
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 2-amino-9H-chromeno[2,3-d]thiazol-9-ones with anti-inflammatory activity via cascade reactions of 2-amino-3 iodochromones with amines and carbon disulfide†
Jiangtao Tan‡
ab,
Yifan He‡ab,
Yu Lina,
Yuanchen Zhonga,
Shijun He*ac,
Jianping Zuoab and
Chunhao Yang *ab
*ab
aState Key Laboratory of Drug Research, Shanghai Institute of Materia, Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: heshijun@shutcm.edu.cn; chyang@simm.ac.cn
bSchool of Pharmacy, University of Chinese Academy of Sciences, No. 19A YuquanRoad, Beijing 100049, China
cInnovation Research Institute of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China
First published on 19th January 2024
Abstract
A simple and efficient synthetic approach to 2-amino-9H-chromeno[2,3-d]thiazol-9-ones via copper-promoted cascade reactions was developed. The reaction employed easily available 2-amino-3-iodochromones and amines as substrates and the targeting tricyclic compounds could be obtained with moderate to good yields. Even more important, several synthesized compounds exhibited potent anti-inflammatory activities, which suggested that this protocol may provide valuable hits for drug development in the future.
Introduction
Chromones (4H-chromen-4-ones) are an important class of oxygen-containing heterocyclic compounds with a benzopyrone scaffold that are ubiquitous in nature, particularly in plants.1,2 Chromone derivatives exhibit various pharmacological activities, such as anti-inflammatory,3,4 antimicrobial,5 antifungal,6 antiviral,7 anti-cancer,8 anti-oxidant,9 and so on. Chromones are also useful building blocks for constructing diverse and complex heterocycles.10–17 Among the chromone containing scaffolds, tricyclic chromone fused heterocycle compounds are quite important. Some of the tricyclic compounds are recurrent drugs or drug candidates, for example, the anti-inflammatory and anti-allergic drug amlexanox (1),18 the anti-tubercular compound 2,19 the antimicrobial compound 3,20 and the antitumor compounds 4 (ref. 21) and 5 (ref. 22) (Fig. 1). Although there has been extensive research on chromone-fused six-membered heterocycles, the synthesis and pharmaceutical activities of five-membered heterocycle fused chromones have been seldomly reported so far.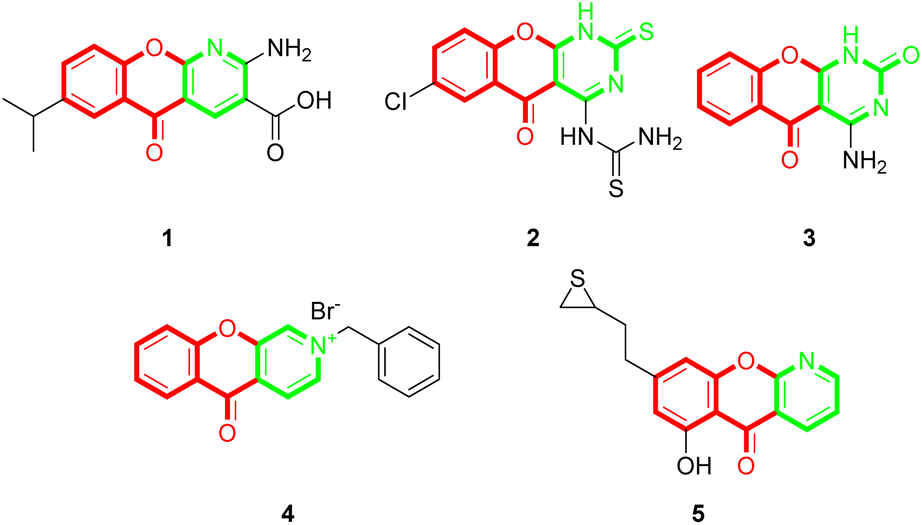 | ||
| Fig. 1 Representative examples of chromone-fused tricyclic derivatives with pharmaceutical activities. | ||
Thiazole is a common heterocyclic skeleton in drug molecules that exhibits various biological activities and excellent physical and chemical properties.23 Among the thiazole derivatives, 2-aminothiazoles are unique and significant in the field of bio-active compounds and drugs, such as the antibacterial drug sulfathiazole,24 the antifungal drug dimazole,25 the antitumor drug dasatinib,26 and the non-steroidal anti-inflammatory drug meloxicam27 (Fig. 2). Based on the broad applications of tricyclic heterocycle-fused chromone and 2-aminothiazole scaffolds in pharmaceutical researches, we aimed to synthesize novel chromone-fused 2-aminothiazole compounds and tried to discover their pharmacological activity from these 2-amino-9H-chromeno[2,3-d]thiazol-9-ones (Scheme 1).
