
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric Michael addition catalysed by copper–amyloid complexes†
Nobutaka Fujieda *a,
Atsushi Tonomurab,
Tomofumi Mochizukic and
Shinobu Itoh*b
*a,
Atsushi Tonomurab,
Tomofumi Mochizukic and
Shinobu Itoh*b
aDepartment of Applied Biological Chemistry, Graduate School of Agriculture, Osaka Metropolitan University, 1-1 Gakuen-cho, Naka-ku, Sakai-shi, Osaka 599-8531, Japan. E-mail: fujieda@omu.ac.jp
bDepartment of Molecular Chemistry, Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
cDepartment of Agricultural Biology, Graduate School of Agriculture, Osaka Metropolitan University, 1-1 Gakuen-cho, Naka-ku, Sakai-shi, Osaka 599-8531, Japan
First published on 2nd January 2024
Abstract
We developed self-assembled peptides containing a partial amyloid β protein sequence and a metal-coordination site. The amyloid fibril–copper complexes exhibited excellent reactivity and moderate enantioselectivity in Michael addition reactions with 2-azachalcone and dimethylmalonate. The catalytic amyloids were characterized using various measurements to confirm their amyloid-like nanofibre structures.
Amyloid fibrils are insoluble fibrous aggregates formed from certain proteins.1 These complexes are historically found in the brains of patients with neurodegenerative diseases and have been deemed partially responsible for these conditions. The formation of amyloid fibrils has been ascribed to protein misfolding;2 however, these fibrils have stable and highly organised deposit cross-β structures,3 and their highly repetitive and ordered architecture, particularly evident in short peptide fibrils, exhibits favourable properties. These include high thermal stability and stiffness, biocompatibility, controllable self-assembly, surface patterning and functional integration, and inexpensive production through chemical synthesis.4,5 Amyloid fibrils functioning as competent catalytic scaffolds are called catalytic amyloids.6–8 Unlike other artificial supramolecular compounds, catalytic amyloid activities can be controlled by the amino acid side chains that construct the self-assembled peptides, which is similar to the case with enzymes.9 It has been reported that metal complexes as well as metal ions can bind to amyloid-β-peptides, whose usefulness has been thoroughly investigated as potential therapeutics in neurodegenerative diseases.10
Synthesized peptides containing core sequences of natural amyloid proteins have been reported to spontaneously assemble into amyloid-like fibrils in vitro.4,11–13 In particular, the core sequence of Aβ (Aβ:16–21 KLVFFA) can self-assemble into amyloid-like fibrils with antiparallel β-sheet structures.14–16 Recently, Das et al. developed catalytic amyloids by introducing functional groups into the edges of the core peptide.17–19 Additionally, based on de novo synthetic peptides, catalytic amyloids containing metal ions as cofactors have been used to mimic enzymes in several hydrolysis and oxidation reactions. Korendovych et al. showed that an amyloid-forming de novo peptide (Ac-IHIHIQI-NH2) can coordinate with metal ions, such as Co2+, Cu2+, or Zn2, after which the Zn2+-loaded amyloid fibrils catalyse acyl ester hydrolysis20 whereas the Cu2+-loaded structures activate oxygen molecules to catalyse the oxidation of dimethoxyphenol.21 However, there are no reports of catalytic amyloids with metal cofactors catalysing enantioselective reactions, other than the study involving the hemin–amyloid complex.22
In this study, we attempted to construct new catalytic scaffolds based on amyloid fibrils to create versatile coordination spheres for stereoselective reactions, focusing on the core sequences of Aβ (KLVFFA). As the N- and C-terminal peptide residues are alternately arranged in this folding pattern, they could be employed as metal-binding sites and functional groups, respectively (Fig. 1A and B). Consequently, altering the N- and C-terminal residues could allow versatile coordination spheres to be readily constructed (Fig. 1C). The amino acid residues at the N- and C-terminals are displayed as two functional groups on the surface of the amyloid fibrils as the catalytic centre. The C-terminal residue and the side chain can influence the substrate through various effects and interactions, such as steric effects, π–π stacking interactions, hydrogen bonds, and electrostatic interactions.
