
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcise synthesis of deuterium-labelled proanthocyanidin metabolite 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone as an analytical tool†
Angelica
Artasensi
 a,
Sarah
Mazzotta
b,
Giovanna
Baron
a,
Giancarlo
Aldini
a and
Laura
Fumagalli
*a
a,
Sarah
Mazzotta
b,
Giovanna
Baron
a,
Giancarlo
Aldini
a and
Laura
Fumagalli
*a
aDepartment of Pharmaceutical Sciences, University of Milan, 20133 Milan, Italy. E-mail: laura.fumagalli@unimi.it
bDepartment of Chemistry, University of Milan, 20133 Milan, Italy
First published on 20th February 2024
Abstract
Deuterated proanthocyanidin metabolite 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone has been successfully produced. This metabolite is responsible for several proanthocyanidin protective effects in the field of cancer chemoprevention, skin wrinkle-prevention, and antimicrobials. The synthetic approach applied employs a short reaction sequence and allows the incorporation of four deuterium atoms on non-exchangeable sites, making it an attractive strategy to produce a stable isotopically labeled internal standard for quantitative mass spectrometry isotope dilution-based methods, as demonstrated by developing an LC-MS/MS method to quantify DHPV in urine samples. Overall, this efficient synthesis provides a valuable analytical tool for the study of the metabolic conversion of proanthocyanidins thus helping to investigate the biological effect and establishing the active dose of the key catabolite 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone.
1. Introduction
In the quest for healthier and more sustainable lifestyles, scientific research has turned its focus towards the remarkable potential of bioactive compounds found in various food sources. (Poly)phenol-rich foods are well-known to exert protective effects against chronic diseases like neurodegeneration, cancer, and cardiovascular disease. Proanthocyanidins (PACs) are a complex and abundant class of (poly)phenols found in foods including fruits, vegetables, cereals, and beverages like tea, coffee and wine, and in botanicals.1 Their structural complexity arises from a diverse range of monomeric units, mainly flavan-3-ols, including (+)-catechin and (−)-epicatechin. The degree of polymerization varies, giving rise to a spectrum of oligomers and polymers with distinctive physicochemical properties. Even though PACs are abundant in nature, they present poor absorption in the small intestine. As a matter of fact, when they reach the colon, they are catabolized by the microbiota into other compounds that could exert health effects after their absorption.2–4 As a result, the bioactivity of PACs is generally strictly correlated with the activity of the microbiota. In recent years, PACs have gained considerable scientific interest due to their potent antioxidant and free radical scavenging abilities. Several studies have shown that a higher dietary intake of PACs is associated with a reduced risk of chronic diseases such as cardiovascular disease, cancer, and neurodegenerative disorders.5–7 The beneficial effects on the human organism are mainly attributed not to PACs themselves, but rather to the corresponding catabolites.8–10 Metabolomics analyses have identified 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone (DHPV, 5) which maintains intact the catechol moiety, as the major catabolite in the human body among the several ones derived from PACs (Fig. 1).11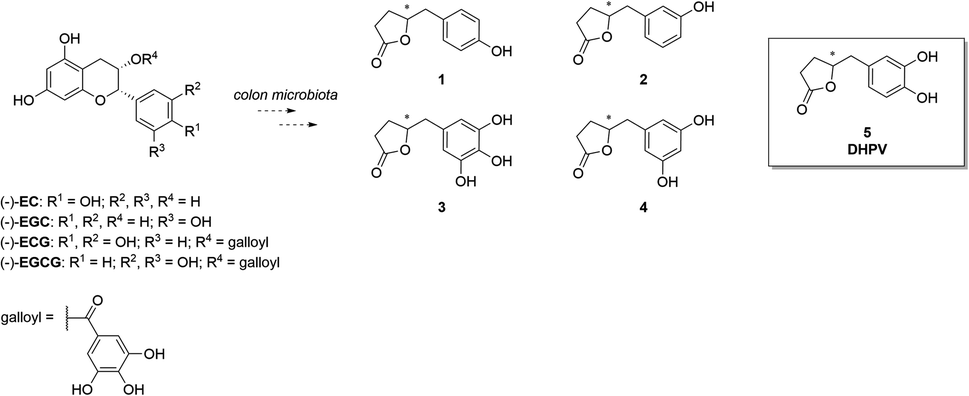 | ||
| Fig. 1 Major polyphenolic catechins and some of their metabolites. | ||
DHPV (5) is well-known for its antioxidant properties, such as cancer chemopreventive, skin wrinkle-reducing effects, and for its anti-adhesive activity.1,12–15 DHPV has been also associated with anti-inflammatory effects, inhibiting the production of pro-inflammatory mediators, and modulating immune response.16–18 Furthermore, DHPV has exhibited promising cardiovascular benefits by enhancing endothelial function and reducing platelet aggregation, suggesting its potential role in preventing atherosclerosis.19 Understanding the bioavailability of DHPV after PACs oral intake is of paramount importance for harnessing their health benefits and designing functional food products.20–24 In this regard, quantitative bioanalytical methods based on stable isotopically labelled (SIL) analogs represent a valuable tool to pursue these goals. In particular, the use of isotopic labelled internal standard in mass spectrometry allows to have the best results in terms of precision and accuracy when quantifying the unknown amount of molecule of interest in a biological matrix. Furthermore, a suitable analytical method could resolve the dilemma of whether DHPV exists only as sulfate conjugate or also in the free form resulted from hydrolysis operated by sulfatase which subsequently undergoes glucuronidation. Several groups1,12,25–27 reported the synthesis of racemic or optically active DHPV for further biological studies or testing their own catalytic system. However, no one, to the best of our knowledge, ever reported the preparation of stable isotopically labelled (SIL) analogs that could be extremely useful for isotope dilution methods in quantitative bioanalyses.