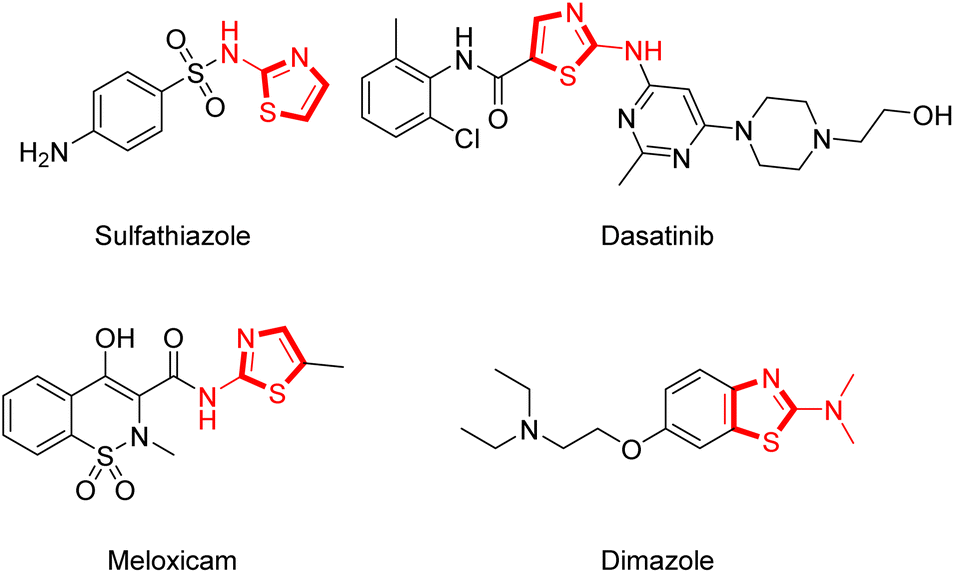 | ||
| Fig. 2 Representative drugs with 2-aminothiazole scaffold. | ||
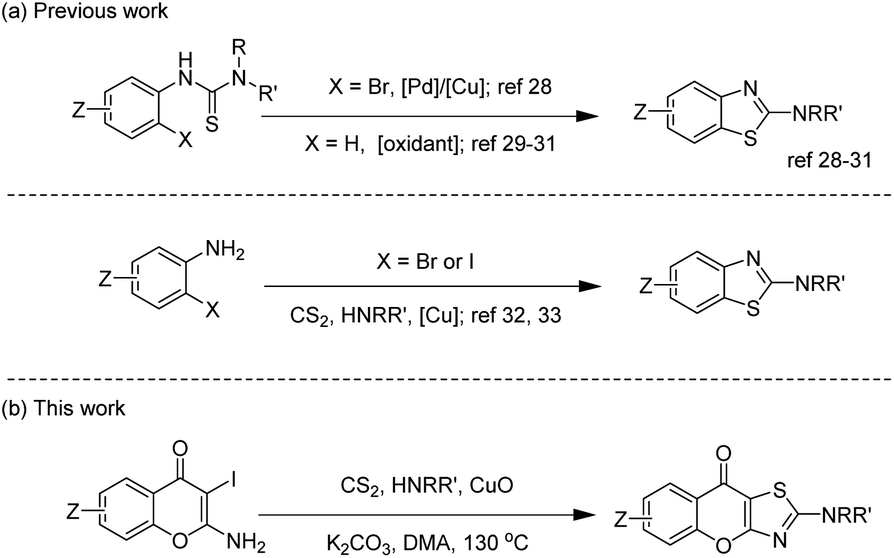 | ||
| Scheme 1 (a) Synthetic strategies of 2-aminobenzothiazole derivatives. (b) Our strategy for synthesis of 2-amino-9H-chromeno[2,3-d]thiazol-9-ones. | ||
2-Aminobenzothiazoles, as simple 2-aminothiazole-fused derivatives, are commonly synthesized in two ways, employing phenylthiourea28–31 and 2-halogenated aniline32,33 as starting materials. The synthesis of 2-amino-9H-chromeno[2,3-d]thiazol-9-ones using the first method requires the prior synthesis of the starting materials 1-(4-oxo-4H-chromen-2-yl)thioureas, which involves tedious procedures. In 2011, Ma's group32 reported an efficient method for preparing 2-N-substituted benzothiazole derivatives through a copper-mediated three-component reaction of ortho-iodoanilines with carbon disulfide and amines. In 2014, inspired by Ma's work, our group reported the synthesis of 2-C-substituted benzothiazoles via a copper-promoted domino reaction.34 Based on the research mentioned above, we proposed using 2-amino-3-iodo-4H-chromen-4-ones which are easy for preparation as substrates to synthesize 2-amino-9H-chromeno[2,3-d]thiazol-9-ones. After successfully constructing this privileged scaffold, we conducted anti-inflammatory tests due to the similar structure of amlexanox. Actually, the results showed several 2-amino-9H-chromeno[2,3-d]thiazol-9-ones had good anti-inflammatory activity.
