
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-synthesis, α-amylase inhibition, and kinetic and molecular docking studies of arylidene-based sesquiterpene coumarins isolated from Ferula tunetana Pomel ex Batt†
Wiem Baccaria,
Ilyes Saidia,
Insaf Filali*b,
Mansour Znatia,
Houda Lazragc,
Moncef Tounsid,
Axel Marchalef,
Pierre Waffo-Teguoef and
Hichem Ben Jannet *a
*a
aUniversity of Monastir, Faculty of Science of Monastir, Laboratory of Heterocyclic Chemistry, Natural Products and Reactivity (LR11ES39), Team: Medicinal Chemistry and Natural Products, Avenue of Environment, 5019 Monastir, Tunisia. E-mail: hichem.bjannet@gmail.com
bDepartment of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
cUniversity of Monastir, Higher Institute of Biotechnology of Monastir, Laboratory of Genetics, Biodiversity and Bioresources Valuation LR11S41, 5019 Monastir, Tunisia
dPreparatory Year Deanship, Basic Science Department, Prince Sattam Bin Abdulaziz University, Alkharj 11942, Saudi Arabia
eUniversité de Bordeaux, Institut des Sciences de la Vigne et du Vin, EA 4577, Unité de Recherche Œnologie 210 Chemin de Leysotte, CS50008, 33882 Villenave d’Ornon, France
fUniversité de Bordeaux, Bordeaux INP, Bordeaux Sciences Agro, UMR 1366 OENOLOGIE, ISVV, 33140 Villenave d’Ornon, France
First published on 5th February 2024
Abstract
Despite all the significant progresses made to enhance the efficacy of the existing bank of drugs used to manage and cure type II diabetes mellitus, there is still a need to search and develop novel bioactive compounds with superior efficacy and minimal adverse effects. This study describes the valorization of the natural bioactive sesquiterpene coumarin via the semi-synthesis of new analogs and the study of their α-amylase inhibition activity. The sesquiterpene coumarin named coladonin (1) was quantitatively isolated from the chloroform extract of endemic Ferula tunetana roots. Subsequently, the oxidation of 1 via the Jones oxidation reaction, used as a key reaction, afforded precursor 2. The condensation of oxidized coladonin (2) with various aryl aldehydes provided a series of new arylidene-based sesquiterpene coumarin derivatives (3a–m), which were characterized by NMR and ESI-HRMS experiments. All derivatives evaluated in vitro for their α-amylase inhibitory potential showed interesting α-amylase inhibition with IC50 values ranging from 7.24 to 28.98 μM. Notably, compounds 3k and 3m exhibited lower IC50 values (7.24 μM and 8.38 μM, respectively) compared to the standard (acarbose: IC50 = 9.83 μM). In addition, the structure–activity relationship (SAR) for all the compounds was studied. The most active compounds were found to be mixed-type inhibitors, which was revealed by kinetic studies. Furthermore, molecular in silico docking studies were established for all synthesized analogs with the binding site for the α-amylase enzyme.
1 Introduction
According to the WHO, with more than 578 million cases expected worldwide by 2030, diabetes mellitus (DM) is considered the most chronic metabolic disease having a high mortality rate and huge health costs.1,2 DM is characterized by abnormal blood-glucose levels (hyperglycemia) due to the developed insulin resistance (T2DM) and/or insufficient secretion of insulin (T1DM), as well as other complications such as nephropathy.3,4 One of the main approaches to managing DM involves inhibiting the α-amylase enzyme, which is responsible for cleaving carbohydrates (starch and oligosaccharides) into small sugar units, ultimately leading to hyperglycemia and the subsequent development of diabetes.5 In this regard, the inhibition of the α-amylase enzyme plays an important role in the treatment of diabetes. In fact, targeting α-amylase inhibitors with small molecules is one of the methods to find new antidiabetic drugs.6,7 In recent years, there has been increasing interest in the use of natural products as potential antidiabetic therapeutic agents.8–10Traditional medicines involve the utilization of plants belonging to the Apiaceae family, especially the genus Ferula (170 species),11 in the treatment of various diseases. Different parts of Ferula (the extracts of F. persica, F. mongolica, F. sinaica and F. ferulago) have a reputation in the treatment of diabetic diseases.12 In Tunisia, only one endemic species of the Ferula genus has been identified: F. tunetana Pomel ex Batt.13 The phytochemical features of the roots of F. tunetana include the presence of various compounds, particularly sesquiterpene coumarins (six compounds) including tunetacoumarin A, coladin, coladonin (major compound), isosamarcandin angelate, 13-hydroxyfeselol, and umbelliprenin.14 The same work reported the significant cytotoxic activity of coladonin (the major compound) against two human colon cancer cell lines: HT-29 and HCT-116. Sesquiterpene coumarins, which consist of a coumarin moiety attached to a sesquiterpene moiety, are almost exclusively found in the roots of the genus Ferula.15 In fact, sesquiterpene coumarins are the most important owing to their wide range of promising biological properties. Numerous studies have shown that this type of natural products exhibit excellent anti-diabetic effects (Fig. 1).16,17
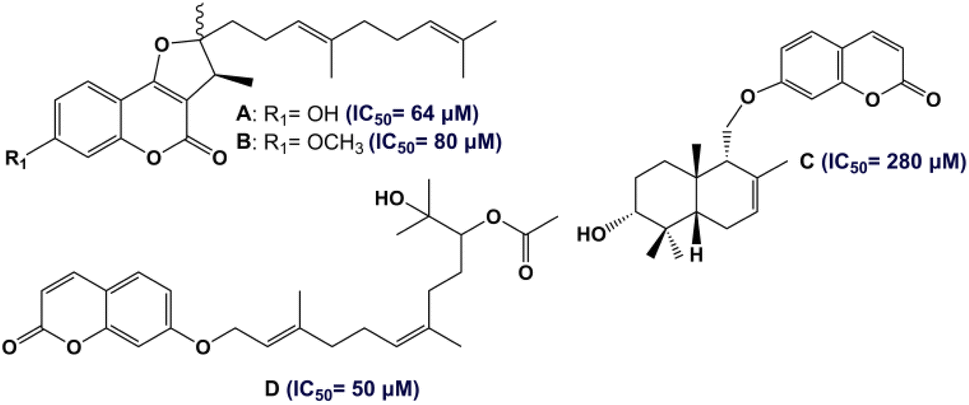 | ||
| Fig. 1 Structures of antidiabetic (anti-α-glucosidase) sesquiterpene coumarins. | ||
Recently, numerous studies have demonstrated that the pharmacophore arylidene fragment has attracted significant attention due to its broad spectrum of pharmacological and biological effects. Similarly, arylidenes are known as antidiabetic agents (Fig. 2).18,19 Moreover, interest in α,β-unsaturated ketones has heightened in drug discovery, as they hold promise for pharmaceutical applications. In particular, the antidiabetic drug Epalrestat is among the remarkable compounds derived from α,β-unsaturated ketones (G in Fig. 2).20
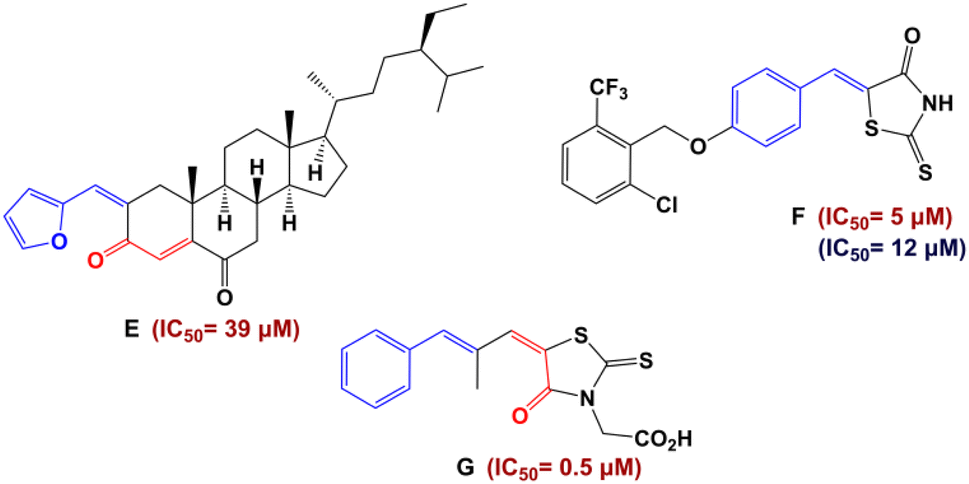 | ||
| Fig. 2 Structures of antidiabetic arylidene and α,β-unsaturated ketone derivatives. Red color indicates IC50 values against α-glucosidase and blue color IC50 values against α-amylase. | ||
Encouraged by the significant antidiabetic potential of sesquiterpene coumarins, arylidene and α,β-unsaturated ketones, and with the aim of accessing new arylidene compounds, we sought to exploit coladonin (1) isolated in sufficient quantity from the roots of F. tunetana to prepare a new series of structural analogs of arylidenes (Fig. 3).
