
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-glycosyl amides: conformational analysis and evaluation as inhibitors of β-galactosidase from E. coli†
Miqueas G. Traverssiab,
Verónica E. Manzano cd,
Oscar Varelacd and
Juan P. Colomer*ab
cd,
Oscar Varelacd and
Juan P. Colomer*ab
aDepartamento de Química Orgánica, Universidad Nacional de Córdoba, Facultad de Ciencias Químicas, Ciudad Universitaria, Edificio de Ciencias II, Córdoba, Argentina. E-mail: jpcolomer@unc.edu.ar
bInstituto de Investigaciones en Fisico-Química de Córdoba (INFIQC), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), UNC, Argentina
cDepartamento de Química Orgánica, Universidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Ciudad Universitaria, Pab. 2, C1428EHA, Buenos Aires, Argentina
dCentro de Investigación en Hidratos de Carbono (CIHIDECAR), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), UBA, Argentina
First published on 16th January 2024
Abstract
The synthesis of N-glycosyl amides typically involves the use of glycosyl amines as direct precursors, resulting in low yields due to hydrolysis and the loss of stereocontrol through anomerization processes. In this study, a sequential synthesis of N-glycosyl amides is proposed, employing glycosyl amines as intermediates obtained from glycosyl azides. Derivatives with gluco, galacto, or xylo configurations were synthesized. Hexose derivatives were obtained under stereocontrol to give only the β anomer, while the xylo derivatives provided a mixture of α and β anomers. Conformational analysis revealed that all β anomers adopted the 4C1 conformation, while α anomers were found in the 1C4 chair as the major conformer. After de-O-acetylation, the derivatives containing a galactose unit were evaluated as inhibitors of β-galactosidase from E. coli and were found to be moderate inhibitors.
Introduction
Glycomimetics are molecules that have structures similar to carbohydrates, but with some chemical modifications. These modifications often involve the glycosidic linkage, for example, replacing the oxygen atom with nitrogen, sulfur, or even carbon to modify the properties of that bond. The modified glycosidic linkage usually shows enhanced stability towards enzyme degradation and, in many cases, the glycomimetics act as enzyme inhibitors, being able to regulate the activity of glycosidases or glycosyltransferases. This has led to a growing interest in obtaining new synthetic sugar analogs for use as tools in glycobiology and medicinal chemistry.1–5N-Glycosyl amides, and more importantly N-glycopeptides, are known to be involved in numerous cell recognition processes, such as in inflammation,6 immune response,7 tumor proliferation and metastasis.8,9 In particular, N-glycosylation of peptides and proteins is one of the most complex and abundant types of co-translational modifications in nature.10 Glycosyl amides have been tested as inhibitors of glycogen phosphorylase, enzyme that catalyzes the rate-limiting step in glycogenolysis. Modulating the activity of this enzyme is believed to be essential for developing a treatment for type 2 diabetes.11–15 Furthermore, it is known that these compounds can also act as inhibitors of glycosidases,16 lectins17,18 or glycosyltransferases,19 displaying a wide range of biological activities.20
Since N-glycosyl amides 1 are involved in many functions and processes, the synthesis of this type of compounds, which can serve as glycomimetics, is a current objective. Formation of the amide bond can be carried out by coupling a N-glycosyl amine 2 with the corresponding acyl derivative 3. However, glycosyl amines 2 are known to anomerize when long reaction times are required (Scheme 1), thus losing selectivity towards the desired anomer. Furthermore, compounds 2 are susceptible to hydrolysis to give hemiacetals 4 as reaction by-products.21,22
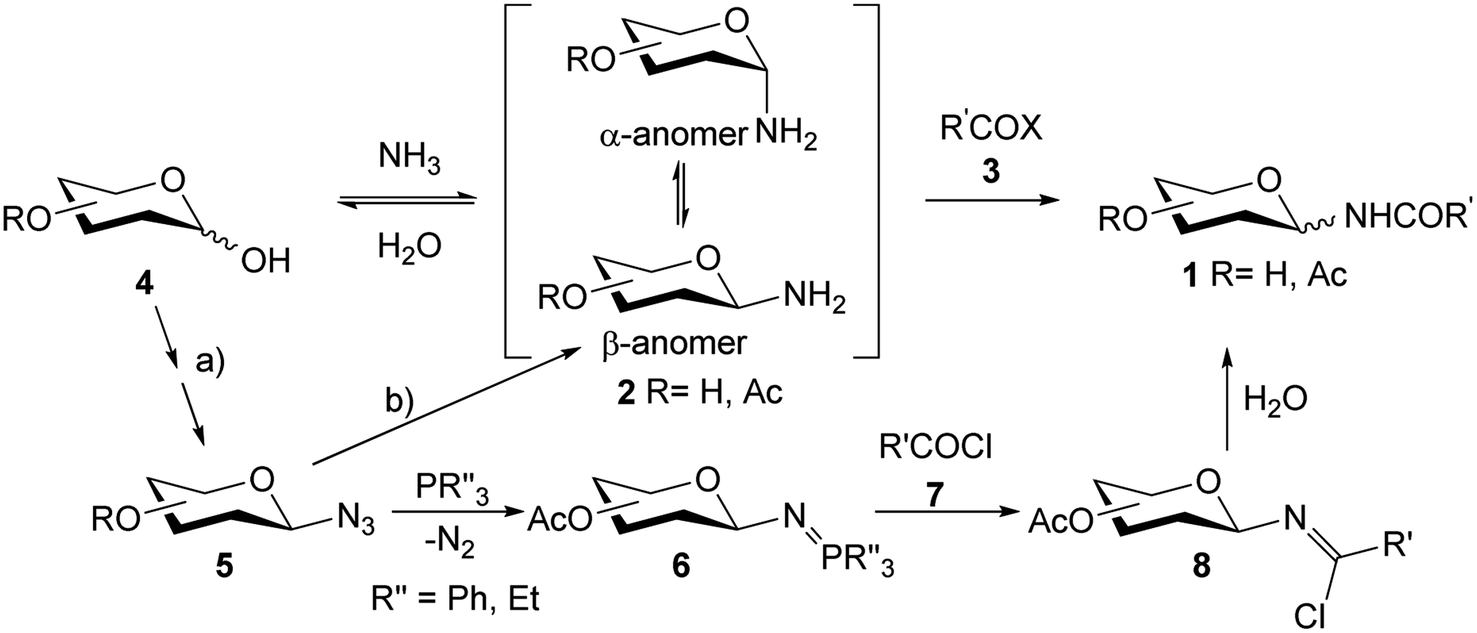 | ||
| Scheme 1 Some methodologies described for the synthesis of N-glycosil amides. | ||
For this reason, glycosyl azides 5 (easily accessible from hemiacetals 4) are used as precursors for glycosyl amides 1, as they have shown to be chemically and configurationally stable at their anomeric centre.23 Compounds 5 can be reduced by various methods prior to coupling with the acyl derivatives 3.24 For example, the modified Staudinger protocol involves coupling of the iminophosphorane intermediate 6 with an acyl halide 7 to afford the β-chloroimine 8, which is hydrolyzed to the desired amides 1 (Scheme 1).22 This methodology has been developed as an alternative to avoid the anomerization of amine 2. However, examples of anomeric interconversion have been reported, mostly for extended reaction times25 and when α-glycopyranosyl azides are employed.26 Additionally, the phosphine oxide obtained as a by-product of Staudinger reactions is difficult to separate, decreasing the yields of the products in the purification steps. Another disadvantage lies in the use of activated carboxylic acids, such as acid halides 7, which are highly reactive and susceptible to hydrolysis.
Taking into account the interesting biological properties of N-glycosyl amides, we describe herein the synthesis of this type of compounds using the hydrogenolysis of glycosyl azides with gluco, galacto, or xylo configuration as the key step. The catalytic hydrogenation of glycosyl azide 5 in the presence of Pd/C constitutes a cleaner alternative to the Staudinger procedure, as this catalyst can be readily removed by filtration to yield the respective glycosyl amine 2. To avoid hydrolysis and anomerization of glycosylamines 2, their derivatization with activated carboxylic acids 3 was carried out immediately after obtaining them. Furthermore, to prevent the use of acyl halides, the amidation of carboxylic acids was performed using DCC. The per-O-acetyl derivatives of glycosyl amides were de-O-acetylated and those containing a galactose residue were tested as inhibitors of the of E. coli β-galactosidase.
Results and discussion
The synthesis of the glycosyl azides of gluco (5a), galacto (5b), or xylo (5c) configuration was performed as shown in Scheme 2.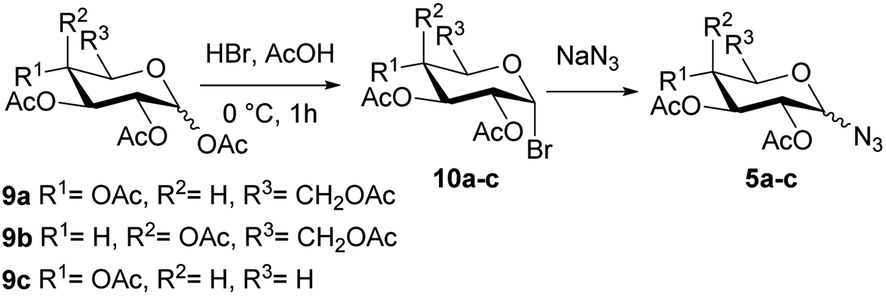 | ||
| Scheme 2 Synthesis of glycosyl azides of gluco (5a), galacto (5b), or xylo (5c) configuration. | ||
The per-O-acetyl monosaccharides 9a–c were treated with a mixture of hydrogen bromide in acetic acid to yield the α-glycosyl bromides 10a–c. These compounds 10a–c were substituted with sodium azide in DMF using protocols similar to those described in the literature,27–29 to afford diastereoselectively the glycosyl azides 5a and 5b of β configuration (Table 1, entry 1 and 2). On the other hand, a mixture of α/β (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1) anomers was obtained in the case of xylo derivate 5c (Table 1, entry 3). Ibatulin and coworkers (2000)30 reported that β-glycosyl azides can be selectively obtained using a water/acetone mixture as a solvent, without significant losses due to hydrolysis of bromide 10. Thereby, employing these conditions, the xylo derivate 5c was obtained exclusively with the β configuration (Table 1, entry 4).
:1.1) anomers was obtained in the case of xylo derivate 5c (Table 1, entry 3). Ibatulin and coworkers (2000)30 reported that β-glycosyl azides can be selectively obtained using a water/acetone mixture as a solvent, without significant losses due to hydrolysis of bromide 10. Thereby, employing these conditions, the xylo derivate 5c was obtained exclusively with the β configuration (Table 1, entry 4).
| Entry | Substrate | Reaction conditions | Isolated yield of 5 |
|---|---|---|---|
| 1 | 10a | NaN3, DMF 70 °C, 5 h | 73% (β only) |
| 2 | 10b | NaN3, DMF 70 °C, 5 h | 71% (β only) |
| 3 | 10c | NaN3, DMF, 70 °C, 5 h | 72% (α/β; 1:1.2) |
| 4 | 10c | NaN3, acetone/H2O, 40 °C, 3 h | 63% (β only) |
The β-configuration of the glycosyl azides was stablished according to the large coupling constant values in the 1H NMR spectra (J1,2 ≈ 9–10 Hz) of 10a–c, which is indicative of a trans diaxial orientation for H-1 and H-2.
With the glycosyl azides in hand, we proceed to select the carboxylic acids 11–13β to obtain the corresponding amides (Chart 1). The carboxylic acid derivatives 11 and 12 were selected since they are commercially available and are also stable solids that can be easily handled. Furthermore, the aromatic moiety of these compounds could contribute to the interaction with the active site of the enzyme, since an aromatic hydrophobic residue in galactose derivatives is usually relevant in the recognition process by E. coli β-galactosidase.31,32
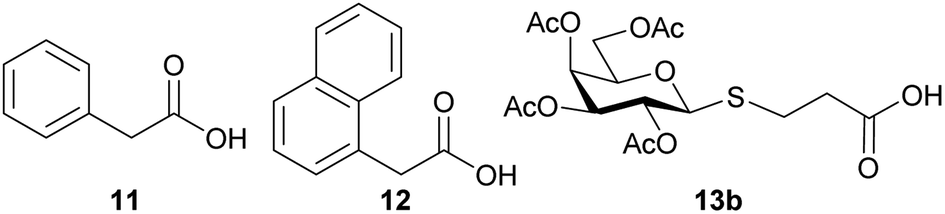 | ||
| Chart 1 Carboxylic acids selected for amidation reactions with glycosyl amines 2a–c. | ||
In order to obtain bridged disaccharides containing the thio and amide functionalities, the thioglyco acid 13 was synthesized. The thioglycosylation of per-O-acetyl galactose 9b with 3-mercaptopropionic acid (14) was conducted in the presence of a Lewis acid. The reaction conditions have been evaluated, as shown in Table 2.
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Lewis acid | Isolated yield | α/β ratio | |
| 13α (%) | 13β (%) | ||||
| a Reaction conditions: 9b (2.5 mmol), 14 (1.1 equiv.), Lewis acid (1.1 equiv.), solvent (30 mL), reflux, 2 h.b Reaction conditions: 9b (2.5 mmol), 14 (4.4 equiv.), Lewis acid (4.4 equiv.), solvent (30 mL), 0 °C, 9 h. | |||||
| 1 | CH3CN | SnCl4 | 21 | 34 | 1:1.6 |
| 2 | CH2Cl2 | SnCl4 | 20 | 32 | 1:1.6 |
| 3 | CH3CN | BF3·OEt2 | 25 | 43 | 1:1.7 |
| 4 | CH2Cl2 | BF3·OEt2 | 14 | 31 | 1:2.4 |
| 5b | CH2Cl2 | BF3·OEt2 | 17 | 23 | 1:1.4 |
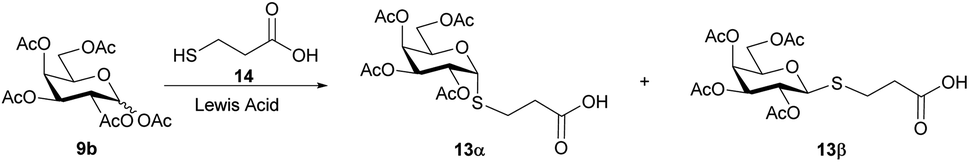
The reaction performed in anhydrous acetonitrile or dichloromethane led to 13 as a mixture of anomers, which could be separated by column chromatography, to afford the β anomer as main product. The compound previously reported in the literature33 as α-anomer had in fact the β-configuration, according to the analysis of the 1H NMR spectrum. The use of SnCl4 in acetonitrile (ESI Table 4.2,† entry 1) or in dichloromethane (ESI Table 4.2,† entry 2) lead to almost identical results in terms of isolated yield and selectivity towards the anomer of interest (≈50–55%; α/β, 1:1.6). When using BF3 OEt2 as Lewis acid, an improvement in the yield was observed in acetonitrile (ESI Table 4.2,† entry 3, 68%; α/β, 1:1.7), while a lower yield was obtained in dichloromethane, compared to that obtained using SnCl4. However, the latter conditions (ESI Table 4.2,† entry 4, 45%; α/β, 1:2.4) led to a higher selectivity toward the β anomer. Additionally, in an attempt to enhance the selectivity, the reaction was carried out at 0 °C (ESI Table 4.2,† entry 5), unfortunately, a decrease in overall yield and selectivity (40%; α/β, 1:1.4) was observed, and a longer reaction time was required.