Results and discussion
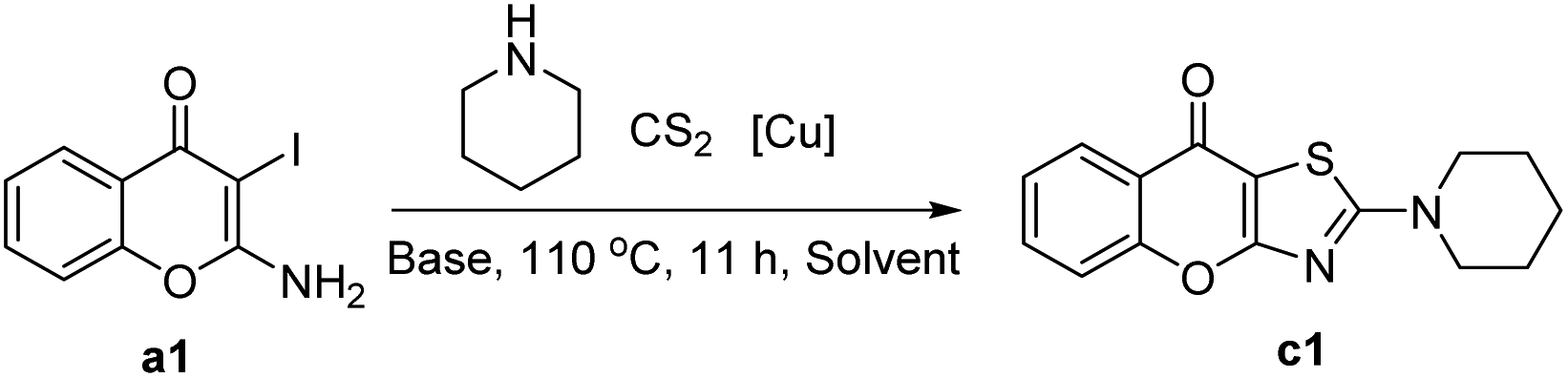
We initially chose 2-amino-3-iodo-4H-chromen-4-one a1 and piperidine b1 as the model substrates using optimal reaction condition reported by Ma.32 Although we were able to detect the target product c1, the yield was only 25% (Table 1, entry 1). Later, several copper salts were screened, namely CuCl2·2H2O, CuSO4·5H2O, CuO, CuBr2, Cu(AcO)2·H2O, Cu2O and CuI (Table 1, entries 2–7). Among these copper salts, CuO was optimal and provided a 50% yield of the desired compound c1. Subsequently, we investigated the influence of several common inorganic bases (Table 1, entries 8–10) in the reaction under the CuO condition. Compared to potassium carbonate, it was found that stronger and weaker bases all gave negative impacts on the reaction. Therefore, potassium carbonate was chosen to be the optimal base. There was only a slight change in yield when the reaction time was reduced from 11 hours to 1 hour (Table 1, entries 11). Notably, increasing the reaction temperature to 130 °C resulted in an increased yield of 56% (Table 1, entries 12). Moreover, by increasing the equivalent of carbon disulfide and piperidine (Table 1, entries 13), the yield was also obviously improved. We next screened several polar aprotic solvents (entries 14–16) and observed a slight reduction when using N-methylpyrrolidone (NMP) and dimethyl sulfoxide (DMSO) as solvents. In contrast, the yield was improved when the N,N-dimethylacetamide (DMA) was used. After combining these optimal conditions, the yield of target compound c1 reached 81% (Table 1, entries 17).
