
14.9 ± 4.6 |
5i |
21.8 ± 5.1 |
| 5e |
24.5 ± 3.5 |
5j |
120.2 ± 12.2 |
| Doxorubicin |
9.8 ± 5.9 |
|
|
3. Structure–activity relationship (SAR)
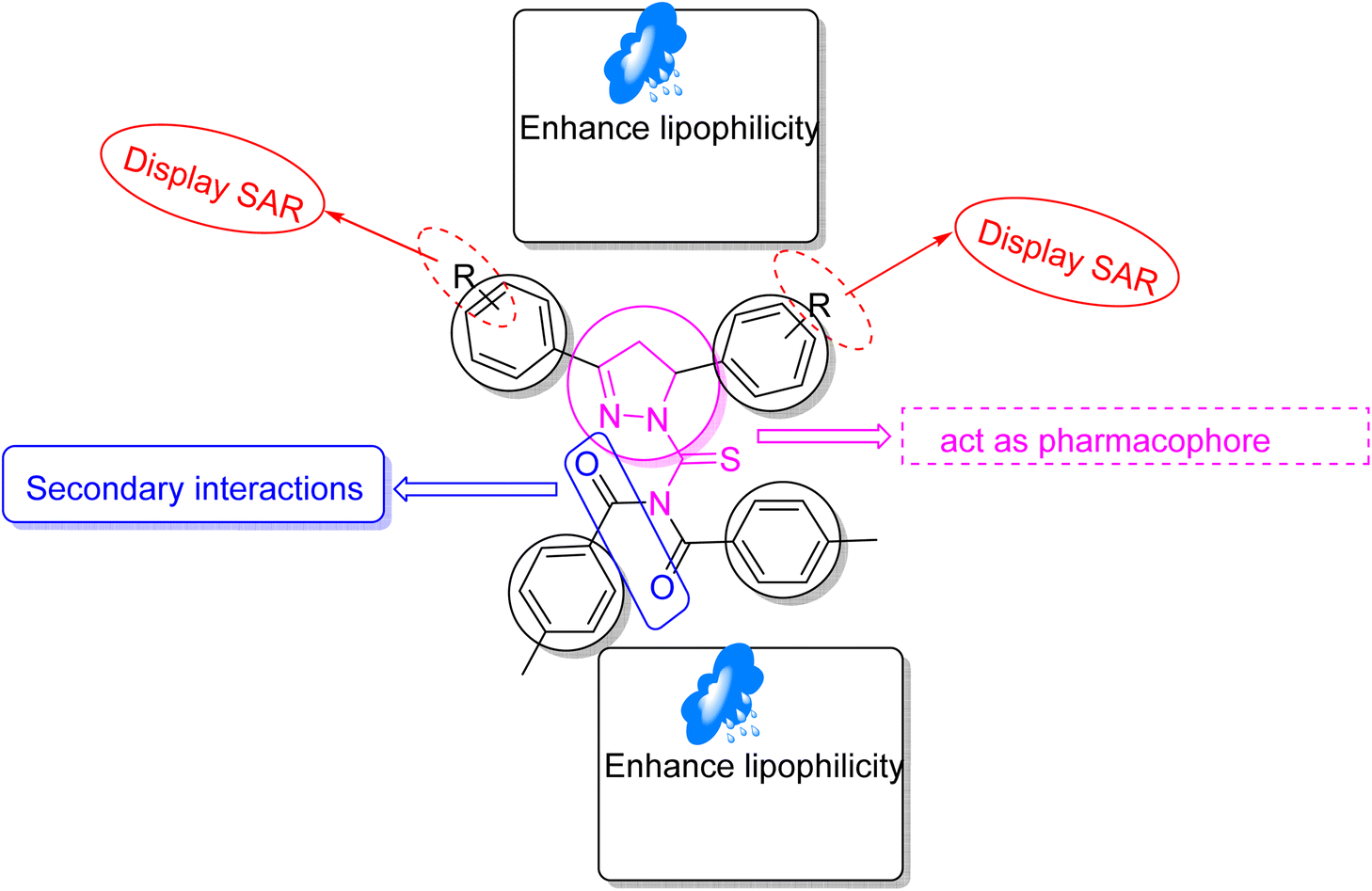
Structure–activity relationship (SAR) represents a fundamental framework in the realm of drug design and molecular activity characterization. The SAR allows the design and modification of the structure to obtain effective drugs. A series of pyrazoline hybrid substituted acyl thiourea derivatives were designed and synthesized as potent inhibitors of urease, α-glucosidase, and α-amylase. These compounds serve as promising candidates for inhibition of critical enzymatic processes such as urease, α-glucosidase, and α-amylase, because the scaffolds of these hybrids consist of various active sites (Fig. 6).