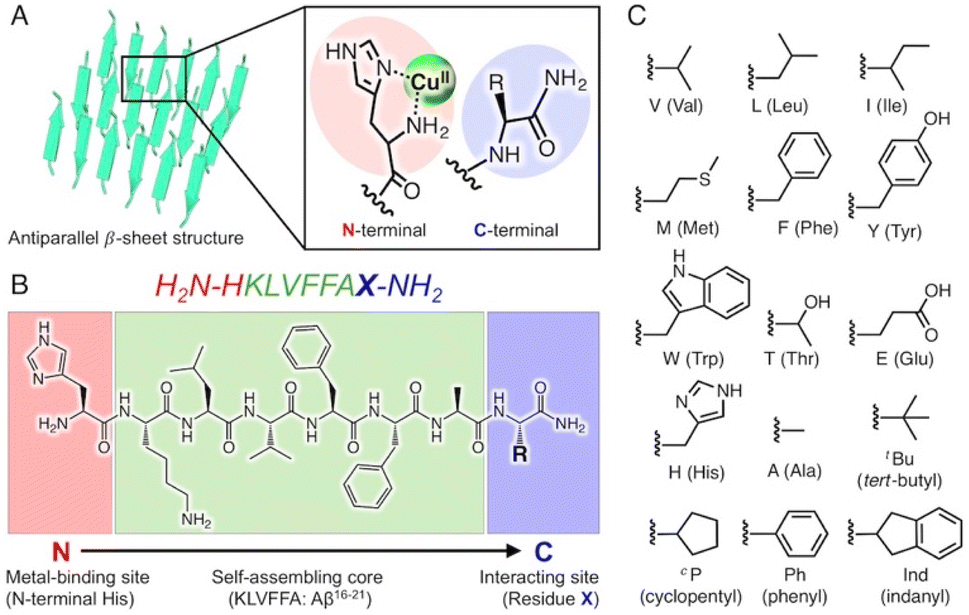 | ||
| Fig. 1 (A) Schematic representation of an amyloid fibril–copper complex. The fibril structures are derived from the X-ray crystal structure of an amyloid-forming peptide (PDB: 3OW9). (B) Peptide sequence of self-assembled peptides developed in this study consisting of three parts (metal-binding site, self-assembling core, and interacting site). (C) Chemical structures of side chain R of amino acid X, which are introduced in the C-terminal position. | ||
At the N-terminus, a histidine residue is introduced as the metal-binding residue. By deprotecting the N-terminal amino group during peptide synthesis, the N-terminal amino group and the imidazole group of the N-terminal histidine can function as metal ligands, similar to the ‘histidine brace’ found in enzymes such as lytic polysaccharide monooxygenase23 and particulate methane monooxygenase.24 Moreover, amino acid residues possessing various side chains are placed at the C-terminus as substrate-interacting sites (Fig. 1B and C).
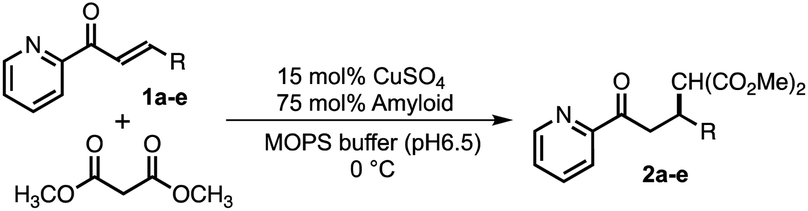
Based on the aforementioned strategies, 12 self-assembling peptides (P01–P12) with different C-terminal residues were synthesized (Table 1 and Fig. 1C). The catalytic activities of the copper–amyloid complexes were investigated in terms of the asymmetric Michael additions between 2-azachalcone 1a and dimethyl malonate (Table 1). The reaction proceeded slowly to result in a low yield of a racemic mixture of products, even when using free Cu2+ (Table 1, entry 1). Although the free amino acid L-His and its derivatives showed poor enantioselectivity (Table 1, entries 2 and 3), the copper–amyloid complexes provided moderate-to-good ee values and good-to-excellent yields under optimized conditions (temperature and amyloid amount, Table S1†). The amino acid residues introduced into the C-terminus of the peptides likely contributed significantly to these ee values. Furthermore, aliphatic residues (Val, Leu, Ile, and Met) exhibited higher enantioselectivity and better yields than those of L-His alone (Table 1 entries 4–7), suggesting that steric repulsion could contribute to selectivity. Similarly, the bulky aromatic amino acids Tyr and Trp showed relatively high enantioselectivities with lower yields, whereas Phe produced a racemic product (Table 1, entries 8–10). The hydrophilic residues Thr, Glu, and His exhibited lower enantioselectivities (Table 1, entries 11–13). In this context, C-terminal Gly generally did not cause enantioselectivity, whereas Ala produced enantioselectivity with a good yield (entries 14 and 15), demonstrating that a certain sized amino acid residue is required for both reactivity and stereoselectivity. The importance of the N-terminal residue as a metal-coordination moiety was then proven using ligand moiety-lacking variants (P13 and P14), in which His was replaced with Ala or the N-terminal amine was acetylated, respectively. The mixture of self-assemblies of the peptides and CuSO4 showed similar yields but lower ee values than those of the peptide P01 (Table 1, entry 4), indicating that the N-terminal His residue acts as a coordination moiety for binding the metal ion and is responsible for enantioselectivity (Table 1, entries 16 and 17).
