
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermal synthesis and electrochemical properties of Sn-based peanut shell biochar electrode materials
Yujie
Wang
a,
Hui
Wang
 *a,
Jiangtao
Ji
b,
Tianyan
You
b,
Chang
Lu
c,
Cuiyun
Liu
a,
Yang
Song
b,
Zhi
Chen
d and
Shufa
Zhu
a
*a,
Jiangtao
Ji
b,
Tianyan
You
b,
Chang
Lu
c,
Cuiyun
Liu
a,
Yang
Song
b,
Zhi
Chen
d and
Shufa
Zhu
a
aDepartment of Environmental Engineering, Henan University of Science and Technology, Luoyang 471023, China. E-mail: wanghui79@haust.edu.cn
bCollege of Agricultural Equipment Engineering, Henan University of Science and Technology, Luoyang 471003, China
cSchool of Materials Science and Engineering, Henan University of Science and Technology, Luoyang 471023, Canada
dDepartment of Building, Civil and Environmental Engineering, Concordia University, Montreal H3G 1M8, Canada
First published on 20th February 2024
Abstract
Using activated-carbon-based electrodes derived from waste biomass in super-capacitor energy technologies is an essential future strategy to achieve sustainable energy and environmental protection. Biomass feed-stocks such as bamboo and straw have been used to prepare activated carbon-based electrodes. This experiment used peanut shells (waste biomass) as carbon precursors. Peanut shell-activated biochar materials were prepared using KOH activation and heat treatment, and SnO2@KBC-CNTs, a composite electrode material of biochar loaded with tin oxide. It was produced through hydrothermal synthesis, utilizing SnCl4–5H2O as the tin precursor. The application of KOH activators during pyrolysis markedly enhanced the porosity and specific surface area of the resultant activated biochar, due to effective dispersion and degradation of pyrolytic products. Characterized by a micro–mesoporous structure, the composite's pores boasted a specific surface area of 158.69 m2 g−1. When tested at a density of current of 0.5 A g−1, the specific capacitance of SnO2@KBC-CNTs reached 198.62 F g−1, nearly doubling the performance of the KBC electrode material alone. Moreover, the composite demonstrated a low ion transfer resistance of 0.71 Ω during charge–discharge cycles.
1 Introduction
In the context of societal advancements, there is an escalating demand for cost-effective electrochemical energy storage devices. This trend is catalyzing the progress and implementation of advanced systems of energy storage.1–3 In various emerging electrical and electronic equipment, supercapacitors can satisfy this requirement in many fields due to their high power, high reliability, high security, low cost, and other advantages, with continuing growth in the market size and application areas, such as rail transportation, wind power, photovoltaic, and aerospace.4 Supercapacitors are mainly composed of electrodes, fluid collectors, diaphragms, and electrolytes, with the primary factor influencing both their performance and production expense having the electrode material. The quest for high-performance, economical electrode materials constitutes a primary research objective in the field of supercapacitors.Among the several available electrode materials, SnO2 has attracted considerable attention for use in lithium-ion batteries, supercapacitors,5 solar cells,6 biosensors,7,8 and catalysts9 because of its high theoretical specific capacitance, high electrochemical activity, low cost, non-toxicity, and good chemical stability.10 However, although conventional SnO2 has a higher specific capacitance (782 mA h g−1), it has a drastically reduced capacitance due to its poor conductivity, which limits the rate of transfer between the electrode and the electrolyte. During charging and discharging, SnO2 undergoes significant volume expansion, leading to potential pulverization or agglomeration of the electrode material. This phenomenon restricts the material's specific capacity from reaching its anticipated effectiveness, thereby limiting its practical application.11,12 Therefore, integrating metal oxides with carbon-based materials can enhance electrode conductivity. Carbon, as an electrode material, offers high stability, excellent electrical conductivity, cost-effectiveness, abundant availability, and ease in pore size control.13 Activated carbon was the first material used in electric double-layer capacitor electrodes, with the advantages of simple preparation, adjustable pore size, low cost, and the easy availability of raw materials. Currently, it is a hot-spot for scientific research and commercial applications.14 The enhancement of activated carbon's microstructure and surface chemical properties is crucial for advancing the electrochemical reaction of supercapacitors.15 In the field of activated carbon electrodes, the selection of carbonaceous precursors and the activation methodology significantly impact their surface and structural features. Peanut is an important oil and cash crop in China, and its subsidiary product, peanut shells, accounts for 30% of its total output.16 Peanut shells are abundant, with huge annual production; however, they have limited comprehensive utilization rates.17 If the low-value waste peanut shells can be converted into useful carbon materials for preparing supercapacitor electrodes, the treatment and disposal problems caused by the incineration of a large amount of waste biomass can be effectively alleviated, and the synthesized materials are abundant in availability, low in cost, and have sustainable characteristics to achieve social, economic and environmental benefits. Due to its elevated carbon and low ash contents, it can be used as a feed-stock for producing carbon materials, providing a viable opportunity for high-value recovery. In order to overcome the carbon material is lower than the capacitor, SnO2 is easy to expand the shortcomings of charge and discharge, in the context of energy storage mechanisms of EDLC and pseudo-capacitors, porous carbon with a substantial specific surface area is essential. It could be utilized to provide the spatial location for the metal nanoparticles on the one hand, and their introduction can provide the pseudo-capacitance on the other, which couples the performance of EDLC with that of the pseudo-capacitor, and describes a new electrode material with high performance and satisfactory stability. Synthesized composites of SnO2 nano-materials and a biochar matrix can further improve the cyclability of SnO2. After the SnO2 nano-materials are tightly composited with carbon, the resilient carbon matrix serves as a buffer for SnO2's volumetric changes during the charge/discharge cycles. It prevents the agglomeration of SnO2 nanoparticles and augments the conductivity of the electrode materials; moreover, the composites themselves have nano-structures, which can shrink the ionic transport and diffusion distance, enhancing the electrochemical performance of SnO2 is imperative.18,19
Several SnO2@C composite materials have been synthesized, each demonstrating superior electrochemical characteristics. SnO2/Cotton Cellulose Nano-fiber (CNF) composites have been prepared by electrostatic spinning, and their specific capacitance was reported as 187 F g−1 at 20 mV s−1 because of the high coverage of the composite surface by the SnO2 nanoparticles.20 SnO2/GO composite nano-materials have been prepared using one-pot synthesis under microwave irradiation with a better specific capacitance of 100 F g−1.21 PANI/SnO2 NRA was synthesized by a one-step electrochemical stripping and electrode-position process, depositing polyaniline nano-wires on SnO2 NRA. This method achieved a specific capacitance of 367.5 F g−1.22 The aniline monomers's in situ polymerization on SnO2 surfaces produced a PANI/SnO2 composite with a specific capacitance of 175 F g−1 at 1 A g−1 density of current.23 Hierarchical SnO2 nano-clusters wrapped around functionalized carbonized cotton cloth were prepared using a two-step method, where the FCC was first subjected to acidification and hierarchical nitrogen dioxide nano-clusters were grown as a flexible substrate using solvent-thermal reaction and calcination processes to prepare SnO2@FCC composite electrode materials, with the carbon cloth acting as a conductive channel for rapid electron and ion transfer and SnO2 nano-clusters promoting charge storage. The specific capacitance at 1 A g−1 was 197 F g−1.24
Therefore, in this experiment, activated carbon with a porous structure was prepared using KOH activation method with peanut shell biochar as the precursor, which was loaded with nano-SnO2 to enhance the specific capacitance of the activated carbon electrode materials; moreover, novel SnO2-loaded graded porous carbon composite electrode materials (SnO2@KBC-CNTs) were synthesized, which encourages further research on the use of abundant agricultural biomass for preparing cost-reducing and efficiency-enhancing materials of super-capacitor electrode.
2 Experiment section
2.1 Preparation of KBC
Before the experiment, the collected peanut shells were soaked and cleaned three to five times, dried in an electric blast drying oven, and the obtained peanut shells were broken up and set aside. An appropriate amount of spare peanut shell powder was placed in a crucible in a pyrotube furnace; under vacuum protection, the temperature was set to rise to a carbonization temperature of 600 °C under constant-temperature carbonization, durations for the carbonization was 1 h. The produced peanut shell biochar was combined with KOH at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 weight ratio until it was all dissolved in KOH; the mixture was then moved into a shaking box to shake for 2 h, filtered by extraction with a Buchholz funnel, and dried. The dried mixture was heated to a temperature of 700 °C in a pyrotube furnace program under vacuum protection, the KOH activation was 2 h. After activation at a constant temperature, the mixture was removed. Following production, the porous peanut shell biochar materials were sequentially cleansed with HCl and deionized water, then dried in a convection oven, milled, and prepared for use, and were named KOH–Biochar (KBC).
:2 weight ratio until it was all dissolved in KOH; the mixture was then moved into a shaking box to shake for 2 h, filtered by extraction with a Buchholz funnel, and dried. The dried mixture was heated to a temperature of 700 °C in a pyrotube furnace program under vacuum protection, the KOH activation was 2 h. After activation at a constant temperature, the mixture was removed. Following production, the porous peanut shell biochar materials were sequentially cleansed with HCl and deionized water, then dried in a convection oven, milled, and prepared for use, and were named KOH–Biochar (KBC).