The reported synthesis of racemic or optically active DHPV (5) are represented in Fig. 2.
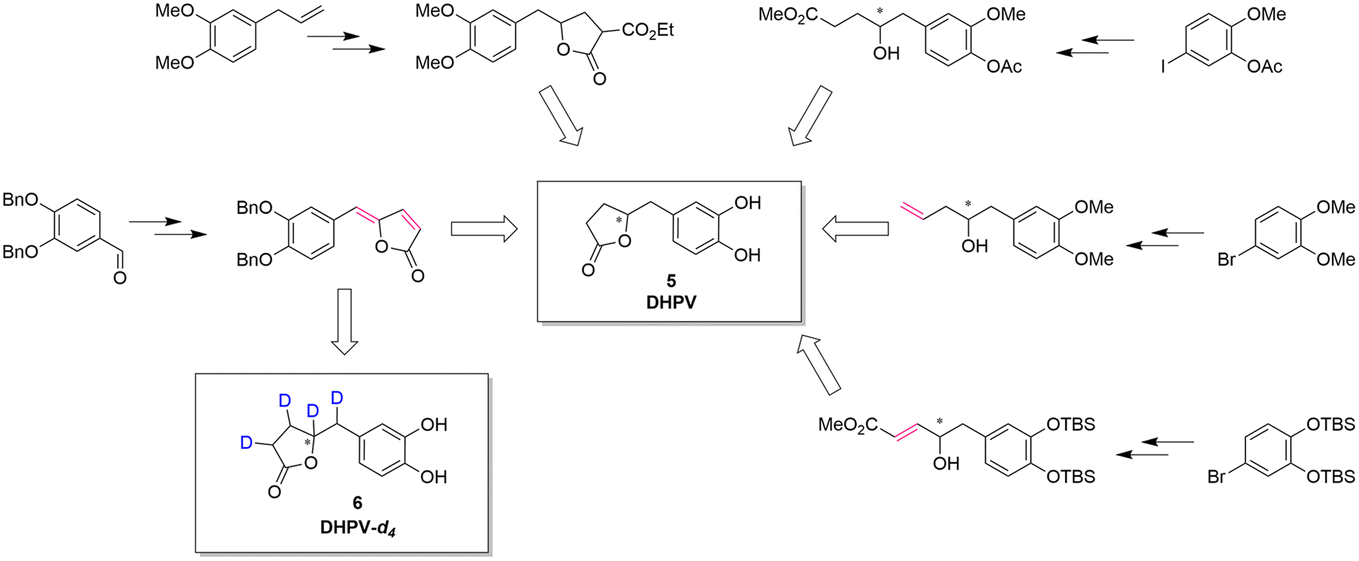 | ||
| Fig. 2 Synthetic approaches toward DHPV. In red, the possible deuteration sites.1,11,19–21 | ||
Generally speaking, these synthetic pathways build the γ-valerolactone structure after adding the necessary carbons to the aromatic portion. However, despite the excellence or the elegance of the previous synthetic pathways, only the one exploiting the vinylogous Mukaiyama aldol reaction incorporates the γ-lactone moiety at benzylic position obtaining a compound with two contiguous alkenes. This intermediate can directly undergo the deuteration step allowing the insertion of four deuterium atoms via a late-stage functionalization.
Herein we report a concise synthesis that allows the obtainment of 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone-d4 (DHPV-d4) (6) which is a useful SIL internal standard to quantify the concentration of the metabolite in biological samples,28 as already shown with other compounds29 and demonstrated by the developed and here reported new method. In fact, this kind of SIL belongs to the preferred internal standards for spectroscopic detection, since it is chemically identical to the analyte thus avoiding variability due to any kind of derivatization that influence, in turn, the chemical and physical properties. Furthermore, the incorporation of the deuterium atoms on non-exchangeable sites and the mono-isotopic mass 4 Da higher than that of the natural compound, make the standard much more suitable for LC/MS-MS analysis.