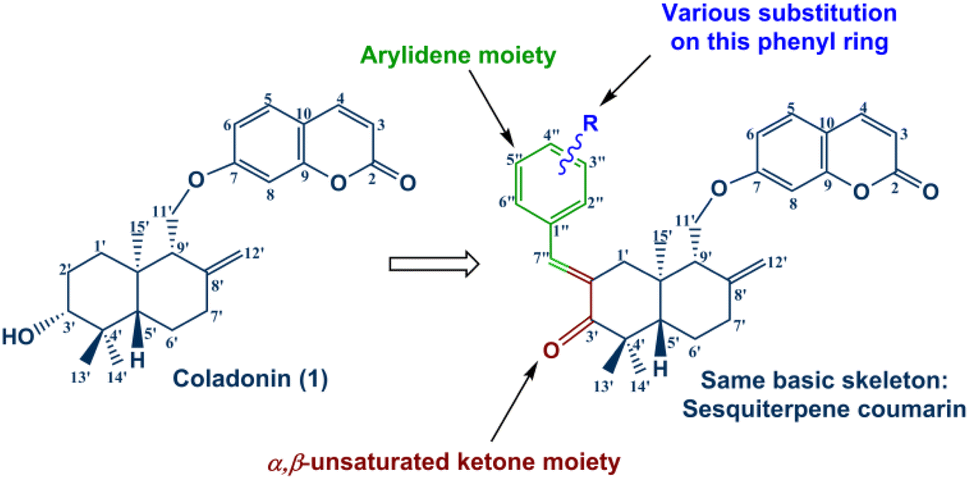 | ||
| Fig. 3 General structure of the new sesquiterpene-coumarin arylidenes. | ||
The evaluation of the α-amylase inhibition of all the synthesized compounds was studied and is reported herein. The mechanism inhibition of the most active compounds was determined via a kinetic study. Furthermore, the structure–activity relationship (SAR) was in silico approved by the molecular docking analysis.
2 Results and discussion
2.1. Chemistry
Coladonin (1) was isolated from the CHCl3 extract of F. tunetana roots. It was identified with the help of its spectroscopic data (NMR spectra (1H, HSQC and HMBC)) and by comparison with the reports in the literature.11 Indeed, analysis of the 1H NMR and HSQC spectra of this compound reveals, among other correlations, the presence of the correlation cross-peaks H-3 (δH 6.25)/C-3 (δc 113.3), H-4 (δH 7.64)/C-4 (δc 143.8), H-5 (δH 7.36)/C-5 (δc 129.0), H-6 (δH 6.83)/C-6 (δc 113.4) and H-8 (δH 6.82)/C-8 (δc 101.6), suggesting the presence of the umbelliferone skeleton, previously identified in the roots of this plant.14 The remaining signals are assigned through HSQC, COSY, HMBC, and ROESY experiments to a sesquiterpene, confirming that compound (1) is a derivative of the sesquiterpene-coumarin nature. Indeed, the complete analysis of all 1D and 2D NMR spectra, supported by bibliographic data on sesquiterpene-coumarins previously isolated from this plant, allowed us to propose the structure of coladonin. The absolute configurations were deduced from dipolar correlations observed in the ROESY spectrum between H-3′/H-13′, H-3′/H-5′, H-5′/H-9′, and H-11′/H-15′, indicating the β orientation of protons H-3′, H-5′, H-9′, and methyl 13′, and the α orientation of the methylene (CH2)-11′, as well as methyl 14′ and 15′.In our approach to access to the desired compounds 3a–m, we started first by the oxidation of the coladonin (1), via the Jones oxidation reaction. The oxidized coladonin (2) was obtained in good yields (92%) (Scheme 1).
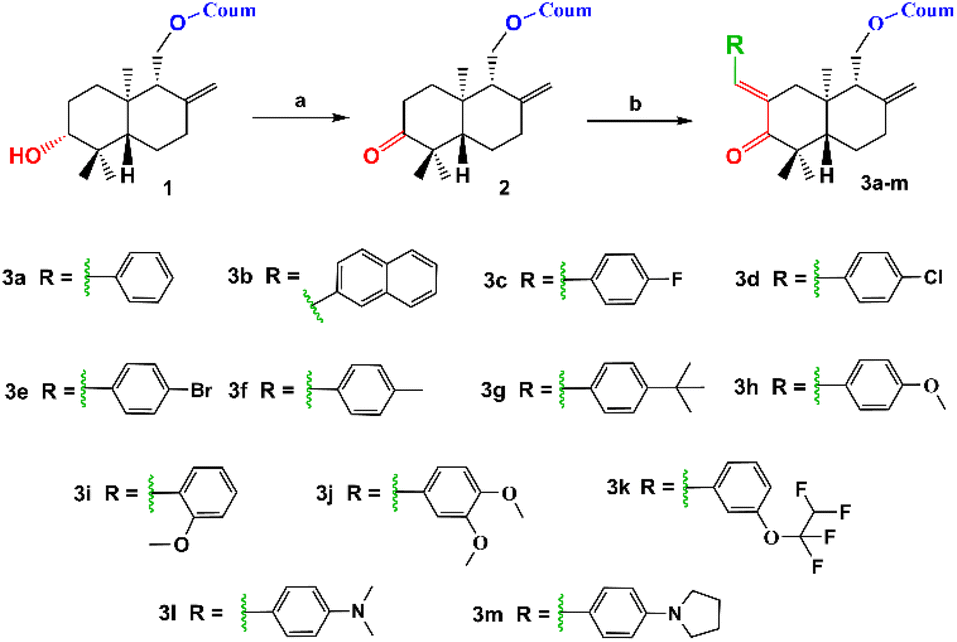 | ||
| Scheme 1 Synthetic route of 3a–m derivatives. (a) Jones reagent, acetone, 0–5 °C, 3 h and (b) RCHO, KOH, ethanol, RT, 24 h. | ||
The structure of compound 2 was established according to its spectral data (NMR spectra (1H, HSQC and HMBC) and ESI-HRMS) (Fig. 4). Indeed, the 1H NMR spectrum of compound 2 showed the absence of the signal at δH 3.31 related to the H-3′ proton compared to the 1H NMR spectrum of coladonin (1). The 13C NMR confirmed the oxidation of the secondary alcohol at C-3′ by the observation of a new signal at δC 215.9 attributable to the carbonyl group.
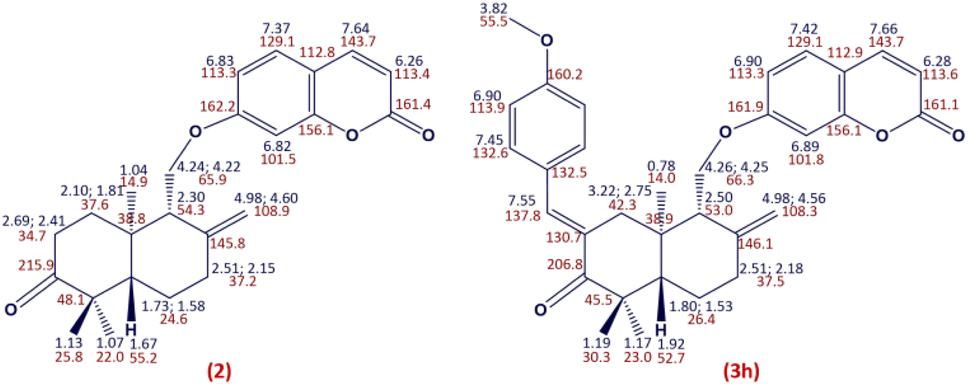 | ||
| Fig. 4 1H (blue) and 13C (red) NMR chemical shift assignments for compounds 2 and 3h. | ||
α-Methylene ketones constitute an important intermediate for the formation of diverse arylidene derivatives known for their high pharmacological and biological properties.21–23 This attracted our attention towards the use of the oxidized coladonin (2) to synthesize the new target compounds 3a–m by using the α-methylene ketone function in 2 to attach an arylidene moiety to coladonin.
