DOI:
10.1039/D3RA07816G
(Paper)
RSC Adv., 2024,
14, 1445-1458
Toxic gas sensing performance of arsenene functionalized by single atoms (Ag, Au): a DFT study†
Received
15th November 2023
, Accepted 21st December 2023
First published on 3rd January 2024
Abstract
The detection and removal of toxic gases from the air are imminent tasks owing to their hazards to the environment and human health. Based on DFT calculations with VdW correction, adsorption configurations, adsorption energies, and electronic properties were compared for the adsorption of toxic gas molecules (CO, NO, NO2, SO2, NH3 and H2S) on pure arsenene (p-arsenene) and Ag/Au-doped arsenene (Ag/Au-arsenene). Our calculations show that all molecules considered to chemisorb on Ag/Au-arsenene and the substitution of noble metal, particularly Ag, could remarkably enhance the interactions and charge transfer between the gas molecules and Ag/Au-arsenene. Thus, Ag/Au-arsenene is expected to show higher sensitivity in detecting CO, NO, NO2, SO2, NH3 and H2S molecules than p-arsenene. Furthermore, the changes in the vibrational frequencies of gas molecules and the work functions of Ag/Au-arsenene substrates upon adsorption are shown to be closely related to the adsorption energies and charge transfer between the molecules and Ag/Au-arsenene, which is dependent on the molecules. Therefore, Ag/Au-arsenene-based gas sensors are expected to show good selectivity of molecules. The analysis of theoretical recovery time suggested that Ag-arsenene shows high reusability while detecting H2S, CO, and NO, whereas Au-arsenene has high selectivity to sensing NO at room temperature. With the increase in work temperature and decrease in recovery times, Ag/Au-arsenene could be used to detect NH3 and NO2 from factory emission and automobile exhaust with quite good reusability. The above results indicated that Ag/Au-arsenene shows good performance in toxic gas sensing with high sensitivity, selectivity, and reusability at different temperatures.
1. Introduction
With the economic development and improvement of living standards, the emission of poisonous gases during industrial production processes, including smelting, chemical synthesis, and fossil fuel processing, worsens air pollution. Most toxic and harmful gases are corrosive and can cause acute poisoning and irreversible damage when they enter the human body through the respiratory tract.1–3 Major toxic gases in the air mainly include sulfides (such as SO2 and H2S), ammonia (NH3), CO, and nitrogen oxides (NOx). For instance, H2S as a nerve poison is harmful when inhaled and generally causes headaches and even suffocation at high concentrations (1000 ppm), leading to unconsciousness in humans.4 The concentration of NH3 in the air should be below 15 ppm; otherwise, exposure to high amounts of NH3 can cause lung damage, even blindness or death.5 When the concentration of CO gas in the air reaches a certain level, CO poisoning is harmful to all forms of life as its inhalation could cause oxygen deprivation in the tissues and organs of bodies.6 Besides, NO2 and SO2 are released into the atmosphere from fossil fuel combustion and vehicle exhaust emissions and fall back to the earth as acid rain, which usually results in heavy economic losses for agriculture, forestry and aquaculture industries.7,8 Above all, these five toxic gases discharged into the air seriously threaten human life, health, and the earth's ecological environment. Therefore, detecting and removing the aforementioned harmful gases from the environment is an important and critical issue in maintaining public health and the agricultural industry because they are primarily colorless, odorless, flammable, corrosive and heavier than air.
Benefitting from high carrier mobility, rich surface chemistry, and low electrical noises, especially a large surface-to-volume ratio, two-dimensional (2D) nanomaterials have been widely studied in the development of next-generation gas sensors.9–17 Similar to graphene, silicone,9 germanene,10 and transition-metal dichalcogenides (TMDs), such as WS2 or WSe2,11,12 SiAs,13 stanine,14 arsenene15,16 and bismuthene17 have recently received great attention owing to their fascinating structures, which could provide more active sites and subsequently make these materials effective for gas detection, especially in harsh environments because of their advantages of desirable sensitivity, reversibility and rapid recovery time. Recently, Jiang et al. reported that penta-BeP2 monolayer strongly interacts with toxic gas molecules (including CO, NH3, NO, and NO2) but weakly interacts with ambient gas molecules (including H2, N2, H2O, O2, and CO2), suggesting that penta-BeP2 monolayer is a superior candidate for detecting toxic gases in air.18 Previous theoretical and experimental studies have confirmed that most gas molecules favor the physical adsorbing of 2D materials. For example, Safari and co-workers predicted that pristine blue phosphorene exhibits a weak sensitivity to NH3, CO and NO molecules with low adsorption energies and negligible charge transfer.19 Pan et al. studied the adsorption behavior of common gas molecules on buckled bismuthene, and their results suggested SO2, NO and NO2 chemisorb on buckled bismuthene, whereas CO, O2, H2O and NH3 molecules prefer physisorb on bismuthene.20 The monolayer of arsenene is an indirect-bandgap semiconductor of 1.62 eV.21 The high carrier mobility, on/off ratio of 104 at room temperature, and the response of arsenene to photons with wavelengths less than 620 nm make it promising for applications in electronics and optoelectronics.22 Previous studies15,16,23 also confirmed that the electronic and transport properties of arsenene are remarkably affected by the adsorption of gas molecules, such as CO, CO2, NO2, NO, N2, NH3, and SO2, which makes it a worthy candidate for applications in gas sensors.
It is known that defects including vacancy, doping and decorating of impurities atoms are effective strategies to modulate the electronic properties of materials and can provide abundant active sites for gas molecule adsorption and catalysts.5,24–31 For instance, the vacancy-crafted bismuthene shows enhanced sensitivity towards sulfur- and nitrogen-rich gases compared with pure bismuthene, which is explained by the considerable alteration in the electrostatic potential difference between the toxic gas and the bismuthene with or without vacancies.5 Furthermore, MoS2 functionalized by doping of V, Nb, or Ta shows high sensitivity to detect CO, H2O and NH3 molecules, which provide appropriate chemical modification methods to enhance the sensing and catalytic performance of MoS2.24 Recently, Zhang et al. proved that the Pd-decorated WTe2 monolayer shows promising applications as a resistance-type gas sensor for the selective detection of SO2 and SOF2 in SF6 insulation devices.25 The theoretical calculations uncovered that transition-metal (TM) (Rh, Au, Pd, and Ag)-doped GeTe monolayers not only have superior adsorption properties compared with pure GeTe for vented gases emitted from Lithium-Ion Batteries, e.g. CO2, CO, C2H2, and C2H4, but also achieve good selectivity to detect and monitor these gases at various temperatures.26 Subsequently, Wang et al. investigated the sensing properties of transition metal (TM) atoms (Mo, Ni, Pd, Ti and Zr) that adsorbed arsenene into toxic nitrogenous gas molecules, including NH3, NO and NO2. The results show that the adsorbed transition metal (TM) atoms not only significantly enhance the interactions between toxic nitrogenous gas molecules and substrate but also effectively improve the adsorption capacity of arsenene.27 Recently, Tian and co-workers also theoretically predicted that TM (from Ti to Ni) dopants increase the sensing performance of arsenene to H2S gas more effectively than the introduction of point defects; particularly, Ti- and Ni-doped arsenenes are particularly prominent for the detection and capture of H2S.28 Fe-decorated antimonene is highly selective and sensitive to CO, COS, NO, NO2, NH3, and SO2 and can be used as candidate materials in toxic gas sensors.29 Owing to the strong and admirable catalytic properties, the decoration and substitution of noble metals (such as Pd, Pt, Ag and Au) in common catalysts are usually utilized for gas adsorption, monitoring and sensing of systems. Ma et al.30 studied the interactions between CO and NO gases and TM-(Au, Pt, Pd and Ni) doped MoS2 monolayer, and it was demonstrated that the substitution of appropriate atoms could significantly improve the adsorption behavior and capture performance of MoS2. Our recent study reported that embedded Pt in arsenene can remarkably enhance the adsorption of gas molecules of CO, NO, NO2, O2, NH3, H2O, H2 and N2 on Pt-doped arsenene, which can be used as a gas (such as O2, CO, and NH3) sensor, or even as a scavenger for NO and NO2, with higher sensitivity than those of pure arsenene owing to the large charge transfer between the molecules and substrate.31 Above all, the decoration and substitution of Ag or Au provide abundant electrons for 2D materials and display strong and admirable catalytic properties, rendering them advantageous for gas adsorption and sensing applications. Nonetheless, there are few studies about arsenene substituted by Ag and Au atoms as toxic gas sensing materials, which are expected to be an efficient approach for widening the sensing application of arsenene. In this work, we investigated the adsorption behaviors of toxic gases (CO, NO, NO2, NH3, SO2 and H2S) adsorbed on perfect and defective arsenene monolayers by the substitution of As with Ag or Au atom and thus explored the potential applications of arsenene monolayers in gas sensing based on density functional theory (DFT) calculations. The structural stability, adsorption energy, electronic band structure, density of states (DOS), Bader charge analysis, charge density difference (CDD), and vibrational frequencies of molecules adsorbed on Ag- and Au-doped arsenene were also analyzed to investigate the influences of toxic gas adsorption. Both Ag- and Au-doped arsenene exhibit a high adsorption capability of NO2 with the largest adsorption energies among all the molecules considered. In addition, arsenene doped with Ag or Au could distinguish between different toxic gases with appropriate adsorption energies and charge transfer, indicating that Ag- or Au-doped arsenene shows high selectivity to gases. The recovery time analysis showed that the recovery times for CO, NO and H2S on Ag-arsenene and NO on Au-doped arsenene are short at room temperature (about 300 K), showing high reusability of Ag/Au-arsenene-based gas sensor when sensing these molecules. The recovery times for NO2 molecules (the recovery times are extremely long at 300 K) on Ag/Au-arsenene are shortened rapidly with the increase in temperature (up to 500 K), indicating that Ag/Au-arsenene based gas sensors have potential applications in sensing NO2 from factory emission and automobile exhaust with quite good reusability.
2. Computational methods
First-principles calculations were performed based on the density functional theory (DFT) using the Vienna Ab initio Simulation Package (VASP).32,33 We chose the Perdew–Burke–Ernzerhof (PBE) functional34 within the generalized gradient approximation (GGA) to handle the exchange correlation interaction between electrons. The projector augmented wave (PAW) potentials35 were selected to describe the electron–ion interactions. The energy cutoff for the plane wave basis was set to 400 eV. We built 7 × 7 × 1 and 9 × 9 × 1 Monkhorst–Pack grids of K-point grids to sample the Brillouin zones (BZs) for geometry optimization and structural calculations, respectively. We chose a 4 × 4 × 1 supercell to simulate the periodic structure of arsenene monolayers, and a vacuum spacing of 20 Å was added along the direction perpendicular to the substrate surface (z direction) to avoid the interactions between adjacent layers. The DFT-D3 scheme proposed by Grimme36 was used to describe the van der Waals interaction between gas molecules and substrate sheets. Spin polarization calculations have been considered for systems adsorbed by paramagnetic molecules, such as NO and NO2. We considered various initial adsorption configurations with different adsorption sites and molecule orientations of the gas molecules to obtain the most stable configurations for molecules adsorbed on arsenene. All the geometry structures are fully relaxed until the total energy converges to 1.0 × 10−5 eV, and the force on each atom is smaller than 0.01 eV Å−1. Good convergence is achieved with the cutoff energy, number of k points and vacuum height for the various adsorption structures considered (details are shown in Tables S1–S3†). We also enlarge the supercell to 5 × 5 × 1 and 6 × 6 × 1 of arsenene with one arsenic atom substituted by an Ag atom to compare the adsorption properties of the NO molecule. Our calculation results show that the most stable configurations of NO/Ag-arsenene are almost identical to those for 4 × 4 × 1. The variations in the adsorption energies of NO molecules on different substrates are also found to be less at 0.03 eV (as shown in Table S4†). Therefore, the 4 × 4 × 1 supercell we used is suitable as a platform to investigate the adsorption performance of the considered toxic gases.