2. Materials and methods
2.1 General
Merck KGaA, Darmstadt, Germany supplied all reagents and solvents. HyperSep™ C18 cartridges were purchased from Thermo Scientific™ (Milan, Italy). Chemical abbreviations: diisopropylethylamine (DIPEA); 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU); tetrahydrofuran (THF); deuterated methanol (CD3OD).2.2 Instrument parameters for compound characterization
Varian 300 Mercury spectrometer at 300 MHz for 1H NMR, and at 75.43 MHz for 13C NMR was used to record NMR spectra. The signal multiplicities are reported as s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet), or br (broad signal).LTQ Orbitrap XL (Thermo Scientific, Bremen, Germany) equipped with an ESI source, operating in negative ion mode was used for the high-resolution MS analysis by direct infusion. The mass spectrometer acquired in full mass scan mode and resolution 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHM at m/z 400.
000 FWHM at m/z 400.
2.3 Synthesis of 5-(3′,4′-bis(benzyloxy)benzylidene)furan-2(5H)-one (II)
Under nitrogen atmosphere in oven dried glassware DIPEA (0.54 mL, 3.06 mmol) was added to 2(5H)-furanone (85.76 mg, 1.02 mmol) dissolved in dry THF (7.0 mL). The reaction mixture was kept at room temperature and stirred for 30 min. After cooling down the reaction flask to −10 °C, 3,4-bis(benzyloxy)benzaldehyde I (282.0 mg, 1.12 mmol) and tert-butyldimethylsilyltrifluoromethanesulfonate (0.26 mL, 1.12 mmol) were added dropwise. The reaction mixture was kept for 2 h under stirring at the same temperature and DBU (0.30 mL, 2.04 mmol) was added. During the overnight period at room temperature the reaction mixture was stirred and then solvent was removed. The crude residue was dissolved in a mixture made by ethyl acetate (40 mL), ethanol (10 mL) and saturated solution of sodium bisulfite (10 mL) and maintained under stirring overnight at 40 °C. The aqueous phase was separated, and ethyl acetate (30 mL) was used to dilute the organic layer which subsequently underwent a treatment with 10% aqueous HCl (3 × 40 mL), washed with saturated NaCl aqueous solution (40 mL), dried over anhydrous sodium sulfate and concentrated to obtain a sticky black oil. The title compound was obtained by purification on silica gel (75:25 cyclohexane/ethyl acetate) as an orange oil (200.0 mg, 0.52 mmol, 52% yield). 1H-NMR, 13C-NMR and HRMS match literature data.30
2.4 Synthesis of 5-(3′,4′-dihydroxyphenyl)-γ-valerolactone-d4 (6)
Compound II (60 mg, 0.16 mmol) was dissolved in CD3OD (3.6 mL, >99.8 atom% D, <0.025% water), and 3% Pd/C (30 mg) was added in an oven-dried vial that was subsequently sealed. After performing vacuum/inert gas cycles to remove air and moisture, deuterium gas was inflated at 1.8 bar. The reaction mixture was stirred overnight at room temperature under deuterium atmosphere, then the catalyst was removed by filtration. The filtrate was concentrated in vacuo to afford the mixture of DHPV-d3 and DHPV-d4 as an orange oil (125 mg, 0.60 mmol, 83% yield). 1H NMR (300 MHz, CD3OD) and 13C NMR (75 MHz, CD3OD) are reported in ESI Fig. 1 and 2† respectively. HRMS characterization is reported in Fig. 4.2.5 Sample preparation for the quantitative analysis of DHPV in urine
A calibration curve was built by spiking DHPV, previously synthesized by our group,31 in artificial urine in five concentrations in the range 0.1–10 μM. Samples were also added with the internal standard DHPV-d4 2.5 μM. After mixing, an aliquot (1 mL) of each sample was extracted on Thermo Scientific™ HyperSep™ C18 cartridges, dried under vacuum and resuspended in 200 μL of 95% mobile phase A (H2O-0.2% HCOOH, % v/v) and 5% mobile phase B (CH3CN-0.2% HCOOH, % v/v), and then transferred in vials for the analysis. QC samples were prepared for intra-day and inter-day precision and accuracy evaluation at 0.1, 1 and 10 μM and processed as previously described.2.6 LC-MS/MS method development and validation for DHPV quantitation in urine
