
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBimetallic metal organic framework/Ni doped ZnO nanomaterials modified carbon paste electrode for selective electrochemical determination of ciprofloxacin†
Susan Sadeghi * and
Javad Javanshiri-Ghasemabadi
* and
Javad Javanshiri-Ghasemabadi
Department of Chemistry, University of Birjand, P. O. Box 97175-615, Birjand, Iran. E-mail: ssadeghi@birjand.ac.ir
First published on 5th March 2024
Abstract
In this work, an efficient and sensitive electrochemical sensor for the determination of ciprofloxacin (CIP) is reported. The sensor was prepared by using a carbon paste electrode (CPE) modified with a combination of bimetallic copper/cerium-based metal organic framework (Cu/Ce-MOF) and nickel doped zinc oxide nanoparticles (NZP). The modifiers were characterized by Brunauer–Emmett–Teller (BET) analysis, Fourier-transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX), and elemental mapping analysis (EDS). The electrochemical behavior of the modified electrode was studied by electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). The developed electrode was employed for the detection of CIP by differential pulse voltammetry (DPV). Under optimal conditions, the anodic peak current response of the electrode was linearly correlated with CIP concentration in the range of 0.75–100 μmol L−1 with a sensitivity of 1.29 μA μmol−1 L−1. The limit of detection and reproducibility of the method were 0.142 μmol L−1 and 2.7%, respectively. The developed sensor showed good selectivity to CIP against possible interferents. The method was applied to determine CIP in water, milk and urine samples. The results indicated that this method has potential to be applied in the analysis of residue CIP in complex matrices with high selectivity, and good reproducibility.
1. Introduction
Along with the rapid growth of the world's population, the demand for the use of antibiotics has increased significantly. As a result, soil, surface water, and underground water are severely affected by residues of antibiotics in human waste disposal, hospital and domestic wastewater effluents.1 Even trace amounts of antibiotics in the environment harm the ecosystem and the health of animals and humans, including interrupting photosynthetic cycles and biological functions of plants and increasing resistance to antibiotics. This global threatening problem led the United Nations to declare in 2017 that the discharge of antibiotics into environmental waters may host wild microorganisms that are completely resistant to the drugs.1 Among broad-spectrum antibiotics, fluoroquinolones with a quinolone ring structure show more activity against Gram-positive and Gram-negative aerobic pathogens, due to the substitution of the fluorine atom at the C-6 position.2 Ciprofloxacin (CIP) is a second-generation fluoroquinolone and the most effective antimicrobial and anti-inflammatory antibiotic widely used in veterinary medicine and the treatment of various bacterial infections in humans. CIP affects bacterial growth and reproduction by inhibiting the function of an essential enzyme that maintains superhelical turns in bacterial DNA.3 Due to the poor metabolizing process and widespread use of CIP, its high concentration in municipal and hospital wastewater has been reported between 2.5 and 245 μg L−1.4 It has also been detected at higher concentrations of 4.9 to 50 mg L−1 in freshwater near pharmaceutical factories. Also, CIP has been found in cow's milk at concentrations of 0.1 to 100 ng mL−1.3,5 CIP shows low biodegradability and high solubility in water at different pH values.6 Considering the residue of CIP in various media such as water and food samples, it is necessary to develop new strategies or methods that are simple, accurate, highly selective, and sensitive for its monitoring. So far, several analytical techniques have been employed to determine CIP in different matrices including liquid chromatography-mass spectrometry,7 high-performance liquid chromatography,8 spectrophotometry,9 spectrofluorometry,10 capillary electrophoresis,11 immunoassay,12 and chemiluminescence,13 photoelectrochemical detection,14 and electrochemical techniques.15–17 Although these techniques are reliable and sensitive, most of them require several time-consuming treatment steps for sample preparation, expensive instruments, or an expert operator. Among the above techniques, electrochemical techniques are characterized by the inherent advantages of high sensitivity and selectivity, ease of use, fast response, easy miniaturization, cost-effective analysis, and enable real-time monitoring of the target analyte.18 Various working electrodes are based on boron-doped diamond,4,19 carbon paste electrode,20 glassy carbon electrode,21 pencil graphite electrode,22 screen printed electrodes,23,24 and etc. have been used to determine different classes of antibiotics. In recent years, carbon paste electrodes have been widely used for the monitoring of fluoroquinolones due to their low cost, wide potential window, rapid fabrication and regeneration of the electrode surface. Modification of the electrode surface with different electroactive materials increases electroactive sites for accumulation of analyte on the electrode surface and facilitates electron transfer. Up to now, various materials including surfactants, polymer layers, inorganic, organic and metal–organic framework materials, biomaterials, metal and semiconductor nanoparticles, and nanocomposites have been used to improve the electrochemical response of electrodes to the target analyte.25,26 An ideal electrocatalyst should generally possess characteristics such as reasonable cost, high conductivity for charge transfer, and stability at high potentials.27 Recently, organic non-precious metals such as transition metal nitrides, carbides, and phosphides have been considered as electrocatalysts, because of their variable oxidation states, presence of 3d electrons, stability, and morphological properties. However, low conductivity, aggregation and instability during electrocatalytic process are drawbacks of these materials.28 Graphitic carbon nitrides, carbon nanotubes, or carbon-based nanocomposites have been considered as metal-free catalysts as alternatives to conventional nobel metal catalysts owing to their high surface area, low cost, robustness, and tunable structures.29 Nonetheless, hybrid systems of inorganic and organic nanomaterials through covalent or non-covalent bonding have opened a new perspective to electrocatalysts. These combinations modify the morphology, improve the efficiency of electrocatalysts, and enhance the accessible catalytic active sites.30,31Metal–organic frameworks (MOFs) consisting of a metal ion that connected with an organic linker represent a very important class of hybrid materials. Compared with other inorganic porous materials, MOFs can provide a sensing platform for electrocatalysis owing to their properties such as intrinsic porosity, tunable pore sizes, and high surface area.32 Howbeit, most MOFs have relatively low stability in aqueous solutions and poor electrical conductivity due to the reversible nature of the coordination bonds.33 Several strategies have been developed to overcome these drawbacks. One common approach is to combine MOFs with other highly conductive functional materials such as carbon or metal oxide nanomaterials.34 On the other hand, to improve high stability, metals with high oxidation states are used to form strong coordination bonds with the linker.
Recently, bimetallic MOFs have received much attention in various fields.35 The incorporation of a second metal with the similar electronic property of the metal in the framework may affect the electronic property of the bimetallic MOF.36 Nonetheless, most of the metal sites in the bulk bimetallic MOF are not accessible to the electrolyte and buried within the framework. To overcome this issue, several protocols have been suggested, including increasing the surface area with increasing porous structure, selection of adequate bimetallic centers, and appropriate MOF substrate.37 Among the synthesized MOFs, cerium-based metal–organic frameworks (Ce-MOFs) have attracted attention due to their unique optical and catalytic properties including low energy 4f orbitals, low cost, remarkable thermal and chemical stability, redox activity, and permanent porosity.38 Although Ce-MOF materials are promising electrocatalysts, but due to poor conductivity, the catalytic activity of Ce-MOF needs further improvement.39 Copper benzene-1,3,5-tricarboxylate MOF has been one of the most studied MOFs because of its large surface area, high pore volume, high chemical stability, and also the ability to bind small molecules.40 It has proven that MOFs with different metal centers exhibit higher conductivity compared to the analogous structures and provide excellent electrochemical activity due to the synergistic electronic effect of the redox sites.41 In addition, metal oxide/bimetal-MOF composites can synergistically enhance activation with higher catalytic performance.42
Herein, we prepared a bimetallic MOF (Cu/Ce MOF) with benzene-1,3,5-tricarboxylic acid (H3BTC) as an organic linker through a facile precipitation approach. Nickel doped zinc oxide nanoparticles (NZP) are non-toxic and facilitate electron transfer.43 MOF is a promising candidate for supporting material in the fabrication of the electrochemical sensors. Then, a combination of Cu/Ce-MOF and nickel doped zinc oxide nanoparticles (NZP) was incorporated into carbon paste to fabricate modified carbon paste electrode for the detection of ciprofloxacin. The synergistic effect of Cu/Ce-MOF and NZP helped improve the ability of the sensor for the selective determination of ciprofloxacin under optimal conditions. The resulting material showed a large specific area, and enhanced electrocatalytic activity. The prepared electrode was characterized and the parameters affecting the response of the developed sensor were investigated and discussed. The electrochemical behavior of the sensor for CIP detection is reported in terms of detection limit and dynamic range. The fabricated electrode was applied for the detection of CIP in water, milk and urine samples with satisfactory results.
2. Materials and methods
2.1 Reagents and materials
Ciprofloxacin was purchased from Darupaksh pharmaceutical company (Karaj, Iran). Graphite powder was obtained from Fluka. Nitrate salts of zinc, nickel, cerium and copper metal ions, as well as 1,3,5-benzenetricarboxylic acid (H3BTC), citric acid, NH3, sodium phosphate mono and dibasic salts, and ethanol were all prepared from Merck company (Darmstadt, Germany). All solutions were prepared with ultrapure water provided by AquaMaX water production system (Young Lin, South Korea).2.2 Apparatus
All electrochemical experiments were performed on an AutoLab potenstiostate PGS302N (Metrohm, Utrecht, Netherlands) equipped with a three-electrode configuration cell in which a Cu/Ce-MOF modified carbon paste electrode, Ag/AgCl (3 M), and Pt rod were used as the working, reference, and counter electrodes, respectively. EIS measurements were carried out in the AutoLab potentiostate. The powder X-ray diffraction (XRD) pattern was obtained by Phillips Xpert X-ray diffractometer with copper X-ray source. Morphology, mapping, and elemental analysis of the synthesized nanomaterials were studied by field emission-scanning electron microscopy (FE-SEM, MRA3 TESCAN, Czech) with an accelerating voltage of 15 kV. Functional groups of the synthesized nanomaterials were studied by recording the absorption spectra in wavenumber range of 400–4000 cm−1 using a AVATAR 370 Fourier transform infrared spectrophotometer (FTIR, Thermo Nicolet, USA). The surface area and pore size of the synthesized MOF modifier were calculated from N2 gas adsorption/desorption isotherm by BET and BJH analysis at 77 K (Micromeritics ASAP 2020, USA). A professional Benchtop pH meter BP3001 (Trans Instruments, Singapore) was used to adjust the pH of solutions. For comparison, determination of CIP in real samples was performed by fluorometry with excitation/emission wavelength at 275/452 nm. The fluorescence spectra were recorded on a RF-5301PC spectrofluorometer (Shimadzu, Japan) equipped with a 10 mm path-length quartz cell and a xenon discharge lamp with an excitation/emission slit width of 5.0 nm.2.3 Synthesis of the modifier
The bimetallic organic framework was prepared according to a previous report.42 Briefly, solution A was prepared by dissolving 1 mmole of each Ce(NO3)3·6H2O and Cu(NO3)2·3H2O in 30 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ethanol/water solvent. Solution B contained 3.0 mmol linker H3BTC in 70 mL of 1:1 (v:v) ethanol/water mixture. Next, solution A was slowly added to solution B under stirring for 30 min until a white precipitate appeared. Then, it was aged for 24 h at room temperature. The precipitate was washed with ethanol/water mixture several times, and then dried in an oven at 60 °C. The resultant is named as Cu/Ce-MOF.
:1 (v/v) ethanol/water solvent. Solution B contained 3.0 mmol linker H3BTC in 70 mL of 1:1 (v:v) ethanol/water mixture. Next, solution A was slowly added to solution B under stirring for 30 min until a white precipitate appeared. Then, it was aged for 24 h at room temperature. The precipitate was washed with ethanol/water mixture several times, and then dried in an oven at 60 °C. The resultant is named as Cu/Ce-MOF.
Nickel doped zinc oxide nanoparticles was prepared by sol gel method using citrate precursor according to our previous report.43 First, Zn(NO3)2·6H2O and Ni(NO3)2·6H2O salts were mixed thoroughly and dissolved in 100 mL of deionized water with a stoichiometric ratio of 0.98:0.02. After addition of citric acid as gelling agent to the mixture solution, ammonium hydroxide solution was slowly added. The pH of the mixture was adjusted to 7 and stirred for 3 h to form a homogeneous gel. The mixture was dried at room temperature to form a xerogel. Then, it was ground and sintered at 600 °C with a heating rate of 10 °C min−1 for 2 h. The greenish-gray powder (Ni0.02Zn0.98O) abbreviated as NZP was used in subsequent experiments.
2.4 Fabrication of Cu/Ce-MOF/NZP/CPE
The carbon paste electrodes were prepared using graphite powder and paraffin oil (2 drops) as binder. The components of 0.15 g in total weight in the absence and presence of modifiers (1.3% NZP, and 2.7% Cu/Ce-MOF) were mixed thoroughly in a mortar for at least 25 min to obtain homogeneous paste. Then, the paste was packed into one end of a polyethylene insulin syringe in 0.4 cm deep, and a copper wire passing through the center of the electrode body for electrical contact. The surface of each electrode was polished using a suitable paper to produce a smooth, shiny and reproducible working surface. Then, the prepared electrode was washed with deionized water to remove impurities. The Cu/Ce-MOF/NZP/CPE electrodes were fabricated by employing different amounts of Cu/Ce-MOF and NZP modifiers in paste composition.2.5 Analytical procedure
The selective detection of CIP by Cu/Ce-MOF/NZP/CPE was conducted by the following procedure: the electrode was first activated in phosphate buffer at pH 3.0. Then, cyclic voltammetry (CV) was carried out in the potential range of 0–1.6 V (vs. SCE) at a scan rate of 50 mV s−1. Quantification of CIP was carried out in 0.1 mol L−1 phosphate buffer at pH 3 by differential pulse voltammetry with a pulse amplitude of 100 mV, a pulse width of 0.4 ms, and a potential scan speed of 100 mV s−1.2.6 Preparation of real samples
3. Results and discussion
3.1 Characterization
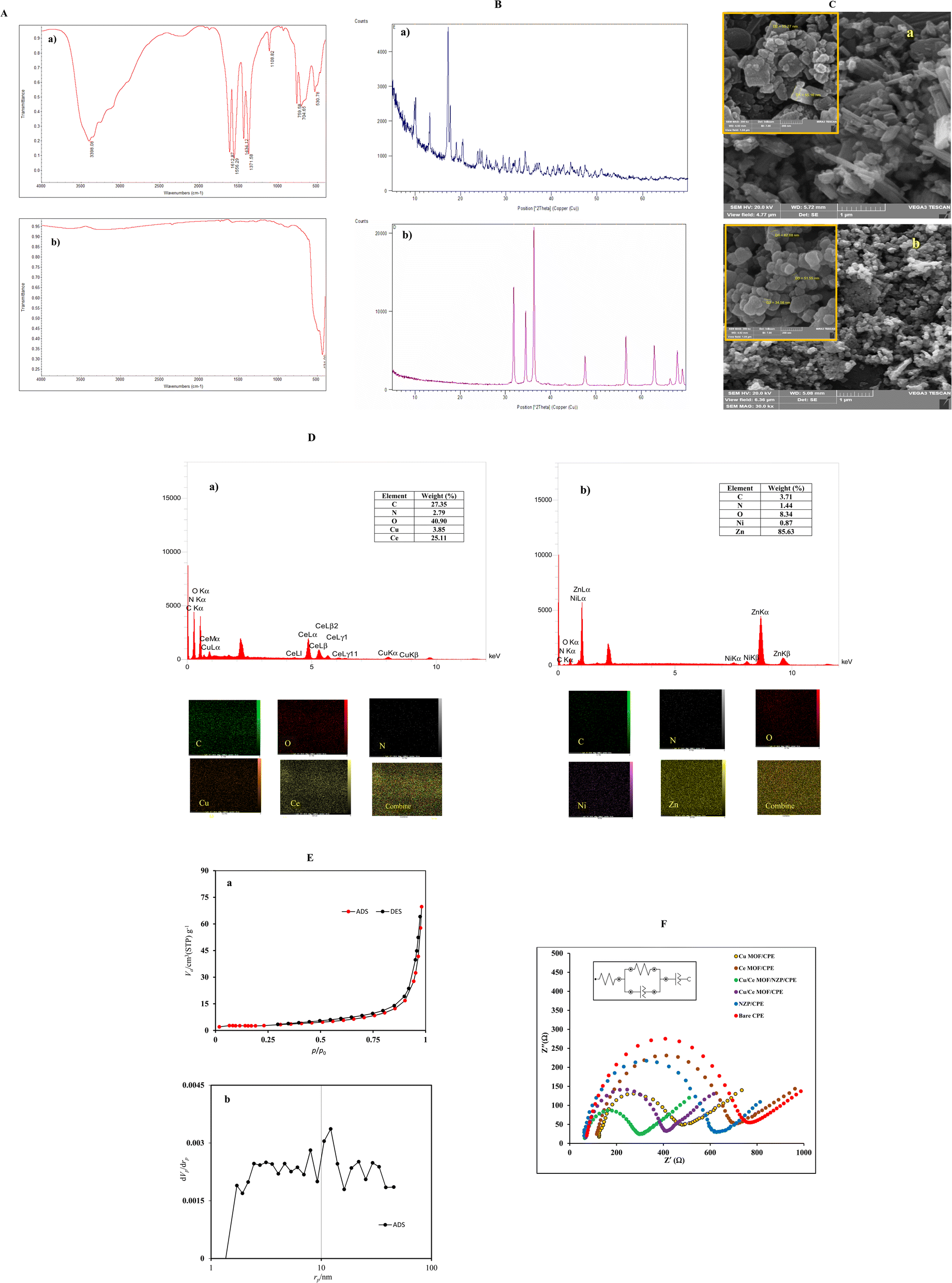 | ||
| Fig. 1 (A) FT-IR spectra of (a) Cu/Ce-MOF, and (b) NZP; (B) XRD patterns for (a) Cu/Ce-MOF, and (b) NZP; (C) SEM images at two magnifications of (a) Cu/Ce-MOF, and (b) NZP; (D) EDX spectra of (a) Cu/Ce-MOF, and (b) NZP (inset shows the EDX and mapping analysis); (E) BET analyses (a) N2 adsorption/desorption isotherm and (b) the BJH pore volume distribution of the synthesized Cu/Ce-MOF, and (F) the Nyquist plots of the bare CPE, NZP/CPE, Cu/Ce-MOF/CPE and Cu/Ce-MOF/NZP/CPE in 5.0 mmol L−1 [Fe(CN)6]3−/4− solution containing 0.10 mol L−1 KCl (Inset shows the equivalent Randles circuit). | ||
The FTIR spectrum of NZP (Fig. 1A(b)) shows high intensity peaks at 550 and 434 cm−1 which are related to the stretching vibrations of Zn–O.43 The broad absorption peak located at 3458 cm−1 corresponds to the O–H stretching vibration of H2O absorbed on the surface of the Ni-doped ZnS nanocomposite. The absorption peak at 2340 cm−1 is attributed to the presence of CO2 in air. The stretching vibrations of Ni–O bond at 870 cm−1 and 434 cm−1 in NZP confirm the inclusion of nickel ions in ZnO matrix.
:1. Mapping analysis in Fig. 1D(a) revealed that Ce and Cu elements are homogeneously distributed in MOF nanostructure.Elemental mapping analysis of pristine NZP in Fig. 1D(b) also revealed that N, O, C, Ni, Zn elements were homogeneously distributed throughout in the nanoparticles structure.
3.2 Electrochemical characterization of modified electrodes
The electroactivity of synthetic materials was tested by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The electrochemical active surface areas of the electrodes were explored in electrochemical probe solution of 5 mM K3Fe(CN)6/K4Fe(CN)6 containing 0.1 M KCl by CV. Fig S1† shows the CV curves recorded on bare CPE, NZP/CPE, Cu/Ce-MOF/CPE and Cu/Ce-MOF/NZP/CPE in the potential window range of −1000 mV to 1000 mV with different potential scan rates (ν) from 5 to 200 mV s−1. The voltammograms show a pair of well redox peaks corresponding to the redox reaction between K3Fe(CN)6 and K4Fe(CN)6. It is obvious that after modification of the bare CPE with Cu/Ce-MOF/NZP material, peak-to-peak separation potential decreased, while the current intensity of the redox peaks increased, indicating the enhanced electron transfer ability in the modified electrodes. In addition, the effect of the scan rate on the peaks' current was studied to evaluate the electrochemical active surface areas of the modified electrodes according to the Randles–Sevcik eqn (1):| ip = (2.65 × 105)AD1/2n3/2ν1/2C* | (1) |
The linearly dependence of the redox peak currents (ip) on the square root of scan rates (ν1/2) indicates the diffusion controlled nature of the reaction occurred at modified CPEs. The electroactive surface area of bare CPE, NZP/CPE, Cu/Ce-MOF/CPE, and Cu/Ce-MOF/NZP/CPE was evaluated from the slope of the corresponding linear plots ip–ν1/2, and found to be 0.184 cm2, 0.197 cm2, 0.215 cm2 and 0.292 cm2, respectively, which are larger than the geometric area (0.125 cm2) of bare CPE. These observations implied that incorporation of Cu/Ce-MOF and NZP modifiers into the CPE provides more active sites, thereby improving the effective surface of the modified CPEs compared to the bare electrode, and enhancing the electrochemical activity of the Cu/Ce-MOF/NZP. Therefore, Cu/Ce-MOF/NZP/CPE was used for subsequent electrochemical measurements of CIP.
The interfacial behavior of electrodes has been extensively studied by EIS analysis. Commonly, an electrochemical system can be described by an analog Rundles circuit containing resistance, capacitance, and inductance. Commercial software is used to fit the experimental impedance data to a simple circuit model. The impedance of an electrochemical cell is examined with the Nyquist plot, which is a plot of the imaginary impedance against real impedance. In order to further prove the benefit of the Cu/Ce-MOF/NZP as an electrocatalyst in the electrooxidation of CIP into CPE, EIS measurements were performed for bare CPE, NZP/CPE, Cu-MOF/CPE, Ce-MOF/CPE, Cu/Ce-MOF/CPE, and Cu/Ce-MOF/NZP/CPE. The fitted corresponding Nyquist plots and equivalent circuit are illustrated in Fig. 1F. The values of the fitting parameters are summarized in Table 1, where Rs represents the electrolyte resistance, Rct is the charge transfer resistance, and constant phase elements CPE1 and CPE2 are the respective capacitances at the electrode/electrolyte interface and faradaic processes. As shown in Table 1, among the modified electrodes, the lowest Rct value obtained for CPE modified with NZP and Cu/Ce-MOF (Cu/Ce-MOF/NZP/CPE), indicating a decrease in resistance to the charge transport of redox probe at the Cu/Ce-MOF/NZP/CPE and the increase in electron-transfer kinetics at this electrode surface.
| Electrode | Rs (Ω) | Rct (Ω) | CPE1 (μMho) | CPE2 (mMho) |
|---|---|---|---|---|
| Bare CPE | 55 | 607 | Y0 = 1.53 n1 = 0.921 | Y0 = 3.05 n2 = 0.28 |
| Ce-MOF/CPE | 98.9 | 514 | Y0 = 2.61 n1 = 0.894 | Y0 = 3.21 n2 = 0.274 |
| NZP/CPE | 53 | 509 | Y0 = 1.53 n1 = 0.944 | Y0 = 4.63 n2 = 0.322 |
| Cu-MOF/CPE | 55 | 262 | Y0 = 3.91 n1 = 0.918 | Y0 = 2.98 n2 = 0.233 |
| Cu/Ce-MOF/CPE | 52 | 300 | Y0 = 2.49 n1 = 0.941 | Y0 = 4.26 n2 = 0.33 |
| Cu/Ce-MOF/NZP/CPE | 53 | 184 | Y0 = 3.67 n1 = 0.926 | Y0 = 4.14 n2 = 0.282 |
The inhomogeneity of the electrode surface often causes heterogeneous electron transfer and deviation from the ideal response of the resistance and capacitor, resulting in frequency dispersion of the interfacial capacitance and the appearance of a constant phase element (CPE). The impedance of CPE (ZQ) can be expressed by eqn (2):
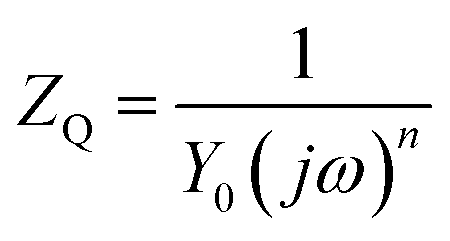 | (2) |
3.3 Electrochemical behavior of target analyte on modified electrodes
Cyclic voltammograms of 1 × 10−4 mol L−1 CIP in 0.1 mol L−1 phosphate buffer solution (pH = 3) at bare CPE, NZP/CPE, Cu/Ce-MOF/CPE, and Cu/Ce-MOF/NZP/CPE were recorded (Fig. 2). The voltammograms for CIP at Cu/Ce-MOF/CPE, and Cu/Ce-MOF/NZP/CPE showed two well defined anodic peaks situated at 125 mV and 486 mV vs. Ag/AgCl corresponding to the oxidation of Cu to Cu+ and Cu2+, respectively, and two cathodic peaks in the reverse scan located at 273 mV and −44 mV, corresponding to the reduction of Cu2+ to Cu+ and Cu, respectively (Fig. 3). These peaks didn't observe in voltammograms for CIP at bare CPE and NZP/CPE. This redox event suggests that the Cu/Ce-MOF is electroactive due to the presence of the Cu2+ state in this framework with a partially metallic state at the Fermi level.51 A new peak for CIP oxidation (vs. Ag/AgCl) was observed at 1.25 V in all voltammograms which can be attributed to the irreversible redox behavior of CIP. The highest anodic peak current of CIP oxidation on the electrode surface of Cu/Ce-MOF/NZP/CPE compared to other CPEs clearly indicates that the combination of Cu/Ce-MOF and NZP nanoparticles improve the electron transfer for CIP oxidation and revealing the catalytic effect of the modifiers on the oxidation of target analyte.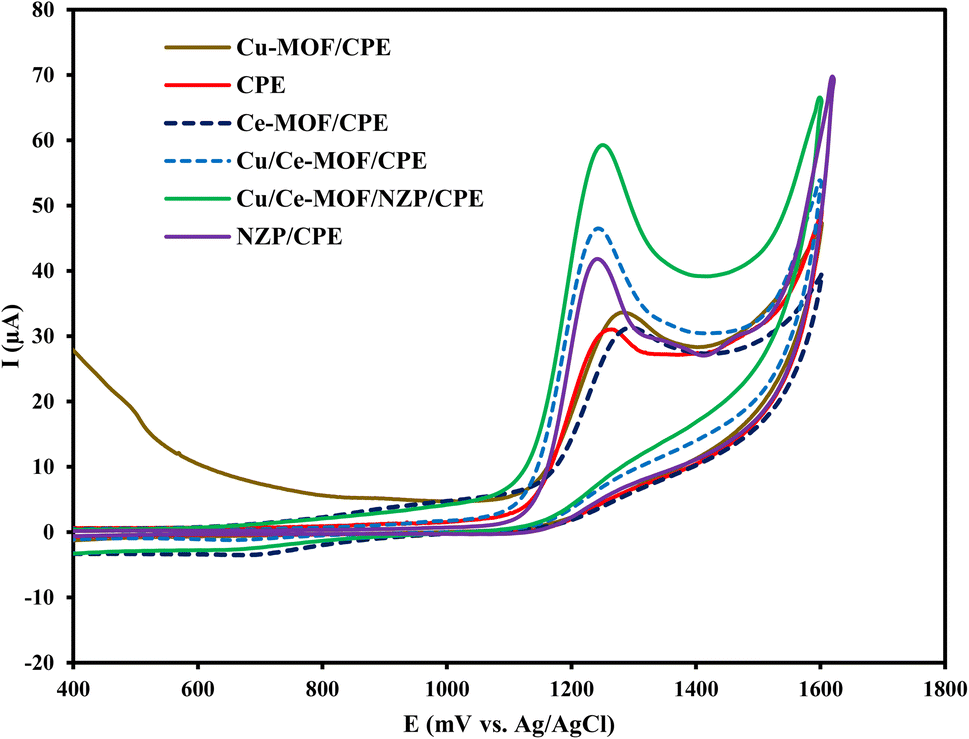 | ||
| Fig. 2 Cyclic voltammograms of 50 μmol L−1 of target analyte in 0.10 mol L−1 phosphate buffer solution at the bare CPE, NZP/CPE, Ce-MOF/CPE, Cu-MOF/CPE, Cu/Ce-MOF/CPE and Cu/Ce-MOF/NZP/CPE. | ||
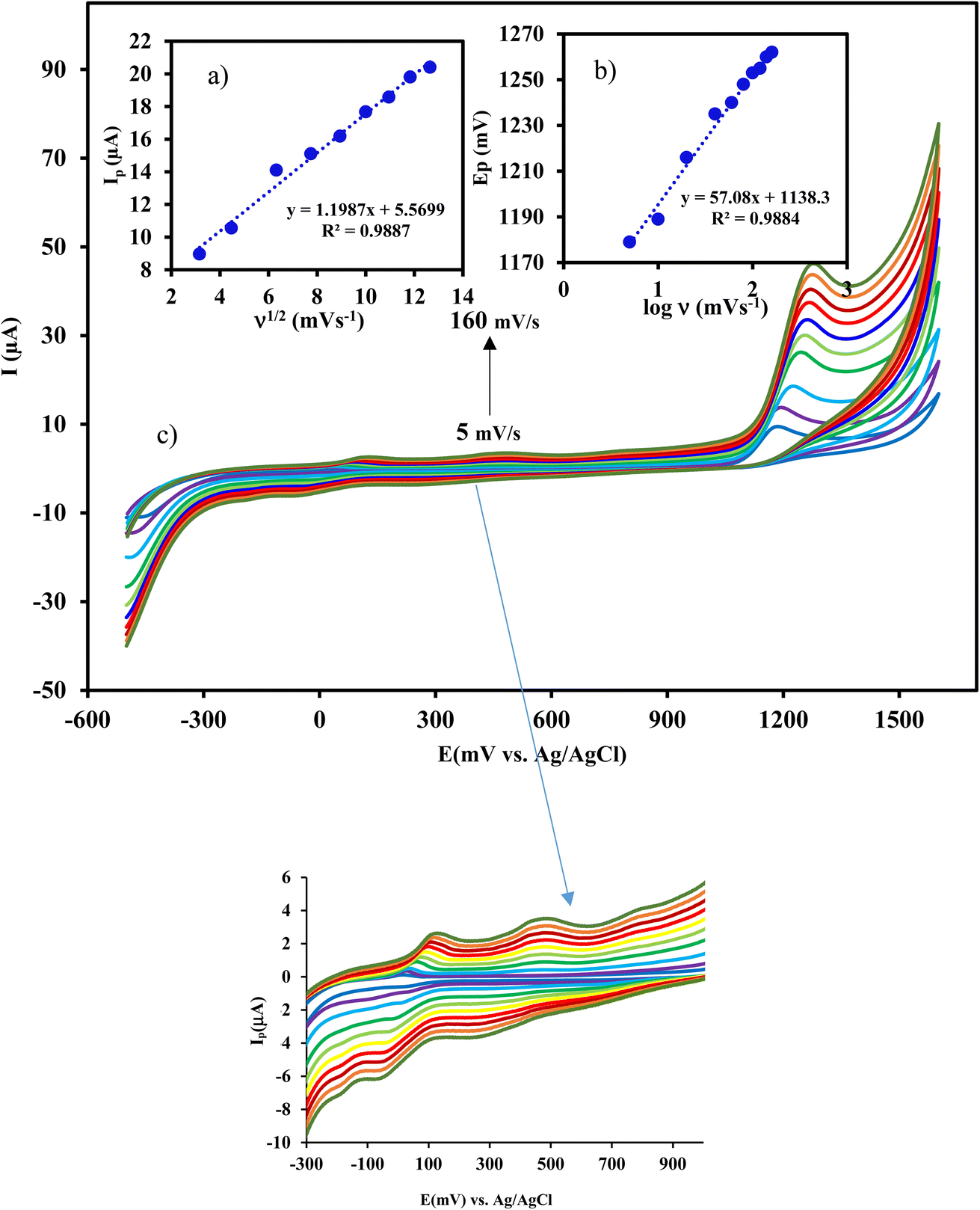 | ||
| Fig. 3 Effect of scan rate (from 5 to 160 mV s−1) on the response of CIP at Cu/Ce-MOF/NZP/CPE. Inset shows the plot of (a) Ip vs. ν1/2, and (b) Ep vs. logν. | ||
3.4 Effect of scan rate and kinetics of oxidation process
Study the effect of scan rate on the peak current can be useful for elucidating the electrochemical mechanism. Therefore, cyclic voltammograms of 50 μmol L−1 CIP at Cu/Ce-MOF/NZP/CPE were recorded at different scan rates from 5 to 160 mV s−1. From Fig. 3, it is evident that the current gradually increases with the increase of the scan rates along with the shift of the oxidation peak potential to positive values. The anodic peak current (ip) showed a linear relationship with the square root of scan rates (ν1/2) by a regression equation of ip (μA) = 1.20 ν1/2 (mV s−1)1/2 + 5.57 and the correlation coefficient R2 = 0.9887. In addition, the plot of logip against logν showed a linearity with a slope of 0.32, which is very close to the theoretical value of 0.5, confirming that the oxidation of CIP on the surface of Cu/Ce-MOF/NZP/CPE is a diffusion-controlled process.
The value of the electron transfer coefficient (α) in an irreversible system is between 0.3 and 0.7. For an anodic reaction, α can be calculated from the slope of Tafel plot using data from the initial part of the voltammetric wave of the current–potential curve for the oxidation of 50 μmol L−1 CIP at a potential scan rate of 20 mV s−1. Considering an arbitrary value of 0.5 for α, the number of electrons in the rate-determining step can be evaluated from the eqn (3):
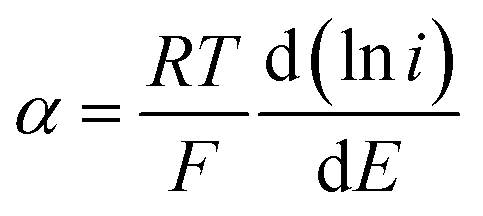 | (3) |
Taking into account that 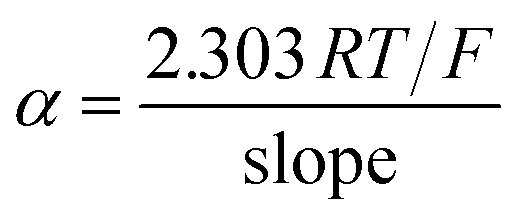 with the slope of the linear regression of the Tafel plot in Fig. S2,† the value of α is estimated to be 0.476. The anodic peak potential (mV) of cyclic voltammogram varies linearly as a function of the logarithm of the scan rate (mV s−1) with the regression equation Epa (mV) = 57.1logν + 1138.3 (R2 = 0.9884) (inset of Fig. 3), according to Laviron equation for irreversible redox process (4):
with the slope of the linear regression of the Tafel plot in Fig. S2,† the value of α is estimated to be 0.476. The anodic peak potential (mV) of cyclic voltammogram varies linearly as a function of the logarithm of the scan rate (mV s−1) with the regression equation Epa (mV) = 57.1logν + 1138.3 (R2 = 0.9884) (inset of Fig. 3), according to Laviron equation for irreversible redox process (4):
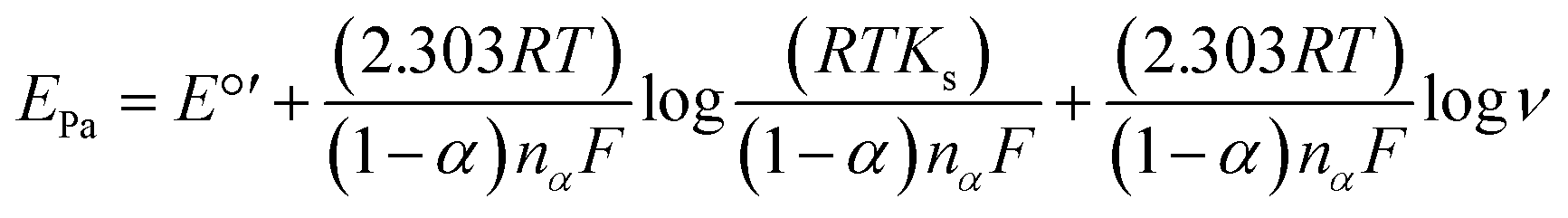 | (4) |
ν plot, the nα value of 1.976 was obtained, suggesting that two electrons are involved in the rate determining step of the CIP electro-oxidation reaction at Cu/Ce-MOF/NZP/CPE. Also, the value of rate constant (ks) at a scan rate of 100 mV s−1 was calculated from eqn (5), and obtained 1.9 × 10−3 s−1.
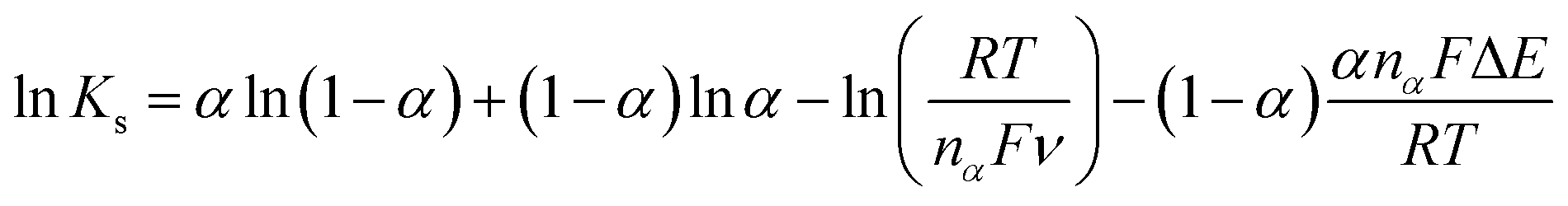 | (5) |
3.5 Chronoamperometric study
Chronoamperometric measurement was carried out for the determination of diffusion coefficient (D) of the target analyte by setting Cu/Ce-MOF/NZP/CPE at a fixed potential of 1.1 V in 50 s for different CIP concentrations in phosphate buffer (Fig. 4). Using the slope of the straight line of the currents against t−1/2 plot at each concentration, and the linear relationship of the slopes with the CIP concentrations (insets of Fig. 4), the value of the diffusion coefficient (D) of CIP was calculated (D = 7.68 × 10−4 cm2 s−1) according to the Cottrell eqn (6):| I = nFACD1/2π−1/2t−1/2 | (6) |
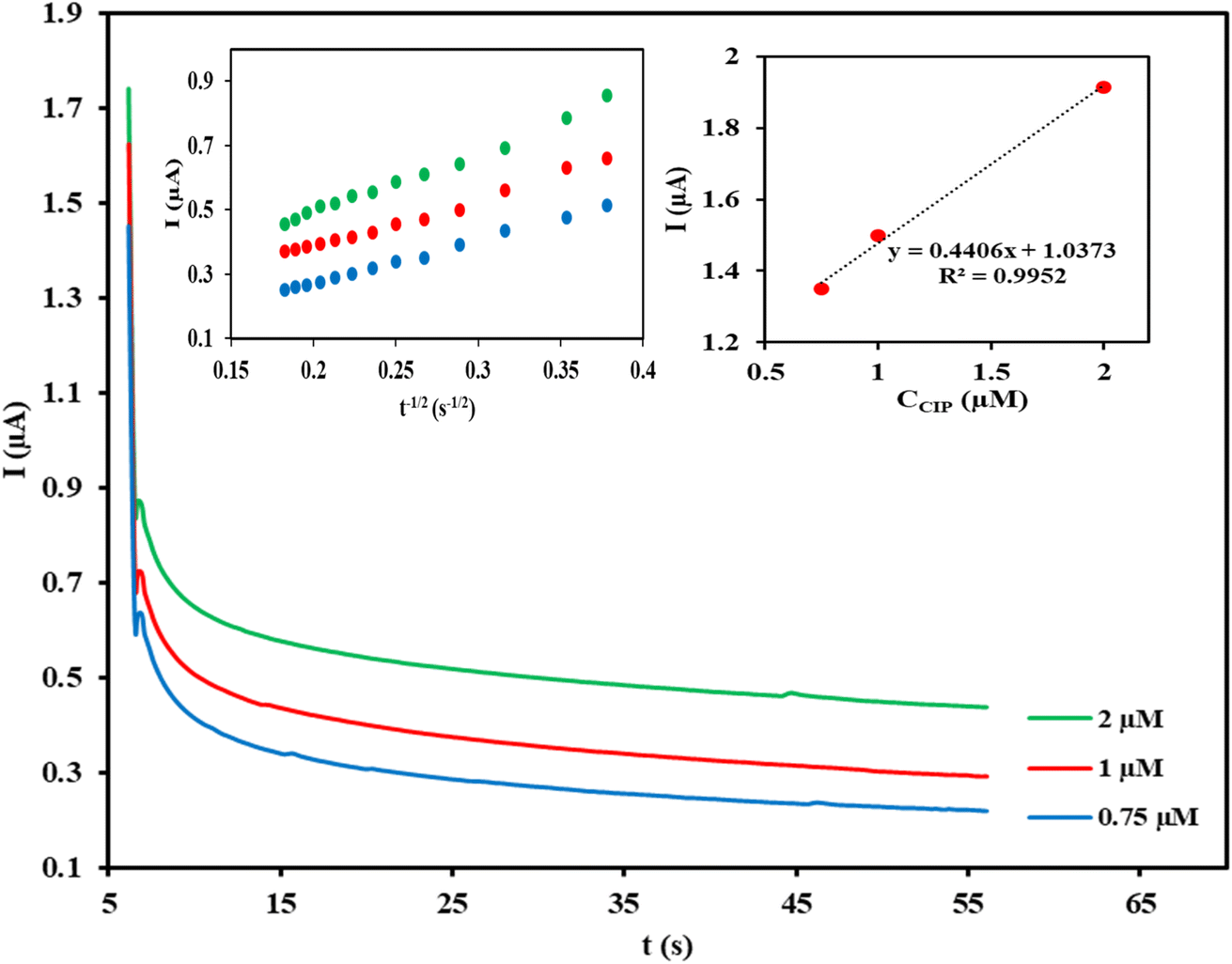 | ||
| Fig. 4 Chronoamperometry of CIP in 0.1 mol L−1 phosphate buffer (pH = 3) at three different concentrations (0.75, 1.0, 2.0 μmol L−1) with a potential step at 1.1 V at Cu/Ce-MOF/NZP/CPE. Insets show the plot of I (μA) vs. t−1/2 (s−1/2) obtained from chronoamperograms, and plot of slopes of the linear lines versus CIP concentrations. | ||
3.6 Effect of pH
The effect of pH on the response of Cu/Ce-MOF/NZP/CPE electrode in CIP solution (10 μmol L−1) was investigated by cyclic voltammetry and the results are shown in Fig. 5. It can be seen that the oxidation current of CIP reached to the highest value at pH 3 and then decreased with increasing pH which might be due to the participation of protons in the oxidation process. Moreover, the anodic peak potential shifted towards less positive values with increasing solution pH. The relationship between the anodic peak potentials and the pH values of the CIP solution (inset of Fig. 5) is linear with the regression equation Epa (V) = −0.0561 pH + 1.4165 (R2 = 0.9882). The closeness of the slope value to the theoretical value of 59 mV per pH decade, implies that the number of protons and electrons in CIP oxidation at Cu/Ce-MOF/NZP/CPE is equal. Thus, the mechanism of CIP oxidation at Cu/Ce-MOF/NZP/CPE is probably through the oxidation of the –NH group to hydroxylamine (–N–OH) pathway by two protons and two electrons as presented in Fig. S3.† CIP contains a carboxylic acid and an amine functional group in piperazinyl ring (pKa1 = 6.1, pKa2 = 8.7), indicating that at pH below 6.1 and above 8.7, cationic and anionic forms of CIP predominate, respectively. Therefore, the electrostatic repulsive interactions between anionic form of CIP and carboxylate group on the electrode surface reduce oxidation peak current. Further, π–π interactions between the linker and the CIP molecule may facilitate the diffusion of CIP to the electrode surface and enhance the response of the electrode to the target analyte.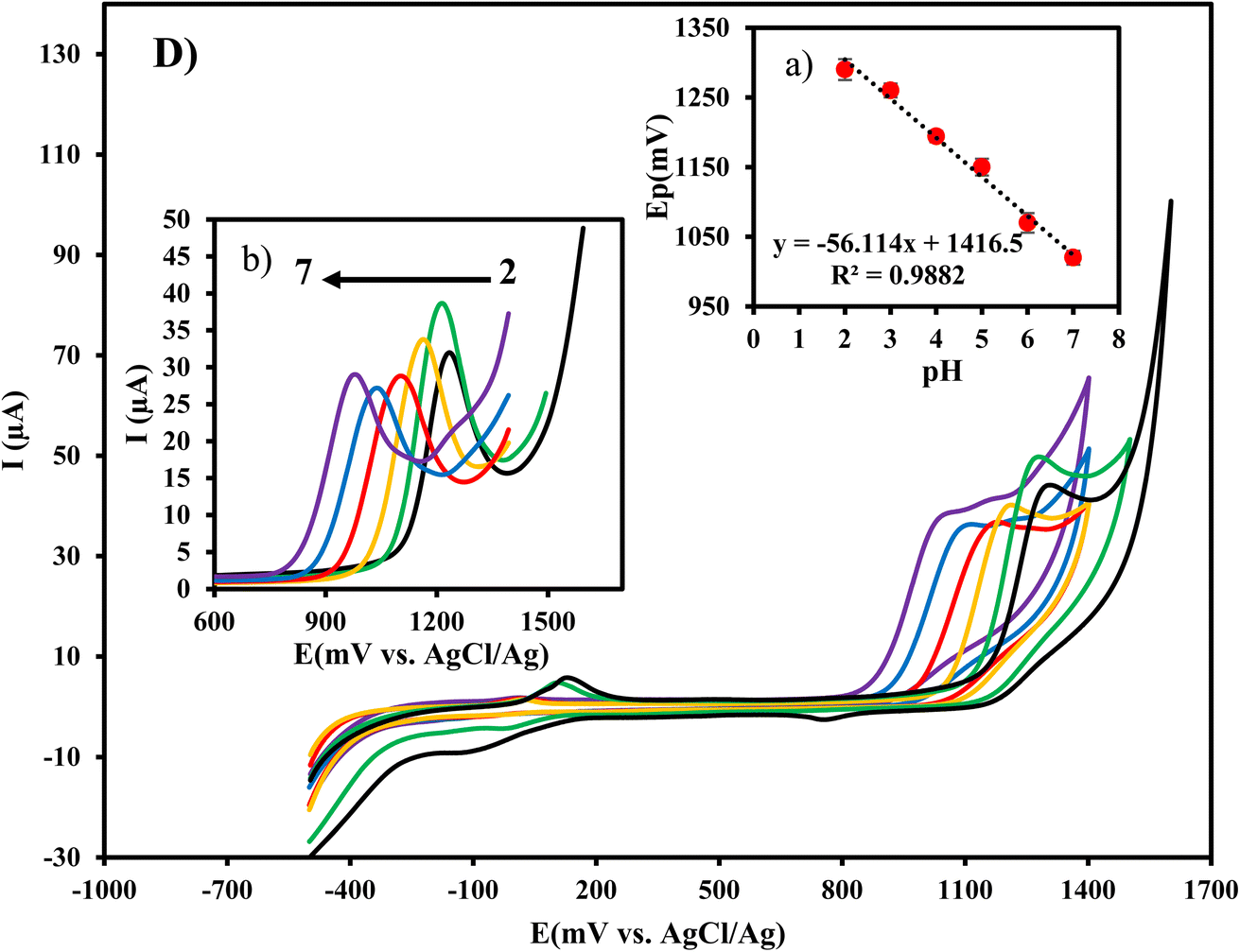 | ||
| Fig. 5 Effect of pH on the response of Cu/Ce-MOF/NZP/CPE for CIP detection. | ||
3.7 Optimization
Since a high electrochemical oxidation peak current is necessary to determine the CIP at Cu/Ce-MOF/NZP/CPE, voltammetric techniques including LSV, SWV, and DPV were evaluated. DPV was chosen because of its higher signal and smaller peak width than the other studied techniques (Fig. 6A).
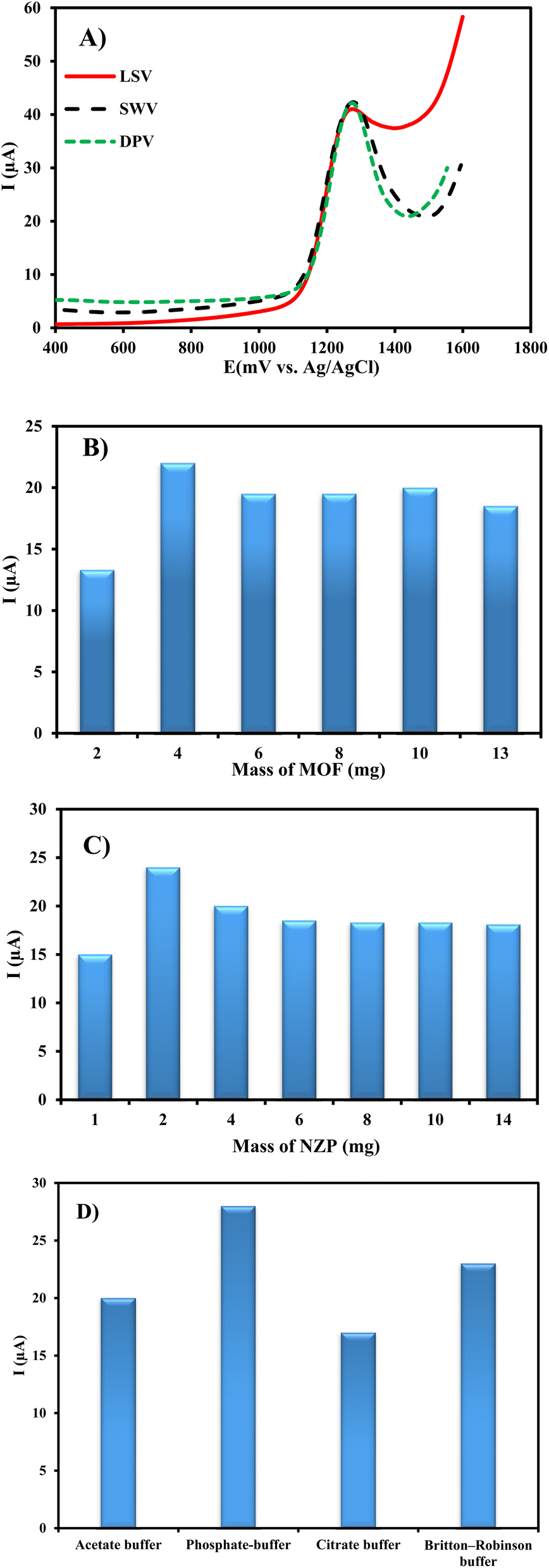 | ||
| Fig. 6 Influence of the (A) type of technique, (B) Cu/Ce-MOF mass, (C) NZP mass, and (D) type of buffer on the anodic response of the Cu/Ce-MOF/NZP/CPE to CIP detection. | ||
The content of Cu/Ce-MOF in the CPE composition affects the oxidation current of CIP. For this reason, six electrodes containing different amounts of Cu/Ce-MOF (2, 4, 6, 8, 10 and 13 mg) were prepared and investigated for the detection of 50 μmol L−1 CIP under the same conditions. As can be seen from Fig. 6B, the peak current increases up to 4 mg (2.7%, w/w) of Cu/Ce-MOF in the CPE composition and decreases with further increase of the modifier amount.
In addition, the effect of the mass of NZP nanoparticles on the anodic current of CIP oxidation was investigated. Seven electrodes containing different amounts of NZP (1, 2, 4, 6, 8, 10 and 14 mg; from 0.7% to 9.0% w/w of the CPE composition) were prepared and examined for the detection of 50 μmol L−1 CIP. It was observed that the peak current increases up to 1.3% of NZP in CPE composition, and then decreases (Fig. 6C). As a result, NZP and Cu/Ce-MOF with 1.3% (w/w) of and 2.7% (w/w) in CPE mass, respectively, were considered as the optimal amounts of modifiers for CIP quantification in subsequent studies.
To investigate the maximum response of the target analyte at pH 3, various buffers such as Britton–Robinson, acetate, citrate and phosphate buffers at 0.1 mol L−1 ionic strength were used. The maximum current response with a well-defined peak shape was obtained in phosphate buffer, which was used to detect the target analyte in subsequent studies (Fig. 6D).
The effect of potential scan rate (ν) on the performance of Cu/Ce-MOF/NZP/CPE was also studied in the range of 5–160 mV s−1. Increasing the scan rate resulted in a higher oxidation peak current without changing the peak width, so 100 mV s−1 was chosen as the optimal scan rate in further studies.
3.8 Electrochemical detection of CIP
The electrochemical response of Cu/Ce-MOF/NZP/CPE to CIP with different concentrations in PB (pH 3) under optimal conditions at an oxidation potential of 1.25 V was determined by DPV technique. As shown in Fig. 7, the oxidation peak current increases with increasing CIP concentration, so that the plot of current responses against CIP concentrations is bilinear within the ranges from 0.75 μmol L−1 to 10 μmol L−1 and from 10 μmol L−1 to 100 μmol L−1 with linear regressions of Ipa = 1.2864CCIP + 1.4663 (R2 = 0.9921) and Ipa = 0.4349CCIP + 11.564 (R2 = 0.9900), respectively. According to the slopes of the calibration curves, the sensitivity of the method in the low dynamic range is 1.2864 μA μmol−1 L, which is about 3 times higher than that of the higher concentration range. According to the IUPAC guideline, the limit of detection (LOD = 3σ/m) and limit of quantitation (LOQ = 10σ/m) of the proposed method are estimated to be 0.142 μmol L−1 and 0.47 μmol L−1, respectively.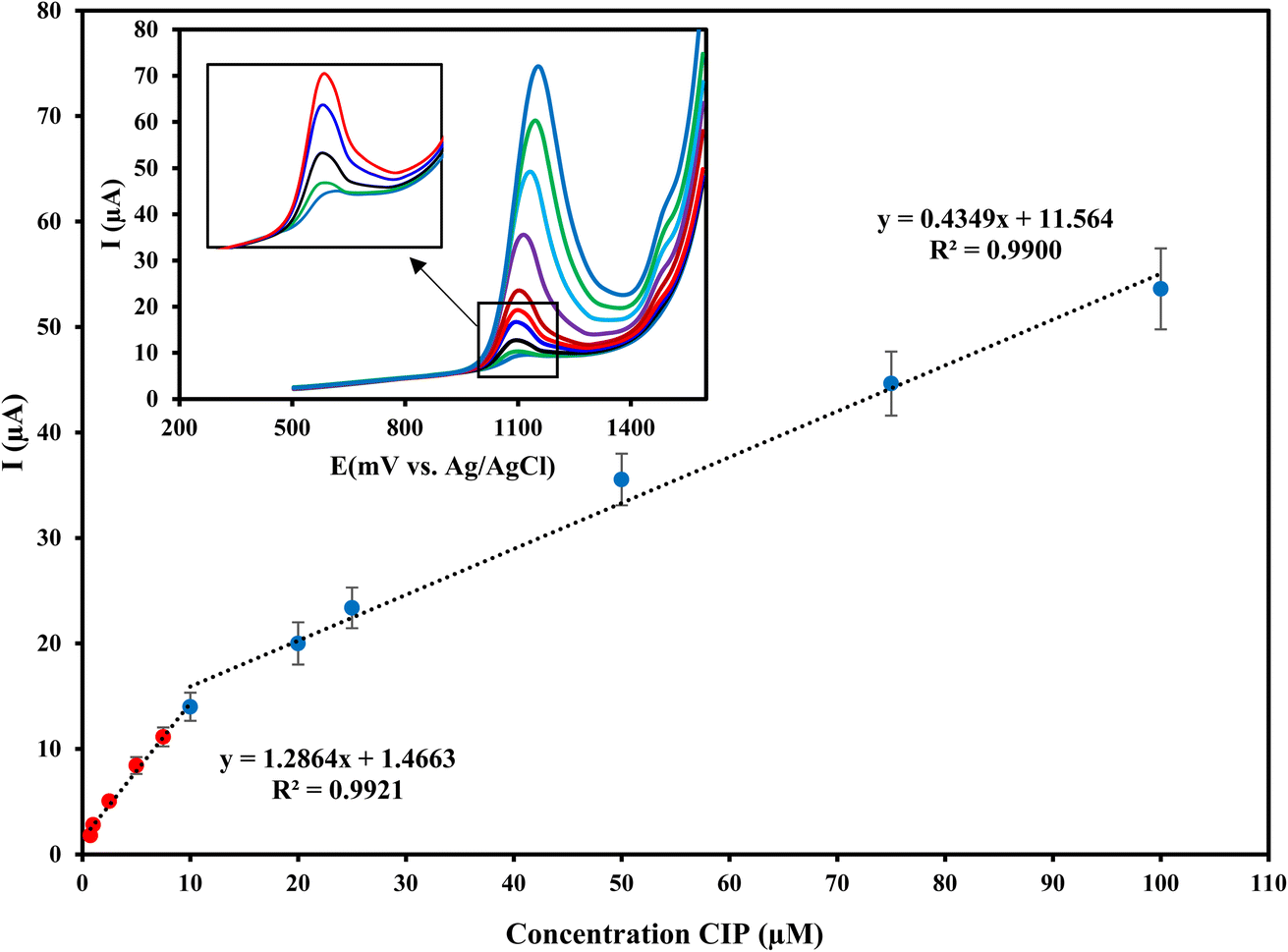 | ||
| Fig. 7 The calibration curve and linear range of Cu/Ce-MOF/NZP/CPE for determining CIP in 0.1 mol L−1 phosphate buffer (pH = 3) under optimal conditions (scan rate: 100 mV s−1, pulse amplitude: 100 mV). Inset shows the differential pulse voltammograms of CIP. | ||
The present method is compared with reported electrochemical methods for the CIP quantification.19,52–57 As can be seen in Table 2, the performance of the designed electrode is comparable or better than other reported electrodes for the detection of CIP in terms of linear range, detection limit, and type of real sample tested. Besides, the proposed electrochemical electrode has a renewable surface, simple preparation and cost-effective compared to some reported electrodes that provide almost the same analytical characteristics, making it a desirable sensing platform for the target analyte.
| Electrode | Linear range (μM) | Technique | LOD (μM) | Real sample | Ref. |
|---|---|---|---|---|---|
| a CPE – carbon paste electrode; GCE – glassy carbon electrode; GE – graphite electrode, GO – graphene oxide, NiONP – nickel oxide nanoparticles; SWV – square wave voltammetry; CRGO – chemically-reduced graphene-oxide. | |||||
| ds-DNA-BDD | 0.5–60 | DPV | 0.44 | Urine | 19 |
| MWCNT/GCE | 3–1200 | LSV | 0.9 | Urine, plasma, tablet | 52 |
| MWCNT-GCE | 40–1000 | Chronoamperometry | 6 | Urine, serum | 53 |
| Boron-doped diamond electrode | 0.74–20 | DPV | 0.6 | Urine | 54 |
| DNA/GCE | 1–10 | DPV | 0.12 | — | 55 |
| TiO2/PB/AuNPs/CMK-3/Nafion/GE | 1–10 | CV | 0.10 | Environmental water | 56 |
| CRGO/GC | 6–40 | SWV | 0.21 | Drugs and milk | 57 |
| Cu/Ce-MOF/NZP/CPE | 0.75–100 | DPV | 0.14 | Water, milk, urine | This work |
3.9 Repeatability, reproducibility, stability and selectivity of the method
The repeatability of Cu/Ce-MOF/NZP/CPE was evaluated in terms of relative standard addition (RSD%) for seven replicated CIP response measurements at a concentration of 10 μmol L−1 and was found to be 2.7%. The reproducibility of the sensor was assessed by measuring the oxidation peak current using seven individual sensors and was 3.6%. The results confirm the efficient performance of the sensor with satisfactory reproducibility. The long-term stability of the developed sensor toward CIP was studied for one month and found that the initial response reached 90% after 2 weeks, which suggests that the developed sensor is stable over a long period.The selectivity of Cu/Ce-MOF/NZP/CPE was evaluated using the DPV technique by measuring the electrode response to CIP in the presence of interfering substances commonly present in water, urine and milk samples such as ions, amino acids, sugars and drugs. In this study, the tolerance limit is defined as the maximum concentration of the coexisting substance that affects the response of Cu/Ce-MOF/NZP/CPE to CIP (10 μmol L−1) by more or less than 5%. The results in Table 3 indicate that most of the examined substances did not interfere in CIP measurement. Among them, nalidixic acid, glutamic acid, naproxen and ofloxacin probably interfere with analyte measurement due to their similar structures.
| Interferents | Tolerance limit (mol mol−1) |
|---|---|
| Ca2+, NO3−, Mg2+, Cl−, Na+, PO43−, K+, Cu2+, NH4+, urea, lysine, creatinine | 1000 |
| SO42− | 800 |
| Citric acid, sucrose, glucose | 500 |
| Ascorbic acid, histidine | 100 |
3.10 Analysis of CIP in urine, water, and milk
Under optimum conditions, the Cu/Ce-MOF/NZP/CPE was applied to determine CIP in urine, water, and milk samples by employing DPV. The recoveries of CIP in the samples were calculated using standard addition method. From the results in Table 4, the recoveries obtained for analysis of CIP in urine, water, and milk samples were found in the ranges of 93–109%, 96–105%, and 98–114%, respectively. The precision of the method as percent relative standard deviation (RSD%) for each sample is summarized in Table 4. The RSD of each sample is less than 5%, implying that the practical ability of the developed sensor for rapid detection of CIP in complex matrices with high precision. Also, the results obtained by the proposed method were compared with fluorometric method due to fluorescence property of CIP. Although the results showed low recoveries of analyte in the studied samples by flourometry, but statistical analysis using Student's t-test and variance ratio F-test at 95% confidence level (p = 0.05) indicated no significant difference between the performance of the two methods (Table 4).| Sample | Amount added (μmol L−1) | Suggested method | Fluorometric method | ||||
|---|---|---|---|---|---|---|---|
| Amount found (μmol L−1) | Recovery RSDb (%) | Amount found (μmol L−1) | Recovery RSDb (%) | ||||
| a tcrit. (p = 0.05, n − 1 = 3) = 3.18; texp. milk = 2.57; texp. urine = 1.36; texp. water = 1.73; Fcrit, 2, 2 = 19.b Based on triplicate analysis. | |||||||
| Milk | 0 | — | — | — | — | — | — |
| 2.5 | 2.9 | 114 | 3.3 | 2.6 | 102.2 | 2.5 | |
| 5.0 | 5.4 | 107 | 0.4 | 4.5 | 90.2 | 1.9 | |
| 7.5 | 7.4 | 98.0 | 3.7 | 6.4 | 85.1 | 1.2 | |
| 10 | 9.9 | 99.0 | 3.8 | 8.2 | 82.3 | 5.6 | |
| Urine | 0 | — | — | — | |||
| 2.5 | 2.4 | 94.0 | 1.8 | 2.6 | 105.6 | 3.3 | |
| 5.0 | 5.5 | 109 | 2.5 | 4.8 | 96.5 | 12.6 | |
| 7.5 | 7.5 | 99.0 | 1.9 | 6.9 | 91.6 | 8.5 | |
| 10 | 9.4 | 93.0 | 4.1 | 8.7 | 86.7 | 8.4 | |
| Water | 0 | — | — | — | — | — | — |
| 2.5 | 2.6 | 104 | 2.2 | 3.3 | 134 | 9.4 | |
| 5.0 | 5.4 | 108 | 2.9 | 5.3 | 106 | 8.1 | |
| 7.5 | 7.4 | 98.0 | 2.8 | 8.4 | 112 | 3.3 | |
| 10 | 9.7 | 96.0 | 2.5 | 10.5 | 105 | 0.6 | |
4. Conclusions
Cu/Ce-MOF and NZP were used as modifiers to fabricate a selective and robust modified electrode for CIP detection by differential pulse voltammetric technique. Characterization of Cu/Ce-MOF modifier showed a relatively high surface area and porous structure which provided more reaction sites and facilitated electron transfer and analyte diffusion to the electrode surface. The carbon paste electrode modified with Cu/Ce-MOF and NZP modifiers showed significant sensitivity and selectivity for the electrochemical determination of CIP. In comparison with other electrochemical sensors for detection of CIP, the proposed sensor showed a large linear concentration range (0.75–100 μmol L−1), acceptable limit of detection (0.142 μmol L−1; 47 ng mL−1), good reproducibility (RSD% < 5), and high selectivity. It was successfully applied to determine CIP in serum, urine, and water samples. This sensor has been shown to be promising for the determination of CIP in complex matrices.Conflicts of interest
There are no conflicts to declare.References
- D. G. Joakim Larsson and C.-F. Flach, Antibiotic resistance in the environment, Nat. Rev. Microbiol., 2022, 20, 257–269 CrossRef.
- D. Fatta-Kassinos, S. Meric and A. Nikolaou, Pharmaceutical residues in environmental waters and wastewater: current state of knowledge and future research, Anal. Bioanal. Chem., 2011, 399, 251–275 CrossRef CAS.
- Z. Zhang, Q. Liu, M. Zhang, F. You, N. Hao, C. Ding and K. Wang, Simultaneous detection of enrofloxacin and ciprofloxacin in milk using a bias potentials controlling-based photoelectro- chemical aptasensor, J. Hazard. Mater., 2021, 125988 CrossRef CAS , https://www.sciencedirect.com/journal/journal-of-hazardous-materials/vol/416/suppl/C416.
- A. J. dos Santos, G. V. Fortunato, M. S. Kronka, L. G. Vernasqui, N. G. Ferreira and M. R. V. Lanza, Electrochemical oxidation of ciprofloxacin in different aqueous matrices using synthesized boron-doped micro and nano-diamond anodes, Environ. Res. Part A, 2022, 204, 112027 CrossRef CAS.
- M. Tumini, O. Nagel, M. P. Molin and R. Althaus, Microbiological assay with Bacillus licheniformis for the easy detection of quinolones in milk, Int. Dairy J., 2017, 64, 9–13 CrossRef CAS.
- S. Mohan and P. Balakrishnan, Kinetics of ciprofloxacin removal using a sequential two-step ozonation-biotreatment process, Environ. Technol. Innov., 2021, 21, 101284 CrossRef CAS.
- S. W. Hyung, J. Lee, S. Y. Baek, S. Lee, J. Han, B. Kim, K. Choi, S. Ahn, D. K. Lim and H. Lee, Method improvement for analysis of enrofloxacin and ciprofloxacin in chicken meat: application of in-sample addition of trace ethylenediaminetetraacetic acid to isotope dilution ultra-performance liquid chromatography-mass spectrometry, Chromatographia, 2022, 85, 35–45 CrossRef CAS.
- S. N. Muchohi, N. Thuo, J. Karisa, A. Muturi, G. O. Kokwaro and K. Maitland, Determination of ciprofloxacin in human plasma using high-performance liquid chromatography coupled with fluorescence detection: application to a population pharmacokinetics study in children with severe malnutrition, J. Chromatogr. B, 2011, 879, 146–152 CrossRef CAS.
- A. Czyrski, The spectrophotometric determination of lipophilicity and dissociation constants of ciprofloxacin and levofloxacin, Spectrochim. Acta, Part A, 2022, 265, 120343 CrossRef CAS.
- Y. Sun, P. Dramou, Z. Song, L. Zheng, X. Zhang, X. Ni and H. He, Lanthanide metal doped organic gel as ratiometric fluorescence probe for selective monitoring of ciprofloxacin, Microchem. J., 2022, 179, 107476 CrossRef CAS.
- K. Michalska, G. Pajchel and S. Tyski, Determination of ciprofloxacin and its impurities by capillary zone electrophoresis, J. Chromatogr. A, 2004, 1051, 267–272 CrossRef CAS.
- G. Tsekenis, G.-Z. Garifallou, F. Davis, P. A. Millner, D. G. Pinacho, F. Sanchez-Baeza, M. P. Marco, T. D. Gibson and S. P. J. Higson, Detection of fluoroquinolone antibiotics in milk via a labeless immunoassay based upon an alternating current impedance protocol, Anal. Chem., 2008, 80, 9233–9239 CrossRef CAS.
- L. Bu, Q. Song, D. Jiang, X. Shan, W. Wang and Z. Chen, A novel molecularly imprinting polypyrrole electrochemiluminescence sensor based on MIL-101-g-C3N4 for supersensitive determination of ciprofloxacin, Microchim. Acta, 2023, 190, 373 CrossRef CAS.
- D. Fumagalli, S. Comis, V. Pifferi and L. Falciola, Gold nanoparticles-titania heterojunction: photoelectrochemical detection of Ciprofloxacin, ChemElectroChem, 2023, 10, e202201136 CrossRef CAS.
- M. Azriouil, M. Matrouf, F. E. Ettadili, F. Laghrib, A. Farahi, S. Saqrane, M. Bakasse, S. Lahrich and M. A. El Mhammedi, Recent trends on electrochemical determination of antibiotic Ciprofloxacin in biological fluids, pharmaceutical formulations, environmental resources and foodstuffs: Direct and indirect approaches, Food Chem. Toxicol., 2022, 168, 113378 CrossRef CAS.
- P. Gayen and B. P. Chaplin, Selective electrochemical detection of ciprofloxacin with a porous nafion/multi-walled carbon nanotube composite film electrode, ACS Appl. Mater. Interfaces, 2016, 8, 1615–1626 CrossRef CAS.
- X. Hu, K. Y. Goud, V. S. Kumar, G. Catanante, Z. Li, Z. Zhu and J. L. Marty, Disposable electrochemical aptasensor based on carbon nanotubes-V2O5-chitosan nanocomposite for detection of ciprofloxacin, Sens. Actuators B Chem., 2018, 268, 278–286 CrossRef CAS.
- S. Bagyalakshmi, A. Sivakami, K. Pal, R. Sarankumar and C. Mahendran, Manufacturing of electrochemical sensors via carbon nanomaterials novel applications: a systematic review, J. Nanopart. Res., 2022, 24, 201 CrossRef CAS.
- G. S. Garbellini, R. C. Rocha-Filho and O. Fatibello-Filho, Voltammetric determination of ciprofloxacin in urine samples and its interaction with dsDNA on a cathodically pretreated boron-doped diamond electrode, Anal. Methods, 2015, 7, 3411–3418 RSC.
- W. D. Adane, B. S. Chandravanshi and M. Tessema, A simple, ultrasensitive and cost-effective electrochemical sensor for the determination of ciprofloxacin in various types of samples, Sens. Bio-Sens. Res., 2023, 39, 100547 CrossRef.
- A. M. Santos, A. Wong, A. A. Almeida and O. Fatibello-Filho, Simultaneous determination of paracetamol and ciprofloxacin in biological fluid samples using a glassy carbon electrode modified with graphene oxide and nickel oxide nanoparticles, Talanta, 2017, 174, 610–618 CrossRef.
- G. F. Alves, T. P. Lisboa, L. V. de Faria, D. M. de Farias, M. A. C. Matos and R. C. Matos, Disposable pencil graphite electrode for ciprofloxacin determination in pharmaceutical formulations by square wave voltammetry, Electroanalysis, 2021, 33, 543–549 CrossRef CAS.
- T. Matsunaga, T. Kondo, T. Osasa, A. Kotsugai, I. Shitanda, Y. Hoshi, M. Itagaki, T. Aikawa, T. Tojo and M. Yuasa, Sensitive electrochemical detection of ciprofloxacin at screen-printed diamond electrodes, Carbon, 2020, 159, 247–254 CrossRef CAS.
- M. Pan, P. Guo, H. Liu, J. Lu and Q. Xie, Graphene oxide modified screen-printed electrode for highly sensitive and selective electrochemical detection of ciprofoxacin residues in milk, J. Anal. Sci. Technol., 2021, 12, 55 CrossRef CAS.
- L. Qian, S. Durairaj, S. Prins and A. Chen, Nanomaterial-Based electrochemical sensors and biosensors for the detection of pharmaceutical compounds, Biosens. Bioelectron., 2021, 175, 112836 CrossRef CAS.
- N. S. Alsaiari, K. M. M. Katubi, F. M. Alzahrani, S. M. Siddeeg and M. A. Tahoon, The application of nanomaterials for the electrochemical detection of antibiotics: A review, Micromachines, 2021, 12, 308 CrossRef.
- Y. Zhou, R. Abazari, J. Chen, M. Tahir, A. Kumar, R. R. Ikreedeegh, E. Rani, H. Singh and A. M. Kirillov, Bimetallic metal–organic frameworks and MOF-derived composites: Recent progress on electro-and photoelectrocatalytic applications, Coord. Chem. Rev., 2022, 451, 214264 CrossRef CAS.
- M. Zhang, X. Li, J. Zhao, X. Han, C. Zhong, W. Hu and Y. Deng, Surface/interface engineering of noble-metals and transition metal-based compounds for electrocatalytic applications, J. Mater. Sci. Technol., 2020, 38, 221–236 CrossRef CAS.
- O. D. Okechukwu, E. Joseph, U. C. Nonso and N. O. Kenechi, Improving heterogeneous catalysis for biodiesel production process, Chem. Eng. Technol., 2022, 3, 100038 Search PubMed.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Organic–inorganic hybrid nanomaterials for electrocatalytic CO2 reduction, Small, 2020, 16, 2001847 CrossRef CAS.
- S. Zhang, S. Malik, N. Ali, A. Khan, M. Bilal and K. Rasool, Covalent and non-covalent functionalized nanomaterials for environmental restoration, Top. Curr. Chem., 2022, 380, 44 CrossRef CAS.
- N. Kajal, V. Singh, R. Gupta and S. Gautam, Metal organic frameworks for electrochemical sensor applications, Environ. Res., 2022, 204, 112320 CrossRef CAS.
- X. Fang, B. Zong and S. Mao, Metal–organic framework-based sensors for environmental contaminant sensing, Nano-Micro Lett., 2018, 10, 64 CrossRef.
- X. Zhang, S. Zhang, Y. Tang, X. Huang and H. Pang, Recent advances and challenges of metal–organic framework/graphene-based composites, Compos. B: Eng., 2022, 230, 109532 CrossRef CAS.
- N. Raza, T. Kumar, V. Singh and K.-H. Kim, Recent advances in bimetallic metal organic framework as a potential candidate for supercapacitor electrode material, Coord. Chem. Rev., 2021, 430, 213660 CrossRef CAS.
- A. Kumari, S. Kaushal and P. P. Singh, Bimetallic metal organic frameworks heterogeneous catalysts: Design, construction, and applications, Mater, Today Energy, 2021, 20, 100667 CrossRef CAS.
- J. Nicks, K. Sasitharan, R. R. R. Prasad, D. J. Ashworth and J. A. Foster, Metal–Organic Framework Nanosheets: Programmable 2D Materials for Catalysis, Sensing, Electronics, and Separation Applications, Adv. Funct. Mater., 2021, 31, 2103723 CrossRef CAS.
- J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, The chemistry of Ce-based metal–organic frameworks, Dalton Trans., 2020, 49, 16551–16586 RSC.
- Y. Cheng, X. Xiao, X. Guo, H. Yao and H. Pang, Synthesis of quasi-Ce-MOF electrocatalysts for enhanced urea oxidation reaction performance, ACS Sustain. Chem. Eng., 2020, 8, 8675–8680 CrossRef CAS.
- R. Rani, A. Deep, B. Mizaikoff and S. Singh, Copper based organic framework modified electrosensor for selective and sensitive detection of ciprofloxacin, Electroanalysis, 2020, 32, 2442–2451 CrossRef CAS.
- L. Tang, S. Zhang, Q. Wu, X. Wang and H. Wu, Heterobimetallic metal–organic framework nanocages as highly efficient catalysts for CO2 conversion under mild conditions, J. Mater. Chem. A, 2018, 6, 2964–2973 RSC.
- S. Wang, Z. Li, F. Duan, B. Hu, L. He, M. Wang, N. Zhou, Q. Jia and Z. Zhang, Bimetallic cerium/copper organic framework-derived cerium and copper oxides embedded by mesoporous carbon: Label-free aptasensor for ultrasensitive tobramycin detection, Anal. Chim. Acta, 2019, 1047, 150–162 CrossRef CAS.
- D. Rostamzadeh and S. Sadeghi, Ni doped zinc oxide nanoparticles supported bentonite clay for photocatalytic degradation of anionic and cationic synthetic dyes in water treatment, J. Photochem. Photobiol., A, 2022, 431, 113947 CrossRef CAS.
- J. Hua, Y. Jiao, M. Wang and Y. Yang, Determination of norfloxacin or ciprofloxacin by carbon dots fluorescence enhancement using magnetic nanoparticles as adsorbent, Microchim. Acta, 2018, 185, 1–9 CrossRef CAS.
- H. Bagheri, H. Khoshsafar, S. Amidi and Y. H. Ardakani, Fabrication of an electrochemical sensor based on magnetic multi-walled carbon nanotubes for the determination of ciprofloxacin, Anal. Methods, 2016, 8, 3383–3390 RSC.
- X. Zhang, F. Hou, H. Li, Y. Yang, Y. Wang, N. Liu and Y. Yang, A strawsheave-like metal organic framework Ce-BTC derivative containing high specific surface area for improving the catalytic activity of CO oxidation reaction, Microporous Mesoporous Mater., 2018, 259, 211–219 CrossRef CAS.
- Z. Guo, L. Song, T. Xu, D. Gao, C. Li, X. Hu and G. Chen, CeO2-CuO bimetal oxides derived from Ce-based MOF and their difference in catalytic activities for CO oxidation, Mater. Chem. Phys., 2019, 226, 338–343 CrossRef CAS.
- P.-P. Sun, Y.-M. Li, Y.-H. Zhang, H. Shi and F.-N. Shi, Preparation and application of Ce-Cu based metal organic framework/biomass carbon composites in energy storage, J. Alloys Compd., 2022, 896, 163081 CrossRef CAS.
- S. Rana and R. P. Singh, Investigation of structural, optical, magnetic properties and antibacterial activity of Ni-doped zinc oxide nanoparticles, J. Mater. Sci.: Mater. Electron., 2016, 27, 9346–9355 CrossRef CAS.
- P. Dong, L. Zhu, J. Huang, J. Ren and J. Lei, Electrocatalysis of cerium metal-organic frameworks for ratiometric electrochemical detection of telomerase activity, Biosens. Bioelectron., 2019, 138, 111313 CrossRef CAS.
- S. Li, N. Wang, Y. Yue, G. Wang, Z. Zu and Y. Zhang, Copper doped ceria porous nanostructures towards a highly efficient bifunctional catalyst for carbon monoxide and nitric oxide elimination, Chem. Sci., 2015, 6, 2495–2500 RSC.
- A. A. Ensafi, M. Taei, T. Khayamian and F. Hasanpour, Simultaneous voltammetric determination of enrofloxacin and ciprofloxacin in urine and plasma using multiwall carbon nanotubes modified glassy carbon electrode by least-squares support vector machines, Anal. Sci., 2010, 26, 803–808 CrossRef CAS.
- L. Fotouhi and M. Alahyari, Electrochemical behavior and analytical application of ciprofloxacin using a multi-walled nanotube composite film-glassy carbon electrode, Colloids Surf., B, 2010, 81, 110–114 CrossRef CAS PubMed.
- K. Cinková, D. Andrejčáková and Ľ. Švorc, Electrochemical method for point-of-care determination of ciprofloxacin using boron-doped diamond electrode, Acta Chim. Slovaca, 2016, 9, 146–151 CrossRef.
- N. Diab, I. Abu-Shqair, R. Salim and M. Al-Subu, The behavior of ciprofloxacin at a DNA modified glassy carbon electrodes, Int. J. Electrochem. Sci., 2014, 9, 1771–1783 CrossRef.
- A. Pollap, K. Baran, N. Kuszewska and J. Kochana, Electrochemical sensing of ciprofloxacin and paracetamol in environmental water using titanium sol based sensor, J. Electroanal. Chem., 2020, 878, 114574 CrossRef CAS.
- L. V. Faria, J. F. Pereira, G. C. Azevedo, M. A. Matos, R. A. Munoz and R. C. Matos, Square-wave voltammetry determination of ciprofloxacin in pharmaceutical formulations and milk using a reduced graphene oxide sensor, J. Braz. Chem. Soc., 2019, 30, 1947–1954 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08174e |
| This journal is © The Royal Society of Chemistry 2024 |