|
||||
|---|---|---|---|---|
| Entry | [Cu] | Base | Solvent | Yieldb (%) |
| a Reaction conditions: a (0.35 mmol), b (0.52 mmol), CuO (0.35 mmol), K2CO3 (1.05 mol), and CS2 (0.42 mmol) at 110 °C for 11 h.b Isolated yields.c T = 110 °C, time = 1 h.d T = 130 °C, time = 1 h.e CS2 (0.7 mmol), piperidine (1.05 mmol). | ||||
| 1 | CuCl2·2H2O | K2CO3 | DMF | 25 |
| 2 | CuSO4·5H2O | K2CO3 | DMF | 15 |
| 3 | CuO | K2CO3 | DMF | 50 |
| 4 | CuBr2 | K2CO3 | DMF | 24 |
| 5 | Cu(AcO)2·H2O | K2CO3 | DMF | 18 |
| 6 | Cu2O | K2CO3 | DMF | 28 |
| 7 | CuI | K2CO3 | DMF | 0 |
| 8 | CuO | CS2CO3 | DMF | 27 |
| 9 | CuO | Na2CO3 | DMF | 36 |
| 10 | CuO | KHCO3 | DMF | 39 |
| 11c | CuO | K2CO3 | DMF | 49 |
| 12d | CuO | K2CO3 | DMF | 56 |
| 13e | CuO | K2CO3 | DMF | 59 |
| 14 | CuO | K2CO3 | NMP | 43 |
| 15 | CuO | K2CO3 | DMSO | 39 |
| 16 | CuO | K2CO3 | DMA | 55 |
| 17d,e | CuO | K2CO3 | DMA | 81 |
After determining the optimal reaction conditions, we investigated the scope of the substrates by employing various amines (Table 2). From the results, most target compounds could be obtained with moderate to good yields. Compared with piperidine, the yields of pyrrolidine and alkyl amines were decreased to moderate level (Table 2, c2–c4). For thiomorpholine, the yield was significantly reduced to 41%. While most piperazines were smoothly transferred under the reaction condition with acceptable results (Table 2, c6–c10). For substituted piperidine, 4,4-difluoropiperidine gave an excellent yield of 80% (Table 2, c11). Meanwhile, most substituents on piperidine like amino, dimethyl hydroxyl, and phenyl resulted in moderate yields (Table 2, c12–c14, c17, c19–c21). However, the spiro compounds were obtained with relatively low yields (Table 2, c15 and c16). This may be attributed to the instability of spiro oxacyclobutyl and azacyclobutyl groups under high temperatures with transition metal. Notably, when pyrrole was employed as the substrate, the desired compound could not be obtained. Our LC-MS analysis revealed that amount of pyrrole did not react, leading us to speculate that the relatively low density of electron clouds on the nitrogen atom of pyrrole resulted in decreased nucleophilic performance. After the replacement of potassium carbonate to sodium hydroxide, compound c24 was synthesized with a yield of 38%. We tried to expand substrates from secondary amines to primary amines, but only a trace amount of the target compound was detected. Nevertheless, the tandem reaction proceeded smoothly when p-methoxybenzyl (–PMB) was utilized as protecting group (Table 2, c25). After removing –PMB, the target compound c25 was finally obtained with a total yield of 32%. In summary, the yields of different substrates in Table 2 varied due to many factors including nucleophilicity, steric hindrance and stability of the amines.

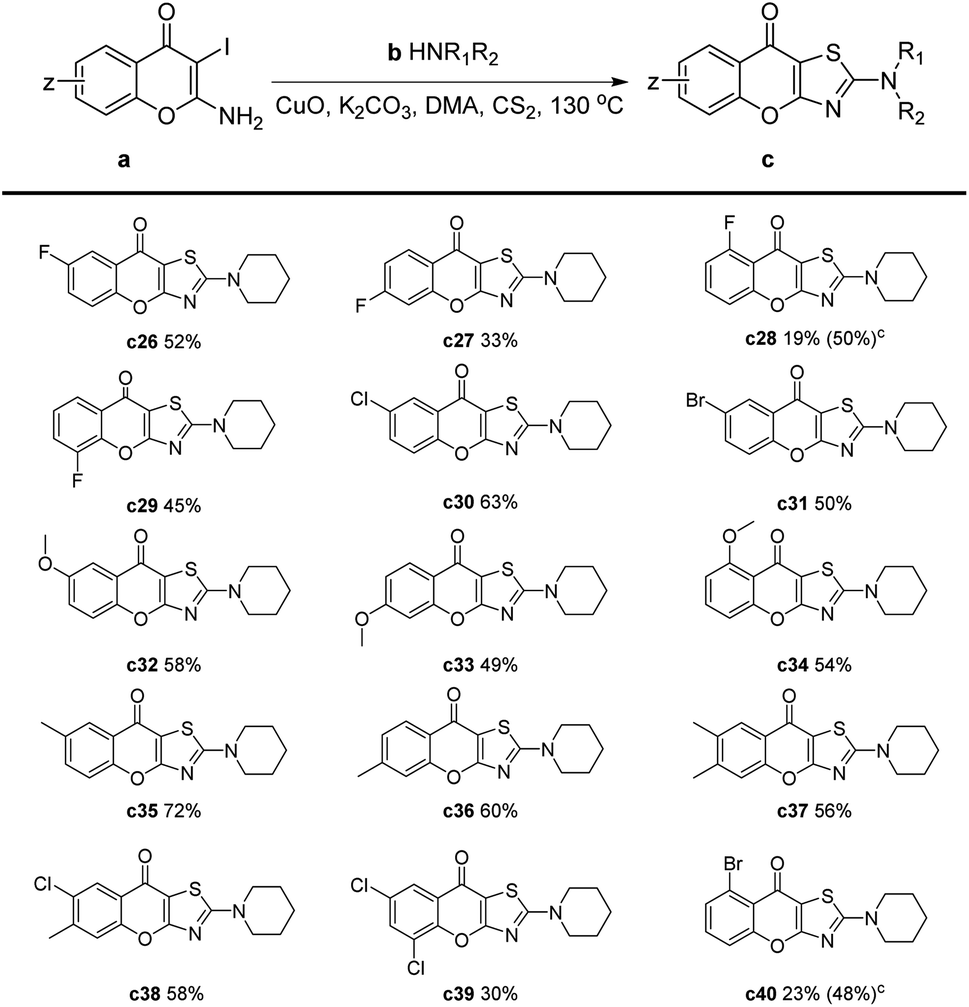
Later, we investigated the impact of different substituents on the chromones for this reaction (Table 3). The results showed that electron-withdrawing substituents, such as fluorine, chlorine, and bromine, had an adverse effect on the reaction (Table 3, c26–c31, c38–c40). Specifically, when these substituents were located at the C-5 position, the yield of –F and –Br substitution was only 19% and 23%, respectively (Table 3, c28 and c40). This may be attributed to the side reaction of the amines with halogen atoms which were on the ortho-position of a ketone. Therefore, we performed the reaction at 90 °C and extended the reaction time to 4 hours, the yields of c28 and c40 were significantly increased to 50% and 48%, respectively. Substrates with an electron-donating group reacted smoothly to give products in moderate to good yields. In the cases of methyl or methoxy substituents, slightly decreased yields could be obtained compared to the unsubstituted chromones (Table 3, c32–c37).
According to the available literature,32,33 a plausible reaction mechanism was outlined in Scheme 2. Initially, the amines react with carbon disulfide in the presence of base, resulting in the formation of dithiocarbamate salts 4. Subsequently, the dithiocarbamate salts act as a coupling agent and participate in Ullmann coupling reaction with 2-amino-3-iodo-4H-chromen-4-ones a mediated by copper(II) oxide, leading to the generation of dithiocarbamates 5. Then, the amino at the C-2 position of the chromones undergoes intramolecular nucleophilic addition to form intermediates 6. Finally, the target product, chromone[2,3-d]thiazole derivatives c, are obtained by intramolecular elimination of hydrogen sulfide. To verify this hypothesis, we conducted a series of stepwise experiments. Initially, we employed piperidine, carbon disulfide, and potassium carbonate in DMA at room temperature for 0.5 hours under Ar to verify the formation of intermediate 4-1 by LC-MS. After that, we added copper oxide and a1 to the mixture and kept the reaction at room temperature for another 12 hours. The intermediate 5-1 was also successfully detected by LC-MS.