Each sample (5 μL) was injected into a reversed-phase Agilent Zorbax SB-C18 (150 × 2.1 mm, i.d. 3.5 μm, CPS analitica, Milan, Italy) protected by an Agilent Zorbax guard column, kept at 40 °C. The chromatographic separation was performed by a Exion LC 100 system (AB Sciex, Milan, Italy) equipped with a quaternary pump, working at a constant flow rate of 0.4 mL min−1 of mobile phase A (H2O-0.2% HCOOH, % v/v) and mobile phase B (CH3CN-0.2% HCOOH, % v/v) with the following multi-step gradient program: from 5% B to 55% B in 20 min, from 55% B to 75% B in 1 min, isocratic of 75% B for 4 min, then isocratic of 5% B for 6 min. The LC was connected to an API 4000 triple quadrupole mass spectrometer (AB Sciex, Milan, Italy), equipped with a TurboV electrospray interface (AB Sciex, Milan, Italy), operating in negative ion mode by applying −4.5 kV ionization potential, 25 units of curtain gas, 40 units of gas 1, 10 units of gas 2 heated at 400 °C, declustering potential (DP) −50 V, entrance potential (EP) −10 V, collision energy (CE) −25 eV, collision cell exit potential (CXP) −8 V. The mass spectrometer conditions for the multiple reaction monitoring (MRM) analysis were optimized by direct infusion of a standard solution (10 μM) of DHPV and DHPV-d4 into the source. Two transitions for each analyte were selected, one for the quantitative analysis and the second one as qualitative confirmation of the compound (Table 1). The developed method was validated in terms of linearity, precision (CV%) and accuracy (bias) according to FDA guidelines for bioanalytical method validation. Calibration curves were built by linear regression using the method of least squares by placing the ratio of peak area of the analyte/peak area of the internal standard on the y-axis and the nominal analyte concentration on the x-axis. The limit of quantitation (LOQ) was determined as the lowest concentration at which the precision (CV%) and bias values fall in the range ±20%. Intra-day and inter-day precision and accuracy were calculated for the LOQ and the concentrations 1 and 10 μM.| Compound | Parent ion [M–H]− (m/z) | Product ions (m/z) | |
|---|---|---|---|
| DHPV | 207 | 163 | 122 |
| DHPV-d4 | 211 | 167 | 122 |
3. Results and discussion
As the scientific community continues to unravel the complexities of PACs and specifically their metabolites like DHPV, it becomes increasingly evident that these natural compounds hold great promise in improving human health and well-being. Thus, the possibility to have the SIL standard analogs is necessary to confirm the potential of the DHPV as bioactive metabolite and to understand its active dose. Our synthetic approach derived from the route of non-deuterated DHPV developed by our group1,32 (Scheme 1) that allows the simultaneous insertion of four deuterium atoms.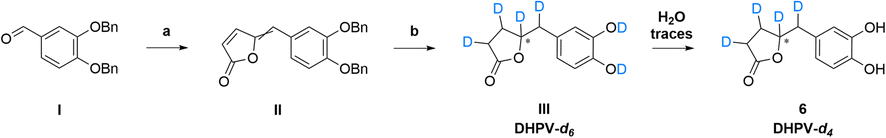 | ||
| Scheme 1 Synthetic route to DHPV-d4 (6) starting from 3,4-dibenzyloxybenzaldehyde (I). | ||
The first step is based on the vinylogous Mukaiyama aldol reaction that allowed to obtain 5-(3,4-bis(benzyloxy)benzylidene)furan-2(5H)-one (II) whose configuration can be influenced according to the added type of base, as previously published.32 Nevertheless, in this case we did not care about the base since the subsequent step did not consider any stereoselective reaction. In fact, compound II underwent a catalytic deuteration that one pot deprotected the catechol moiety and saturated the conjugated diene system affording the DHPV-d4 in good yield. For sake of clarity, the obtained product should be DHPV-d6 since also the two hydroxy groups of catechol moiety should carry deuterium atom. Nevertheless, the discussion will consider DHPV-d4 because of the mobility of the two phenolic protons that makes them undetectable either during 1H-NMR or HRMS analyses. The reaction was set in a sealed vial using deuterated methanol as solvent, palladium on activated charcoal as catalyst and deuterium was inflated at 1.8 bar. Fig. 3 reports the comparison of the 1H-NMR spectra of compounds 5 and its isotopologue 6.
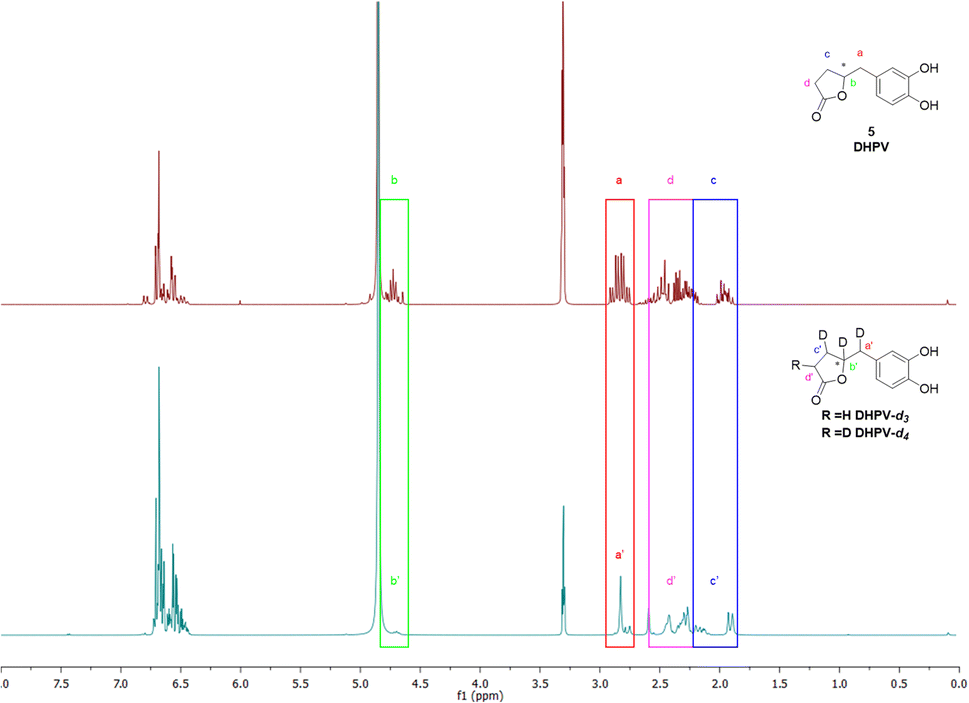 | ||
| Fig. 3 Comparison of 1H NMR spectra of DHPV (5) and its isotopologues DHPV-d3 + DHPV-d4 (6). | ||
From the spectrum of compound 6, some differences are clearly visible: the first concerns the disappearance of the signal that rises from the chiral proton (green box); the second relates to the doublet of doublet generated by the benzylic methylene that becomes a singlet (red box); the third involves the right side of the spectrum where, surprisingly, multiplicity produced by the c′ and d′ protons of the lactone ring become broader but not simpler as expected (purple and blue boxes). As a matter of fact, the d4 isotopologue should give, in this range of ppm, a more clearer spectrum due to the presence of two deuterium in c′ and d′ respectively.
The risen signals and their broad shape suggest the insertion of a proton in position d′ in place of a deuterium. In fact, the signal shape is due to a very low coupling constant between deuterium and the vicinal proton. Furthermore, this hypothesis was confirmed by the MS analysis that detected the 81.29% of DHPV-d4 (6) and the 17.29% of the isotopologue d3 (Fig. 4).
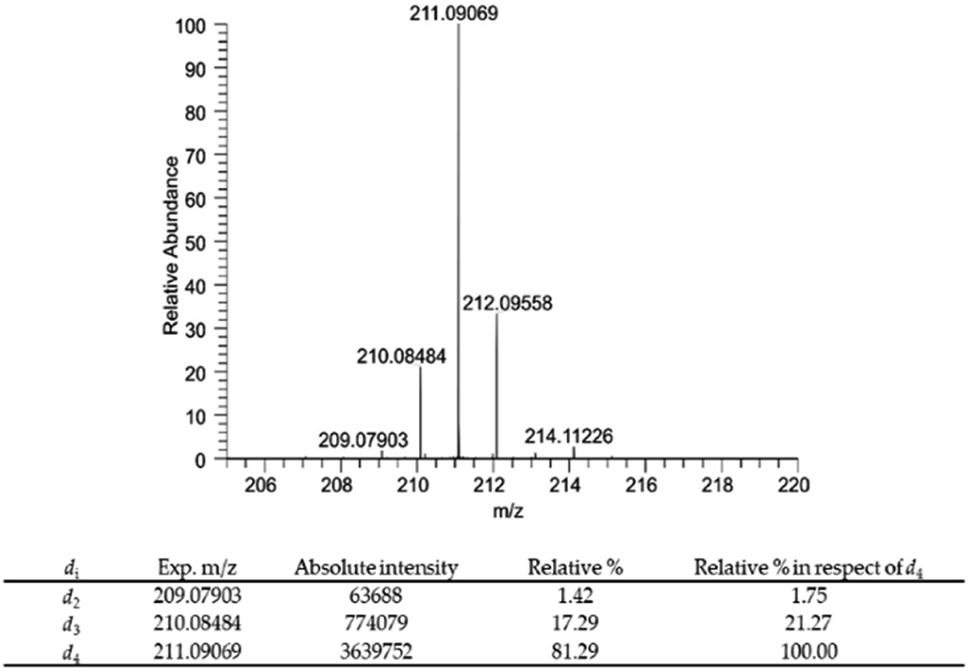 | ||
| Fig. 4 Mass spectrum of compound 6 and relative percentage of deuterium atoms in compound 6 expressed as relative% and relative% in respect to the most abundant deuterium labelled derivative DHPV-d4. | ||
In particular compound 6 isotopic purity was calculated using ESI-(−)-HRMS and 3 monoisotopic peaks [M–Di–H]− (i = number of deuterium atoms = 2–4) were detected (Fig. 4), which were C11H9D2O4− (calc. m/z 209.07863, found m/z 209.07903; delta ppm 1.913), C11H8D3O4− (calc. m/z 210.08481, found m/z 210.08484, delta ppm 0.143), and C11H7D4O4− (calc. m/z 211.09098, found m/z 211.09069, delta ppm −1.374) all assigned by their accurate mass values (mass tolerance of 5 ppm). The relative% of each isotopic peak was calculated using the absolute intensity of d4 monoisotopic peak as 100%. Results are reported in Fig. 4.