|
|||
|---|---|---|---|
| Entry | Ligand | Yield (%) | ee (%) |
| a Reaction conditions: 475 μM amyloid-like fibrils and 95 μM CuSO4 in 20 mM MOPS buffer (pH 6.5) for 1 day at 0 °C. The yields and ee values are the average from 3 experiments. The absolute configuration of product 2 is determined based on a previous report.25 | |||
| 1 | No peptide | 26 | n.d. |
| 2 | L-His-OH | 61 | 16 (S) |
| 3 | L-His-OCH3 | 84 | n.d. |
| 4 | (P01) H2N-HKLVFFAV-NH2 | 78 | 38 (S) |
| 5 | (P02) H2N-HKLVFFAL-NH2 | 64 | 22 (S) |
| 6 | (P03) H2N-HKLVFFAI-NH2 | 92 | 37 (S) |
| 7 | (P04) H2N-HKLVFFAM-NH2 | 78 | 19 (S) |
| 8 | (P05) H2N-HKLVFFAF-NH2 | 79 | 15 (S) |
| 9 | (P06) H2N-HKLVFFAY-NH2 | 39 | 43 (S) |
| 10 | (P07) H2N-HKLVFFAW-NH2 | 54 | 33 (S) |
| 11 | (P08) H2N-HKLVFFAT-NH2 | 54 | 10 (S) |
| 12 | (P09) H2N-HKLVFFAE-NH2 | 78 | 13 (S) |
| 13 | (P10) H2N-HKLVFFAH-NH2 | 44 | 14 (S) |
| 14 | (P11) H2N-HKLVFFAA-NH2 | 78 | 21 (S) |
| 15 | (P12) H2N-HKLVFFAG-NH2 | 46 | 15 (S) |
| 16 | (P13) H2N-AKLVFFAV-NH2 | 78 | 15 (S) |
| 17 | (P14) Ac-AKLVFFAV-NH2 | 67 | n.d. |
| 18 | (P15) H2N-DHDKDLDVDFDFDADV-NH2 | 92 | 38 (R) |
| 19 | (P16) H2N-HKLVFFAtBu-NH2 | 85 | 47 (S) |
| 20 | (P17) H2N-HKLVFFAcP-NH2 | 54 | 12 (S) |
| 21 | (P18) H2N-HKLVFFAPh-NH2 | 79 | 24 (S) |
| 22 | (P19) H2N-HKLVFFAInd-NH2 | 63 | 34 (S) |
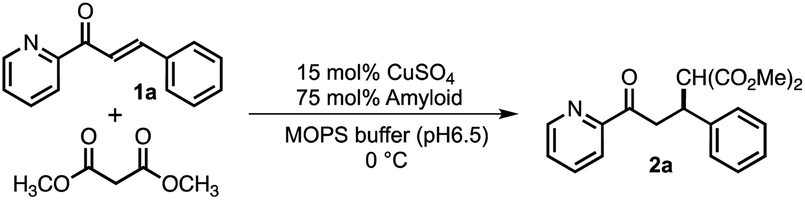
To understand the details of the relative reactivity and enantioselectivity of the copper–amyloid complexes, we examined the aggregation behaviour of P01, which showed good ee values. First, self-assembled P01 showed fluorescence enhancement in the presence of Thioflavin T, which is often used to verify the presence of amyloid-like β-sheet structures (Fig. S1†), suggesting that the newly designed peptides self-assembled to form amyloid-like fibrils. Circular dichroism (CD) spectroscopy was used to comprehensively evaluate the β-sheet structure of the peptide self-assemblies (Fig. 2A). Secondary structure contents were then analysed based on the specific spectrum of the self-assembly of peptide P01. The percentage of secondary structure content was determined to be 46% β-sheets in the peptide assembly (Table S2†). This value is similar to that previously reported for peptide nanofibres.5 Moreover, Fourier transform infrared (FT-IR) spectroscopy showed that the self-assembly of peptide P01 generated two specific peaks at 1626 and 1694 cm−1 (Fig. 2B), which are assigned to the amide I band and consistent with the vibrations of antiparallel β-sheet structures.26,27 To further confirm the fibril structures, the morphologies of the peptide self-assemblies were examined using transmission electron microscopy (TEM), whereby stained complexes showed fibrous structures with lengths of 1–5 μm and widths of 50–200 nm (Fig. 2C). This result indicated that the amyloid-like fibrils likely underwent the Michael addition reaction described, and the catalytic activities of the copper–amyloid complexes could be attributed to the properties of the side chains of the terminal residues.
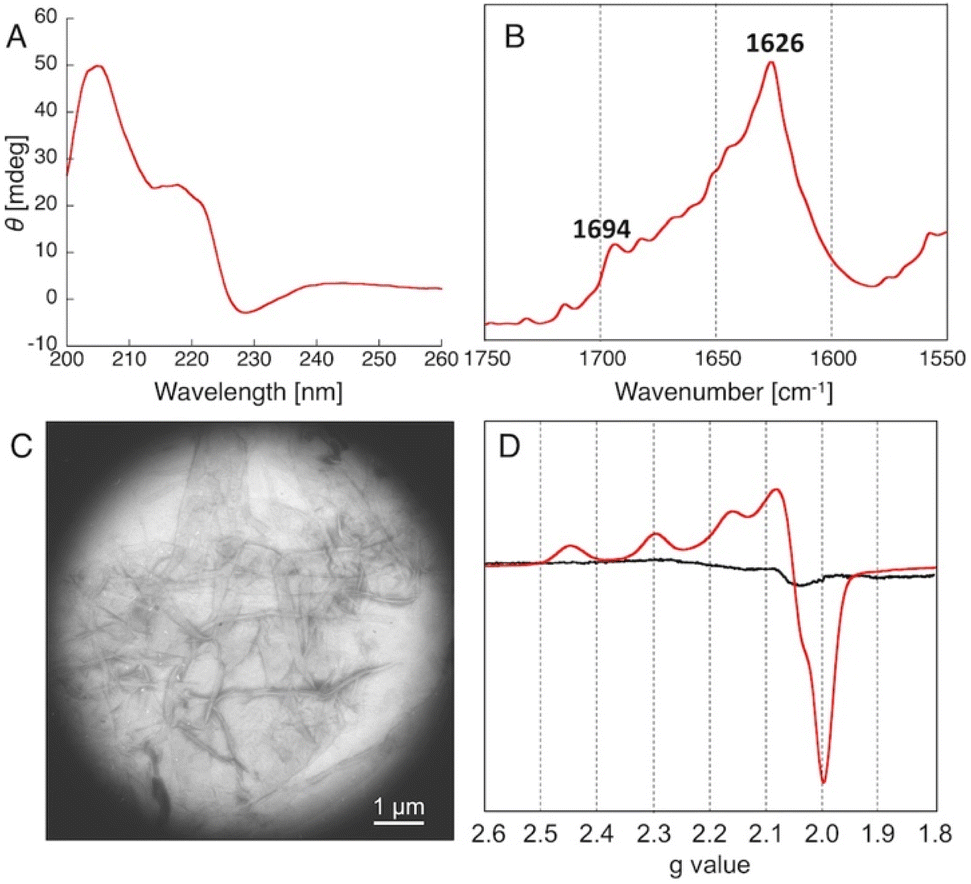 | ||
| Fig. 2 Characterization of the self-assembly of peptide P01. (A) CD spectrum, (B) FT-IR spectrum, (C) TEM image, (D) X-band ESR spectrum of Cu2+ in the absence (black) and presence (red) of the self-assembly. | ||
We then conducted ESR measurements to determine the Cu2+ coordination environments of the copper–amyloid complexes. Upon the addition of CuSO4, the peptide P01 self-assembly exhibited typical axial signals due to a single tetragonal copper ion, while CuSO4 did not show any characteristic signals in the absence of the amyloid-like fibrils. The g∥, g⊥, and A∥ parameters were determined to be 2.23, 2.06, and 188, respectively, based on the spectrum, assuming that the copper–amyloid complex had a single tetragonal coordination structure. The other peptide self-assemblies were structurally analysed as having the same behaviour as that of peptide P01, confirming their capability to form amyloid fibrils (Table S2 and Fig. S1–S4†).
To determine the stereochemical mechanism responsible for enantioselectivity, we manually constructed a simple molecular model of a copper–amyloid complex. The reported X-ray crystal structure of the KLVFFA peptide (PDB entry: 3OW9) was used as a starter model, which was extended by adding histidine and valine residues to the N- and C-terminals, respectively, while retaining the β-strand structure in silico (Fig. 3). As the N-terminal histidine residue acts as a bidentate metal ligand, 2-azachalcone 1a can coordinate with Cu2+ to complete the square planar geometry. The pyridine moiety of 1a appears to bind to copper from a position trans to the amino and imidazole groups to form two coordination structures: mode A (Fig. 3, yellow) and mode B (Fig. 3, pink). In mode A, 1a is inserted into the small cavity formed by the self-assembled peptides, and the unsaturated β-carbon atom is sterically hindered by the C-terminal Val8 residue to prevent easy access by the dimethyl malonate from the Re-face (Fig. 3D). Furthermore, Val8 may interact with the phenyl moiety of 1a via the CH–π interaction, which governs the binding mode. In mode B, the β-carbon atom is distant from the surface of the copper–amyloid complex, and the reaction is allowed to proceed with little interference from the C-terminal Val8 residue (Fig. 3E). This model could lead to the preferential generation of an (S)-form product by the copper–amyloid complex of P01.
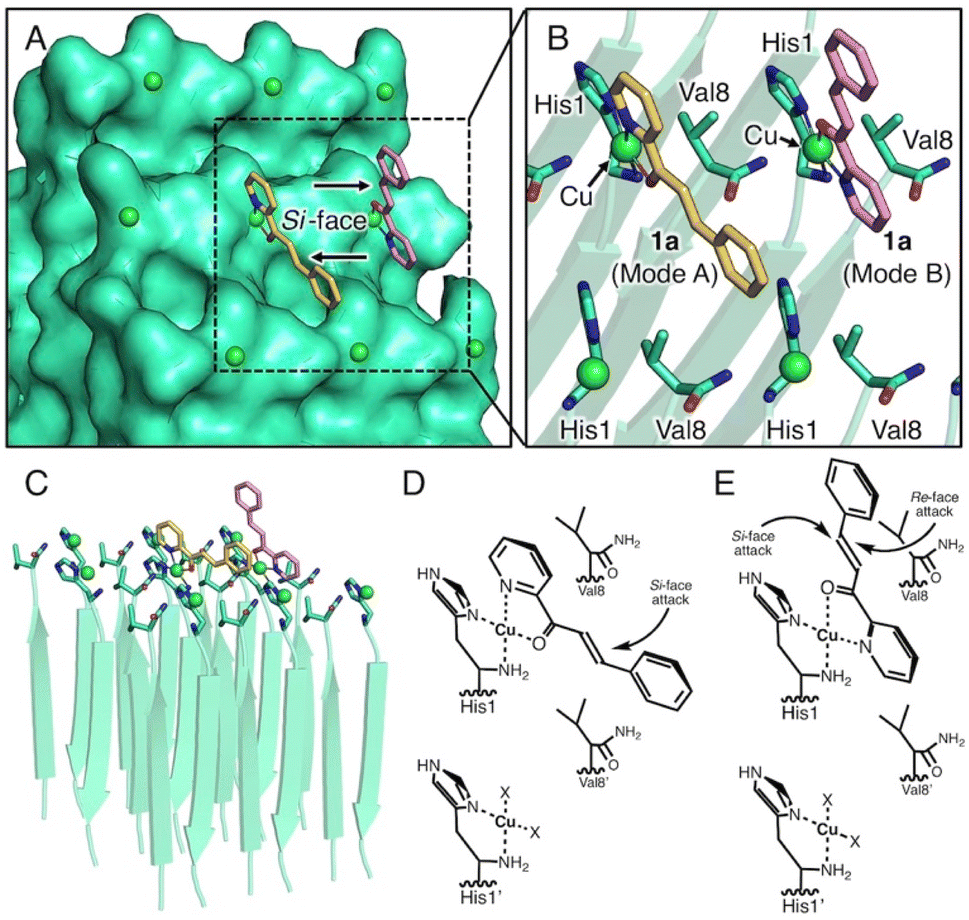 | ||
| Fig. 3 Simple molecular model of self-assembly of the peptide P01. (A) Top view of the surface model, (B) top view of the ribbon model, (C) side view of the ribbon model, (D and E) plausible schematic representation of the ternary complex of amyloid, Cu, and 2-azachalcone, 1 in binding modes A (D) and B (E). X indicates a water molecule. | ||
With intent to explore the catalytic performance of such copper–amyloid complexes (P01), Michael addition was examined using azachalcone derivatives 1b–e as the substrates (Table 2). As compared to the substrate with the electron-donating group (1b, Table 2, entry 2), the substrates having electron-withdrawing group (1c and 1d, Table 2, entries 3 and 4) appeared to be negative effect on the product yields. In the case of naphthyl group (1e, Table 2, entry 5), steric effect is likely to govern the reaction rather than electronic effect. Nevertheless, the ee values retained among these substrates, promising the asymmetric pocket of copper–amyloid complexes.
|
||||
|---|---|---|---|---|
| Entry | R | 2 | Yield (%) | ee (%) |
| a Reaction conditions: 475 μM amyloid-like fibrils and 95 μM CuSO4 in 20 mM MOPS buffer (pH 6.5) for 1 day at 0 °C. The yields and ee values are the average from 3 experiments. | ||||
| 1 | Ph | 2a | 78 | 38 |
| 2 | 4-CH3-C6H5 | 2b | 75 | 31 |
| 3 | 4-NO2-C6H5 | 2c | 29 | 29 |
| 4 | 4-Br-C6H5 | 2d | 26 | 31 |
| 5 | 2-Naphthyl | 2e | 41 | 37 |
To further fine-tune the enantioselectivity of copper–amyloid complexes, we synthesized non-natural amino acid-bearing self-assembling peptides. To explore the potential of catalytic amyloids, we attempted to reverse the enantioselectivity of this reaction, as the (S)-product was preferentially obtained using self-assembled peptides with only L-amino acids. We focused on D-amino acids, which are amino acid enantiomers of L-amino acids that are generally incorporated into proteins. Two enantiomers can traditionally be obtained with enantioselectivities using two opposite chiral sources;28 therefore, peptide self-assemblies containing D-amino acids were prepared, which showed the inverted enantioselectivity preferring (S)-form (Table 1, P15, entry 18).
To obtain higher ee values, we attempted to incorporate non-natural amino acids by screening the C-terminal residues. Val and Ile showed very good yields and higher ee values; therefore, the C-terminal residues were changed to non-natural amino acids containing t-butyl, cyclopentyl, phenyl, and indanyl groups, which have β-position carbons that are sterically crowded (Fig. 1C and Table 1, entries 19–22). Consequently, non-natural amino acids bearing t-butyl moieties that have quaternary carbon atoms at the β-position produced good yields and higher ee values compared to those of the other peptides screened (Table 1, entries 19–22). This suggests that the C-terminal residue plays a significant role in determining the enantioselectivity of asymmetric Michael addition.
Conclusions
Amyloid fibrils have potential for use in various applications as novel soft bionanomaterials owing to their thermal stability and highly ordered structure. However, few examples have been reported regarding their use as metal–amyloid hybrids for catalysis, despite the flexibility and diversity of their molecular design. In this report, we described novel copper–amyloid complexes that act as asymmetric catalysts in the Michael addition reaction between 2-azachalcone and dimethyl malonate to generate (S)-form products. Furthermore, (R)-form products were obtained enantioselectively using peptides containing D-amino acids as the metal ligand. Moreover, in this peptide design, self-assembled core sequence residues were placed at the centre of the peptide sequence to allow for the addition of various amino acid residues at the N- and C-terminal positions, thereby leading to diverse primary and secondary coordination spheres. These design principles can be applied to create novel heterogeneous catalysts that combine a controlled coordination sphere with high thermal stability.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
N. F. would like to thank JSPS and MEXT, Japan (JSPS KAKENHI Grant Number JP21H01954 and MEXT KAKENHI Grant Numbers JP15H01066 and 17H05233 in Hadean Bioscience and JP16H01025 and 18H04270 in Precisely Designed Catalysts with Customized Scaffolding). S. I. would like to thank JST (CREST Program, 161052502).Notes and references
- G. B. Irvine, O. M. El-Agnaf, G. M. Shankar and D. M. Walsh, Mol. Med., 2008, 14, 451–464 CAS.
- S. T. Ferreira, M. N. N. Vieira and F. G. De Felice, IUBMB Life, 2007, 59, 332–345 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- D. Li, E. M. Jones, M. R. Sawaya, H. Furukawa, F. Luo, M. Ivanova, S. A. Sievers, W. Wang, O. M. Yaghi, C. Liu and D. S. Eisenberg, J. Am. Chem. Soc., 2014, 136, 18044–18051 CrossRef CAS PubMed.
- B. Dai, D. Li, W. Xi, F. Luo, X. Zhang, M. Zou, M. Cao, J. Hu, W. Wang, G. Wei, Y. Zhang and C. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2996–3001 CrossRef CAS PubMed.
- L. R. Marshall and I. V. Korendovych, Curr. Opin. Chem. Biol., 2021, 64, 145–153 CrossRef CAS PubMed.
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 3047–3070 RSC.
- Q. Liu, A. Kuzuya and Z.-G. Wang, iScience, 2023, 26, 105831 CrossRef CAS PubMed.
- C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. G. Pappas, V. Narang, T. Wang, C. Maldarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS PubMed.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412, 213255 CrossRef CAS.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- M. I. Ivanova, S. A. Sievers, M. R. Sawaya, J. S. Wall and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18990–18995 CrossRef CAS PubMed.
- J.-P. Colletier, A. Laganowsky, M. Landau, M. Zhao, A. B. Soriaga, L. Goldschmidt, D. Flot, D. Cascio, M. R. Sawaya and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16938–16943 CrossRef CAS PubMed.
- L. O. Tjernberg, D. J. E. Callaway, A. Tjernberg, S. Hahne, C. Lilliehöök, L. Terenius, J. Thyberg and C. Nordstedt, J. Biol. Chem., 1999, 274, 12619–12625 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- Z. Bu, Y. Shi, D. J. E. Callaway and R. Tycko, Biophys. J., 2007, 92, 594–602 CrossRef CAS PubMed.
- B. Sarkhel, A. Chatterjee and D. Das, J. Am. Chem. Soc., 2020, 142, 4098–4103 CrossRef CAS PubMed.
- C. Mahato, S. Menon, A. Singh, S. P. Afrose, J. Mondal and D. Das, Chem. Sci., 2022, 13, 9225–9231 RSC.
- S. Roy, A. Chatterjee, S. Bal and D. Das, Angew. Chem., Int. Ed., 2022, 61, e202210972 CrossRef CAS PubMed.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- O. V. Makhlynets, P. M. Gosavi and I. V. Korendovych, Angew. Chem., Int. Ed., 2016, 55, 9017–9020 CrossRef CAS PubMed.
- O. Zozulia and I. V. Korendovych, Angew. Chem., Int. Ed., 2020, 59, 8108–8112 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton, K. S. Johansen, K. O. Hodgson, B. Hedman and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8797–8802 CrossRef CAS PubMed.
- R. A. Himes, K. Barnese and K. D. Karlin, Angew. Chem., Int. Ed., 2010, 49, 6714–6716 CrossRef CAS PubMed.
- R. Matsumoto, S. Yoshioka, M. Yuasa, Y. Morita, G. Kurisu and N. Fujieda, Chem. Sci., 2023, 14, 3932–3937 RSC.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- N. Yamada, K. Ariga, M. Naito, K. Matsubara and E. Koyama, J. Am. Chem. Soc., 1998, 120, 12192–12199 CrossRef CAS.
- N. Molleti, N. K. Rana and V. K. Singh, Org. Lett., 2012, 14, 4322–4325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07900g |
| This journal is © The Royal Society of Chemistry 2024 |