Once all the precursors were obtained, the synthesis of glycosyl amides 1 was carried out through a two-step reaction sequence as illustrated in Scheme 3.
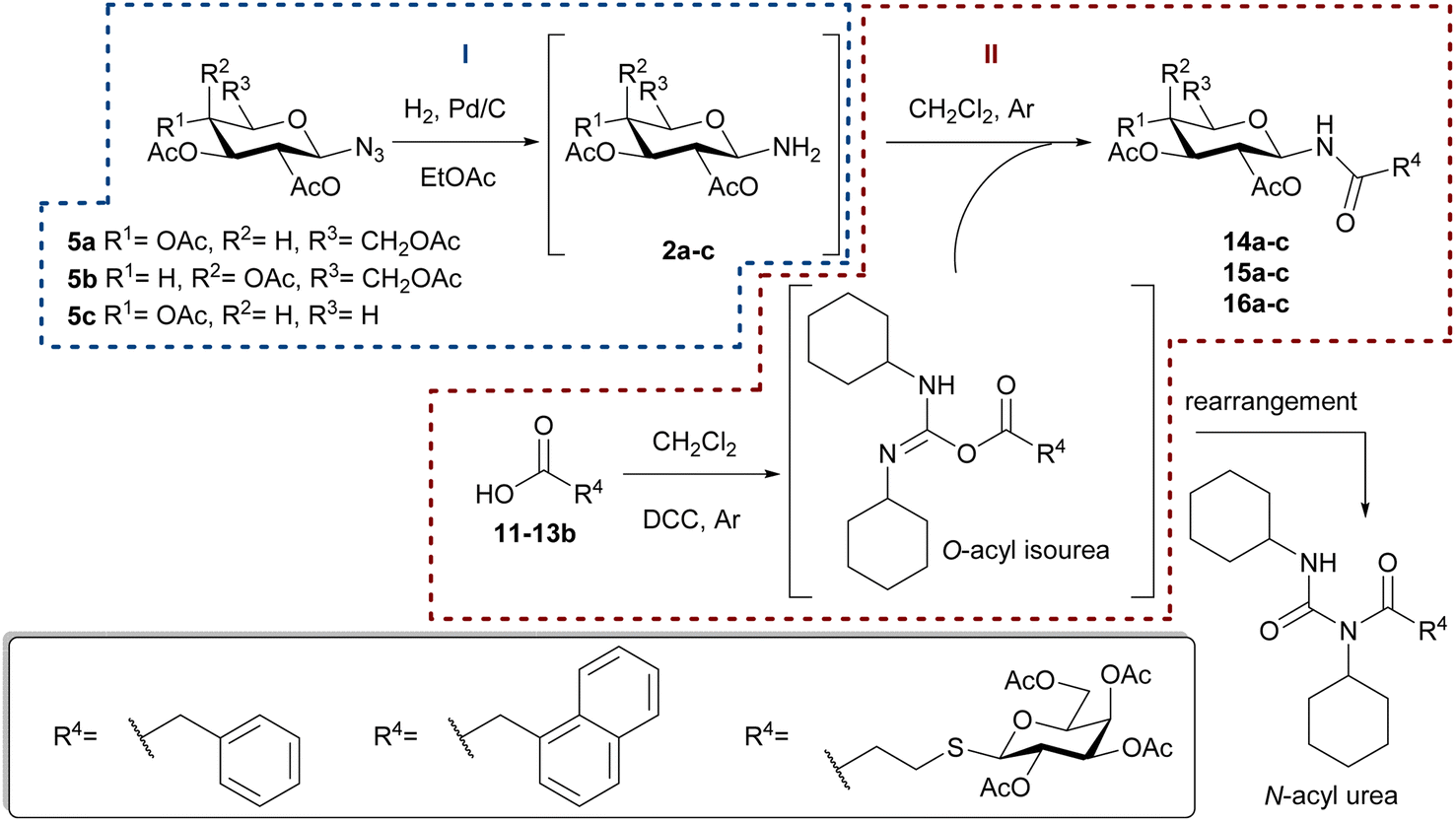 | ||
| Scheme 3 Synthesis of N-glycosylamides 14a–c, 15a–c, and 16a–c. | ||
The catalytic hydrogenation of glycosyl azides 5a–c was performed using H2 in the presence of Pd/C as the catalyst, in anhydrous ethyl acetate (Scheme 3, Reaction I). The temperature control is a key factor, as below 40 °C the substrate conversion is very low. Upon complete consumption of the substrate, the catalyst was filtered, and the solvent evaporated to afford quantitatively the glycosyl amines 2a–c. Immediately after the hydrogenation, and to prevent the anomerization, the amino group of 2a–c was acylated with carboxylic acids 11–13β, with DCC as coupling agent, to yield the desired glycosyl amides 1 (Scheme 3, Reaction II). The products having gluco (14a, 15a), galacto (14b, 15b) or xylo (14c, 15c) configurations were obtained in good yields after the two-step synthetic sequence (Chart 2). Somewhat lower yields were obtained for N-glycosyl amides 16a–c, which were accompanied by significant amounts of the N-acyl urea, derived from acid 13β. The formation of this byproduct was attributed to the rearrangement of the O-acyl isourea intermediate34 that, in this case, derived from 13β and DCC. Additionally, lower yields have also been reported in similar coupling reactions using bulky acids or amines derivatives.35,36
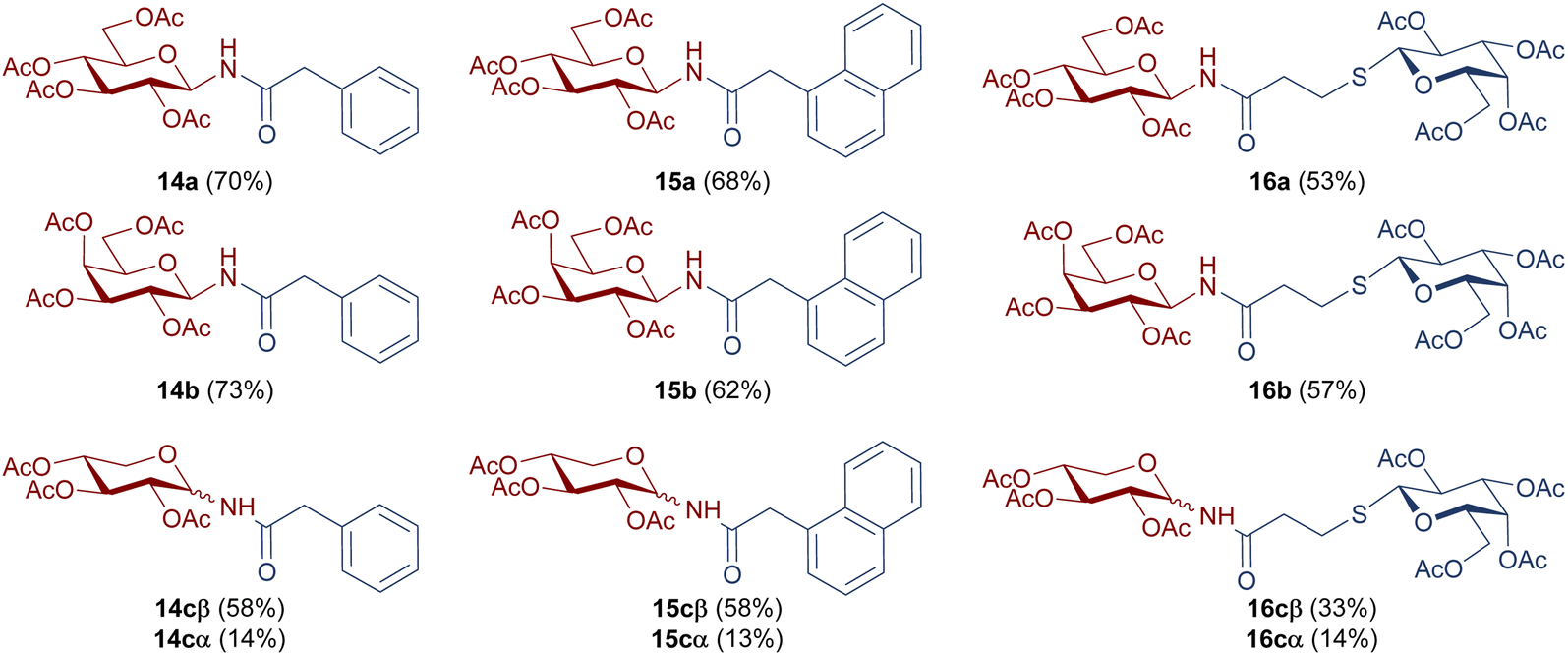 | ||
| Chart 2 Structure and isolated yields of per-O-acetylated N-glycosyl amides 14a–c, 15a–c, and 16a–c. | ||
To improve the yields in the production of bridged disaccharide derivatives, alternative conditions were evaluated using as a model the synthesis of 16a from the glycosyl azide 5a and the thioglyco acid 13β (Table 3). The Steglich amidation was attempted, since the use of N,N-dimethylaminopyridine (DMAP) and carbodiimides was reported to facilitate the formation of esters37–39 or amides40,41 of sterically hindered acids or low-reactivity nucleophiles (alcohols or amines).
|
|||
|---|---|---|---|
| Entry | Additive | Temperature (°C) | Isolated yield of 16a (%) |
| a Reaction conditions: (I) 5a (0.33 mmol), Pd/C 10% (18.6 mg), EtOAc(anh) (3 mL), H2(atm), 40 °C, 1 h. (II) 13β (0.36 mmol, 1.1 equiv.), DCC (0.5 mmol, 1.5 equiv.), CH2Cl2(anh) (3 mL), Ar(atm), 16 h. | |||
| 1 | DMAP (5 mol%) | 30 | 46 |
| 2 | DMAP (10 mol%) | 30 | 45 |
| 3 | DMAP (1 equiv.) | 30 | 51 |
| 4 | — | Reflux | 47 |
| 5 | DMAP (1 equiv.) | Reflux | 54 |
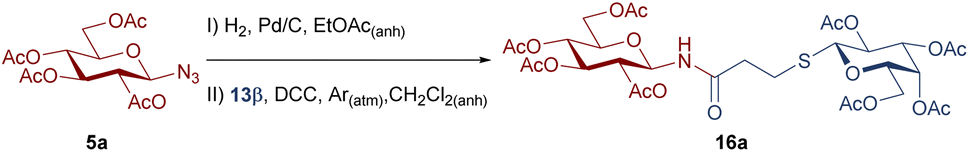
The addition of catalytic amounts of DMAP (Table 3, entries 1 and 2) led to a slightly decreased in yield, compared to the conditions initially employed for the production of 16a (53%). When the quantity of DMAP was increased to 1 equiv., the yield underwent a subtle improvement (Table 3, entry 3), approaching the initial value. Finally, the reaction was also carried out under reflux in the absence or presence of 1 equiv. of DMAP (Table 3, entries 4 and 5). However, even under these conditions, the yields were similar to those obtained initially.
It should be noted that glycosyl amides with gluco (14a–16a) or galacto configuration (14b–16b) were diastereoselectivity obtained, with exclusive formation of β anomers. Therefore, no anomerization of the glycosyl amine 2a, 2b took place. In contrast, the glycosyl amides with the xylo configuration (14c–16c) were obtained as a mixture of α and β anomers (Scheme 4), which were separated by column chromatography (the β anomer was the main product). In these cases, an anomerization process occurred in glycosyl amine 2c prior to amidation.
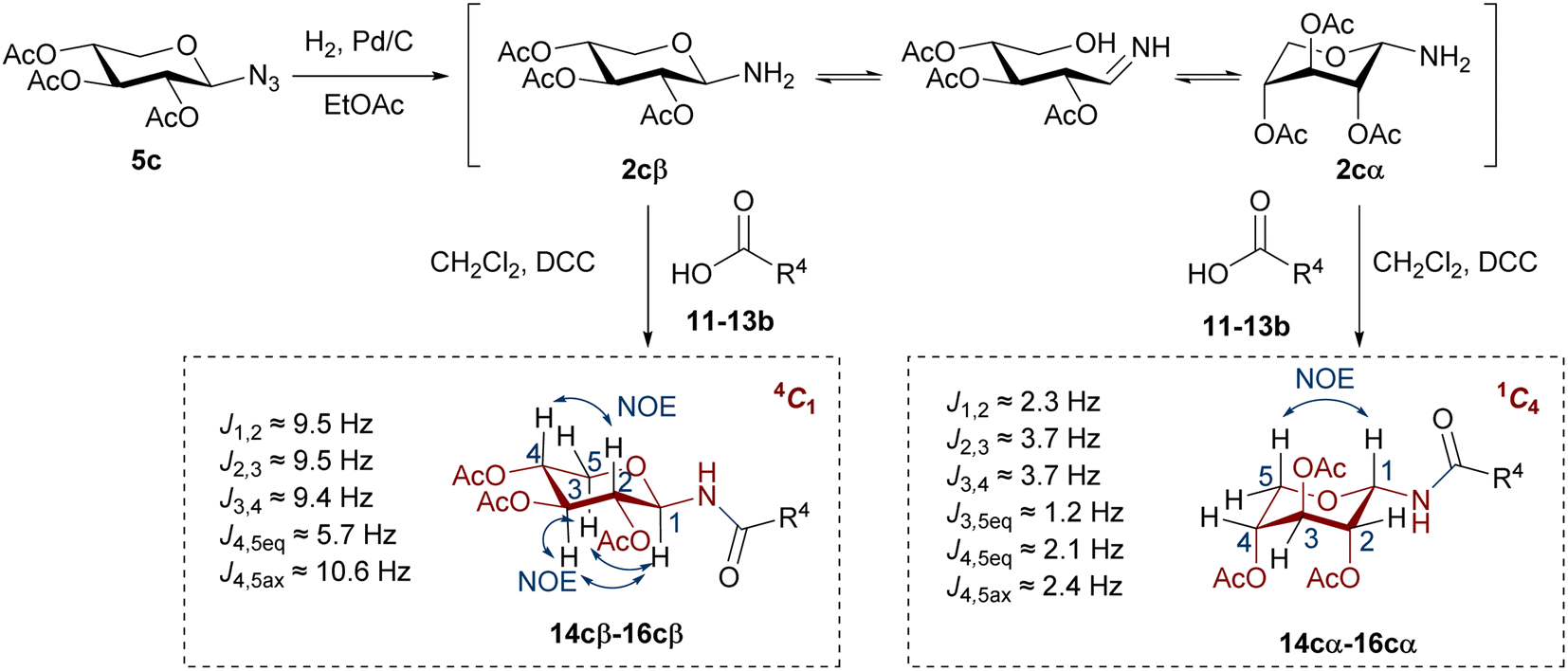 | ||
| Scheme 4 Synthesis of glycosyl amides with the xylo configuration. | ||
The configuration of the anomeric centre of the amides was established according to the value of the coupling constant J1,2, determined from the 1H NMR spectra. The configuration β was assigned for the derivatives with higher values (J1,2 ∼ 9–10 Hz), and α to those with lower values (J1,2 ∼ 2–3 Hz). However, the analysis of the coupling constants of the remaining ring protons in the xylo derivatives 14c–16c (J2,3, J3,4, J4,5eq, J4,5ax) showed significant differences in the respective J values determined for the α or β anomers. The amides with β configuration showed high J values (∼9–10 Hz) for the protons of the pyranose ring, indicative of a trans-diaxial arrangement characteristic of the 4C1 chair conformation (Scheme 4). In contrast, α anomers exhibited smaller J values (J ∼ 2–4 Hz), consistent with a trans-diequatorial arrangement of the coupled protons, and hence a preferential 1C4 chair. Additionally, long-range coupling constants (4J) between diequatorial protons in a W disposition (H2, H4 or H3, H5eq) support the 1C4 conformation for α anomers. Finally, 2D-NOESY spectra of the β glycosyl amides confirmed the assigned conformations. Thus, NOE contacts were observed between the 1,3-diaxial protons (H1, H3, and H5ax), as well as between H2 and H4, justifying the 4C1 conformation. These NOE contacts were absent in the α anomers, which instead exhibited a spatial interaction between H1 and H5ax, as expected for the 1C4 chair. In this conformation of α anomers the O-acetyl substituents are found in an axial arrangement, with those at C-2 and C-4 displaying a repulsive 1,3-diaxial interaction. However, the 1C4 chair is stabilized by the equatorial orientation of the bulky anomeric substituent, which in turn leads to the exo-anomeric effect, characteristic of N-glycosydic linked derivatives.42
To verify the anomerization of glycosyl amine 2cβ, this compound was synthesized from 5c (Scheme 5). Thus, the mixture of hydrogenation of 5c was subjected to the usual work-up and the resulting product was immediately analyzed by NMR spectroscopy. The 1H NMR spectrum (Fig. 1) showed, as expected, a mixture of 2cβ and 2cα (β/α = 5:1). Similar to hemiacetals the aminoacetals undergo rapid anomerization through the open-chain intermediate, as shown in Scheme 4. In the 4C1 conformation the β anomer is stabilized by the exo-anomeric effect, while the α anomer should be stabilized by both, the anomeric and exo-anomeric effects. However, in the 4C1 conformation of the α anomer the nN → *σCO orbital interaction, associated with the exo-anomeric effect, necessarily points the hydrogen atom on nitrogen under the pyranose ring, resulting in a destabilizing steric effect.43 Thus, the major anomer 2cβ adopted the 4C1 conformation, similar to 14cβ–16cβ, which is stabilized by the exo-anomeric effect and the equatorial orientation of the ring substituents. This conformation was evidenced by the large coupling constant values for the ring protons.