The presence of DHPV-d3 could be due to the insertion of a hydrogen that comes from the humidity present in the system.
The insertion of hydrogen at d′ (see Fig. 3) may be justify by the fact that the saturation of the double bond between a′ and b′ is favored because of b′ corresponds to the position 6 of the α-β-unsaturated system. On the contrary the deuteration of the second double bond between c′ and d′ is more complicated thus the first deuterium attacks at c′ that corresponds to the position 4 of the α-β-unsaturated system while, even in a low relative percentage, the insertion of hydrogen occurs in position d′ (Fig. 5).
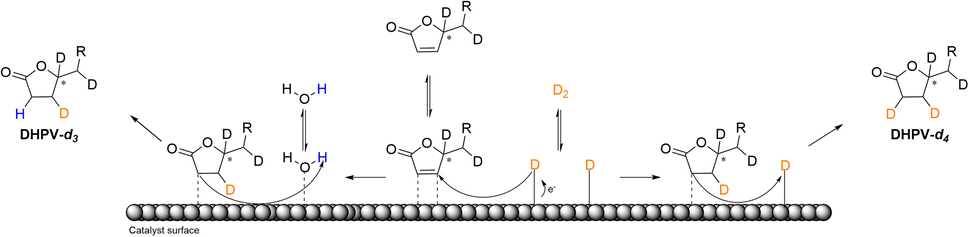 | ||
| Fig. 5 Schematic illustration of the proposed reaction pathway of the deuterium insertion for the obtainment of DHPV-d4 (6) and DHPV-d3. | ||
To validate, as analytical tool, the synthesized DHPV-d4 a new method was developed. Fig. 6 shows the XICs (extracted ion chromatograms) of DHPV and DHPV-d4, both eluting at 6.7 minutes.
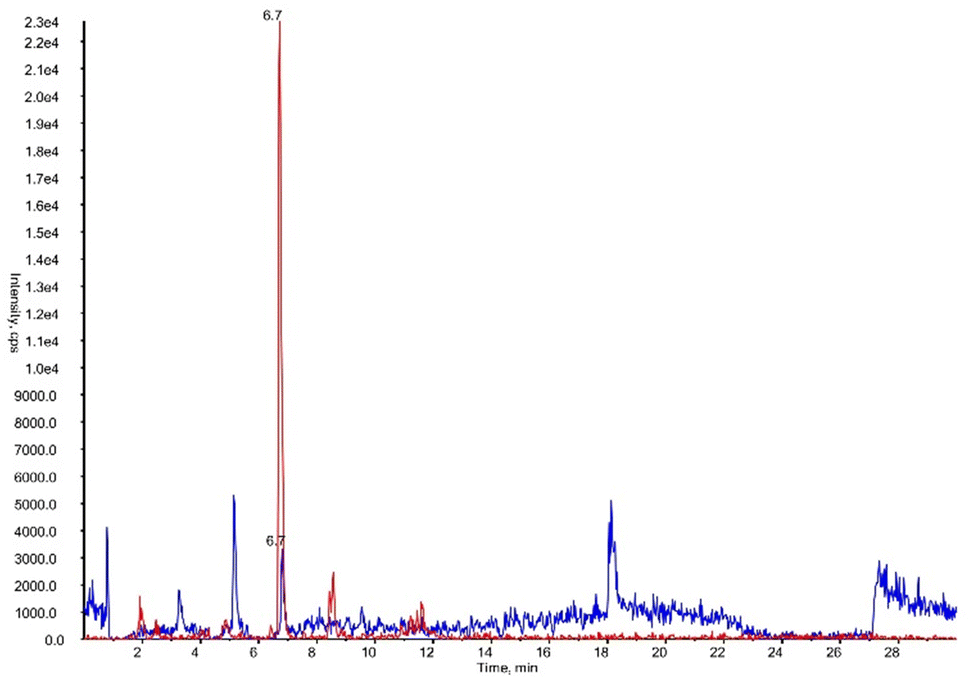 | ||
| Fig. 6 XICs (extracted ion chromatograms) of DHPV at LOQ (blue) and DHPV-d4 (red) contained in artificial urine both eluting at 6.7 minutes. | ||
The linearity was validated in the concentration range 0.1–10 μM (as reported in Fig. 3 of ESI†), where the equation of the curve is y = 0.7359x + 0.04657 (r2 = 0.9990). The LOQ was set at 0.1 μM with a CV% of 7.97% and a bias value of 1.14%. Intra-day and inter-day precision and accuracy values are reported in Table 2.
| Concentration (μM) | Intra-day precision (CV%) | Intra-day accuracy (bias) | Inter-day precision (CV%) | Inter-day accuracy (bias) |
|---|---|---|---|---|
| 0.1 | 7.97 | 1.14 | 12.34 | −9.87 |
| 1 | 8.71 | −7.31 | 10.43 | −11.28 |
| 10 | 1.3 | 0.83 | 1.59 | 2.98 |
All the values fall within the required criteria of FDA guidelines. Thus, the use of the isotope dilution mass spectrometric approach based on DHPV-d4 allowed to obtain a high selective, precise and accurate method that can be applied to quantify DHPV in urine samples in metabolomics studies concerning the oral intake of natural extracts containing PACs.
4. Conclusion
To conclude, the present research describes a concise method to synthetize stable isotopically labeled DHPV (6), one of the most interesting microbial metabolites of PACs. The synthetic strategy here proposed allowed the 100% of isotopic insertion. In particular, the isotopologues DHPV-d3 and DHPV-d4 were in 17.19% and 81.29% respectively, as validated by 1H NMR and HMRS studies. This is the first reported synthesis of deuterium labeled DHPV, that allows to obtain the necessary mass difference required for an internal standard for MS quantitative analysis. Indeed, the use of the synthesized labeled DHPV allowed to develop a precise and accurate isotope dilution mass spectrometric method that can be applied in in vivo studies for the quantitation of DHPV in urine. Furthermore, the same synthesis approach on different aromatic moiety allows the obtainment of other metabolites of PACs.Author contributions
Angelica Artasensi and Sarah Mazzotta share the first author. Angelica Artasensi: investigations, visualization, methodology for the synthetic part. Sarah Mazzotta: investigations and methodology for the synthetic part. Giovanna Baron: investigations, methodology and visualization for the analytical application. Giancarlo Aldini: project administration for the analytical part, funding acquisition, writing review and editing. Laura Fumagalli: project administration for the synthetic part, funding aquisition, conceptualization, writing original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is part of the project “MIND FoodS HUB (Milano Innovation District Food System Hub): Innovative concept for the ecointensification of agricultural production and for the promotion of dietary patterns for human health and longevity through the creation in MIND of a digital Food System Hub”, cofunded by POR FESR 2014-2020_BANDO Call HUB Ricerca e Innovazione, Regione Lombardia. The authors aknowledge support from the University of Milan through the APC initiative.Notes and references
- G. Baron, A. Altomare, L. Regazzoni, L. Fumagalli, A. Artasensi, E. Borghi, E. Ottaviano, C. Del Bo, P. Riso, P. Allegrini, G. Petrangolini, P. Morazzoni, A. Riva, L. Arnoldi, M. Carini and G. Aldini, Biochem. Pharmacol., 2020, 173, 113726 CrossRef CAS PubMed.
- M. V Selma, J. C. Espín and F. A. Tomás-Barberán, J. Agric. Food Chem., 2009, 57, 6485–6501 CrossRef.
- G. Williamson and M. N. Clifford, Br. J. Nutr., 2010, 104, S48–S66 CrossRef CAS.
- J. Rubert, P. Gatto, M. Pancher, V. Sidarovich, C. Curti, P. Mena, D. Del Rio, A. Quattrone and F. Mattivi, Mol. Nutr. Food Res., 2022, 66, 2101043 CrossRef CAS PubMed.
- W. Fan, H. Zong, T. Zhao, J. Deng and H. Yang, Crit. Rev. Food Sci. Nutr., 2022, 1–17 Search PubMed.
- J. Wieczfinska, P. Sitarek, T. Kowalczyk, E. Skała and R. Pawliczak, Curr. Pharm. Des., 2020, 26, 2876–2884 CrossRef CAS.
- T. K. Wang, S. Xu, S. Li and Y. Zhang, Molecules, 2020, 25, 5971 CrossRef CAS.
- M. M. Appeldoorn, J.-P. Vincken, A.-M. Aura, P. C. H. Hollman and H. Gruppen, J. Agric. Food Chem., 2009, 57, 1084–1092 CrossRef CAS.
- M. Schantz, T. Erk and E. Richling, Biotechnol. J., 2010, 5, 1050–1059 CrossRef CAS PubMed.
- J. van Duynhoven, E. E. Vaughan, F. van Dorsten, V. Gomez-Roldan, R. de Vos, J. Vervoort, J. J. J. van der Hooft, L. Roger, R. Draijer and D. M. Jacobs, Am. J. Clin. Nutr., 2013, 98, 1631S–1641S CrossRef CAS PubMed.
- P. Mena, L. Bresciani, N. Brindani, I. A. Ludwig, G. Pereira-Caro, D. Angelino, R. Llorach, L. Calani, F. Brighenti, M. N. Clifford, C. I. R. Gill, A. Crozier, C. Curti and D. Del Rio, Nat. Prod. Rep., 2019, 36, 714–752 RSC.
- J. Hur, A.-R. Kim, H. S. Kim, C. Lim, T. Kim, T.-A. Kim, J. Sim and Y.-G. Suh, Molecules, 2020, 25, 1970 CrossRef CAS.
- E. Ottaviano, G. Baron, L. Fumagalli, J. Leite, E. A. Colombo, A. Artasensi, G. Aldini and E. Borghi, Microorganisms, 2021, 9, 1492 CrossRef CAS PubMed.
- C. C. Lee, J. H. Kim, J. S. Kim, Y. S. Oh, S. M. Han, J. H. Y. Park, K. W. Lee and C. Y. Lee, Int. J. Mol. Sci., 2017, 18, 1363 CrossRef PubMed.
- P. Mena, D. González de Llano, N. Brindani, A. Esteban-Fernández, C. Curti, M. V. Moreno-Arribas, D. Del Rio and B. Bartolomé, J. Funct. Foods, 2017, 29, 275–280 CrossRef CAS.
- C. Liu, S. Boeren and I. M. C. M. Rietjens, Front. Nutr., 2022, 9, 910785 CrossRef PubMed.
- T. Grimm, A. Schäfer and P. Högger, Free Radical Biol. Med., 2004, 36, 811–822 CrossRef CAS PubMed.
- L. Mele, S. Carobbio, N. Brindani, C. Curti, S. Rodriguez-Cuenca, G. Bidault, P. Mena, I. Zanotti, M. Vacca, A. Vidal-Puig and D. Del Rio, Mol. Nutr. Food Res., 2017, 61, 1700074 CrossRef.
- C. C. Lee, J. H. Kim, J. S. Kim, Y. S. Oh, S. M. Han, J. H. Y. Park, K. W. Lee and C. Y. Lee, Int. J. Mol. Sci., 2017, 18, 1363 CrossRef.
- M. Makarewicz, I. Drożdż, T. Tarko and A. Duda-Chodak, Antioxidants, 2021, 10, 188 CrossRef CAS.
- Z. Li, Z. Ren, L. Zhao, L. Chen, Y. Yu, D. Wang, X. Mao, G. Cao, Z. Zhao and H. Yang, Food Chem., 2023, 399, 133959 CrossRef CAS PubMed.
- R. Jia, Y. Hou, W. Feng, M. Nomingerel, B. Li and J. Zhu, Antioxidants, 2023, 12, 2095 CrossRef CAS PubMed.
- X. Zhang, X. Song, X. Hu, F. Chen and C. Ma, Food Chem., 2023, 404, 134596 CrossRef CAS PubMed.
- P. A. Vlaicu, A. E. Untea, I. Varzaru, M. Saracila and A. G. Oancea, Foods, 2023, 12, 4001 CrossRef CAS PubMed.
- H. Watanabe, Bull. Agric. Chem. Soc. Jpn., 1959, 23, 257–271 CAS.
- J. D. Lambert, J. E. Rice, J. Hong, Z. Hou and C. S. Yang, Bioorg. Med. Chem. Lett., 2005, 15, 873–876 CrossRef CAS PubMed.
- M. Hamada, A. Furuno, S. Nakano, T. Kishimoto and N. Nakajima, Synthesis, 2010, 2010, 1512–1520 CrossRef.
- G. Baron, A. Altomare, L. Della Vedova, F. Gado, O. Quagliano, S. Casati, N. Tosi, L. Bresciani, D. Del Rio, G. Roda, A. D'Amato, C. Lammi, A. Macorano, S. Vittorio, G. Vistoli, L. Fumagalli, M. Carini, A. Leone, M. Marino, C. Del Bo', G. Miotto, F. Ursini, P. Morazzoni and G. Aldini, Redox Biol., 2024, 69, 102981 CrossRef CAS PubMed.
- S. Mazzotta, G. Baron and L. Fumagalli, Food Chem.: X, 2022, 13, 100227 CAS.
- A. Artasensi, G. Baron, G. Vistoli, G. Aldini and L. Fumagalli, Molbank, 2021, 2021, M1193 CrossRef.
- G. Baron, A. Altomare, L. Regazzoni, L. Fumagalli, A. Artasensi, E. Borghi, E. Ottaviano, C. Del Bo, P. Riso, P. Allegrini, G. Petrangolini, P. Morazzoni, A. Riva, L. Arnoldi, M. Carini and G. Aldini, Biochem. Pharmacol., 2020, 173, 113726 CrossRef CAS PubMed.
- A. Artasensi, G. Baron, G. Vistoli, G. Aldini and L. Fumagalli, Molbank, 2021, 2021, M1193 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08665h |
| This journal is © The Royal Society of Chemistry 2024 |