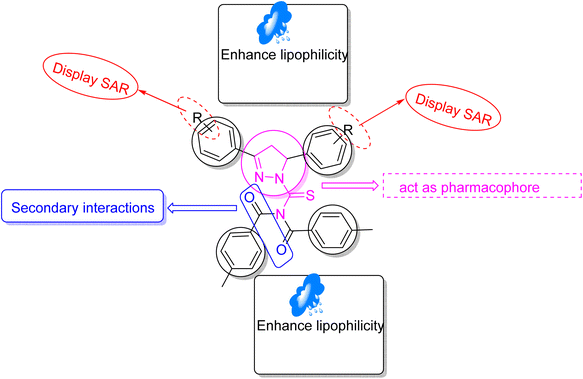 |
| | Fig. 6 Structure–activity relationship of synthesized pharmacophores. | |
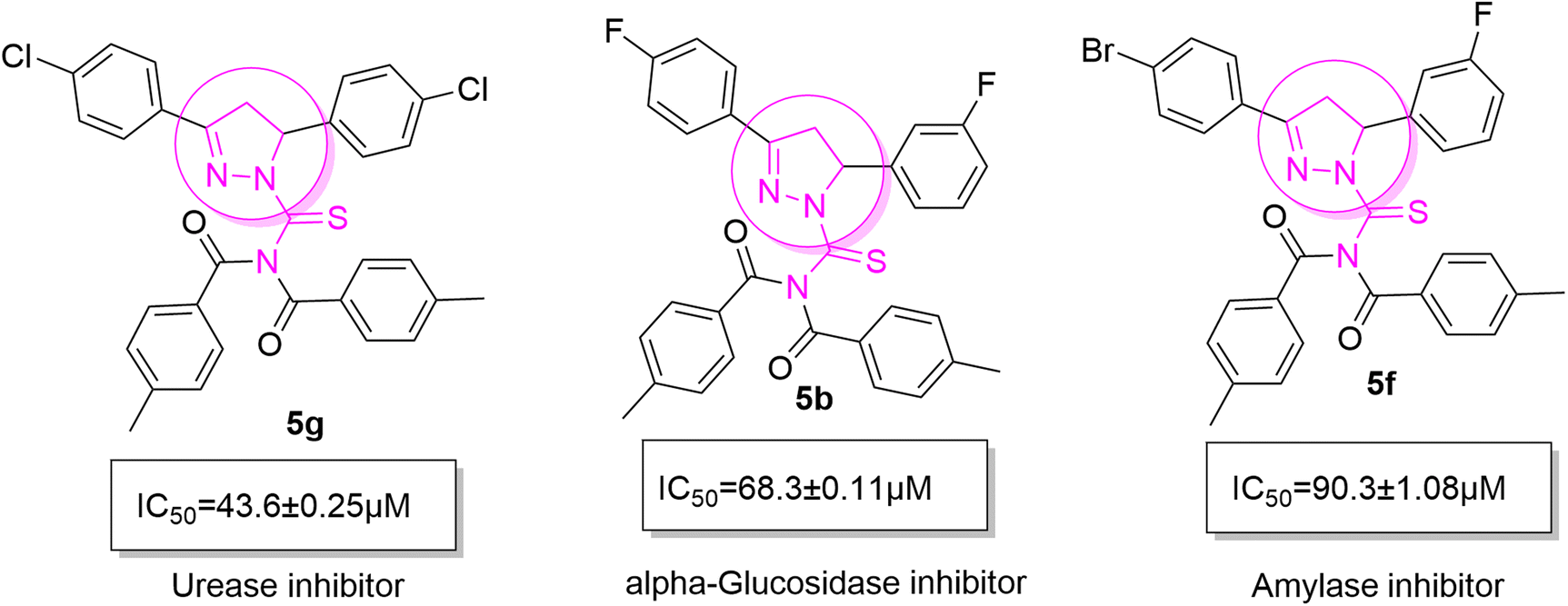
The resulting urease inhibitory effects of compound 5g revealed that the compound 5g was most potent in the series and exhibited the lowest IC50 values amongst the series. In compounds 5g, 5b and 5f, the chloro, fluoro, and bromo electron withdrawing groups at meta and para positions are responsible for the urease, α-glucosidase, and amylase inhibitory activity (Fig. 7). The urease inhibitory effect of compound 5g is due to strong hydrophobic interaction of the two-chlorine group at the para position. Compound 5b shows strong alpha glucosidase inhibitory effect due to the strong polar interaction and small size of fluorine at meta and para positions, respectively, while compound 5f shows potent amylase inhibitory activity due to hydrophobic interaction of bromine at the para position and polar interaction, and small size of fluorine at the meta position.
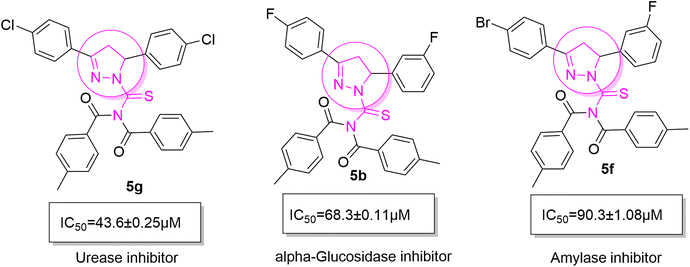 |
| | Fig. 7 Derivatives 5g, 5b and 5f along with IC50 indicating significant biological potency against urease, α-glucosidase and amylase. | |
4. Experimental
4.1. Materials and methods
All reagents were used without further purification from Sigma Aldrich. Melting points were recorded by using StuartSMP3 melting point apparatus. The NMR spectra were recorded on a Bruker 1H-NMR at 300 MHz and for 13C-NMR at 75 MHz. The chemical shifts were described in parts per million (ppm) versus TMS or the residual solvent resonance as an internal reference standard. The reactions were monitored by using thin layer chromatography (TLC), which was performed using pre-coated plates with silica gel Kiesel 60 F254.254 and 360 nm UV light was used for detection of the separated spots on the chromatogram.
4.2. Synthetic approach for pyrazoline hybrid acyl thiourea analogs (5a–j) and (5k–o)
Chalcones (3) were synthesized by reaction of acetophenone 1 (0.006 mol) and aldehyde 2 (0.006 mol) derivatives with KOH (0.012 mol) as a base using a pestle and mortar, which were then converted to pyrazoline (4) derivatives by reacting 3 (0.002 mol) with thiosemicarbazide (0.002 mol) in dry ethanol under reflux for 6–8 hours along with KOH (0.004 mol). The completion of reaction was indicated by the analytical TLC. Upon cooling the formed precipitate was filtered, dried and recrystallized from ethanol. Compound 4 (0.0004 mol) was then reacted with 2 equivalents (0.0008 mol) of p-tolyl chloride to furnish pyrazoline hybrid acyl thiourea pharmacophores (5a–j) and one equivalent (0.0004 mol) to obtain (5k–o) in dry acetone as a solvent under reflux for 1 hour with triethyl amine (few drops) as a base and quencher under a nitrogen atmosphere. Upon completion of the reaction the mixture was cooled to room temperature and water was added, and the reaction mixture was stirred to obtain ppt, which were filtered, dried, and recrystallized using ethanol. Finally the pure desired product was obtained.
4.3. Experimental data
4.3.1 N-(3,5-Bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5a).
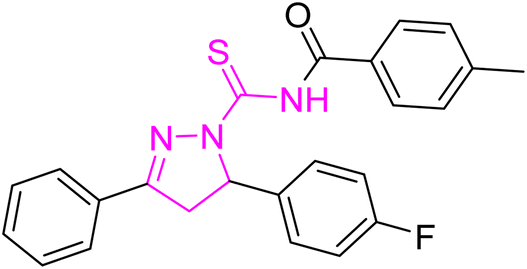
Yellow crystal; melting point: 205–207 °C; yield: 75%; Rf: 0.66 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3050 (C–H Ar), 2948 (sp3 C–H), 1694 (CO), 1601 (CN), 1504 (CC Ar-ring), 1288 (CS), 1145 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.81–7.78 (d, 4H, J = 8.1 Ar–CH), 7.60–7.55 (m, 2H, Ar–CH), 7.21–6.95 (m, 10H, Ar–CH), 6.01–5.96 (dd, J = 3.6, 3.9 Hz, 1H), 3.93–3.84 (dd, J = 11.1, 11.1 Hz, 1H), 3.24–3.17 (dd, J = 3.9, 3.6 Hz, 1H), 2.40 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.3 (CS), 171.8 (CO), 157.7 (CN), 166.4, 163.8, 160.6, 143.6, 134.8, 134.8, 131.9, 129.6, 129.5, 129.4, 129.2, 128.2, 128.1, 126.3, 126.2, 116.4, 116.1, 115.8, 115.6, (Ar–C), 64.7, (CH), 43.0, (CH2), 21.7, (CH3). Anal. calcd for C32H25F2N3O2S (553.63): C, 69.42; H, 4.55; N, 7.59; S, 5.79%. Found: C, 69.41; H, 4.54; N, 7.58; S, 5.78%.
4.3.2 N-(5-(3-Fluorophenyl)-3-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5b).
Off-white powder; melting point: 203–205 °C; yield: 65%; Rf: 0.71 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3055 (C–H Ar), 2950 (sp3 C–H), 1689 (CO), 1602 (CN), 1520 (CC Ar ring), 1280 (CS), 1150 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.82–7.77 (d, 4H, J = 8.1 Ar–CH), 7.61–7.54 (m, 2H, Ar–CH), 7.29–6.94 (m, 10H, Ar–CH), 6.02–5.95 (dd, J = 3.6, 3.9 Hz, 1H), 3.94–3.83 (dd, J = 11.1, 11.1 Hz, 1H), 3.25–3.16 (dd, J = 3.9, 3.6 Hz, 1H), 2.40 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.3 (CS), 171.8 (CO), 157.7 (CN), 166.4, 162.8, 160.6, 141.6, 134.8, 134.8, 131.9, 129.6, 129.5, 129.4, 129.2, 128.2, 128.2, 128.1, 126.3, 126.2, 116.4, 116.1, 115.8, 113.6, (Ar–C), 64.7, (CH), 43.0, (CH2), 21.7, (CH3). Anal. calcd for C32H25F2N3O2S (553.63): C, 69.42; H, 4.55; N, 7.59; S, 5.79%. Found: C, 69.41; H, 4.54; N, 7.58; S, 5.78%.
4.3.3 N-(3-(4-Chlorophenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5c).
Greenish yellow powder; melting point: 140–142 °C; yield: 71%; Rf: 0.68 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3037 (C–H Ar), 2918 (sp3 C–H), 1709 (CO), 1607 (CN), 1510 (CC Ar-ring), 1284 (CS), 1158 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.80–7.76 (d, 4H, J = 7.7 Ar–CH), 7.48–6.95 (m, 12H, Ar–CH), 6.00–5.95 (dd, J = 3.7, 3.7 Hz, 1H), 3.92–3.84 (dd, J = 11.3, 11.3 Hz, 1H), 3.23–3.17 (dd, J = 3.7, 3.8 Hz, 1H), 2.48 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 176.4 (CS), 172.6 (CO), 158.6 (CN), 144.5, 137.5, 133.2, 131.8, 129.4, 129.3, 128.8, 128.6, 128.2, 128.1, 126.4, 116.4, 115.6, (Ar–C), 64.7, (CH), 42.8, (CH2), 21.7, (CH3). Anal. calcd for C23H25ClFN3O2S (570.08): C, 67.42; H, 4.42; N, 7.37; S, 5.62%. Found: C, 67.41; H, 4.41; N, 7.36; S, 5.61%.
4.3.4 N-(5-(4-Fluorophenyl)-3-phenyl-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5d).
Greenish yellow crystal; melting point: 195–197 °C; yield: 80%; Rf: 0.75 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3049 (C–H Ar), 2945 (sp3 C–H), 1695 (CO), 1606 (CN), 1505 (CC Ar-ring), 1287 (CS), 1173 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.86–7.84 (d, 4H, J = 8.1 Ar–CH), 7.62–7.60 (m, 2H, Ar–CH), 7.49–6.96 (m, 11H, Ar–CH), 6.02–5.97 (dd, J = 3.9, 3.6 Hz, 1H), 3.95–3.86 (dd, J = 11.1, 11.1 Hz, 1H), 3.27–3.17 (dd, J = 3.9, 3.9 Hz, 1H), 2.40 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.3 (CS), 171.8 (CO), 163.7 (CN), 160.8, 159.2, 143.6, 135.0, 135.0, 132.0, 131.7, 129.9, 129.4, 129.2, 129.0, 128.3, 128.2, 127.4, 115.8, 115.5, (Ar–C), 64.6, (CH), 43.0, (CH2), 21.8, (CH3). Anal. calcd for C32H26FN3O2S (535.64) C, 71.76; H, 4.89; N, 7.85; S, 5.99%. Found: C, 71.75; H, 4.88; N, 7.84; S, 5.98%.
4.3.5 N-(3-(4-Bromophenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5e).
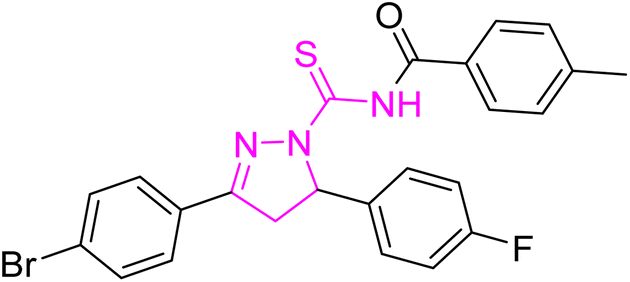
Yellow powder; melting point: 125–127 °C; yield: 83%; Rf: 0.75 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3051 (C–H Ar), 2915 (sp3 C–H), 1692 (CO), 1606 (CN), 1509 (CC Ar-ring),1239 (CS), 1175 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.80–7.77 (d, 4H, J = 7.8 Ar–CH), 7.49–6.96 (m, 12H, Ar–CH), 6.00–5.96 (dd, J = 3.3, 3.3 Hz, 1H), 3.93–3.83 (dd, J = 11.1, 11.1 Hz, 1H), 3.23–3.16 (dd, J = 3.6, 3.6 Hz, 1H), 2.47 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.5 (CS), 171.8 (CO), 157.6 (CN), 143.7, 134.8, 132.2, 131.8, 129.4, 129.2, 128.9, 128.6, 128.2, 128.0, 126.3, 115.9, 115.6, (Ar–C), 64.7, (CH), 42.8, (CH2), 21.7, (CH3). Anal. calcd for C32H25BrFN3O2S (614.53) C, 62.54; H, 4.10; N, 6.84; S, 5.22%. Found: C, 62.53; H, 4.09; N, 6.83; S, 5.21%.
4.3.6 N-(3-(4-Bromophenyl)-5-(3-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5f).
Yellow powder; melting point: 120–122 °C; yield: 68%; Rf: 0.68 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3150 (C–H Ar), 2938 (sp3 C–H), 1684 (CO), 1601 (CN), 1504 (CC Ar ring), 1266 (CS), 1127 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.82–7.79 (d, 4H, J = 8.4, Ar–CH), 7.49–6.80 (m, 12H, Ar–CH), 6.01–5.96 (dd, J = 3.9, 3.9 Hz, 1H), 3.93–3.83 (dd, J = 10.8, 11.1 Hz, 1H), 3.23–3.16 (dd, J = 3.9, 3.9 Hz, 1H), 2.46 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.5 (CS), 171.4 (CO), 157.5 (CN), 143.7, 134.8, 132.2, 131.8, 129.4, 129.2, 128.6, 128.6, 128.2, 128.0, 126.3, 115.9, 115.6, (Ar–C), 64.4, (CH), 42.7, (CH2), 21.7, (CH3). Anal. calcd for C32H25BrFN3O2S (614.53) C, 62.54; H, 4.10; N, 6.84; S, 5.22%. Found: C, 62.53; H, 4.09; N, 6.83; S, 5.21%.
4.3.7 N-(3,5-Bis(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5g).
Yellow crystal; melting point: 215–217 °C; yield: 74%; Rf: 0.73 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3029 (C–H Ar), 2972 (sp3 C–H), 1698 (CO), 1610 (CN), 1491 (CC Ar ring), 1290 (CS), 1170 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.86–7.82 (d, 4H, J = 8.4 Ar–CH), 7.70–7.66 (m, 2H, Ar–CH), 7.42–7.21 (m, 10H, Ar–CH), 6.19–6.14 (dd, J = 3.9, 3.9 Hz, 1H), 3.97–3.85 (dd, J = 11.1, 11.1 Hz, 1H), 3.28–3.18 (dd, J = 3.9, 3.9 Hz, 1H), 2.47 (s, 6H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.3 (CS), 171.8 (CO), 157.7 (CN), 166.4, 163.8, 160.6, 143.6, 134.8, 133.6, 131.3, 129.6, 129.5, 129.3, 129.2, 128.2, 128.1, 127.3, 127.2, 116.4, 116.1, 115.8, 115.6, (Ar–C), 62.7, (CH), 42.4, (CH2), 21.6, (CH3). Anal. calcd for C32H25C12N3O2S (586.53) C, 65.53; H, 4.30; N, 7.16; S, 5.47%. Found: C, 65.52; H, 3.29; N, 7.16; S, 5.46%.
4.3.8 4-Methyl-N-(4-methylbenzoyl)-N-(3-phenyl-5-(p-tolyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)benzamide (5h).
White powder; melting point: 206–208 °C; yield: 85%; Rf: 0.71 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3120 (C–H Ar), 2946 (sp3 C–H), 1701 (CO), 1602 (CN), 1504 (CC Ar-ring), 1267 (CS), 1140 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.85–7.83 (d, 4H, J = 5.4 Ar–CH), 7.61–7.59 (m, 2H, Ar–CH), 7.48–6.95 (m, 11H, Ar–CH), 6.01–5.96 (dd, J = 3.8, 3.8 Hz, 1H), 3.95–3.85 (dd, J = 11.1, 11.1 Hz, 1H), 3.26–3.16 (dd, J = 3.8, 3.8 Hz, 1H), 2.39 (s, 9H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 174.3 (CS), 170.4 (CO), 155.7 (CN), 160.8, 159.1, 143.4, 134.0, 135.0, 132.0, 131.6, 129.7, 129.3, 129.1, 129.0, 128.2, 128.2, 127.3, 115.6, 115.4, (Ar–C), 64.4, (CH), 42.3, (CH2), 21.4, (CH3). Anal. calcd for C33H29N3O2S (531.67) C, 74.55; H, 5.50; N, 7.90; S, 6.03%. Found: C, 74.54; H, 5.49; N, 7.89; S, 6.02%.
4.3.9 N-(5-(4-Fluorophenyl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5i).
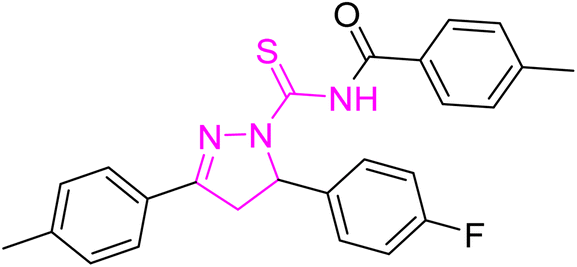
White powder; melting point: 175–177 °C; yield: 80%; Rf: 0.73 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3031 (C–H Ar), 2912 (sp3 C–H), 1712 (CO), 1608 (CN), 1491 (CC Ar ring), 1290 (CS), 1186 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.81–7.78 (d, 4H, J = 8.1 Ar–CH), 7.50–7.47 (d, 2H, J = 8.4, Ar–CH), 7.20–6.94 (m, 10H, Ar–CH), 5.99–5.94 (dd, J = 3.9, 3.6 Hz, 1H), 3.93–3.83 (dd, J = 11.1, 11.1 Hz, 1H), 3.25–3.18 (dd, J = 3.9, 3.6 Hz, 1H), 2.40–2.37 (s, 9H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.1 (CS), 171.7 (CO), 158.9 (CN), 143.4, 142.4, 134.9, 132.0, 129.6, 129.3, 129.2, 128.2, 128.1, 127.3, 127.1, 115.8, 115.5, (Ar–C), 64.5, (CH), 42.4, (CH2), 21.7, 21.6 (CH3). Anal. calcd for C33H28FN3O2S (549.66) C, 72.11; H, 5.13; N, 7.64; S, 5.83%. Found: C, 72.10; H, 5.12; N, 7.63; S, 5.82%.
4.3.10 N-(5-(4-Chlorophenyl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methyl-N-(4-methylbenzoyl)benzamide (5j).
White powder; melting point: 220–222 °C; yield: 71%; Rf: 0.67 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3041 (C–H Ar), 2916 (sp3 C–H), 1719 (CO), 1697 (CN), 1606 (CC Ar-ring), 1248 (CS), 1174 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 7.81–7.78 (d, 4H, J = 8.1 Ar–CH), 7.50–7.47 (d, 2H, J = 8.4, Ar–CH), 7.21–6.95 (m, 10H, Ar–CH), 5.99–5.93 (dd, J = 3.9, 3.6 Hz, 1H), 3.93–3.82 (dd, J = 11.1, 11.1 Hz, 1H), 3.25–3.17 (dd, J = 3.9, 3.6 Hz, 1H), 2.40–2.36 (s, 9H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.1 (CS), 170.6 (CO), 157.9 (CN), 143.4, 140.7, 133.4, 132.6, 129.6, 129.4, 129.2, 128.2, 128.1, 127.4, 127.2, 115.5 (Ar–C), 62.4, (CH), 42.3, (CH2), 21.6, 21.3 (CH3). Anal. calcd for C33H28ClN3O2S (566.12) C, 70.01; H, 4.99; N, 7.42; S, 5.66%. Found: C, 70.00; H, 4.98; N, 7.41; S, 5.65%.
4.3.11 N-(3,5-Bis(4-chlorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methylbenzamide (5k).
Yellow crystal; melting point: 215–217 °C; yield: 70%; Rf: 0.73 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3355 (NH), 3028 (C–H Ar), 2970 (sp3 C–H), 1737 (CO), 1712 (CN), 1552 (CC Ar ring), 1290 (CS), 1144 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 10.44 (s, 1H, NH), 7.86–7.83 (d, 2H, J = 8.4, Ar–CH), 7.70–7.67 (q, 2H, J = 1.8, 1.8 Hz, Ar–CH), 7.48–7.20 (m, 8H, Ar–CH), 6.18–6.130 (dd, J = 3.9, 3.9 Hz, 1H), 3.96–3.862 (dd, J = 11.4, 11.4 Hz, 1H), 3.27–3.19 (dd, J = 3.9, 3.9 Hz, 1H), 2.47 (s, 3H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 170.5 (CS), 163.9 (CO), 155.3 (CN), 143.7, 138.9, 137.8, 133.6, 131.3, 129.7, 129.4, 129.2, 128.4, 128.3, 127.5, 127.2, (Ar–C), 62.7, (CH), 42.4, (CH2) 21.6, (CH3). Anal. calcd for C24H19C12N3OS (468.40) C, 61.54; H, 4.09; N, 8.97; S, 6.84%. Found: C, 61.53; H, 4.08; N, 8.97; S, 6.83%.
4.3.12 N-(3,5-Bis(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methylbenzamide (5l).
White powder; melting point: 220–222 °C; yield: 65%; Rf: 0.68 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3340 (NH), 3040 (C–H Ar), 2960 (sp3 C–H), 1712 (CO), 1690 (CN), 1574 (CC Ar ring), 1292 (CS), 1147 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 10.35 (s, 1H, NH), 7.81–7.78 (d, 2H, J = 8.3, Ar–CH), 7.60–7.57 (m, 2H, Ar–CH), 7.22–6.94 (m, 8H, Ar–CH), 6.01–5.95 (dd, J = 3.9, 3.9 Hz, 1H), 3.93–3.83 (dd, J = 11.4, 11.4 Hz, 1H), 3.24–3.16 (dd, J = 3.9, 3.9 Hz, 1H), 2.48 (s, 3H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.5 (CS), 171.9 (CO), 157.3 (CN), 160.9, 141.8, 130.2, 129.5, 129.1, 128.4, 128.3, 127.2, 120.4, 115.6, 115.6, 115.3, (Ar–C), 71.4, (CH), 42.4, (CH2), 21.6, (CH3). Anal. calcd for C24H19F2N3OS (435.49) C, 66.19; H, 4.40; N, 9.65; S, 7.36%. Found: C, 66.18; H, 4.39; N, 9.64; S, 7.35%.
4.3.13 N-(5-(4-Fluorophenyl)-3-phenyl-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methylbenzamide (5m).
White powder; melting point: 180–182 °C; yield: 71%; Rf: 0.71 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3367 (NH), 3036 (C–H Ar), 2917 (sp3 C–H), 1708 (CO), 1606 (CN), 1509 (CC Ar ring), 1283 (CS), 1176 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 10.25 (s, 1H, NH), 7.86–7.84 (d, 2H, J = 8.4, Ar–CH), 7.62–7.60 (m, 2H, Ar–CH), 7.49–6.95 (m, 9H, Ar–CH), 6.02–5.96 (dd, J = 3.9, 3.9 Hz, 1H), 3.95–3.85 (dd, J = 11.4, 11.4 Hz, 1H), 3.27–3.18 (dd, J = 3.9, 3.9 Hz, 1H), 2.42 (s, 3H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 175.3 (CS), 170.4 (CO), 155.3 (CN), 157.4, 143.9, 142.8, 135.1, 131.6, 129.2, 129.0, 128.8, 128.3, 128.2, 127.4, 115.5, (Ar–C), 66.4, (CH), 43.4, (CH2), 21.7, (CH3). Anal. calcd for C24H20FN3OS (417.50) C, 69.04; H, 4.83; N, 10.06; S, 7.68%. Found: C, 69.03; H, 4.54; N, 10.05; S, 7.67%.
4.3.14 N-(3-(4-Bromophenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methylbenzamide (5n).
Greenish yellow crystal; melting point: 195–197 °C; yield: 70%; Rf: 0.77 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3329 (NH), 3040 (C–H Ar), 2961 (sp3 C–H), 1723 (CO), 1609 (CN), 1513 (CC Ar ring), 1174 (C–N), 1282 (CS); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 10.37 (s, 1H, NH), 7.80–7.77 (d, 2H, J = 8.3, Ar–CH), 7.49–7.40 (m, 2H, Ar–CH), 7.39–6.95 (m, 8H, Ar–CH), 6.00–5.95 (dd, J = 3.9, 3.9, 1H), 3.93–3.82 (dd, J = 11.4, 11.4 Hz, 1H), 3.24–3.15 (dd, J = 3.9, 3.9 Hz, 1H), 2.46 (s, 3H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 177.5 (CS), 171.4 (CO), 154.3 (CN), 143.7, 134.8, 132.6, 131.3, 129.4, 129.4, 128.7, 128.5, 128.4, 125.2, 115.8, 115.5, (Ar–C), 64.6, (CH), 42.7, (CH2), 21.7, (CH3). Anal. calcd for C24H19BrFN3OS (496.40) C, 58.07; H, 3.86; N, 8.47; S, 6.46%. Found: C, 58.06; H, 3.85; N, 8.46; S, 6.45%.
4.3.15 N-(5-(4-Fluorophenyl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazole-1-carbonothioyl)-4-methylbenzamide (5o).
Yellow powder; melting point: 205–207 °C; yield: 77%; Rf: 0.69 (n-hexane:ethyl acetate 8:2); FT-IR (ATR) in cm−1, 3427 (NH), 3143 (C–H Ar), 2951 (sp3 C–H), 1704 (CO), 1601 (CN), 1515 (CC Ar ring), 1280 (CS), 1173 (C–N); 1H-NMR (CDCl3, 300 MHz); δ (ppm): 10.44 (s, 1H, NH), 7.81–7.78 (d, 2H, J = 8.4, Ar–CH), 7.51–6.90 (m, 2H, Ar–CH), 7.20–6.95 (m, 8H, Ar–CH), 5.99–5.95 (dd, J = 3.9, 3.9 Hz, 1H), 3.93–3.84 (dd, J = 11.4, 11.4 Hz, 1H), 3.24–3.19 (dd, J = 3.9, 3.9 Hz, 1H), 2.36 (s, 3H, methyl); 13C-NMR (75 MHz CDCl3); δ (ppm): 181.5 (CS), 175.4 (CO), 158.3 (CN), 151.7, 142.9, 141.2, 132.0, 129.5, 129.4, 129.2, 128.4, 128.3, 127.4, 127.2, 115.5, (Ar–C), 64.3, (CH), 42.7, (CH2), 21.6, (CH3). Anal. calcd for C25H22FN3OS (431.53) C, 69.58; H, 5.14; N, 9.74; S, 7.43%. Found: C, 69.57; H, 5.13; N, 9.73; S, 7.42%.
5. Protocol for activity and docking
5.1. DPPH assay
DPPH assay as reported earlier was used to determine the antioxidant potential of the newly synthesized compounds.34 For experiment, 195 DPPH (100 μM) solution was mixed with 5 μl of sample compounds and ascorbic acid (positive control) at concentrations of 100, 50 and 25 μM in a 96-well plate, respectively. The plate was incubated in the dark for 30 minutes at 37 °C and then absorbance was measured at 515 nm using an Elx800 microtiter plate reader. The experiment was performed in triplicate and percentage scavenging was calculated as follows:
| Percentage scavenging (%) = [1 − absorbance of sample/absorbance of control] × 100 |
5.2. In vitro α-glucosidase inhibition assay protocol
α-Glucosidase inhibitory activities were determined in a 96-well microtiter plate based on p-nitrophenyl-α-D-glucopyranoside (PNPG) as a substrate.35 The α-glucosidase enzyme solution (2 μl), 10 μl PNPG substrate solution (20 mM in phosphate buffer), test sample (5 μl) or acarbose (positive control) at concentrations of 100, 50 and 25 μM and buffer (68 μl) were added and mixed in a 96 well microtiter plate and incubated at 37 °C for 30 min. Then after incubation 100 μl of 0.5 mM sodium bicarbonate solution was added to stop the reaction and absorbance was measured at the wavelength of 405 nm with a microtiter plate reader. The experiments were performed in triplicate and percentage inhibition was calculated using the following formula and IC50 was calculated using GraphPad PrismV8.
| % inhibition = (absorbance of control − absorbance of sample/absorbance of control) × 100 |
5.3. In vitro α-amylase inhibition assay protocol
The α-amylase inhibition assay was performed to calculate the enzyme inhibition potential of the newly synthesized compounds.36 For the experiment, 40 μl starch (0.5 mg ml−1), 10 μl enzyme (0.1 U), 40 μl phosphate buffer (pH 6.8) and 10 μl sample compounds and acarbose (positive control) at concentrations of 100, 50 and 25 μM were added in a 96-well plate. For the blank sample, 40 μl starch and 50 μl phosphate buffer were added. The plate was incubated for 30 minutes at 50 °C, followed by addition of 20 μl HCL (1 M) as the stopping reagent and 100 μl of iodine reagent (5 mM KI + 5 mM I2). Then absorbance was measured at 540 nm using an Elx800 microtiter plate reader. The experiment was performed in triplicate and percentage inhibition was calculated using the following formula and IC50 was calculated using GraphPad PrismV8.
| % inhibition = 100 − ((absorbance of blank − absorbance of sample/absorbance of blank) × 100) |
5.4. In vitro anti urease inhibition protocol
The anti-urease activity of the compounds was measured by determining the amount of free ammonia produced as described earlier.37 The experiment was performed by mixing 10 μl enzyme (0.1 U per reaction), 30 μl of each concentration (100, 50 and 25 μM) of compound and 50 μl buffer of pH 8.2 consisting of 100 mM urea, 0.01 M LiCl2, 1 mM EDTA and 0.01 M K2HPO4. Reaction mixtures were incubated at 37 °C for 15 min in a 96-well plate. Then 50 μl of phenol reagent (0.005% sodium nitroprusside and 1% phenol) and 50 μl of alkali reagent (0.1% NaOCl and 0.5% NaOH) were added to each well and plates were incubated at 37 °C for 50 min. The assay was performed in triplicate and absorbance was recoded at 625 nm using a microplate reader. The anti-urease activity of the compounds was calculated in percentage using the following formula and IC50 was calculated using GraphPad PrismV8.
| % inhibition = (absorbance of control − absorbance of sample/absorbance of control) × 100 |
5.5. Antibacterial assay protocol
Disc diffusion assay was used to determine the antibacterial potential of the newly synthesised compounds as reported earlier.38 For experiment, 1% inoculum of Bacillus subtilis, Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa was transferred to autoclaved nutrient agar media (23 g l−1), respectively. Then media were poured in respective plates and allowed to solidify. Samples (100 μM) and kanamycin drug (10 μM) were loaded on sterile paper discs (5 mm) and these discs were placed on Petri plates at appropriate distances. The plates were incubated for 24 hours at 37 °C, and zones of inhibition were measured using a Vernier calliper.
5.6. Antifungal assay protocol
Antifungal potential of the newly synthesised compounds was determined by disc diffusion assay as reported earlier.39 For experiment, 1% inoculum of Candida albicans and Aspergillus fumigatus was transferred to autoclaved Sabouraud dextrose agar (65 g l−1), respectively. Then media were poured in respective plates and allowed to solidify. Samples (100 μM) and terbinafine drug (10 μM) were loaded on sterile paper discs (5 mm) and these discs were placed on Petri plates at appropriate distances. The plates were incubated for 24–48 hours at 28 °C and zones of inhibition were measured using a Vernier calliper.
5.7. Docking analysis
The ligand was developed by using Chemdraw from which it was converted to SDF file. The receptors alpha amylase, alpha glucosidase and urease were downloaded from the RCSB site as PDB files having codes 2QMK, 5NN3 and 4AC7, respectively. These receptors and ligand files were prepared using the software BIOVIA DISCOVERY STUDIO Client 2020 and MGL Tools 1.5.6 and saved as PDBQT file. The docking was performed via AUTODOCK VINA. The results were analyzed by PyMOL. Docking results were interpreted in terms of binding energies, number of binding interactions, binding amino acids and distance of the ligand from the protein during interactions.40
6. Conclusions
Pyrazoline hybrid acyl thioureas (5a–j) were synthesized and evaluated biologically for α-glucosidase, amylase, and urease inhibition activities along with antioxidant studies. The synthesized compounds were also screened for potential antibacterial and anti-fungal inhibition zones. The compound 5d has an excellent bacterial inhibition zone against Escherichia coli, Bacillus subtilis and Staphylococcus aureus as compared to the standard. Compound 5i is the only compound which showed antifungal activity against Aspergillus fumigatus. All the synthesized analogs showed moderate activity against amylase, glucosidase, urease, and DPPH. From the corresponding synthesized compounds 5a, 5b, 5g, 5f and 5j demonstrate potential activities as compared to the standard. Based on the structure–activity relationship (SAR) studies it was found that the analogs having electron withdrawing groups, such as chlorine, fluorine and bromine, display potent α-glucosidase, urease, and α-amylase inhibition as well as antioxidant activity. The compounds having electron donating groups such as methyl were found to have a decreased activity. Furthermore, molecular docking was used for examining the many forms of binding interactions that may occur during the interaction of the synthetic compounds within the active site.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- G. Shabir, I. Shafique and A. Saeed, J. Heterocycl. Chem., 2022, 59(10), 1669–1702 CrossRef CAS.
- A. Saeed, R. Qamar, T. A. Fattah, U. Flörke and M. F. Erben, Res. Chem. Intermed., 2017, 43, 3053–3093 CrossRef CAS.
- B. Varghese, S. N. Al-Busafi, F. O. Suliman and S. M. Al-Kindy, RSC Adv., 2017, 7(74), 46999–47016 RSC.
- A. Saeed, P. A. Mahesar, P. A. Channar, Q. Abbas, F. A. Larik, M. Hassan and S. Y. Seo, Bioorg. Chem., 2017, 74, 187–196 CrossRef CAS PubMed.
- A. Saeed, P. A. Mahesar, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan and S. Y. Seo, Chem. Biodiversity, 2017, 14(8), 1700035 CrossRef PubMed.
- M. Faisal, A. Saeed, S. Hussain, P. Dar and F. A. Larik, J. Chem. Sci., 2019, 131, 1–30 CrossRef CAS.
- M. Balkenhohl, S. Kölbl, T. Georgiev and E. M. Carreira, JACS Au, 2021, 1(7), 919–924 CrossRef CAS PubMed.
- O. Unsal-Tan, T. T. Küçükkılınç, B. Ayazgök, A. Balkan and K. Ozadali-Sari, MedChemComm, 2019, 10(6), 1018–1026 RSC.
- M. J. Ahsan, J. G. Samy, H. Khalilullah, M. A. Bakht and M. Z. Hassan, Eur. J. Med. Chem., 2011, 46(11), 5694–5697 CrossRef CAS PubMed.
- W. Akhtar, A. Marella, M. M. Alam, M. F. Khan, M. Akhtar, T. Anwer and M. Shaquiquzzaman, Arch. Pharm., 2021, 354(1), 2000116 CrossRef CAS PubMed.
- M. J. Ahsan, J. Govindasamy, H. Khalilullah, G. Mohan, J. P. Stables, C. Pannecouque and E. De Clercq, Bioorg. Med. Chem. Lett., 2012, 22(23), 7029–7035 CrossRef CAS PubMed.
- Q. ul Ain, A. Saeed, A. Khan, A. Ahmed, S. Ullah, S. A. Halim and A. Al-Harrasi, Bioorg. Chem., 2023, 131, 106302 CrossRef CAS PubMed.
- R. Singh, S. Thota and R. Bansal, ACS Chem. Neurosci., 2018, 9(2), 272–283 CrossRef CAS PubMed.
- P. A. Channar, A. Saeed, M. F. Erben, F. A. Larik, S. Riaz, U. Flörke and M. Arshad, J. Mol. Struct., 2019, 1191, 152–157 CrossRef CAS.
- B. Nehra, S. Rulhania, S. Jaswal, B. Kumar, G. Singh and V. Monga, Eur. J. Med. Chem., 2020, 205, 112666 CrossRef CAS PubMed.
- A. Mumtaz, A. Majeed, S. Zaib, S. U. Rahman, S. Hameed, A. Saeed and J. Iqbal, Bioorg. Chem., 2019, 90, 103036 CrossRef CAS PubMed.
- N. Gökhan-Kelekçi, S. Yabanoğlu, E. Küpeli, U. Salgın, Ö. Özgen, G. Uçar and A. A. Bilgin, Bioorg. Med. Chem., 2007, 15(17), 5775–5786 CrossRef PubMed.
- M. Johnson, B. Younglove, L. Lee, R. LeBlanc, H. Holt Jr, P. Hills and M. Lee, Bioorg. Med. Chem. Lett., 2007, 17(21), 5897–5901 CrossRef CAS PubMed.
- M. Faisal, A. Saeed, D. Shahzad, T. A. Fattah, B. Lal, P. A. Channar and F. A. Larik, Eur. J. Med. Chem., 2017, 141, 386–403 CrossRef CAS PubMed.
- A. Samrot and A. Vijay, Int. J. Microbiol., 2008, 6(2), 1–6 Search PubMed.
- X. Qin, L. Ren, X. Yang, F. Bai, L. Wang, P. Geng and Y. Shen, J. Struct. Biol., 2011, 174(1), 196–202 CrossRef CAS PubMed.
- A. Saeed, P. A. Mahesar, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan and S. Y. Seo, Chem. Biodiversity, 2017, 14(8), 1700035 CrossRef PubMed.
- P. A. Channar, R. D. Alharthy, S. A. Ejaz, A. Saeed and J. Iqbal, Molecules, 2022, 27(24), 8723 CrossRef CAS PubMed.
- A. Saeed, M. Bolte, M. F. Erben and H. Pérez, CrystEngComm, 2015, 17(39), 7551–7563 RSC.
- A. Saeed, S. Zaib, A. Pervez, A. Mumtaz, M. Shahid and J. Iqbal, Med. Chem. Res., 2015, 22, 3653–3662 CrossRef.
- A. Saeed, S. Zaib, A. Pervez, A. Mumtaz, M. Shahid and J. Iqbal, Med. Chem. Res., 2013, 22, 3653–3662 CrossRef CAS.
- A. Saeed, H. Rafique, A. Hameed and S. Rasheed, J. Pharm. Chem., 2008, 42, 191–195 CrossRef CAS.
- N. Arshad, A. Saeed, F. Perveen, R. Ujan, S. I. Farooqi, P. A. Channar and T. Hökelek, Bioorg. Chem., 2021, 109, 104707 CrossRef CAS PubMed.
- A. Ahmed, I. Shafique, A. Saeed, G. Shabir, A. Saleem, P. Taslimi and M. Z. Hashmi, Eur. J. Med. Chem., 2022, 6, 100082 CAS.
- A. Saeed, R. Qamar, T. A. Fattah, U. Flörke and M. F. Erben, Res. Chem. Intermed., 2017, 43, 3053–3093 CrossRef CAS.
- A. Saeed, M. F. Erben and M. Bolte, Spectrochim. Acta, Part A, 2013, 102, 408–413 CrossRef CAS PubMed.
- M. Shabbir, I. Ahmad, H. Ismail, S. Ahmed, V. McKee, Z. Akhter and B. Mirza, Polyhedron, 2017, 133, 270–278 CrossRef CAS.
- B. Iftikhar, K. Javed, M. S. U. Khan, Z. Akhter, B. Mirza and V. Mckee, J. Mol. Struct., 2018, 1155, 337–348 CrossRef CAS.
- S. Naseem and H. Ismail, BMC Complementary Med. Ther., 2022, 22(1), 30 CrossRef CAS PubMed.
- M. Sajid, M. R. Khan, H. Ismail, S. Latif, A. A. Rahim, R. Mehboob and S. A. Shah, J. Ethnopharmacol., 2020, 251, 112544 CrossRef CAS PubMed.
- A. Mushtaq, S. Ali, M. N. Tahir, H. Ismail, B. Mirza, M. Saadiq and M. Iqbal, Acta Chim. Slov., 2017, 64(2), 397–408 CrossRef CAS PubMed.
- N. Arshad, U. Parveen, P. A. Channar, A. Saeed, W. S. Saeed, F. Perveen and I. Khan, Molecules, 2023, 28(6), 2707 CrossRef CAS PubMed.
- M. Shabbir, I. Ahmad, H. Ismail, S. Ahmed, V. McKee, Z. Akhter and B. Mirza, Polyhedron, 2017, 133, 270–278 CrossRef CAS.
- F. A. Larik, A. Saeed, M. Faisal, P. A. Channar, S. S. Azam, H. Ismail and B. Mirza, Comput. Biol. Chem., 2018, 77, 193–198 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Atteeque Ahmeda,
Main Bilal Haidera,
Hammad Ismailb,
Khizar Hayatc,
Ghulam Shabira and
Hesham R. El-Seedide
*a,
Atteeque Ahmeda,
Main Bilal Haidera,
Hammad Ismailb,
Khizar Hayatc,
Ghulam Shabira and
Hesham R. El-Seedide