2.2 Preparation of SnO2@KBC-CNT composite
A solution of SnCl4–5H2O in 50 mL of anhydrous ethanol was prepared, to which KBC and CNTs were added in equal proportions. The solution was ultrasonicated for 30 min using a cell crusher and then magnetically stirred for 2 h until it became a homogeneous and dispersed suspension, which was then transferred to the polytetrafluoroethylene inner liner of a high-pressure reactor (the working pressure is 3 M pa). The resulting mixture was thermally treated at 180 °C for a duration of 8 hours to obtain a black solid. The samples were centrifugally washed to neutralize the pH and freeze-dried to obtain the KOH–Biochar–SnO2 (SnO2@KBC-CNTs) composites.2.3 Preparation of electrode materials
Nickel foam, with a thickness of 1 mm, was uniformly cut into squares measuring 1 cm × 1 cm to be used as the substrate of the electrode sheet. The cut nickel foam was immersed in an ethanol solution for ultrasonication, and the uniformly sized cleaned nickel mesh was then dried at a constant temperature in a blower drying oven and stored for later use.The binder polyvinylidene fluoride (PVDF, 10 wt%; 0.01 g) was pipetted into a 10 mL centrifuge tube, 20 μL of N-methylpyrrolidone (NMP) was pipetted into it as a solvent with a pipette gun, and the mixture was vibrated on an oscillator to completely dissolve the NMP.
The weight ratio of PVDF:electrode active material:acetylene carbon black was 8:1:1. The prepared super-capacitor electrode material (biochar) (0.08 g) was added to acetylene black (0.01 g) in a mortar, ground evenly, and transferred to a 25 mL beaker; the fully dissolved PVDF mixture was added into the 25 mL beaker, and an appropriate amount of NMP was pipetted for “slurry” viscosity adjustment, followed by ultrasonic dispersion treatment.
The spare nickel mesh was weighed with an electronic balance (recorded as m1), and a pipette gun was used to inject drops of the “sample mixture” into the weighed mass of the nickel foam, restricting them to an area of one centimeter × one centimeter. Subsequently, the mesh underwent drying in a convection drying oven, and a manual tablet press was then used to obtain electrodes, with the weight of the electrodes after tablet pressing recorded as m2; the electrode on the loaded material quality is (m2 − m1). The mass loading of active materials for all three electrodes tested in this work were 2 mg. The specific flow chart is shown in Fig. 1.
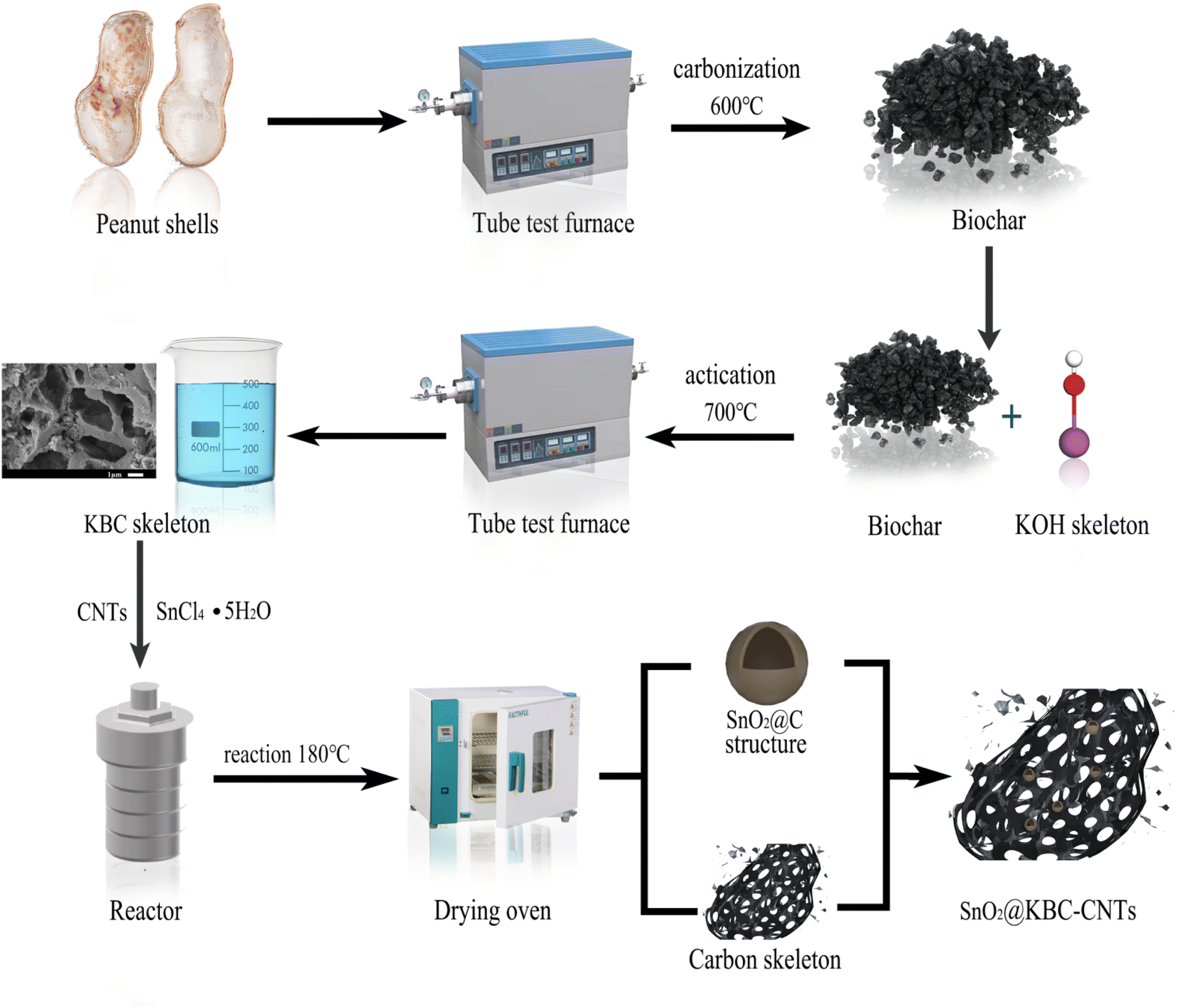 | ||
| Fig. 1 Diagram of SnO2@KBC-CNTs preparation. | ||
2.4 Structural and physical features
A powder diffractometer functioning under copper target circumstances was used to analyze the physical phases of the samples. Scanning electron microscopy (SEM) was used to analyze surface morphologies. Employing an automated surface and porosity analyzer for eight hours at 120 °C degassing temperature, we assessed the specific surface area and pore size distribution. X-ray photo-electron spectroscopy was used to determine the elemental composition and chemical states on the sample surfaces.2.5 Electrochemical characterizations
Electrochemical assessments for the three-electrode setup were conducted employing electrochemical workstation, at ambient temperature. This configuration included KBC, CNTs, and SnO2@KBC-CNTs as working electrodes, a platinum sheet electrode (S = 1 × 1 cm2) as the counter electrode, Hg/HgO as the reference electrode, as well as 6 M KOH as the electrolyte.Cyclic voltammetry (CV) experiments were performed at scanning speeds of 10–100 mV s−1 throughout a potential range of −1 to 0 V; constant-current charge/discharge (GCD) tests were performed from 0 V to 0.5 V, with current densities ranging from 0.5 to 2 A g−1. Electrochemical impedance spectroscopy (EIS) experiments ranged from 0.01 to 105 Hz. Eqn (1), derived from the GCD curve, was used to compute the electrode-specific capacitance.