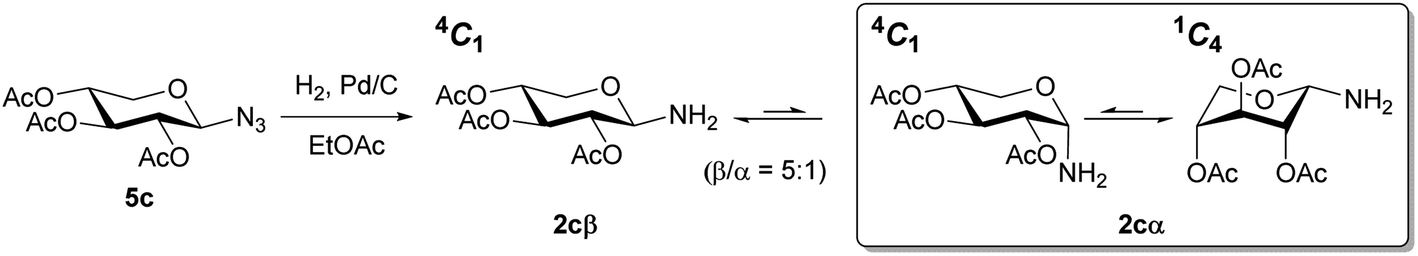 | ||
| Scheme 5 Hydrogenation of azide 5c to afford N-glycosyl amine 2cβ and anomerization to 2cα. | ||
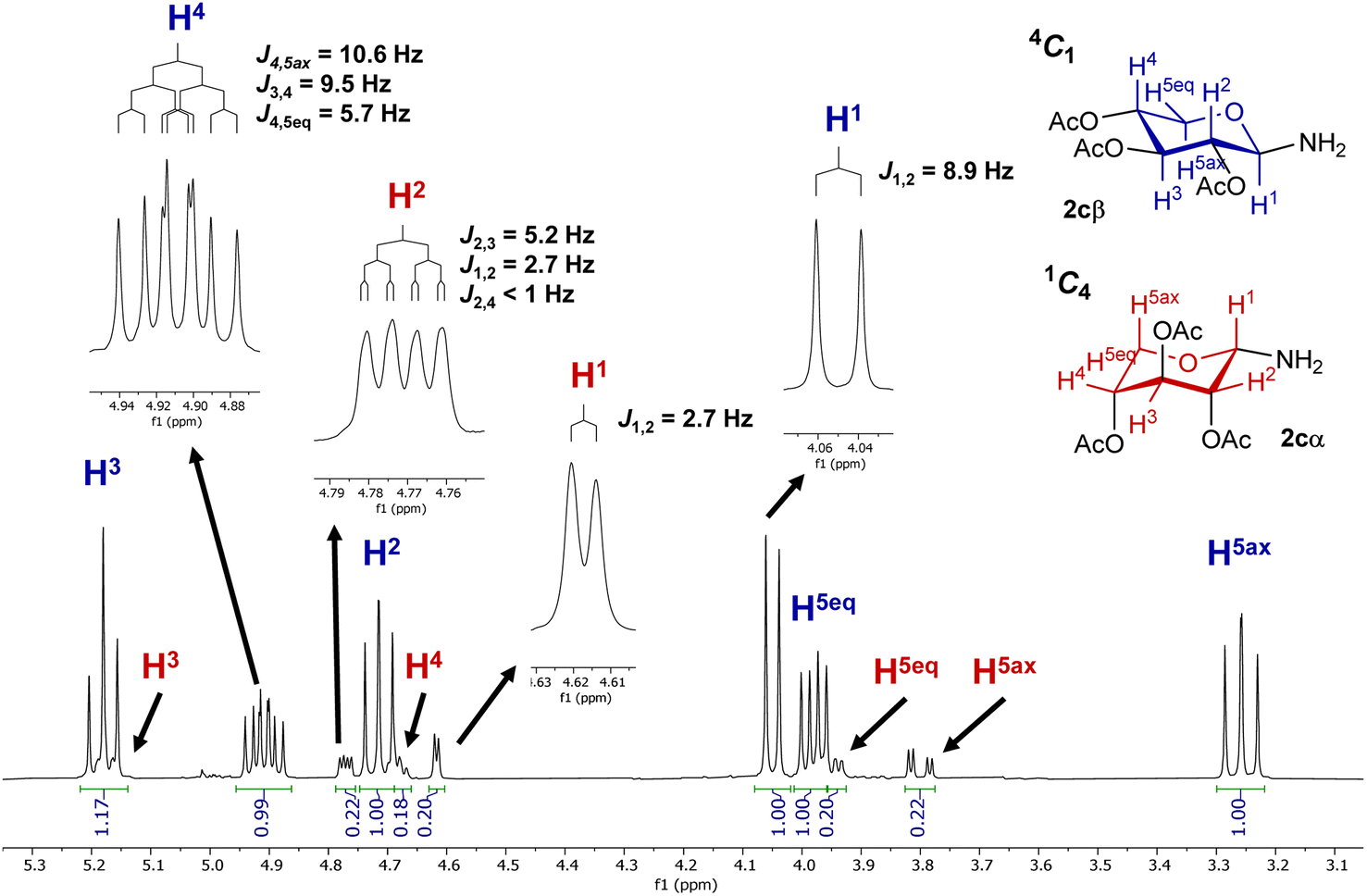 | ||
| Fig. 1 1H NMR spectrum of the mixture of N-glycosyl amines 2cβ and 2cα. | ||
In contrast, glycosyl amine 2cα was found preferentially in the 1C4 conformation, as dictated by the small coupling constant values and the presence of W-type coupling (4J) between H2 and H4 (as was observed for 14cα–16cα). However, the values of J2,3 (5.2 Hz) and J4,5eq (4.4 Hz) are rather large for diequatorially arranged protons (in 1C4) and suggest some contribution of the 4C1 chair to the conformational equilibrium. The fact that the 2cβ structure is free of the 1,3-diaxial interactions observed for 2cα, is in agreement with the general observation that for D-glycosyl amines the α configuration is less stable than the β configuration.23 To support the findings on the conformational behaviour of N-glycosyl amides having the xylo configuration, a conformational search was performed using Molecular Mechanics and MMFF94 as the force field. All conformations up to 3.00 kcal mol−1 above the conformer with the minimum energy were analysed.
As a selection criterion, among the various conformations obtained, only those that showed chair forms (1C4 and 4C1) were considered.
The conformational search revealed that for all β anomers the 4C1 chair was the lowest energy conformer (Fig. 2), and that was the one exclusively populated within the energy window of 3.00 kcal mol−1 (ESI Table 1,† entries 1–3). On the other hand, for α N-glycosyl amides the 4C1 and 1C4 chairs were found to have similar energy (except for 16cα, which was determined to populate only the 1C4 form). However, in all cases, the 1C4 chair was the lowest energy conformation (ESI Table 1,† entries 4–6). These results agree completely with those obtained from the analysis of 1H NMR and 2D NOESY spectra.
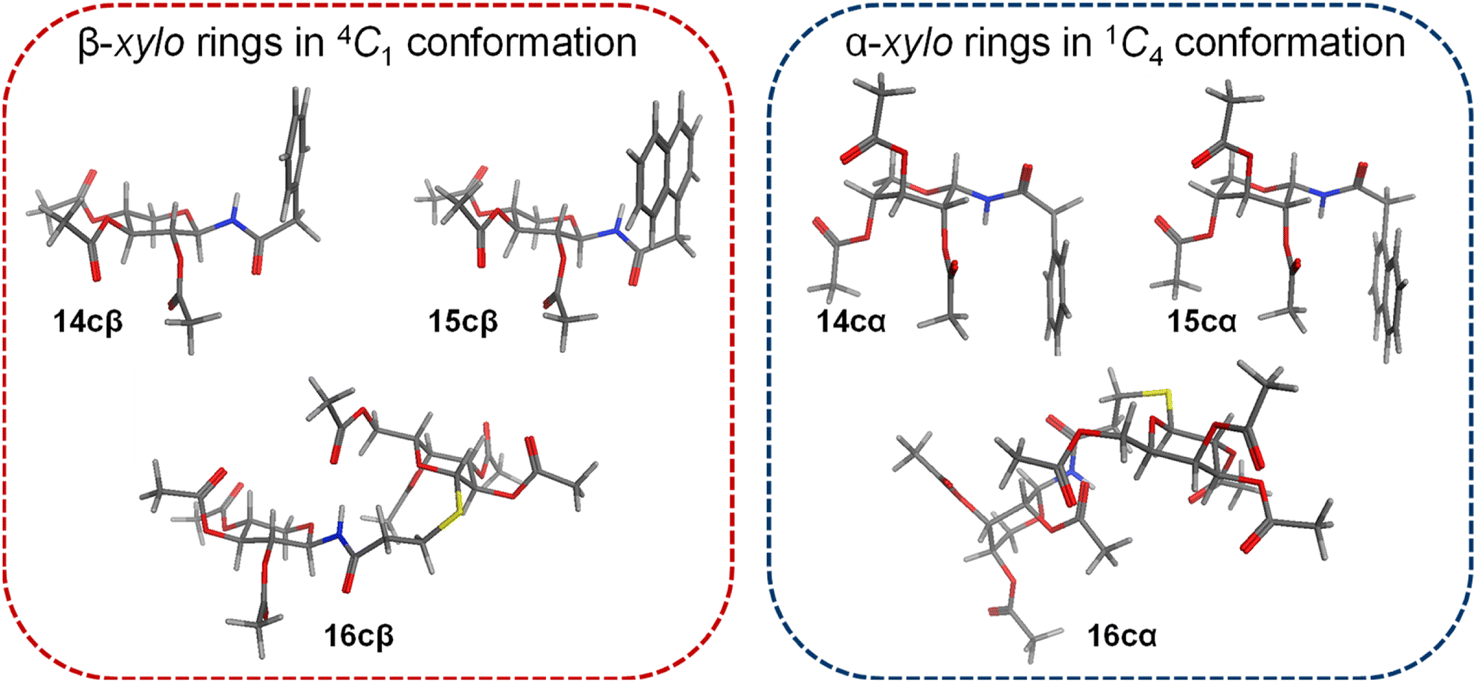 | ||
| Fig. 2 Stable conformations of xylopyranosyl amides, determined by NMR and MM calculations. | ||
For the evaluation of the inhibitory activity, OH free compounds were required. For this reason, the de-O-acetylation of 14a-cβ, 15a-cβ, and 16a-cβ was carried out under mild conditions, as shown in Scheme 6. The hydrolysis of acetoxy functionalities was performed with a mixture of CH3OH/Et3N/H2O (4:1:5), to afford the hydroxyl free derivatives 17a-cβ, 18a-cβ, and 19a-cβ with very good to excellent yields.