The doping of noble metal (NM) of Ag and Au atoms in arsenene is investigated by the formation energy, which is defined as follows:
| | |
Ef = ENM-arsenene + EAs − Earsenene − ENM,
| (1) |
where
Ef is the formation energy;
ENM-arsenene and
Earsenene are the total energies of arsenene with and without Ag or Au doping, respectively; and
EAs and
ENM represent the total energy of an isolated As and Ag or Au atom in a big box with a size of 20 Å × 20 Å × 20 Å, respectively. To analyze the adsorption properties (sensing capability) of toxic gas molecules on Ag/Au-doped arsenene, the adsorption energies evaluating the interactions between the gas molecule and arsenene are calculated using the following equation:
| | |
Eads = Egas/surface − Egas − Esurface,
| (2) |
where
Egas/surface and
Esurface are the total energy of pristine arsenene (denoted as p-arsenene) or Ag/Au-arsenene with and without gas adsorption, respectively, and
Egas is the total energy of a gas molecule.
3. Results and discussion
3.1 Structures and electronic properties of pristine and Ag/Au-arsenene
To investigate the adsorption behaviors of toxic molecules on arsenene nanosheets, it is essential to explore the structural and electronic properties of p-arsenene and arsenene with one As atom substituted by Ag/Au (denoted as Ag/Au-arsenene) for comparison. The optimized structures, band structures and density of states for p-arsenene are shown in Fig. S1.† The calculated lattice constants, buckling parameters and As–As bond length upon full relaxation are 3.61 Å, 1.40 Å and 2.51 Å, respectively. P-arsenene displays a semiconductor feature with an indirect band gap of about 1.61 eV, which is in agreement with other previous theoretical studies.37
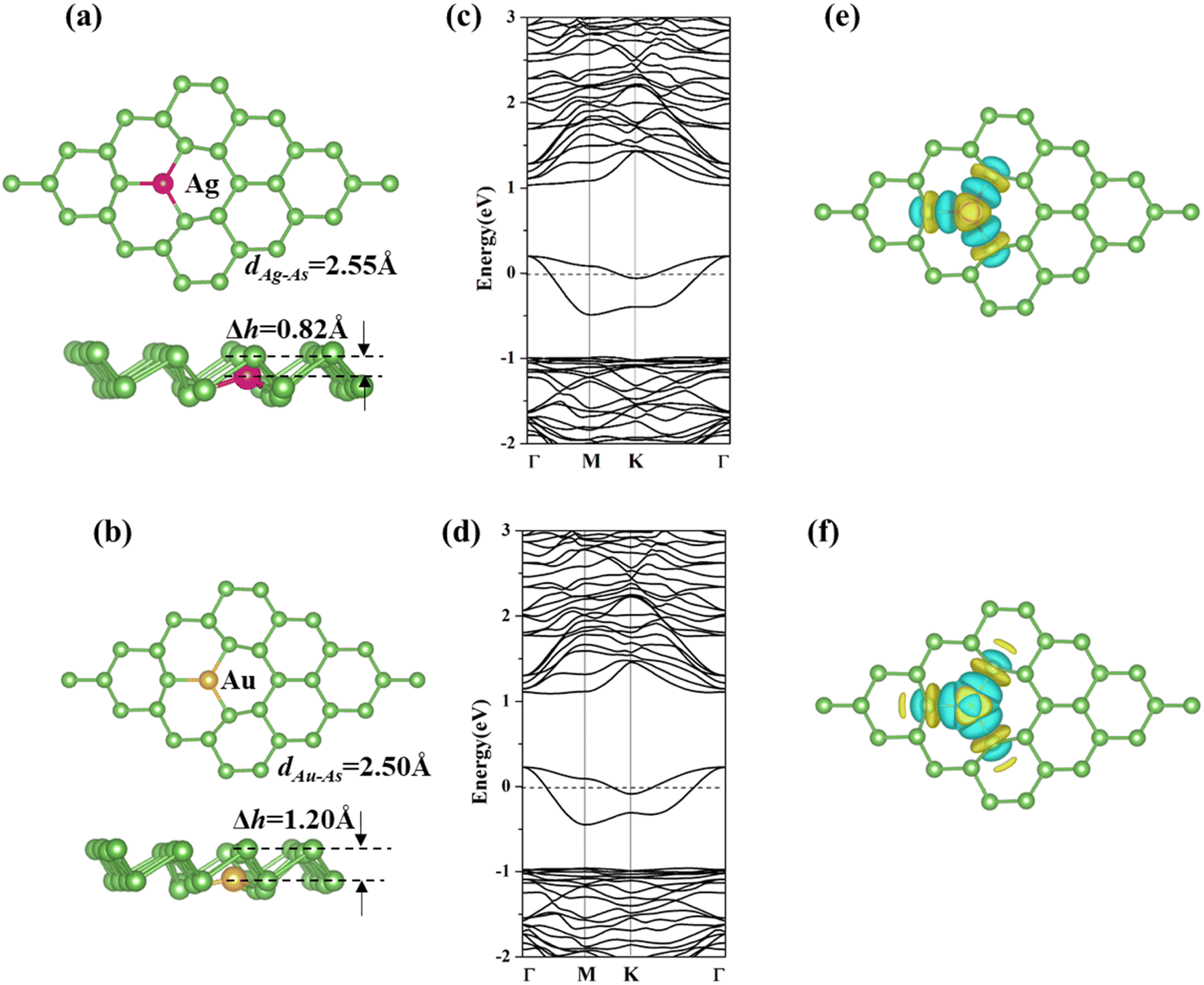
We displaced one arsenic atom with an Ag or Au atom in a (4 × 4 × 1) arsenene supercell to simulate the doping process (as shown in Fig. 1a and b). After optimization, the bond angles of As–X–As (X = Ag and Au) are enlarged from 92.00° for p-arsenene to 110.03° and 116.55° for Ag-arsenene and Au-arsenene, respectively. The dopant atoms (X = Ag and Au) moved downwards by 0.82 and 1.20 Å related to the upper As layer, respectively. We also found that the bond angles of three As–X–As and the bond length of X–As (X = Ag and Au) are equivalent, implying that the doping of Ag/Au atoms does not break the C3v symmetry, which is similar to TM-doped arsenene systems.38 The calculated formation energies (Ef) of Ag- and Au-arsenene are about −3.04 and −4.18 eV, respectively, indicating that the considered Ag/Au-arsenene systems show relatively high stability compared with TM (Cr, Mn, Fe, Co, Ni, and Cu)-doped arsenene, in which Cr-arsenene has the lowest formation energy of −0.45 eV.38
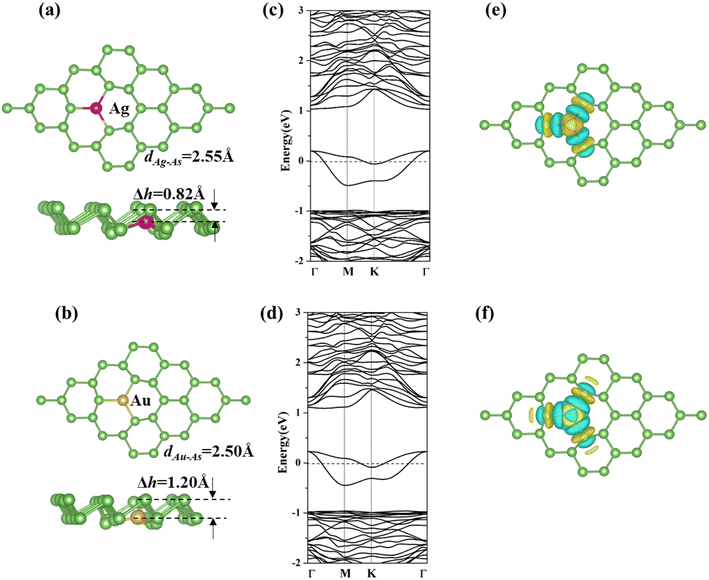 |
| | Fig. 1 (a and b) Top and side views, (c and d) band structures, and (e and f) charge density difference (CDD) for Ag and Au-arsenene systems. The rosy, golden and green balls represent Ag, Au and As atoms, respectively. The Fermi level is set to 0 eV, which is donated by dash lines in (b) and (c). | |
We further investigated the spin-polarized electronic band structures of Ag- and Au-doped arsenene monolayers, as depicted in Fig. 1c and d. The majority and minority states overlap completely, indicating that the considered Ag/Au-doped arsenene displays nonmagnetic states. For both cases considered, 2-fold degenerate impurity states below the Fermi level and another 2-fold degenerate impurity states crossing the Fermi energy appeared in the density of states (DOS), as shown in Fig. S2,† which are mainly contributed by strong hybridization between 4d states of Ag or 5d states for Au and 4p states of surrounding As atoms, implying that Ag/Au-doped systems exhibit metallic characteristics. We also presented the charge density differences (CDD) of Ag/Au-arseneneas, as shown in Fig. 1e and f. Upon substitution, the electron densities mainly accumulated on the dopants (Ag and Au) and surrounding As atoms, which indicated the formation of covalent bonds between them. Furthermore, the CCD distributions are obviously identical along the three Ag/Au–As bonds, confirming the C3v symmetry of Ag/Au-arsenene. The calculated DOS, band structure and CCD distributions are almost identical for Ag-arsenene and Au-arsenene, which could be explained by similar shell electron configurations of Ag (4d105s1) and Au (5d106s1).
3.2 Adsorption behaviors of gas molecules on Ag and Au-arsenene
To investigate the influences of various gas molecules (CO, NO, NO2, NH3, SO2 and H2S) adsorption on the structural and electronic properties of Ag and Au-arsenene, gas molecules were initially placed at different adsorption sites with various orientations and different heights above Ag and Au-arsenene and then relaxed all the structures to obtain the most favorable binding configurations. For comparison, the adsorption behaviors of the considered gases on p-arsenene and the interactions between molecules and p-arsenene are also explored in this work, as shown in Table 1 and Fig. S3.† Table 1 summarizes the adsorption energies (Eads), the distance from the adsorbed molecule to pristine arsenene or Ag/Au-arsenene surfaces (d) and the amount of charge transfer (Δq). It is clear that all considered molecules tend to chemically adsorb at Ag/Au-arsenene with adsorption energies in the range of −0.39 to −1.20 eV, especially NO2 on Ag/Au-arsenene with the largest adsorption energies of −1.12 eV and −0.92 eV, respectively. However, CO, H2S and NH3 prefer to adopt physical adsorption on p-arsenene via Van der Waals interactions accompanied by tiny charge transfer. NO, NO2 and SO2 molecules weakly chemisorb at p-arsenene with a relatively small adsorption energy of about −0.252, −0.326, and −0.214 eV and low charge transfer of about 0.126, 0.254 and 0.216e, respectively. The distances between gas molecules and Ag-arsenene are 1.80, 1.86, 2.02, 1.86, 1.99 and 2.03 Å for CO, NO, NO2, SO2, NH3 and H2S, respectively, while they are 1.99, 1,99, 2.00, 1.58, 1.98 and 1.79 Å for Au-arsenens, as listed in Table 1. Compared with the gas adsorbed on p-arsenene (Fig. S3†), the remarkable reduction of the distance between gas and Ag/Au-arsenene indicated that the introduction of Ag and Au enhances the interaction between gas and arsenene to some extent. Because of Ag/Au doping, the adsorption energy for the molecule adsorbed on Ag/Au-arsenene is larger than the corresponding adsorption energy on p-arsenene. Above all, the higher adsorption energies and the shorter distance between molecules and Ag/Au-arsenene indicated that the introduction of Ag and Au dopants in arsenene can effectively enhance the interactions between molecules and arsenene substrate.
Table 1 Distance from the adsorbed molecule to the substrate surface. Adsorption energy Eads, charge transfer Δq between the gas molecules and arsenene, as well as the total magnetic moment (Mtot) of the adsorbed systems. Note that a positive (negative) Δq indicates a loss (gain) of electrons from each molecule to the substrate
| Molecule |
Substrate |
d (Å) |
Δq (e) |
Style |
Eads (eV) |
Mtot (μB) |
Mgas (μB) |
| CO |
Arsenene |
3.10 |
−0.022 |
Acceptor |
−0.085 |
0 |
0 |
| Ag-arsenene |
1.85 |
−0.073 |
Acceptor |
−0.606 |
0 |
0 |
| Au-arsenene |
1.96 |
−0.107 |
Acceptor |
−0.517 |
0 |
0 |
| NO |
Arsenene |
2.35 |
−0.126 |
Acceptor |
−0.252 |
1.00 |
0.99 |
| Ag-arsenene |
1.94 |
−0.154 |
Acceptor |
−0.724 |
1.00 |
0.70 |
| Au-arsenene |
1.96 |
−0.267 |
Acceptor |
−0.607 |
1.00 |
0.56 |
| NO2 |
Arsenene |
2.59 |
−0.254 |
Acceptor |
−0.326 |
1.00 |
0.98 |
| Ag-arsenene |
2.08 |
−0.648 |
Acceptor |
−1.120 |
0 |
0 |
| Au-arsenene |
2.04 |
−0.584 |
Acceptor |
−0.923 |
0 |
0 |
| SO2 |
Arsenene |
2.68 |
−0.216 |
Acceptor |
−0.214 |
0 |
0 |
| Ag-arsenene |
1.99 |
−0.199 |
Acceptor |
−0.394 |
0 |
0 |
| Au-arsenene |
1.73 |
−0.210 |
Acceptor |
−0.429 |
0 |
0 |
| H2S |
Arsenene |
2.56 |
−0.021 |
Acceptor |
−0.170 |
0 |
0 |
| Ag-arsenene |
2.14 |
0.100 |
Donor |
−0.638 |
0 |
0 |
| Au-arsenene |
2.06 |
0.102 |
Donor |
−0.391 |
0 |
0 |
| NH3 |
Arsenene |
2.69 |
0.021 |
Donor |
−0.225 |
0 |
0 |
| Ag-arsenene |
2.08 |
0.123 |
Donor |
−0.846 |
0 |
0 |
| Au-arsenene |
1.91 |
0.138 |
Donor |
−0.528 |
0 |
0 |
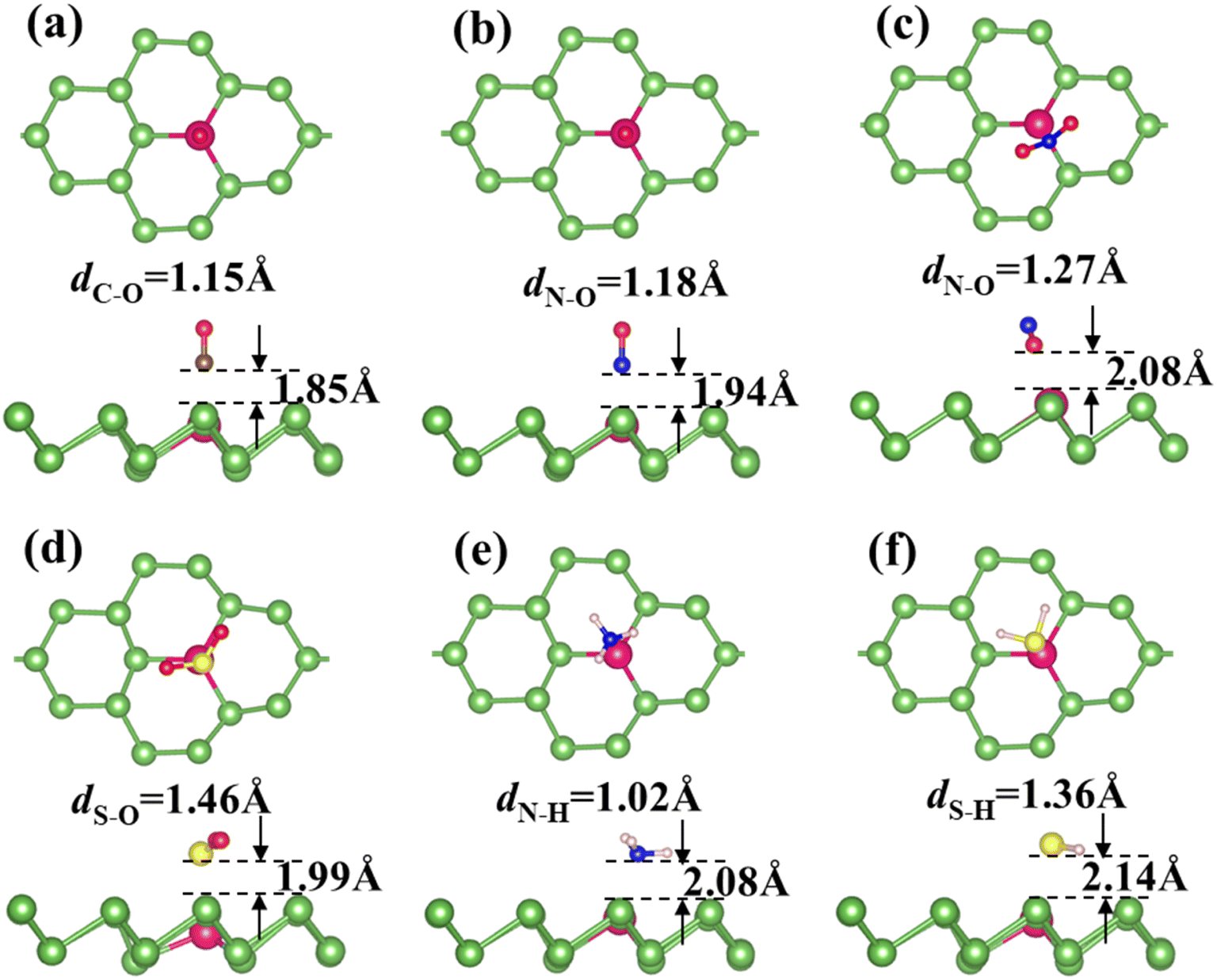
Fig. 2 and S4† display the most favorable configurations for CO, NO, NO2, NH3, SO2 and H2S adsorbed on Ag-arsenene and Au-arsenene, respectively. It is not surprising that the adsorption configurations are almost identical for all the toxic molecules adsorbed on both Ag-arsenene and Au-arsenene; this is because Ag and Au possess similar valence electron distribution and similar atomic radius. For CO and NO adsorptions (Fig. 2a, b and S4a, b†), they both prefer to adopt a nearly perpendicular alignment with C or N towards the As surface, resulting in C–Ag/Au or N–Ag/Au bond with bond lengths of 2.12/2.02 Å and 2.06/2.00 Å, which is different from the adsorption configurations of CO and NO on p-arsenene (Fig. S3a and b†), in which NO and CO are inclined along As–As bonds. Interestingly, it can be observed from Fig. 2c that NO2 favors adopting an inverted V-type bend structure with both oxygen atoms connected to the Ag/Au atom, and Ag/Au–O distance of 2.36/2.39 Å, which could be explained by the interaction between the electron-rich oxygen atoms and Ag/Au dopant. A similar adsorption configuration was observed for NO2 on p-arsenene, as shown in Fig. S3c.† SO2 diametrically adopted a V-type orientation with S actively participating with Ag/Au-arsenene surface with Ag/Au–S distance of 2.68/2.80 Å (Fig. 2d), while SO2 adopted a flat configuration almost parallel to the p-arsenene surface (Fig. S3d†) accompanied by an S bond to the underlying As atom. In the case of NH3 adsorption (Fig. 2e), the N atom points towards Ag/Au dopant with hydrogen atoms on the next end, while H2S favors lying nearly parallel to the surface where the S atom captured by Ag/Au and S–Ag/Au bond length is about 2.63/2.78 Å, as shown in Fig. 2f and S4f.† However, NH3 (Fig. S3e†) and H2S (Fig. S3f†) are favored to adsorb at the hollow sites of p-arsenene. Furthermore, the bond length stretching for all the toxic molecules adsorbed on Ag-arsenene and Au-arsenene was found to be identical. Upon adsorption on Ag/Au-arsenene, the bond lengths of CO and H2S show tiny elongations, such as 1.15 and 1.36 Å for C–O and H–S bonds, respectively, compared with the cases of p-arsenene (1.14 Å for C–O and 1.35 Å for H–S bond). N–O bonds in NO2 elongate remarkably from 1.23 Å for NO2 adsorbed on p-arsenene to 1.27 Å for NO2 adsorbed on Ag/Au-arsenene. For the cases of NO, SO2 and NH3 molecules, negligible changes in the bond length of gas molecules are observed upon the adsorption of NO, SO2 and NH3 on both Ag/Au-arsenene and p-arsenene. Thus, the calculated results show that the introduction of Ag and Au could effectively stretch the bond length of NO2 with the highest adsorption energies, implying that Ag/Au-arsenene are suitable candidates for capturing NO2 molecules. We also found that the distances between SO2 (and H2S) and Au-arsenene are shorter than those of SO2/Ag-arsenene and H2S/Ag-arsenene, which are consistent with the results of adsorption energies, as listed in Table 1.
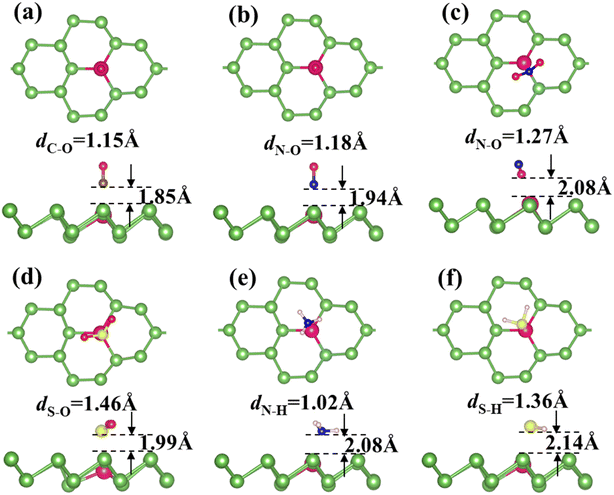 |
| | Fig. 2 (a–f) Top (upper panel) and side views (lower panel) of the most favorable configurations of Ag-arsenene with the adsorption of CO, NO, NO2, SO2, NH3 and H2S. | |
It is clear from Table 1 that the adsorption energy for each molecule on Ag-arsenene is generally larger than that for the corresponding molecule on Au-arsenene (the adsorption energy for SO2 is slightly larger on Au-arsenene than on Ag-arsenene by 0.035 eV). Such phenomena may be related to the atomic structures of Ag/Au-arsenene, as shown in Fig. 1a and b, showing that Au moved downwards related to the upper As layer by 0.38 Å more than that for Ag. Among all the molecules considered, the NO2 molecule shows the highest adsorption energy on both Ag/Au-arsenene, which is due to the inverted V-type structure of NO2. In such a unique configuration, two O atoms may lose electrons easily to the bellowing Ag/Au atoms by forming new bonds. We also found that Ag/Au dopants move upwards to the upper As layer to some extent upon the adsorption of molecules, as shown in Fig. 2 and S4.† In the case of Ag-arsenene, the movement of Ag upon adsorption is the least for the SO2 molecule with the lowest adsorption energy. For the case of Au-arsenene, the movement of Au upon adsorption is slight for both SO2 and H2S with an adsorption energy difference of only 0.038 eV, as shown in Table 1.
3.3 Electronic and magnetic properties of gas adsorption systems
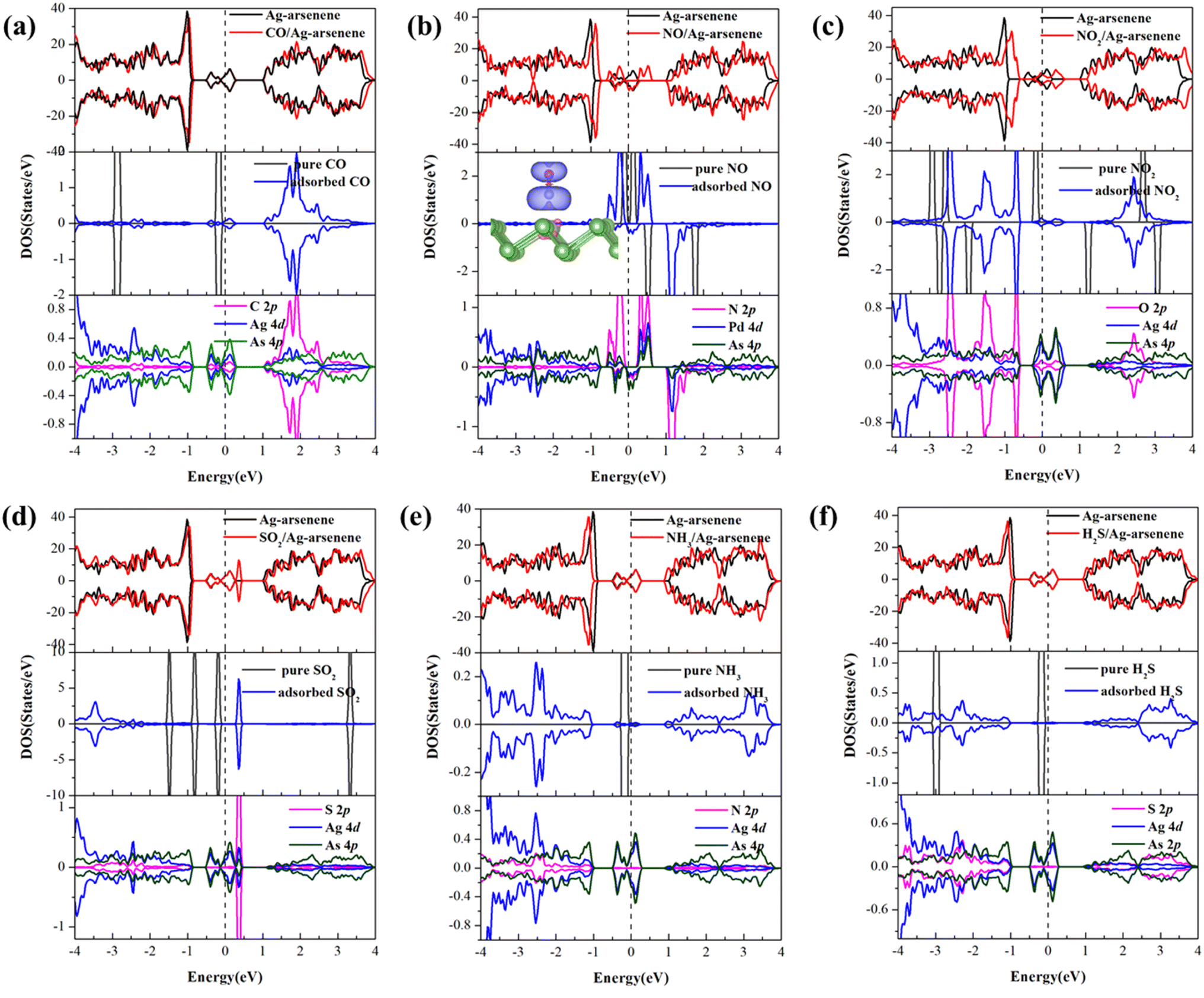
To develop an understanding of the effects of gas molecule adsorption on the electronic properties of Ag/Au-arsenene, we calculated the density of states (DOS) for Ag-arsenene and Au-arsenene upon gas adsorption, as shown in Fig. 3 and S5,† respectively. The identical DOS in the spin up channel and spin down channel indicated that Ag/Au-arsenene with gas adsorptions exhibit nonmagnetic ground states, except for the case of NO adsorbed on Ag/Au-arsenene (as shown in Fig. 3b and S5b†) with a magnetic moment of 1.0 μB. Interestingly, we previously reported that the ground state is nonmagnetic upon NO adsorbed at Pt-doped arsenene.31 This difference in magnetic characteristics could be explained by different adsorption configurations of NO and different shell layer electronic arrangements of Ag/Au and Pt atom, such as NO favors bonding with Pt atom by N forming a tilted configuration to the surface, which caused relatively strong orbital hybridization between NO and the underlying Pt atom. The NO molecule is vertical to the As surface on top of the Ag or Au dopant with Eads of −0.72 eV and −0.61 eV, which is much less than that of NO/Pt-arsenene (−1.48 eV). According to the DOS results, the corresponding magnetization density distributions were analysed to explain the origin of magnetism, as shown by the inset of Fig. 3b and S5b† for NO/Ag-arsenene and NO/Au-arsenene, respectively. This confirmed that the origin of magnetism mainly results from the contributions of NO with unpaired electrons; meanwhile, underlying Ag or Au atoms have certain contributions to total magnetism. For CO adsorbed on Ag/Au-arsenene, as shown in Fig. 3a and S5a,† it is clear that the density distribution of states has slight change except that the bandgap states (in the range of −0.5 to 0.4 eV) are enhanced by CO introduction. Interestingly, the total magnetic moment of NO2/p-arsenene is about 1.0 μB, while the magnetism disappeared upon NO2 adsorption on Ag/Au-arsenene, as shown in Fig. 3c and S5c.† As shown in Fig. 3d and S5d,† valence-band maximum (VBM) and conduction band minimum (CBM) slightly shifted up to about 0.05 eV for SO2 adsorbed on the Ag/Au-arsenene surface, respectively. The new flat impurity states emerged at 0.59 eV and 0.34 eV above EF, respectively, which is caused by strong hybridizations between the 2p states of S and the 5d (4d) states of Au (Ag) dopant. For the adsorption of H2S and NH3 on Ag/Au-arsenene, the density distributions display a downward trend, as shown in Fig. 3e, f and S5e, f.† However, it is found that the gap states around EF basically remain unchanged, which are still mainly contributed by the orbital hybridization of Au or Ag dopant and surrounding As atoms for most of the gas adsorbed on Ag/Au-arsenene substrates.
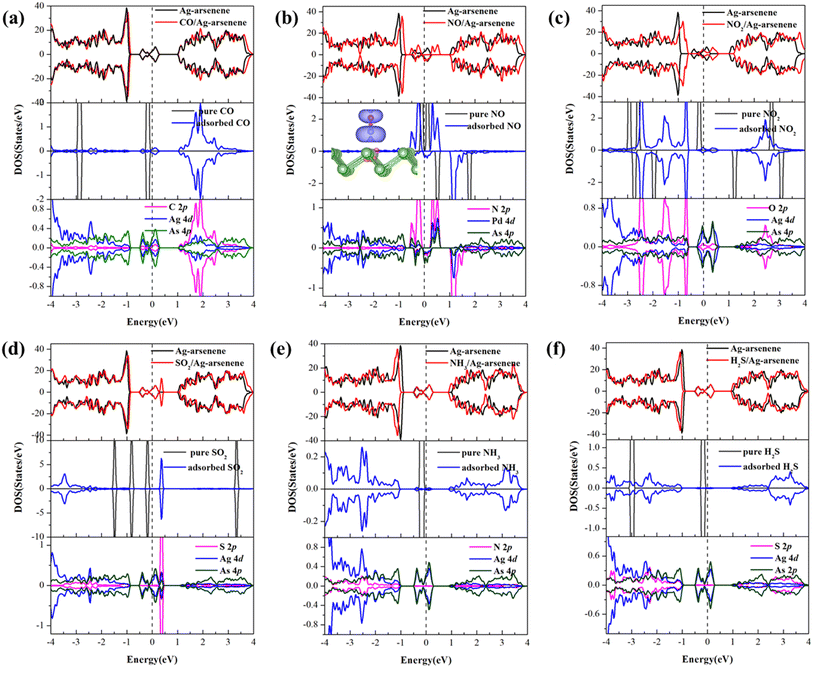 |
| | Fig. 3 (a)–(f) Density of states (PDOS) for considered gas molecules of CO, NO, NO2, SO2, NH3 and H2S adsorbed at Ag-arsenene. EF is set to be 0 eV and is denoted by dash lines. The magnetization charge density difference for NO/Au-arsenene is plotted in the inset of (b) and the magnetization charge density isosurface value is 0.001 e Å−3. | |
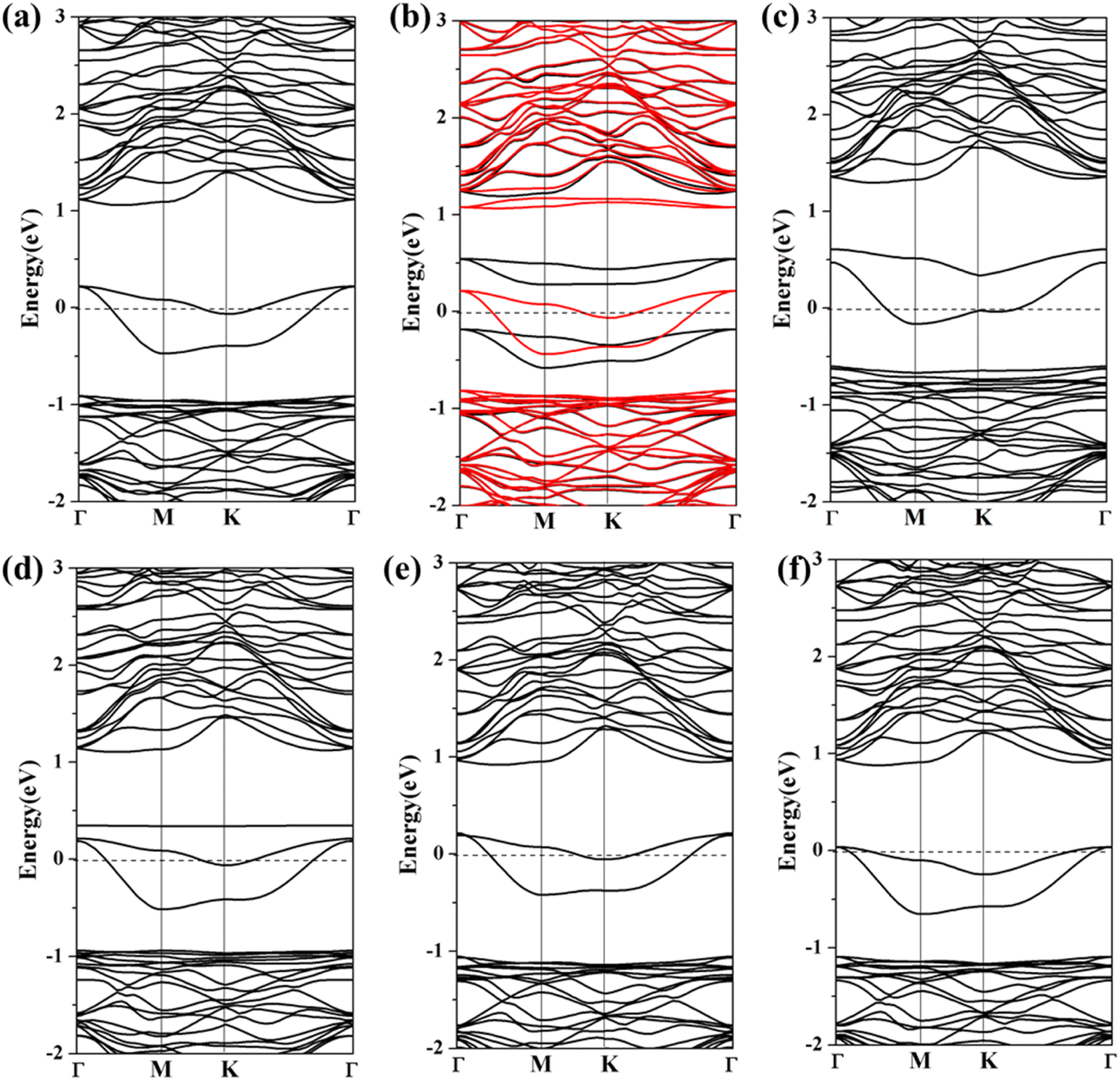
To explore the influence of toxic gas molecule adsorption on the electronic properties of Ag/Au-arsenene, the band structures for gas adsorption on Ag/Au-arsenene systems are shown in Fig. 4 and S6,† respectively. For CO and SO2 adsorbed on Ag-arsenene, as shown in Fig. 4a and d, respectively, the band gap states near the Fermi level (EF) have no significant change. However, the valence-band maximum (VBM) and conduction band minimum (CBM) overall increased by about 0.10 eV upon the adsorption of CO and SO2. By analyzing the DOS of pure and adsorbed CO (Fig. 3a), it can be observed that the molecule orbital of adsorbed CO is delocalized compared to the isolated CO, which is caused by a violent mixture between 2π* antibonding of the CO molecule and 4d of Ag in the energy range of 1.0–3.5 eV. For NO/Ag-arsenene, NO-induced bands near EF were observed, as shown in Fig. 3b, which originated from orbital splitting of NO and overlapping between the 2p states of the N and 4d states of underlying Ag, as shown in Fig. 4b. Upon the NO2 adsorption, the symmetrical DOS (Fig. 3c) and overlapping band structure (Fig. 4c) indicated the nonmagnetic ground state, which was quite different from NO2/p-arsenene with 1.0 μB caused by unpaired electrons in NO2 and weaker interaction between gas and pristine substrate. The disappearance of magnetism is a consequence of the hybridization of the 4d states of Ag and the 2p states of N, as shown in Fig. 4c. For SO2/Ag-arsenene (Fig. 4d), the VBM and CBM shift up slightly owing to the weak loss electron of the substrate; remarkably, a new flat band state emerges at about 0.34 eV above EF. Combined with the PDOS results (shown in Fig. 3d), it is found that the flat band mainly originates from the contributions of strong hybridization between the 2p orbital of S attached to Ag and the 4d orbital of Ag. The two bands across EF are still caused by hybridization between Ag and surrounding As atoms. Concerning NH3 and H2S adsorption, the bands of the two systems except for impurity states around EF move down slightly with respect to that of the unabsorbed surface, as shown in Fig. 4e and f, which is related to the direction and strength of charge transfer in Ag-arsenene. In addition, one interesting phenomenon was observed: the adsorption of H2S and NH3 narrowed the energy range of impurities states near EF.
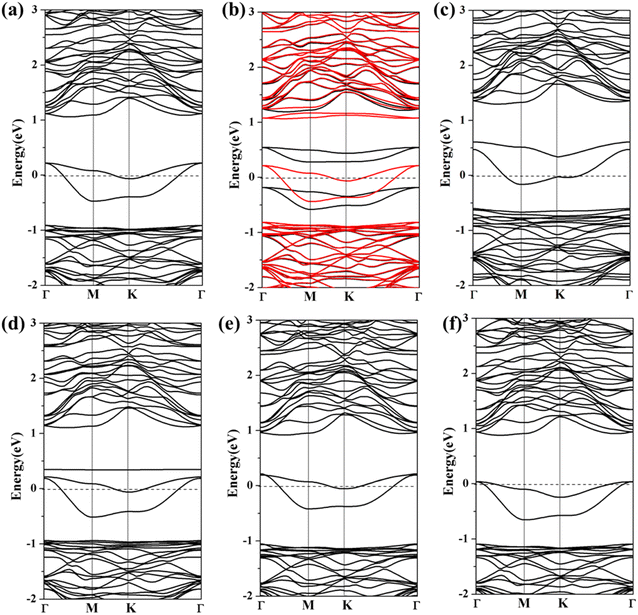 |
| | Fig. 4 (a)–(f) Spin-polarized band structures of CO, NO, NO2, SO2, NH3 and H2S adsorbed at Ag-arsenene. The black and red lines show spin up and spin down band structures, respectively. EF is set to be 0 eV and is denoted by the black dash lines. | |
Similar results regarding the density of states and band structures are observed when toxic gas is adsorbed on Au-arsenene, as shown in Fig. S5 and S6.† Generally, the bands of Au-arsenene after adsorption moved up entirely when substrates acted as donors to supply electrons, such as the adsorptions of CO, NO, NO2 and SO2, while adsorptions of H2S and NH3 resulted in shifting down of band structures for Au-arsenene when the substrate obtained charge from adsorbed gas molecules. Concerning NO adsorption systems, the Au-arsenene systems exhibited 100% spin polarization near EF, which caused semiconducting and metallic characteristics for spin-up and spin-down channels, respectively. Upon the adsorption of NO2, Au-arsenene remains metallic, similar to the cases without gas adsorption. For H2S, the impurity states near EF have no significant change, corresponding to the low adsorption energy.
3.4 Charge transfer
To quantitatively determine the amount of charge transfer from the molecule to Ag/Au-arsenene systems, we performed Bader charge analysis and summarized the results, as listed in Table 1. We found that H2S and NH3 behaved the same as weak donors by supplying about 0.100e and 0.123e to Ag-doped substrates, respectively; similarly, H2S and NH3 denote 0.100e and 0.123e to Au-arsenene, respectively. When NH3 was adsorbed at p-arsenene, NH3 acting as a weak provider only donated about 0.021e to the substrate, while H2S acted as an acceptor gained about 0.216e from p-arsenene. About 0.073 and 0.107e are transferred from Ag-arsenene and Au-arsenene substrates to adsorbed CO molecules, respectively, indicating that the introduction of Ag/Au dopant does not significantly enhance charge transfer compared with those adsorbed at p-arsenene (CO obtained about −0.022e). SO2 as acceptors obtained about 0.199 and 0.210e from Ag- and Au-arsenene, respectively. Upon adsorption on Ag-arsenene, NO and NO2 acting as strong acceptors obtained 0.154e and 0.648e from underlying substrates, respectively, which is similar to the cases of Pt-doped arsenene. The charge transfer between NO and Au-arsenene (NO obtained about 0.267e) is stronger than that of NO/Ag-arsenene, while NO2 exhibits a slightly weaker electron accepting ability (obtained 0.584e) from Au-arsenene compared with that of Ag-arsenene (obtained 0.648e). Although charge transfers in SO2/Ag-arsenene and SO2/Au-arsenene systems are almost identical to those in SO2/p-arsenene system, the adsorption energies are enhanced upon the doping of Ag or Au to some extent, indicating that the doping of Ag or Au can slightly enhance the interactions between SO2 and arsenene. Viewed collectively, the introduction of Ag and Au could enhance the adsorption energies or charge transfer between molecules and the arsenene support. Therefore, Ag/Au-arsenene can be expected to show higher sensitivity in detecting such molecules compared with pristine arsenene.
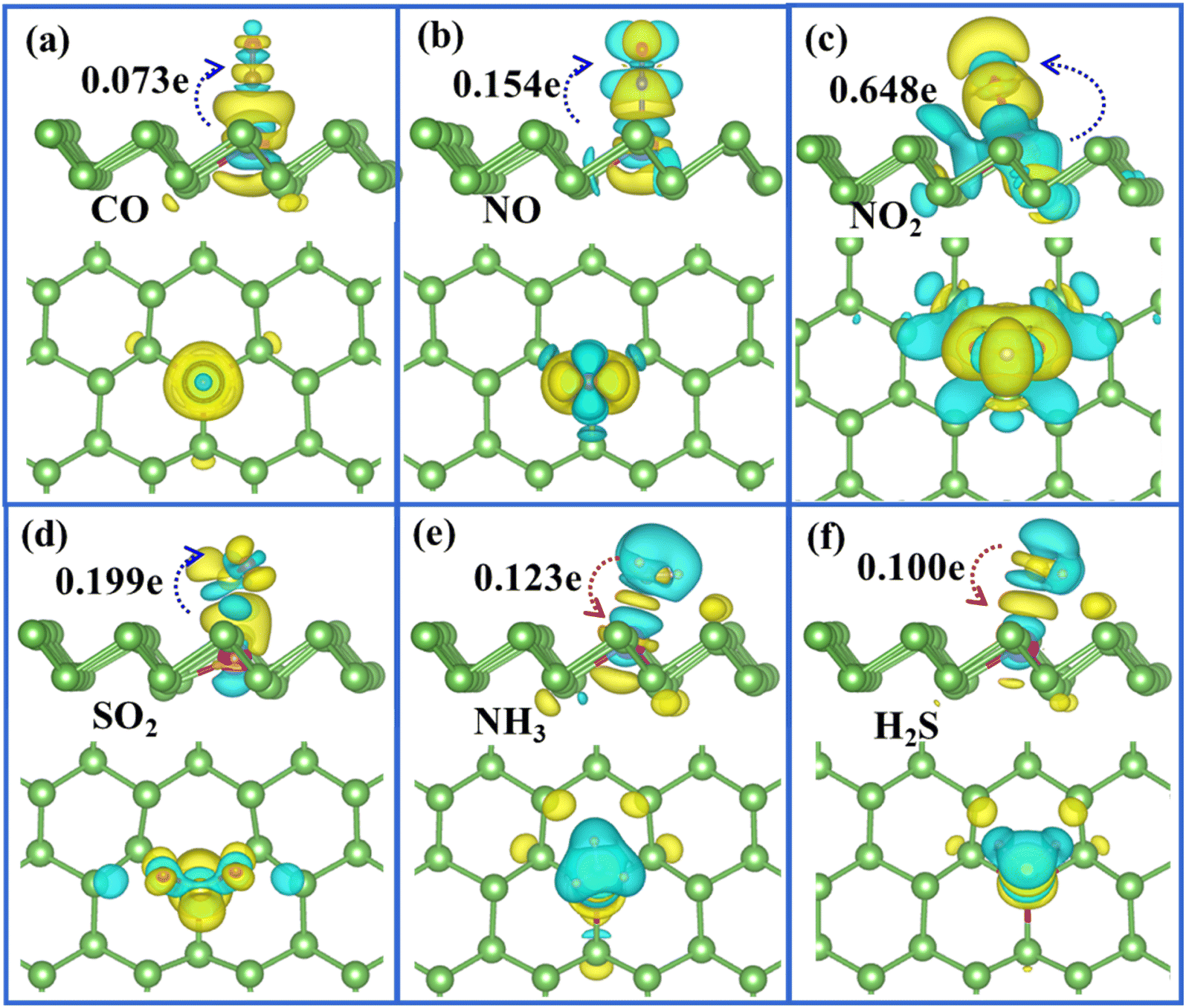
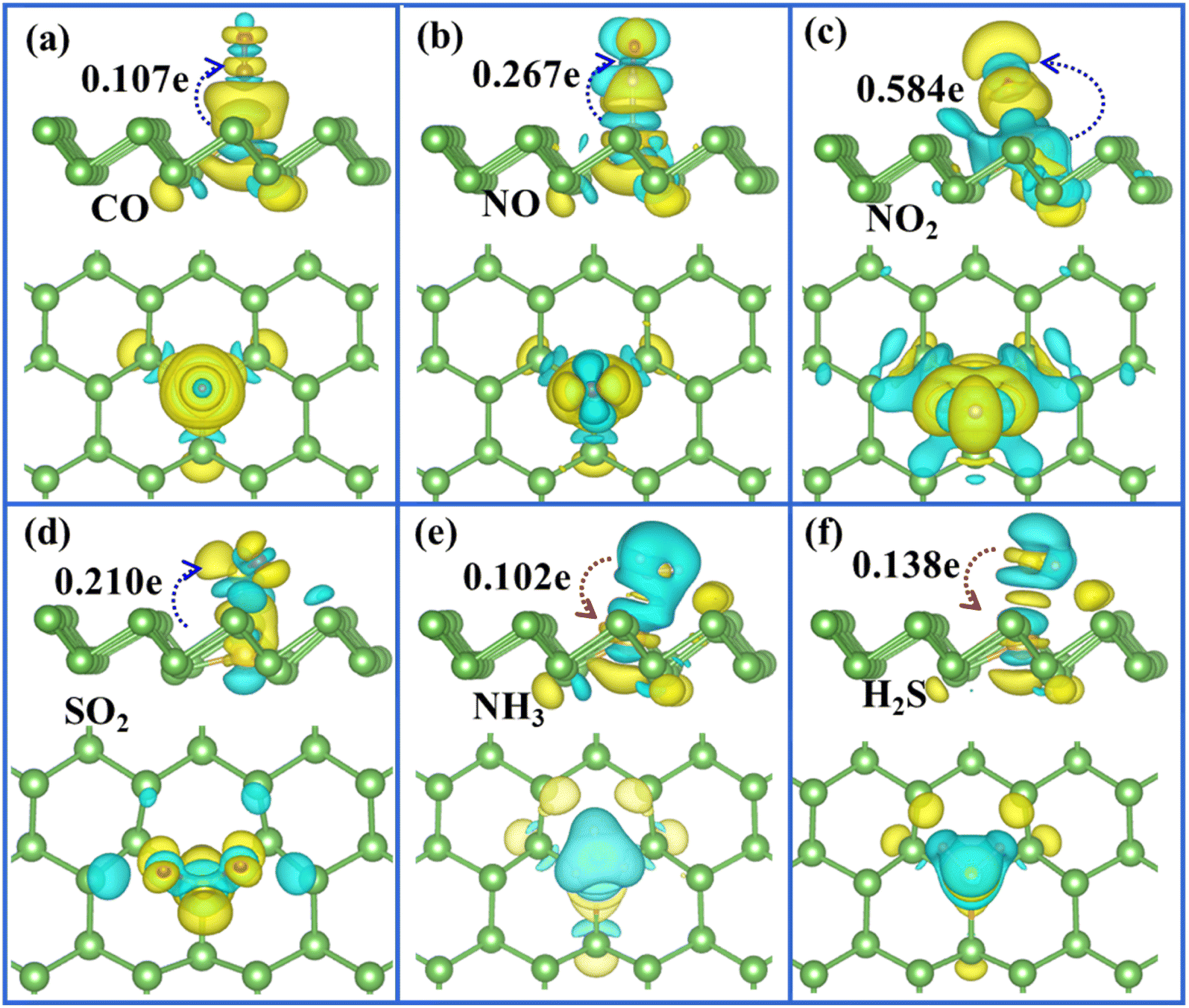
To clearly visualise the charge transfer and the bonding mechanism between gas molecules and arsenene substrate, we also plotted the corresponding charge density difference images for Ag/Au-doped arsenene (Fig. 5 and 6) as well as pristine arsenene (Fig. S7†) with molecules adsorbed, respectively. According to Fig. 5a–d, 6a–d and S7a–d,† the remarkable charge redistribution between molecules and Ag/Au-arsenene is observed. The adsorbed CO, NO, NO2 and SO2 molecules obtained charge from underlying Ag or Au atom and surrounding As atoms, corresponding to the noticeable change in the DOS near EF, as shown in Fig. 3a–d and S5a–d.† Such strong interactions between the above four molecules and the substrate are identified as the main reason for the significant elongation of bond length and enhancement of the adsorption behavior of gas molecules on Ag/Au-arsenene. Distinctly, it has been observed that charge accumulation occurs around Ag (or Au) and surrounding As atoms, whereas charge depletion appears near NH3 and H2S molecules for NH3 and H2S adsorbed on Ag/Au-arsenene, as shown in Fig. 5e, f and 6e, f. The significant charge redistribution upon NH3 adsorption indicated that the N atom participated in strong hybridization with the underlying Ag or Au and formed chemical bonds with ionic and covalent bond characteristics, arising mainly from the ionization of the lone electron pair in the N atom. Instead of SO2 adsorbed at the hollow site of p-arsenene (Fig. S7†), a shorter distance between S and underlying Ag (or Au) results in a strong overlapping of the electron density distribution for S and Ag (or Au), which has been proved by the results of DOS, as shown in Fig. 3e and S5e.†
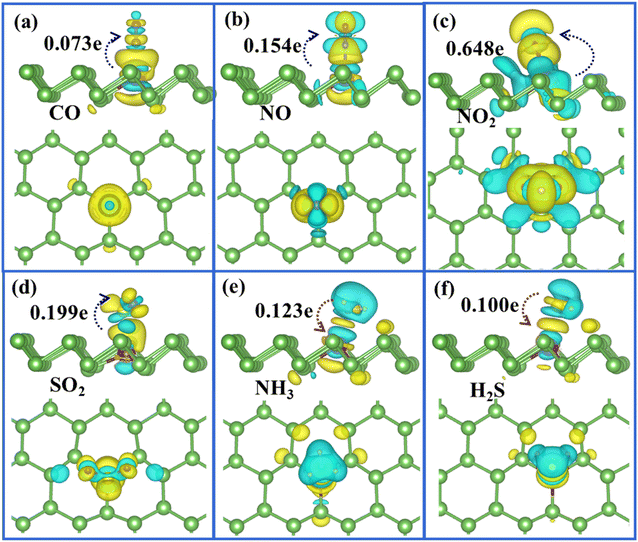 |
| | Fig. 5 (a)–(f) Top (upper panel) and side views (lower panel) of the charge density difference plots for CO, NO, NO2, SO2, NH3 and H2S adsorbed at Ag-arsenene, in which yellow and green regions refer to the charge accumulation and depletion, respectively. The isosurface level is in the unit of 0.001 e Å−3. | |
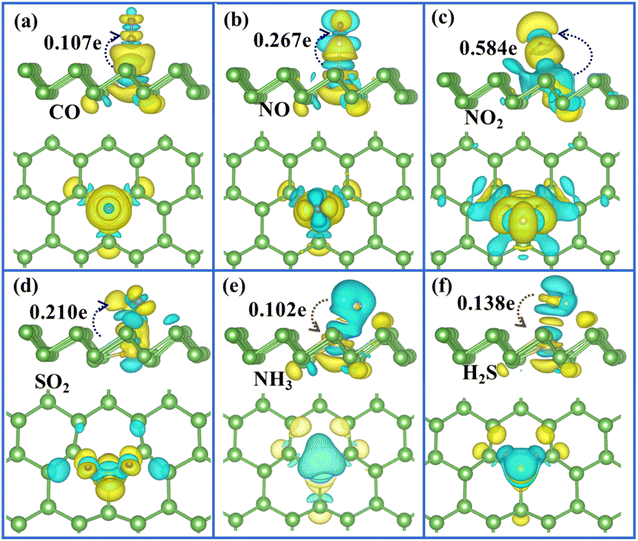 |
| | Fig. 6 (a)–(f) Top (upper panel) and side views (lower panel) of the charge density difference plots for CO, NO, NO2, SO2, NH3 and H2S adsorbed at Au-arsenene, respectively. | |
3.5 Vibrational frequency analysis
To further confirm the configuration structures obtained by the minima of the potential energy surface, we also calculated the harmonic vibrational frequencies for gas molecules before and after adsorption on Ag/Au-arsenene monolayers. The strength of the interaction between gas and substrates could be qualitatively reflected by the changes in the vibrational frequency of gas molecules before and after adsorption on arsenene surfaces. Table 2 summarizes the vibrational frequency distributions for gas molecules of CO, NO, NO2, SO2, H2S and NH3 in their absorbed complex configurations compared with those in the gas phase. We found that vibrational frequencies of considered gas molecules upon adsorption on Ag/Au-arsenene decreased to some extent compared with those of free standing molecules. Combined with the adsorption energy results, the results in Table 2 further confirm that the vibrational frequencies of gas molecules prominently depend on the strength of the interactions between gas molecules and the Ag/Au-arsenene surface. For the CO molecule, the computed stretching frequency of CO shifts from 2127 cm−1 for free standing CO to 2061 and 2029 cm−1 when binding with Ag-arsenene and Au-arsenene, respectively. The calculated frequencies of the NO molecule are 1923 cm−1 and 1785/1770 cm−1 before and after adsorption on Ag/Au-arsenene, respectively. In this case, the symmetric/asymmetric vibrational frequencies of NO2 decrease sharply to 1197/1245 cm−1 and 1201/1238 cm−1 upon adsorption on Ag-arsenene and Au-arsenene from 1345/1686 cm−1 for free NO2, respectively. Similar phenomena were also observed by Kumar et al., where the decrement in frequency is very high, about 22.95%, after NO2 was adsorbed on the GeBi nanosheet.39 This demonstrates the robust interaction between NO2 and Ag/Au-arsenene, which corresponds with the highest Eads of −1.12 eV and −0.92 eV and the largest charge transfer (NO2 gained 0.648e and 0.584e from underlying Ag/Au-arsenene, respectively). SO2 gas is stretched from 1089/1275 cm−1 to about 1031/1228 cm−1 and 1038/1216 cm−1 after adsorption on Ag and Au-arsenene, respectively. The symmetric/asymmetric vibrational frequencies of H2S decreased from 2648/2663 cm−1 to 2561/2557 cm−1 after adsorption on Ag-arsenene, showing a higher decrement than those of H2S/Au-arsenene (decrement in symmetric/asymmetric frequency of about 2.49%/3.27%). The lower frequency change (H2S of 3.29%/3.98% and CO of 3.10%) refers to the relatively weaker interaction between H2S or CO and the underlying Ag-arsenene substrate compared to the case of NO2/Ag-arsenene, which is confirmed by the calculated results of charge transfer and adsorption energy, as listed in Table 1. For polyatomic NH3 molecules, the potential normal modes are six (3n − 6 is equal to 6, where n is the number of atoms). The calculated vibrational frequencies range from 1000 cm−1 to 3546 cm−1. The calculated stretching frequencies are 1621 cm−1 and 1605/1597 cm−1 for NH3 in free and adsorbed Ag/Au-arsenene, respectively. Although the adsorption energy and charge transfer amount of NH3 on Ag/Au-arsenene are higher than those of H2S adsorption cases, a smaller frequency change reveals that NH3 is more weakly interacting with underlying Ag/Au-arsenene compared to H2S adsorbed at Ag/Au-arsenene. These results could be explained by the fact that the electronegativity of S is stronger than that of N when bonding to underlying Ag/Au dopants. Above all, we can conclude that more significant changes in the vibrational properties of NO, SO2 and NO2 have been observed compared with those of CO, NH3 and H2S, which is consistent with the relatively higher adsorption energies and larger charge transfers for the adsorption of NO, SO2 and NO2 on Ag/Au-arsenene compared with the cases of CO, NH3 and H2S.
Table 2 Vibrational frequency distributions for gas molecules of CO, NO, NO2, SO2, H2S and NH3 in free phase and absorbed complex configurations
| Molecules |
Vibrational frequency (cm−1) |
| Free gas molecules |
Adsorbed gas molecules |
| Our work |
Previous studiesb |
Ag-arsenene |
Au-arsenene |
| Two values correspond to symmetric and asymmetric vibrational frequencies of polyatomic molecules. Obtained from the NIST database (website: https://cccbdb.nist.gov). |
| CO |
2127 |
2143 |
2061 |
2029 |
| NO |
1923 |
1876 |
1785 |
1770 |
| NO2 |
1345/1686a |
1318/1618a |
1197/1245a |
1201/1238a |
| SO2 |
1089/1275a |
1151/1362a |
1031/1228a |
1038/1216a |
| H2S |
2648/2663a |
2615/2626a |
2561/2557a |
2582/2576a |
| NH3 |
1621 |
1627 |
1605 |
1597 |
3.6 Work function
To identify the adsorption activity of different gas molecules on the Ag and Au-doped arsenene surface, we compute the work function (ϕ) and evaluate the work function change (Δϕ) resulting from molecule adsorption by considering the vacuum level as a reference. The work function is represented as follows:where Evac and EF denote the vacuum level of electrostatic potential energies and the Fermi level for the considered arsenene systems, respectively. The calculated work functions for six toxic gases adsorbed on Ag/Au-arsenene as well as on p-arsenene are summarized in Table 3. The work functions of NM-arsenene upon adsorption of molecules increase to some extent. The increase in work function follows the order of NO2 > SO2 > NO > CO > H2S > NH3 for Ag-arsenene, and NO2 > NO > SO2 > CO > H2S > NH3 for Au-arsenene, which are consistent with the order of charge transfer between adsorbates and substrate, as listed in Table 1.
Table 3 Work function (ϕ) for Ag/Au-doped arsenene and p-arsenene with and without the adsorption of gas. The values in parentheses are the work function changes (Δϕ) of the substrates upon the adsorption of molecules
| Molecules |
p-arsenene |
Ag-arsenene |
Au-arsenene |
| Substrate |
5.169 |
4.783 |
4.802 |
| CO |
5.136 (−0.033) |
4.800 (0.017) |
4.840 (0.038) |
| NO |
4.399 (−0.770) |
4.891 (0.108) |
4.984 (0.182) |
| NO2 |
5.326 (0.157) |
5.278 (0.495) |
5.111 (0.309) |
| SO2 |
5.091 (−0.078) |
4.951 (0.168) |
4.968 (0.166) |
| H2S |
5.297 (0.128) |
4.672 (−0.111) |
4.651 (−0.151) |
| NH3 |
5.167 (−0.002) |
4.594 (−0.189) |
4.556 (−0.246) |
In general, the work function change is proportionally associated with the adsorption energy Eads and the charge transfer between the absorbed molecule and Ag/Au-arsenene. In the cases of CO molecules adsorbed on Ag- and Au-arsenene, the work function varies slightly, which is due to the relatively low adsorption energy or small charge transfer in these adsorbed systems. In contrast, the work function is prominently increased when the NO2 molecule is chemically absorbed on the two considered surfaces, corresponding to the strongest charge transfer and the largest adsorption energy among all considered molecules. It is noteworthy that NO, SO2 and H2S adsorptions on Ag-/Au-arsenene lead to moderate work function change in the range of 0.10–0.20 eV. It is also found that the work function is remarkably reduced when NH3 was adsorbed on considered Ag/Au-arsenene owing to the shift up of the EF. Previous studies have also reported that the work function variation in the 2D material upon the adsorption of gas molecules can be used to design novel gas sensors with superior sensitivity to that of the common resistance-type sensor.40 Therefore, the different work function responses to the different adsorbed gas types confirm that Ag- and Au-doped arsenene possesses a possible sensing application for detecting toxic gas molecules with high selectivity based on work function changes.
3.7 Recovery time analysis
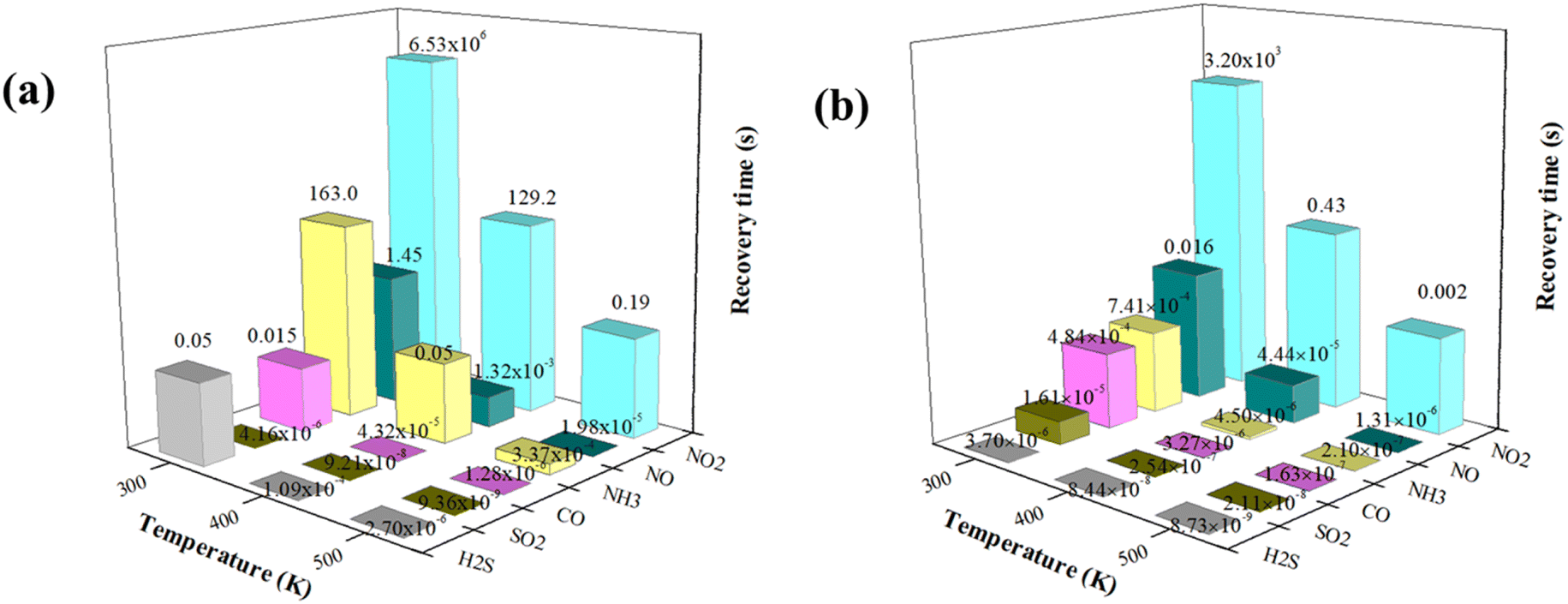
It is known that the recovery time is the required time for gas desorbing from the surface of materials, which is another important target for evaluating the sensitivity and reusability of sensing materials. It becomes difficult to remove the adsorbed gases from the surface when gas molecules form strong chemical bonds with the underlying substrates, indicating that the gas sensor requires a lengthy recovery time. According to the transition state theory,41,42 the recovery time (τ) could be computed using the following formula:where A is the operational frequency of about 1012 s−1 according to previous studies,43,44 kB is the Boltzmann's constant (8.62 × 10−5 eV K−1) and T is the working temperature. Given that desorption could be regarded as the inverse process of adsorption, we assumed the value of Eads to be the barrier energy (E*) of the corresponding desorption process. From the formula, the more negative the adsorption energy, the longer the recovery time. In other words, smaller values for adsorption energy actually mean more reversible gas sensors. To explore the effect of metal-doped arsenene systems on the sensing of these six toxic gas molecules, we calculated the recovery time (τ) of the considered gas molecules in the Ag- and Au-arsenene systems at various temperatures (300 K, 400 K and 500 K), as depicted in Fig. 7. The calculated τ at 300 K for CO, NO, NO2, SO2, NH3 and H2S adsorbed on Ag-arsenene (Fig. 7a) are 0.02, 1.45, 6.53 × 106, 4.16 × 10−6, 163 and 0.05 seconds, respectively. The ideal recovery times (in the order of 10−2 or 10−1 seconds) indicated that Ag-arsenene can be a reversible sensor to detect CO, NO and H2S in an environment at room temperature. However, the high adsorption energy of NO2 on Ag-arsenene results in a very long recovery time (6.53 × 106 s) at room temperature, demonstrating their strong interaction with Ag-arsenene. This feature indicates that it is difficult for NO2 gas molecules to be desorbed from the surface, implying that arsenene defected by Ag is more applicable to utilize as NO2 capture with non-reversibility at room temperature. We also calculated the recovery times of the molecules on the Au-arsenene systems, as shown in Fig. 7b. For Au-arsenene, the recovery times for CO, NO, NO2, SO2, NH3 and H2S are estimated to be 4.84 × 10−4, 0.016, 3.20 × 103, 1.61 × 10−5, 7.41 × 10−4 and 3.70 × 10−6 seconds at 300 K, respectively. The extremely short recovery times for CO, SO2, NH3 and H2S on Au-arsenene as well as SO2 on Ag-arsenene indicated that these gases easily detached from the arsenene surfaces at room temperature. It is clear that similar to the case of Ag-arsenene, the NO2 molecule has a long recovery time on Au-arsenene at room temperature, indicating that NO2 could be stably adsorbed on the surface of arsenene modified by Au and Ag at room temperature. Therefore, arsenene doped by Ag and Au is expected to be an excellent candidate for NO2 captured at room temperature. Our results also show that Ag-arsenene can operate as reversible CO, NO and H2S sensors with moderate recovery times, while Au-arsenene only shows high reversibility in sensing NO gas at 300 K.
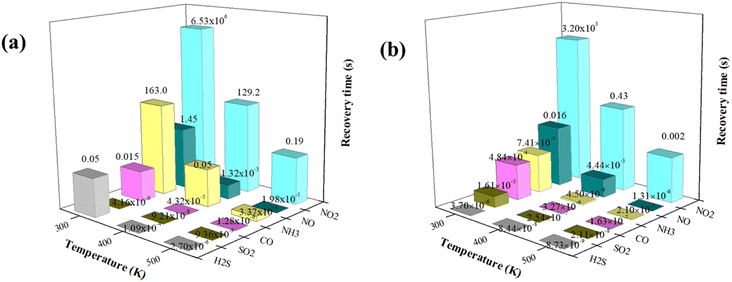 |
| | Fig. 7 Recovery times of gases adsorbed at (a) Ag-arsenene and (b) Au-arsenene at different temperatures. | |
Notably, the recovery times of some adsorbed systems were extremely short (such as τ lower than the response time of the measuring instrument) or extremely long (implying that the repeated utilization factor of the sensor is very low owing to the long desorption time for sensing gas), indicating that Ag/Au-arsenene could not be used to detect some specific gases. For Ag-doped arsenene, it is unsuitable to sense SO2 owing to very short recovery times and unsuitable to detect NO2 owing to the long recovery time at 300 K. For Au-doped arsenene, it is not suitable to sense H2S, SO2, CO and NH3 with very short recovery times and not suitable to detect NO2 owing to the long recovery time at 300 K. The above results also suggested that for a promoting gas sensor, the foreign gas should show a chemisorption process with suitable adsorption energy in the range of −0.60 to −0.80 eV at around room temperature. According to eqn (4), the recovery time decreased exponentially with an increase in temperature up to 500 K, as shown in Fig. 7, implying that more gas molecules could be effectively desorbed and sensed at high temperatures by Ag/Au-arsenene. Therefore, the resolution process is completed in the order of 10−2 or 10−4 s. For example, NH3 can be easily desorbed from an Ag-arsenene-based gas sensor simply by heating to 400 K (recovery time reduced from 163 s at 300 K to 0.05 s at 400 K). The recovery time for NO2 on Ag- and Au-doped arsenene decrease to 129.2 s and 0.43 s at a temperature of 400 K and further decrease to 0.19 and 0.002 seconds at 500 K, respectively, indicating that Ag- and Au-arsenene systems have potential applications in sensing NH3 and NO2 from factory emission and automobile exhaust. Consequently, it is reasonable to expect that Ag-arsenene could achieve good reusability to detect and monitor CO, NO and H2S gas molecules, while the Au-arsenene system may exhibit good performance in sensing NO at room temperature. The relatively long desorption time and high adsorption energies revealed that Ag- and Au-arsenene are appropriate for use as adsorbents to eliminate NO2 at low temperatures.
To illustrate the superior sensing capacity of Au/Ag functionalized arsenene, we compared the adsorption performance of the considered toxic gases on different 2D materials by summarizing the adsorption energies, as listed in Table 4. It is clear that the doping or adsorption of metal atoms (including Sc, Ti, Fe, Ag, Pt and Au) can enhance the adsorption energies of all toxic gas molecules considered in 2D materials, further confirming that the doping of metal atoms is an effective strategy for enhancing the adsorption performance of toxic gas molecules. Previous studies show CO, NO, NO2, NH3 or SO2 and H2S favor to physisorb on pristine 2D materials, including graphene, phosphorene, arsenene and MoS2.19,45–53 On the contrary, all the molecules chemisorb on metal-doped 2D materials owing to the significant electron hybridization between metal atoms and toxic gases. It is noteworthy that the adsorption energies for the toxic gases adsorbed on Sc-arsenene, Ti-arsenene and Pt-arsenene are higher than those for the corresponding gas adsorbed on Ag-arsenene and Au-arsenene. Such excessive adsorption can elongate the recovery time and cause it to lose its reusability. Therefore, Ag/Au doping on arsenene enhances the adsorption of most considered toxic gases with moderate adsorption energies (in the range of −0.60 to 0.80 eV), making it more favorable for application in gas sensing. Our results also confirmed that the scope of detectable gas could be extended when gas sensors based on Ag/Au-arsenene operate at higher temperatures.
Table 4 Comparison of the adsorption energies (eV) for CO, NO, NO2, SO2, H2S and NH3 molecules adsorbed on various 2-dimensional materials
| Literature/gases |
CO |
NO |
NO2 |
SO2 |
H2S |
NH3 |
| Ag-arsenene[this work] |
−0.606 |
−0.724 |
−1.120 |
−0.394 |
−0.638 |
−0.846 |
| Au-arsenene[this work] |
−0.517 |
−0.607 |
−0.923 |
−0.429 |
−0.391 |
−0.528 |
| Blue phosphorene19 |
−0.065 |
−0.216 |
−0.484 |
|
|
−0.131 |
| Graphene45 |
−0.01 |
−0.02 |
−0.06 |
|
|
−0.03 |
| Black-arsenene46 |
−0.14 |
−0.33 |
−0.34 |
|
|
−0.26 |
| MoS2 (ref. 47) |
|
|
|
−0.14 |
−0.048 |
|
| Ag-bismuthene48 |
−1.42 |
|
|
−3.48 |
−2.55 |
−3.45 |
| Fe-antimonene33 |
−2.35 |
−3.69 |
−2.19 |
−1.54 |
|
−0.24 |
| Au-stanene49 |
−1.22 |
−1.64 |
−2.32 |
−2.36 |
|
−1.45 |
| Au–MoS2 (ref. 50) |
−0.91 |
−1.08 |
|
|
|
|
| Sc-arsenene51 |
|
|
|
−1.71 |
|
|
| Pt-arsenene52 |
−1.12 |
−1.02 |
−1.72 |
−1.84 |
|
−1.06 |
| Ti-arsenene53 |
|
−1.77 |
−2.57 |
|
|
−1.36 |
4. Conclusions
Using density-functional theory (DFT) with Vdw correction, we investigated the adsorption behaviors, and electronic and magnetic properties of toxic molecules (including CO, NO, NO2, SO2, NH3 and H2S) on pristine and Ag/Au-doped arsenene systems. A transformation from semiconducting to the metallic characteristic of arsenene upon the introduction of the considered Ag/Au dopants was observed, which was due to the emergence of impurities states derived from the hybridization between Ag/Au dopants and surrounding As atoms. Generally, the introduction of metal dopants can increase the adsorption energies and interactions between gas and arsenene surfaces. For example, all considered toxic gases favor chemisorb on Ag/Au-arsenene with NO2 having the highest adsorption energies. According to the charge transfer analysis, we found that CO, NO, NO2 and SO2 gas molecules acting as accepter capture charge from arsenene through underlying metal dopants as an intermediary, while H2S and NH3 act as a weaker donor to donate charge to the substrates. The vibrational frequencies of molecules adsorbed on Ag/Au-arsenene are calculated and compared with the cases for molecules in a freestanding state. Our calculations revealed that the changes in the vibrational frequencies of NO, NO2 and SO2 are more significant compared with those of CO, NH3 and H2S, which is consistent with the relatively higher adsorption energies and larger charge transfers for the adsorption of NO, SO2 and NO2 on Ag/Au-arsenene compared with the cases of CO, NH3 and H2S. The introduction of Ag or Au could enhance the sensitivity of detecting such molecules compared with pristine arsenene owing to higher adsorption energies or charge transfer for Ag/Au-arsenene surfaces. Furthermore, the adsorptions of molecules modify the work functions of Ag/Au-arsenene to some extent. The increase in the work function follows the order of NO2 > SO2 > NO > CO > H2S > NH3 for Ag-arsenene, and NO2 > NO > SO2 > CO > H2S > NH3 for Au-arsenene, which are consistent with the order of charge transfer between molecules and Ag/Au-arsenene. It is expected that Ag/Au-doped arsenene possesses possible sensing applications for detecting toxic gas molecules with high selectivity because of the different work function responses to the different gas types adsorbed on Ag/Au-arsenene. Analysis of the recovery time revealed that toxic gas molecules could be detected by Ag/Au-arsenene at different working temperatures. The recovery times for CO, NO and H2S on Ag-arsenene and NO on Au-arsenene are short at room temperature (about 300 K), showing high reusability of Ag/Au-arsenene-based gas sensors when sensing these molecules. In addition, the recovery times of molecules on Ag/Au-arsenene are shortened rapidly with the increase in temperature (up to 500 K). The moderate recovery time for NH3 on Ag-arsenene (about 163 seconds) at 300 K can be reduced to 0.05 seconds simply by heating to 400 K. The recovery times for NO2 on Ag/Au-arsenene (extremely long at 300 K) decrease rapidly to 0.19 and 0.002 seconds at 500 K, respectively, indicating Ag/Au-arsenene-based gas sensors have potential applications in sensing NO2 from factory emission or automobile exhaust with quite good reusability. Consequently, the phenomena analyzed and described above indicate that Ag/Au-doped arsenene can achieve high sensitivity, selectivity and reusability to detect and monitor toxic gases at different operating temperatures.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the High Education Key Program of Henan Province of China (No. 192102210205 and 20A140022).
References
- X. Han and L. P. Naeher, Environ. Int., 2006, 32, 106–120 CrossRef CAS.
- K.-C. Hsu, T.-H. Fang, Y.-J. Hsiao and Z.-J. Li, J. Alloys Compd., 2021, 852, 157014 CrossRef CAS.
- J. Chakraborty, Ann. Am. Assoc. Geogr., 2009, 99, 674–697 CrossRef.
- C. Wang, X. Chu and M. Wu, Sens. Actuators, B, 2006, 113(1), 320–323 CrossRef CAS.
- P. Panigrahi, P. K. Panda, Y. Pal, H. Bae, H. Lee, R. Ahuja and T. Hussain, ACS Appl. Nano Mater., 2022, 5, 2984–2993 CrossRef CAS.
- A. Ernst and J. Zibrak, N. Engl. J. Med., 1998, 339, 1603–1608 CrossRef CAS PubMed.
- A. Ngoipala, T. Kaewmaraya, T. Hussain and A. Karton, Appl. Surf. Sci., 2021, 537, 147711 CrossRef CAS.
- I. Mochida, Y. Kawabuchi, S. Kawano, Y. Matsumura and M. Yoshikawa, Fuel, 1997, 76(6), 543–548 CrossRef CAS.
- J. Prasongkit, R. G. Amorim, S. Chakraborty, R. Ahuja, R. H. Scheicher and V. Amornkitbamrung, J. Phys. Chem. C, 2015, 119, 16934–16940 CrossRef CAS.
- W. Xia, W. Hu, Z. Li and J. Yang, Phys. Chem. Chem. Phys., 2014, 16, 22495 RSC.
- J. Ni, W. Wang, M. Quintana, F. Jia and S. Song, Appl. Surf. Sci., 2020, 514, 145911 CrossRef CAS.
- Z. Cui, K. Yang, Y. Shen, Z. Yuan, Y. Dong and E. Li, Appl. Surf. Sci., 2023, 613, 155978 CrossRef CAS.
- J. Zhao, X. Cui, Q. Huang and H. Zeng, Appl. Surf. Sci., 2023, 613, 156010 CrossRef CAS.
- A. Zhang, H. Yang, Q. Liu, W. Li and Y. Wang, Synth. Met., 2020, 266, 116441 CrossRef CAS.
- J. Mao and Y. Chen, J. Mater. Chem. C, 2020, 8, 4073 RSC.
- F. Ersan, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2016, 120, 14345–14355 CrossRef CAS.
- S. Batool, M. Idrees, S. -T. Han and Y. Zhou, Adv. Sci., 2023, 10, 2203956 CrossRef CAS.
- X. Jiang, G. Zhang, W. Yi, T. Yang and X. Liu, ACS Appl. Mater. Interfaces, 2023, 14, 35229–35236 CrossRef.
- F. Safaria, M. Moradinasabb, M. Fathipourb and H. Kosina, Appl. Surf. Sci., 2019, 464, 153–161 CrossRef.
- W. F. Pan, N. Qi, B. Zhao, S. Chang, S. Z. Ye and Z. Q. Chen, Phys. Chem. Chem. Phys., 2019, 21, 11455–11463 RSC.
- C. Kamal and M. Ezawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 85423 CrossRef.
- J. Du, C. Xia, T. Wang, X. Zhao, X. Tan and S. Wei, Appl. Surf. Sci., 2016, 378, 350–356 CrossRef CAS.
- C. Liu, C. Liu and X. Yan, Phys. Lett. A, 2017, 381, 1092–1096 CrossRef CAS.
- J. Zhu, H. Zhang, Y. Tong, L. Zhao, Y. Zhang, Y. Qiu and X. Lin, Appl. Surf. Sci., 2017, 419, 522–530 CrossRef CAS.
- Z. Xu, H. Cui and G. Zhang, ACS Omega, 2023, 8, 4244–4250 CrossRef CAS PubMed.
- W. Zhang, J. Tang, H. Yuan, K. Li, H. Liu, H. Zhu, T. Zhang and T. Jiang, ACS Appl. Nano Mater., 2023, 6, 4056–4066 CrossRef CAS.
- Y. Wang, Y. Zheng, J. Xiao, L. Xu, X. Dai and Z. Wang, Vacuum, 2023, 210, 111845 CrossRef CAS.
- Y. Tian, C. Wang and W. Gong, RSC Adv., 2023, 13, 2234 RSC.
- M. Isa Khan, M. Hassan, A. Majid, M. Shakil and M. Rafique, Appl. Surf. Sci., 2023, 616, 156520 CrossRef.
- D. Ma, W. Ju, T. Li, X. Zhang, C. He, B. Ma, Z. Lu and Z. Yang, Appl. Surf. Sci., 2016, 383, 98–105 CrossRef CAS.
- P. Wu and M. Huang, RSC Adv., 2013, 13, 3807 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef.
- S. Zhang, S. Guo, Z. Chen, Y. Wang, H. Gao, J. Gomez-Herrero, P. Ares, F. Zamora, Z. Zhu and H. Zeng, Chem. Soc. Rev., 2018, 47, 982–1021 RSC.
- P. Wu and M. Huang, Appl. Surf. Sci., 2020, 506, 144660 CrossRef CAS.
- V. Kumar, K. Rajput and D. R. Roy, Appl. Surf. Sci., 2022, 606, 154741 CrossRef CAS.
- S. Yang, C. Jiang and S.-H. Wei, Appl. Phys. Rev., 2017, 4(2), 021304 Search PubMed.
- Y.-H. Zhang, Y.-B. Chen, K.-G. Zhou, C.-H. Liu, J. Zeng, H.-L. Zhang and Y. Peng, Nanotech, 2009, 20, 185504 CrossRef PubMed.
- T. Kaewmaraya, L. Ngamwongwan, P. Moontragoon, W. Jarenboon, D. Singh, R. Ahuja, A. Karton and T. Hussain, J. Hazard. Mater., 2020, 401, 123340 CrossRef PubMed.
- X. Wang, G. Fan, X. Tu, X. Liu, C. Li and H. Xu, Appl. Surf. Sci., 2021, 552, 149449 CrossRef CAS.
- V. Kumar, K. Rajput and D. R. Roy, Appl. Surf. Sci., 2020, 534, 147609 CrossRef CAS.
- O. Leenaerts, B. Partoens and F. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77(12), 125416 CrossRef.
- J. Mao and Y. Chen, J. Mater. Chem. C, 2020, 8, 4073 RSC.
- M. Huang, A. Dinesh and S. Wu, Phys. Chem. Chem. Phys., 2021, 23, 15364 RSC.
- M. Isa, I. Ashfaq, A. Majid, M. Shakil and T. Iqbal, Mater. Sci. Semicond. Process., 2022, 145, 106635 CrossRef CAS.
- A. Zhang, H. Yang, Q. Liu, W. Li and Y. Wang, Synth. Met., 2020, 266, 116441 CrossRef CAS.
- D. Ma, W. Ju, T. Li, X. Zhang, C. He, B. Ma, Z. Lu and Z. Yang, Appl. Surf. Sci., 2016, 383, 98–105 CrossRef CAS.
- X. Wang, G. Fan, X. Tu, X. Liu, C. Li and H. Xu, Appl. Surf. Sci., 2021, 552, 149449 CrossRef CAS.
- J. Liu, C. Liu and J. Yu, Comput. Theor. Chem., 2020, 1190, 112977 CrossRef CAS.
- Y. Wang, Y. Zheng, J. Xiao, L. Xu, X. Dai and Z. Wang, Vacuum, 2023, 210, 111845 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b