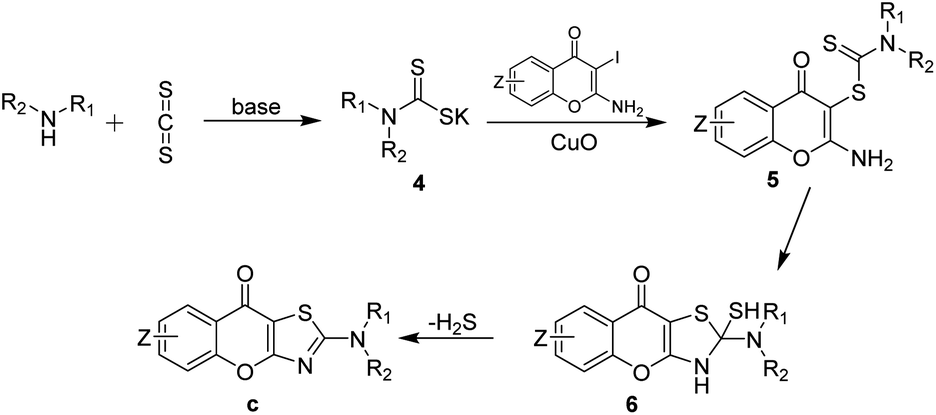 | ||
| Scheme 2 Proposed mechanism. | ||
We next tested the anti-inflammatory activity of these compounds as planned. Lipopolysaccharide (LPS) is widely used to establish inflammation models due to the ability of stimulating various cell types to release inflammatory cytokines, such as IL-1β, IL-6, and TNF-α.35 We assessed our compounds on the level of inflammatory cytokine IL-1β on LPS induced RAW264.7 cells. Among these compounds, c1, c3, c12, c14, c30, and c39 showed good anti-inflammatory activity (Table 4). The IC50 values of these compounds were lower than the positive control diacerein which was known as a typical IL-1β inhibitor. Compound c12 exhibited the most potent activity (IC50 = 8.19 μM) and showed excellent safety with extremely low cytotoxicity (CC50 = 84.64 μM, Fig. 3a and b). Subsequently, we further deepened the investigation of the anti-inflammatory mechanism of compound c12. The results showed that c12 also performed potent suppression on the production of IL-6 (IC50 = 12.62 μM) but weaker inhibition on TNF-α (IC50 = 38.62 μM). Furthermore, since both NF-κB and c-Jun N-terminal kinase (JNK) signallings are involved in regulating immune responses and the secretion of inflammatory cytokines,36 we analysed the impact of c12 on these two pathways. As shown in Fig. 3c and d, c12 significantly inhibited the phosphorylation of NF-κB and IκB proteins rather than JNK. It meant that, compound c12 suppressed the release of IL-1β by acting on a specific target through the NF-κB pathway and this result may provide a clue for drug development.
| Compounds | CC50 (μM) | IC50 (μM) | |
|---|---|---|---|
| IL-1β | SI | ||
| c1 | 51.40 | 9.64 | 5.33 |
| c3 | 47.81 | 16.98 | 2.82 |
| c12 | 84.64 | 8.19 | 10.33 |
| c14 | 107.50 | 24.40 | 4.41 |
| c30 | >200 | 35.43 | >5.64 |
| c39 | >200 | 22.22 | >9.00 |
| Diacerein | 151.4 | 26.57 | 5.70 |
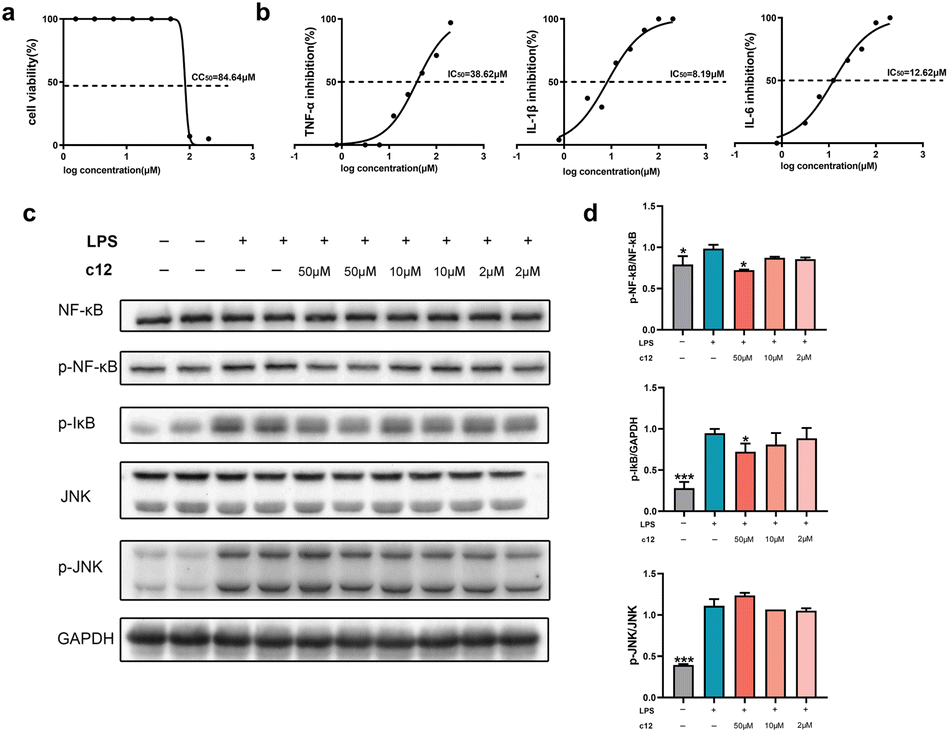 | ||
| Fig. 3 c12 inhibits the secretion of IL-1β and IL-6 in LPS-stimulated RAW264.7 cells by suppressing phosphorylation of NF-κB/IκB. (a) Cell viability of RAW264.7 cells cultured with c12 for 48 h. CC50 represented the concentration of c12 that reduced cell viability to 50%. (b) Inhibition of cytokines production by c12 in the 48 h culture of LPS-stimulated RAW264.7 cells. IC50 value represented the concentration of c12 that reduced the cytokine production by 50%. (c) Immunoblotting of NF-κB, p-NF-κB, p-IκB, JNK, p-JNK in RAW264.7 cells following LPS stimulation with or without c12 treatment. (d) Protein expression levels were normalized against the indicated protein. Data are given as mean values of two independent experiments ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 vs. cells cultured with LPS only. | ||