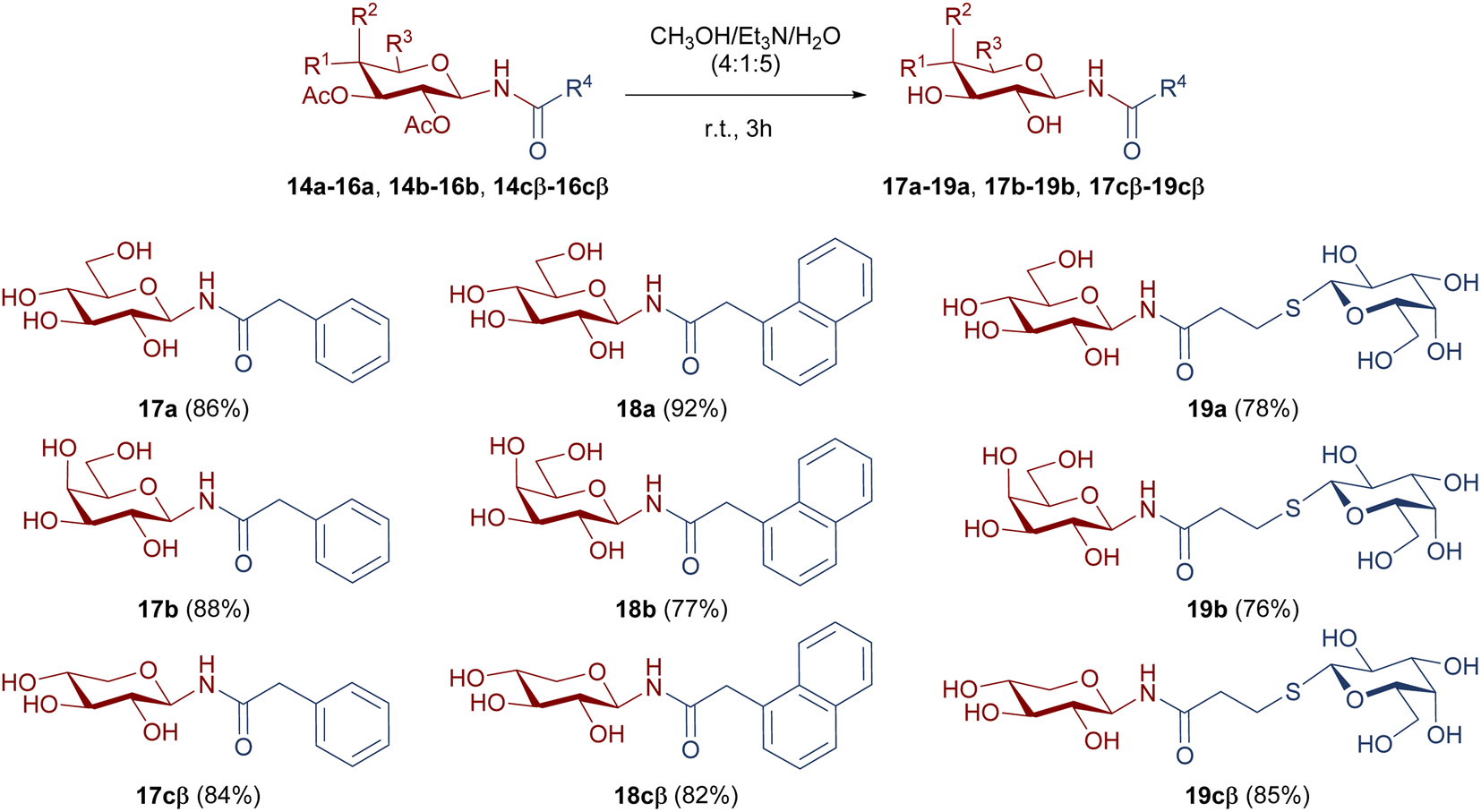 | ||
| Scheme 6 de-O-Acetylation of N-glycosyl amides 14a-cβ, 15a-cβ, and 16a-cβ. | ||
Biological evaluation
The N-glycosyl amides 17b, 18b, 19a-cβ, containing a galactopyranose residue, were evaluated as inhibitors of β-galactosidase from E. coli. Thus, the enzyme was incubated in a solution of o-nitrophenyl β-D-galactopyranoside as a substrate and with increasing concentrations of the glycomimetics. After 10 min at 37 °C, the enzyme was denatured upon addition of a 0.2 M sodium borate buffer and the released o-nitrophenol was quantified spectrophotometrically. All compounds were inhibitors of β-galactosidase from E. coli, as shown in the inhibition diagram in (Fig. 3).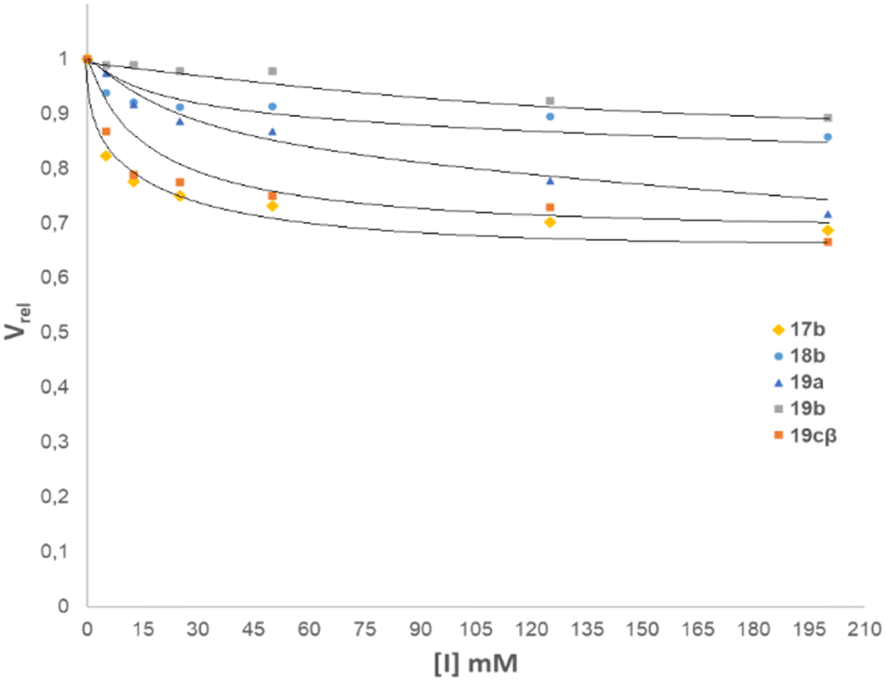 | ||
| Fig. 3 Effect of the concentration of N-glycosyl amides 17b, 18b, 19a, 19b, and 19cβ on the enzymatic activity of the β-galactosidase from E. coli. | ||
The inhibition studies showed that the N-glycosylamides investigated were moderate inhibitors of E. coli β-galactosidase. The compounds that displayed a better inhibition profile were the thio-bridged N-glycosyl amide disaccharide 19cβ and the N-glycosyl amide 17b (Fig. 3). Even in these cases, the concentration of inhibitor needed to decrease the relative velocity by 30% was greater than 2.5 mM, for a substrate concentration of 1 mM. It is clear that these compounds have an effect on the inhibition of the enzyme, but this effect is moderate.
Since these glycomimetics exhibit low structural similarity to the natural enzyme substrate (lactose), it is not surprising that they display moderate inhibition profiles. Nevertheless, this study represents a significant contribution as an initial starting point for the design and development of novel and improved inhibitors, as this work constitutes the first report of N-glycosyl amides with inhibitory activity against the β-galactosidase from E. coli.
Experimental
Materials and methods
Column chromatography was carried out with silica gel 60 (230–400 mesh). Analytical thin layer chromatography (TLC) was carried out on silica gel 60 F254 aluminium-backed plates (layer thickness 0.2 mm). The spots were visualized by charring with a solution of (NH4)6Mo7O24·4H2O 25 g L−1, (NH4)4Ce(SO4)4·2H2O 10 g L−1 and 10% H2SO4 in H2O. Nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz (1H) or 100 MHz (13C). Chemical shifts were calibrated to tetramethylsilane or to a residual solvent peak (CHCl3: 1H: δ 7.26 ppm, 13C: δ 77.2 ppm or H2O: δ 4.79 ppm). Assignments of 1H and 13C NMR spectra were assisted by 2D 1H-COSY and 2D 1H–13C HSQC and HMBC experiments. 2D NOESY Spectra were also recorded. To assign the NMR signals of bridged disaccharides, the H and C atoms of the thiogalactopyranose residue have been labeled with primed locators. The coupling constants values are reported in Hz and resonance multiplicities abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sx = sextet, m = multiplet, br = broad. High-resolution mass spectra (HRMS) were obtained using the electrospray ionization (ESI) technique and Q-TOF detection.Synthetic procedures
General procedure for the synthesis of per-O-acetyl-β-D-pyranosyl-1-azides27–29
:1 → 2:1) to afford the per-O-acetyl-β-D-pyranosyl-1-azides 5a–c.:1). [α24D] = −31.3 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.20 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 5.08 (t, J3,4 = J4,5 = 9.7 Hz, 1H, 4-H), 4.93 (t, J1,2 = J2,3 = 9.2 Hz, 1H, 2-H), 4.63 (d, J1,2 = 8.9 Hz, 1H, 1-H), 4.25 (dd, J6a,6b = 12.5, J5,6a = 4.8 Hz, 1H, 6a-H), 4.15 (dd, J6a,6b = 12.5, J5,6b = 2.2 Hz, 1H, 6b-H), 3.78 (ddd, J4,5 = 10.0, J5,6a = 4.7, J5,6b = 2.3 Hz, 1H, 5-H), 2.08, 2.05, 2.01, 1.99 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) = 170.7, 170.2, 169.4, 169.3 (COCH3), 88.0 (C-1), 74.2 (C-5), 72.7 (C-3), 70.8 (C-2), 68.0 (C-4), 61.8 (C-6), 20.8, 20.6 (×3) (COCH3) ppm. HRMS (ESI): calcd for C14H19N3NaO9 396.1013 [M + Na]+; found 396.1014.:1). [α25D] = −15.9 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.40 (br. d, J3,4 = 3.2, J4,5 < 1 Hz, 1H, 4-H), 5.14 (dd, J2,3 = 10.2, J1,2 = 8.9 Hz, 1H, 2-H), 5.02 (dd, J2,3 = 10.4, J3,4 = 3.3 Hz, 1H, 3-H), 4.59 (d, J1,2 = 8.7 Hz, 1H, 1-H), 4.17 (dd, J6a,6b = 11.3, J5,6a = 6.9 Hz, 1H, 6a-H), 4.13 (dd, J6a,6b = 11.3, J5,6b = 6.2 Hz, 1H, 6b-H), 4.00 (br. t, J5,6a = J5,6b = 6.5, J4,5 < 1 Hz, 1H, 5-H), 2.15, 2.07, 2.04, 1.97 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 170.4, 170.2, 170.1, 169.4 (COCH3), 88.4 (C-1), 73.0 (C-5), 70.9 (C-3), 68.2 (C-2), 67.0 (C-4), 61.3 (C-6), 20.7 (×3), 20.6 (COCH3) ppm. HRMS (ESI): calcd for C14H19N3NaO9 396.1013 [M + Na]+; found 396.1006.When the general procedure (method A) was applied to bromide 10c, both anomers of azide 5c were isolated as an anomeric mixture (α/β = 1:1.2) that could not be separated by column chromatography. Yield: 73%. Rf = 0.61, pentane/EtOAc (2:1).
:1.2). 1H NMR (400 MHz, CDCl3): δ = 5.49 (d, J1,2 = 4.0 Hz, 1H, 1-H), 5.34 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 4.92 (td, J3,4 = J4,5ax = 9.6, J4,5eq = 5.9 Hz, 1H, 4-H overlapped with 4-H of 5cβ), 4.86 (dd, J2,3 = 9.2, J1,2 = 4.0 Hz, 1H, 2-H overlapped with 2-H of 5cβ), 3.91 (dd, J5ax,5eq = 11.3, J4,5eq = 5.8 Hz, 1H, 5eq-H), 3.75 (t, J4,5ax = J5ax,5eq = 10.8 Hz, 1H, 5ax-H), 2.07, 2.01 (×2) (3s, 9H, COCH3 overlapped with COCH3 of 5cβ) ppm. 13C NMR (100 MHz, CDCl3) δ = 170.0, 169.9, 169.8 (COCH3), 86.5 (C-1), 70.2 (C-2), 69.0 (C-3), 68.7 (C-4), 60.6 (C-5), 20.7 (×2), 20.6 (COCH3 overlapped with COCH3 of 5cβ) ppm.The β anomer was obtained diastereoselectively using Method B, as mentioned below.
2,3,4,-Tri-O-acetyl-β-D-xylopyranosyl-1-azide (5cβ)30,55,56. To a solution of glycosyl bromide 10c (4.32 g, 10.5 mmol) in acetone (38 mL), a solution of NaN3 (0.82 g, 12.6 mmol) in H2O (9.5 mL) was added, and the mixture was stirred at room temperature for 3 h, when TLC showed complete consumption of the starting bromide. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography using pentane/EtOAc (4
:1 → 2:1) to afford the glycosyl azide 5c (β anomer) as a white solid. M.p. 83.9–85.5 °C. Yield: 63% (1.986 g). Rf = 0.61; pentane/EtOAc (2:1). [α24D] = −82.7 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.18 (t, J2,3 = J3,4 = 8.9 Hz, 1H, 3-H), 4.97 (td, J3,4 = J4,5ax = 9.3, J4,5eq = 5.3 Hz, 1H, 4-H), 4.86 (t, J1,2 = J2,3 = 8.5 Hz, 1H, 2-H), 4.62 (d, J1,2 = 8.1 Hz, 1H, 1-H), 4.20 (dd, J5ax,5eq = 11.7, J4,5eq = 5.3 Hz, 1H, 5eq-H), 3.43 (dd, J5ax,5eq = 11.7, J4,5ax = 9.6 Hz, 1H, 5ax-H), 2.07, 2.04, 2.03 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 170.1, 169.8, 169.4 (COCH3), 88.4 (C-1), 71.6 (C-3), 70.5 (C-2), 68.5 (C-4), 64.4 (C-5), 20.8, 20.7 (×2) (COCH3) ppm. HRMS (ESI): calcd for C11H15N3NaO7 324.0802 [M + Na]+; found 324.0793.
3-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl) thiopropionic acid (13β) and 3-(2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl) thiopropionic acid (13α). To a solution of penta-O-acetyl galactopyranose 9b (0.976 g, 2.5 mmol) in anhydrous CH3CN (30 mL), mercaptopropionic acid (265 μL, 2.75 mmol) was added, followed by the slow addition of BF3·OEt2 (348 μL, 2.75 mmol). The reaction mixture was refluxed for 2 hours, cooled to room temperature, and stirred overnight. At this time, complete conversion of the starting material was confirmed by TLC. The solvent was removed under reduced pressure and the products were purified by column chromatography using hexane/EtOAc (8
:1 → 1:1) with 1% v/v AcOH.The major product was isolated as a clear yellow syrup and was identified as thioglyco acid 13β.57–59 Yield: 43% (469 mg, β-anomer). Rf = 0.25; hexane/EtOAc (1:1; 1% v/v AcOH). [α24D] = −9.3 (c = 1.1, CHCl3).·1H NMR (400 MHz, CDCl3): δ = 5.41 (d, J3,4 = 2.9 Hz, 1H, 4-H), 5.20 (t, J1,2 = J2,3 = 10.0 Hz, 1H, 2-H), 5.03 (dd, J2,3 = 9.9, J3,4 = 3.3 Hz, 1H, 3-H), 4.54 (d, J1,2 = 9.9 Hz, 1H, 1-H), 4.16 (dd, J6a,6b = 11.3, J5,6a = 6.7 Hz, 1H, 6a-H), 4.07 (dd, J6a,6b = 11.3, J5,6b = 6.5 Hz, 1H, 6b-H), 3.92 (t, J5,6a = J5,6b = 6.4 Hz, 1H, 5-H), 2.97 (dt, Jgem = 13.9, JCH2S,CH2COOH = 7.1 Hz, 1H, CH2S), 2.89 (dt, Jgem = 13.7, JCH2S,CH2COOH = 6.8 Hz, 1H, CH2S), 2.73 (t, JCH2S,CH2COOH = 6.9 Hz, 2H, CH2COOH), 2.14, 2.04 (×2), 1.97 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 176.5 (COOH), 170.7, 170.4, 170.2, 169.8 (COCH3), 84.7 (C-1), 74.6 (C-5), 72.0 (C-3), 67.4 (C-4), 67.3 (C-2), 61.7 (C-6), 35.4 (CH2COOH), 25.5 (CH2S), 20.9, 20.7 (×3) (COCH3) ppm. HRMS (ESI): calcd for C17H24NaO11S 459.0932 [M + Na]+; found 459.0916.
The minor product was isolated as a light-yellow syrup and identified as the thioglyco acid 13α. The structure of this compound has been misassigned33 as the signals reported agreed with those of the β-anomer. Yield: 25% (273 mg, α-anomer). Rf = 0.30; hexane/EtOAc (1:1; 1% v/v AcOH). [α18D] = +127.3 (c = 1.1, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.77 (d, J1,2 = 5.5 Hz, 1H, 1-H), 5.44 (d, J3,4 = 2.4 Hz, 1H, 4-H), 5.26 (dd, J2,3 = 10.9, J1,2 = 5.5 Hz, 1H, 2-H), 5.17 (dd, J2,3 = 10.9, J3,4 = 3.1 Hz, 1H, 3-H), 4.57 (t, J5,6 = 6.3 Hz, 1H, 5-H), 4.12 (d, J5,6 = 6.3 Hz, 2H, 6a-H and 6b-H), 2.88 (dt, Jgem = 14.0, JCH2S,CH2COOH = 7.1 Hz, 1H, CH2S), 2.82–2.76 (m, 1H, CH2S), 2.71 (t, JCH2S,CH2COOH = 6.9 Hz, 2H, CH2COOH), 2.14, 2.07, 2.06, 1.99 (4s, 12H, COCH3) ppm. 13C NMR (101 MHz, CDCl3) δ = 175.8 (COOH), 170.7, 170.3 (×2), 170.0 (COCH3), 83.1 (C-1), 68.3 (C-3), 68.1 (×2, C-2, C-4), 67.0 (C-5), 62.1 (C-6), 34.6 (CH2COOH), 25.2 (CH2S), 20.9, 20.8, 20.7 (×2) (COCH3) ppm. HRMS (ESI): calcd for C17H24NaO11S 459.0932 [M + Na]+; found 459.0917.
Meanwhile, to a solution of carboxylic acid 11–13β (0.36 mmol, 1.1 equiv.) in anhydrous CH2Cl2 (3 mL) was added DCC (103 mg, 0.5 mmol, 1.5 equiv.), under Argon atmosphere. To the resulting suspension, the glycosyl amine 2a–c (obtained in the previous step), was added and stirred overnight under Argon. The mixture was cooled in a freezer for 2 h, filtered (to separate the dicyclohexylurea byproduct) and washed with cold CH2Cl2. The resulting solution was evaporated under reduced pressure and the crude was purified by column chromatography using hexane/EtOAc (4:1 → 1:1) for amides 14a–c and 15a–c, and toluene/EtOAc (4:1 → 1:1) for amides 16a–c.
Only the glycosyl amines derived from xylose 2c were characterized by NMR, to perform a conformational analysis of the product obtained (2cβ and 2cα).
:5). Yield: 90%. Rf = 0.13; hexane/EtOAc (1:1).1H NMR (400 MHz, CDCl3) of 2cβ55 δ = 5.17 (t, J2,3 = J3,4 = 9.6 Hz, 1H, 3-H), 4.91 (ddd, J4,5ax = 10.6, J3,4 = 9.5, J4,5eq = 5.7 Hz, 1H, 4-H), 4.71 (dd, J2,3 = 9.7, J1,2 = 8.9 Hz, 1H, 2-H), 4.05 (d, J1,2 = 8.9 Hz, 1H, 1-H), 3.98 (dd, J5ax,5eq = 11.4, J4,5eq = 5.7, 1H, 5eq-H), 3.26 (dd, J5ax,5eq = 11.5, J4,5ax = 10.5 Hz, 1H, 5ax-H), 2.02, 1.98, 1.98 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) of 2cβ δ = 170.3, 170.2, 170.0 (COCH3), 85.5 (C-1), 72.7 (C-3), 72.3 (C-2), 69.5 (C-4), 63.8 (C-5), 20.9, 20.7 (×2) (COCH3) ppm.
1H NMR (400 MHz, CDCl3) of 2cα δ = 5.19–5.16 (m, 1H, 3-H overlapped with 3-H of 2cβ), 4.77 (ddd, J2,3 = 5.2, J1,2 = 2.7, J2,4 < 1 Hz, 1H, 2-H), 4.70–4.67 (m, 1H, 4-H overlapped with 2-H of 2cβ), 4.62 (d, J1,2 = 2.7 Hz, 1H, 1-H), 3.95 (ddd, J5ax,5eq = 12.7, J4,5eq = 4.4, J3,5eq < 1.0 Hz, 1H, 5eq-H overlapped with 5eq-H of 2cβ), 3.80 (dd, J5ax,5eq = 12.8, J4,5ax = 3.2 Hz, 1H, 5ax-H), 2.09, 2.05, 2.05 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) of 2cα δ = 169.8, 169.7, 169.0 (COCH3), 79.6 (C-1), 69.6 (C-2), 67.8 (C-3), 67.2 (C-4), 62.4 (C-5), 21.0, 20.8 (×2) (COCH3) ppm.
:1). [α24D] = +1.0 (c = 1.1, CHCl3)·1H NMR (400 MHz, CDCl3) δ = 7.37–7.21 (m, 5H, PhCH2CONH), 6.33 (d, JNH,1 = 9.1 Hz, 1H, CONH), 5.27 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 5.20 (t, JNH,1 = J1,2 = 9.3 Hz, 1H, 1-H), 5.03 (t, J3,4 = J4,5 = 9.7 Hz, 1H, 4-H), 4.83 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-H), 4.30 (dd, J6a,6b = 12.5, J5,6a = 4.3 Hz, 1H, 6a-H), 4.07 (dd, J6a,6b = 12.4, J5,6b = 1.5 Hz, 1H, 6b-H), 3.80 (ddd, J4,5 = 10.0, J5,6a = 3.9, J5,6b = 2.0 Hz, 1H, 5-H), 3.58 (d, Jgem = 15.2 Hz, 1H, PhCH2CONH), 3.50 (d, Jgem = 15.2 Hz, 1H, PhCH2CONH), 2.07, 2.02, 1.98, 1.83 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.4 (CONH), 170.7 (×2), 169.9, 169.6 (COCH3), 133.9, 129.3, 129.1, 127.6 (PhCH2CONH), 78.4 (C-1), 73.7 (C-5), 72.7 (C-3), 70.3 (C-2), 68.2 (C-4), 61.7 (C-6), 43.9 (PhCH2CONH), 20.8, 20.6 (×2), 20.4 (COCH3) ppm. HRMS (ESI): calcd for C22H27NNaO10 488.1527 [M + Na]+; found 488.1483.:1). [α26D] = +17.2 (c = 1.0, CHCl3)·1H NMR (400 MHz, CDCl3) δ = 7.38–7.23 (m, 5H, PhCH2CO), 6.31 (d, JNH,1 = 9.2 Hz, 1H, NH), 5.41 (br. d, J3,4 = 2.9 Hz, J4,5 < 1 Hz, 1H, 4-H), 5.20 (t, JNH,1 = J1,2 = 9.2 Hz, 1H, 1-H), 5.09 (dd, J2,3 = 10.3, J3,4 = 3.3 Hz, 1H, 3-H), 5.01 (t, J1,2 = J2,3 = 9.8 Hz, 1H, 2-H), 4.14–4.03 (m, 2H, 6b-H and 6a-H), 4.02 (br. t, J5,6a = J5,6b = 6.6, J4,5 < 1 Hz, 1H, 5-H), 3.58 (d, Jgem = 15.3 Hz, 1H, NHCOCH2Ph), 3.50 (d, Jgem = 15.2 Hz, 1H, NHCOCH2Ph), 2.12, 2.03, 1.96, 1.85 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.3 (CONH), 170.9, 170.4, 170.1, 169.8 (COCH3), 134.0, 129.3, 129.1, 127.6 (PhCH2CO), 78.7 (C-1), 72.5 (C-5), 70.9 (C-3), 68.0 (C-2), 67.3 (C-4), 61.2 (C-6), 43.9 (NHCOCH2Ph), 20.7, 20.6 (×2), 20.5 (COCH3) ppm. HRMS (ESI): calcd for C22H27NNaO10 488.1527 [M + Na]+; found 488.1503.:1). [α23D] = −3.6 (c = 1.0, CHCl3).·1H NMR (400 MHz, CDCl3) δ = 7.35–7.23 (m, 5H, PhCH2CO), 6.71 (d, JNH,1 = 9.2 Hz, 1H, NH), 5.25 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 5.15 (t, JNH,1 = J1,2 = 9.3 Hz, 1H, 1-H), 4.95 (ddd, J4,5ax = 10.6, J3,4 = 9.4, J4,5eq = 5.7 Hz, 1H, 4-H), 4.83 (t, J1,2 = J2,3 = 9.5 Hz, 1H, 2-H), 4.05 (dd, J5ax,5eq = 11.6, J4,5eq = 5.7 Hz, 1H, 5eq-H), 3.57 (d, Jgem = 15.0 Hz, 1H, NHCOCH2Ph), 3.51 (d, Jgem = 14.9 Hz, 1H, NHCOCH2Ph), 3.42 (dd, J5ax,5eq = 11.6, J4,5ax = 10.6 Hz, 1H, 5ax-H), 2.01, 2.00, 1.86 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.7 (CONH), 170.6, 169.9, 169.8 (COCH3), 133.2, 129.2, 129.0, 127.4 (PhCH2CO), 78.7 (C-1), 72.3 (C-3), 70.4 (C-2), 69.0 (C-4), 64.5 (C-5), 43.7 (NHCOCH2Ph), 20.7, 20.6, 20.4 (COCH3) ppm. HRMS (ESI): calcd for C19H23NNaO8 416.1316 [M + Na]+; found 416.1298.:1). [α18D] = −22.6 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.37–7.21 (m, 5H, PhCH2CO), 6.17 (d, JNH,1 = 9.0 Hz, 1H, NH), 5.54 (dd, JNH,1 = 9.3, J1,2 = 2.3 Hz, 1H, 1-H), 5.05 (td, J2,3 = J3,4 = 3.8, J3,5eq = 1.5 Hz, 1H, 3-H), 4.69–4.68 (m, 1H, 2-H), 4.66 (br. q, J3,4 = J4,5ax = J4,5eq = 2.6, J2,4 < 1 Hz, 1H, 4-H), 3.98 (dd, J5ax,5eq = 13.4, J4,5ax = 2.4 Hz, 1H, 5ax-H), 3.91 (dt, J5ax,5eq = 13.5, J4,5eq = J3,5eq = 1.9 Hz, 1H, 5eq-H), 3.64 (d, Jgem = 16.5 Hz, 1H, NHCOCH2Ph), 3.59 (d, Jgem = 16.4 Hz, 1H, NHCOCH2Ph), 2.10, 2.05, 1.90 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 170.7 (CONH), 169.6, 169.3, 168.7 (COCH3), 134.0, 129.7, 129.3, 127.8 (PhCH2CO), 74.2 (C-1), 67.9 (C-3), 67.0 (C-2), 66.1 (C-4), 65.0 (C-5), 43.7 (NHCOCH2Ph), 21.0, 20.8, 20.5 (COCH3) ppm. HRMS (ESI): calcd for C19H23NNaO8 416.1316 [M + Na]+; found 416.1294.:1). [α25D] = +6.6 (c = 1.1, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.91 (dd, Jortho = 6.4, Jmeta = 2.7 Hz, 1H, NpthCH2CO), 7.90 (dd, Jortho = 6.3, Jmeta = 2.6 Hz, 1H, NpthCH2CO), 7.87 (br. d, Jortho = 8.1 Hz, 1H, NpthCH2CO), 7.53 (td, Jortho = Jortho = 7.1, Jmeta = 2.3 Hz, 1H, NpthCH2CO), 7.53–7.49 (m, 1H, NpthCH2CO), 7.49 (dd, Jortho = 8.1, Jortho = 7.2 Hz, 1H, NpthCH2CO), 7.40 (dd, Jortho = 6.9, Jmeta = 1.3 Hz, 1H, NpthCH2CO), 6.20 (d, JNH,1 = 8.9 Hz, 1H, NH), 5.16 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 5.13 (t, JNH,1 = J1,2 = 9.3 Hz, 1H, 1-H), 4.96 (dd, J4,5 = 10.1, J3,4 = 9.3 Hz, 1H, 4-H), 4.60 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-H), 4.29 (dd, J6a,6b = 12.5, J5,6b = 4.2 Hz, 1H, 6a-H), 4.12 (d, Jgem = 16.1 Hz, 1H, NHCOCH2), 4.06 (dd, J6a,6b = 12.5, J5,6b = 2.2 Hz, 1H, 6b-H), 3.91 (d, Jgem = 16.1 Hz, 1H, NHCOCH2), 3.78 (ddd, J4,5 = 10.1, J5,6a = 4.3, J5,6b = 2.2 Hz, 1H, 5-H), 2.07, 1.99, 1.90, 1.27 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.5 (CONH), 170.7, 170.3, 169.9, 169.6 (COCH3), 134.3, 132.2, 130.1, 129.1, 128.9, 128.7, 127.0, 126.3, 125.9, 123.7 (NpthCH2CO), 78.5 (C-1), 73.7 (C-5), 72.7 (C-3), 69.9 (C-2), 68.3 (C-4), 61.7 (C-6), 42.2 (NHCOCH2), 20.8, 20.7, 20.6, 19.6 (COCH3) ppm. HRMS (ESI): calcd for C26H29NNaO10 538.1684 [M + Na]+; found 538.1671.:1). [α26D] = +18.7 (c = 1.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.93–7.89 (m, 2H, NpthCH2CO), 7.87 (br. d, Jortho = 8.5 Hz, 1H, NpthCH2CO), 7.55–7.48 (m, 3H, NpthCH2CO), 7.41 (br. d, Jortho = 6.8 Hz, 1H, NpthCH2CO), 6.23 (d, JNH,1 = 8.9 Hz, 1H, NHCO), 5.36 (br. d, J3,4 = 2.8, J4,5 < 1 Hz, 1H, 4-H), 5.12 (t, JNH,1 = J1,2 = 9.2 Hz, 1H, 1-H), 4.99 (dd, J2,3 = 10.3, J3,4 = 3.3 Hz, 1H, 3-H), 4.79 (t, J1,2 = J2,3 = 9.9 Hz, 1H, 2-H), 4.11 (d, Jgem = 16.7 Hz, 1H, NHCOCH2), 4.08–4.06 (m, 2H, 6b-H and 6a-H), 3.98 (br. t, J5,6a = J5,6b = 6.6, J4,5 < 1 Hz, 1H, 5-H), 3.91 (d, Jgem = 16.0 Hz, 1H, NHCOCH2), 2.07, 2.02, 1.89, 1.30 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.4 (NHCO), 170.5 (×2), 170.1, 169.8 (COCH3), 134.3, 132.2, 130.2, 129.0, 128.9, 128.7, 127.0, 126.3, 125.9, 123.7 (NpthCH2CO), 78.8 (C-1), 72.4 (C-5), 70.8 (C-3), 67.6 (C-2), 67.2 (C-4), 61.1 (C-6), 42.3 (NHCOCH2), 20.8, 20.7, 20.5, 19.7 (COCH3) ppm. HRMS (ESI): calcd for C26H29NNaO10 538.1684 [M + Na]+; found 538.1633.:1). [α22D] = +1.11 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.92–7.87 (m, 2H, NpthCH2CO), 7.86 (dd, Jortho = 8.2, Jmeta = 1.2 Hz, 1H, NpthCH2CO), 7.55–7.50 (m, 2H, NpthCH2CO), 7.48 (dd, Jortho = 8.1, Jortho = 7.0 Hz, 1H, NpthCH2CO), 7.39 (dd, Jortho = 7.0, Jmeta = 1.2 Hz, 1H, NpthCH2CO), 6.20 (d, JNH,1 = 8.8 Hz, 1H, NHCO), 5.15 (t, J2,3 = J3,4 = 9.5 Hz, 1H, 3-H), 5.04 (t, JNH,1 = J1,2 = 9.1 Hz, 1H, 1-H), 4.84 (ddd, J4,5ax = 10.5, J3,4 = 9.3, J4,5eq = 5.7 Hz, 1H, 4-H), 4.53 (t, J1,2 = J2,3 = 9.5 Hz, 1H, 2-H), 4.09 (d, Jgem = 16.2 Hz, 1H, NHCOCH2), 4.01 (dd, J5ax,5eq = 11.6, J4,5eq = 5.7 Hz, 1H, 5eq-H), 3.92 (d, Jgem = 16.2 Hz, 1H, NHCOCH2), 3.39 (dd, J5ax,5eq = 11.7, J4,5ax = 10.5 Hz, 1H, 5ax-H), 1.99, 1.91, 1.36 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.7 (NHCO), 170.4, 169.9, 169.8 (COCH3), 134.3, 132.2, 130.1, 129.1, 128.9, 128.7, 127.0, 126.3, 125.9, 123.7 (NpthCH2CO), 79.0 (C-1), 72.1 (C-3), 70.1 (C-2), 69.1 (C-4), 64.5 (C-5), 42.1 (NHCOCH2), 20.8, 20.6, 19.7 (COCH3) ppm. HRMS (ESI): calcd for C23H25NNaO8 466.1472 [M + Na]+; found 466.1467.:1). [α22D] = −43.59 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.89–7.88 (m, 2H, NpthCH2CO), 7.85 (dd, Jortho = 8.1, Jmeta = 1.1 Hz, 1H, NpthCH2CO), 7.57–7.51 (m, 2H, NpthCH2CO), 7.46 (dd, Jortho = 8.1, Jortho = 6.9 Hz, 1H, NpthCH2CO), 7.41 (dd, Jortho = 7.4, Jmeta = 1.1 Hz, 1H, NpthCH2CO), 6.06 (d, JNH,1 = 9.0 Hz, 1H, NHCO), 5.54 (dd, JNH,1 = 9.2, J1,2 = 2.3 Hz, 1H, 1-H), 4.94 (td, J3,4 = J2,3 = 3.6, J3,5eq = 1.6 Hz, 1H, 3-H), 4.60 (qd, J4,5eq = J4,5ax = J3,4 = 3.5, J2,4 < 1 Hz, 1H, 4-H), 4.49 (ddd, J2,3 = 3.5, J1,2 = 2.3, J2,4 < 1 Hz, 1H, 2-H), 4.14 (d, Jgem = 16.9 Hz, 1H, NHCOCH2), 4.01 (d, Jgem = 16.9 Hz, 1H, NHCOCH2), 3.94 (dd, J5ax,5eq = 13.4, J4,5ax = 2.4 Hz, 1H, 5ax-H), 3.85 (dt, J5ax,5eq = 13.6, J4,5eq = J3,5eq = 2.0 Hz, 1H, 5eq-H), 2.10, 1.99, 1.50 (3s, 9H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 170.5 (NHCO), 169.6, 168.9, 168.6 (COCH3), 134.1, 132.0, 130.2, 129.0, 129.0, 128.9, 127.4, 126.6, 125.8, 123.5 (NpthCH2CO), 74.3 (C-1), 67.9 (C-2), 66.9 (C-3), 66.0 (C-4), 65.1 (C-5), 41.8 (NHCOCH2), 21.0, 20.9, 19.9 (COCH3) ppm. HRMS (ESI): calcd for C23H25NNaO8 466.1472 [M + Na]+; found 466.1457.:2). [α22D] = +5.61 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.56 (d, JNH,1 = 9.3 Hz, 1H, NHCO), 5.44 (dd, J3′,4′ = 3.4, J4′,5′ = 1.2 Hz, 1H, 4′-H), 5.29 (t, J2,3 = J3,4 = 9.4 Hz, 1H, 3-H), 5.28 (t, JNH,1 = J1,2 = 9.4 Hz, 1H, 1-H), 5.24 (t, J1′,2′ = J2′,3′ = 10.1 Hz, 1H, 2′-H), 5.06 (t, J3,4 = J4,5 = 9.8 Hz, 1H, 4-H), 5.04 (dd, J2′,3′ = 10.0, J3′,4′ = 3.5 Hz, 1H, 3′-H), 4.95 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-H), 4.47 (d, J1′,2′ = 9.9 Hz, 1H, 1′-H), 4.30 (dd, J6a,6b = 12.5, J5,6a = 4.3 Hz, 1H, 6a-H), 4.20 (dd, J6′a,6′b = 11.5, J5′,6′a = 6.5 Hz, 1H, 6′a-H), 4.13 (dd, J6′a,6′b = 11.2, J5′,6′b = 6.1 Hz, 1H, 6′b-H), 4.08 (dd, J6a,6b = 12.6, J5,6b = 2.0 Hz, 1H, 6b-H), 3.94 (td, J5′,6a′ = J5′,6b′ = 6.2, J4′,5′ = 1.2 Hz, 1H, 5′-H), 3.82 (ddd, J4,5 = 10.2, J5,6a = 4.3, J5,6b = 2.2 Hz, 1H, 5-H), 3.05 (dt, Jgem = 13.5, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.7 Hz, 1H, CH2S), 2.86 (dt, Jgem = 13.9, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.3 Hz, 1H, CH2S), 2.58–2.53 (m, 2H, CH2CONH), 2.16, 2.08, 2.07, 2.07, 2.05, 2.03, 2.01, 1.98 (8s, 24H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.4 (NHCO), 171.0, 170.7, 170.7, 170.3, 170.1, 170.0, 169.9, 169.7 (COCH3), 84.1 (C-1′), 78.2 (C-1), 75.1 (C-5′), 73.7 (C-5), 73.0 (C-3), 71.9 (C-3′), 70.6 (C-2), 68.2 (C-4), 67.6 (C-4′), 66.8 (C-2′), 61.8 (C-6), 61.7 (C-6′), 37.4 (CH2CONH), 25.2 (CH2S), 20.9 (×2), 20.9, 20.9, 20.8, 20.7 (×3) (COCH3) ppm. HRMS (ESI): calcd for C31H43NNaO19S 788.2042 [M + Na]+; found 788.2056.:2). [α20D] = +12.02 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.52 (d, JNH,1 = 9.2 Hz, 1H, NHCO), 5.44–5.42 (m, 2H, 4-H, 4′-H), 5.25 (t, JNH,1 = J1,2 = 9.2, 1H, 1-H), 5.24 (t, J1′,2′ = J2′,3′ = 10.0 Hz, 1H, 2′-H), 5.13 (dd, J2,3 = 9.5, J3,4 = 3.2 Hz, 1H, 3-H), 5.11 (t, J1,2 = J2,3 = 9.3 Hz, 1H, 2-H), 5.04 (dd, J2′,3′ = 10.0, J3′,4′ = 3.3 Hz, 1H, 3′-H), 4.48 (d, J1′,2′ = 9.9 Hz, 1H, 1′-H), 4.22 (dd, J6a′,6b′ = 11.3, J5′,6a′ = 6.4 Hz, 1H, 6a′-H), 4.11 (dd, J6a′,6b′ = 11.4, J5′,6b′ = 6.4 Hz, 1H, 6b′-H), 4.14–4.07 (m, 2H, 6b-H, 6a-H), 4.03 (td, J5,6a = J5,6b = 6.6, J4,5 = 1.1 Hz, 1H, 5-H), 3.94 (td, J5′,6a′ = J5′,6b′ = 6.4, J4′,5′ = 1.2 Hz, 1H, 5′-H), 3.03 (dt, Jgem = 13.7, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.8 Hz, 1H, CH2S), 2.87 (dt, Jgem = 13.7, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.3 Hz, 1H, CH2S), 2.59–2.50 (m, 2H, CH2CONH), 2.16, 2.13, 2.07 (×2), 2.06, 2.03, 1.98, 1.98 (8s, 24H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.3 (NHCO), 171.3, 170.6, 170.5, 170.3, 170.1 (×2), 169.9, 169.8 (COCH3), 84.2 (C-1′), 78.5 (C-1), 74.9 (C-5′), 72.5 (C-5), 71.9 (C-3′), 71.0 (C-3), 68.4 (C-2), 67.5 (C-4′), 67.3 (C-4), 66.9 (C-2′), 61.6 (C-6), 61.2 (C-6′), 37.5 (CH2CONH), 25.2 (CH2S), 21.0, 20.9, 20.8, 20.8 (×2), 20.7 (×2), 20.7 (COCH3) ppm. HRMS (ESI): calcd for C31H43NNaO19S 788.2042 [M + Na]+; found 788.2045.:2). [α20D] = −5.02 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.63 (d, JNH,1 = 9.2 Hz, 1H, NHCO), 5.43 (dd, J3′,4′ = 3.4, J4′,5′ = 1.1 Hz, 1H, 4′-H), 5.28 (t, J2,3 = J3,4 = 9.6 Hz, 1H, 3-H), 5.23 (t, J1′,2′ = J2′,3′ = 10.0 Hz, 1H, 2′-H), 5.17 (t, JNH,1 = 9.3, J1,2 = 9.5 Hz, 1H, 1-H), 5.03 (dd, J2′,3′ = 10.0, J3′,4′ = 3.3 Hz, 1H, 3′-H), 4.96 (ddd, J4,5ax = 10.5, J3,4 = 9.5, J4,5eq = 5.7 Hz, 1H, 4-H), 4.88 (t, J1,2 = J2,3 = 9.5 Hz, 1H, 2-H), 4.47 (d, J1′,2′ = 9.9 Hz, 1H, 1′-H), 4.19 (dd, J6a′,6b′ = 11.5, J5′,6a′ = 6.6 Hz, 1H, 6a′-H), 4.12 (dd, J6a′,6b′ = 11.5, J5′,6b′ = 6.0 Hz, 1H, 6b′-H), 4.05 (dd, J5ax,5eq = 11.6, J4,5eq = 5.7 Hz, 1H, 5eq-H), 3.93 (td, J5′,6a′ = J5′,6b′ = 6.2, J4′,5′ = 1.1 Hz, 1H, 5′-H), 3.43 (dd, J5ax,5eq = 11.6, J4,5ax = 10.5 Hz, 1H, 5ax-H), 3.04 (dt, Jgem = 13.7, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.8 Hz, 1H, CH2S), 2.85 (dt, Jgem = 14.0, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.2 Hz, 1H, CH2S), 2.54 (t, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.0 Hz, 2H, CH2CONH), 2.15, 2.07, 2.05, 2.04, 2.02, 2.02, 1.97 (7s, 21H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.6 (NHCO), 171.0, 170.7, 170.3, 170.1, 170.0, 169.9, 169.8 (COCH3), 84.1 (C-1′), 78.7 (C-1), 75.0 (C-5′), 72.6 (C-3), 71.9 (C-3′), 70.8 (C-2), 69.1 (C-4), 67.6 (C-4′), 66.9 (C-2′), 64.6 (C-5), 61.8 (C-6′), 37.4 (CH2CONH), 25.3 (CH2S), 20.9 (×2), 20.9, 20.8 (×2), 20.7, 20.7 (COCH3) ppm. HRMS (ESI): calcd for C28H39NNaO17S 716.1831 [M + Na]+; found 716.1835.:2). [α21D] = +1.89 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.77 (d, JNH,1 = 8.9 Hz, 1H, NHCO), 5.65 (dd, JNH,1 = 8.8, J1,2 = 2.9 Hz, 1H, 1-H), 5.44 (dd, J3′,4′ = 3.4, J4′,5′ = 1.1 Hz, 1H, 4′-H), 5.21 (t, J1′,2′ = J2′,3′ = 10.0, 1H, 2′-H), 5.22–5.19 (m, 1H, 3-H), 5.04 (dd, J2′,3′ = 10.1, J3′,4′ = 3.4 Hz, 1H, 3′-H), 4.84 (dd, J2,3 = 5.1, J1,2 = 2.9 Hz, 1H, 2-H), 4.76 (br. q, J3,4 = J4,5ax = J4,5eq = 4.1, J2,4 < 1 Hz, 1H, 4-H), 4.51 (d, J1′,2′ = 10.0 Hz, 1H, 1′-H), 4.33 (dd, J6a′,6b′ = 11.6, J5′,6a′ = 7.0 Hz, 1H, 6a′-H), 4.07 (dd, J6a′,6b′ = 11.6, J5′,6b′ = 5.3 Hz, 1H, 6b′-H), 3.98 (dd, J5ax,5eq = 12.9, J4,5ax = 3.1 Hz, 1H, 5ax-H), 3.93 (ddd, J5′,6a′ = 6.9, J5′,6b′ = 5.4 Hz, J4′,5′ = 1.2 Hz, 1H, 5′-H), 3.89 (dd, J5ax,5eq = 13.0, J4,5eq = 4.2 Hz, 1H, 5eq-H), 3.04 (dt, Jgem = 14.2, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.5 Hz, 1H, CH2S), 2.91 (dt, Jgem = 14.0, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.6 Hz, 1H, CH2S), 2.65–2.55 (m, 2H, CH2CONH), 2.16, 2.15, 2.11, 2.08, 2.07, 2.05, 1.98 (7s, 21H, COCH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 171.4 (NHCO), 170.8, 170.3, 170.1, 169.8, 169.7, 169.7, 169.1 (COCH3), 85.0 (C-1′), 75.2 (C-5′), 74.4 (C-1), 71.8 (C-3′), 68.4 (C-2), 67.6 (C-4′), 67.6 (C-3), 67.0 (C-2′), 66.9 (C-4), 63.9 (C-5), 62.0 (C-6′), 37.4 (CH2CONH), 26.4 (CH2S), 21.0, 20.9, 20.9 (×2), 20.9, 20.8, 20.7 (COCH3) ppm. HRMS (ESI): calcd for C28H39NNaO17S 716.1831 [M + Na]+; found 716.1850.:1:5, 1.12 mL) was stirred at room temperature. The solid gradually dissolved, and TLC (hexane/EtOAc, 1:1 or 1:1.5) indicated the complete consumption of the starting material after 3 h. The solution was concentrated, and the residue was dissolved in water (1 mL) and eluted through a column packed with Dowex MR-3C mixed bed ion-exchange resin. The eluate was concentrated and additionally purified by silica gel column chromatography using CH2Cl2/CH3OH (8:1 → 4:1) for 17a-cβ and 18a-cβ; and CH3CN/H2O (9:1 → 3:1) for 19a-cβ. Evaporation of the solvent afforded the free N-glycosylamides, which showed a single spot by TLC. The respective Rf values are given in each individual case.:1). [α18D] = +7.2 (c = 1.0, CH3OH). 1H NMR (400 MHz, D2O) δ = 7.44–7.33 (m, 5H, PhCH2CO), 4.97 (br. d, J1,2 = 9.2 Hz, 1H, 1-H), 3.87 (br. dd, J6a,6b = 12.4, J5,6a = 1.5 Hz, 1H, 6a-H), 3.73–3.69 (m, 3H, NHCOCH2 and 6b-H), 3.55 (t, J2,3 = J3,4 = 9.0 Hz, 1H, 3-H), 3.53–3.49 (m, 1H, 5-H), 3.42 (dd, J4,5 = 9.7, J3,4 = 9.1 Hz, 1H, 4-H), 3.41 (t, J1,2 = J2,3 = 9.2 Hz, 1H, 2-H) ppm. 13C NMR (100 MHz, D2O) δ = 175.9 (CONH), 134.5, 129.3, 128.9, 127.4 (PhCH2CO), 79.4 (C-1), 77.6 (C-5), 76.5 (C-3), 71.8 (C-2), 69.3 (C-4), 60.6 (C-6), 42.2 (NHCOCH2Ph) ppm. HRMS (ESI): calcd for C14H19NNaO6 320.1105 [M + Na]+; found 320.1083.:1). [α19D] = +22.5 (c = 1.0, CH3OH). 1H NMR (400 MHz, D2O) δ = 7.43–7.33 (m, 5H, PhCH2CO), 4.91 (d, J1,2 = 8.7 Hz, 1H, 1-H), 3.96 (br. d, J3,4 = 3.2 Hz, 1H, 4-H), 3.77–3.74 (m, 1H, 5-H), 3.73–3.68 (m, 5H, 3-H, NHCOCH2, 6b-H, 6a-H), 3.66 (t, J1,2 = J2,3 = 8.7 Hz, 1H, 2-H) ppm. 13C NMR (100 MHz, D2O) δ = 176.0 (CONH), 134.5, 129.3, 128.9, 127.4 (PhCH2CO), 79.9 (C-1), 76.7 (C-5), 73.4 (C-3), 69.3 (C-2), 68.7 (C-4), 60.9 (C-6), 42.2 (NHCOCH2Ph) ppm. HRMS (ESI): calcd for C14H19NNaO6 320.1105 [M + Na]+; found 320.1109.:1). [α19D] = +22.0 (c = 1.0, CH3OH). 1H NMR (400 MHz, D2O) δ = 7.39–7.27 (m, 5H, PhCH2CO), 4.85 (d, J1,2 = 9.0 Hz, 1H, 1-H), 3.87 (dd, J5ax,5eq = 11.5, J4,5eq = 5.4 Hz, 1H, 5eq-H), 3.63 (br. s, 2H, NHCOCH2), 3.57 (ddd, J4,5b = 10.3, J3,4 = 9.3, J4,5a = 5.4 Hz, 1H, 4-H), 3.44 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-H), 3.36 (t, J1,2 = J2,3 = 9.0 Hz, 1H, 2-H), 3.32 (t, J4,5ax = J5ax,5eq = 11.0 Hz, 1H, 5ax-H) ppm. 13C NMR (100 MHz, D2O) δ = 175.9 (CONH), 134.3, 129.2, 128.9, 127.3 (PhCH2CO), 80.0 (C-1), 76.5 (C-3), 71.6 (C-2), 68.9 (C-4), 66.7 (C-5), 42.1 (NHCOCH2Ph) ppm. HRMS (ESI): calcd for C13H17NNaO5 290.0999 [M + Na]+; found 290.0994.:1). [α19D] = +8.8 (c = 1.0, CH3OH). 1H NMR (400 MHz, D2O) δ = 8.00–7.85 (m, 2H, NpthCH2CO), 7.92 (br. d, Jortho = 7.9 Hz, 1H, NpthCH2CO), 7.61 (td, Jortho = Jortho = 6.8, Jmeta = 1.7 Hz, 1H, NpthCH2CO), 7.58 (td, Jortho = Jortho = 6.8, Jmeta = 1.6 Hz, 1H, NpthCH2CO), 7.52 (dd, Jortho = 7.9, Jortho = 7.1 Hz, 1H, NpthCH2CO), 7.48 (dd, Jortho = 6.9, Jmeta = 1.1 Hz, 1H, NpthCH2CO), 4.97 (d, J1,2 = 9.2 Hz, 1H, 1-H), 4.20 (d, Jgem = 16.5 Hz, 1H, NHCOCH2), 4.15 (d, Jgem = 16.5 Hz, 1H, NHCOCH2), 3.84 (dd, J6a,6b = 12.3, J5,6a = 2.1 Hz, 1H, 6a-H), 3.70 (dd, J6a,6b = 12.4, J5,6b = 5.2 Hz, 1H, 6b-H), 3.52 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-H), 3.48 (ddd, J4,5 = 9.7, J5,6b = 5.1, J5,6a = 2.1 Hz, 1H, 5-H), 3.40 (dd, J3,4 = 9.6, J4,5 = 9.1 Hz, 1H, 4-H), 3.39 (t, J1,2 = J2,3 = 9.2 Hz, 1H, 2-H) ppm. 13C NMR (100 MHz, D2O) δ = 175.8 (CONH), 133.5, 131.7, 130.5, 128.7, 128.6, 128.2, 126.7, 126.2, 125.8, 123.6 (NpthCH2CO), 79.4 (C-1), 77.5 (C-5), 76.5 (C-3), 71.8 (C-2), 69.2 (C-4), 60.5 (C-6), 39.8 (NHCOCH2) ppm. HRMS (ESI): calcd for C18H21NNaO6 370.1261 [M + Na]+; found 370.1245.:1). [α19D] = +14.1 (c = 0.6, CH3OH). 1H NMR (400 MHz, D2O) δ = 8.04–8.00 (m, 2H, NpthCH2CO), 7.96 (dd, Jortho = 7.9, Jmeta = 0.8 Hz, 1H, NpthCH2CO), 7.65 (td, Jortho = Jortho = 6.7, Jmeta = 1.7 Hz, 1H, NpthCH2CO), 7.62 (td, Jortho = Jortho = 6.8, Jmeta = 1.6 Hz, 1H, NpthCH2CO), 7.57 (dd, Jortho = 7.9, Jortho = 7.1 Hz, 1H, NpthCH2CO), 7.53 (dd, Jortho = 7.0, Jmeta = 1.5 Hz, 1H, NpthCH2CO), 4.96 (d, J1,2 = 8.6 Hz, 1H, 1-H), 4.25 (d, Jgem = 16.5 Hz, 1H, NHCOCH2), 4.19 (d, Jgem = 16.5 Hz, 1H, NHCOCH2), 3.99 (br. d, J3,4 = 3.1 Hz, 1H, 4-H), 3.79–3.73 (m, 3H, 6b-H, 6a-H, and 5-H), 3.72 (dd, J2,3 = 10.1, J3,4 = 3.0 Hz, 1H, 3-H), 3.67 (dd, J2,3 = 9.6, J1,2 = 8.7 Hz, 1H, 2-H) ppm. 13C NMR (100 MHz, D2O) δ = 178.4 (CONH), 136.1, 134.3, 133.1, 131.3, 131.2, 130.7, 129.3, 128.8, 128.4, 126.1 (NpthCH2CO), 82.4 (C-1), 79.3 (C-5), 75.9 (C-3), 71.9 (C-2), 71.2 (C-4), 63.5 (C-6), 42.3 (NHCOCH2) ppm. HRMS (ESI): calcd for C18H21NNaO6 370.1261 [M + Na]+; found 370.1241.:1). [α25D] = +11.64 (c = 1.0, CH3OH). 1H NMR (400 MHz, D2O) δ = 7.96–7.89 (m, 2H, NpthCH2CO), 7.88 (dd, Jortho = 8.0, Jmeta = 1.1 Hz, 1H, NpthCH2CO), 7.58–7.52 (m, 2H, NpthCH2CO), 7.48 (dd, Jortho = 8.1, Jortho = 7.0 Hz, 1H, NpthCH2CO), 7.43 (dd, Jortho = 7.0, Jmeta = 1.1 Hz, 1H, NpthCH2CO), 4.86 (d, J1,2 = 8.9 Hz, 1H, 1-H), 4.15 (d, Jgem = 16.6 Hz, 1H, NHCOCH2), 4.10 (d, Jgem = 16.5 Hz, 1H, NHCOCH2), 3.85 (dd, J5ax,5eq = 11.5, J4,5eq = 5.3 Hz, 1H, 5eq-H), 3.56 (ddd, J4,5ax = 10.6, J3,4 = 8.9, J4,5eq = 5.3 Hz, 1H, 4-H), 3.42 (t, J2,3 = J3,4 = 9.0 Hz, 1H, 3-H), 3.34 (t, J1,2 = J2,3 = 9.0 Hz, 1H, 2-H), 3.29 (dd, J5ax,5eq = 11.3, J4,5ax = 10.8 Hz, 1H, 5ax-H) ppm. 13C NMR (100 MHz, D2O) δ = 175.8 (CONH), 133.4, 131.6, 130.4, 128.7, 128.6, 128.2, 126.7, 126.2, 125.8, 123.4 (NpthCH2CO), 80.0 (C-1), 76.5 (C-3), 71.5 (C-2), 68.9 (C-4), 66.7 (C-5), 39.7 (NHCOCH2) ppm. HRMS (ESI): calcd for C17H19NNaO5 340.1155 [M + Na]+; found 340.1143.:4:4). [α25D] = +6.84 (c = 0.6, CH3OH). 1H NMR (400 MHz, D2O) δ = 4.95 (d, J1,2 = 9.1 Hz, 1H, 1-H), 4.48 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 3.94 (dd, J3′,4′ = 3.4, J4′,5′ = 0.8 Hz, 1H, 4′-H), 3.85 (dd, J6a,6b = 12.4, J5,6a = 2.2 Hz, 1H, 6a-H), 3.77–3.66 (m, 4H, 6a′-H, 6b′-H, 5′-H, 6b-H), 3.62 (dd, J2′,3′ = 9.5, J3′,4′ = 3.4 Hz, 1H, 3′-H), 3.52 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 3.51 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-H), 3.49 (ddd, J4,5 = 9.7, J5,6b = 5.3, J5,6a = 2.2 Hz, 1H, 5-H), 3.38 (dd, J4,5 = 9.7, J3,4 = 9.1 Hz, 1H, 4-H), 3.36 (dd, J1,2 = J2,3 = 9.2 Hz, 1H, 2-H), 3.02 (dt, Jgem = 13.8, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.2 Hz, 1H, CH2S), 2.96 (dt, Jgem = 13.7, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.8 Hz, 1H, CH2S), 2.75–2.65 (m, 2H, CH2CONH) ppm. 13C NMR (100 MHz, D2O) δ = 175.7 (CONH), 86.1 (C-1′), 79.2 (C-1), 78.9 (C-5′), 77.5 (C-5), 76.4 (C-3), 73.8 (C-3′), 71.7 (C-2), 69.5 (C-2′), 69.2 (C-4), 68.8 (C-4′), 61.1 (C-6′), 60.5 (C-6), 36.4 (CH2CONH), 25.7 (CH2S) ppm. HRMS (ESI): calcd for C15H27NNaO11S 452.1197 [M + Na]+; found 452.1193.:4:4). [α22D] = +4.87 (c = 0.8, CH3OH). 1H NMR (400 MHz, D2O) δ = 4.89 (d, J1,2 = 9.0 Hz, 1H, 1-H), 4.48 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 3.94–3.92 (m, 2H, 4-H, 4′-H), 3.74 (dt, J5,6a = 5.0, J5,6b = J4,5 = 1.0 Hz, 1H, 5-H), 3.72–3.67 (m, 5H, 6a-H, 6b-H, 5′-H, 6a′-H, 6b′-H), 3.67 (dd, J2,3 = 9.1, J3,4 = 3.3 Hz, 1H, 3-H), 3.61 (dd, J2′,3′ = 9.5, J3′,4′ = 3.4 Hz, 1H, 3′-H), 3.59 (t, J1,2 = J2,3 = 9.1 Hz, 1H, 2-H), 3.51 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 3.03 (dt, Jgem = 14.2, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.2 Hz, 1H, CH2S), 2.95 (dt, Jgem = 13.6, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.8 Hz, 1H, CH2S), 2.73–2.65 (m, 2H, CH2CONH) ppm. 13C NMR (100 MHz, D2O) δ = 175.7 (CONH), 86.0 (C-1′), 79.7 (C-1), 78.9 (C-5′), 76.7 (C-5), 73.8 (C-3′), 73.3 (C-3), 69.5 (C-2′), 69.3 (C-2), 68.8, 68.6 (C-4 and C-4′), 61.1 (C-6′), 60.9 (C-6), 36.4 (CH2CONH), 25.6 (CH2S) ppm. HRMS (ESI): calcd for C15H27NNaO11S 452.1197 [M + Na]+; found 452.1194.:4:4). [α22D] = +1.09 (c = 1.0, H2O). 1H NMR (400 MHz, D2O) δ = 4.90 (d, J1,2 = 9.1 Hz, 1H, 1-H), 4.51 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 3.97 (dd, J3′,4′ = 3.4, J4′,5′ = 0.8 Hz, 1H, 4′-H), 3.93 (dd, J5ax,5eq = 11.6, J4,5eq = 5.7 Hz, 1H, 5eq-H), 3.77–3.71 (m, 3H, 6a′-H, 6b′-H, 5′-H), 3.65 (dd, J2′,3′ = 9.6, J3′,4′ = 3.4 Hz, 1H, 3′-H), 3.61 (ddd, J4,5ax = 10.3, J3,4 = 9.2, J4,5eq = 5.3 Hz, 1H, 4-H), 3.55 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 3.49 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-H), 3.38 (t, J1,2 = J2,3 = 9.1 Hz, 1H, 2-H), 3.37 (dd, J5ax,5eq = 11.6, J4,5ax = 10.4 Hz, 1H, 5ax-H), 3.05 (dt, Jgem = 13.9, JCH2S,CH2CONH = JCH2S,CH2CONH = 7.3 Hz, 1H, CH2S), 2.99 (dt, Jgem = 13.9, JCH2S,CH2CONH = JCH2S,CH2CONH = 6.6 Hz, 1H, CH2S), 2.77–2.65 (m, 2H, CH2CONH) ppm. 13C NMR (100 MHz, D2O) δ = 175.8 (CONH), 86.2 (C-1′), 80.0 (C-1), 79.0 (C-5′), 76.6 (C-3), 73.9 (C-3′), 71.7 (C-2), 69.6 (C-2′), 69.0 (C-4), 68.9 (C-4′), 66.8 (C-5), 61.2 (C-6′), 36.4 (CH2CONH), 25.9 (CH2S) ppm. HRMS (ESI): calcd for C14H25NNaO10S 422.1091 [M + Na]+; found 422.1063.Inhibition of β-galactosidase from E. coli enzymatic assays
Compounds 17b, 18b, 19a-cβ were evaluated as inhibitors against E. coli β-galactosidase (grade VIII, Sigma, EC 3.2.1.23, 1019 U mg−1 protein). One unit of the enzyme hydrolyzes 1 μmol of o-nitrophenyl β-D-galactopyranoside per minute. Thus, the enzyme (0.08 U) was incubated in a solution of o-nitrophenyl β-D-galactopyranoside (concentration range: 0.4–4.5 mM) in sodium phosphate buffer (100 mM, pH 7.3, MgCl2 1.2 mM, 2-mercaptoethanol 130 mM) in the absence or presence of N-glycosyl amides. The final volume was adjusted to 0.5 mL, and after incubation at 37 °C for 10 minutes, the enzyme was denatured upon the addition of 0.2 M sodium borate buffer (4.0 mL, pH 10.0). The concentration of the o-nitrophenol released was measured by absorption spectroscopy at 410 nm, in the visible region.Conclusions
β-N-glycosyl amides were obtained through a direct sequence from per-O-acetyl pyranoses, via the intermediate glycosyl azides. Hydrogenation of these compounds to the corresponding glycosyl amine derivatives and subsequent acylation, with carboxylic acids in the presence of DCC, led to the glycosyl amides. Those having the gluco or galacto configuration were diastereoselectively obtained as the desired β-anomer, while those with xylo configuration gave also α-anomers as minor products.Spectroscopic analysis of the xylo derivatives revealed that the amides having α anomeric configuration exclusively populate the 1C4 conformation; in contrast to the β-anomers, which adopt the 4C1 chair. This result was supported by conformational search calculations. Additionally, a similar behavior was observed for the xylopyranosyl amine, since the α-anomer predominantly adopted the 1C4 chair, while the β-anomer exclusively populated the 4C1 chair. These findings are consistent with literature reports42 describing that anomeric nitrogen substituents preferentially adopt conformations where they are found in equatorial orientation, even at the expense of positioning axially the rest of the substituents. This phenomenon was primarily attributed to the high stabilization generated by the exo-anomeric effect and steric factors.
Deprotection of the per-O-acetyl-β-N-glycosyl amides with gluco, galacto, or xylo configurations provided the compounds with free OH groups in very good yields. Inhibition studies showed that amides containing a D-galactopyranose residue exhibited a moderate inhibitory effect on E. coli β-galactosidase. However, it should be noted that this is the first report of N-glycosyl amide derivatives exhibiting this activity. These findings are significant as they serve as a starting point for the design and development of future N-glycosyl amides with improved inhibition against this enzyme.
Author contributions
Conceptualization and methodology were executed by J. P. C. Synthesis, NMR analysis, and calculations were performed by M. G. T. and J. P. C. The inhibition/kinetic studies were carried out by V. E. M. Funding acquisition, project administration, resources provision, supervision, and validation were made by O. V. and J. P. C. All authors contributed to the writing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to the Secretaría de Ciencia y Tecnología (SECyT), Universidad Nacional de Córdoba (UNC), Universidad de Buenos Aires (UBA), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Fondo para la investigación Científica y Tecnológica Argentina (FONCyT) for financial support. M. G. T. gratefully acknowledges the receipt of a fellowship from CONICET. V. E. M., O. V., and J. P. C. are research members from CONICET.Notes and references
- A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS PubMed.
- A. Mazzoleni, J.-M. Mallet, P. Rovero and A. M. Papini, Arch. Biochem. Biophys., 2019, 663, 44–53 CrossRef CAS PubMed.
- P. Compain, Molecules, 2018, 23, 1658 CrossRef PubMed.
- J. Rojo, A. Sousa-Herves and A. Mascaraque, in Comprehensive Medicinal Chemistry III, ed. S. Chackalamannil, D. Rotella and S. E. Ward, Elsevier, Oxford, 2017, pp. 577–610 Search PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS PubMed.
- D. M. Moncada, S. J. Kammanadiminti and K. Chadee, Trends Parasitol., 2003, 19, 305–311 CrossRef CAS PubMed.
- J. W. Dennis, M. Granovsky and C. E. Warren, Biochim. Biophys. Acta, 1999, 1473, 21–34 CrossRef CAS PubMed.
- Y. J. Kim and A. Varki, Glycoconj. J., 1997, 14, 569–576 CrossRef CAS PubMed.
- E. Weerapana and B. Imperiali, Glycobiology, 2006, 16, 91R–101R CrossRef CAS PubMed.
- L. Somsak, K. Czifrak, M. Toth, E. Bokor, D. E. Chrysina, K. M. Alexacou, M. J. Hayes, C. Tiraidis, E. Lazoura, D. D. Leonidas, E. S. Zographos and G. N. Oikonomakos, Curr. Med. Chem., 2008, 15, 2933–2983 CrossRef CAS PubMed.
- K. Czifrák, Z. Hadady, T. Docsa, P. Gergely, J. Schmidt, L. Wessjohann and L. Somsák, Carbohydr. Res., 2006, 341, 947–956 CrossRef PubMed.
- T. Hadjiloi, C. Tiraidis, E. D. Chrysina, D. D. Leonidas, N. G. Oikonomakos, P. Tsipos and T. Gimisis, Bioorg. Med. Chem., 2006, 14, 3872–3882 CrossRef CAS.
- Z. Györgydeák, Z. Hadady, N. Felföldi, A. Krakomperger, V. Nagy, M. Tóth, A. Brunyánszki, T. Docsa, P. Gergely and L. Somsák, Bioorg. Med. Chem., 2004, 12, 4861–4870 CrossRef PubMed.
- K. A. Watson, E. P. Mitchell, L. N. Johnson, G. Cruciani, J. C. Son, C. J. F. Bichard, G. W. J. Fleet, N. G. Oikonomakos, M. Kontou and S. E. Zographos, Acta Crystallogr. D, 1995, 51, 458–472 CrossRef CAS PubMed.
- M. Bols, Ó. López and F. Ortega-Caballero, in Comprehensive Glycoscience, ed. H. Kamerling, Elsevier, Oxford, 2007, pp. 815–884 Search PubMed.
- M. E. Cano, O. Varela, M. I. García-Moreno, J. M. García Fernández, J. Kovensky and M. L. Uhrig, Carbohydr. Res., 2017, 443–444, 58–67 CrossRef CAS.
- J. Ohlsson, A. Larsson, S. Haataja, J. Alajääski, P. Stenlund, J. S. Pinkner, S. J. Hultgren, J. Finne, J. Kihlberg and U. J. Nilsson, Org. Biomol. Chem., 2005, 3, 886–900 RSC.
- S. Peluso, M. d. L. Ufret, M. K. O'Reilly and B. Imperiali, Chem. Biol., 2002, 9, 1323–1328 CrossRef CAS.
- D. Chennamadhavuni and A. R. Howell, Tetrahedron Lett., 2015, 56, 3583–3586 CrossRef CAS PubMed.
- M. A. Rapp, O. R. Baudendistel, U. E. Steiner and V. Wittmann, Chem. Sci., 2021, 12, 14901–14906 RSC.
- V. Maunier, P. Boullanger and D. Lafont, J. Carbohydr. Chem., 1997, 16, 231–235 CrossRef CAS.
- K. Villadsen, M. C. Martos-Maldonado, K. J. Jensen and M. B. Thygesen, ChemBioChem, 2017, 18, 574–612 CrossRef CAS PubMed.
- P. Norris, Curr. Top. Med. Chem., 2008, 8, 101–113 CrossRef CAS PubMed.
- T. Inazu and K. Kobayashi, Synlett, 1993, 1993, 869–870 CrossRef.
- P. Boullanger, V. Maunier and D. Lafont, Carbohydr. Res., 2000, 324, 97–106 CrossRef CAS PubMed.
- E. Ay, Nucleosides, Nucleotides Nucleic Acids, 2023, 42, 191–205 CrossRef CAS PubMed.
- G. Gonzalez, J. Hodoň, A. Kazakova, C. W. D'Acunto, P. Kaňovský, M. Urban and M. Strnad, Eur. J. Med. Chem., 2021, 213, 113168 CrossRef CAS PubMed.
- A. K. Agrahari, M. K. Jaiswal, M. S. Yadav and V. K. Tiwari, Carbohydr. Res., 2021, 508, 108403 CrossRef CAS PubMed.
- F. M. Ibatullin and K. A. Shabalin, Synth. Commun., 2000, 30, 2819–2823 CrossRef CAS.
- A. J. Cagnoni, M. L. Uhrig and O. Varela, Bioorg. Med. Chem., 2009, 17, 6203–6212 CrossRef CAS PubMed.
- A. J. Steiner, G. Schitter, A. E. Stütz, T. M. Wrodnigg, C. A. Tarling, S. G. Withers, K. Fantur, D. Mahuran, E. Paschke and M. Tropak, Bioorg. Med. Chem., 2008, 16, 10216–10220 CrossRef CAS PubMed.
- A. Thomas, A. Shukla, S. Sivakumar and S. Verma, Chem. Commun., 2014, 50, 15752–15755 RSC.
- M. C. Sheikh, S. Takagi, T. Yoshimura and H. Morita, Tetrahedron, 2010, 66, 7272–7278 CrossRef CAS.
- M. Mart, I. Karakaya and J. Jurczak, ChemistrySelect, 2022, 7, e202202436 CrossRef CAS.
- A. Jordan, K. D. Whymark, J. Sydenham and H. F. Sneddon, Green Chem., 2021, 23, 6405–6413 RSC.
- M. H. Bilal, R. Alaneed, J. Steiner, K. Mäder, M. Pietzsch and J. Kressler, in Meth. Enzymol., ed. N. Bruns and K. Loos, Academic Press, 2019, vol. 627, pp. 57–97 Search PubMed.
- G. Zong and W. Q. Shi, in Strateg. Tactics Org. Synth., ed. M. Harmata, Academic Press, 2017, vol. 13, pp. 81–110 Search PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- S. Traboni, F. Esposito, M. Ziaco, E. Bedini and A. Iadonisi, Tetrahedron, 2023, 133, 133291 CrossRef CAS.
- A. K. Ghosh and D. Shahabi, Tetrahedron Lett., 2021, 63, 152719 CrossRef CAS PubMed.
- M. Monsigny, C. Quétard, S. Bourgerie, D. Delay, C. Pichon, P. Midoux, R. Mayer and A. C. Roche, Biochimie, 1998, 80, 99–108 CrossRef CAS PubMed.
- R. J. Batchelor, D. F. Green, B. D. Johnston, B. O. Patrick and B. M. Pinto, Carbohydr. Res., 2001, 330, 421–426 CrossRef CAS PubMed.
- S. M. Piazza, M. R. Reynolds, J. Chiaramonte, P. Xu, F. A. Chapa-Villarreal and J. F. Trant, New J. Chem., 2021, 45, 19224–19227 RSC.
- X. Meng, X. Lian, X. Li, Q. Ya, T. Li, Y. Zhang, Y. Yang and Y. Zhang, Carbohydr. Res., 2020, 493, 108034 CrossRef CAS PubMed.
- E. W. Mora Flores, M. L. Uhrig and A. Postigo, Org. Biomol. Chem., 2020, 18, 8724–8734 RSC.
- S. Singh, S. Singh, R. K. Sharma, A. Kaul, R. Mathur, S. Tomar, R. Varshney and A. K. Mishra, New J. Chem., 2020, 44, 3062–3071 RSC.
- N. D. Thanh, D. S. Hai, V. T. Ngoc Bich, P. T. Thu Hien, N. T. Ky Duyen, N. T. Mai, T. T. Dung, V. N. Toan, H. T. Kim Van, L. H. Dang, D. N. Toan and T. T. Thanh Van, Eur. J. Med. Chem., 2019, 167, 454–471 CrossRef CAS PubMed.
- L. Goswami, S. Paul, T. K. Kotammagari and A. K. Bhattacharya, New J. Chem., 2019, 43, 4017–4021 RSC.
- J. Rajput, S. Hotha and M. Vangala, Beilstein J. Org. Chem., 2018, 14, 682–687 CrossRef CAS PubMed.
- G. Wang, D. Wang, J. Bietsch, A. Chen and P. Sharma, J. Org. Chem., 2020, 85, 16136–16156 CrossRef CAS PubMed.
- H. Song, S. J. Allison, V. Brabec, H. E. Bridgewater, J. Kasparkova, H. Kostrhunova, V. Novohradsky, R. M. Phillips, J. Pracharova, N. J. Rogers, S. L. Shepherd and P. Scott, Angew. Chem., Int. Ed., 2020, 59, 14677–14685 CrossRef CAS PubMed.
- A. Brinkø, C. Risinger, A. Lambert, O. Blixt, C. Grandjean and H. H. Jensen, Org. Lett., 2019, 21, 7544–7548 CrossRef PubMed.
- Z. Györgydeák and L. Szilágyi, Liebigs Ann. Chem., 1986, 1986, 1393–1397 CrossRef.
- C. Badía, F. Souard and C. Vicent, J. Org. Chem., 2012, 77, 10870–10881 CrossRef PubMed.
- A. Bertho, Adv. Cycloaddit., 1949, 562, 229–239 CAS.
- P. R. Ashton, S. E. Boyd, C. L. Brown, S. A. Nepogodiev, E. W. Meijer, H. W. I. Peerlings, H. W. I. Peerlings and J. F. Stoddart, Chem.–Eur. J., 1997, 3, 974–984 CrossRef CAS.
- M. Elofsson, B. Walse and J. Kihlberg, Tetrahedron Lett., 1991, 32, 7613–7616 CrossRef CAS.
- L. Moroder, Biol. Chem. Hoppe-Seyler, 1988, 369, 381–386 CrossRef CAS PubMed.
- L. Silva, R. F. Affeldt and D. S. Lüdtke, J. Org. Chem., 2016, 81, 5464–5473 CrossRef CAS PubMed.
- F. Damkaci and P. DeShong, J. Am. Chem. Soc., 2003, 125, 4408–4409 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07763b |
| This journal is © The Royal Society of Chemistry 2024 |