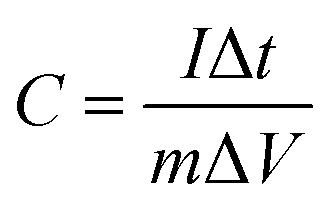 | (1) |
3 Results and discussion
3.1 Structural and physical characterizations
SEM images of KBC are presented in Fig. 2(a) and (b). The KOH-activated peanut-shell biochar exhibits many micro-porous structures, and the skeleton shows an ordered gully and loose porous structure. During KOH activation, the produced K2CO3 attaches to the carbon skeleton and further decomposes to form K2O, CO, and CO, which disappear in the form of gases, leaving many micro-pores. The subsequently generated K2O reacts with C at 700 °C, and part of the K2O is reduced to K vapor. In contrast, a part of it is embedded in the carbon skeleton, and K2O and K2CO3 are removed in the subsequent pickling process to form a large number of micro-porous structures.25,26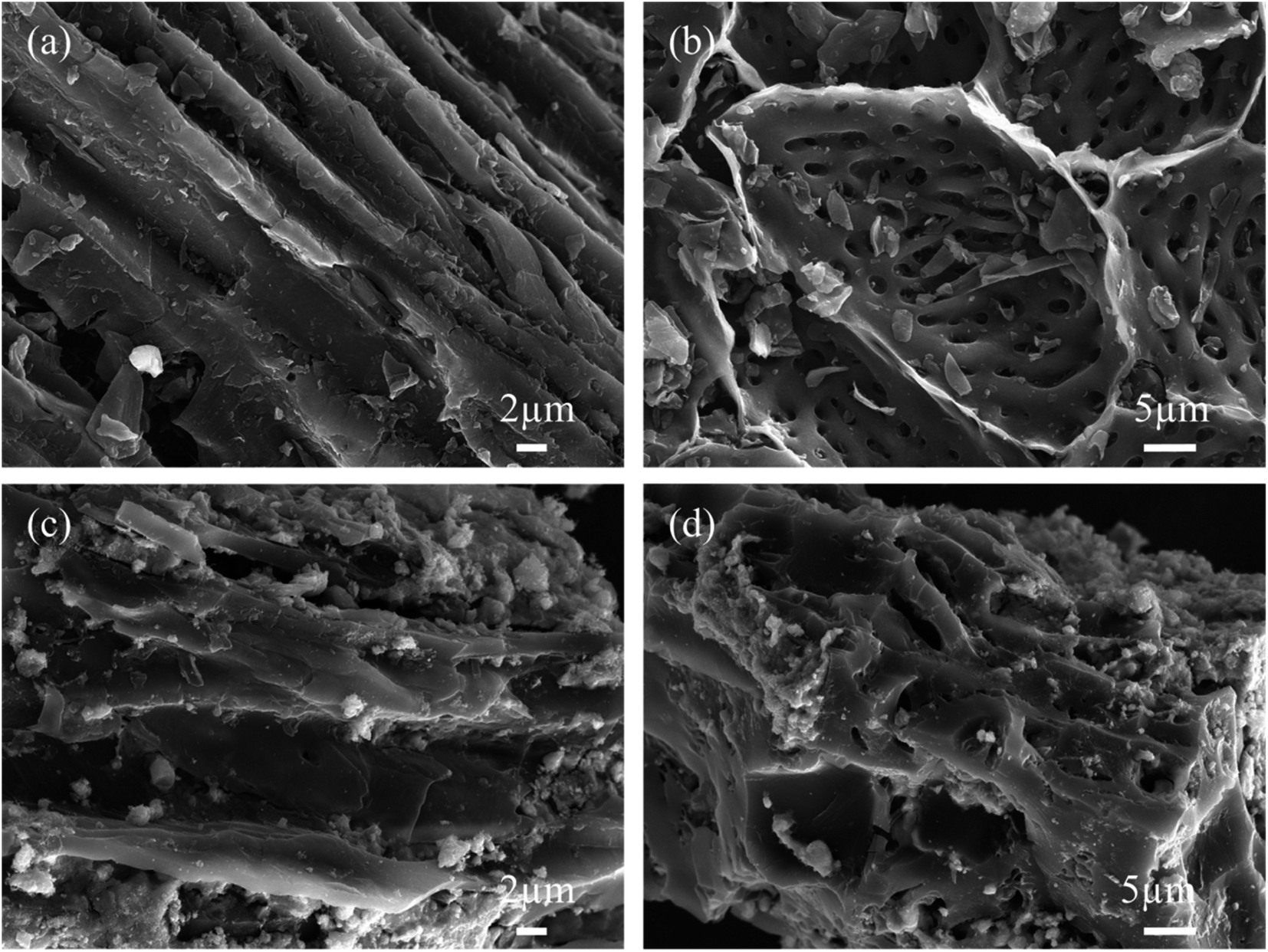 | ||
| Fig. 2 SEM images for (a and b) KBC; (c and d) SnO2@KBC-CNTs. | ||
Fig. 2(c) and (d) describes SEM photos of the SnO2@KBC-CNTs. The SnO2 nanoparticles have a white spherical structure that is uniformly dispersed within the inner wall vesicles of the KBC. During both charging and discharging cycles, this design efficiently limits the volumetric growth of SnO2. Meanwhile, the pore structure of KBC did not change significantly, indicating that the loading of SnO2 nanoparticles retained the excellent spatial structure of KBC, providing a good basis for inter-ion transport.27
In order to analyze the material morphology and structure more clearly and intuitively, TEM tests were done focusing on SnO2@KBC-CNTs as shown in Fig. 3. Fig. 3(a) and (b) reveals agglomeration in the SnO2-carbon composite, attributable to the substantial surface free energy of the nano SnO2 synthesized via hydrothermal method. High-resolution TEM imaging, as depicted in Fig. 3(c), displays a detailed view of a single SnO2 nano-particle. This particle has significant lattice fringe and a measured inter-planar separation of 0.337 nm, which aligns with the SnO2 (110) crystalline plane. This finding corroborates the initial formation of SnO2, consistent with XRD results. The carbon layer lattice spacing observed in Fig. 2(d) measures 0.340 nm and 0.211 nm, equivalent to the (002) and (110) crystal planes of carbon, respectively. This indicates the presence of ordered layered structures within the carbon matrix,28 similar to the graphite (002) crystal plane in the amorphous carbon substrate. This structure enhances inter-layer connectivity, thereby improving the conductivity of the composite electrodes.
 | ||
| Fig. 3 TEM images (a–d) of SnO2@KBC-CNTs. | ||
XRD patterns of the three samples, presented in Fig. 4(a), exhibit two prominent diffraction peaks at 26° and 44°. These peaks are indicative of the (002) and (101) crystal planes of graphitic carbon in KBC and CNTs, respectively.29 The peanut-shell-based porous carbon has a small grain size, defective crystal structure, and anisotropic lattice, which were proved by two diffraction peaks with low intensity and large width.30 SnO2@KBC-CNTs show seven diffraction peaks at 26.5°, 33.74°, 37.54°, 51.86°, 54.7°, 61.8°, and 65.26°, which correspond to the (110), (101), (200), (211), (220), (310), and (301) crystallographic surfaces of SnO2@KBC-CNTs, respectively.31 The diffraction peaks all matched exactly when compared with the SnO2 standard map (JCPDS of za41-1445),32 affirming that SnO2 with tetragonal rutile phase structure was synthesized. No additional diffraction peaks were detected in the spectrum; the peaks presented are all narrow and sharp, which prove that SnO2@KBC-CNTs had complete crystallinity and high purity and that no other impurities were introduced in the hydrothermal reaction.33
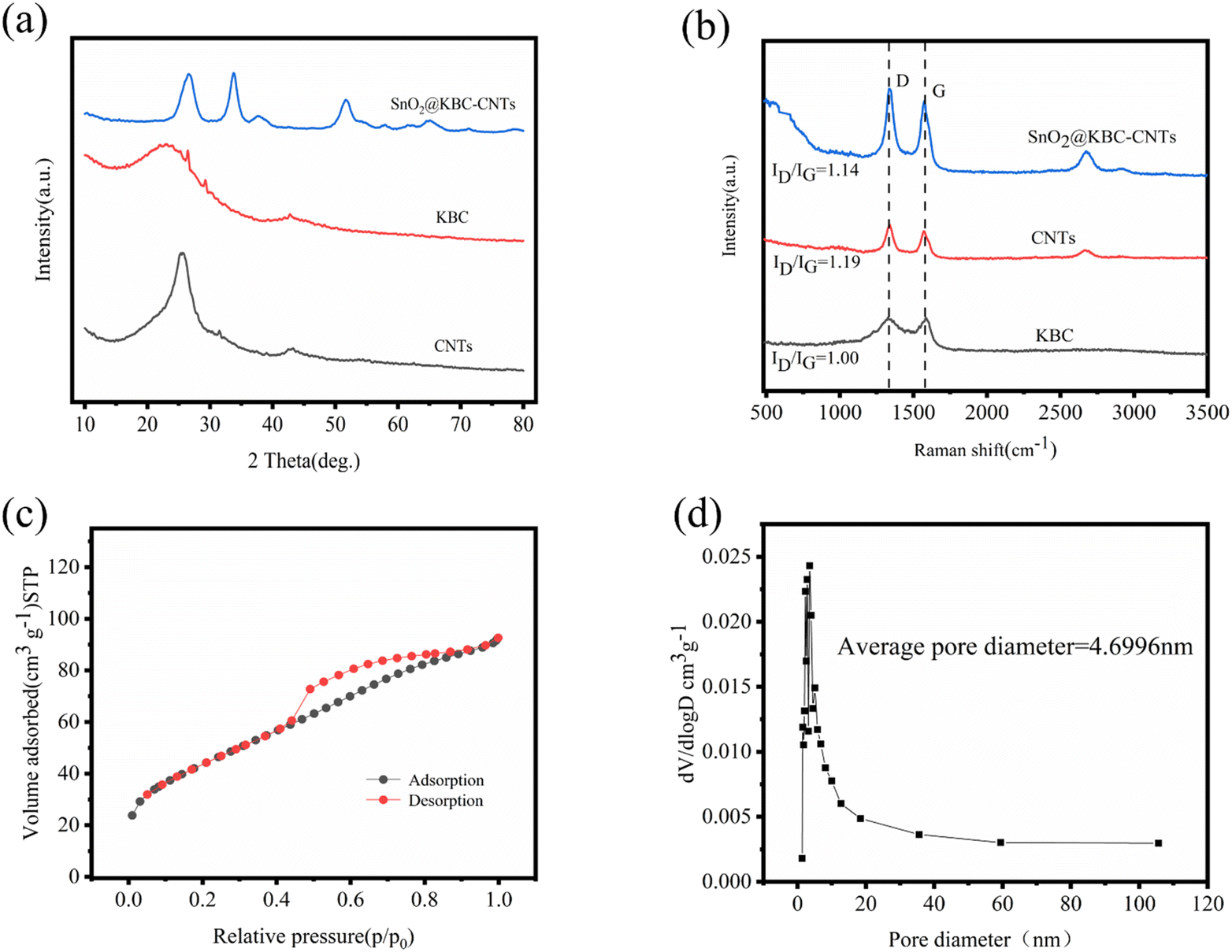 | ||
| Fig. 4 (a) XRD patterns of CNTs, KBC, and SnO2@KBC-CNTs; (b) Raman spectra of the same; and (c and d) isotherms of nitrogen adsorption and desorption and the distribution of pore size for SnO2@KBC-CNTs. | ||
Fig. 4(b) depicts the Raman spectra of the three samples, showcasing two prominent peaks: the D peak at 1335 cm−1 and the G peak at 1577 cm−1. The D peak, indicative of defects within the carbon lattice, becomes more pronounced with an increase in lattice irregularities. Conversely, the G peak arises from the first-order scattering of the E2g mode in the carbon material at the Gamma point, reflecting the in-plane vibrational modes of sp2-hybridized carbon atoms. The intensity ratio of these peaks, ID/IG, serves as an indicator of carbon material graphitization: a lower ID/IG ratio signifies higher graphitization and, consequently, improved electrical conductivity. The ID/IG values of KBC, CNTs, and SnO2@KBC-CNTs were 1.00, 1.19, and 1.14, respectively. The increased ID/IG value of SnO2@KBC-CNTs compared with that of KBC indicates that the sp2 domain size of KBC decreases during the reduction process. The graphitization and electrical conductivity are higher than those of CNTs and SnO2@KBC-CNTs. The formation of nano-SnO2 further destroys the graphitic structure of carbon and generates more disordered carbon. It provides more active sites for electrolyte ion transfer, promotes the ion transfer between electrode and electrolyte, and improves the electrochemical performance of the composite electrode.