Conclusions
In conclusion, we have developed a novel copper(II) oxide promoted cascade reaction of 2-amino-3-iodo-4H-chromen-4-ones with secondary amines and carbon disulfide in the presence of bases, leading to the formation of 2-amino-9H-chromeno[2,3-d]thiazol-9-ones. The reaction is simple and efficient, and moderate to good yields can be obtained for most tested substrates. In addition, several compounds with good anti-inflammatory activities were obtained from the synthesized molecules, especially compound c12. Therefore, this method can be used for constructing a compound library of natural-like molecules with pharmacological activity.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from the Science and Technology Commission of Shanghai Municipality (18431907100), the State Key Laboratory of Drug Research Program (SIMM2103ZZ-03).Notes and references
- A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS PubMed
.
- R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS PubMed
- A. E. Sherif, Y. Amen, K. Shimizu and S. Afr, J. Bot., 2022, 147, 467–471 CAS
- Y. Liu, X. Yu, W. Zhang, T. Wang, B. Jiang, H. Tang, Q. Su and Y. Fu, Bioorg. Chem., 2020, 101, 104030 CrossRef CAS PubMed
- M. Hiruy, D. Bisrat, A. Mazumder and K. Asres, Nat. Prod. Res., 2021, 35, 1052–1056 CrossRef CAS PubMed
- O. Prakash, R. Kumar and V. Parkash, Eur. J. Med. Chem., 2008, 43, 435–440 CrossRef CAS PubMed
- M. K. Kim, H. Yoon, D. L. Barnard and Y. Chong, Chem. Pharm. Bull., 2013, 61, 486–488 CrossRef CAS PubMed
- A. Kantankar, Y. Jayaprakash Rao, G. Mallikarjun, Y. Hemasri and R. R. Kethiri, J. Mol. Struct., 2021, 1239, 130502 CrossRef CAS
- M. Kuroda, S. Uchida, K. Watanabe and Y. Mimaki, Phytochemistry, 2009, 70, 288–293 CrossRef CAS PubMed
- P. Tong, Z. Sun, S. Wang, Y. Zhang and Y. Li, J. Org. Chem., 2019, 84, 13967–13974 CrossRef CAS
- T. Dai, C. Cui, X. Qi, Y. Cheng, Q. He, X. Zhang, X. Luo and C. Yang, Org. Biomol. Chem., 2020, 18, 6162–6170 RSC
- D. R. Joshi and I. Kim, J. Org. Chem., 2021, 86, 10235–10248 CrossRef CAS PubMed
- S. Li, L. Zhang, Q. He, X. Zhang and C. Yang, Org. Biomol. Chem., 2021, 19, 5348–5352 RSC
- W. Zhu, Y. Fang, W. Han, F. Li, M. Yang and Y. Chen, Org. Chem. Front., 2021, 8, 3082–3090 RSC
- L. Meng, H. Liu, Z. Lin and J. Wang, Org. Lett., 2022, 24, 5890–5895 CrossRef CAS PubMed