By application of the CLAISENE-SCHMIDT reaction, the oxidized coladonin (2) was reacted with a series of aryl aldehydes at room temperature in ethanol for 24 hours, to give 3a–m in good yields (72–93%) (Scheme 1). The structures of 3a–m were established by their spectral data (NMR spectra (1H, HSQC and HMBC) and ESI-HRMS). In fact, the 1H NMR spectra of these derivatives showed, in addition to the signals of the oxidized coladonin 2 moiety, the appearance of a new doublet at δH 7.45–7.75 due to ethylenic proton H-7′′ and also other signals at δH 6.70–7.90 attributable to aromatic protons introduced by the used aryl aldehydes. As an example, consider the compound (3h); the analysis of its 1H NMR spectrum primarily revealed the appearance of a doublet at δH 7.55 (1H, d, J = 1.8 Hz), attributable to the ethylenic proton H-7′′. Additionally, two doublets at δH 6.90 (2H, d, J = 8.8 Hz) and 7.45 (2H, d, J = 8.8 Hz) were assigned to the aromatic protons of the para-substituted aromatic ring, and a singlet at δH 3.82 (3H, s) was identified for the methoxy group protons. The HSQC spectrum confirms these spectral data by revealing a new correlation cross-peak with signals at δC 113–138, assigned to the carbons of the methine groups of the used aryl aldehyde (Fig. 4).
The stereochemistry (E) of the exocyclic double bond Δ2′,7′′ thus adopted in compounds 3a–m was confirmed by ROESY spectra showing a NOE between H-1′ and H-2′′,6′′-atom (Fig. S1†).
2.2. Evaluation of α-amylase inhibition
Among the therapeutic strategies to reduce hyperglycemia is the retarded glucose absorption by inhibiting the activity of α-amylase.24,25 Meanwhile, all compounds 1, 2 and 3a–m were subjected to the evaluation of their in vitro α-amylase inhibition. Acarbose served as a standard in this investigation for comparison. The results are expressed in IC50 (μM) and reported in Fig. 5.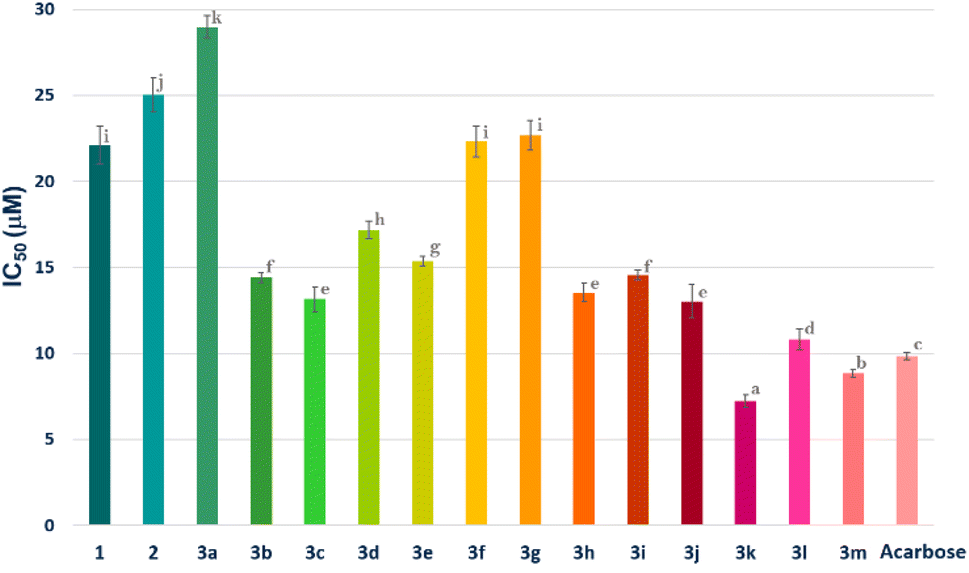 | ||
| Fig. 5 α-Amylase inhibition (IC50 μM) of compounds 1, 2, 3a–m and acarbose. Compounds with the same letter do not have significantly different IC50 values at a p-value less than 0.05. | ||
According to the results, all compounds (1, 2 and 3a–m) showed promising activity against the α-amylase enzyme with IC50 values ranging from 7.24 ± 0.36 to 28.98 ± 0.65 μM. In fact, the derivatives 3k, 3l and 3m (IC50 = 7.24 ± 0.36, 10.82 ± 0.60 and 8.83 ± 0.22 μM, respectively), followed by 3c, 3h, 3b, 3i, 3e and 3d (IC50 = 13.16 ± 0.73, 13.54 ± 0.54, 14.41 ± 0.30, 14.55 ± 0.30, 15.37 ± 0.29 and 17.17 ± 0.50 μM, respectively), showed the highest inhibition potential. Moreover, compounds 3f, 3g and 3a (IC50 = 22.31 ± 0.90, 22.67 ± 0.86 and 28.98 ± 0.65 μM, respectively) exhibited moderate activity. It is notable that parent molecules 1 and 2 did not offer remarkable α-amylase inhibition potential (IC50 = 22.08 ± 1.10 and 25.03 ± 0.99 μM, respectively) (Fig. 5).
2.3. Structure–activity relationship (SAR) analysis
It is clear that the α-amylase inhibition potential of the newly synthesized compounds depends on the structural factors in relation to the nature and position of substituents on the phenyl rings attached to the arylidene moiety.As can be seen in Fig. 6, an unsubstituted phenyl ring attached to the arylidene moiety (3a, R = Ph) exhibits a weak inhibition potential (IC50 = 28.98 ± 0.65 μM) compared to the other derivatives. However, the compound 3b with a non-substituted naphthalene system was found to be twice as active as compound 3a, with an IC50 value of 14.41 ± 0.30 μM. The increase in the inhibitory activity of compound 3b (R = naphthalene) compared to 3a (R = Ph) may be due to the extent of conjugation in the two aromatic rings of naphthalene (Fig. 5). These results are in good agreement with those reported in the literature, showing that the presence of the naphthalene ring in the structure can contribute to inhibit the α-amylase enzyme.26 Moreover, the alkyl group at the p-position of the phenyl group introduced by the aryl aldehyde in 3f (R = p-CH3C6H4) and 3g (R = p-tert-BuC6H4) allowed to reach a moderate activity with IC50 values of 22.31 ± 0.90 and 22.67 ± 0.86 μM, respectively compared to 3a (R = Ph). This finding allows noticing that the alkyl groups with a donor inductive (+I) effect did not improve significantly the anti-α-amylase activity.