Porous carbon's effectiveness as an electrode material is inextricably tied to its pore size distribution, particular area of surface, as well as surface functional groups. Micro-pores provide abundant active sites for ion storage and diffusion and exhibit a large specific surface area, which is conducive to the distribution of active sites, increasing the contact between active sites and the substrate and contributing to the improvement of specific capacitance. The mesopores can provide channels for ion storage and transport, which are conducive to ion transfer.34 In order to obtain a more comprehensive understanding of the pore size distribution and specific surface area of SnO2@KBC-CNTs, nitrogen adsorption and desorption tests were performed; the outcomes are illustrated in Fig. 4(c). Characteristic of mesoporous structures, the N2-adsorption–desorption isotherm displays type-IV isotherm featuring a distinctive H4 hysteresis loop in the high P/P0 range.35 When hysteresis commences at a specific threshold of pore width, it indicates that the material contains an abundance of micro-pores and mesopores. Its pore size distribution of SnO2@KBC-CNTs predominantly ranges from 2 nm to 50 nm, averaging at 4.6996 nm in Fig. 4(d). Mesoporous structures are advantageous for electrolyte ion migration. Achieving an ideal number of mesopores facilitates inter-ion exchange rates, shortens ion diffusion paths to the electrode surface, and lowers material internal resistance.36 Micro-pores have a high charge accumulation capacity at the electrode–electrolyte interface and provide a large specific surface area. The specific surface areas of CNTs, KBC and SnO2@KBC-CNTs composites were respectively 240.43 m2 g−1, 176.35 m2 g−1, 158.69 m2 g−1. Because SnO2 were loaded on the surface of peanut shell biochar, the specific surface area of SnO2@KBC-CNTs was reduced. The pore volume of SnO2@KBC-CNTs was 0.14 cm3 g−1. The large specific surface area can promote mutual contact within the electrode material and the KOH electrolyte and increase the contact area within ions during the impregnation process. In contrast, the rich porous structure provides more active sites that facilitates the swift movement of the electrolyte and diminishes the distance at which ions diffuse, and provides favorable conditions for charge transfer in the electrochemical process.
Fig. 5 illustrates the XPS pattern of SnO2@KBC-CNTs, revealing simultaneous signals for Sn 3d, O 1s, and C 1s, as well as O KLL, indicating the co-presence of Sn, O, and C in the composite. High-resolution XPS patterns for Sn, C, and O are shown in Fig. 5(b)–(d). In the Sn 3d peak map, two peaks at 486.9 eV and 495.3 eV are observed, corresponding to Sn 3d5/2 and Sn 3d3/2 binding energies in SnO2; both these belong to the +4 valence binding energies of Sn,37 and the absence of any other simultaneous oxidation state peak of Sn proves the generation of SnO2 in agreement with the findings in the XRD patterns. The peaking separation of 8.5 eV aligns with the binding energy for Sn 3d5/2 and Sn 3d3/2. The C 1s and O 1s splitting curves, depicted in Fig. 5(c) and (d), show peaking at 284.8 eV, 285.85 eV, as well as 288.57 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,38 C–O, and O–CO bonds.39,40 The pronounced CC peaks in SnO2@KBC-CNTs indicate a higher degree of graphitization, which significantly lowers charge-transfer resistance during charge and discharge cycles. In Fig. 5(d), peaks at 531.49 eV, 532.8 eV, as well as 533.41 eV represent, C–O–Sn/C–O–C/C–OH, as well as O–CO bonds.41,42 The carbon materials with oxygen-containing groups have increased hydrophilicity, which lowers ion transfer resistance and increases adsorption ability. In addition, the presence of Sn–O further confirmed the presence of SnO2 in the SnO2@KBC-CNTs.
C,38 C–O, and O–CO bonds.39,40 The pronounced CC peaks in SnO2@KBC-CNTs indicate a higher degree of graphitization, which significantly lowers charge-transfer resistance during charge and discharge cycles. In Fig. 5(d), peaks at 531.49 eV, 532.8 eV, as well as 533.41 eV represent, C–O–Sn/C–O–C/C–OH, as well as O–CO bonds.41,42 The carbon materials with oxygen-containing groups have increased hydrophilicity, which lowers ion transfer resistance and increases adsorption ability. In addition, the presence of Sn–O further confirmed the presence of SnO2 in the SnO2@KBC-CNTs.
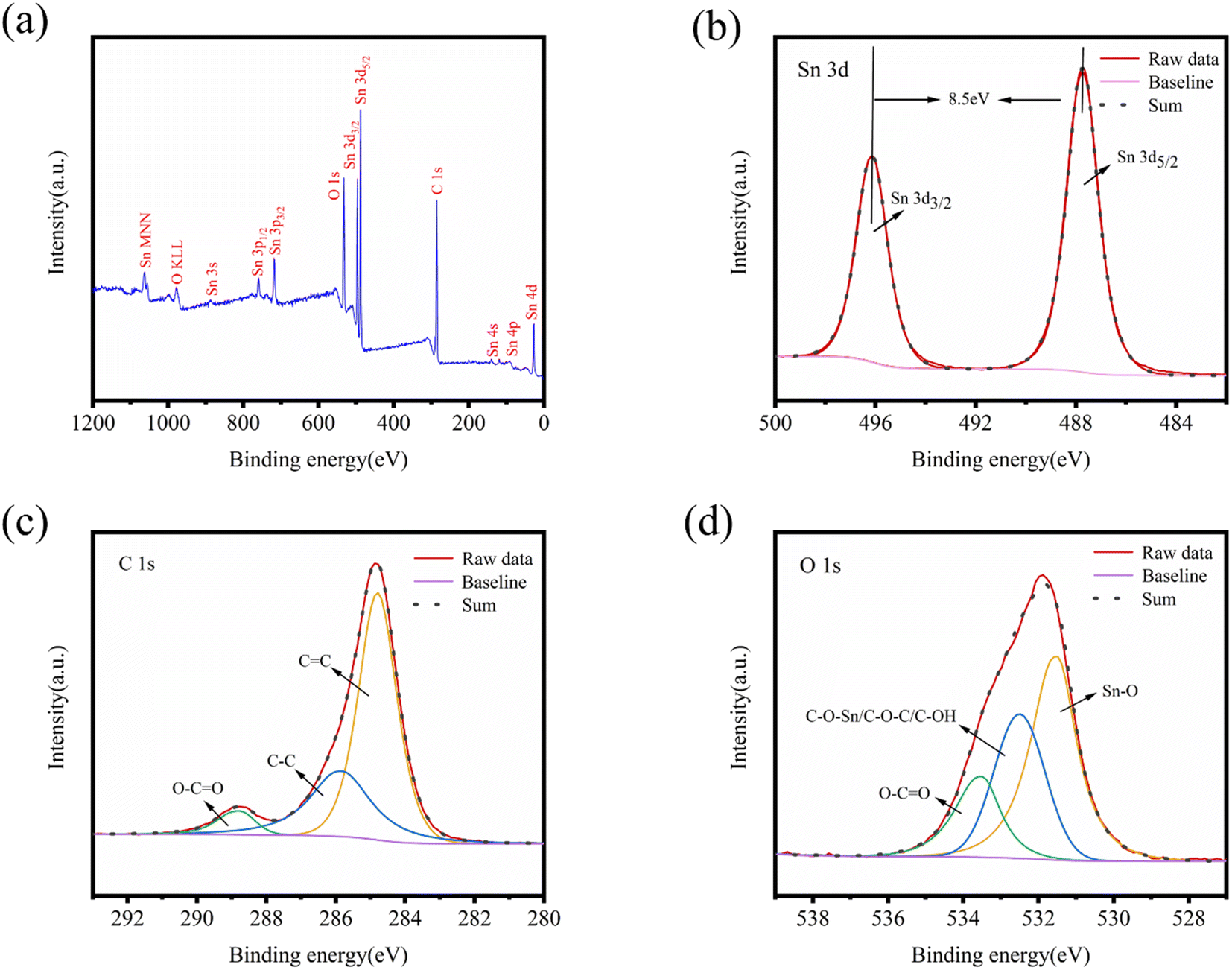 | ||
| Fig. 5 (a) XPS spectra involving all peaks; (b–d) Sn 3d, C 1s, and O 1s peaks of SnO2@KBC-CNTs. | ||
3.2 Electrochemical performance studies
Employing an aqueous-phase three-electrode structure, the electrochemical characteristics of KBC, CNTs, as well as SnO2@KBC-CNTs were assessed. The results are displayed in Fig. 6. The CNTs' CV curves roughly resemble a rectangle, indicating that porous carbon mainly provides double-layer capacitance when used as a super-capacitor electrode, which satisfies the energy storage principle of double-layer capacitance. At various scanning rates, the CV curves of the CNTs exhibit no redox peaks; that is, the capacitance obtained by the electrodes is high compared with the theoretical capacitance of the contained active substances, and the substance utilization is sufficiently high to fully charge and discharge the CNTs. Compared with CNTs, KBC and SnO2@KBC-CNTs had a distinct irreversible reduction peak at −1 V, and an irreversible reduction reaction was presumed to have occurred. The CV curves of KBC and SnO2@KBC-CNTs deviated from the rectangular shape, suggesting that the activation and loading of metal oxides introduced pseudo-capacitance.43 SnO2@KBC-CNTs electrode has a distinct oxidation peak in the −0.1 to −0.2 V voltage interval and a distinct reduction peak around −0.6 V. When the density of current increased, K+ in the electrolyte migrated toward the outer surface of the electrode. K+ migrated more readily to the interior of the electrode when the scanning rate decreased. Pseudo-capacitance within a 6 M KOH alkaline electrolyte results from K+ and SnO2 ion adsorption–desorption responses:| (SnO2)surface + K+ + e− ↔ (SnO2−K+)surface | (2) |
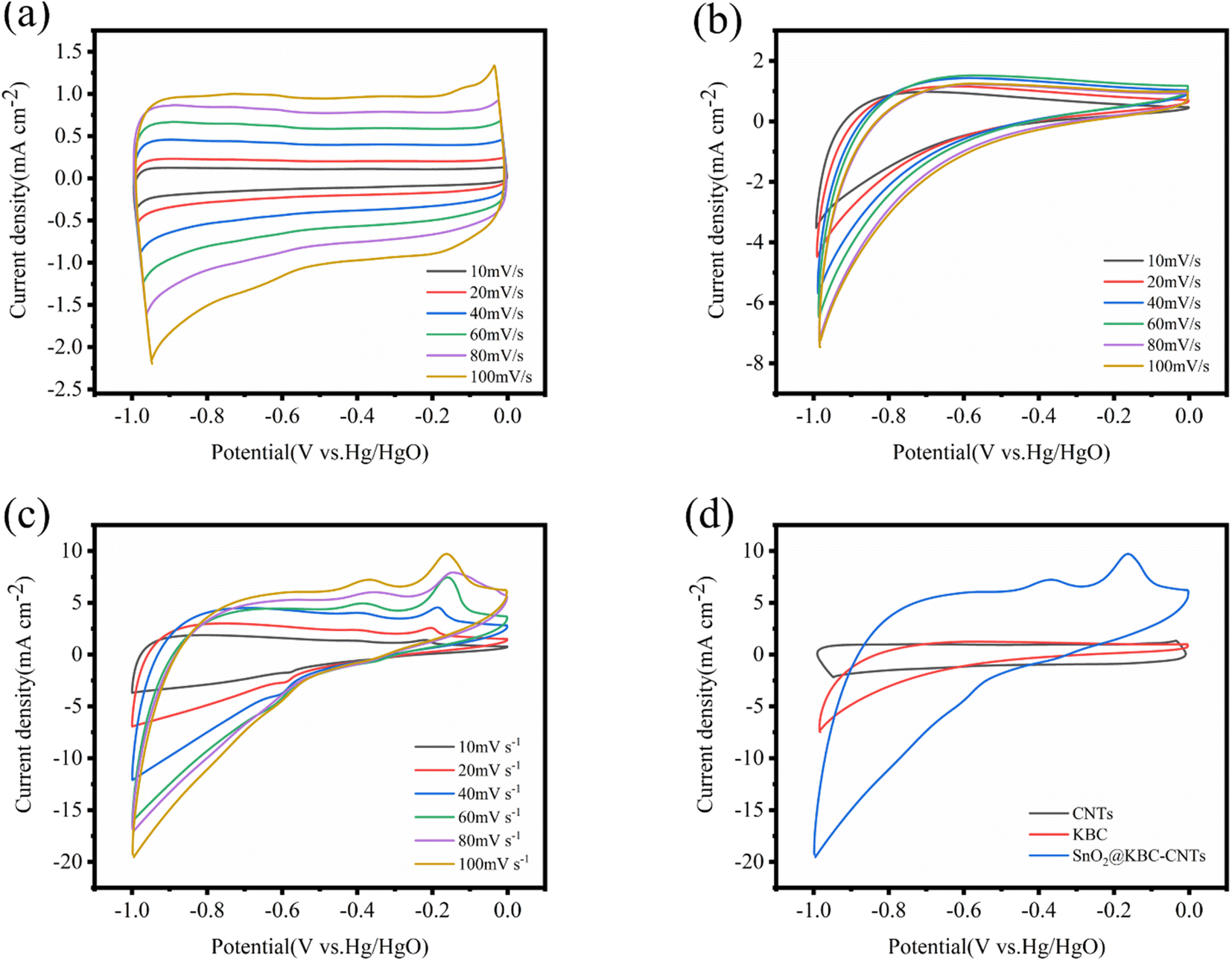 | ||
| Fig. 6 CV curves of (a) CNTs; (b) KBC; (c) SnO2@KBC-CNTs within 6 M KOH at varying scanning rates; (d) shows CV curves of CNTs, KBC, and SnO2@KBC-CNTs at a scanning rate (100 mV s−1). | ||
As seen in the Fig. 6, the peak current escalates with scanning rate, while the curve shape remains consistent. This indicates electrochemical stability and excellent reversibility of the three electrodes in the −1 to 0 V range. A comparison of the CV curves of the three electrodes at the identical 100 mV s−1 scanning rate shows that the CV curves of SnO2@KBC-CNTs have the largest area of the closed graph surrounded by the CV curves, suggesting that the SnO2@KBC-CNTs may exhibit the highest mass ratio capacitance in these samples. This is because SnO2@KBC-CNTs have more micro-pores than in KBC and CNTs. Micro-pores significantly contribute to the electrode material's particular surface area, with larger micro-pores providing a more specific surface area and adsorbing more ions.
Fig. 7(a)–(c) illustrates the charge–discharge curves of CNTs, KBC, and SnO2@KBC-CNTs with different electrical densities in 6 M KOH. As could be observed in the figure, all three samples show low voltage drop (IR); the GCD curves of CNTs exhibit a symmetric isosceles triangle shape, while those of KBC and SnO2@KBC-CNTs deviate from the isosceles triangle shape and are asymmetric, which suggests a redox reaction, exhibiting their typical pseudo-capacitance characteristics. Fig. 7(d) indicates that the charging and discharging times of SnO2@KBC-CNTs are longer than those of CNTs and KBC. These curves indicate larger specific capacitances, aligning with the CV curve results. Calculated using eqn (1), the specific capacitances of CNTs, KBC, and SnO2@KBC-CNTs are 37.92 F g−1, 92.19 F g−1, as well as 198.62 F g−1, at a density of current (0.5 A g−1). The specific capacitances of these composite electrodes surpass those of CNTs and KBC alone, and are superior to many reported SnO2@C composites (as listed in Table 1). This suggests that the synergistic interaction among nano-SnO2, CNTs, and KBC markedly increases the composite electrode's specific capacitance.44 Particular capacitance diminishes with increased density of current, attributed to insufficient time for positive ions to penetrate the internal voids at high densities of current, leading to adsorption predominantly on the material's external surface. When positive ions have low densities of current, they can be efficiently transported to each pore of the electrode material, generating a large number of adsorption–desorption reactions, leading to a high specific capacitance.45 In addition, combined with the results of BET pore size analysis, SnO2@KBC-CNTs have a more developed microporous–mesoporous structure compared with that in CNTs and KBC, which is favorable for the adsorption and transport of the particles, leading to smaller resistance and improved capacitance performance.46
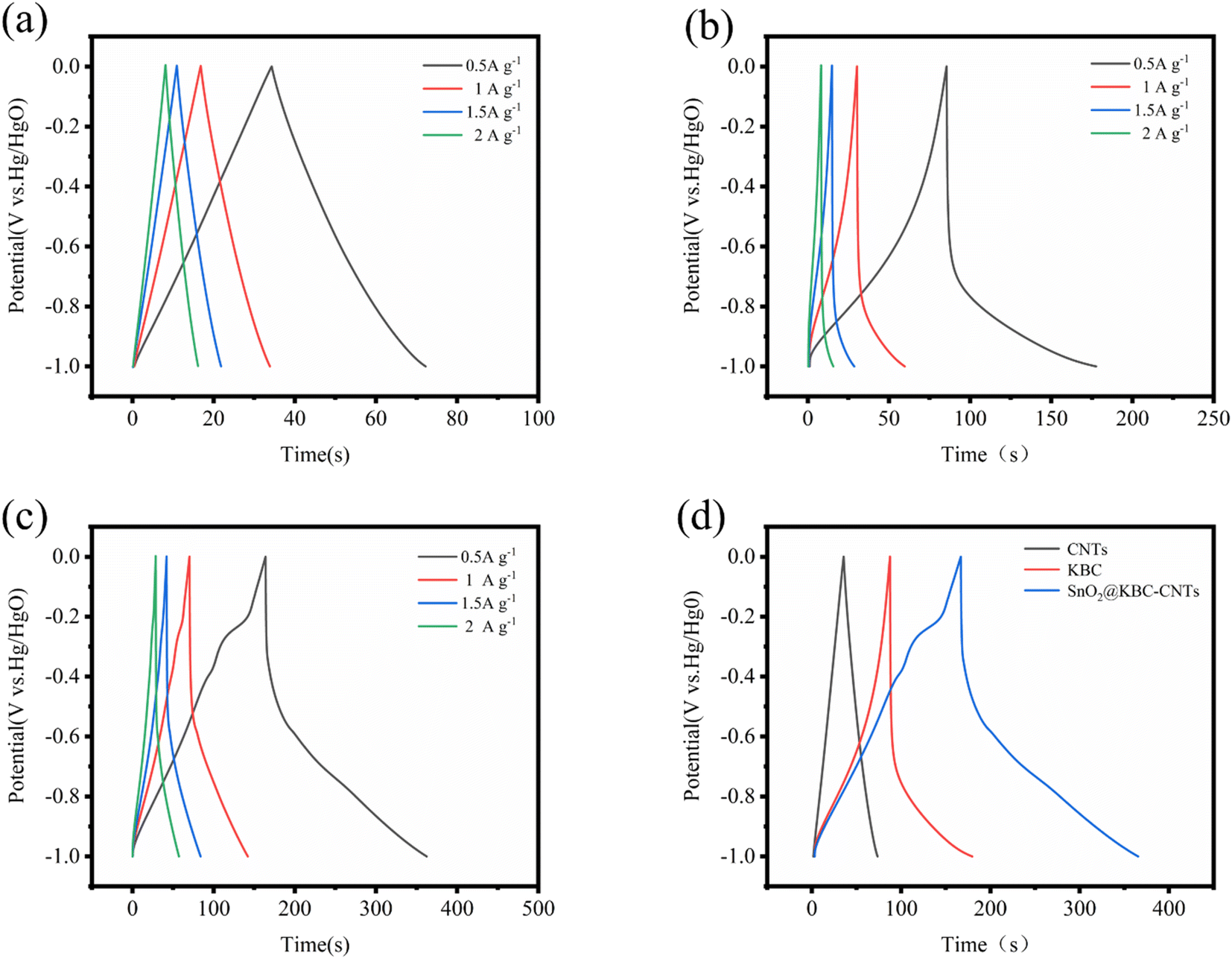 | ||
| Fig. 7 GCD curves of (a) CNTs; (b) KBC; (c) SnO2@KBC-CNTs within 6 M KOH at various densities of current; (d) presents GCD curves for CNTs, KBC, and SnO2@KBC-CNTs at a density of current of 0.5 A g−1. | ||
| Composite | Scanning rate/current density | Electrolyte | Specific capacitance (F g−1) | References |
|---|---|---|---|---|
| SnO2@carbon aerogel | 10 mA g−1 | 1 M H2SO4 | 69.8 | 47 |
| SnO2/GO | 50 mV s−1 | 1 M H2SO4 | 99.7 | 21 |
| SnO2@Graphene | 0.2 A g−1 | 1 M H2SO4 | 126 | 48 |
| SnO2@MWCNT | 1 A g−1 | 4.5 M H2SO4 | 133.33 | 49 |
| SnO2@CNF | 1 A g−1 | 1 M H2SO4 | 118 | 50 |
| C@SnO2 | 2 A g−1 | 4.5 M H2SO4 | 150 | 51 |
| SnO2@FCC | 1 A g−1 | 1 M Na2SO4 | 197.7 | 52 |
| SnO2@KBC-CNTs | 0.5 A g−1 | 6 M KOH | 198.62 | This work |
To identify the electrochemical differences, investigations using electrochemical impedance spectroscopy were conducted among CNTs, KBC, and SnO2@KBC-CNTs. EIS curves typically bifurcate to regions of high and low frequencies. In the realm of high frequencies, the semicircular arc signifies charge transfer resistance (Rct),53 where a smaller arc indicates reduced Rct, particularly relevant for activated carbon electrodes. Conversely, the low-frequency region is characterized by a linear segment, indicative of the material's ion diffusion resistance; a steeper gradient denotes a reduced resistance to ion diffusion. Its intercept on the Z′-axis corresponds to the electrolyte resistance (Rs).54Fig. 8(a) shows that SnO2@KBC-CNTs possess smaller semicircular arcs, suggesting lower internal resistance compared to KBC, thereby facilitating charge transfer and enhancing conductivity. The linear segment in the low-frequency range of SnO2@KBC-CNTs, being closer to the y-axis than KBC, implies that the micro–mesoporous structure of SnO2@KBC-CNTs fosters charge transport and electrolyte ion permeation. EIS results were modeled using an equivalent circuit, displayed in Fig. 8's inset. Zview fitting calculations yielded Rs values of 0.08 Ω, 0.27 Ω, as well as 0.32 Ω, and Rct values of 0.2 Ω, 0.60 Ω, and 0.71 Ω, for CNTs, KBC, and SnO2@KBC-CNTs respectively. After the charge and discharge cycle test on the electrodes, the Rs values of CNTs, KBC and SnO2@KBC-CNTs were calculated by Zview fitting to be 0.09 Ω, 0.31 Ω and 0.34 Ω, and the Rct values were 0.25 Ω, 0.63 Ω and 0.73 Ω, respectively. The relatively lower resistance values of SnO2@KBC-CNTs, influenced by their high micropore content and moderate graphitization, align with the reduced IR drop seen in the GCD as well as CV curves.
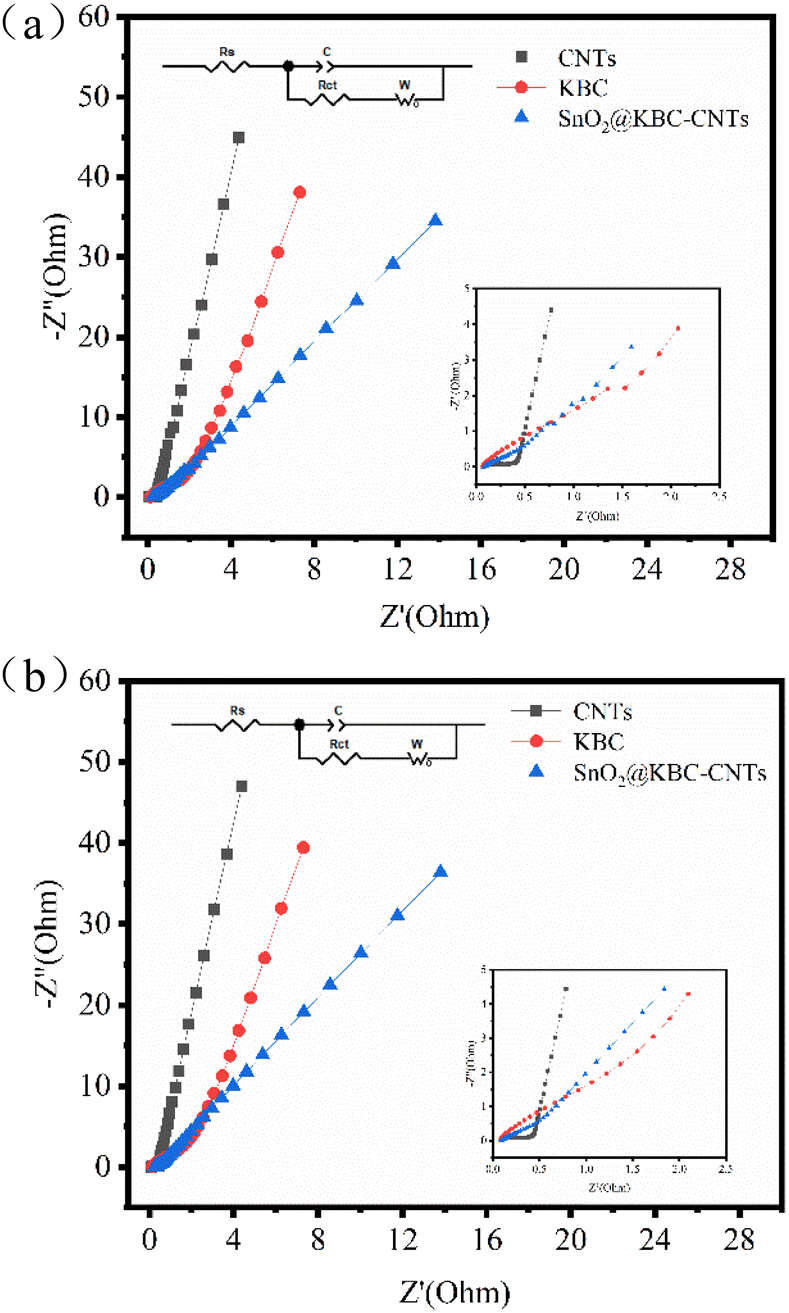 | ||
| Fig. 8 EIS of CNTs, KBC, and SnO2@KBC-CNTs within 6 M KOH ((a) before charge–discharge; (b) after charge–discharge). | ||
The exceptional electrochemical performance of SnO2@KBC-CNTs stems chiefly from the synergistic interplay between SnO2 nanoparticles and carbon, coupled with the composite's distinctive microstructure. This configuration effectively counters the volumetric expansion of SnO2 during charge–discharge cycles. Additionally, incorporating carbon partially addresses the SnO2 matrix's inherent electrical conductivity limitations, thereby augmenting the overall electrode conductivity.
The evaluation of electrode performance includes the assessment of cycling stability, which is a crucial parameter. In this study, the composite electrodes were put through 2000 cycles of charging and discharging at a density of current of 0.5 A g−1 in order to test the endurance during cycling of the materials. As depicted in Fig. 9(a), at the end of the 2000th cycle, the capacity retention rate of the SnO2@KBC-CNTs, KBC, CNTs were 91.2%, 81.03%, 53.9%, respectively. The SnO2@KBC-CNTs electrode exhibited excellent cycling stability. The observed minimal capacitance decay during charge and discharge cycles is ascribed to the volumetric expansion and contraction of SnO2. Such ultra-thin interface layer between nano-SnO2 and carbon in SnO2@KBC-CNTs acts as a buffer, mitigating SnO2's volumetric changes during repeated cycling.
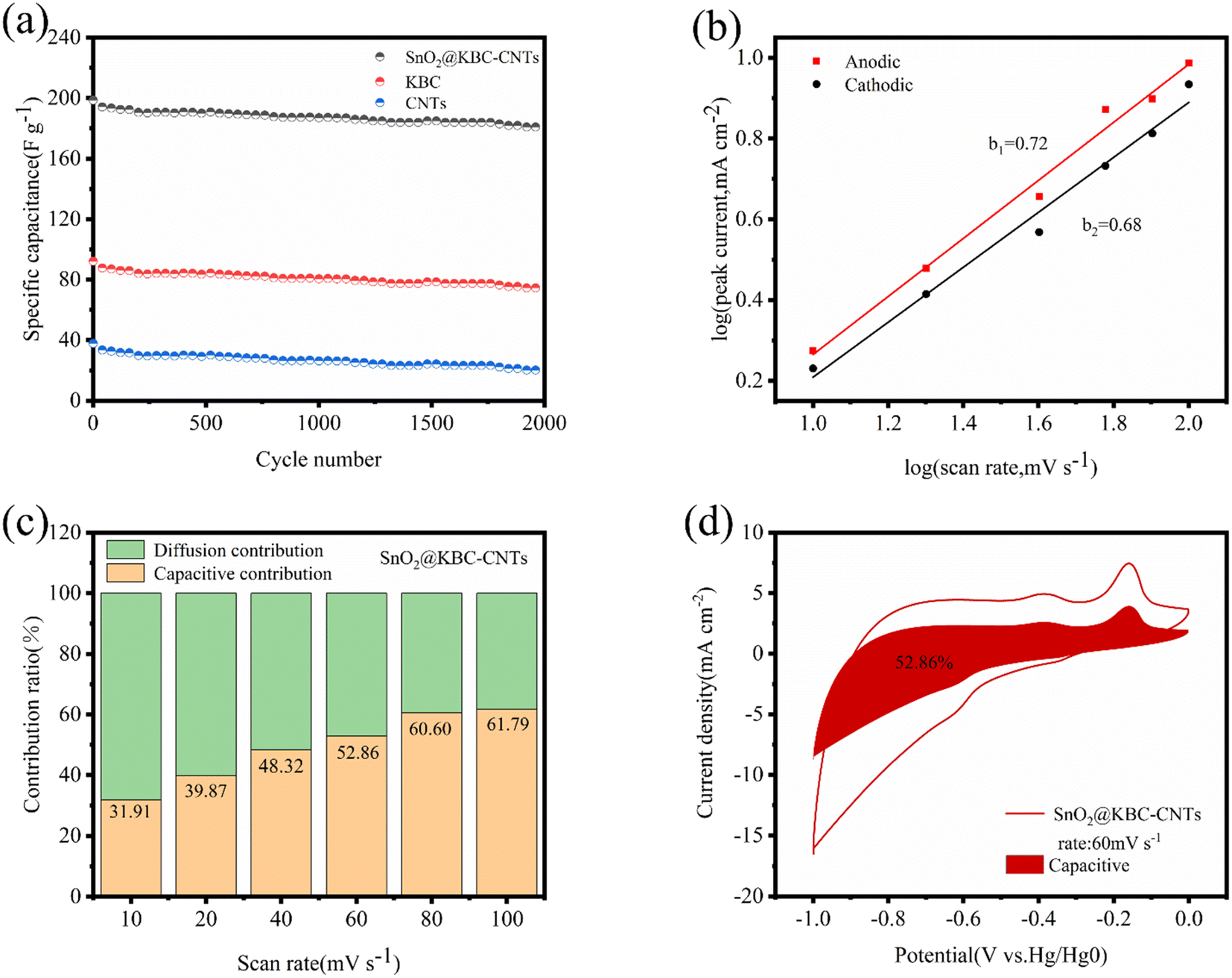 | ||
| Fig. 9 (a) The cycling effectiveness of three electrodes were evaluated at a density of current of 0.5 A g−1; (b) linear fits of logi and logv for SnO2@KBC-CNTs at different sweep rates; (c) capacitive contributions of SnO2@KBC-CNTs at various scan speeds, including 10 mV s−1, 20 mV s−1, 40 mV s−1, 60 mV s−1, 80 mV s−1, & 100 mV s−1; (d) capacitive contributions of SnO2@KBC-CNTs at 60 mV s−1 sweep rate. | ||
To elucidate the mechanism of charge storage of the SnO2@KBC-CNTs porous composite, pseudocapacitive behavior was analyzed. Fig. 6(c) displays that cyclic voltammetry (CV) measurements were conducted at scanning rates varying from 10 mV s−1 to 100 mV s−1, showing the ion diffusion kinetics of the SnO2@KBC-CNTs porous composites during the charging and discharging processes. Eqn (3) was employed to interpret the response mechanism of different peaks in the CV curves, fitting the association involving scanning rates (v) and corresponding peak currents (i) to identify capacitive behaviors during the charge and discharge processes.
| i = avb | (3) |
Considering the logarithms on both sides of eqn (3) yields eqn (4):
| logi = logv + loga | (4) |
i as well as logv are linearly related to each other, and b is the slope of the line; b ≥ 1 denotes capacitively controlled storage of charge; b, equaling to 0.5 represents typical diffusion-controlled storage of charge; a value of b between 0.5 and 1 indicates two types of charge storage behaviors: capacitive and diffusion-controlled.55
Fig. 9(b) describes the linear fits of log i and logv for SnO2@KBC-CNTs at various rates of scan (b values of 0.72 as well as 0.68), indicating that the porous electrode material of SnO2@KBC-CNTs is controlled by two types of charge storage behaviors, namely, diffusion-controlled and capacitive, during the charging and discharging processes. The capacitive contribution was quantified using eqn (4):
| i = k1v + k2v1/2 | (5) |
According to Fig. 9(d), the contribution of the capacitive behavior is 52.86% at 60 mV s−1 scanning rate, and the capacitance-controlled region exhibits a profile similar to that of the CV curve, indicating capacitance-controlled behavior in the SnO2@KBC-CNTs composite electrode material and that the primary source of pseudocapacitive charge storage is predominantly derived from the surface reaction occurring on SnO2@KBC-CNTs electrode. Therefore, the SnO2@KBC-CNTs composite electrode materials are controlled by two charge-storage behaviors, diffusion-controlled and capacitance-controlled, during charging and discharging, which is one of the reasons for the excellent electrochemical performance of the SnO2@KBC-CNTs.
4 Conclusions
The precursor material utilized in the present research consisted of peanut shells, while KOH was used as the activator. And directionally impregnated with SnCl4 to ultimately obtain the nano-SnO2-loaded peanut shell-based porous capacitor carbon. SnO2@KBC-CNTs had a well-developed pore structure. The incorporation of SnO2 inside the porous carbon matrix effectively combined pseudo-capacitance with bi-layer capacitance, preserving the original pore structure while achieving a specific surface area of 158.69 m2 g−1. The composite electrode consisting of SnO2@KBC-CNTs exhibited a specific capacitance of 198.62 F g−1 when subjected to a density of current of 0.5 A g−1 in a 6 M KOH electrolyte solution. Notably, after 2000 cycles, the electrode maintained 91.2% of its initial capacity, demonstrating remarkable cycling stability.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. The authors declare no competing financial interests.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (41471256); Project for the Training of Young Backbone Teachers of Higher Education Institutions in Henan Province (2018GGJS047); Henan Provincial Science and Technology Research Project (192102110050); National Natural Science Foundation of China (22374061).References
- X. Wei and J. Zhang, IOP Conf. Ser. Earth Environ. Sci., 2016, 52, 012079 CrossRef.
- Y. Gu, L.-Q. Fan, J.-L. Huang, C.-L. Geng, J.-M. Lin, M.-L. Huang, Y.-F. Huang and J.-H. Wu, J. Power Sources, 2019, 425, 60–68 CrossRef CAS.
- W. Shen, J. Zang, H. Hu, J. Xu, Z. Zhang, R. Yan and S. Dai, Mater. Des., 2020, 195, 108992 CrossRef CAS.
- S. Huang, X. Zhu, S. Sarkar and Y. Zhao, APL Mater., 2019, 7, 7–10 Search PubMed.
- F. Wang, X. T. Zhou, J. Zhou, T. K. Sham and Z. Ding, J. Phys. Chem. C, 2007, 111, 18839–18843 CrossRef CAS.
- X. Li, T. Li, Q. Zhong, X. Zhang, H. Li and J. Huang, Mater. Lett., 2014, 130, 232–235 CrossRef CAS.
- X. W. Lou, Y. Wang, C. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325–2329 CrossRef CAS.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 RSC.
- Z. Dong, H. Ren, C. M. Hessel, J. Wang, R. Yu, Q. Jin, M. Yang, Z. Hu, Y. Chen, Z. Tang, H. Zhao and D. Wang, Adv. Mater., 2014, 26, 905–909 CrossRef CAS PubMed.
- C.-M. Chen, in Surface Chemistry and Macroscopic Assembly of Graphene for Application in Energy Storage, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 123–141, DOI:10.1007/978-3-662-48676-4_6.
- J. Yang, S. Wang, R. Dong, L. Zhang, Z. Zhu and X. Gao, Mater. Lett., 2016, 184, 9–12 CrossRef CAS.
- W. Wang, Y. Tian, X. Li, X. Wang, H. He, Y. Xu and C. He, Appl. Surf. Sci., 2012, 261, 890–895 CrossRef CAS.
- P. Forouzandeh, V. Kumaravel and S. C. Pillai, Catalysts, 2020, 10, 969 CrossRef CAS.
- N. Manikandan, B. Lakshmi and S. Shivakumara, J. Mater. Sci.: Mater. Electron., 2022, 33, 7109–7118 CrossRef CAS.
- Y. Tang, Y. Liu, S. Yu, F. Gao and Y. Zhao, Russ. J. Electrochem., 2015, 51, 77–85 CrossRef CAS.
- S. S. Arya, A. R. Salve and S. Chauhan, J. Food Sci. Technol., 2016, 53, 31–41 CrossRef CAS.
- F. Duan, J.-P. Zhang, C.-S. Chyang, Y.-J. Wang and J. Tso, Fuel Process. Technol., 2014, 128, 28–35 CrossRef CAS.
- H. Xiao, F. Qu, A. Umar and X. Wu, Mater. Res. Bull., 2016, 74, 284–290 CrossRef CAS.
- Y. Yang, S. Ren, S. Ma, C. Hao and M. Ji, Electrochim. Acta, 2015, 155, 437–446 CrossRef CAS.
- J. Mu, B. Chen, Z. Guo, M. Zhang, Z. Zhang, C. Shao and Y. Liu, J. Colloid Interface Sci., 2011, 356, 706–712 CrossRef CAS PubMed.
- S. Wang, S. P. Jiang and X. Wang, Electrochim. Acta, 2011, 56, 3338–3344 CrossRef CAS.
- Y. Xie and F. Zhu, J. Solid State Electrochem., 2017, 21, 1675–1685 CrossRef CAS.
- X. Shen, L. Ma, M. Gan, Z. Li, J. Yan, S. Xie, H. Yin and J. Zhang, Synth. Met., 2014, 196, 20–26 CrossRef CAS.
- X. Hong, S. Li, R. Wang and J. Fu, J. Alloys Compd., 2019, 775, 15–21 CrossRef CAS.
- F. Guo, X. Jia, S. Liang, X. Jiang, K. Peng and L. Qian, J. Electrochem. Soc., 2019, 166, A3109–A3118 CrossRef.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- R. E. A. Ardhi, G. Liu, J. Park and J. K. Lee, Energy Storage Mater., 2023, 54, 863–874 CrossRef.
- M. X. Tran, R. E. A. Ardhi, G. Liu, J. Y. Kim and J. K. Lee, Chem. Eng. J., 2020, 401, 126075 CrossRef CAS.
- Y. Zhang, S. Liu, X. Zheng, X. Wang, Y. Xu, H. Tang, F. Kang, Q.-H. Yang and J. Luo, Adv. Funct. Mater., 2017, 27, 1604687 CrossRef.
- A. Y. Kim, R. E. A. Ardhi, G. Liu, J. Y. Kim, H.-J. Shin, D. Byun and J. K. Lee, Carbon, 2019, 153, 62–72 CrossRef CAS.
- P. Wu, N. Du, H. Zhang, J. Yu and D. Yang, J. Phys. Chem. C, 2010, 114, 22535–22538 CrossRef CAS.
- M. Gao, X. Chen, H. Pan, L. Xiang, F. Wu and Y. Liu, Electrochim. Acta, 2010, 55, 9067–9074 CrossRef CAS.
- R. E. A. Ardhi, G. Liu, M. X. Tran, C. Hudaya, J. Y. Kim, H. Yu and J. K. Lee, ACS Nano, 2018, 12, 5588–5604 CrossRef CAS PubMed.
- Y. Wen, L. Chi, X. Wen, X. Chen and E. Mijowska, Adv. Electron. Mater., 2020, 6, 2000450 CrossRef CAS.
- F. L. Braghiroli, V. Fierro, A. Szczurek, N. Stein, J. Parmentier and A. Celzard, Carbon, 2015, 90, 63–74 CrossRef CAS.
- S. R. S. Prabaharan, R. Vimala and Z. Zainal, J. Power Sources, 2006, 161, 730–736 CrossRef CAS.
- J. Guo, P. Li, L. Chai, Y. Su, J. Diao and X. Guo, RSC Adv., 2017, 7, 30070–30079 RSC.
- R. E. A. Ardhi, G. C. Liu and J. K. Lee, ACS Energy Lett., 2021, 6, 1432–1442 CrossRef CAS.
- A. Bhaumik, A. Haque, M. F. N. Taufique, P. Karnati, R. Z. Patel, M. Nath and K. Ghosh, J. Mater. Sci. Eng., 2017, 6, 1–11 Search PubMed.
- Q. B. Bui, D. M. Nguyen, T. M. L. Nguyen, L. K. Kwac, H.-G. Kim, S. C. Ko and H. Jeong, J. Electrochem. Sci. Technol., 2018, 9, 229–237 CrossRef CAS.
- Y. Luan, G. Nie, X. Zhao, N. Qiao, X. Liu, H. Wang, X. Zhang, Y. Chen and Y.-Z. Long, Electrochim. Acta, 2019, 308, 121–130 CrossRef CAS.
- Z. Li, C. Tong-qin, G. Yun, J. Guo and B.-c. Yang, J. Alloys Compd., 2014, 586, 353–359 CrossRef CAS.
- M. X. Tran, G. Liu, R. E. A. Ardhi, S.-W. Lee and J. K. Lee, Energy Storage Mater., 2024, 65, 103145 CrossRef.
- M. Halim, G. C. Liu, R. E. A. Ardhi, C. Hudaya, O. Wijaya, S. H. Lee, A. Y. Kim and J. K. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 20566–20576 CrossRef CAS.
- M. U. Rani, V. Naresh, D. Damodar, S. Muduli, S. K. Martha and A. S. Deshpande, Electrochim. Acta, 2021, 365, 137284 CrossRef CAS.
- A. A. Arie, L. Hadisaputra, R. F. Susanti, H. Devianto, M. Halim, R. Enggar and J. K. Lee, J. Phys.: Conf. Ser., 2016, 877, 012020 CrossRef.
- S.-W. Hwang and S.-H. Hyun, J. Power Sources, 2007, 172, 451–459 CrossRef CAS.
- J. Li, X. Zhang, J. Guo, R. Peng, R. Xie, Y. Huang and Y. Qi, J. Alloys Compd., 2016, 674, 44–50 CrossRef CAS.
- V. Vinoth, J. J. Wu, A. M. Asiri, T. Lana-Villarreal, P. Bonete and S. Anandan, Ultrason. Sonochem., 2016, 29, 205–212 CrossRef CAS PubMed.
- J. Ge, Y. Qu, L. Cao, F. Wang, L. Dou, J. Yu and B. Ding, J. Mater. Chem. A, 2016, 4, 7795–7804 RSC.
- V. Naresh and S. K. Martha, J. Electrochem. Soc., 2019, 166, A551–A558 CrossRef CAS.
- X. Hong, S. Li, R. Wang and J. Fu, J. Alloys Compd., 2019, 775, 15–21 CrossRef CAS.
- C. Gong, X. Wang, D. Ma, H. Chen, S. Zhang and Z. Liao, Electrochim. Acta, 2016, 220, 331–339 CrossRef CAS.
- H. Jia, J. Sun, X. Xie, K. Yin and L. Sun, Carbon, 2019, 143, 309–317 CrossRef CAS.
- J. Zhao and A. F. Burke, Energy Storage Mater., 2021, 36, 31–55 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |