
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, biological activities, and evaluation molecular docking-dynamics studies of new phenylisoxazole quinoxalin-2-amine hybrids as potential α-amylase and α-glucosidase inhibitors†
Siti Nurshahira Mohd Radzuan a,
Lacksany Phongphanea,
Mohamad Hafizi Abu Bakarb,
Mohammad Tasyriq Che Omarc,
Nor Shafiqah Nor Shahrilb,
Unang Supratmand,
Desi Harnetid,
Habibah A. Wahabe and
Mohamad Nurul Azmi*a
a,
Lacksany Phongphanea,
Mohamad Hafizi Abu Bakarb,
Mohammad Tasyriq Che Omarc,
Nor Shafiqah Nor Shahrilb,
Unang Supratmand,
Desi Harnetid,
Habibah A. Wahabe and
Mohamad Nurul Azmi*a
aSchool of Chemical Sciences, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia. E-mail: shiraradzuan@gmail.com; lacksany.phongphane@gmail.com
bBioprocess Technology Division, School of Industrial Technology, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia. E-mail: mhafizi88@usm.my; shafiqah17@gmail.com
cBiological Section, School of Distance Education, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia. E-mail: mtasyriq@usm.my
dDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, 45363 Jatinangor, Indonesia. E-mail: unang.supratman@unpad.ac.id; desi.harneti@unpad.ac.id
eSchool of Pharmaceutical Sciences, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia. E-mail: habibahw@usm.my
First published on 5th March 2024
Abstract
New phenylisoxazole quinoxalin-2-amine hybrids 5a–i were successfully synthesised with yields of 53–85% and characterised with various spectroscopy methods. The synthesised hybrids underwent in vitro α-amylase and α-glucosidase inhibitory assays, with acarbose as the positive control. Through the biological study, compound 5h exhibits the highest α-amylase inhibitory activity with IC50 = 16.4 ± 0.1 μM while compounds 5a–c, 5e and 5h exhibit great potential as α-glucosidase inhibitors, with 5c being the most potent (IC50 = 15.2 ± 0.3 μM). Among the compounds, 5h exhibits potential as a dual inhibitor for both α-amylase (IC50 = 16.4 ± 0.1 μM) and α-glucosidase (IC50 = 31.6 ± 0.4 μM) enzymes. Through the molecular docking studies, the inhibition potential of the selected compounds is supported. Compound 5h showed important interactions with α-amylase enzyme active sites and exhibited the highest binding energy of −8.9 ± 0.10 kcal mol−1, while compound 5c exhibited the highest binding energy of −9.0 ± 0.20 kcal mol−1 by forming important interactions with the α-glucosidase enzyme active sites. The molecular dynamics study showed that the selected compounds exhibited relative stability when binding with α-amylase and α-glucosidase enzymes. Additionally, compound 5h demonstrated a similar pattern of motion and mechanism of action as the commercially available miglitol.
1. Introduction
Diabetes mellitus, commonly known as diabetes, is a metabolic disorder caused by the defects of insulin production, insulin secretion, or both.1 Chronic cases of diabetes are often associated with long-term complications, dysfunction, or damage of internal organs.1 Generally, diabetes cases are often categorized into two types: type 1 and type 2 diabetes (T2DM). T2DM is the most common type of diabetes, where it accounts for around 90% of all diabetes patients.2 Due to the heterogeneous nature and inconsistency of patient response to the different T2DM medications, the care and treatment approach of this disease can be complex. The main medical treatments for T2DM include metformin, a first line T2DM drug, as well as sulfonylureas, DPP-4 inhibitors, GLP-1 receptor agonists, meglitinides, α-amylase inhibitors and α-glucosidase inhibitors.3Nitrogen-containing aromatic heterocyclic compounds have been researched to have great applications in various fields. Quinoxaline is a type of aromatic heterocyclic compound, with its structure composed of a benzene ring and a pyrazine ring condensed together.4,5 Quinoxaline derivatives have been researched to have numerous biological activities including antituberculosis, antibacterial, anticancer, anti-inflammatory, anti-malarial and anti-hyperglycaemic activities.5 Quinoxaline derivatives exhibit great potential as T2DM treatment which includes DPP-4 inhibitors, GLP-1 receptor agonists, PPARγ and SUR agonists, α-amylase inhibitors, and α-glucosidase inhibitors.4–11 In addition, isoxazoles are a class of azoles, with their structure containing a nitrogen and an oxygen atom in a five-membered aromatic ring.12 This class of compound has been proven to play an important role in medicinal chemistry, exhibiting great biological activity as antimicrobial, antibacterial, antiviral, anticancer, anti-inflammatory and antidiabetic agents.13 As T2DM treatments, isoxazoles can be seen as a major component for various types of T2DM drugs such as protein tyrosine phosphatase 1B (PTP1B) inhibitors, GPR40 agonists, as well as α-amylase and α-glucosidase inhibitors.12–17
α-Glucosidase inhibitors are a type of digestive enzyme inhibitor for T2DM treatment that works to inhibit the glucosidase enzyme complexes, which are important for the digestion of carbohydrates. The inhibition of these enzymes causes a delay in carbohydrate absorption, which leads to the reduction of postprandial hyperglycaemia and lower blood glucose levels.18 Meanwhile, α-amylase inhibitors are also a type of digestive enzyme inhibitor, which inhibits the α-amylase enzymes in charge of breaking down dietary carbohydrates. These inhibitors, however, are less common compared to α-glucosidase inhibitors.19 Fig. 1 shows the structures of some quinoxaline and isoxazole-containing compounds that have been reported to have the potential as α-glucosidase and α-amylase inhibitors.8,10,11,16,17 Through this, we can observe the potential of the hybridisation of quinoxaline and isoxazole groups to synthesise potent α-glucosidase and α-amylase inhibitors.
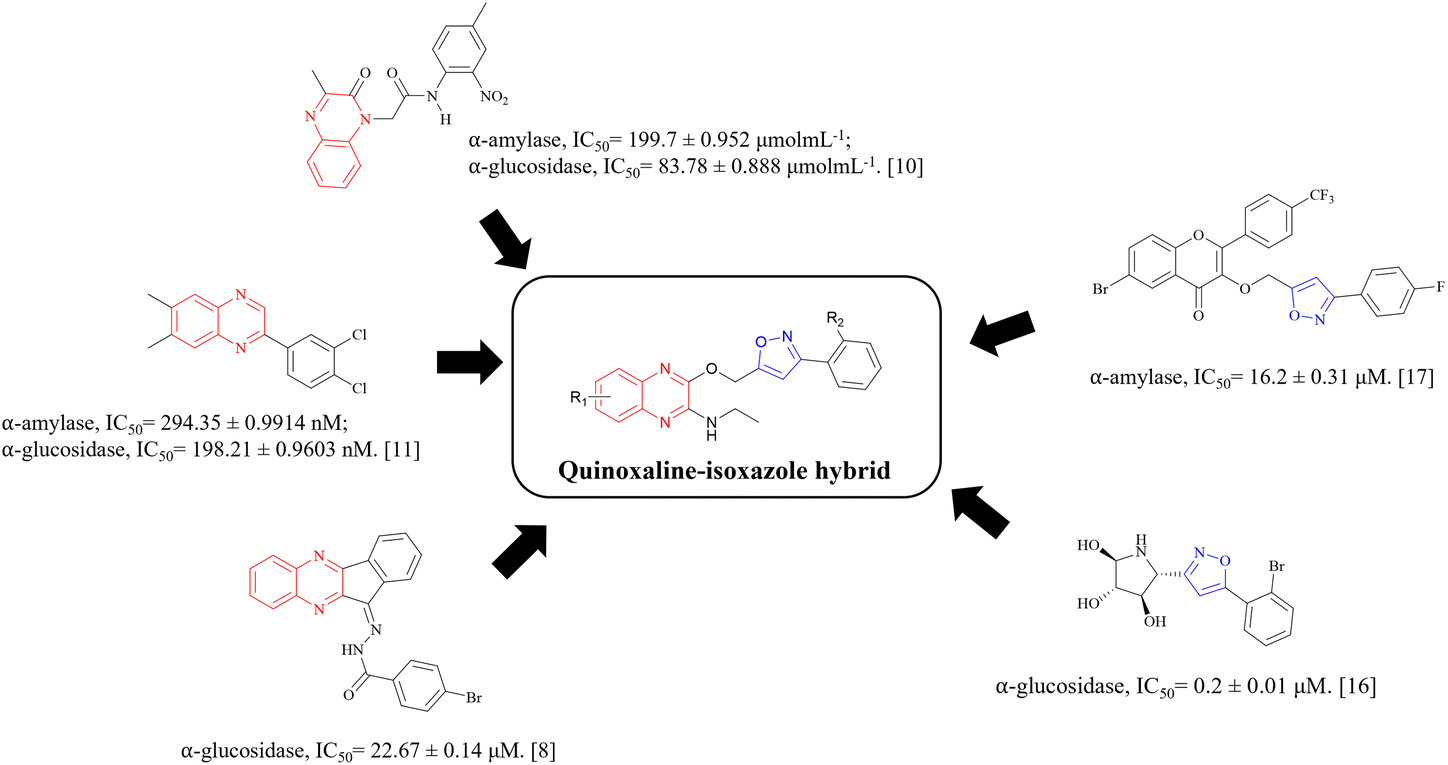 | ||
| Fig. 1 Structures of potential α-glucosidase and α-amylase inhibitors bearing quinoxaline and isoxazole groups. | ||
Some of the approved α-glucosidase and α-amylase inhibitors clinically used include acarbose, voglibose and miglitol, where they mainly inhibit α-glucosidase enzymes and only weakly inhibit α-amylase enzymes.20 Generally, the side effects of these drugs can vary by patient and can include abdominal pain, diarrhoea, bloating, fluctuance, nausea and constipation. Compared to other T2DM treatments, this class of treatment still has a limit of usage for monotherapy due to its low efficacy and is in need of more research. Hence, taking the factors of the obvious potential of quinoxaline and isoxazole moieties towards the research for the treatment of T2DM, the lack of choice as well as the adverse side effects of this type of inhibitors into consideration, new quinoxaline–isoxazole derivatives are synthesised and evaluated for their α-amylase and α-glucosidase inhibitory activities. Through these biological studies, the quinoxaline–isoxazole derivatives show mild to outstanding potential as antidiabetic agents when compared to acarbose, and the inhibitory activities of these compounds are further confirmed with molecular docking and dynamic studies.
2. Results and discussion
2.1 Chemistry
The quinoxaline–isoxazole derivatives (5a–i) were synthesised via a 5-step reaction involving different reactions with several substituents, as illustrated in Scheme 1. The details of the synthesised compounds are depicted in the ESI.† All the reported compounds (5a–i) were synthesised in satisfactory to good yields (53 to 85%) and were characterised by spectroscopic methods including 1H-NMR, 13C-NMR, FTIR and HRMS analysis.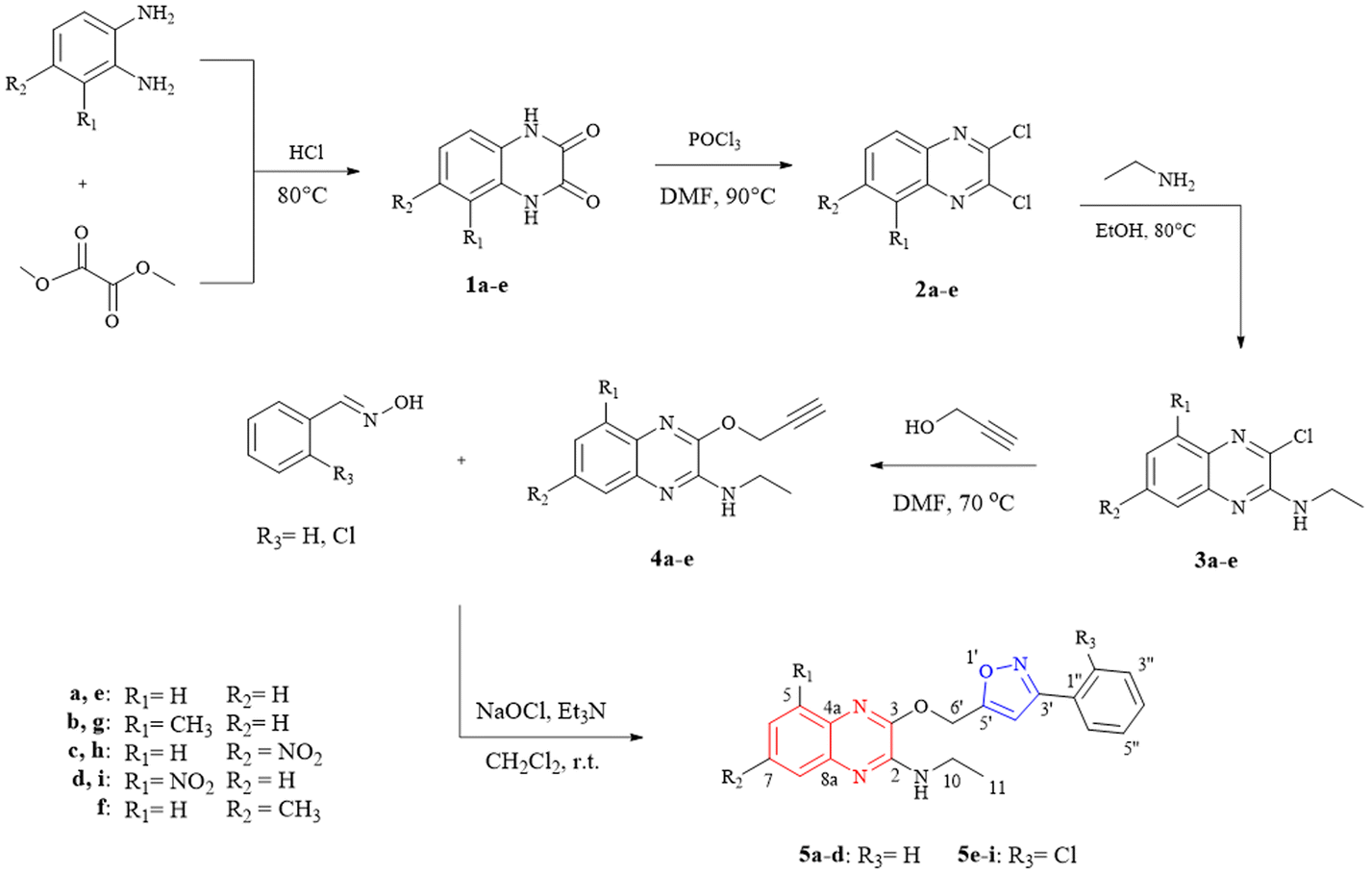 | ||
| Scheme 1 Synthetic pathway of quinoxaline–isoxazole derivatives 5a–i. | ||
Compound 5a exhibits peaks at 3431, 3088, 2978, 1531, and 1195 cm−1 of FTIR spectrum corresponding to the peaks of NH stretching, aromatic C–H stretching, C–H stretching for –CH2CH3, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching for isoxazole and quinoxaline groups, and C–O stretching for isoxazole group.
N stretching for isoxazole and quinoxaline groups, and C–O stretching for isoxazole group.
In the 1H-NMR spectrum of compound 5a, a total of 18 protons were integrated within the range of 1.24–7.75 ppm. A downfield singlet peak at 6.73 ppm corresponds to proton H-4′ attached to the isoxazole group. The integrated singlet peak for the two protons at H-6′ can be seen at 5.70 ppm, while the broad peak at 5.41 ppm corresponds to the NH-9 proton, with one proton integration. In the upfield region, a multiplet peak with 2 proton integration at the 3.66–3.58 ppm range was assigned to the protons at H-2, and a triplet peak at 1.33 ppm (J = 7.2 Hz) with 3 proton integration corresponds to the C–H3 protons at H-11. The 13C-NMR was also examined, and the structure of the compound is further confirmed. Within the 14.6–167.5 ppm range, a total of 20 carbon signals were observed. The signals for the quaternary and aromatic carbons are expected to be within the 167.5–124.1 ppm range. A carbon signal at 102.8 corresponds to carbon C-4′ in the isoxazole group. The signals 58.2 and 35.7 ppm in the upfield region are assigned to carbons at C-6′ and C-10 respectively, while the remaining signal at 14.7 ppm corresponds to the CH3 carbon at C-11. The structure of the compound was further confirmed by running DEPT-135, COSY and HMBC NMR spectroscopy. The regioselectivity of the synthesis of compounds 3a–e, 4a–e and 5a–i were also determined by previously reported literatures21,22 and was further confirmed with X-ray previously reported crystallography data.21
2.2 Biology
All synthesised quinoxaline–isoxazole derivatives (5a–i) were tested for their in vitro α-amylase and α-glucosidase inhibitory activity. Fig. S68 and S69† visualises the graphs of inhibitory activity of the compounds with α-amylase and α-glucosidase enzymes respectively. The probability of the compounds to be toxic at certain concentrations may cause the curve to deviate in a concentration-dependent manner and not follow the dose–response curve following a phenomenon termed as hormesis.23,24 Table 1 demonstrates the inhibitory potential of the compounds against α-amylase and α-glucosidase activity with their calculated IC50 values. For α-amylase, compound 5h displayed the most significant inhibitory potential (IC50 = 16.4 ± 0.1 μM) compared to acarbose (IC50 = 24.0 ± 0.9 μM). Compound 5i also exhibits good inhibitory activity (IC50 = 34.4 ± 0.36 μM) comparable to acarbose. As for α-glucosidase enzyme, compounds 5a–c, 5e and 5h exhibits significant inhibitory potential. Compound 5c in particular, exhibits the highest inhibitory potential (IC50 = 15.2 ± 0.3 μM) compared to acarbose (IC50 = 49.3 ± 1.1 μM). The rest of the compounds also exhibit great potential, with IC50 values in the range of 31.6–46.6 μM. Furthermore, from the IC50 values calculated, it can be observed that compound 5h exhibits potential as both α-amylase and α-glucosidase inhibitors, in comparison to acarbose. The compound which exhibits the greatest α-amylase inhibitory activity and 2nd highest α-glucosidase activity, can be deduced to have the potential as a dual inhibitor.| Compound | IC50 (μM) | |
|---|---|---|
| α-Amylase | α-Glucosidase | |
| 5a | >125 | 41.9 ± 0.78 |
| 5b | >125 | 46.6 ± 0.1 |
| 5c | >125 | 15.2 ± 0.3 |
| 5d | >125 | 54.9 ± 0.9 |
| 5e | 121.2 ± 1.4 | 42.8 ± 0.2 |
| 5f | 42.1 ± 0.2 | >125 |
| 5g | >125 | >125 |
| 5h | 16.4 ± 0.1 | 31.6 ± 0.4 |
| 5i | 34.4 ± 0.36 | 66.9 ± 0.5 |
| Acarbose | 24.0 ± 0.9 | 49.3 ± 1.1 |
2.3 Molecular docking study
Through the α-amylase and α-glucosidase inhibition assays performed on the quinoxaline–isoxazole derivatives, several potent compounds were selected for molecular docking analysis. The molecular docking analysis was done by using the crystal structures of the human pancreatic α-amylase complexed with nitrite and acarbose (PDB ID: 2QV4)25 and the C-terminal of human maltase glucoamylase (CtMGAM, α-glucosidase) complexed with acarbose (PDB ID: 3TOP)26 as reference complexes for the docking process. The MGAM protein is a type of α-glucosidase enzyme that is in charge of hydrolysing sugars like oligosaccharides and maltose into glucose, which makes them vital in production of glucose for human beings.26 The C-terminal MGAM (PDB ID: 3TOP) is selected as the docking protein for α-glucosidase as it has been reported to be preferred in inhibition with acarbose, compared to the N-terminal.26 This is also supported by our previous study reported by Phongphane et al. in 2023.27 The molecular docking results of the selected quinoxaline–isoxazole compounds and acarbose are indicated via binding energy with targeted enzymes and are tabulated in Table 2.With α-amylase, compounds 5h and 5i were selected to perform docking analysis, and it can be observed that both compounds exhibit higher binding energy with enzyme compared to acarbose (−7.7 ± 0.11 kcal mol−1). Compound 5h exhibits the highest binding energy of −8.9 ± 0.10 kcal mol−1 and forms hydrogen bonds with amide group (ASP300), nitrogen atom of isoxazole (HIS101), and NO2 substituent (LYS200). With the quinoxaline group, two π–alkyl bonds were formed with LEU162 and ILE235. More π–alkyl bonds were formed between the phenyl group with protein residue LEU165 and Cl substituent respectively with proteins LEU162 and LEU165. A π–π stacked interaction was also formed between the isoxazole group and TYR62 residue.
For compound 5i, similar bond interactions are also seen between ASP300 residue with the amide group to form a hydrogen bond, residues TRP58 and TRP59 with quinoxaline group to form π–π stacked, as well as an π–alkyl bond formed between the isoxazole group and residue LEU162. The three-dimensional (3-D) binding interaction of these compounds with the active site of α-amylase is exhibited in Fig. 2A and B. From the docking performed, it can be said that the presence of the quinoxaline, isoxazole and amide groups are essential, whereas the type of substituent, such as the –Cl substituent, also plays a great role in increasing the binding energy of the compounds with the target protein, α-amylase. The details of the binding interactions of these compounds with α-amylase have been tabulated in Table 3.
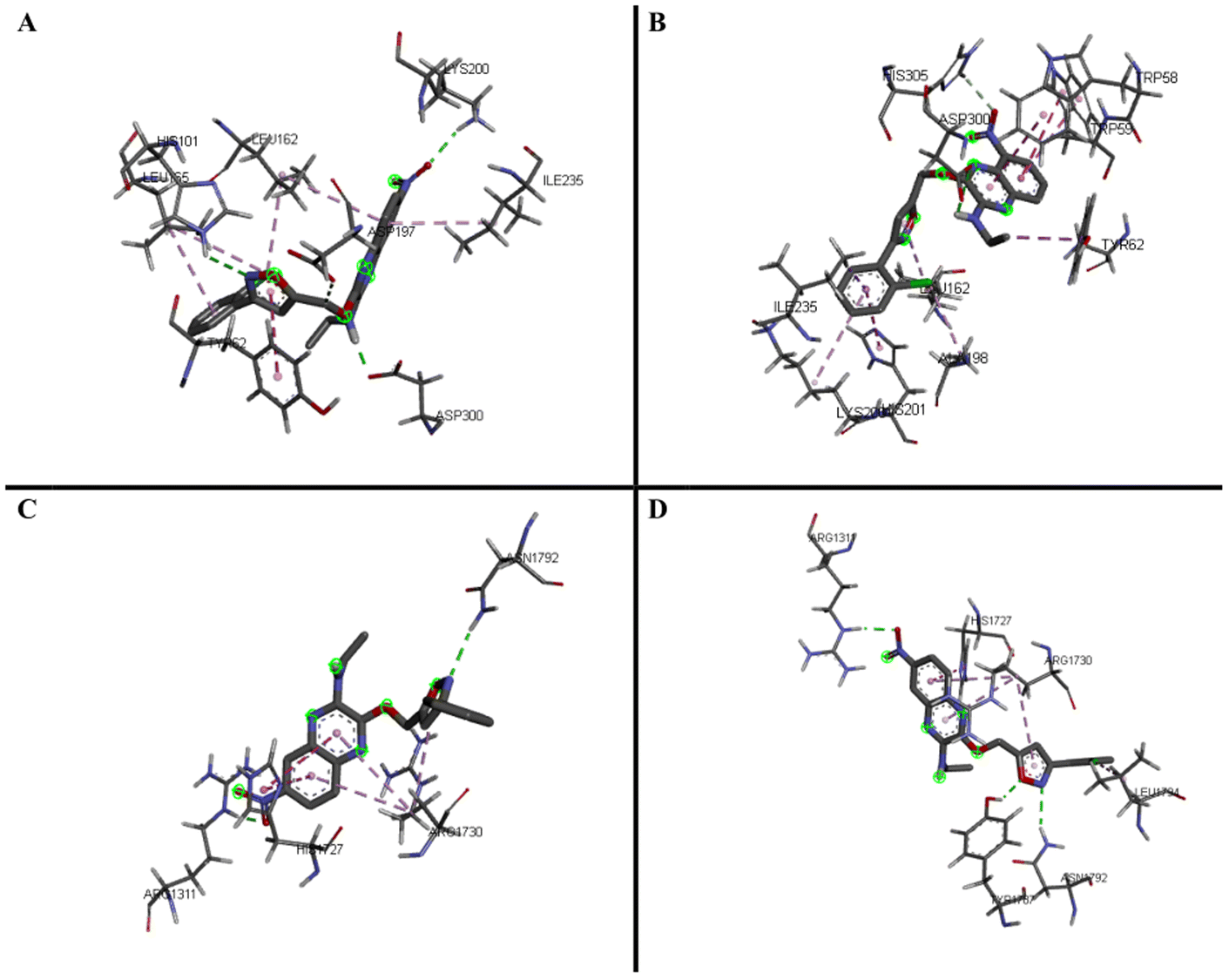 | ||
| Fig. 2 3D predicted binding modes of compounds 5h (A) and 5i (B) with modelled α-amylase and compounds 5c (C) and 5h (D) with modelled α-glucosidase (CtMGAM). | ||
| Protein | Compound | Protein residue | Interaction unit of compounds | Type of interaction |
|---|---|---|---|---|
| α-Amylase (2QV4) | 5h | ASP300 | NH of amide | H-Bond |
| HIS101 | N of isoxazole | H-Bond | ||
| LYS200 | NO2 | H-Bond | ||
| ASP197 | CH2 | Carbon H-bond | ||
| TYR62 | Isoxazole | π–π stacked | ||
| LEU162 | Quinoxaline, –Cl | π–alkyl | ||
| LEU165 | Phenyl, quinoxaline | π–alkyl | ||
| ILE235 | NH of amide | π–alkyl | ||
| 5i | ASP300 | NH of amide | H-Bond | |
| HIS305 | NO2 | Carbon H-bond | ||
| ILE235 | Phenyl | π–sigma | ||
| TYR62 | CH2CH3 | π–sigma | ||
| HIS201 | Phenyl | π–π T-shaped/π–π stacked | ||
| TRP58 | Quinoxaline | π–π T-shaped/π–π stacked | ||
| TRP59 | Quinoxaline | π–π T-shaped/π–π stacked | ||
| ALA198 | –Cl | π–alkyl/alkyl | ||
| LYS200 | Phenyl | π–alkyl/alkyl | ||
| LEU162 | Isoxazole | π–alkyl/alkyl | ||
| –Cl | π–alkyl/alkyl |
For α-glucosidase enzyme, five of the most potent inhibitors (5a–c, 5e, and 5h) were selected based on their IC50 values, and all the compounds exhibit higher binding energy when compared to acarbose (−7.5 ± 0.00 kcal mol−1). Among the five compounds, 5c exhibits the highest binding energy (−9.0 ± 0.20 kcal mol−1), followed by 5h (−8.7 ± 0.15 kcal mol−1). Both compounds exhibit hydrogen bond interactions, with the bonds in 5c forming via the nitro substituent (ARG1311) and the nitrogen atom of isoxazole (ASN1792), while for 5h, the bonds formed via the oxygen atom (TYR1787) and the nitrogen atom (ASN1792) of the isoxazole group. The rest of the interactions in compound 5c include the π–π T-shaped bond forming via the quinoxaline group (HIS1727) as well as π–alkyl interactions with the quinoxaline and isoxazole group with protein residue ARG1730.
In 5h, similar interactions are also observed, with π–π T-shaped interaction forming via the quinoxaline group (HIS1727). Three π–alkyl bonds were also formed via the Cl substitution at the phenyl group (LEU1794), the quinoxaline and isoxazole group (ARG1730), and the phenyl group (ARG1311). The three-dimensional (3-D) binding interaction of these compounds with the active site of α-glucosidase (CtMGAM) is exhibited in Fig. 2C and D and the details of the binding interactions of the selected compounds with α-glucosidase have been tabulated in Table 4. From this analysis, it can be said that the presence of the quinoxaline and isoxazole groups are necessary, while the presence of certain substituents such as chloro and nitro substituents also play an important role to achieve high binding energy with the target protein, α-glucosidase.
| Protein | Compound | Protein residue | Interaction unit of compounds | Type of interaction |
|---|---|---|---|---|
| α-Glucosidase (3TOP) | 5c | ARG1311 | –NO2 | H-Bond |
| ASN1792 | N of isoxazole | H-Bond | ||
| ARG1730 | Quinoxaline, isoxazole | π–alkyl | ||
| ARG1311 | –NO2 | H-Bond | ||
| 5h | ASN1792 | O of isoxazole | Carbon H-bond | |
| ARG1311 | Phenyl | π–alkyl | ||
| TYR1787 | O of isoxazole | H-Bond | ||
| HIS1727 | Quinoxaline | π–π T-shaped | ||
| LEU1794 | –Cl | π–alkyl | ||
| ARG1730 | Quinoxaline, isoxazole | π–alkyl |
2.4 Structure–activity relationship (SAR)
The SAR studies of the in vitro tested compounds 5a–i were done by considering the types and positions of the substituents in the compounds and their potential as α-amylase and α-glucosidase inhibitors. For α-amylase inhibition, the compounds with unsubstituted R3 site (5a–d) display weaker inhibitory activity when compared to compounds with chloro substitution at R3 (5e–i) and acarbose. Through this, it can be concluded that the presence of Cl substituent as an electronegative atom at site R3 is necessary to achieve higher α-amylase inhibitory potential. This can be further solidified by the in silico molecular docking studies, where the Cl substituent is observed to form multiple interaction bonds such as π–alkyl and alkyl bonds with the enzyme.From the binding energy of the compounds exhibited in the in silico study, it can be said that substitution at position C-7 may be essential to achieve greater α-amylase inhibitory activity compared to substitution at C-5. It can also be concluded that when the type of substituents at the quinoxaline moiety is compared, the compounds with nitro substituent exhibit the most potent α-amylase inhibitory activity. These trends can be observed with the comparison of compounds 5h and 5i; 5h exhibits the greatest α-amylase inhibitory potential among all the quinoxaline–isoxazole compounds synthesised, followed by 5i, where both compounds are nitro substituted at C-7 (R2) and C-5 (R1) respectively.
As for α-glucosidase inhibition, among all the quinoxaline–isoxazole compounds synthesised, compound 5c with nitro substitution at C-7 and unsubstituted R3 site, exhibits the most potent inhibition activity (IC50 = 15.2 ± 0.3 μM), enhanced to about three-folds, compared to acarbose (IC50 = 49.3 ± 1.1 μM). However, when a nitro group is substituted at C-5 for compound 5d, the inhibition activity decreased over 3 folds (IC50 = 54.9 ± 0.9 μM). The same trend can be observed for compounds with chloro substitution at site R3, where the inhibitory activity of compound 5h (IC50 = 31.6 ± 0.4 μM) bearing nitro substitution at C-7 was enhanced to two folds compared to its counterpart 5i (IC50 = 66.9 ± 0.5 μM) that bears a nitro substitution at C-5. From this, it can be concluded that among the nitro-substituted compounds synthesised, substitution at C-7 is favoured to produce greater α-glucosidase inhibitory potential compared to substitution at C-5. Moreover, it can be observed that overall, the unsubstituted R3 compounds (5a–d) exhibit more potent α-glucosidase inhibitory activity compared to the chloro-substituted compounds (5e–f). For instance, compound 5c exhibits a more potent α-glucosidase inhibitory activity compared to 5h, where both compounds are nitro-substituted at C-7. The same can also be said for the other substituents: 5-CH3 (5b and 5g), 5-NO2 (5d and 5i) as well as the unsubstituted derivatives (5a and 5e). These trends are further solidified by the in silico study conducted, where compound 5c exhibited the highest binding energy, followed by 5h, and compounds 5a, 5b and 5e. From this overall analysis, it can be also concluded that compound 5h exhibits a potential to act as dual inhibitors for both α-amylase and α-glucosidase enzymes. The summary of the SAR study of these compounds as potential α-amylase and α-glucosidase inhibitors is illustrated in Fig. 3.
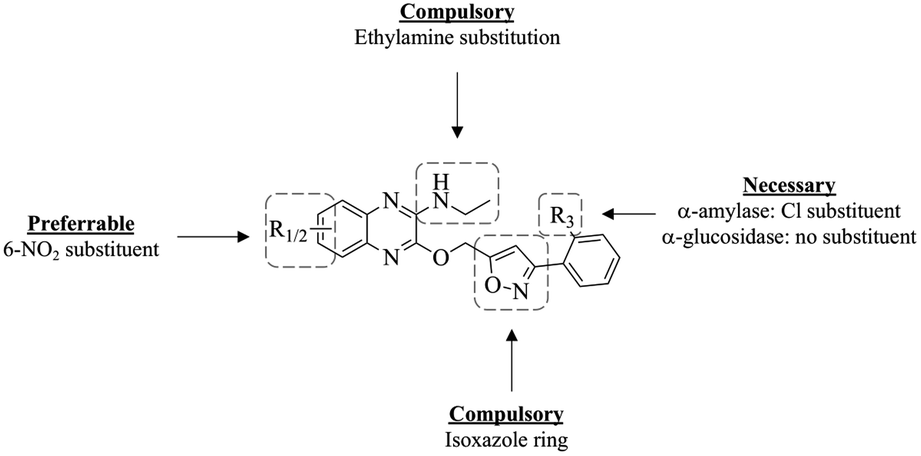 | ||
| Fig. 3 SAR study of quinoxaline–isoxazole compounds. | ||
2.5 Molecular dynamics study
The simulations of four enzyme–compound complexes (5c–glucosidase, 5h–glucosidase, 5h–amylase, 5i–amylase) and two control enzyme–compound complexes (miglitol–glucosidase, and miglitol–amylase) were carried out with the Amber program for 5000 picoseconds. To evaluate the stability and dynamics of each complex, various analyses were performed on the resulting MD trajectories, including root mean square deviation (RMSD) profile, Cα root mean square fluctuation (RMSF), radius of gyration (Rg), interaction energy, binding free energy, and residue interaction, using the PyTraj and ProLIF tools integrated in a python notebook.28,29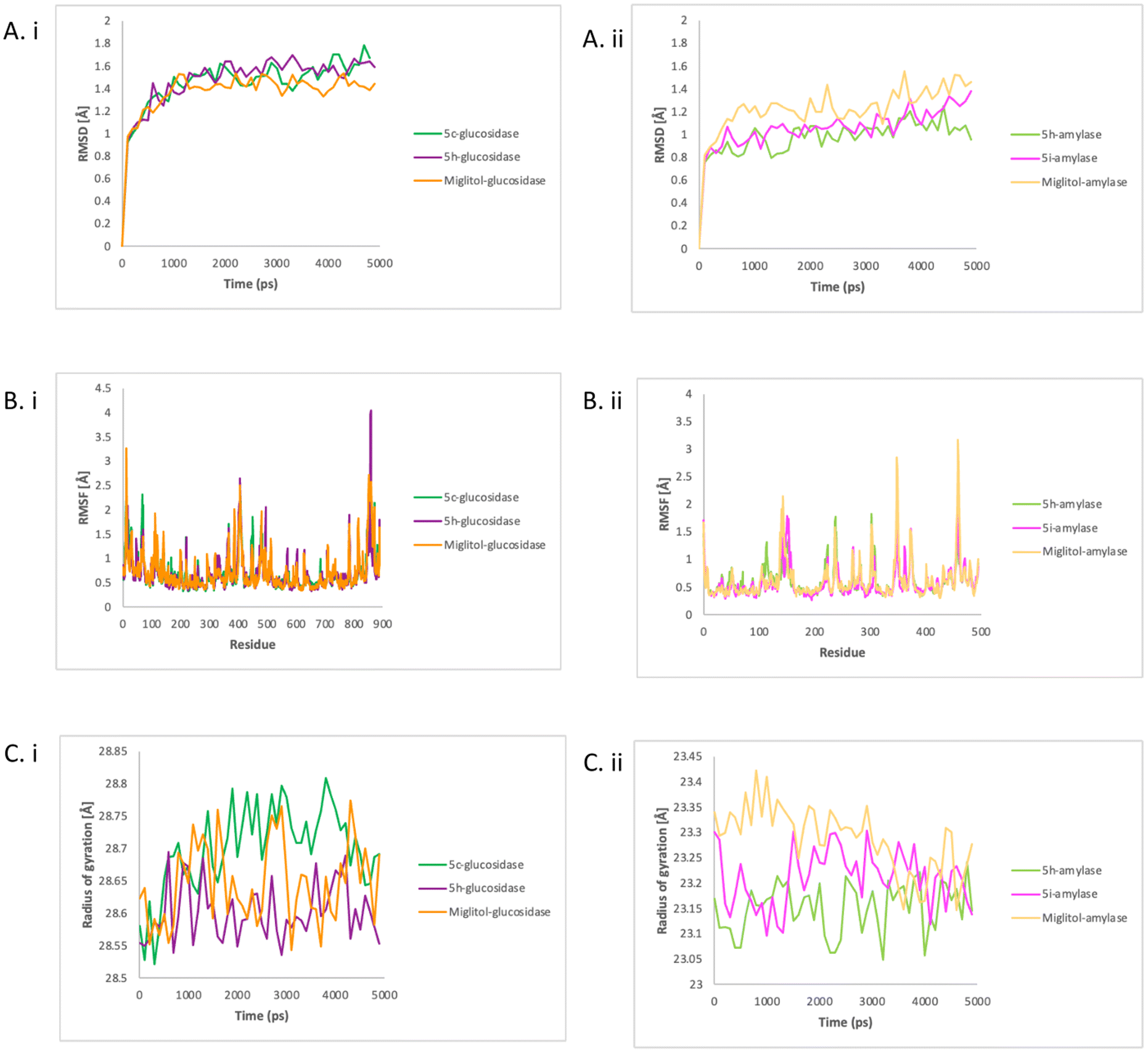 | ||
| Fig. 4 The RMSD (A), RMSF (B) and Rg (C) of glucosidase with compounds 5c, 5h, and miglitol (i) and amylase with compounds 5h, 5i, and miglitol (ii). | ||
In contrast with glucosidase, the miglitol–amylase had higher values, indicating a greater deviation from the reference set of values. All the systems showed a relatively stable in RMSD values over time, indicating a stable dynamics simulation between enzymes and compounds was achieved. To assess the flexibility and stiffness of different residues in amylase and glucosidase when complexed with compounds, RMSF plots were created from the simulation trajectories of the enzyme's dynamics. As shown in Fig. 4Bi, the highest fluctuations were detected in 5h–glucosidase at residue SER1366, followed by miglitol–glucosidase also at residue SER1366 and 5c–glucosidase at residue SER1440.
As shown in Fig. 4Bii, significant fluctuation patterns were observed in miglitol–amylase and 5i–amylase, indicating restricted movement during the simulation. The greatest deviation in the Cα atom of PHE348 in miglitol–amylase was detected throughout the simulation, indicating that this residue was crucial for ligand binding. Binding with 5i resulted in a greater deviation of the amylase residue ASP153 than the effect of 5h binding on residue LEU237 and GLN302 in the Cα atom. In both enzymes, 5h showed a similar fluctuation pattern with miglitol and might share the similar mode of action to miglitol. The compactness of both enzymes when bound with tested compounds and control was analysed by calculating the Rg values, which represent the square root of the average of the squared distances of the atoms or particles from the centre of mass. As shown in Fig. 4Ci, 5h–glucosidase and miglitol–glucosidase had a relatively steady Rg value over time, with an average of 28.6 angstroms. However, 5c–glucosidase had higher values on average (28.7 angstroms), indicating that it was becoming less compact when bound with 5c. In contrast, Fig. 4Cii showed that after amylase was complexed with 5h, 5i, and miglitol, different Rg values were observed, indicating different levels of compactness. Despite this, there was a relatively similar average value observed from 3500 ps to the end of the simulation time. The decreasing values of the miglitol–amylase complex indicated that the amylase enzyme changed from rigid to less compact when bound with miglitol, while the size and shape of amylase enzymes remained stable when complexed with 5h and 5i. In glucosidase, 5h showed a similar compactness pattern as miglitol while in amylase, 5i showed a similar compactness pattern as miglitol.
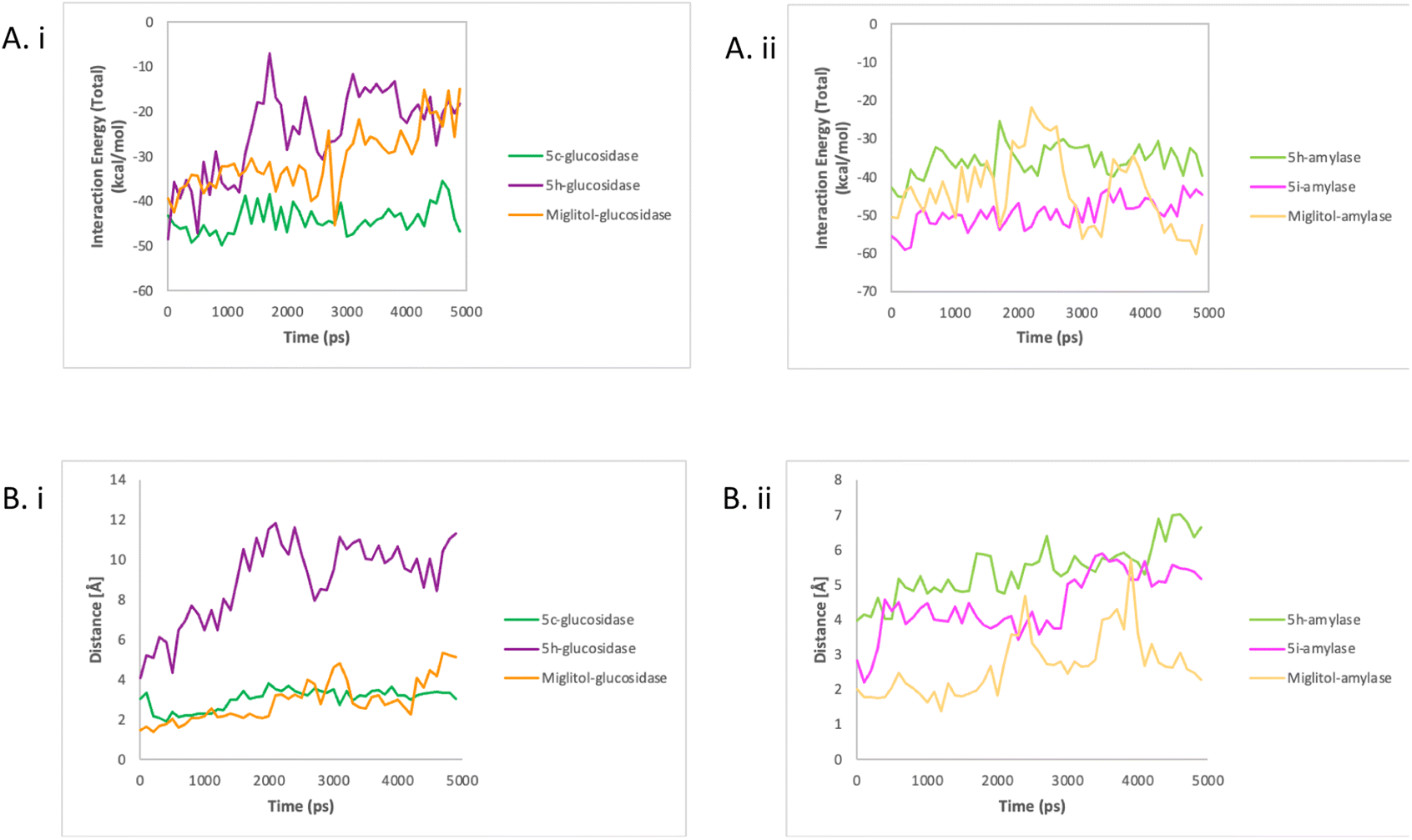 | ||
| Fig. 5 The interaction energy (A) and enzyme–compound distance (B) of glucosidase with compounds 5c, 5h, and miglitol (i) and amylase with compounds 5h, 5i, and miglitol (ii). | ||
These findings also proved that nonbonded interactions are important in compound–enzyme interaction and stability as previously reported.30 The positioning of the compounds in the active site of enzymes was one of the key factors that led to a strong interaction energy. As shown in Fig. 5Bi, there was a short average distance of 2 angstroms observed between miglitol and the residues in the active site of glucosidase. The distance between 5c and residues in the active site was only slightly higher, with an average of 3 angstroms. On the other hand, longer distances ranging from 4 to 12 angstroms were recorded between 5h and the active site residues. A similar trend was seen in amylase, as shown in Fig. 5Bii, with initially short distances between miglitol and the active site residues, but with higher distances observed after 2000 ps reaching up to 5 angstroms. Unlike glucosidase, 5i maintained a stable distance of 4.5 angstroms. As for 5h, it displayed longer distances with the active site residues, which ranged from 4 to 7 angstroms. These findings support the impact of compound position on the interaction energy between enzymes and compounds.
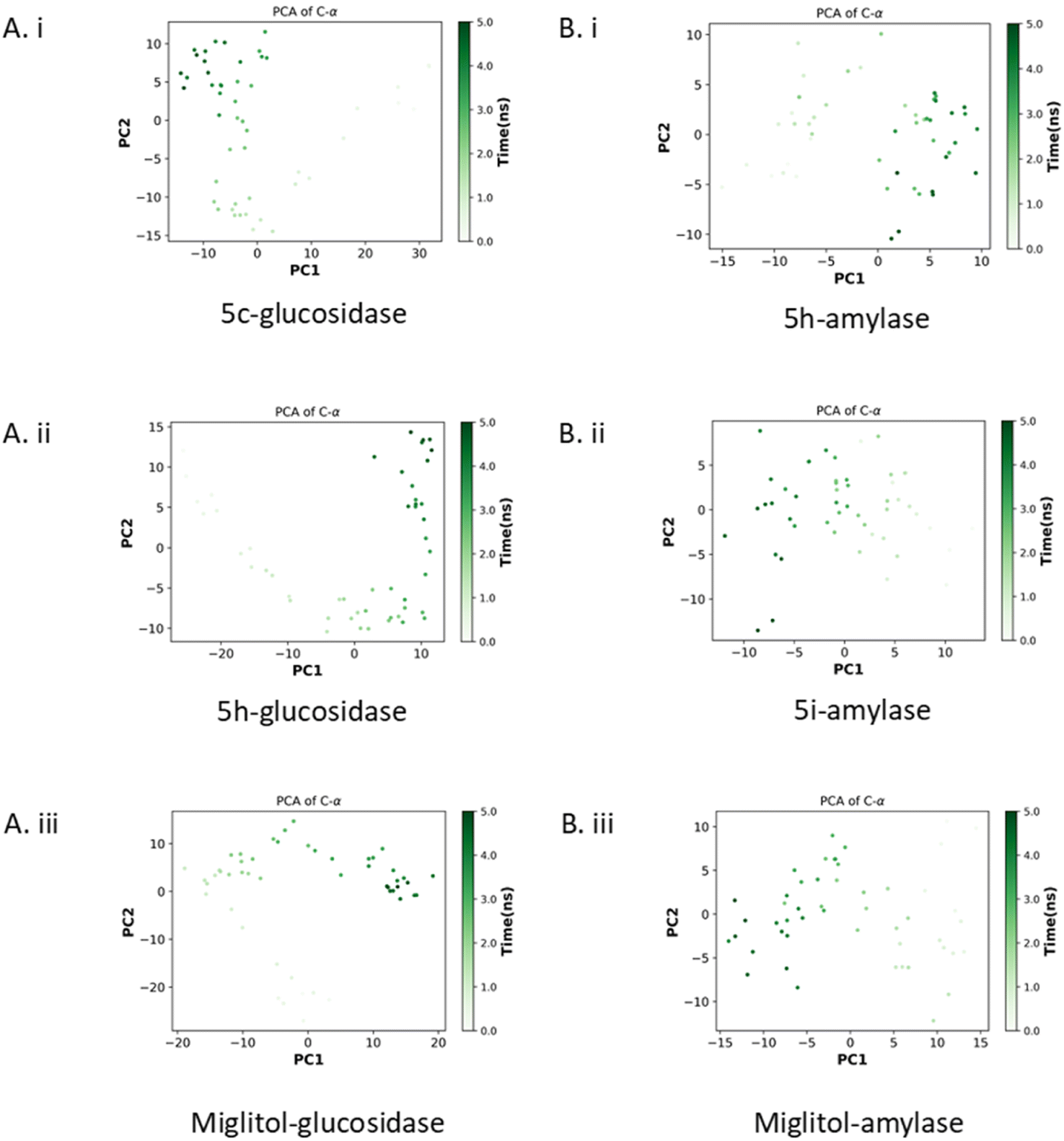 | ||
| Fig. 6 The principal component analysis. Glucosidase with compounds 5c, 5h, and miglitol (Ai–iii) and amylase with compounds 5h, 5i, and miglitol (Bi–iii). | ||
As shown in Fig. 6Bi–iii, the eigenvalues of the 5h–amylase complex fluctuated between −13 to 10 for PC1 and −10 to 10 for PC2. Meanwhile, the 5i–amylase complex had eigenvalues ranging from 14 to −12 for PC1 and −14 to 10 for PC2. The miglitol-bound glucosidase had eigenvalues that ranged from 13 to −14 for PC1 and −13 to 10 for PC2. The PCA results indicated that the binding of the compounds to glucosidase created stable enzyme–compound complexes. The 5h–glucosidase complex showed a similar pattern of motion to the miglitol–glucosidase complex, especially at the end of the simulation, while the 5c–glucosidase complex showed an opposing motion. However, all glucosidase complexes were very stable and occupied the least phase space compared to the amylase complexes. Additionally, the 5i–amylase complex showed a similar pattern of motion to the miglitol–amylase complex, suggesting that the compound may have the same mechanism of action as miglitol.
The binding free energy values for the complexes of 5c–glucosidase, 5h–glucosidase, and miglitol–glucosidase are displayed in Table 5. The 5c–glucosidase complex had the lowest binding free energy at −25.13 kcal mol−1, followed by 5h–glucosidase complex with −15.73 kcal mol−1, and miglitol–glucosidase complex with −14.68 kcal mol−1. Analysis of the simulation revealed the interactions that occurred in more than 70% of frames. For the 5c–glucosidase complex, five residues showed interaction as displayed in molecular docking, with the exception of TRP1749's hydrogen bond. Additionally, six other residues formed hydrophobic interactions. A similar pattern was observed in the 5h–glucosidase complex where five residues maintained their hydrophobic interactions, except for LYS1460, and four other residues generated additional hydrophobic interactions. The 5000 ps trajectories analysis showed that both the 5c–glucosidase and 5h–glucosidase complexes mainly formed hydrophobic interactions and π-stacking.
| Complex | MM-GBSA | Residues with interaction occur more 70% of frames | Type of interaction |
|---|---|---|---|
| Generalized Born | |||
| (Δ total ± std. dev.) | |||
| 5c–Glucosidase | −25.1252 ± 1.7469 | TYR1618 | Hydrophobic |
| THR1621 | Hydrophobic | ||
| LEU1622 | Hydrophobic | ||
| LYS1625 | Hydrophobic | ||
| VAL1631 | Hydrophobic | ||
| PRO1658 | Hydrophobic | ||
| TYR1715 | Hydrophobic | ||
| π-Stacking | |||
| Hydrophobic | |||
| ALA1746 | Hydrophobic | ||
| GLY1747 | Hydrophobic | ||
| GLY1748 | Hydrophobic | ||
| TRP1749 | π-Stacking | ||
| 5h–Glucosidase | −15.7285 ± 6.4800 | TYR1251 | Hydrophobic |
| π-Stacking | |||
| TRP1355 | Hydrophobic | ||
| π-Stacking | |||
| Hydrophobic | |||
| ASP1368 | Hydrophobic | ||
| TRP1369 | π-Stacking | ||
| Hydrophobic | |||
| Hydrophobic | |||
| ASP1370 | Hydrophobic | ||
| GLN1372 | Hydrophobic | ||
| PHE1427 | Hydrophobic | ||
| PHE1559 | π-Stacking | ||
| PHE1560 | |||
| Miglitol–glucosidase | −14.6832 ± 4.1961 | PHE1289 | Hydrophobic |
| THR1290 | Hydrophobic | ||
| PRO1329 | Hydrophobic | ||
| GLU1400 | Hydrophobic | ||
| ASN1404 | Hydrophobic | ||
| PRO1405 | H-Bond | ||
| GLN1406 | Hydrophobic | ||
| ARG1410 | Hydrophobic | ||
| H-Bond |
On the other hand, the miglitol–glucosidase complex formed both hydrophobic interactions and hydrogen bonds. The depiction of the binding free energy of 5h–amylase, 5i–amylase, and miglitol–amylase complexes are shown in Table 6. The binding free energy value was recorded as −23.12 kcal mol−1 for the 5h–amylase complex, −28.87 kcal mol−1 for the 5i–amylase complex, and −22.25 kcal mol−1 for the miglitol–amylase complex. Out of the three compounds, 5i showed the lowest total binding free energy value. The simulation analysis revealed that interactions occurred in more than 70% of frames. In the 5h–amylase complex, seven residues that demonstrated interaction as presented in the molecular docking were maintained in the simulation, except for the hydrogen bond of residue THR163 which was replaced with a hydrophobic interaction. Furthermore, a hydrogen bond was formed at SER108, and hydrophobic interactions were formed by two adjacent residues. A similar pattern was observed in the 5i–amylase complex. Eight residues maintained their hydrophobic interactions as shown in the molecular docking. However, ASP300 and HIS305 formed a hydrophobic interaction instead of a hydrogen bond as demonstrated by molecular docking. Additional hydrophobic interactions were generated by four adjacent residues. Similar to glucosidase, 5000 ps of trajectories analysis showed that both 5h–amylase and 5i–amylase complexes mostly formed hydrophobic interactions and π-stacking. In contrast, the miglitol–glucosidase complex formed hydrophobic interactions and hydrogen bonds.
| Complex | MM-GBSA | Residues with interaction occur more 70% of frames | Type of interaction |
|---|---|---|---|
| Generalized Born | |||
| (Δ total ± std. dev.) | |||
| 5h–Amylase | −23.1177 ± 2.5202 | PRO54 | Hydrophobic |
| TRP58 | Hydrophobic | ||
| π-Stacking | |||
| TRP59 | Hydrophobic | ||
| π-Stacking | |||
| TYR62 | Hydrophobic | ||
| GLN63 | Hydrophobic | ||
| ALA106 | Hydrophobic | ||
| VAL107 | Hydrophobic | ||
| SER108 | H-Bond | ||
| THR163 | Hydrophobic | ||
| LEU165 | Hydrophobic | ||
| 5i–Amylase | −28.8653 ± 2.0533 | TRP58 | Hydrophobic |
| TRP59 | Hydrophobic | ||
| π-Stacking | |||
| π–Cation | |||
| TYR62 | Hydrophobic | ||
| GLN63 | Hydrophobic | ||
| TYR151 | Hydrophobic | ||
| π-Stacking | |||
| LEU162 | Hydrophobic | ||
| THR163 | Hydrophobic | ||
| LEU165 | Hydrophobic | ||
| ASP197 | Hydrophobic | ||
| ALA198 | Hydrophobic | ||
| LYS200 | Hydrophobic | ||
| HIS201 | Hydrophobic | ||
| π-Stacking | |||
| ILE235 | Hydrophobic | ||
| HIS299 | Hydrophobic | ||
| ASP300 | Hydrophobic | ||
| HIS305 | Hydrophobic | ||
| π-Stacking | |||
| Miglitol–amylase | −22.2484 ± 7.9166 | ARG267 | H-Bond |
| Hydrophobic | |||
| ASN301 | H-Bond | ||
| Hydrophobic | |||
| GLN302 | Hydrophobic | ||
| ARG303 | Hydrophobic | ||
| GLY304 | Hydrophobic | ||
| GLY309 | Hydrophobic | ||
| ALA310 | Hydrophobic | ||
| ILE312 | Hydrophobic | ||
| THR314 | Hydrophobic | ||
| TRP316 | Hydrophobic | ||
| ASP317 | H-Bond | ||
| Hydrophobic | |||
| ARG346 | H-Bond | ||
| PHE348 | Hydrophobic |
3. Conclusion
To summarise, new quinoxaline–isoxazole hybrids 5a–i were successfully synthesised with moderate to high yield of range 53–85%. The synthesised compounds underwent in vitro α-amylase and α-glucosidase inhibitory assays, and several compounds exhibit potential. Compound 5h, bearing 6-NO2 and 2-Cl substituents, exhibits the most potential as α-amylase inhibitor (IC50 = 16.39 ± 0.1 μM) while compounds 5a–c, 5e and 5h all exhibit potential as α-glucosidase inhibitor, in which compound 5c, bearing 6-NO2 substituent, is the most potent inhibitor with IC50 = 15.15 ± 0.3 μM. From this, it can be observed that compound 5h exhibits the potential to act as dual inhibitors for both α-amylase and α-glucosidase enzymes. Based on the SAR studies conducted, nitro substitution at C-6 (R1/2) is the most preferred substituent to achieve the most potent inhibitory activity for both enzymes, while the substitution at R3 varies between enzymes. Chloro substitution at site R3 is only necessary to achieve more potent α-amylase inhibitory activity, while unsubstituted R3 compounds exhibit more potent inhibitory activity for α-glucosidase. The molecular docking and dynamic studies conducted also further confirm the inhibitory activity of the selected compounds, where compound 5h exhibited the greatest binding energy of −8.9 ± 0.10 kcal mol−1 with α-amylase, while compound 5c exhibited the greatest binding energy of −9.0 ± 0.20 kcal mol−1 with α-glucosidase. The molecular dynamics study revealed that the selected compounds displayed relative stability when binding with α-amylase and α-glucosidase enzymes. The formation of hydrophobic interactions between the compounds and residues within the active site of enzymes played a role in influencing the enzyme's compactness and affinity. In addition, 5h exhibited a comparable pattern of motion and mechanism of action to the commercially available miglitol inhibitor. Overall, compound 5h exhibits promising potential as dual inhibitor for α-amylase and α-glucosidase enzymes and serve as the steppingstone towards the research for more effective T2DM treatments.4. Materials and methods
4.1 Chemistry
All chemicals and materials purchased from Sigma Aldrich Co. and Merck Chemical Co. and used without purification. DMF and DCM solvents were dried over 4 Å molecular sieves. The purification of synthesised compounds via column chromatography was performed using Merck silica gel (0.040–0.063 mm), while the thin-layer chromatography (TLC) was performed using silica-coated aluminium sheets (silica gel 60 F254) and the chromatograms were visualized under UV 254–366 nm Fourier-Transform Infrared (FTIR) spectra were obtained using a PerkinElmer 2000 FTIR Spectrum spectrometer (PerkinElmer, Waltham, MA, USA). The nuclear magnetic resonance (NMR) spectra were obtained using 500 MHz Bruker Advance NMR (500 MHz for 1H-NMR, 125 MHz for 13C-NMR) spectrometer system and the data was analysed using Topspin 4.1.4 software (Bruker Bioscience, Billerica, MA, USA). The chemical shifts were internally calibrated using the residual DMSO peak (1H: 2.50 ppm, 13C: 39.5 ppm), the CDCl3 peak (1H: 7.26 ppm, 13C: 77.0 ppm) or the tetramethylsilane (TMS) signal at 0.00 ppm for both 1H and 13C-NMR. The high-resolution mass spectroscopy (HRMS) was recorded by Waters Xevo QTOF MS (Milford, Massachusetts, United States), and reported in m/z. The synthesis method for the intermediates, as well as the NMR, FTIR and HRSM spectra of all synthesised compounds are presented in ESI.†4.1.2.1 N-Ethyl-3-((3-phenylisoxazol-5-yl)methoxy)quinoxalin-2-amine (5a). IR (neat) ν: 3431 (m, N–H), 3088 (w, aromatic C–H), 2978 (w, Csp3–H), 1531 (s, aromatic C
C), 1447 (s, CN), 1378 (m, C–N), 1195 (s, C–O); 1H-NMR (500 MHz, CDCl3) δ: 7.80–7.82 (m, 2H), 7.68 (d, J = 8.0 Hz, 2H), 7.44–7.47 (m, 4H), 7.31–7.34 (m, 1H), 6.73 (s, 1H), 5.70 (s, 2H), 5.41 (s), 3.58–3.66 (m, 2H), 1.33 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 167.5, 162.7, 146.9, 144.4, 139.6, 134.4, 130.2, 129.0, 129.0, 128.7, 127.1, 126.9, 126.9, 126.4, 125.5, 124.1, 102.8, 58.2, 35.7, 14.7; HRMS (+ESI) [M + H]+: 347.1504, C20H19N4O2, requires 347.1508.
4.1.2.2 N-Ethyl-5-methyl-3-((3-phenylisoxazol-5-yl)methoxy)quinoxalin-2-amine (5b). IR (neat) ν: 3445 (m, N–H), 3062 (w, aromatic C–H), 2963 (w, Csp2–H), 2826 (w, Csp3–H), 1529 (s, aromatic C
C), 1476 (m, CN), 1298 (m, C–N), 1203 (s, C–O); 1H-NMR (500 MHz, CDCl3) δ: 7.80–7.82 (m, 2H), 7.54 (d, J = 7.6 Hz, 1H), 7.45–7.46 (m, 3H), 7.31 (d, J = 7.6 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 6.76 (s, 1H), 5.73 (s, 2H), 5.44 (s, 1H), 3.65–3.72 (m, 2H), 2.67 (s, 3H), 1.38 (t, J = 7.2, 3H); 13C-NMR (125 MHz, CDCl3) δ: 167.7, 162.7, 146.6, 143.4, 138.3, 134.3, 133.8, 130.2, 128.9, 128.9, 128.7, 127.4, 126.9, 124.2, 123.7, 102.7, 58.0, 35.7, 17.4, 14.5; HRMS (+ESI) [M + H]+: 361.1678, C21H21N4O2, requires 361.1650.
4.1.2.3 N-Ethyl-7-nitro-3-((3-phenylisoxazol-5-yl)methoxy)quinoxalin-2-amine (5c). IR (neat) ν: 3379 (w, N–H), 3057 (w, aromatic C–H), 2926 (w, Csp3–H), 1579 (s, aromatic C
C), 1546 (s, NO2), 1502 (m, CN), 1327 (m, C–N), 1201 (s, C–O); 1H-NMR (500 MHz, CDCl3) δ: 8.57 (d, J = 2.5 Hz, 1H), 8.25 (dd, J = 9.0, 2.5 Hz, 1H), 7.80–7.82 (m, 2H), 7.68 (d, J = 9.0 Hz, 1H), 7.46–7.47 (m, 3H), 6.78 (s, 1H), 5.84 (s, 1H), 5.72 (s, 2H), 3.64–3.69 (m, 2H), 1.35 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 166.5, 162.8, 148.1, 145.8, 144.4, 143.6, 133.4, 130.3, 129.1, 128.5, 127.4, 126.9, 126.3, 125.9, 122.7, 121.7, 103.3, 58.5, 35.9, 14.4; HRMS (+ESI) [M + H]+: 392.1357, C20H18N5O4, requires 392.1359.
4.1.2.4 N-Ethyl-5-nitro-3-((3-phenylisoxazol-5-yl)methoxy)quinoxalin-2-amine (5d). IR (neat) ν: 3414 (w, N–H), 3067 (aromatic C–H), 2932 (w, Csp3–H), 1529 (s, NO2), 1297 (m, C–N), 1189 (s, C–O); 1H-NMR (500 MHz, CDCl3) δ: 7.79–7.84 (m, 4H), 7.45–7.46 (m, 3H), 7.31 (t, J = 8.0 Hz, 1H), 6.72 (s, 1H), 5.77 (s, 1H), 5.70 (s, 2H), 3.59–3.62 (m, 2H), 1.30 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 171.3, 166.8, 162.8, 147.6, 145.3, 135.6, 132.7, 130.5, 130.4, 129.1, 128.6, 126.9, 122.3, 122.0, 103.1, 60.5, 58.7, 36.1, 14.4; HRMS (+ESI) [M + H]+: 392.1354, C20H18N5O4, requires 392.1359.
4.1.2.5 3-((3-(2-Chlorophenyl)isoxazol-5-yl)methoxy)-N-ethylquinoxalin-2-amine (5e). IR (neat) ν: 3441 (w, N–H), 3064 (w, aromatic C–H), 2974 (w, Csp3–H), 1531 (s, aromatic C
C), 1442 (m, CN), 1309 (m, C–N), 1196 (m, C–O), 759 (m, Cl); 1H-NMR (500 MHz, CDCl3) δ: 7.74 (dd, J = 7.5, 1.8 Hz, 1H), 7.65–7.69 (m, 2H), 7.50 (dd, J = 7.5, 1.8 Hz, 1H), 7.30–7.45 (m, 4H), 6.92 (s, 1H), 5.71 (s, 2H), 5.43 (s, 1H), 3.57–3.66 (m, 2H), 1.33 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 166.7, 150.5, 147.6, 146.9, 139.6, 134.4, 132.9, 131.0, 130.5, 130.0, 128.8, 127.9, 127.0, 126.4, 125.4, 124.2, 106.2, 58.2, 35.7, 14.6; HRMS (+ESI) [M + H]+: 381.1119, C20H18ClN4O2, requires 381.1118.
4.1.2.6 3-((3-(2-Chlorophenyl)isoxazol-5-yl)methoxy)-N-ethyl-7-methylquinoxalin-2-amine (5f). IR (neat) ν: 3451 (w, N–H), 3056 (aromatic C–H), 2967 (w, Csp2–H), 2928 (w, Csp3–H), 1592 (m, aromatic C
C), 1530 (s, CN), 1317 (m, C–N), 1200 (m, C–O), 763 (m, Cl); 1H-NMR (500 MHz, CDCl3) δ 7.76 (dd, J = 7.6, 1.7 Hz, 1H, H-5), 7.54–7.57 (m, 1H, H-3′′), 7.45–7.50 (m, 2H), 7.34–7.41 (m, 2H, H-7), 7.14 (dd, J = 7.5, 1.7 Hz, 1H), 6.91 (s, 1H′), 5.69 (s, 2H), 5.39 (br. s, 1H), 3.58–3.64 (m, 2H), 2.49 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 166.8, 161.3, 146.5, 144.4, 139.5, 137.1, 132.9, 132.4, 131.1, 130.5, 128.7, 128.0, 127.2, 125.9, 125.8, 125.1, 106.2, 58.1, 35.6, 21.5, 14.7 (C-11); HRMS (+ESI) [M + H]+: 395.1274, C21H20ClN4O2, requires 395.1275.
4.1.2.7 3-((3-(2-Chlorophenyl)isoxazol-5-yl)methoxy)-N-ethyl-5-methylquinoxalin-2-amine (5g). IR (neat) ν: 3445 w, (N–H), 3065 (w, aromatic C–H), 2967 (w, Csp2–H), 2927 (w, Csp3–H), 1529 (s, aromatic C
C), 1298 (m, C–N), 1203 (m, C–O), 762 (m, Cl); 1H-NMR (500 MHz, CDCl3) δ: 7.73 (dd, J = 7.9, 1.8 Hz, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.49 (dd, J = 7.9, 1.2 Hz, 1H), 7.34–7.41 (m, 2H, H-8), 7.30 (d, J = 7.9 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 6.91 (s, 1H, H-4′), 5.70 (s, 2H, H-6′), 5.40 (br. s, 1H, N–H), 3.61–3.66 (m, 2H, H-10), 2.62 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 166.9, 161.2, 146.6, 143.3, 138.3, 134.3, 133.8, 132.9, 131.0, 130.5, 128.0, 127.4, 127.2, 124.2, 123.7, 106.1, 58.0, 35.7, 17.4, 14.5; HRMS (+ESI) [M + H]+: 395.1279, C21H20ClN4O2, requires 395.1275.
4.1.2.8 3-((3-(2-Chlorophenyl)isoxazol-5-yl)methoxy)-N-ethyl-7-nitroquinoxalin-2-amine (5h). IR (neat) ν: 3426 (w, N–H), 3065 (m, aromatic C–H), 2924 (m, Csp3–H), 1544 (m, aromatic C
C), 1500 (CN), 1459 (m, NO2), 1325 (m, C–N), 1080 (m, C–O), 767 (m, Cl); 1H-NMR (500 MHz, CDCl3) δ: 8.57 (d, J = 2.5 Hz, 1H), 8.25 (dd, J = 9.0, 2.5 Hz, 1H), 7.74 (dd, J = 9.0, 2.5 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.51 (d, J = 9.0 Hz, 1H), 7.35–7.43 (m, 2H), 6.96 (s, 1H), 5.85 (s, 1H), 5.74 (s, 2H), 3.65–3.70 (m, 2H), 1.36 (t, J = 7.2, 3H); 13C-NMR (125 MHz, CDCl3) δ: 165.7, 161.3, 148.1, 145.8, 144.4, 143.6, 133.4, 132.9, 131.3, 130.9, 130.5, 127.7, 127.2, 125.9, 122.7, 121.6, 106.7, 58.6, 35.9, 14.4; HRMS (+ESI) [M + H]+: 426.0959, C20H17ClN5O4, requires 426.0969.
4.1.2.9 3-((3-(2-Chlorophenyl)isoxazol-5-yl)methoxy)-N-ethyl-5-nitroquinoxalin-2-amine (5i). IR (neat) ν: 3327 (w, N–H), 3141 (s, aromatic C–H), 2923 (m, Csp3–H), 1594 (m, aromatic C
C), 1532 (s, CN), 1502 (s, NO2), 1224 (m, C–N), 1032 (m, C–O), 767 (m, Cl); 1H-NMR (500 MHz, CDCl3) δ: 7.80–7.84 (m, 2H), 7.74 (dd, J = 7.5, 1.8 Hz, 1H), 7.50 (dd, J = 7.5, 1.8 Hz, 1H), 7.34–7.42 (m, 2H), 7.31 (t, J = 8.0 Hz, 1H) 6.92 (s, 1H), 5.77 (s, 1H), 5.72 (s, 2H), 3.60–3.66 (m, 2H), 1.32 (t, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ: 165.9, 161.3, 147.5, 145.2, 135.5, 132.9, 132.6, 131.2, 131.0, 130.5, 130.4, 129.5, 127.8, 127.2, 122.2, 121.9, 106.4, 58.5, 35.9, 14.3; HRMS (+ESI) [M + H]+: 426.0971, C20H17ClN5O4, requires 426.0969.
4.2 Biology
where Abscontrol represents the absorbance value of the control and Abscommand represents the absorbance value of the compound. The IC50 values of each compound were calculated using the GraphPad Prism 9.0 software (GraphPad Software, La Jolla, CA).
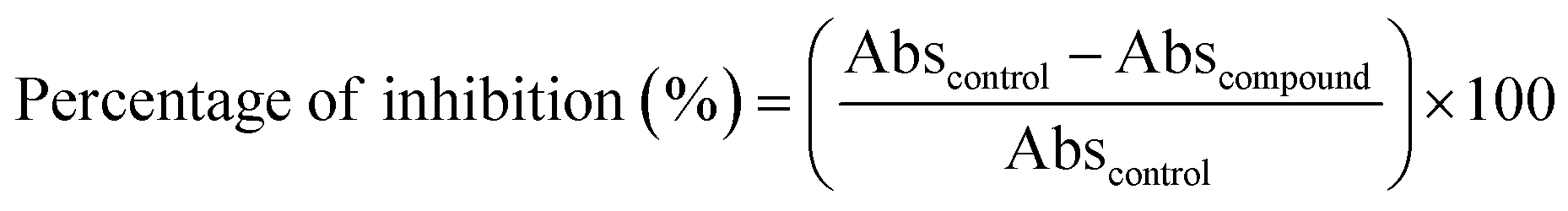
where Abscontrol represents the absorbance value of the control and Abscommand represents the absorbance value of the compound. The IC50 values of each compound were calculated using the GraphPad Prism 9.0 software (GraphPad Software, La Jolla, CA).
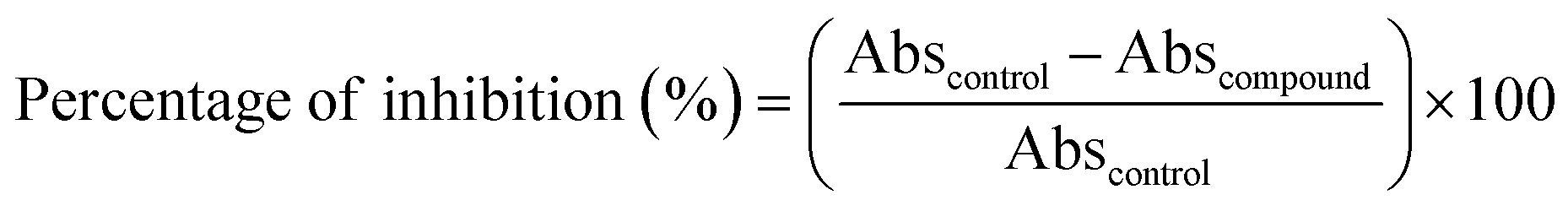
4.3 Molecular docking
Selected quinoxaline compounds were selected to perform molecular docking studies according to their potency as great enzyme inhibitors. For α-amylase enzyme, compounds 5h and 5i, while compounds 5a, 5b, 5c, 5e, and 5h were selected to perform molecular docking studies with α-glucosidase enzyme. The molecular docking studies were performed according to the method reported by Mohamad et al.36 Firstly, the compound structures were drawn using ChemDraw and converted to PDB format. The 3D structures of human pancreatic alpha-amylase complexed with nitrite and acarbose (α-amylase, PDB ID: 2QV4) and C-terminal of human Maltase-Glucoamylase (ctMGAM) complexed with acarbose (α-glucosidase, PDB ID: 3TOP) were fetched by ID via from the protein databank via the UCSF Chimera 1.14. The water molecules and unrelated heteroatoms were removed using the Dock Prep tool and further processed before docking commenced. To recognise the α-amylase enzyme binding sites, the grid box parameters were set to 68, 81 and 77 Å along the X, Y, and Z-axis as grid size and 20, 62 and 16 Å along the X, Y, and Z-axes as the grid centre with 0.375 Å grid spacing. As for α-glucosidase, the grid box parameters were set to 127, 117 and 151 in grid size and −38, 11 and −13 in grid centre along the X, Y, and Z-axis respectively, with 0.375 Å in grid spacing. The docking was performed, and binding energies were calculated via the AutoDock Vina tool. The output results of the docking were further analysed and visualised in 2D and 3D via Discovery Studio Visualizer Client, 2020 (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016).4.4 Molecular dynamics
The molecular dynamics simulations for all the enzyme–compound complexes (5c–glucosidase, 5h–glucosidase, 5h–amylase, and 5i–amylase) and controls (miglitol–glucosidase and miglitol–amylase) were performed using protein_ligand.ipynb37 with the ff19SB force field. The Generalized AMBER Force Field 2 (GAFF2) was used for compounds as characterized using the Antechamber program. For each molecular simulation, the initial conformation for each complex was the docking pose generated from the docking protocol. The complex was solvated in an orthorhombic TIP3P water box (12 nm) with periodic boundary conditions and neutralized with 0.15 molar of NaCl using the AMBER tleap program. Prior to beginning the simulation, each complex was treated with an energy minimization consisting of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of steepest descent algorithm. Next, each molecular model was equilibrated for 1 ns in the isothermal–isobaric (NPT) ensemble (298 K and 1.01325 MPa) with position restraint force constant at 700 kJ mol−1. Finally, the molecular dynamics simulations were carried out in the NPT ensemble (298 K) for 5000 ps with integration timestep of 2 fs. Structural analyses of MD simulation were done by using ProLIF and PyTraj tools integrated in protein_lignad.ipynb. The plots depicting the dynamics stabilities including root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), interaction energy, principal component analysis (PCA), binding free energy, and residue interaction of all systems were generated from the tools.
000 steps of steepest descent algorithm. Next, each molecular model was equilibrated for 1 ns in the isothermal–isobaric (NPT) ensemble (298 K and 1.01325 MPa) with position restraint force constant at 700 kJ mol−1. Finally, the molecular dynamics simulations were carried out in the NPT ensemble (298 K) for 5000 ps with integration timestep of 2 fs. Structural analyses of MD simulation were done by using ProLIF and PyTraj tools integrated in protein_lignad.ipynb. The plots depicting the dynamics stabilities including root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), interaction energy, principal component analysis (PCA), binding free energy, and residue interaction of all systems were generated from the tools.
Author contributions
Conceptualization, M. N. A.; methodology, S. N. M. R., L. P., M. H. A. B., M. T. C. O. and M. N. A.; investigation, S. N. M. R., L. P., and M. T. C. O.; formal analysis, S. N. M. R., L. P., M. T. C. O., U. S., D. H., and N. S. N. S.; data curation, S. N. M. R. and L. P.; validation, M. N. A., M. H. A. B., M. T. C. O., U. S., D. H., and H. A. W.; resources, M. N. A., M. H. A. B., and U. S.; visualization, S. N. M. R., L. P., and M. T. C. O.; writing—original draft preparation, S. N. M. R., M. T. C. O. and M. N. A.; writing—review and editing, M. N. A., M. H. A. B. and M. T. C. O.; supervision, M. N. A., M. H. A. B. and M. T. C. O.; project administration, M. N. A.; funding acquisition, M. N. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the financial support from the Ministry of Higher Education Malaysia (MOHE) under Fundamental Grant Research Scheme (FRGS) – FRGS/1/2019/STG01/USM/01/2 and Graduate Excellence Programme (GrEP) MARA for S. N. M. R.Notes and references
- American Diabetes Association, Diabetes Care, 2014, 37, S81–S90 CrossRef PubMed.
- What is diabetes, https://www.idf.org/aboutdiabetes/what-is-diabetes.html, accessed December 2022.
- D. M. Nathan, Jama, 2015, 314, 1052–1062 CrossRef CAS PubMed.
- J. A. Pereira, A. M. Pessoa, M. N. D. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed.
- S. Tariq, K. Somakala and M. Amir, Eur. J. Med. Chem., 2018, 143, 542–557 CrossRef CAS PubMed.
- M. K. Ibrahim, I. H. Eissa, A. E. Abdallah, A. M. Metwaly, M. M. Radwan and M. A. ElSohly, Bioorg. Med. Chem., 2017, 25, 1496–1513 CrossRef CAS.
- Y. M. Syam, M. M. Anwar, S. S. Abd El-Karim, S. A. Elseginy, B. M. Essa and T. M. Sakr, RSC Adv., 2021, 11, 36989–37010 RSC.
- M. S. Khan, M. A. Munawar, M. Ashraf, U. Alam, A. Ata, A. M. Asiri, S. Kousar and M. A. Khan, Bioorg. Med. Chem., 2014, 22, 1195–1200 CrossRef CAS.
- T. Settypalli, V. R. Chunduri, A. K. Maddineni, N. Begari, R. Allagadda, P. Kotha and A. R. Chippada, New J. Chem., 2019, 43, 15435–15452 RSC.
- M. Missioui, S. Mortada, W. Guerrab, G. Serdaroğlu, S. Kaya, J. T. Mague, E. M. Essassi, M. E. Faouzi and Y. Ramli, J. Mol. Struct., 2021, 1239, 130484, DOI:10.1016/j.molstruc.2021.130484.
- S. Hameed, K. M. Khan, P. Taslimi, U. Salar, T. Taskin-Tok, D. Kisa, F. Saleem, M. Solangi, M. H. U. Ahmed and K. Rani, Int. J. Biol. Macromol., 2022, 211, 653–668 CrossRef CAS.
- F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS.
- A. Sysak and B. Obmińska-Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS.
- H. Zhao, G. Liu, Z. Xin, M. D. Serby, Z. Pei, B. G. Szczepankiewicz, P. J. Hajduk, C. Abad-Zapatero, C. W. Hutchins, T. H. Lubben and M. R. Jirousek, Bioorg. Med. Chem. Lett., 2004, 14, 5543–5546 CrossRef CAS.
- L. Yang, J. Zhang, L. Si, L. Han, B. Zhang, H. Ma, J. Xing, L. Zhao, J. Zhou and H. Zhang, Eur. J. Med. Chem., 2016, 116, 46–58 CrossRef CAS.
- C. K. Lin, L. W. Cheng, H. Y. Li, W. Y. Yun and W. C. Cheng, Org. Biomol. Chem., 2015, 13, 2100–2107 RSC.
- I. Saidi, M. Manachou, M. Znati, J. Bouajila and H. B. Jannet, J. Mol. Struct., 2022, 1247, 131379, DOI:10.1016/j.molstruc.2021.131379.
- J. J. Marín-Peñalver, I. Martín-Timón, C. Sevillano-Collantes and F. J. del Cañizo-Gómez, World J. Diabetes, 2016, 7, 354, DOI:10.4239/wjd.v7.i17.354.
- S. Kumar, S. Narwal, V. Kumar and O. Prakash, Pharmacogn. Rev., 2011, 5, 19, DOI:10.4103/0973-7847.79096.
- A. Bedekar, K. Shah and M. Koffas, Adv. Appl. Microbiol., 2010, 71, 21–73 CAS.
- Y. B. Lee, Y. D. Gong, H. Yoon, C. H. Ahn, M. K. Jeon and J. Y. Kong, Bioorg. Med. Chem., 2010, 18, 7966–7974 CrossRef CAS.
- P. V. Babu, S. Mukherjee, G. S. Deora, K. S. Chennubhotla, R. Medisetti, S. Yellanki, P. Kulkarni, S. Sripelly, K. V. Parsa, K. Chatti, K. Mukkanti and M. Pal, Org. Biomol. Chem., 2013, 11, 6680 RSC.
- G. Nordberg, B. A. Fowler, M. Nordberg, D. B. Moffett, D. B. Mumtaz, D. W. Sullivan Jr and B. Fowler, Handbook on the Toxicology of Metals, Elsevier Academic Press, Netherlands, 2015 Search PubMed.
- S. I. S. Rattan, M. Kyriazis, S. A. Hofbrucker-MacKenzie, I. Sivaprakasam, Y. Ji and M. Manfred Kessels, The Science of Hormesis in Health and Longevity, Elsevier Academic Press, Netherlands, 2019 Search PubMed.
- R. Maurus, A. Begum, L. K. Williams, J. R. Fredriksen, R. Zhang, S. G. Withers and G. D. Brayer, Biochemistry, 2008, 47, 3332–3344 CrossRef CAS PubMed.
- L. Ren, X. Qin, X. Cao, L. Wang, F. Bai, G. Bai and Y. Shen, Protein Cell, 2011, 2, 827–836 CrossRef CAS PubMed.
- L. Phongphane, S. N. Mohd Radzuan, M. H. Abu Bakar, M. T. Che Omar, U. Supratman, D. Harneti, H. A. Wahab and M. N. Azmi, Comput. Biol. Chem., 2023, 106, 107938, DOI:10.1016/j.compbiolchem.2023.107938.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
- C. Bouysset and S. Fiorucci, J. Cheminf., 2021, 13, 1–9 Search PubMed.
- Y. Fu, J. Zhao and Z. Chen, Computational and Mathematical Methods in Medicine, 2018, 3502514, DOI:10.1155/2018/3502514.
- J. Lin, P. Wang, Z. Zhang, G. Xue, D. Zha, J. Wang, X. Xu and Z. Li, Synth. Commun., 2020, 50, 823–830 CrossRef CAS.
- A. Keivanloo, M. Bakherad and A. Rahimi, Synthesis, 2010, 10, 1599–1602 CrossRef.
- A. Keivanloo, M. Bakherad, F. Abbasi, T. Besharati-Seidani and A. H. Amin, RSC Adv., 2016, 6, 105433–105441 RSC.
- J. H. Frederich, J. K. Matsui, R. O. Chang and P. G. Harran, Tetrahedron Lett., 2013, 54, 2645–2647 CrossRef CAS PubMed.
- M. H. A. Bakar, P. Y. Lee, M. N. Azmi, N. Syifa'Lotfiamir, M. S. F. Mohamad, N. S. N. Shahril, K. A. Shariff, H. Ya'akob, K. Awang and M. Litaudon, Biocatal. Agric. Biotechnol., 2020, 25, 101594, DOI:10.1016/j.bcab.2020.101594.
- N. Mohamad, Y. H. Phua, M. H. A. Bakar, M. T. C. Omar, H. A. Wahab, U. Supratman, K. Awang and M. N. Azmi, J. Mol. Struct., 2021, 1245, 131007, DOI:10.1016/j.molstruc.2021.131007.
- P. R. Arantes, M. D. Polêto, C. Pedebos and R. Ligabue-Braun, J. Chem. Inf. Model., 2021, 61, 4852–4856 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08642a |
| This journal is © The Royal Society of Chemistry 2024 |