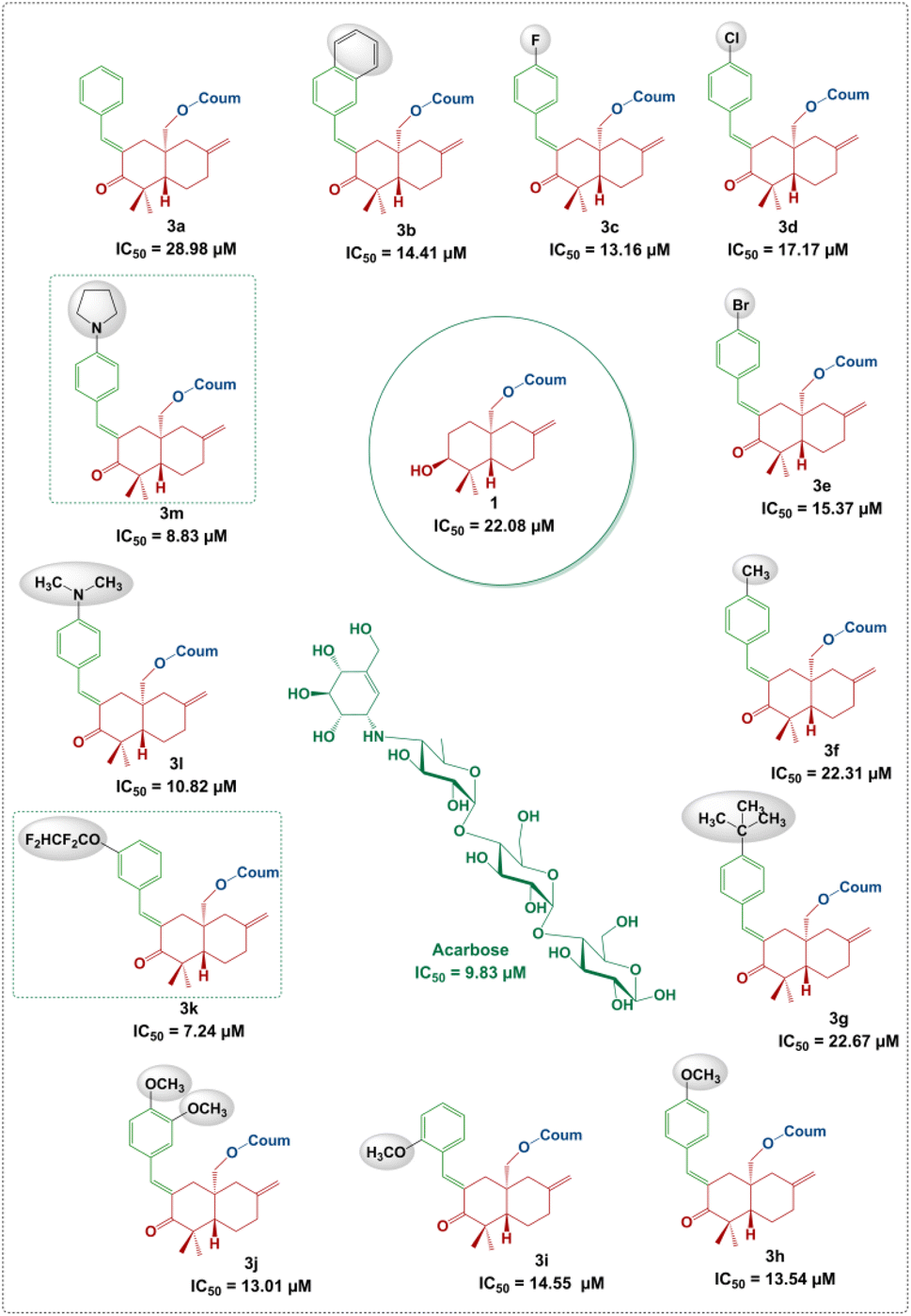 | ||
| Fig. 6 α-Amylase inhibitory activity of the derivatives 3a–m and acarbose. | ||
Furthermore, the incorporation of a halogen atom at the p-position of the phenyl group from the arylidene system in 3c (R = p-FC6H4), 3d (R = p-ClC6H4) and 3e (R = p-BrC6H4) enhances the inhibitory activity compared to 3a (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6H5). Among the above-mentioned halogenated derivatives, the fluorinated one 3c was found to exhibit the highest activity (IC50 = 13.16 ± 0.73 μM) (Fig. 6). This finding is consistent with the literature, showing that the incorporation of halogen atoms (F, Cl and Br) into the structure had an effect on the inhibition of α-amylase.27,28 However, the double-methoxylated compound 3j (R = 3,4-(OCH3)2C6H3) showed a slightly higher activity (IC50 = 13.01 ± 0.96 μM), than that of the mono-methoxylated compounds, 3h (p-OCH3C6H4; IC50 = 13.54 ± 0.54 μM) and 3i (o-OCH3C6H4; IC50 = 14.55 ± 0.30 μM). This finding shows that the more methoxyl groups there are, the more the activity increases even slightly. The electronic effects of methoxy groups (−I and +M) comparable to those of halogens (F, Cl and Br) may explain their significant activity.
C6H5). Among the above-mentioned halogenated derivatives, the fluorinated one 3c was found to exhibit the highest activity (IC50 = 13.16 ± 0.73 μM) (Fig. 6). This finding is consistent with the literature, showing that the incorporation of halogen atoms (F, Cl and Br) into the structure had an effect on the inhibition of α-amylase.27,28 However, the double-methoxylated compound 3j (R = 3,4-(OCH3)2C6H3) showed a slightly higher activity (IC50 = 13.01 ± 0.96 μM), than that of the mono-methoxylated compounds, 3h (p-OCH3C6H4; IC50 = 13.54 ± 0.54 μM) and 3i (o-OCH3C6H4; IC50 = 14.55 ± 0.30 μM). This finding shows that the more methoxyl groups there are, the more the activity increases even slightly. The electronic effects of methoxy groups (−I and +M) comparable to those of halogens (F, Cl and Br) may explain their significant activity.
Interestingly, compound 3k (R = m-OCF2CHF2C6H4; IC50 = 7.24 ± 0.36 μM) displays more interesting activity than acarbose (IC50 = 9.83 ± 0.21 μM) and methoxylated derivatives 3h, 3i and 3j. This result showed the net contribution of the introduced tetrafluoroethoxy fragment (–OCF2CHF2) to enhance the inhibition activity of the α-amylase enzyme (Fig. 6). It should also be mentioned that compound 3m (IC50 = 8.83 ± 0.22 μM) bearing an aryl p-pyrrolidine moiety shows a higher activity than the standard compound (acarbose). This could be due, in part, to the donor (+M) mesomer effect of the nitrogen atom. The results indicated clearly that the α-amylase enzyme is more sensitive towards the p-pyrrolidine aryl moiety in 3m compared to the acyclic amine in 3l (IC50 = 10.82 ± 0.60 μM).
The inhibition potency of our arylidenes may be compared favorably with those of other arylidene compounds investigated by other research teams such as arylidene-linked chromane-2,4-dione analogs (7.7 ± 0.1 μM < IC50 < 60.7 ± 0.1 μM)29 and 4-(arylidene)amino-1,2,4-traizole-5-thiol (IC50 = 1.65 ± 0.05–5.78 ± 0.18 μM).30
2.4. Kinetic studies
Kinetic studies on the parent molecule 1 and all synthesized compounds (3a–m) were performed to explain the mechanism of inhibition. The mechanism of enzyme inhibition was determined by graph fitting analysis using the Sigma-Plot enzyme kinetic software and kinetic parameters (Vmax and KM).In particular, Fig. 7 shows the inhibition kinetic behavior of coladonin (1) and the most active arylidene derivatives (IC50 ≤ 10 μM), 3k, 3l and 3m. As can be seen, the plots gave straight lines with different slopes intersecting one another in different quadrants, showing a mixed-type inhibition. The difference found for these three compounds is that 3k and 3l are mixed competitive-non-competitive inhibitors, while 3m is a mixed non-competitive-uncompetitive inhibitor. Analyzing the arrangement of these three compounds, the only difference observed was in the substituent at the p-position of the benzene ring of the arylidene. Specifically, compound 3k features a m-tetrafluoroethoxyl group, 3l possesses a p-N,N-dimethylamino group, while 3m possesses a p-pyrrolidinyl group. Similarly, with the exception of 3j, which exhibited non-competitive inhibition, all other arylidene derivatives proved to be mixed competitive-noncompetitive inhibitors.
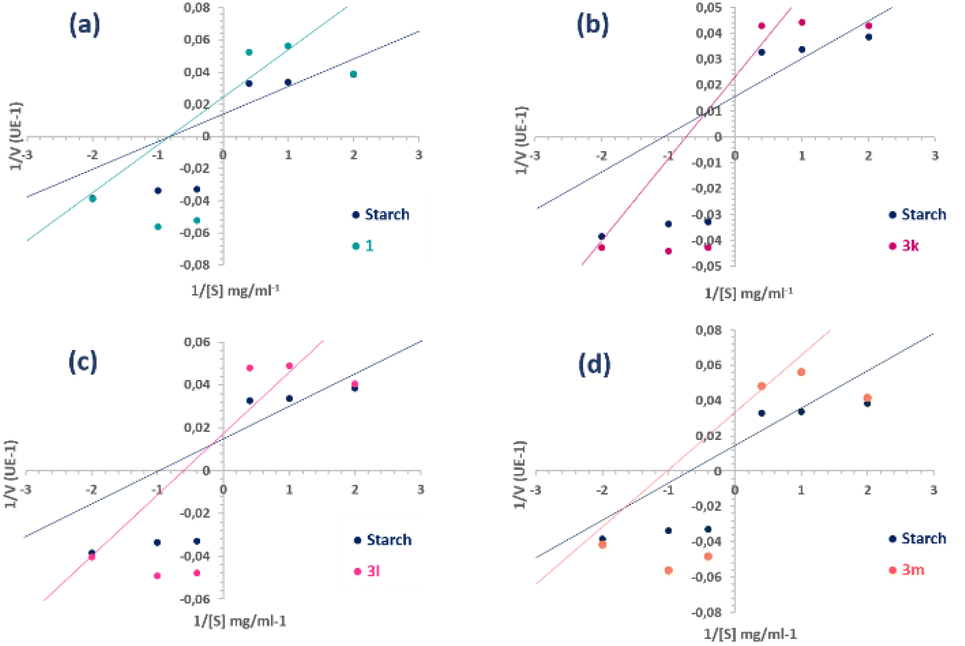 | ||
| Fig. 7 Lineweaver–Burk plots of α-amylase inhibition at different starch (substrate) concentrations [S] in the presence and absence of compounds 1 (a), 3k (b), 3l (c) and 3m (d). | ||
Thus, in comparison with the plots of the semi-synthetic compounds and coladonin (1), we can deduce that the mechanism of inhibition has shifted from non-competitive to various mixed types of inhibition (Fig. 7). The identification of various inhibitor types has the potential to enhance research on α-amylase inhibitors.
Using the Dixon plot (Table S5†), the inhibitory constants (Ki) were determined for the most potent compounds. Compound 3k showed a Ki value of 1.02 μM, indicating the most potent inhibitory activity.
2.5. Molecular docking analysis
Enzyme inhibitors are bioactive molecules capable of inhibiting or limiting the natural functioning of target enzymes by occupying their active pocket and competing with the substrate,28,31 or by occupying distant allosteric sites. Indeed, the binding energy reflects the stability of the molecule-enzyme complex.31,32Chemical compound-based virtual inspection is an effective process for confirming certain enzyme inhibitory activities and predicting molecular interactions.31 Therefore, docking analyses were performed to reveal the mode of molecule-enzyme binding and to better understand the action manner of the novel synthesized compounds in the binding pocket or in other sites of the Aspergillus oryzae α-amylase enzyme using the AutoDock Vina software. In the present study, in addition to “active site docking”, the “blind docking” approach was employed to investigate the possible binding sites of our compounds, distinct from the known binding site, due to their mixed inhibition characteristics against the α-amylase protein. In mixed-type inhibition, the inhibitor can bind to the enzyme whether or not the substrate is already bound.33,34
The binding positions of acarbose (the used standard reference) and the all compounds (in particular 3k, 3l and 3m) in the structure of the α-amylase enzyme are shown in Fig. 8. The 2D-presentation of binding modes of compounds 3k, 3l and 3m, compared to that of acarbose, is shown in Fig. 9.
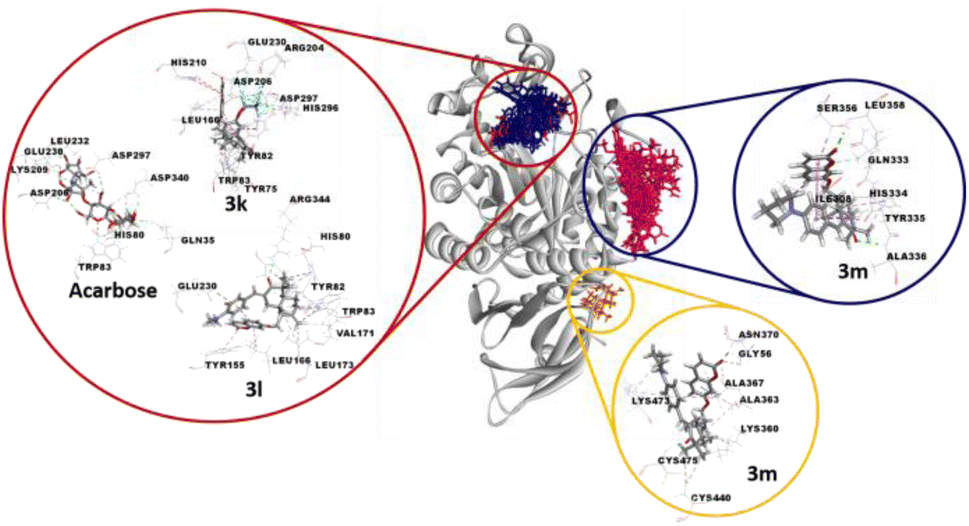 | ||
| Fig. 8 Docking sites and 3D representations of the docked poses of the standard inhibitor (acarbose) and the three most active compounds 3k, 3l and 3m into the α-amylase enzyme. Compounds 3a, 3b, 3c, 3d, 3e, 3f, 3g, 3h, 3i, 3I, 3k, 3l (blue molecules) and acarbose (red molecule) were docked into the active pocket (red circle), while compounds 1 and 3a–m (pink molecules) were docked into an allosteric site (blue circle). In the yellow circle, compound 3m was docked in a second allosteric site. The α-amylase enzyme is represented as a grey ribbon. | ||
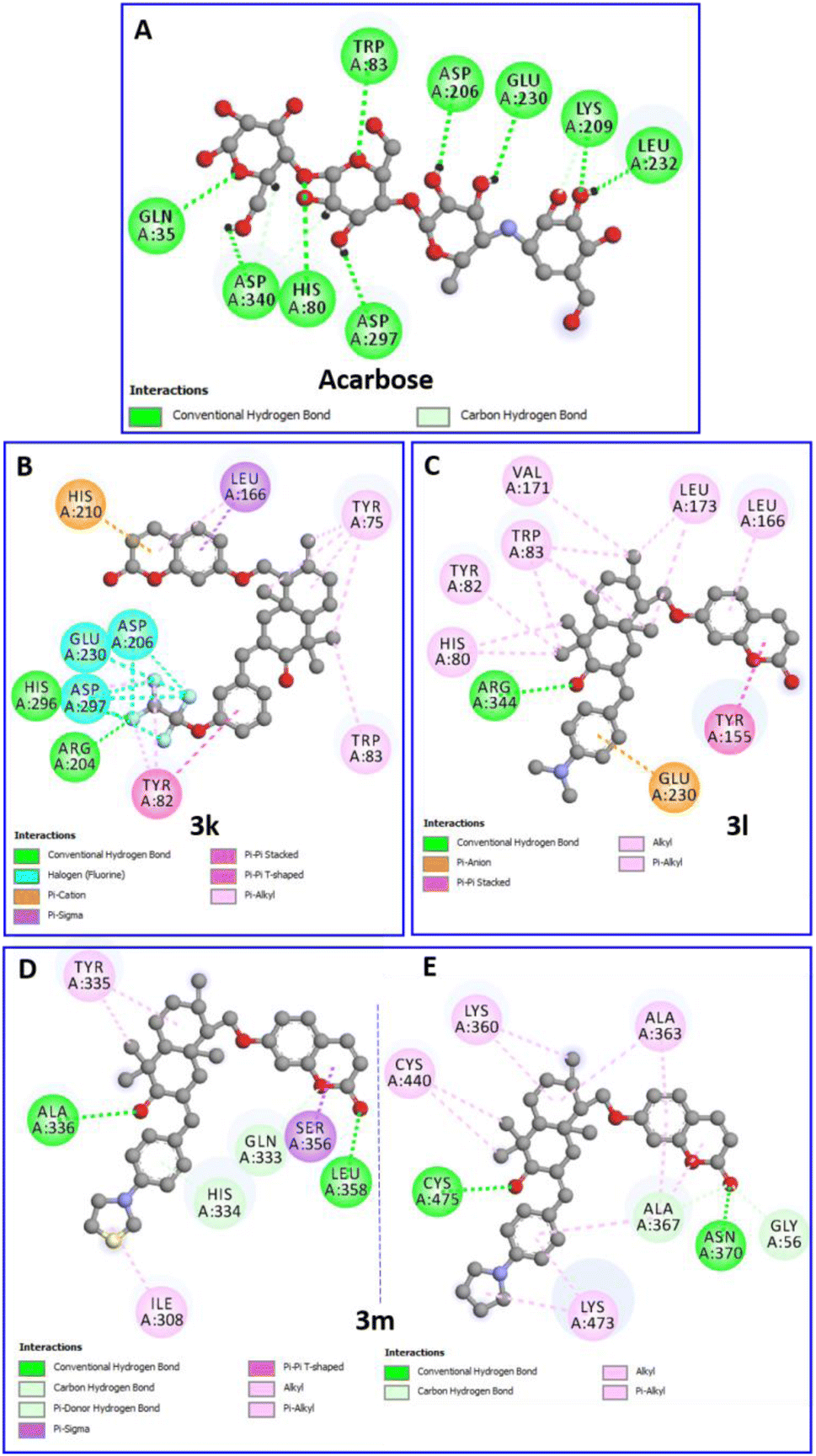 | ||
| Fig. 9 Two-dimensional profiles showing the interaction modes of acarbose (A) and the three most active compounds 3k (B), 3l (C) and 3m (D and E) into the binding pocket or/and allosteric site of the α-amylase enzyme (PDB: 7TAA). | ||
Details of interactions and the binding energies of the selected compounds with the distant allosteric site and the active site residues are listed in Tables 1 and 2, respectively.