- T. Wang, B. Zhang, L. Hu, H. Sun, Y. Wang, H. Zhai and B. Cheng, J. Org. Chem., 2022, 87, 1348–1356 CrossRef CAS PubMed
- J. W. Attard, J. R. Noel, Y. Guan and A. E. Mattson, Org. Lett., 2023, 25, 2450–2455 CrossRef CAS PubMed
- H. Makino, T. Saijo, Y. Ashida, H. Kuriki and Y. Maki, Int. Arch. Allergy Immunol., 1987, 82, 66–71 CrossRef CAS PubMed
- D. D. Haveliwala, N. R. Kamdar, P. T. Mistry and S. K. Patel, J. Sulfur Chem., 2011, 32, 451–462 CrossRef CAS
- D. D. Haveliwala, N. R. Kamdar, P. T. Mistry and S. K. Patel, Nucleosides, Nucleotides Nucleic Acids, 2014, 33, 80–91 CrossRef CAS PubMed
- A. Bizarro, D. Sousa, R. T. Lima, L. Musso, R. Cincinelli, V. Zuco, M. De Cesare, S. Dallavalle and M. H. Vasconcelos, Molecules, 2018, 23, 407 CrossRef PubMed
- H. Cho, M. Jung, Y. Kwon and Y. Na, Bioorg. Med. Chem. Lett., 2009, 19, 6766–6769 CrossRef CAS PubMed
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed, M. H. Abdellattif, M. S. Nafie and M. K. Hassan, Front. Chem., 2021, 9, 694870 CrossRef CAS PubMed
- R. D. Muir, V. J. Shamleffer and L. R. Jones, J. Bacteriol., 1942, 44, 95–110 CrossRef CAS PubMed
- E. Grunberg, G. Soohoo, E. Titsworth, D. Ressetar and R. J. Schnitzer, Trans. N. Y. Acad. Sci., 1950, 13, 22–27 CrossRef CAS
- Q. Yao, J. Choi, Z. Dai, J. Wang, D. Kim, X. Tang and L. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 36642–36654 CrossRef CAS PubMed
- H. Li, Y. Luo, Y. Xu, L. Yang, C. Hu, Q. Chen, Y. Yang, J. Ma, J. Zhang, H. Xia, Y. Li and J. Yang, Mol. Pharm., 2018, 15, 4121–4131 CrossRef CAS PubMed
- L. L. Joyce, G. Evindar and R. A. Batey, Chem. Commun., 2004, 446–447, 10.1039/b311591g
- A. Banerjee, S. K. Santra, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 9096–9104 CrossRef CAS
- M. Gao, J. Li, S. Zhang, L. Chen, Y. Li and Z. Dong, J. Org. Chem., 2019, 85, 493–500 CrossRef PubMed
- M. M. Katiya, M. M. Sontakke and M. G. Dhonde, Results Chem., 2022, 4, 100512 CrossRef CAS
- D. Ma, X. Lu, L. Shi, H. Zhang, Y. Jiang and X. Liu, Angew. Chem., Int. Ed., 2011, 50, 1118–1121 CrossRef CAS PubMed
- G. Satish, K. H. V. Reddy, K. Ramesh, K. Karnakar and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 2518–2521 CrossRef CAS
- H. Xiang, J. Qi, Q. He, M. Jiang, C. Yang and L. Deng, Org. Biomol. Chem., 2014, 12, 4633–4636 RSC
- W. Tong, X. Chen, X. Song, Y. Chen, R. Jia, Y. Zou, L. Li, L. Yin, C. He, X. Liang, G. Ye, C. Lv, J. Lin and Z. Yin, Exp. Ther. Med., 2020, 19, 1824–1834 CAS
- Z. Zhao, Y. Sun and X. Ruan, Phytomedicine, 2023, 114, 154781 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07209f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |