
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling therapeutic efficacy of extract and multi-targeting phytocompounds from Christella dentata (Forssk.) Brownsey & Jermy against multidrug-resistant Pseudomonas aeruginosa†
Md. Mashiar
Rahman
 *a,
Md. Rakibul
Islam
a,
Md. Enamul Kabir
Talukder
a,
Md. Farhan
Atif
a,
Rahat
Alam
a,
A. F. M. Shahab
Uddin
b,
K. M.
Anis-Ul-Haque
c,
Md. Saidul
Islam
d,
Mohammad Jashim
Uddn
e and
Shahina
Akhter
*f
*a,
Md. Rakibul
Islam
a,
Md. Enamul Kabir
Talukder
a,
Md. Farhan
Atif
a,
Rahat
Alam
a,
A. F. M. Shahab
Uddin
b,
K. M.
Anis-Ul-Haque
c,
Md. Saidul
Islam
d,
Mohammad Jashim
Uddn
e and
Shahina
Akhter
*f
aMolecular and Cellular Biology Laboratory, Department of Genetic Engineering and Biotechnology, Jashore University of Science and Technology, Jashore 7408, Bangladesh. E-mail: mm.rahman@just.edu.bd
bDepartment of Computer Science and Engineering, Jashore University of Science and Technology, Jashore 7408, Bangladesh
cDepartment of Chemistry, Jashore University of Science and Technology, Jashore 7408, Bangladesh
dKorea Institute of Radiological & Medical Sciences, 75, Nowon-ro, Nowon-gu, Seoul, South Korea
eDepartment of Pharmacy, Jashore University of Science and Technology, Jashore 7408, Bangladesh
fDepartment of Biochemistry and Biotechnology, University of Science and Technology Chittagong (USTC), Foy's Lake, Chittagong 4202, Bangladesh. E-mail: shahinabtge.med@ustc.ac.bd
First published on 16th February 2024
Abstract
Christella dentata (Forssk.) Brownsey & Jermy has been commonly used in traditional medicinal practices but its effects on multi-drug-resistant (MDR) bacteria have remained unexplored. We aimed to assess the in vitro antibacterial potential of the ethanol extract of Christella dentata (EECD) against MDR Pseudomonas aeruginosa and to identify potential multi-targeting antibacterial phytocompounds through computer-aided drug design focusing on the LasR and LpxC proteins. PPS, FT-IR and GC-MS were used for profiling of the phytocompounds in EECD. The antimicrobial activity of EECD was assessed using in vitro agar well diffusion, disc diffusion, MIC and MBC. Computer-aided drug design was used to identify multi-targeting leads from GC-MS-annotated phytocompounds. EECD exhibited dose-dependent antibacterial activity and revealed the presence of 51 phytocompounds in GC-MS analysis. Among these, three phytocompounds; (2E,4E)-N-isobutylhexadeca-2,4-dienamide (CID 6442402), bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy- (CID 536446) and 1,4-diethylbenzene (CID 7734) were identified as promising antibacterial phytocompounds as they strongly bonded with LasR and LpxC. Of them, CID 536446 and CID 7734 exhibited multiple targeting abilities with LasR and LpxC. On further screening, both CID 536446 and CID 7734 exhibited favorable drug-able, pharmacokinetics and toxicity properties. Finally, molecular dynamics (MD) simulation proved the binding stability of bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy- and 1,4-diethylbenzene to active pockets of LasR and LpxC. The results of this study offer scientific validation for the traditional use of Christella dentata in bacterial infection-related diseases. It also suggests that bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy- and 1,4-diethylbenzene from Christella dentata might be responsible for the antibacterial activity and could act as phytopharmacological leads for the development of LasR and LpxC inhibitors against MDR P. aeruginosa.
1. Introduction
Eliminating Pseudomonas aeruginosa has become progressively challenging owing to its impressive ability to resist antibiotics. Strains of P. aeruginosa effectively employ both inherent and acquired resistance mechanisms to counteract the impact of most antibiotics. Notably, P. aeruginosa has recently displayed adaptive antibiotic resistance mechanisms, encompassing resistance facilitated by biofilm formation and the emergence of multidrug-tolerant persister cells. These mechanisms contribute to the stubborn nature of infections, leading to persistence and recurrence.1 In this situation, current biomedical science requires the urgent need for improved disease-modifying treatments against multidrug-resistant (MDR) microorganisms, especially P. aeruginosa. The World Health Organization prioritizes the development of innovative therapies for P. aeruginosa, while the U.S. Centers for Disease Control considers it a significant issue. Infections caused by P. aeruginosa, including those acquired in healthcare settings, ventilator-related pneumonia, as well as conditions like cystic fibrosis, cancer, trauma, COPD, burns and infections after surgery, make up about 7.1–7.3% of all infections globally and contribute to high mortality rates worldwide.2 Confronted with a range of environmental stimuli, bacteria develop adaptive resistance strategies, leading to increased antibiotic resistance by inducing temporary alterations in gene and/or protein expression. As for P. aeruginosa, this acclimatized recalcitrance involves the development of biofilms. These biofilms act as barriers that impede antibiotic penetration to bacterial cells, consequently augmenting pathogenicity.3P. aeruginosa utilizes quorum sensing to control its virulency and the development of biofilms.1,2 Quorum sensing (QS) enables bacterial communication through signaling molecules, prompting the bacterial population to work together with coordinated metabolic activities. This process produces autoinducers that spread into both bacterial and host cells, triggering transcriptional regulation. This regulation supports the bacteria's survival, reduces the immune response to infections, and enhances resistance to antimicrobial treatments.2,4P. aeruginosa possesses two similar QS systems that are regulated by the genes lasR/lasI and rhlR/rhlI.5,6 The lasI gene produces LasI, accountable for the synthesis of a QS molecule 3-oxo-dodecanoyl homoserine lactone (3OC12-HSL), and the rhlI gene produces RhlI, which produces another QS signaling molecule N-butyryl-l-homoserine lactone (C4-HSL). The LasR and RhlR receptors are produced from the lasR and rhlR genes, respectively. When the QS signaling molecules (3OC12-HSL and C4-HSL) reach a threshold concentration, they bind to their respective receptors, forming complexes known as LasR:3OC12-HSL and RhlR-C4-HSL. The LasR:3OC12-HSL complex controls the activation of genes responsible for hemolysin, protease, elastase, and exotoxin-A production, crucial for the formation of biofilm.7 The RhlR-C4-HSL complex produces virulence factors like pyocyanin, elastases and rhamnolipid and swarming motility factors, crucial for biofilm establishment.6,8 However, these two QS systems are structured hierarchically, with the RhlR/RhlI system sited as subservient to the LasR/LasI system, as the production of rhlR and rhlI relies on LasR.5 QS system studies have shown that P. aeruginosa LasR mutants exhibit significantly reduced virulence and invasiveness in various in vivo infection models. Sitagliptin, a drug used to treat type 2 diabetes, has been found to interact with the LasR receptor in P. aeruginosa and effectively suppress biofilm production at inhibitory concentrations.9 LpxC stands as a pivotal zinc metalloenzyme critical for the survivability of numerous Gram-negative bacteria, including P. aeruginosa. It catalyzes the primary critical stage in the formation of lipid A (endotoxin), the building block of lipopolysaccharides (LPSs), which is an important element of the cell wall, biofilm formation, and antibiotic resistance of the mostly of Gram-negative bacteria, inclusive of P. aeruginosa.3,10–12 LpxC is greatly salvaged in majority of Gram-negative bacterial strains and does not possess its homolog in the human genome.13 Taking into account the interconnected functions of LasR and LpxC in virulence, biofilm formation, and antibiotic resistance, both proteins present themselves as appealing targets for novel antibacterial discovery. It has been indicated that addressing the emergence and propagation of drug-resistant infectious organisms necessitates a multifaceted approach, as no singular or uncomplicated strategy is deemed sufficient.14
Natural plant products have gained significant interest for their capacity to provide a broad spectrum of structurally diverse compounds with multi-targeting antimicrobial functionality, which disrupt essential cellular activities and present potential for developing antibacterial agents.15 Moreover, the traditional use of medicinal plants in folk medicine offers valuable insights and references for developing antimicrobial compounds from natural sources.16 Medicinal plants possess various bioactive phytochemicals that exhibit clinically relevant antimicrobial properties and are less susceptible to the development of bacterial resistance.14 To date, there are no reports of bacteria developing resistance to plant-based antimicrobials. For example, coumarins derived from plants show potent antibacterial action averse to Staphylococcus aureus.17 Berberine, a compound derived from plants, showcases potent antibacterial properties averse to Gram-positive bacteria, encompassing drug-resistant strains of Staphylococcus aureus and Mycobacterium tuberculosis.18,19 Plumbagin, a compound found in plants, serves as a growth inhibitor for pathogenic bacteria such as Escherichia coli, Enterobacter aerogenes, Klebsiella pneumoniae, and P. aeruginosa.20,21
Christella dentata (Forssk.) Brownsey & Jermy, commonly known as the Toothed Cloak Fern, is a medicinal plant classified under the Thelypteridaceae family.22 The Toothed Cloak Fern, native to tropical and subtropical regions, can be found in diverse locations across the globe.23 It is characterized by its distinctive fronds, which have toothed edges, hence the name “dentata.” In traditional medicine systems, Christella dentata has been used to treat skin infections, cuts, and wounds.22,24 Extracts from Christella dentata have shown potential antimicrobial, antifungal, anti-gout and anti-rheumatism properties in various studies.25 These properties make it a promising candidate for the development of natural remedies to fight infection caused by microorganisms. Additionally, the plant's availability and cultural significance in traditional medicine systems highlight its importance as a valuable medicinal plant. Further research and exploration of the bioactive components and potential health benefits of Christella dentata hold promise for the evolution of new therapeutic interventions. Although Christella dentata holds significance in traditional medicine, its antibacterial effects and the molecular mechanisms of its compounds against multidrug-resistant bacteria have yet to be explored.
Our research strives to identify compounds capable of targeting multiple signaling pathways in MDR P. aeruginosa, aiming to effectively thwart its robust resistance mechanisms. It would be promising approach to combined target of LasR and LpxC with a single molecule. Therefore, this study concentrated on examining the antibacterial properties and pharmacological characteristics of phytocompounds from Christella dentata. Specifically, the study targeted P. aeruginosa's virulence and multidrug resistance by focusing on biofilm formation and the synthesis of lipid A in the bacterial outer membrane. This comprehensive approach included lab experiments, computer-based evaluations of pharmacokinetics, toxicity, and drug characteristics, along with MD simulations to validate the efficacy of the identified compounds. The findings from this research are anticipated to offer valuable insights regarding the prospective medicinal applications of Christella dentata in combating MDR P. aeruginosa, and they are likely to contribute to the development of innovative approaches for addressing infections associated with multidrug-resistance.
2. Materials and methods
2.1. Chemicals and reagents
Methanol, HCl, H2SO4, lead acetate, sodium hydroxide and ethanol were acquired from Wako Pure Chemicals Ind., Ltd, Japan. NaCl, LB media, and bacto agar were obtained from Liofilchem in Italy. Fehling's solution A and B, FeCl3, CuSO4, sodium nitroprusside, DMSO, CHCl3 were obtained from Merck in Germany. Ampicillin discs were obtained from Bio-Rad in USA. Anhydrous Na2CO3, ninhydrin and sodium citrate were acquired from Sigma-Aldrich in Germany.2.2. Plant material
Dr Sardar Nasiruddin, a taxonomist from the National Herbarium in Dhaka, Bangladesh, verified the plant's identification and preserved a specimen labeled as DACB 326 in the National Herbarium. After collection, the plant parts of the Christella dentata were cleansed with flowing tap water and then air-dried in a room with air conditioning at around 25 °C. The dried plant material was subsequently finely pulverized into a powder and kept in a tightly sealed container for further use.2.3. Bacterial strains for in vitro activity assessment
A glycerol stock containing a strain of P. aeruginosa (Gene Bank Accession Number: OK355439) that is resistant to several antibiotics. This bacterial strain was isolated and identified from wastewater in a medical facility. Its antibiotic susceptibility was assessed by testing it against various antibiotics, including Amoxicillin, Azithromycin, Ciprofloxacin, Doxycycline, Erythromycin, Gentamicin, Levofloxacin, Metronidazole, Streptomycin, and Tetracycline.262.4. Preparation of plant extract
Following the methodology outlined in a prior investigation with slight changes, an ethanol extract of Christella dentata's aerial parts (EECD) was prepared.27 A total of 100 grams of powdered plant material were split into four separate 500 mL conical flasks. To each flask, 100 mL of ethanol was introduced, and the flasks were then kept within a shaking incubator (JSSI-300T, JSR, South Korea), undergoing agitation at 250 rpm for 72 h at 37 °C. After this agitation period, the mixture underwent centrifugation at 8000 rpm for 15 minutes. The resulting precipitates were mixed again with an equivalent volume of ethanol and placed in a incubator for agitation an additional 48 hours. The afloat part was sieved through filter paper of Whatman no. 1 (Lab Asia Science & Technology Corporation, Bangladesh). The filtrate was then concentrated under vacuum by a rotatory dehydrator (DLAB Scientific Inc., CA, USA) at RT to eliminate any residual solvent. The condensed extract was balanced and stowed within a sterile 50 mL conical tube in a refrigerator at 4 °C for future experimentation. 6.5 grams of crude ethanol extract, equivalent to 6.5% by weight, was obtained from 100 grams of the initial powdered plant material.2.5. In vitro antibacterial activity assay
2.6. Analytical analysis of ethanol extract of aerial parts of Christella dentata
2.7. In silico deciphering of antibacterial activity of EECD
2.8. Statistical analysis
The results of the antibacterial activity tests were presented as the average value along with the standard deviation (STDEV) obtained from three separate replicates. These tests were conducted at different concentrations of EECD.3. Results
3.1. Antibacterial effects of EECD
Due to the challenges posed by multi-drug resistance, we performed an investigation to assess the antibacterial potential of EECD against MDR P. aeruginosa. Our results demonstrated that EECD hindered the proliferation of MDR P. aeruginosa in a dose-proportionate manner, with inhibition zones ranging from 18.66 ± 5.03 to 25.33 ± 6.50 mm in the agar well diffusion assay and from 13.33 ± 2.51 to 19 ± 2.64 mm in the disc diffusion assay when tested at concentrations ranging from 62.5 to 500 μg mL−1 (Fig. 1A). Apparently, the agar well diffusion assay exhibited higher antimicrobial activity for EECD compared to the disc diffusion assay (Fig. 1B). Interestingly, the antibiotic ampicillin, which is ineffective against MDR P. aeruginosa due to its known resistance, showed no antibacterial activity. The MIC of EECD was determined to be 225 μg mL−1, whereas the minimum concentration at which it is bactericidal (MBC) was identified as 500 μg mL−1 (Fig. 1C and D). It is worth noting that a greater amount of EECD was needed in the MBC assay to achieve complete eradication of bacteria compared to the concentration needed to inhibit visible in vitro bacterial growth in the MIC assay.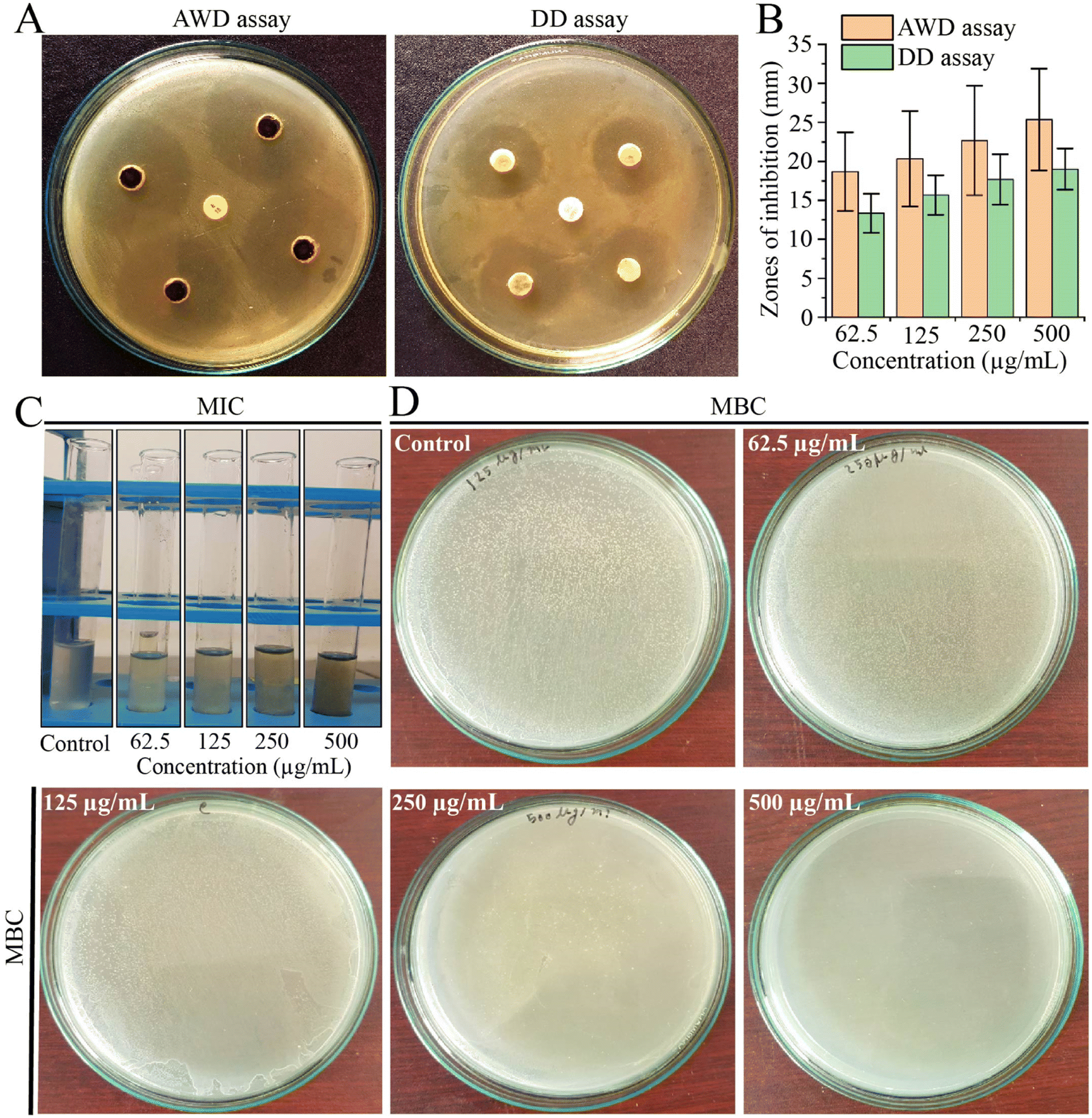 | ||
| Fig. 1 Antibacterial activity of EECD against MDR P. aeruginosa. (A) The AWD and DD assays revealed areas of growth inhibition of MDR P. aeruginosa caused by EECD. (B) The zones of inhibition in the AWD and DD assays were measured in millimeters (mm) to assess the extent of inhibition caused by EECD. (C) The MIC in μg mL−1 and (D) the MBC in μg mL−1 of EECD. AWD = agar well diffusion; DD = disc diffusion. | ||
3.2. Preliminary phytochemical screening of EECD
To assess the nature of phytochemicals in the aerial parts of Christella dentata (Forssk.) Brownsey & Jermy (ESI Fig. S1†), standard color change methods were employed to detect the presence of phytocompounds mentioned in ESI Table S1 and Fig. S2.† Alkaline reagent tests using 5% NaOH solutions indicated the presence of flavonoids, turning the solution light-yellow, which fades to colorless upon adding 10% HCl. Tannins were identified by the formation of a brownish-black color upon treatment with 3 and 4 drops of a 5% FeCl3 solution and an intense yellow color upon treatment with 10% lead acetate solution. Salkowski's test revealed the presence of terpenoids with a greenish-brown layer at the upper surface and the presence of steroids with a greenish-brown layer at the bottom. Foaming and frothing experiments indicated the presence of saponins, showing 10 minutes foaming stability on the tops of the test tubes. Benedict's test displayed a greenish-yellow layer, indicating the presence of traces of reducing sugar. The sodium nitroprusside test exhibited a dark-brown color, signifying the presence of a ketone, while the ninhydrin test produced an intense yellow color instead of a violet-blue color, confirming the absence of amino acids.3.3. Analysis of functional groups in EECD
FTIR spectral analysis aids in the annotation of phytocompounds by detecting characteristic peak positions in the spectrum, corresponding to specific bond vibrations. Recognizing these bonds enables the identification of functional groups and confirms the presence of a compound in a particular natural or synthetic source. As depicted in ESI Fig. 2,† the FTIR spectra results displayed distinct peaks, confirming the presence of various functional groups (ESI Table S2†) in EECD. Notably, EECD exhibited peaks at 3650–3000 (N–H stretch) for primary and secondary amines or amides, 2926.14 and 2853.81 (C–H) for aldehyde, 1737.94 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) for ester, 1638.60 for aromatic, 1456.36 (–CH3 bend) for alkanes, 1402.31 (CC stretch) for aromatic alkene, 1076.33 and 417.57 (C–X) for alkyl halide, and 594.10 (C–Br) for alkyl bromide. In this study, the major peak was found at 1638 and 1632 cm−1 indicating the existence of an aromatic CC bond. This is obvious as most of the phytocompounds contain a lot of aromatic compounds, and this (aromatic CC) bond should be much higher than any other bond in the sample. The broad peak at around 3650–3000 cm−1 may come from primary and secondary amines (N–H) or amides. The trace of carbonyl (CO) bond for the ester functional group was also found in the study of FTIR spectral analysis at around 1737 cm−1. The presence of different hydrocarbons, alkyl halide, aldehyde, and aromatic components was found in the FTIR spectral analysis (Fig. 2).
O) for ester, 1638.60 for aromatic, 1456.36 (–CH3 bend) for alkanes, 1402.31 (CC stretch) for aromatic alkene, 1076.33 and 417.57 (C–X) for alkyl halide, and 594.10 (C–Br) for alkyl bromide. In this study, the major peak was found at 1638 and 1632 cm−1 indicating the existence of an aromatic CC bond. This is obvious as most of the phytocompounds contain a lot of aromatic compounds, and this (aromatic CC) bond should be much higher than any other bond in the sample. The broad peak at around 3650–3000 cm−1 may come from primary and secondary amines (N–H) or amides. The trace of carbonyl (CO) bond for the ester functional group was also found in the study of FTIR spectral analysis at around 1737 cm−1. The presence of different hydrocarbons, alkyl halide, aldehyde, and aromatic components was found in the FTIR spectral analysis (Fig. 2).
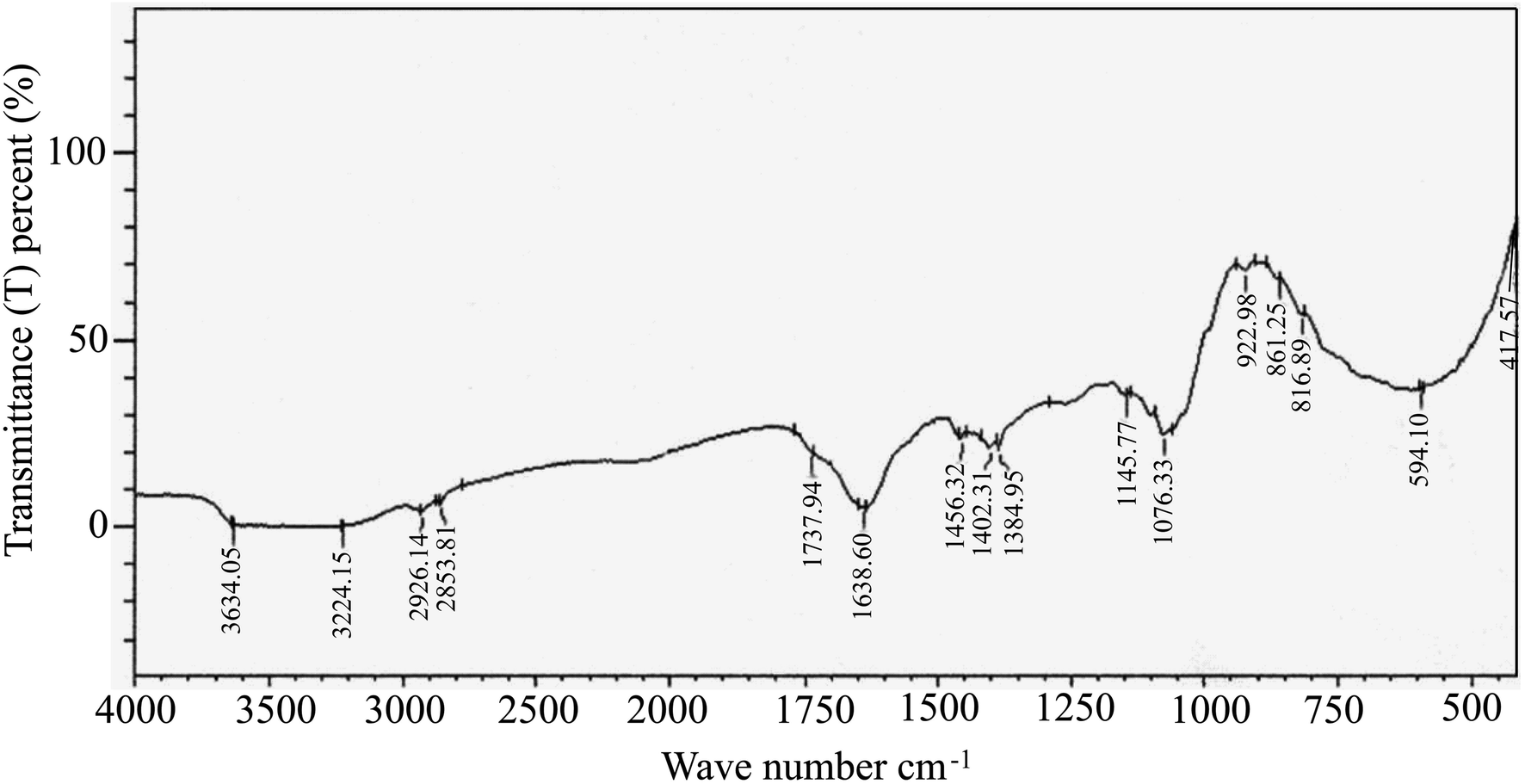 | ||
| Fig. 2 Fourier transform-infrared spectra (FT-IR) of EECD. (A) Peaks at 3650–3000, 2926.14 and 2853.8, 1737.94, 1638.6, 1456.36, 1402.31, aromatic alkene (CC stretch), 1076.33 and 417.57 and 594.10 cm−1 indicate the presence of corresponding functional groups in EECD: primary and secondary amines or amides (N–H stretch), aldehyde (C–H), ester (CO), aromatic (CC), alkanes (–CH3 bend), alkyl halide (C–X), alkyl bromide (C–Br), respectively. | ||
3.4. GC-MS analysis of EECD
The GC-MS chromatogram of metabolite annotation for EECD revealed 52 distinct peaks (Fig. 3). Among them, 18 peaks exhibited a similarity of ≥90% based on the MS fragmentation pattern or mass of the observed compound compared to entries in the NIST library. Two peaks were identified as the same compound, with a similarity to the NIST library of less than 90%. Consequently, the remaining 33 peaks are regarded as unknown compounds. As shown in ESI Table S3,† each of these peaks corresponds to a particular phytocompound, and their relative proportions were calculated by comparing the average area of each peak to the total area under the retention time (RT) curve. The relative content of phytocompounds within EECD was assessed corresponding the total peak area (%), and their order of abundance was as follows: alkane > terpenoids > esters > alkene > phenolics > alkyl halide > aromatic hydrocarbon. Notably, the prominent phytocompounds identified in EECD included bicyclo[4.3.0]nonane (7.88%), 2,2,6,7-tetramethyl-7-hydroxy (5.29%), tetradecane (3.34%), octadecane, 1-chloro (3.24%), and ethyl 13-methyltetradecanoate. | ||
| Fig. 3 The GC-MS chromatogram of metabolite annotation for EECD showing different peaks demonstrating various compounds. | ||
3.5. Molecular docking and MM-GBSA analysis
As shown in Fig. 4, 5 and ESI Table S4,† in molecular docking of 51, three phytocompounds exhibited notable negative binding affinities, ranging from −7.169 to −7.383 kcal mol−1 for LasR (Fig. 4) and −6.826 to −6.984 kcal mol−1 for LpxC (Fig. 5), with higher negative binding affinity compared to control drug, ampicillin (CID 6249) (−6.333 kcal mol−1). During the MM-GBSA analysis of five phytocompounds with higher molecular docking scores compared to the control drug ampicillin, three specific phytocompounds (CID 6442402, CID 7734, and CID 536446) showed higher ΔG binding energies when interacting with LasR, measuring −46.59, −39.48, and −16.56 kcal mol−1, in contrast to ampicillin's binding energy of −7.17 kcal mol−1. CID 536446 and CID 7734 also exhibited greater ΔG binding energies with LpxC of −31.5 and −29.15 kcal mol−1 sequentially than ampicillin (−29.02 kcal mol−1). Additionally, our analysis of LasR–ligand and LpxC–ligand complexes revealed diverse interaction energies, such as Coulomb energy (ΔGBind Coulomb), ΔGBind covalent, hydrogen bond energy (ΔGBind H-bond), lipophilicity energy (ΔGBind Lipo), ΔGBind packing, ΔGBind Solv GB, and van der Waals interaction energy (ΔGBind vdW). These different energy factors works together to collectively improve the overall stability of the complexes. | ||
| Fig. 4 The interaction between LasR and three selected phytocompounds, as well as the control drug ampicillin, is depicted in both 3D (left) and 2D (right) formats. Panels (A–C), and (D) showed CID 6442402, CID 536446, CID 7734, and CID 6249 (control drug, ampicillin), respectively, bound in the active pocket of the LasR protein. | ||
 | ||
| Fig. 5 The molecular docking interactions between LpxC and two specific phytocompounds, along with the control drug ampicillin, are explained in 3D (left) and 2D (right) formats. Panels (A–C) depict CID 536446, CID 7734 and CID 6249 (control drug, ampicillin) respectively, bound within the active pocket of the LpxC protein. | ||
3.6. Multi-targeting capabilities of phytocompounds
Based on molecular docking and MM-GBSA results represented in ESI Table S4,† CID 536446 and CID 7734 showed interaction with LasR and LpxC with higher binding energies with multi-targeting capabilities with LasR and LpxC. CID 6442402 showed only interaction with LasR with higher binding energies. Hence, we ultimately chose CID 536446 and CID 7734 as multi-targeting phytocompounds for advanced in silico analysis within the framework of an ideal drug development strategy.3.7. Examination of molecular characteristics and assessment of toxicity for the chosen phytocompounds
The pharmacokinetics (PK) associated with ADME of drug candidates are significantly influenced by their physicochemical properties. During this study, the SwissADME server was utilized to test the physicochemical properties such as MW, HBA, HBD, RB and TPSA of the selected pair of phytocompounds. As demonstrated in ESI Table S5,† both phytocompounds exhibit molecular weights (≤500 g mol−1), HBA (≤10), HBD (≤5), and TPSA values (≤140 Å2) within the desirable extent, suggesting a potential for high oral bioavailability. The optimal RB range is 0 to 11, and our phytocompounds possess RBs of 0 to 2, aligning well with favorable absorption. A c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP value of 1> to <5 typically signifies strong absorption, and both our phytocompounds meet this criterion with clogP values ranging from 3.26 to 4.47. The logS value, which ideally remains low to enhance drug solubility, conforms to an acceptable range of −4.0 to 0.5. Both selected phytocompounds demonstrate logS values of −3.32 to −3.87. The bioavailability of a drug is significantly influenced by human intestinal absorption (HIA), thus HIA calculations were conducted. CID 536446 exhibited high GI absorption, while CID 7734 and the control drug (ampicillin, CID 6249) displayed low GI absorption. Of the two phytocompounds, CID 536446 adhered to Lipinski's rule of five, whereas CID 7734 violates one of the five rules. Both compounds indicated ease of synthesis within medicinal chemistry. Regarding toxicity, both compounds demonstrated favorable outcomes in computational assessments, indicating they are non-hepatotoxic, non-immunogenic, non-mutagenic, and non-cytotoxic.
logP value of 1> to <5 typically signifies strong absorption, and both our phytocompounds meet this criterion with clogP values ranging from 3.26 to 4.47. The logS value, which ideally remains low to enhance drug solubility, conforms to an acceptable range of −4.0 to 0.5. Both selected phytocompounds demonstrate logS values of −3.32 to −3.87. The bioavailability of a drug is significantly influenced by human intestinal absorption (HIA), thus HIA calculations were conducted. CID 536446 exhibited high GI absorption, while CID 7734 and the control drug (ampicillin, CID 6249) displayed low GI absorption. Of the two phytocompounds, CID 536446 adhered to Lipinski's rule of five, whereas CID 7734 violates one of the five rules. Both compounds indicated ease of synthesis within medicinal chemistry. Regarding toxicity, both compounds demonstrated favorable outcomes in computational assessments, indicating they are non-hepatotoxic, non-immunogenic, non-mutagenic, and non-cytotoxic.
3.8. MD simulation
To evaluate the stability of the top two potential candidates (CID 536446 and CID 7734) within the protein's binding site, we conducted molecular dynamics simulations on the protein–ligand complex structure. The assessment involved analyzing parameters such as RMSD, RMSF, Rg and SASA to evaluate the binding stability of CID 536446 and CID 7734 with both LasR and LpxC. | ||
| Fig. 6 MD simulation of the selected ligand and LasR complexes deliberated from a 100 ns simulation. (A) The extracted RMSD values from Cα atoms of the protein–ligand docked complex. The RMSD of the LasR protein as apoprotein and control drug (ampicillin) are shown in orange and blue color while the selected two compounds CID 536446 and CID 7734 in complex with the LasR protein were characterized by gray and yellow color separately. (B) The RMSF values were derived from the protein Cα atoms of the docked protein–ligand complex. The RMSF of the LasR apoprotein is depicted in orange, the control drug (ampicillin) in blue and the designated two phytocompounds CID 536446 and CID 7734 in complex with the LasR protein in gray and yellow color. (C) The Rg of the protein–ligand complexes. The Rg value of the selected two phytocompounds CID 536446 and CID 7734 and the control drug (ampicillin) in complex with the LasR delineated by orange, gray and yellow color, respectively. (D) A graphic presentation of the protein–ligand complex's SASA. The SASA value of the specific two phytocompounds (CID 536446 and CID 7734) and the control drug (ampicillin) in complex with the LasR denoted by orange, gray, and yellow color, respectively. | ||
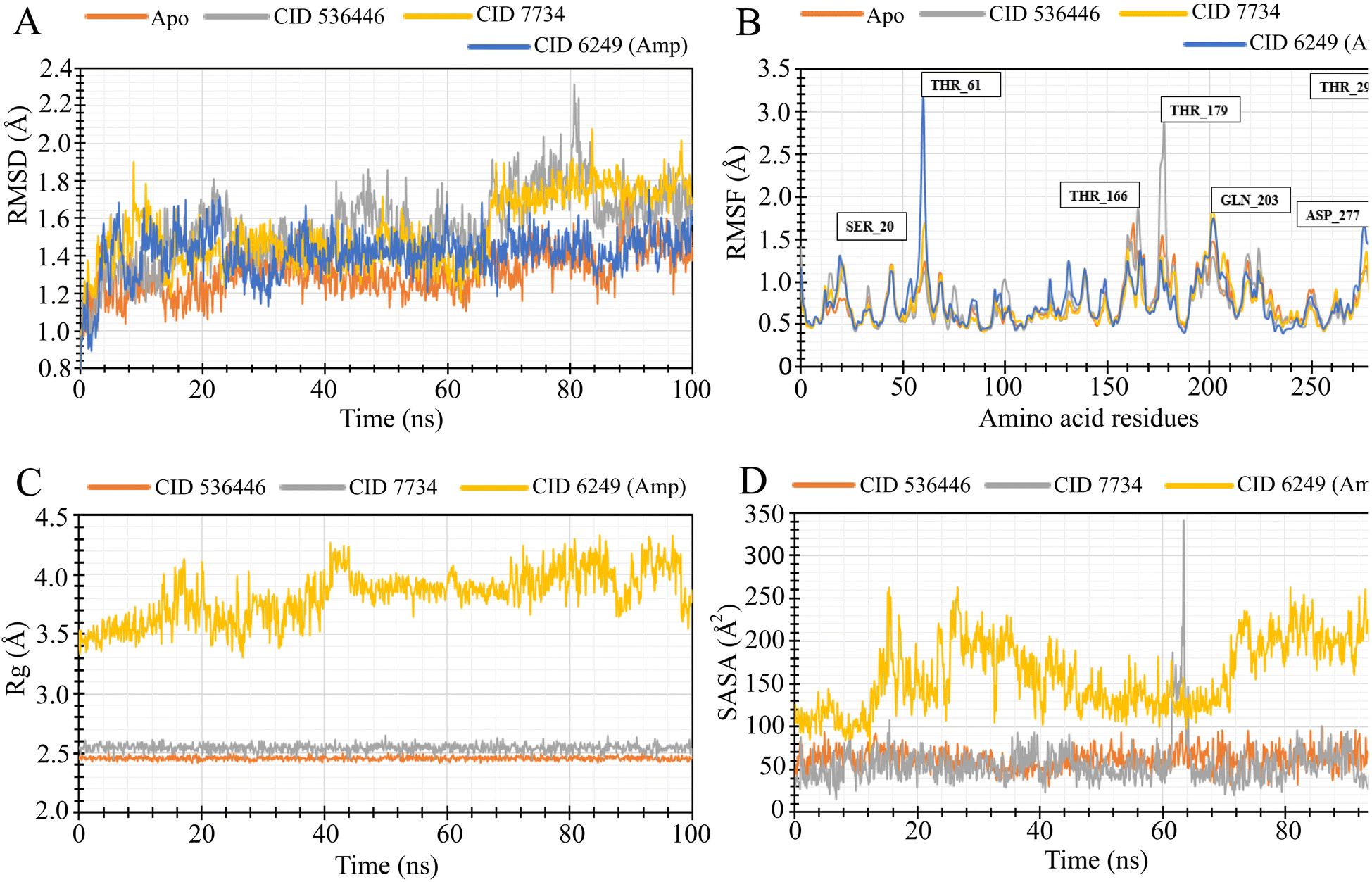 | ||
| Fig. 7 MD simulation of the selected ligand and LpxC complexes deliberated from a 100 ns simulation. (A) The RMSD values obtained from the Cα atoms of the LpxC–ligand docked complex were extracted. The RMSD of the LpxC protein as apoprotein and the control drug (ampicillin) are shown in orange and blue color while the selected two phytocompounds (CID 536446 and CID 7734) in complex with the LpxC protein were signified by gray and yellow color separately. (B) The RMSF values were obtained from the Cα atoms of the protein in the LpxC–ligand docked complex. The RMSF of the LpxC apoprotein is depicted in orange, the control drug (ampicillin) in blue and the designated two phytocompounds CID 536446 and CID 7734 in complex with the LpxC protein in gray and yellow color. (C) The Rg of the LpxC–ligand complexes. The Rg value of the selected two phytocompounds CID 536446 and CID 7734 and the control drug (ampicillin) in complex with the LpxC denoted by orange, gray and yellow color, respectively. (D) A graphic representation of the protein–ligand complex's SASA. The SASA value of the specific two phytocompounds (CID 536446 and CID 7734) and the control drug (ampicillin) in complex with the LpxC denoted by orange, gray, and yellow color, respectively. | ||
3.9. Protein–ligand bonding interactions
The arrangement of proteins bound to the selected ligands and their interactions at the molecular level were observed during a 100 ns simulation, utilizing the simulation interactions diagram. Incorporating factors such as hydrogen bonding, non-covalent interactions (hydrophobic bonding), ionic bonding, and water bridge bonding, the interactions between the proteins (LasR and LpxC) and the designated compounds (CID 536446 and CID 7734) were investigated and depicted in Fig. 8 and ESI Table S6.† Over the duration of the 100 ns simulation, both compounds were observed to partake in a variety of interactions, including hydrogen bonding, hydrophobic interactions, ionic bonding, and water bridge bonding. These interactions persisted throughout the simulation duration, contributing to a stable binding between the target protein and the compounds. CID 536446 established hydrogen bonding, hydrophobic interactions, and water bridge bonding with both LasR and LpxC. In contrast, CID 7734 engaged with LasR and LpxC exclusively through hydrophobic bonding. In contrast, the LasR's native ligand (ampicillin) established connections through hydrogen bonding, hydrophobic interactions, and water bridge bonding. On the other hand, when interacting with LpxC, ampicillin engaged in hydrogen bonding, hydrophobic interactions, water bridge bonding, and ionic bonding.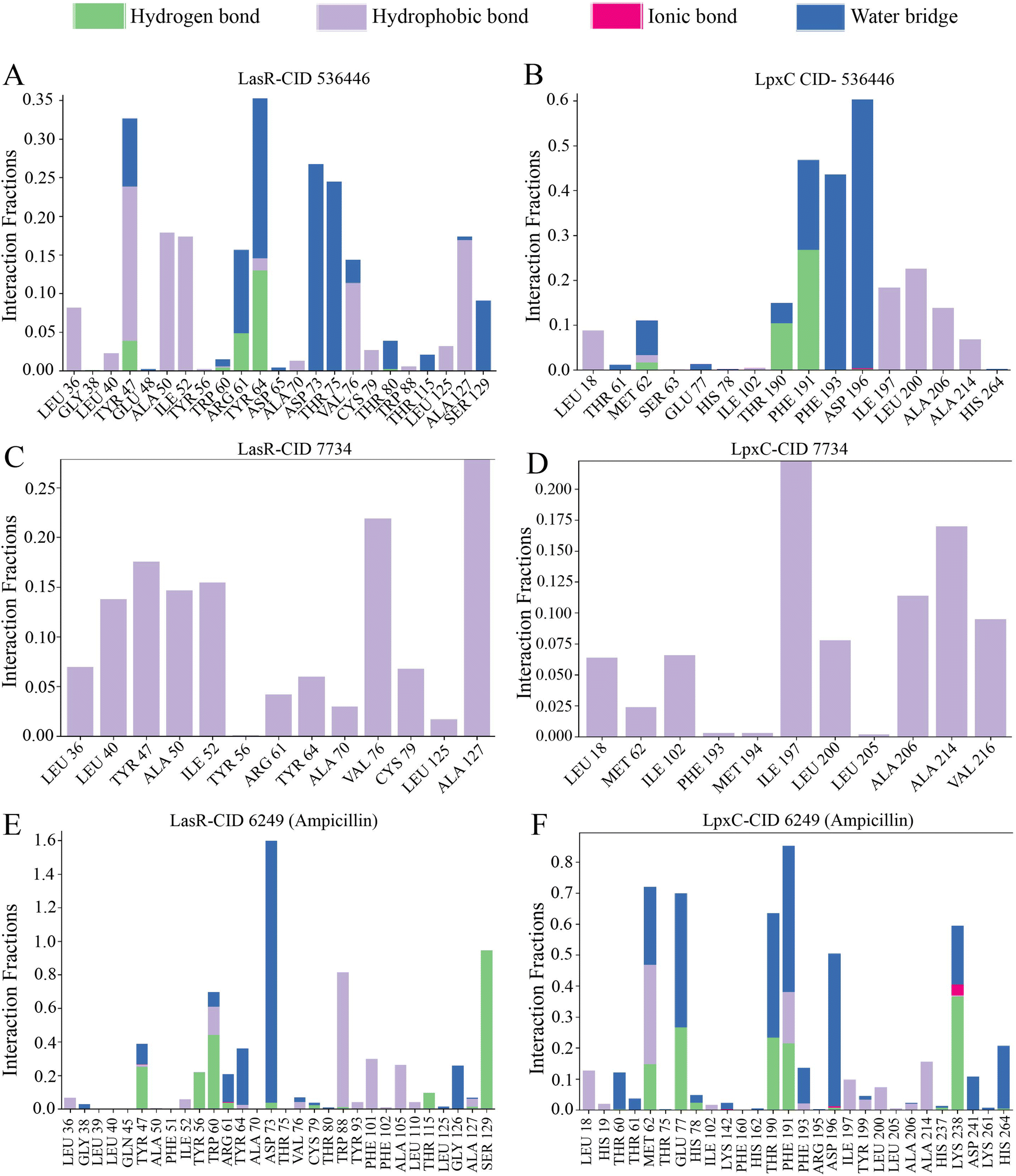 | ||
| Fig. 8 The stacked bar graph represented the interactions between proteins and ligands observed throughout the 100 ns simulation. Here, the interaction between two specific phytocompounds and the native ligand with both LasR and LpxC is demonstrated. (A) LasR-CID 536446, (B) LpxC-CID 536446, (C) LasR-CID 7734, (D) LpxC-CID 7734, (E) LasR-CID 6249 and (F) LpxC-CID 6249. | ||
4. Discussion
Due to multidrug-resistant nature, Pseudomonas aeruginosa offers a significant public health risk, particularly in healthcare settings like hospitals and intensive care units.41 The current methods of treatment require combining antibiotics, but the rise of drug resistance and the occurrence of adverse effects emphasized the necessity for novel antibacterial medications.42 Within this backdrop, there is an increasing fascination with investigating phytocompounds as potential antimicrobial agents, due to their wide range of chemical compositions and biological functions, along with their limited adverse consequences.While previous research has explored the antimicrobial capabilities of extracts against various pathogens, there was a gap of information regarding their effectiveness against MDR bacteria, including P. aeruginosa. Therefore, our study was focused on investigating the lethal impact of extracts obtained from the aerial parts of Christella dentata and their phytochemical components on MDR P. aeruginosa. In this research, we evaluated lethal effect of EECD and subsequently, the phytocompounds in EECD, with a specific focus on targeting LasR and LpxC. LasR plays a key role in drug resistance,43 while LpxC is pivotal for lipid-A biosynthesis, a critical component of bacterial outer membrane formation.13
GC-MS analysis initially annotated 51 distinct phytocompounds. The efficacy of these phytocompounds as antibacterial agents was evaluated using molecular docking and MM-GBSA analysis, with a special emphasis on LasR and LpxC. Phytoligand–protein docking analysis revealed three phytocompounds exhibiting stronger binding affinity. Among these three phytocompounds, two phytocompounds (CID 536446 and CID 7734) demonstrated multi-targeting ability. Subsequently, the physicochemical properties, lipidophilicity, solubility in water, pharmacokinetics, RO5, medicinal chemistry and toxicity profiles of these two phytocompounds were assessed. The results were favorable, except for one violation of RO5 by CID 7734. These findings render these two phytocompounds attractive candidates for antimicrobial therapeutics, leading to their selection for additional MD simulation studies. MD simulations were conducted to evaluate the steadiness and stability in the structure of the protein–ligand complexes. Both phytocompounds CID 536446 and CID 7734 exhibited stable interactions with both LasR and LpxC, as evidenced by the analysis of RMSD, RMSF, Rg, and SASA, affirming their potentiality as effective antibacterial phyto-agent averse to MDR P. aeruginosa.
The lead phytochemical, CID 536446, named bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy-, falls within the category of oxygenated terpenoids and is a major phytocompound in EECD. In accordance with our results, it has been reported that oxygenated terpenoids exhibit strong antibacterial activity, particularly against Gram-negative.44 Our findings align with research demonstrating that carvacrol, thymol and cinnamaldehyde, also classified as oxygenated monoterpenes, inhibit quorum sensing by suppressing the self-inducer of bacterial quorum sensing signaling molecules, namely, acyl homoserine lactone (AHL).45 Both of our lead phytochemicals (CID 536446 and CID 7734) exhibited dual binding affinity and inhibitory activity with LasR and LpxC proteins. LasR is a receptor of quorum sensing signaling molecule in QS system where LpxC is a key enzyme that catalyzes the synthesis of lipopolysaccharides (LPSs) in the bacterial cell wall and cell membrane.6,12 Our results also align with research indicating that terpenoids, due to their lipophilicity, can impair the cell membrane of bacteria.45 Moreover, terpenoids also exhibits various biological functions, ranging from anti-tumor effects and cardiovascular impacts to anti-inflammatory properties.46 Ethyl acetate is known for its effectiveness in extracting terpenoids,47 our research goal extended beyond terpenoids isolation alone. Ethanol, a more polar solvent than ethyl acetate, allows for the extraction of a wider array of bioactive compounds, including polyphenols,48 flavonoids49 and other polar constituents in addition to terpenoids which are of significant interest in our investigation. In accordance with our research, bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy have also been identified in the methanol extract of Dicliptera roxburghiana, demonstrating appreciable anticancer characteristics,50 additionally, this compound has been found in Nicotiana tabacum51 and in the leaf and stem of Marsilea quadrifolia (L.).52 Another promising lead phytochemical, 1,4-diethylbenzene (CID 7734), was exclusively annotated in Christella dentata in our study. Similar to our findings, another research study detected the presence of 1,4-diethylbenzene in the chloroform and ethyl acetate fractions of metabolites from Penicillium species, namely P. italicum, P. expansum, P. simplicissimum, P. oxalicum, and P. citrinum. This compound demonstrated anti-fungal activity against the fungal pathogen Macrophomina phaseolina.53
Considering multiple evaluation factors, bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy-, and 1,4-diethylbenzene have emerged as the most promising phytochemicals for the creation of antibacterial medications targeting MDR P. aeruginosa and related infectious diseases. Further research through human in vivo studies is necessary to confirm the potential of these phytochemicals as effective antimicrobial treatments.
5. Conclusion
In the conducted research, the aerial parts of Christella dentata (Forssk.) Brownsey & Jermy extract displayed antibacterial effectiveness against MDR P. aeruginosa, evident through the presence of inhibition zones in disc diffusion and agar well diffusion assays. Through computer-aided drug design, two prominent phytocompounds bicyclo[4.3.0]nonane, 2,2,6,7-tetramethyl-7-hydroxy- (536446) and 1,4-diethylbenzene (CID 7734) were identified which affordably inhibited LasR, a pivotal signaling receptor accountable for P. aeruginosa's virulence and multidrug resistance, and LpxC, essential for the biosynthesis of lipid A, a vital constituent of bacterial outer membrane. These findings hold promise for the development of novel bioactive compounds to fight antibiotic-resistant infections. To confirm our results, additional in vivo evaluations are required.Author contribution
Md. Mashiar Rahman: resources, investigation, methodology, formal analysis, writing – original draft, supervision. Md. Rakibul Islam: investigation, methodology, formal analysis. Md. Enamul Kabir Talukder: investigation, methodology, formal analysis. Md. Farhan Atif: investigation, methodology, formal analysis. A F M Shahab Uddin: resources, software, writing – review & editing. K. M. Anis-Ul-haque: investigation, methodology, formal analysis. Md. Saidul Islam: conceptualization, writing – review & editing. Mohammad Jashim Uddin: resources, investigation, formal analysis, writing – original draft. Shahina Akhter: conceptualization, data curation, validation, supervision, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work presented in this paper.Acknowledgements
The authors extend their appreciation to Mohammad Abu Hena Mostofa Jamal, Professor, Islamic University, Bangladesh for providing bacterial strain (Pseudomonas aeruginosa). Additionally, the authors acknowledge the Bangladesh Reference Institute for Chemical Measurements (BRICM) for their support in FT-IR and GC-MS analysis. Funding for this study was provided by the Research Cell at Jashore University of Science and Technology in Bangladesh, under Grant ID JUST/Research Cell/22-FoBST-06/2022-2023.References
- Z. Pang, R. Raudonis, B. R. Glick, T. J. Lin and Z. Cheng, Biotechnol. Adv., 2019, 37, 177–192 CrossRef CAS.
- D. Reynolds and M. Kollef, Drugs, 2021, 81, 2117–2131 CrossRef CAS.
- S. Qin, W. Xiao, C. Zhou, Q. Pu, X. Deng, L. Lan, H. Liang, X. Song and M. Wu, Signal Transduction Targeted Ther., 2022, 7, 1–27 CrossRef.
- K. Papenfort and B. L. Bassler, Nat. Rev. Microbiol., 2016, 14, 576–588 CrossRef CAS PubMed.
- G. Rampioni, I. Bertani, E. Zennaro, F. Polticelli, V. Venturi and L. Leoni, J. Bacteriol., 2006, 188, 815–819 CrossRef CAS.
- D. J. Lee, A. R. Jo, M. C. Jang, J. Nam, H. J. Choi, G. W. Choi, H. Y. Sung, H. Bae, Y. G. Ku and Y. T. Chi, Microb. Pathog., 2018, 119, 162–169 CrossRef CAS.
- F. F. Tuon, L. R. Dantas, P. H. Suss and V. S. Tasca Ribeiro, Pathogens, 2022, 11, 300 CrossRef CAS.
- S. Mukherjee, D. Moustafa, C. D. Smith, J. B. Goldberg and B. L. Bassler, PLoS Pathog., 2017, 13, 1–25 Search PubMed.
- H. A. Abbas, M. A. Shaldam and D. Eldamasi, Curr. Microbiol., 2020, 77, 1051–1060 CrossRef CAS.
- S. Reig, A. Le Gouellec and S. Bleves, Front. Cell. Infect. Microbiol., 2022, 12, 1–16 Search PubMed.
- S. M. Huszczynski, J. S. Lam and C. M. Khursigara, Pathogens, 2020, 9, 6 CrossRef CAS PubMed.
- S. Langklotz, M. Schäkermann and F. Narberhaus, J. Bacteriol., 2011, 193, 1090–1097 CrossRef CAS PubMed.
- K. M. Krause, C. M. Haglund, C. Hebner, A. W. Serio, G. Lee, V. Nieto, F. Cohen, T. R. Kane, T. D. Machajewski, D. Hildebrandt, C. Pillar, M. Thwaites, D. Hall, L. Miesel, M. Hackel, A. Burek, L. D. Andrews, E. Armstrong, L. Swem, A. Jubb and R. T. Cirz, Antimicrob. Agents Chemother., 2019, 63, e009777–e119112 CrossRef.
- N. Vaou, E. Stavropoulou, C. Voidarou, C. Tsigalou and E. Bezirtzoglou, Microorganisms, 2021, 9, 1–28 CrossRef.
- M. V. Ashraf, S. Pant, M. A. H. Khan, A. A. Shah, S. Siddiqui, M. Jeridi, H. W. S. Alhamdi and S. Ahmad, Pharmaceuticals, 2023, 16, 881 CrossRef CAS PubMed.
- M. A. Aziz, M. Adnan, A. H. Khan, A. A. Shahat, M. S. Al-Said and R. Ullah, J. ethnobiol. ethnomed., 2018, 14, 1–16 CrossRef.
- K. Lewis and F. M. Ausubel, Nat. Biotechnol., 2006, 24, 1504–1507 CrossRef CAS.
- S. Wu, K. Yang, Y. Hong, Y. Gong, J. Ni, N. Yang and W. Ding, Front. Microbiol., 2022, 13, 1–13 Search PubMed.
- M. Ozturk, J. E. Chia, R. Hazra, M. Saqib, R. A. Maine, R. Guler, H. Suzuki, B. B. Mishra, F. Brombacher and S. P. Parihar, Front. Immunol., 2021, 12, 1–15 Search PubMed.
- N. S. Mone, S. A. Bhagwat, D. Sharma, M. Chaskar, R. H. Patil, P. Zamboni, N. N. Nawani and S. K. Satpute, Coatings, 2021, 11, 434 CrossRef CAS.
- D. Załuski, Ł. Ciessla and Z. Janeczko, Stud. Nat. Prod. Chem., 2015, 45, 217–249 Search PubMed.
- U. Anand, C. K. Tudu, S. Nandy, K. Sunita, V. Tripathi, G. J. Loake, A. Dey and J. Proćków, J. Ethnopharmacol., 2022, 284, 114744 CrossRef CAS.
- G. Xu, C. Zhang, S. Y. Lee, Z. Chen and X. Zeng, Mitochondrial DNA B: Resour., 2023, 8, 181–185 CrossRef PubMed.
- A. J. Raj, S. Biswakarma, N. A. Pala, G. Shukla, M. Kumar, S. Chakravarty and R. W. Bussmann, J. ethnobiol. ethnomed., 2018, 14, 1–28 CrossRef.
- K. Bharathi, V. Thirumurugan, M. Kavitha, G. Muruganandam and M. Sethuraman, Int. J. Pharm. Sci. Rev. Res., 2014, 27, 283–287 Search PubMed.
- M. A. Al Mashud, M. Moinuzzaman, M. S. Hossain, S. Ahmed, G. Ahsan, A. Reza, R. Bin Anwar Ratul, M. H. Uddin, M. A. Momin and M. A. Hena Mostofa Jamal, Heliyon, 2022, 8, e09920 CrossRef CAS PubMed.
- M. Kabir, A. Al-Noman, B. K. Dash, M. Hasan, S. Akhter and M. Rahman, J. Basic Clin. Physiol. Pharmacol., 2020, 31, 1–12 Search PubMed.
- M. M. Rahman, M. M. Kabir, M. A. AL Noman, M. R. Islam, B. K. Dash, S. Akhter, M. J. Uddin and A. Rahman, J. Appl. Pharm. Sci., 2020, 10, 112–122 CrossRef CAS.
- M. M. Rahman, M. A. Al Noman, S. Khatun, R. Alam, M. M. H. Shetu, E. K. Talukder, R. R. Imon, M. Y. Biswas, K. M. Anis-Ul-Haque, M. J. Uddin and S. Akhter, Heliyon, 2023, 9, e12855 CrossRef CAS.
- S. Akhter, M. W. Hossain, S. Sultana, J. Ferdous Jharna, N. Sultana Meghla, R. Alam, K. M. Anis-Ul-Haque and M. Mashiar Rahman, J. Saudi Chem. Soc., 2022, 26, 101401 CrossRef CAS.
- C. M. M. Koh, L. S. Y. Ping, C. H. H. Xuan, L. B. Theng, H. S. San, E. A. Palombo and X. C. Wezen, Bioengineered, 2023, 14, 2243416 CrossRef.
- I. Mochalkin, J. D. Knafels and S. Lightle, Protein Sci., 2008, 17, 450–457 CrossRef CAS PubMed.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- D. Shivakumar, E. Harder, W. Damm, R. A. Friesner and W. Sherman, J. Chem. Theory Comput., 2012, 8, 2553–2558 CrossRef CAS.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS.
- F. Ahammad, R. Alam, R. Mahmud, S. Akhter, E. K. Talukder, A. M. Tonmoy, S. Fahim, K. Al-Ghamdi, A. Samad and I. Qadri, Briefings Bioinf., 2021, 22, 1–15 CrossRef CAS.
- P. D. Lyne, M. L. Lamb and J. C. Saeh, J. Med. Chem., 2006, 49, 4805–4808 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257–W263 CrossRef CAS PubMed.
- K. V. Sukhum, E. P. Newcomer, C. Cass, M. A. Wallace, C. Johnson, J. Fine, S. Sax, M. H. Barlet, C.-A. D. Burnham, G. Dantas and J. H. Kwon, Commun. Med., 2022, 2, 1–15 CrossRef PubMed.
- M. Saha and A. Sarkar, J. Xenobiot., 2021, 11, 197–214 CrossRef CAS PubMed.
- L. Cremet, A. G. Leroy, D. Muller, S. Delanou, A. Burghelea, A. Broquet, A. Roquilly and N. Caroff, Int. J. Antimicrob. Agents, 2021, 57, 106341 CrossRef CAS PubMed.
- R. E. Duval, M. Grare and B. Demore, Molecules, 2019, 24, 1–9 CrossRef.
- W. Huang, Y. Wang, W. Tian, X. Cui, P. Tu, J. Li, S. Shi and X. Liu, Antibiotics, 2022, 11, 1380 CrossRef CAS PubMed.
- N. A. Eddy and P. Ichalkaranje, Molecules, 2016, 21, 1358 CrossRef PubMed.
- A. Masyita, R. Mustika Sari, A. Dwi Astuti, B. Yasir, N. Rahma Rumata, T. Bin Emran, F. Nainu and J. Simal-Gandara, Food Chem.: X, 2022, 13, 100217 CAS.
- D. Palaiogiannis, T. Chatzimitakos, V. Athanasiadis, E. Bozinou, D. P. Makris and S. I. Lalas, Oxygen, 2023, 3, 274–286 CrossRef CAS.
- M. Tourabi, A. Metouekel, A. E. L. ghouizi, M. Jeddi, G. Nouioura, H. Laaroussi, M. E. Hosen, K. F. Benbrahim, M. Bourhia, A. M. Salamatullah, H. A. Nafidi, G. F. Wondmie, B. Lyoussi and E. Derwich, Sci. Rep., 2023, 13, 1–15 CrossRef.
- B. Ahmad, M. R. Khan, N. A. Shah, T. P. Kondratyuk, N. Ahmed, J. M. Pezzuto, A. Gul and A. Khattak, Integr. Cancer Ther., 2022, 21, 1–10 CrossRef.
- Y. S. Ikhsanov, M. Nauryzbaev, A. Musabekova, M. Alimzhanova and E. Burashev, J. Appl. Eng. Sci., 2019, 17, 338–353 CrossRef.
- K. Gopalakrishnan, Int. J. Biochem. Res. Rev., 2014, 4, 517–526 CrossRef PubMed.
- I. H. Khan and A. Javaid, Mycologia, 2022, 114, 24–34 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08367e |
| This journal is © The Royal Society of Chemistry 2024 |