| N° | α-Amylase inhibition | Binding energy | Interaction detail: NI/NIAA: IAA |
|---|---|---|---|
| a NI: Number of interactions, NIAA: number of interacting amino acids, IAA: interacting amino acids.b 1 Hydrogen bond.c α-Amylase inhibitory activity as IC50 ± SD (μM) is the compound concentration providing 50% of inhibition. IC50 values followed by the same letter are not significantly different at p-value less than 0.05. The binding energy is in kcal mol−1. | |||
| 1 | 22.08 ± 1.10 | −7.1 | 9/6: THR:32b, ALA:33b, TYR:335, ALA:336, ALA:342, LEU:349 |
| 3a | 28.98 ± 0.65 | −7.5 | 8/5: TYR:335b, ALA:336b, ALA:342, GLU:345, LEU:349 |
| 3b | 14.41 ± 0.30 | −8.6 | 10/7: TYR:335, ALA:336b, GLY:337, ALA:342, GLU:345, LEU:349, SER:350 |
| 3c | 13.16 ± 0.73 | −8.0 | 9/6: CYS:30, TYR:335b, ALA:336b, ALA:342, GLU:345, LEU:349 |
| 3d | 17.17 ± 0.50 | −7.4 | 7/5: HIS:334, TYR:335, ALA:336, GLU:345, LEU:349 |
| 3e | 15.37 ± 0.29 | −7.6 | 9/5: ASN:31, THR:32b, TYR:335, ALA:336, LEU:349 |
| 3f | 22.31 ± 0.90 | −8.1 | 5/3: TYR:335, ALA:336b, LEU:349 |
| 3g | 22.67 ± 0.86 | −7.9 | 8/5: HIS:334, TYR:335, ALA:336b, GLU:345, LEU:349 |
| 3h | 13.54 ± 0.54 | −8.0 | 6/5: ASN:306, TYR:335, ALA:336b, GLU:357b, LEU:358b |
| 3i | 14.55 ± 0.30 | −7.6 | 6/5: THR:32b, TYR:335, ALA:336, GLU:345, LEU:349 |
| 3j | 13.01 ± 0.96 | −8.0 | 10/7: THR:32b, HIS:334, TYR:335, ALA:336b, GLY:338b, ALA:342, LEU:349 |
| 3k | 7.24 ± 0.36 | −8.3 | 9/8: ILE:308, GLN:333, HIS:334b, TYR:335, ALA:336b, LEU:349, SER:350, GLU:357b |
| 3l | 10.82 ± 0.60 | −9.4 | 8/5: TYR:335, ALA:336b, ALA:342, GLU:345b, LEU:349 |
| 3m | 8.83 ± 0.22 | −8.4 | 7/6: ILE:308, HIS:334, TYR:335, ALA:336b, SER:356, LEU:358b |
| N° | Binding energy | Interaction detail: NI/NIAA: IAA |
|---|---|---|
| a Acr: acarbose (the used standard reference), NI: number of interactions, NIAA: number of interacting amino acids, IAA: interacting amino acids.b 1 Hydrogen bond.c 2 Hydrogen bonds.d 3 Hydrogen bonds, bold type indicates interactions with key amino acids.e The binding energy is in Kcal mol−1. | ||
| 3a | −8.8 | 14/8: HIS:80, TYR:82, HIS:122, TYR:155, ASP:206, HIS:210b, LEU:232, ARG:344d |
| 3b | −9.6 | 13/10: HIS:80, TYR:82, TYR:155, LEU:166, ARG:204c, ASP:206, HIS:210, LEU:232, HIS:296b, ASP:297 |
| 3c | −9.6 | 11/8: TYR:75, TYR:82, TRP:83b, TYR:155b, LEU:166, ASP:206, GLU:230, ARG:344b |
| 3d | −10.1 | 10/7: TYR:75, HIS:80, TYR:82, TRP:83, LEU:166, GLU:230, HIS:296b |
| 3e | −10.0 | 10/7: TYR:75, HIS:80, TYR:82, TRP:83, LEU:166, GLU:230, HIS:296b |
| 3f | −8.7 | 15/10: HIS:80, TYR:82, TRP:83, TYR:155, LEU:166, LEU:173, HIS:210, GLU:230, LEU:232, ARG:344b |
| 3g | −8.2 | 14/13: TYR:75, HIS:80, TYR:82, TRP:83, ILE:152b, GLY:167b, ASP:168, LEU:173, HIS:210, GLU:230, LEU:232, ASP:297, ARG:344b |
| 3h | −8.5 | 14/11: HIS:80, TYR:82, TRP:83, ILE:152b, TYR:155, LEU:166, GLY:167b, LEU:173, HIS:210b, GLU:230, ARG:344b |
| 3i | −9.1 | 15/10: HIS:80, HIS:122, TYR:155, LEU:166, LEU:173, ASP:206, HIS:210b, LEU:232, ASP:340, ARG:344c |
| 3k | −10.2 | 25/10: TYR:75, TYR:82, TRP:83, LEU:166, ARG:204c, ASP:206, HIS:210, GLU:230, HIS:296b, ASP:297 |
| 3l | −8.9 | 14/9: HIS:80, TYR:82, TRP:83, TYR:155, LEU:166, VAL:171, LEU:173, GLU:230, ARG:344b |
| Acr | −7.9 | 11/9: GLN:35b, HIS:80b, TRP:83b, ASP:206b, LYS:209b, GLU:230b, LEU:232b, ASP:297b, ASP:340b |
According to the docking results, all the selected compounds manifest an allosteric site docking, demonstrating a noncompetitive inhibition, which coincided with the results of noncompetitive inhibition for compounds 1 and 3j, mixed non-competitive-uncompetitive inhibition for 3m and mixed competitive-non-competitive inhibition for all remaining compounds.
The compound 3m, among the compounds with the best activity results, showed an excellent affinity (−8.4 kcal mol−1) with the α-amylase enzyme. The pyrrolidinyl moiety formed alkyl interaction with ILE:308 and a π-donor hydrogen bond with the HIS:334 residue. The sesquiterpene system fits by a conventional hydrogen bond with the ALA:336 amino acid and a π-alkyl and π-sigma interactions with TYR:335. The coumarin moiety is involved in a conventional hydrogen bond with the LEU:358 amino acid, and a π-sigma interaction with the SER:356 residue (Fig. 9D). Overall, the blind docking results revealed that most derivatives, in the “non-active site”, commonly adopt conventional hydrogen interactions with the residue ALA:336. In the case of the most active derivatives 3k, 3l and 3m in the series, they adopt similar interactions and have residues in common with which they react, such as ILE:308, HIS 334, TYR:335, and ALA:336 (hydrogen bonds in all 3 cases).
It is worth noting that, in the blind docking process, compound 3m demonstrated a remarkable affinity for a second allosteric site, exhibiting a binding energy value of −8.0 kcal mol−1. This affinity was established through the formation of 14 interactions (2 conventional hydrogen bonds, 1 carbon hydrogen bond, 5 π-alkyl and 6 alkyl bonds) with eight different amino acids (GLY:56, LYS:360, ALA:363, ALA:367, ASN:370, CYS:440, LYS:473 and CYS:475). This observation can be attributed to its partial uncompetitive inhibition mode (Fig. 9E).
However, many considerable results of the selected molecules docked in the active site of the α-amylase enzyme were observed. Based on the binding energies (−8.2 to −10.2 kcal mol−1), all compounds (except 1, 3j and 3m) proved to have high affinity within the active cavity of the α-amylase enzyme, since they exhibit mixed competitive–non-competitive inhibition. Interestingly and as expected, all these compounds are perfectly localized in the active pocket of the target enzyme.
Acarbose (−7.9 kcal mol−1) forms nine hydrogen bonds with nine different residues (Table 1), including the two key amino acids essential for the cleavage of glycosidic bindings, namely, the nucleophilic residue ASP:206 as well as the catalytic acid/base residue GLU:230.31 Therefore, the compound ability to fit with these key residues increases the potency to inhibit polysaccharide hydrolysis.
Interestingly, the complex of the 3k derivative with the target enzyme showed exceptional stability (−10.2 kcal mol−1) superior to that of acarbose. In the binding profile of the compound 3k, the 1,1,2,2-tetrafluoroethoxyl group alone is implicated in fifteen interactions with six amino acids. It formed three hydrogen bonds with ARG:204 and HIS:296, eight halogen (Fluorine) bonds with ASP:206, GLU:230 and ASP:297, and four π-alkyl bonds with TYR:82 and HIS:296. The aromatic ring of the arylidene group fits with a π–π stacked binding with TYR:82. The sesquiterpene moiety performed five π-alkyl bonds with TYR:75 and TRP:83. However, the coumarin moiety forms a π–π T-shaped and a π-cation bond with HIS:210 and two π-alkyl interactions with LEU:166 (Fig. 9B).
Furthermore, the binding profile of the compound 3l, exhibiting an impressive binding energy (−8.9 kcal mol−1), indicates that it is engaged in fourteen non-covalent interactions with nine residues. The arylidene group fits by a π-anion bond with the key residue GLU:230. The carbonyl in C-3′ forms a conventional hydrogen bond with ARG:344. The rest of the sesquiterpene moiety performed seven π-alkyl interactions with HIS:80, TYR:82 and TRP:83 residues and three alkyl bonds with VAL:171 and LEU:173. A π–π stacked binding with TYR:155 and a π-alkyl bond with LEU:166 were formed by the coumarin fragment (Fig. 9C).
2.6. Toxicity analysis
It is crucial to evaluate the toxicity of the synthesized chemical compounds with medical significance. An in silico approach (https://www.organic-chemistry.org/prog/peo/) is indeed available for predicting specific toxicity risks such as mutagenicity, tumorigenicity, irritation and reproductive effects. All the predicted compounds were found to be free from irritation risk. Moreover, the majority of compounds, except 3l and 3m, exhibited no mutagenicity or tumorigenicity risks. Similarly, concerning the reproductive effects, only 3h and 3j presented toxicity risks, while the remaining compounds were deemed safe (Table S4†). The observed toxicity might be attributed to the presence of oxygen or nitrogen, particularly in the para position of the arylidene aromatic ring.3 Experimental
3.1. General procedures
All chemicals were obtained from commercial suppliers and used without further purification. 1H (600 MHz) and 13C (deduced from HSQC and HMBC spectra) NMR spectra were recorded using a Bruker AVANCE III, with CDCl3 as the solvent and TMS as the internal standard. Melting points were measured using a Buchi melting point apparatus, model B-545. ESI-HRMS spectra were recorded with a platform consisting of an HTC PAL® 4 autosampler (CTC Analytics AG, Zwingen, Switzerland), an Accela U-HPLC system with quaternary pumps, and an Exactive Orbitrap mass spectrometer equipped with a heated electrospray ionization (H-ESI) probe (both from Thermo Fisher Scientific, Bremen, Germany). α-Amylase was extracted from porcine pancreas (Sigma Aldrich 6255, 1151 Umg−1 of protein, Sigma Aldrich, Saint Louis, MO).3.2. Plant material
The roots of F. tunetana were collected from the region of Menzel Kamel (Tunisia) (35°38′21.4′′N 10°36′31.2′′E), in April 2019. The botanical identification was carried out by distinguished professor Fethia Harzallah-Skhiri, and a voucher specimen (FT/19) was deposited in the laboratory mentioned above.3.3. Extraction and isolation
First, 5 kg of fresh roots of the F. tunetana plant were macerated at room temperature in a MeOH/water mixture (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3/v:v) for 7 days. The aqueous phase, after evaporation of methanol, was extracted with chloroform (CHCl3). The CHCl3 extract (15 g), namely RC, was separated using a silica gel chromatographic column, eluted initially with a petroleum ether/EtOAc mixture of increasing polarity (20 to 100% EtOAc), and then by adding MeOH to EtOAc (0 to 100% MeOH) in 15 fractions (RC1–RC15 (g): 0.66; 0.79; 0.61; 0.28; 1.5; 0.18; 0.80; 0.32; 0.21; 0.25; 2.80; 1.31; 0.75; 0.35; and 2.4), according to TLC analysis and sulfur vanillin revelation. Coladonin 1 (1.5 g) was isolated from the RC11 fraction following precipitation in n-hexane.
:3/v:v) for 7 days. The aqueous phase, after evaporation of methanol, was extracted with chloroform (CHCl3). The CHCl3 extract (15 g), namely RC, was separated using a silica gel chromatographic column, eluted initially with a petroleum ether/EtOAc mixture of increasing polarity (20 to 100% EtOAc), and then by adding MeOH to EtOAc (0 to 100% MeOH) in 15 fractions (RC1–RC15 (g): 0.66; 0.79; 0.61; 0.28; 1.5; 0.18; 0.80; 0.32; 0.21; 0.25; 2.80; 1.31; 0.75; 0.35; and 2.4), according to TLC analysis and sulfur vanillin revelation. Coladonin 1 (1.5 g) was isolated from the RC11 fraction following precipitation in n-hexane.
3.4. General procedure for oxidation at C-3′–OH
Coladonin 1 (1 g, 2.6 mmol) was dissolved in acetone and 5 drops of the prepared Jones reagent were added to the reaction flask placed in an ice bath, and then the mixture was stirred for 3 h.35 After evaporation of acetone, 100 mL of cold water was added and the mixture was extracted with ethyl acetate (3 × 25 mL), and the organic phase was filtered and dried with Na2SO4. Finally, a white solid 2 (0.92 g, 92%) was obtained by column chromatography (petroleum ether/EtOAc: (80:20)).
3.5. General procedure for the preparation of analogues (3a–m)
A solution of 2 (50 mg, 0.13 mmol) and KOH (1.5 equiv.) in ethanol, aryl-aldehyde (1.5 equiv.) was added, and the mixture was reacted for 24 h.30 The crude was diluted in distilled water, neutralized with H2SO4 solution (2 M) and extracted with EtOAc (3 × 10 mL). After the removal of EtOAc, the resulting mixture was purified by silica gel column chromatography (petroleum ether/EtOAc: (80:20)) to give the derivatives 3a–m.
3.6. α-Amylase inhibition and kinetics Assay
Anti-α-amylase activity was evaluated according to the protocol reported in the literature with slight modifications.36 First, 500 μL of diluted α-amylase from porcine pancreas (1151 U mg−1 of protein: 480 μL of amylase in 0.2 mol L−1 phosphate buffer, pH = 6) and 500 μL of soluble starch solution (1%). To evaluate this activity, the effects of each of the synthesized compounds, the solutions of diluted α-amylase and the different concentrations of compound (6.25–50 μM) were preincubated at 25 °C for 5 min. To start the reaction, 500 μL of substrate (soluble starch) was added to solutions and incubated for 10 min at 25 °C. Finally, 3 mL of 3,5-dinitrosalicylic acid (DNS) was quickly added to the mixture and heated in boiling water for 15 min. Glucose was used to schematize a standard curve. Acarbose was used as the positive control. The absorbance was measured at 540 nm. The increased substrate concentration (from 0.5 to 2.5%) was tested by kinetic analysis to determine the inhibition type exerted on α-amylase (competitive, non-competitive, mixed or uncompetitive) by the line Weaver–Burk plot.3.7. Molecular docking procedure
The chemical structures of all molecules were sketched and optimized using the ACD software (3D Viewer) (http://www.filefacts.com/acd3d-viewer-freeware-info). The 3D-structure of Aspergillus oryzae α-amylase complexed with acarbose (7TAA.pdb) was retrieved from RCSB PDB, Protein Data Bank website (https://www.rcsb.org/). Python Molecule Viewer (version 1.5.6rc3) was employed for enzyme optimization and simplification, by removing water molecules and the co-crystallized inhibitor (acarbose) from the protein file, and then protonated by adding polar hydrogens. Kollman charges were inserted. All structures were saved in PDBQT file format before running the docking using the AutoDock Vina software.37 Hence, for the competitive-type inhibitors, the molecular docking launch was performed in the grid box centered on the enzymatic 3D-structure with the following coordinates: 38.725, 44.127, 25.633 (x, y, z), a spacing of 0.375 Å and a box size of 65 × 52 × 52.28 These parameters, together with the enzyme and ligand pdbqt files and the number of modes (9), were included in the configuration file. The exhaustiveness value was fixed to 8. For the uncompetitive and noncompetitive-type inhibitors, the whole protein was selected as a binding site to dock the selected ligands. The BIOVIA Discovery Studio Visualizer (2017) (BIOVIA, D. S., version 17.2.0.16349) was used to visualize the molecule-enzyme interactions and generate Figures.3.8. Statistical analysis
GraphPad Prism 7.0 (Graph Pad Software Inc., CA and USA) was used to conduct the statistical analysis. The results are given as mean values ± standard deviation (SD). Tukey′s test was applied to determine the difference between the activities of all compounds.4 Conclusions
The natural bioactive coladonin 1 isolated quantitatively from the roots of the Tunisian endemic plant F. tunetana has been oxidized and condensed with different aryl aldehydes via the Claisen–Schmidt reaction to obtain a series of new arylidene-based coladonin conjugates. All synthetic derivatives were evaluated for their potential α-amylase inhibitory activity. The compounds 3k and 3m with a more interesting activity than that of acarbose were used as the standard. Kinetic studies have shown that these compounds act as mixed inhibitors towards α-amylase. From the molecular docking analysis, it can be concluded that the arylidene moiety is essential for the buildup and enhancement of α-amylase inhibitory activity of compounds 3a–m. In silico “SAR” studies were found to be in good agreement with the experimental biological results, showing that the nature of the arylidene moiety is essential to form several important binding interactions with the amino acids of the enzyme. Thus, these arylidene-based coladonin derivatives as valuable candidates may serve as lead for the future development of potential anti-diabetic agents.Author contributions
Wiem Baccari, Ilyes Saidi and Mansour Znati: investigation, methodology, software, writing – original draft. Houda Lazrag, Axel Marchal, Pierre Waffo-Teguo: validation, supervision and conceptualization. Insaf Filali and Moncef Tounsi: visualization. Hichem Ben Jannet: supervision, validation, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported via funding from Prince sattam bin Abdulaziz University project number (PSAU/2023/R/1444). The authors are grateful to Researchers Supporting Project number (PSAU/2023/R/1444) at Prince sattam bin Abdulaziz University, Saudi Arabia.References
- M. A. B. Khan, M. J. Hashim, J. K. King, R. D. Govender, H. Mustafa and J. Al Kaabi, J. Epidemiol. Glob. Health, 2000, 10, 107–111 CrossRef.
- M. E. Febrian, F. X. Ferdinan, G. P. Sendani, K. M. Suryanigrum and R. Yunanda, Procedia Comput. Sci., 2023, 216, 21–30 CrossRef.
- C. Lo, T. Toyama, Y. Wang, J. Lin, Y. Hirakawa, M. Jun, A. Cass, C. M. Hawley, H. Pilmore and S. V. Badve, Cochrane Database Syst. Rev., 2018, 9, CD011798 Search PubMed.
- J. Dowarah and V. P. Singh, Bioorg. Med. Chem., 2020, 28, 115263 CrossRef CAS PubMed.
- S. Khan, M. Khan, W. Rehman, M. Shah, R. Hussain, L. Rasheed, Y. Khan, A. Dera, R. Pashameah and E. Alzahrani, Pharmaceuticals, 2022, 15, 1164–1180 CrossRef CAS PubMed.
- H. Gin and V. Rigalleau, Diabetes Metab., 2000, 26, 265–272 CAS.
- N. Kaur, V. Kumar, S. K. Nayak, P. Wadhwa, P. Kaur and S. K. Sahu, Chem. Biol. Drug Des., 2021, 98, 539–560 CrossRef CAS.
- J. Wang, X. Rong, W. Li, Y. Yang, J. Yamahara and Y. Li, J. Ethnopharmacol., 2012, 142, 782–788 CrossRef CAS PubMed.
- J. P. Nie, Z. N. Qu, Y. Chen, J. H. Chen, Y. Jiang, M. N. Jin, Y. Yu, W. Y. Niu, H. Q. Duan and N. Qin, Fitoterapia, 2020, 142, 104499 CrossRef CAS PubMed.
- H. Kashtoh and K. H. Baek, Plants, 2023, 12, 2944 CrossRef CAS PubMed.
- M. Znati, I. Filali, A. Jabrane, J. Casanova and H. Ben Jannet, Chem. Biodiversity, 2017, 14, e1600116 CrossRef PubMed.
- F. U. Afifi and B. Abu-Irmaileh, J. Ethnopharmacol., 2000, 72, 101–110 CrossRef CAS.
- G. Pottier-Alapetite, ‘Flore de la Tunisie, AngiospermesDicotyledones, Apetales-Dialypetales’, Publications Scientifiques Tunisiennes, Imprimerie Officielle de la Republique Tunisienne, Tunisie, 1979, p. 607 Search PubMed.
- A. Jabrane, O. Duchamp, H. Ben Jannet, Z. Mighri, J. F. Mirjolet, F. Harzallah-Skhiri and M. A. Lacaille-Dubois, Chem. Biodiversity, 2010, 7, 392–399 CrossRef CAS.
- M. Curini, G. Cravotto, F. Epifano and G. Giannone, Curr. Med. Chem., 2006, 13, 199–222 CrossRef CAS PubMed.
- I. Choudhary, M. I. Baig and M. Nur-e-Alam, Helv. Chim. Acta, 2001, 84, 2409–2416 CrossRef.
- A. Amin, M. Hanif, A. Rafey, S. Zaib, S. Bakhsh, M. Ramzan, A. Zaman, F. Ur Rehman, J. Iqbal and L. Pieters, Rev. Bras. Farmacogn., 2020, 30, 12–17 CrossRef CAS.
- A. V. Petrova, E. F. Khusnutdinova, A. N. Lobov, L. M. Zakirova, N. T. T. Ha and D. A. Babkov, Nat. Prod. Res., 2022, 36, 1–8 CrossRef.
- V. Kumar, R. Ramu, P. S. Shirahatti, V. B. C. Kumari, P. Sushma, S. P. Mandal and S. M. Patil, ChemistrySelect, 2021, 6, 9637–9644 CrossRef CAS.
- D. Kumar, S. Narwal and J. S. Sandhu, Int. J. Med. Chem., 2013, 2013, 1–4 Search PubMed.
- S. W. Kahssay, G. S. Hailu and K. T. Desta, Drug Des., Dev. Ther., 2021, 15, 3119–3129 CrossRef PubMed.
- S. Saouli, I. Selatnia, B. Zouchoune, A. Sid, S. M. Zendaoui, C. Bensouici and E. Bendeif, J. Mol. Struct., 2020, 1213, 128203 CrossRef CAS.
- S. Farooq and Z. Ngaini, ChemistrySelect, 2020, 5, 9974–9979 CrossRef CAS.
- K. Kaleshkumar, R. Rajaram, N. Gayathri, T. Sivasudha, G. Arun, G. Archunan, B. Gulyas and P. Padmanabhan, Bioorg. Chem., 2019, 90, 103072 CrossRef CAS.
- V. B. Nguyen, A. D. Nguyen, Y. H. Kuo and S. L. Wang, Int. J. Mol. Sci, 2017, 18, 700–711 CrossRef PubMed.
- B. Somashekara, B. Thippeswamy and G. R. Vijayakumar, J. Chem. Sci., 2019, 131, 1–7 CrossRef CAS.
- M. Nawaz, M. Taha, F. Qureshi, N. Ullah, M. Selvaraj, S. Shahzad, S. Chigurupati, A. Waheed and F. A. Almutairi, BMC Chem., 2020, 14, 43–53 CrossRef PubMed.
- I. Saidi, M. Manachou, M. Znati, J. Bouajila and H. Ben Jannet, J. Mol. Struct., 2022, 1247, 131379 CrossRef CAS.
- I. Ul Haq, M. Ahmad, I. Ali, K. M. Khan, S. Chigurupati, A. Habib, U. Sala, S. Konanki, S. G. Felemban, M. Taha and Z. Ul Haq, Chem. Pap., 2023, 77, 2581–2604 CrossRef.
- E. S. H. El Ashry, M. M. K. Farahat, L. F. Awad, M. Balbaa, H. Yusef, M. E. I. Badawy and M. N. Abd Al Moaty, J. Mol. Struct., 2022, 1259, 132733 CrossRef CAS.
- W. Baccari, I. Saidi, M. Znati, A. M. Mustafa, G. Caprioli, A. H. Harrath and H. Ben Jannet, Process Biochem., 2023, 129, 230–240 CrossRef CAS.
- F. K. Algethami, I. Saidi, H. N. Abdelhamid, M. R. Elamin, B. Y. Abdulkhair, A. Chrouda and H. Ben Jannet, Molecules, 2021, 26, 5214–5231 CrossRef CAS PubMed.
- E. O. Yeye, K. M. Khan, S. Chigurupati, A. Wadood, A. U. Rehman, S. Perveen and M. Taha, Bioorg. Med. Chem, 2020, 28, 115467 CrossRef CAS.
- W. D. Duan, J. Y. Cao, C. Y. Cai, Z. R. Yang, J. F. Cui, T. Lan and B. Wang, J. Mol. Struct., 2022, 1263, 133026 CrossRef CAS.
- Y. Yang, Q. Zhu, Y. Zhong, X. Cui, Z. Jiang, P. Wu and S. Zhao, Bioorg. Chem., 2020, 101, 103985 CrossRef CAS PubMed.
- A. Assel, A. Hajlaoui, H. Lazrag, M. Manachou, A. Romdhane, J. Kraiem and H. Ben Jannet, J. Mol. Struct., 2023, 1271, 134020 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07540k |
| This journal is © The Royal Society of Chemistry 2024 |