
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free synthesis of γ-ketosulfones through Brønsted acid-promoted conjugate addition of sulfinamides†
Nicolas Gigant*,
Sami Kayal,
Emmanuelle Drège and
Delphine Joseph*
and
Delphine Joseph*
Université Paris-Saclay, CNRS, BioCIS, 91400 Orsay, France. E-mail: delphine.joseph@universite-paris-saclay.fr; nicolas.gigant@universite-paris-saclay.fr
First published on 5th February 2024
Abstract
A straightforward and general metal-free method has been developed to add sufinamide-derived sulfone units on Michael acceptors under mild conditions. This reaction enables the preparation of a large variety of original γ-ketosulfones, of which only a few synthetic methods have been reported. The mild reaction conditions used tolerate a wide diversity of functional groups and empower the implementation of a late-stage functionalisation strategy.
Introduction
Sulfur–carbon bond formation belongs to the fundamental transformations in synthetic organic chemistry. Indeed, organosulfur compounds are a relevant class of molecules with applications in various fields of chemistry, including natural products,1 bioactive compounds,2 and functional materials science.3 Among them, sulfones are important molecular frameworks and have drawn much attention for their synthesis.4 Although effective, most of the synthesis methods suffer from drawbacks such as the use of a toxic additive, a metal catalyst, some hazardous raw materials, and harsh reaction conditions, often generating undesired by-products that can pollute the final product and reduce the environmental attractiveness of these approaches.Turning more specifically to the relevant subclasses of ketosulfones,5 while a wide variety of synthetic methods have been developed for the preparation of β-ketosulfones,6 the synthesis of their γ-ketosulfone isomers remains surprisingly underexplored and challenging. Usually, γ-ketosulfones are obtained in two steps by the conjugate addition of thiol derivatives onto various α,β-unsaturated ketones, followed by the oxidation of the corresponding sulfides into sulfones.7 The direct introduction of the sulfone moiety has also been investigated mainly using sulfinates8 or sulfinic acids9 as a source of sulfonyl derivatives. More specifically, complementary approaches have been devised from alternative sources of sulfone motif such as (i) the photoredox-catalyzed10a,b or metal-catalyzed10e hydrosulfonylation of enones with p-tolyl N-sulfonylimine or sulfur dioxide, (ii) a sulfonylation/migration process applied to arylallylic alcohols with arenesulfinic acid in the presence of hypervalent iodine (III) and concentrated sulfuric acid,10c (iii) the direct sulfonylation of acrylates or acrylamides with arenesulfonyl hydrazides,10d (iv) the asymmetric sulfonation of enones with arene sulfonylimines by cooperative organic multicatalysis,10f or (v) the N-heterocyclic carbene-catalyzed Stetter reaction of aldehydes with α,β-unsaturated sulfones.10g To our knowledge, no method has explored the use of sulfinamide as a source of sulfone moiety and there is still a strong demand to develop versatile and atom-economical approaches to produce γ-ketosulfones directly under mild conditions.
Our research group has long been interested in developing original environmentally friendly synthetic methods based on conjugate additions.11 In this context, we have described the preparation of highly substituted sulfonated cyclopentanes via an acid-promoted Rauhut–Currier (RC) cascade12 in the presence of simple and readily available sulfinamides.13 Mechanistically, the cascade is initiated by the nucleophilic addition of a S-nucleophile onto a Michael acceptor playing the role of RC donor. Interestingly, HBF4 proved the best catalyst among a variety of Lewis and Brønsted acids explored, such as In(OTf)3, Bi(OTf)3, BF3·OEt2, TfOH, p-TsOH or Tf2NH, to efficiently promote the conjugate addition of deactivated nucleophiles.12c,13 In our continuing effort to develop eco-friendly synthetic methodologies, we have taken advantage of this unique sulfonamide capability to extend this approach to the synthesis γ-ketosulfones (Scheme 1). Indeed, there are several obvious advantages of the present method: (i) cheap and readily available aqueous HBF4 proves to be an efficient promoter instead of a metal catalyst, which is usually expensive and complicated to remove completely from the target product; (ii) the reaction shows broad substrate scopes: cyclic, linear, aromatic, and heteroaromatic compounds are well tolerated at room temperature and in the open air; (iii) for aprotic polar solvent use, acetonitrile is recognised as a sustainable solvent by the Sanofi's Solvent Selection Guide;14 (iv) the by-product of the reaction is ammonium which is directly captured in situ by an acid–base interaction; (v) in general, reactions are clean, allowing the expected products to be easily purified and isolated in moderate to excellent yields.
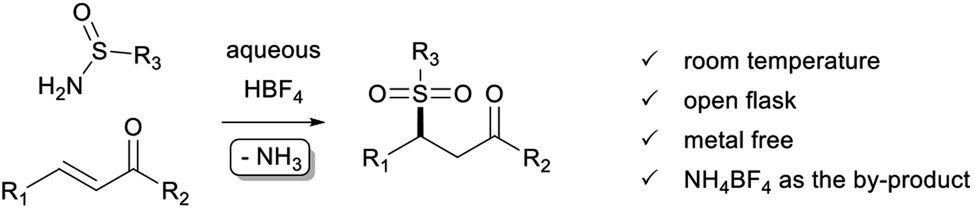 | ||
| Scheme 1 Acid-promoted synthesis of γ-ketosulfones. | ||
Results and discussion
Based on the optimized conditions of our seminal work,13 the preliminary study was performed by reacting cyclohex-2-enone 1a with tert-butylsulfinamide 2a in the presence of one equivalent of aqueous HBF4 at room temperature (Table 1). Pleasingly, in a CH2Cl2/CH3CN 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture, the desired γ-sulfone 3aa was isolated in a 41% yield (entry 1). A subsequent investigation on the solvent effect highlighted that an aprotic polar solvent was essential for the reaction, with CH3CN giving the best results (entries 2–4). The reaction efficiency was not improved by adjusting the amount of HBF4: a stoichiometric amount is required to reach full conversion of the starting material (entries 5–6). Conversely, the yield dropped to 73% starting from 2 equivalents of 2a, whereas the reaction concentration does not influence the reaction efficacy (entries 7–9).
:4 mixture, the desired γ-sulfone 3aa was isolated in a 41% yield (entry 1). A subsequent investigation on the solvent effect highlighted that an aprotic polar solvent was essential for the reaction, with CH3CN giving the best results (entries 2–4). The reaction efficiency was not improved by adjusting the amount of HBF4: a stoichiometric amount is required to reach full conversion of the starting material (entries 5–6). Conversely, the yield dropped to 73% starting from 2 equivalents of 2a, whereas the reaction concentration does not influence the reaction efficacy (entries 7–9).
|
||||
|---|---|---|---|---|
| Entry | Solvent | HBF4 (equiv.) | tBuSONH2 2a (equiv.) | Yieldb (%) |
| a Reaction conditions: cyclohex-2-enone (0.20 mmol, 1 equiv.) under the appropriate reaction conditions (c = 0.4 mol L−1) at room temperature for 24 h.b Isolated yield.c Starting material.d c = 0.8 mol L−1. | ||||
| 1 | CH2Cl2/CH3CN = 1/4 | 1 | 1.6 | 41 |
| 2 | CH2Cl2 | 1 | 1.6 | 32 |
| 3 | MeOH | 1 | 1.6 | s.m.c |
| 4 | CH3CN | 1 | 1.6 | 46 |
| 5 | CH3CN | 0.2 | 1.6 | 16 |
| 6 | CH3CN | 0.5 | 1.6 | 34 |
| 7 | CH3CN | 1 | 1.2 | 25 |
| 8 | CH3CN | 1 | 2 | 73 |
| 9d | CH3CN | 1 | 2 | 71 |
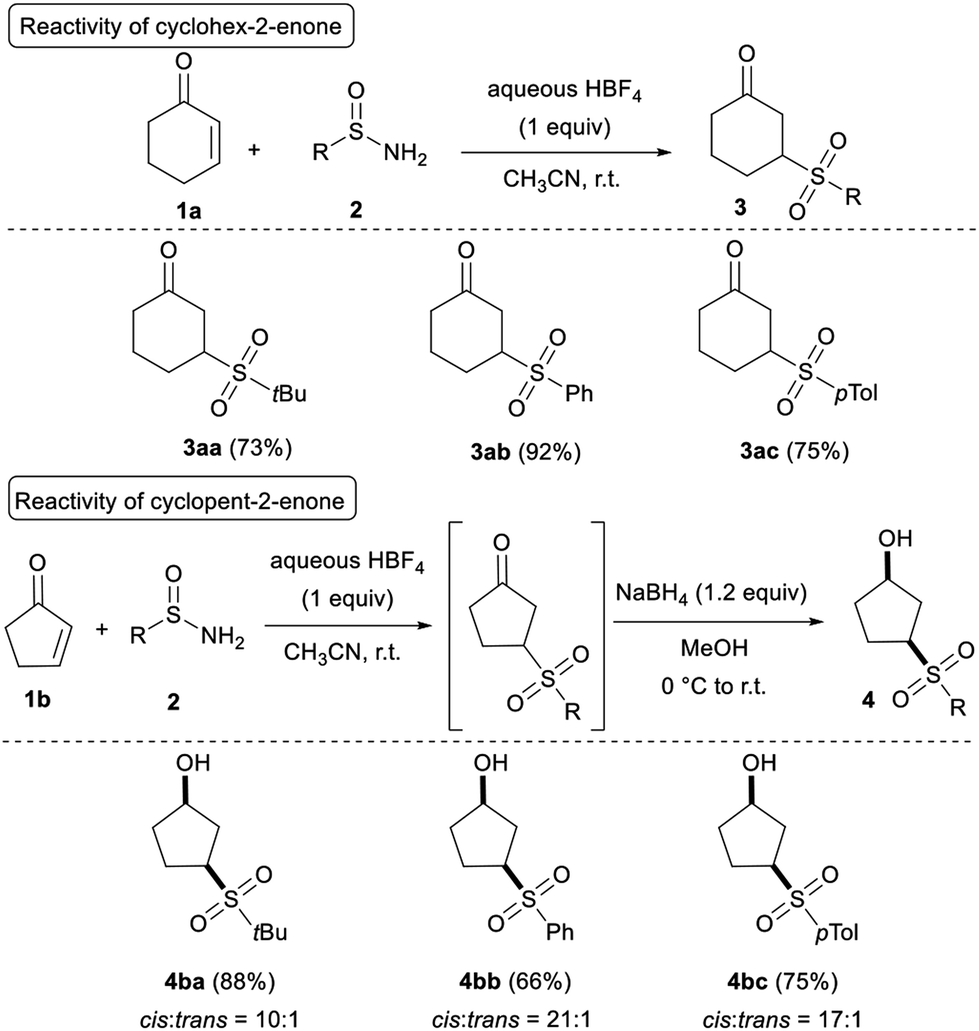
With the optimised reaction conditions established (Table 1, entry 8), the reaction scope was investigated, and Table 2 shows that cyclic enones efficiently participate in the reaction. Good to high yields were obtained for 3aa–3ac by reacting cyclohex-2-enone 1a with both alkyl- and aryl-sulfinamides. By starting from the inferior homolog cyclopent-2-enone 1b, the telescoped reduction of the ketone group was necessary to avoid degradation of the expected adduct. Indeed, during the purification by flash chromatography on silica gel, a retro-Michael reaction was observed restoring the starting material 1b. Notably, sulfinamides 2a–2c worked equally well, forming the desired γ-hydroxy sulfones 4ba, 4bb, and 4bc in high yields. Finally, the cis diastereoselectivity of the reduction turned out to be very satisfying (>10:1).
| a Unless otherwise noted, reaction conditions: 1a or 1b (0.20 mmol, 1 equiv.), 2 (0.40 mmol, 2 equiv.), HBF4 (0.20 mmol, 1 equiv.) in CH3CN (c = 0.4 mol L−1) at r.t. for 24 h. For the ketone reduction: NaBH4 (0.24 mmol, 1.2 equiv.) in MeOH (c = 0.1 mol L−1) at 0 °C to r.t. for 2 h.b Isolated yield.c Diastereomeric ratio and relative configuration were determined by 1H NMR from the isolated product. |
|---|
 |
Next, we sought to expand this transformation to the reactivity of diverse acyclic α,β-unsaturated ketones. We were pleased to find that a range of scaffolds were efficiently converted in the presence of tert-butylsulfinamide 2a (Table 3). Either methyl, ethyl, or thien-2-ylmethyl vinyl ketone is compatible with our procedure, leading to 3ca, 3da or 3ea, respectively, in good to excellent yields. When the conjugate cyclohexenyl methyl ketone 1f was subjected to the procedure, the desired product 3fa was isolated in 67% yield with a complete cis selectivity. Next, we investigated the reactivity of acrolein 1g with 1 equivalent of the sulfinimine, and the targeted sulfonated scaffold 3ga was isolated. When the amount of 2a was increased up to 3 equivalents, we observed the major formation of the corresponding imine 5ga in a 3ga/5ga 36:64 ratio. Adding one more equivalent of acid reverses the reaction chemoselectivity, highlighting the key role of the acid in both the selectivity and the mechanism of the reaction.
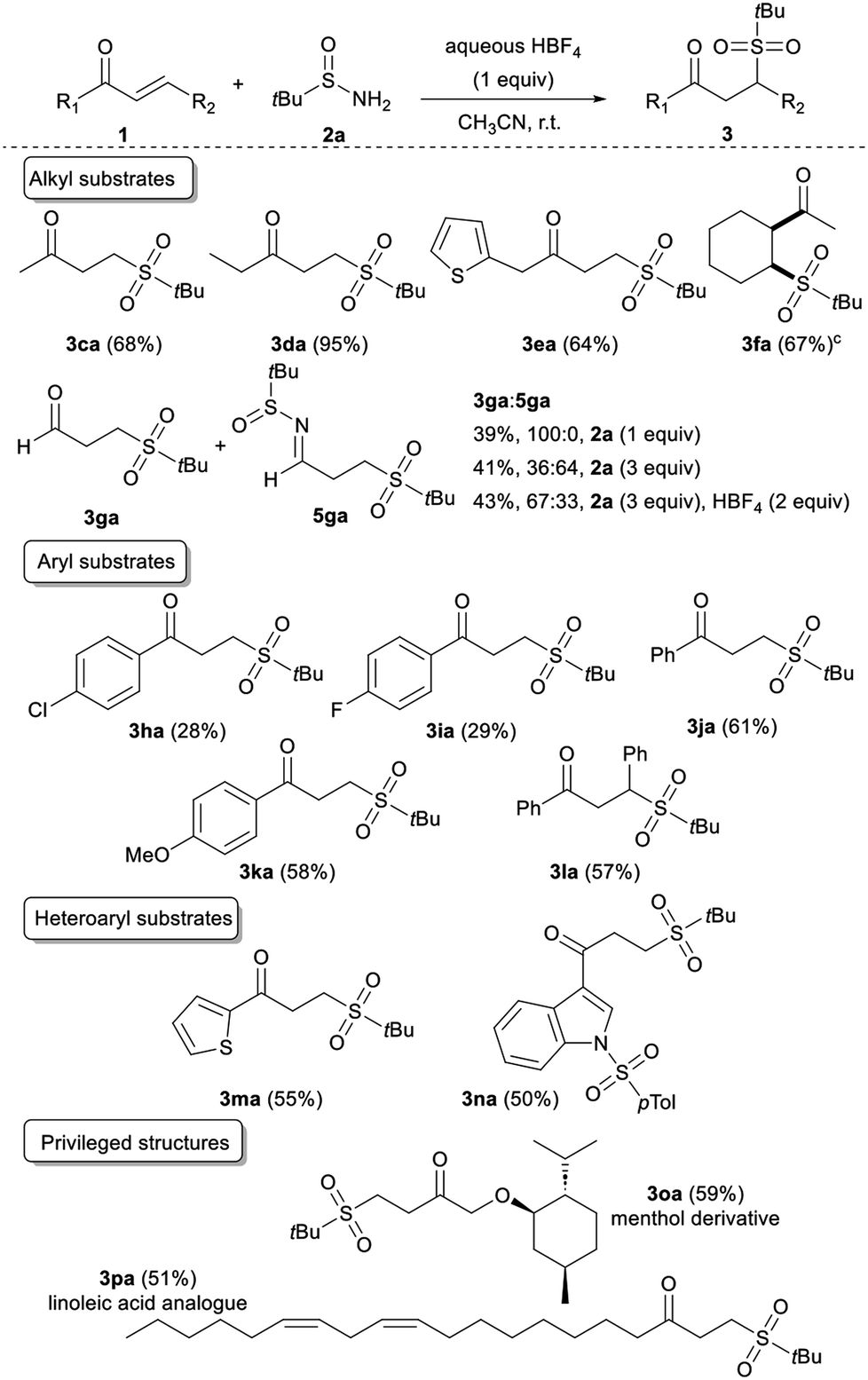
Useful 4-chloro- and 4-fluoro-phenyl vinyl ketones were also accommodated, albeit in lower yields (3ha and 3ia). The reaction is more efficient by using electron-rich or -neutral arenes such as 1j and 1k, respectively giving 3ja and 3ka in good yields. Furthermore, as chalcones have historically received intensive attention from chemists because of their prominent drug activity,15 chalcone 1l was submitted to the reaction conditions affording the sulfone 3la in a 57% yield. Similarly, knowing the widespread presence of heteroarenes in many biologically active natural and synthetic products,16 heteroaryl vinyl ketones, such as 1m and 1n were also evaluated as substrates in our system. They provided access to the corresponding original heterocyclic γ-ketosulfones 3ma and 3na in satisfying yields. Given that this strategy promises to rapidly access sulfonyl compounds to explore structure–activity relationships,17 we speculated that our mild reaction conditions might be compatible with highly functionalised bioactive molecules. Therefore, we applied our approach to more complex structures derived from menthol and linoleic acid. The corresponding sulfonated 3oa and 3pa, respectively, were cleanly isolated, demonstrating the generality of this sulfonyl unit transfer method. Even if most of the synthesised γ-ketosulfones were isolated in good to high yields, 3ha, 3ia, 3ka, and 3ma were obtained in moderate yields. To evaluate the part of instability of the considered γ-ketosulfones in these unsatisfactory results, we studied the effect of telescoping a reduction step to the sulfonylation reaction (Table 4). Unfortunately, the newly formed γ-hydroxy sulfones 4ha, 4ia, 4ka, and 4ma were isolated in similar yields ruling out the involvement of a retro-Michael reaction to explain the yield erosion and questioning the stability of the starting vinyl ketones under acidic reaction conditions.
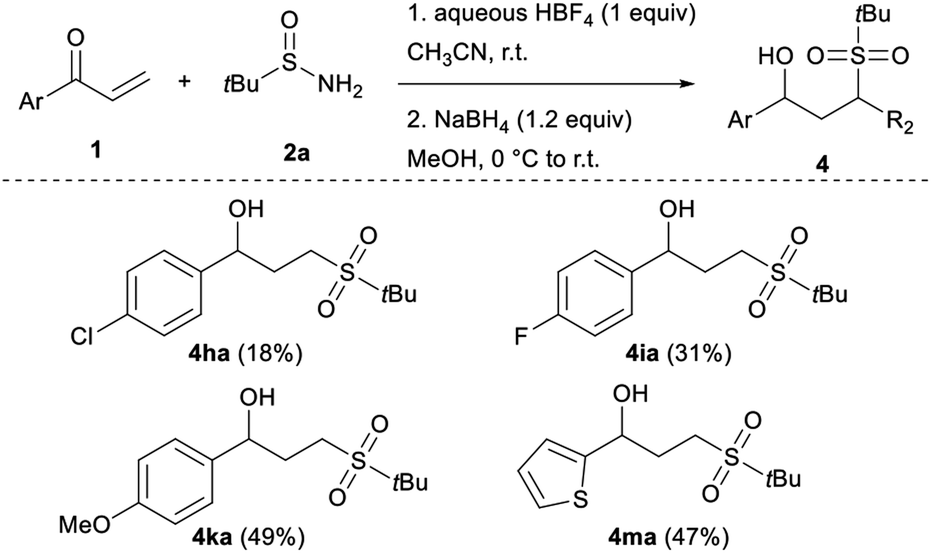
From a mechanistic point of view, in our early work, we showed that prior acid hydrolysis of sulfinamide to sulfinic acid could not be excluded and that the sulfone formation could result from the addition of both proton-activated sulfinamide or sulfinic acid (Scheme 3).13 To further decipher the reaction mechanism, we tried identifying some reaction intermediates by exploring the reactivity of N-octyl tert-butyl sulfinamide 2d with ethyl vinyl ketone 1d (Scheme 2). The sulfinamide 2d was chosen because the possibly expected N-alkylated sulfoximine intermediate should be less sensitive to hydrolytic conditions and thus, easier to characterise. After 5 h of reaction according to our procedure, an aliquot of the crude mixture was directly analysed by electrospray ionisation mass spectrometry analysis (ESI-MS). In positive ion mode, three major cationic intermediates were trapped: the hydrated sulfinyl adduct 6dd (m/z [M + H+ H2O]+), the sulfonated sodium adduct 3da (m/z [M + Na]+) and the octylamine 7d (m/z [M + H]+) (Scheme 2).18 No sulfinic acid was detected in either positive or negative mode. Even if attempts to isolate the sulfinyl adduct 6dd were unsuccessful, these results support the fact that a highly hydrolysable sulfoximine might be a transient intermediate of the reaction. These observations enabled us to revisit our firstly proposed mechanism.13 Indeed, as we demonstrated in our previous work, the nucleophilic strength of the deactivated amine and the acidity of the proton source act as a Lewis pair and are key parameters to control the selectivity of the Michael reaction.11c Thus, in acetonitrile, the protonation of the oxygen atom of the sulfinamide by aqueous HBF4 is preferred due to its stronger basicity,22 which increases the nucleophilicity of the sulfur atom (Scheme 3). The corresponding protonated sulfinamide, acting as a proton shuttle, would favour the addition of the S-nucleophile by promoting enolization of the Michael acceptor. The resulting sulfoximine intermediate would then be rapidly hydrolyzed to sulfone 3 under the reaction conditions (Scheme 3).
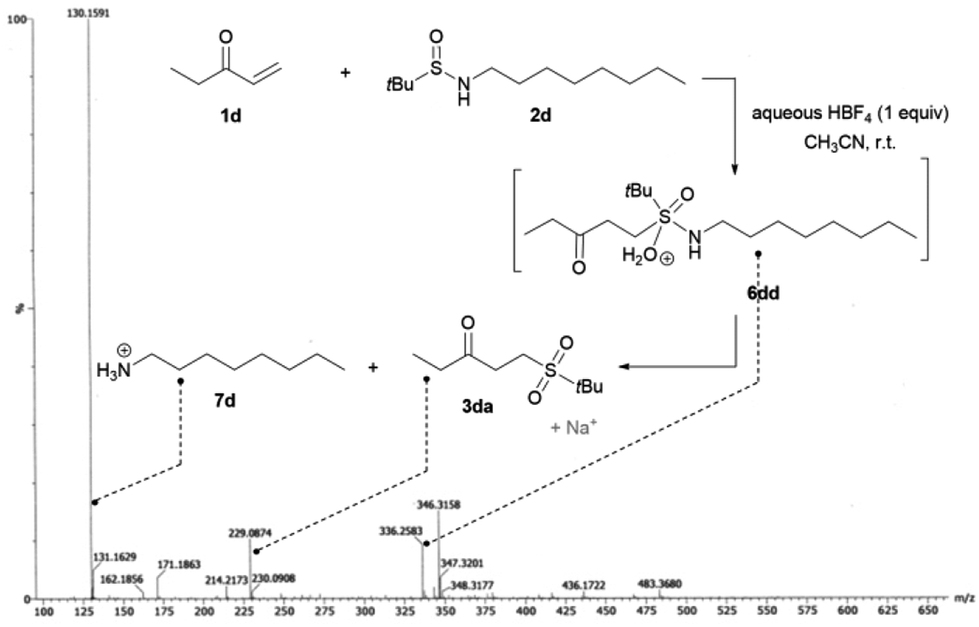 | ||
| Scheme 2 ESI-MS analysis for the crude reaction mixture of 1d with 2d. | ||
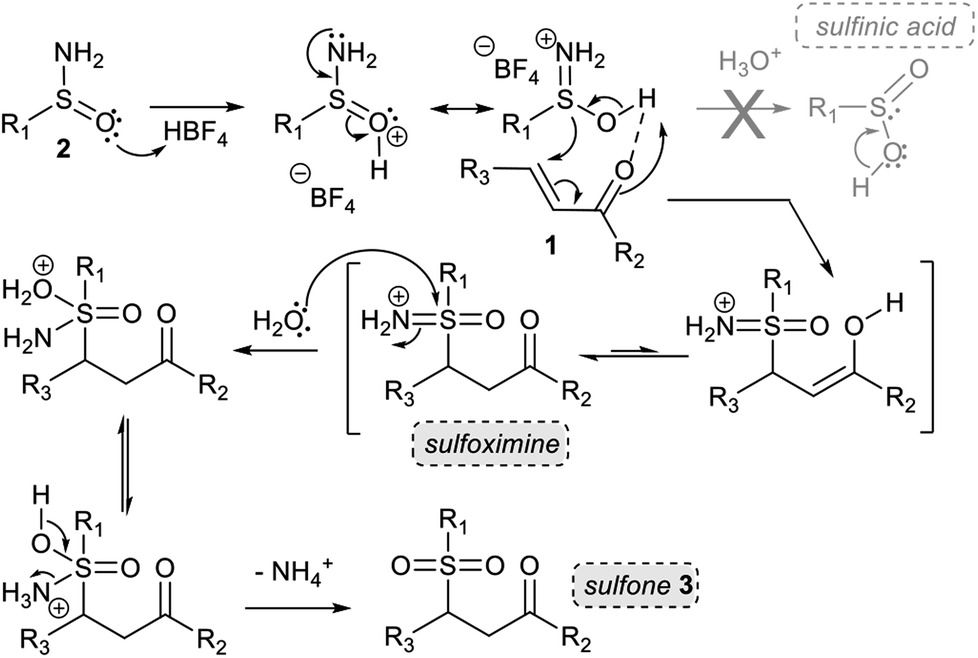 | ||
| Scheme 3 Plausible mechanism for the synthesis of γ-ketosulfone. | ||
Conclusions
We have developed a metal-free, operationally simple, and environmentally friendly method for the synthesis of γ-ketosulfones at ambient temperature by using various readily available sulfinamides as the sulfur source. This easy-to-implement protocol represents not only new routes to the synthesis of original γ-ketosulfones but more importantly is tolerant to a wide range of substrates, including complex molecules. This chemistry should provide access to other sulfone-containing bioactive compound libraries which are difficult to synthesise via other methods.Experimental section
General procedure for the synthesis of ketosulfones 3
To a solution of alkenone 1 (0.20 mmol) in CH3CN (c = 0.4 mol L−1) were successively added sulfinamide 2 (0.40 mmol) and aqueous HBF4 48% wt. (0.20 mmol). The resulting mixture was stirred at room temperature until the disappearance of the starting material (4 to 48 hours) before quenching with the addition of water (2 mL). The solution was then extracted with CH2Cl2 (3 × 10 mL). The organic phase was washed with brine, dried over MgSO4, filtered and concentrated. The crude product was purified by flash chromatography with cyclohexane/ethyl acetate to yield the desired γ-ketosulfone 3.3-(tert-Butylsulfonyl)cyclohexanone (3aa)
3aa was prepared following the general procedure by reacting 1a (19.4 μL, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. Flash chromatography (cyclohexane/ethyl acetate: 4/6) afforded the title compound 3aa as a white solid (32 mg, 73%). M.p.: 108–109 °C; IR (neat) νmax/cm−1 2968, 1705, 1270, 1110, 664; 1H NMR (300 MHz, CDCl3) δ 3.57–3.47 (m, 1H), 2.75 (d, J = 9.1 Hz, 2H), 2.40–2.21 (m, 4H), 2.08–1.97 (m, 1H), 1.75–1.63 (m, 1H), 1.42 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 207.4, 61.4, 55.8, 42.4, 40.5, 25.3, 24.2 (3C), 23.8; HRMS (TOF-ESI) m/z: calcd for C10H18O3NaS [M + Na]+ 241.0869, found 241.0875.3-(Benzenesulfonyl)cyclohexanone (3ab)
3ab was prepared following the general procedure by reacting 1a (19.4 μL, 0.20 mmol), benzenesulfinamide 2b (56 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. Flash chromatography (cyclohexane/ethyl acetate: 7:3 to 6:4) afforded the title compound 3ab as a white solid (44 mg, 92%). M.p.: 87–88 °C; IR (neat) νmax/cm−1 2965, 1706, 1260, 1137, 1084, 1019, 798; 1H NMR (300 MHz, CDCl3) δ 7.85 (br d, J = 7.2 Hz, 2H), 7.68 (br t, J = 7.7 Hz, 1H), 7.57 (br t, J = 7.2 Hz, 2H), 3.33–3.23 (m, 1H), 2.57 (m, 2H), 2.41–2.17 (m, 4H), 1.91 (qd, J = 12.7, 3.1 Hz, 1H), 1.71–1.57 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 206.6, 136.8, 134.4, 129.6 (2C), 129.1 (2C), 62.4, 40.6, 40.5, 23.8, 23.6; HRMS (TOF-ESI) m/z: calcd for C12H14O3NaS [M + Na]+ 261.0556, found 261.0562. The data presented above agrees with that detailed in the literature.19
3-Tosyl cyclohexanone (3ac)
3ac was prepared following the general procedure by reacting 1a (19.4 μL, 0.20 mmol), p-toluenesulfinamide 2c (62 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. Flash chromatography (cyclohexane/ethyl acetate: 7:3 to 6:4) afforded the title compound 3ac as a yellow solid (38 mg, 75%). M.p.: 82–83 °C; IR (neat) νmax/cm−1 2952, 1713, 1283, 1086, 1018, 664; 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 3.30–3.20 (m, 1H), 2.53 (m, 2H), 2.44 (s, 3H), 2.41–2.18 (m, 4H), 1.95–1.73 (m, 1H), 1.67–1.58 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 206.7, 145.4, 133.6, 130.1 (2C), 129.0 (2C), 62.4, 40.6 (2C), 23.8, 23.6, 21.7; HRMS (TOF-ESI) m/z: calcd for C13H16O3NaS [M + Na]+ 275.0712, found 275.0721. The data presented above agree with that detailed in the literature.19
4-(tert-Butylsulfonyl)butan-2-one (3ca)
3ca was prepared following the general procedure by reacting 1c (16.7 μL, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 7 hours. Flash chromatography (cyclohexane/ethyl acetate 3:7 ratio) afforded the title compound 3ca as a white solid (26 mg, 68%). M.p.: 56–57 °C; IR (neat) νmax/cm−1 2978, 1714, 1475, 1300, 1160, 1108 (CH); 1H NMR (300 MHz, CDCl3) δ 3.20 (t, J = 6.9 Hz, 2H), 3.02 (t, J = 7.7 Hz, 2H), 2.24 (s, 3H), 1.41 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 204.7, 59.2, 40.2, 34.1, 30.1, 23.4 (3C); HRMS (TOF-ESI) m/z: calcd for C8H16O3NaS [M + Na]+ 215.0712, found 215.0719. The data presented above agree with that detailed in the literature.20
1-(tert-Butylsulfonyl)pentan-3-one (3da)
3da was prepared following the general procedure by reacting 1d (19.9 μL, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 7 hours. Flash chromatography (cyclohexane/ethyl acetate: 5/5 to 4/6) afforded the title compound 3da as a white solid (39 mg, 95%). M.p.: 55–56 °C; IR (neat) νmax/cm−1 2978, 1714, 1414, 1265, 1114, 976; 1H NMR (300 MHz, CDCl3) δ 3.21 (t, J = 6.9 Hz, 2H), 2.98 (t, J = 7.6 Hz, 2H), 2.51 (q, J = 7.3 Hz, 2H), 1.40 (s, 9H), 1.07 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 207.6, 59.1, 40.1, 36.2, 32.7, 23.4 (3C), 7.9; HRMS (TOF-ESI) m/z: calcd for C9H18O3NaS [M + Na]+ 229.0869, found 229.0865.4-(tert-Butylsulfonyl)-1-(thien-2-yl)butan-2-one (3ea)
3ea was prepared following the general procedure by reacting 1e (30 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 10 hours. Flash chromatography (cyclohexane/ethyl acetate: 75/25) afforded the title compound 3ea as a white solid (35 mg, 64%). M.p.: 84–85 °C; IR (neat) νmax/cm−1 2962, 1717, 1473, 1265, 1108, 1007, 669; 1H NMR (300 MHz, CDCl3) δ 7.22 (dd, J = 5.1, 1.1 Hz, 1H), 6.98 (dd, J = 5.0, 3.6 Hz, 1H), 6.91 (m, 1H), 3.99 (s, 2H), 3.21 (t, J = 6.6 Hz, 2H), 3.08 (t, J = 6.8 Hz, 2H), 1.71 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 203.5, 134.4, 127.5, 127.4, 125.7, 59.3, 43.9, 40.4, 32.5, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C12H18O3NaS2 [M + Na]+ 297.0590, found 297.0592.1-Acetyl-2-tert-butylsulfonyl cyclohexane (3fa)
3fa was prepared following the general procedure by reacting 1f (25.7 μL, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 48 hours. Flash chromatography (cyclohexane/ethyl acetate: 5/5) afforded the title compound 3fa as a brown foam (33 mg, 67%). IR (neat) νmax/cm−1 2944, 1711, 1271, 1110, 681; 1H NMR (300 MHz, CDCl3) δ 3.59 (m, 1H), 2.90 (m, 1H), 2.62–2.51 (m, 1H), 2.23 (s, 3H), 2.02 (m, 2H), 1.81–1.74 (m, 2H), 1.47–1.41 (m, 3H), 1.36 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 208.9, 61.1, 57.1, 48.3, 29.1, 27.6, 26.2, 24.6, 23.5 (3C), 21.9; HRMS (TOF-ESI) m/z: calcd for C12H22O3SNa [M + Na]+ 269.1182, found 269.1180.3-(tert-Butylsulfonyl)propanal (3ga)
3ga was prepared following the general procedure by reacting 1g (13.4 μL, 0.20 mmol), tert-butylsulfinamide 2a (24 mg, 0.20 mmol), HBF4 (26 μL, 0.20 mmol) for 4 hours. Flash chromatography (cyclohexane/ethyl acetate: 3/7) afforded the title compound 3ga as a colourless oil (14 mg, 39%). IR (neat) νmax/cm−1 2988, 1723, 1297, 1110; 1H NMR (300 MHz, CDCl3) δ 9.87 (s, 1H), 3.25 (t, J = 7.4 Hz, 2H), 3.10 (t, J = 7.0 Hz, 2H), 1.44 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 197.8, 59.4, 38.7, 34.8, 23.5 (3C); HRMS (TOF-ESI) m/z: calcd for C7H14O3NaS [M + Na]+ 201.0556, found 201.0564.(E)-N-[3-(tert-Butylsulfonyl)propyliden-1-yl]-tert-butylsulfinamide (5ga)
5ga was prepared following the general procedure by reacting 1g (13.4 μL, 0.20 mmol), tert-butylsulfinamide 2a (73 mg, 0.60 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. Flash chromatography (cyclohexane/ethyl acetate: 3/7) afforded the title compound 5ga as a white solid (14.5 mg, 26%). M.p.: 101–102 °C; IR (neat) νmax/cm−1 2985, 1651, 1300, 1263, 1117, 1078; 1H NMR (300 MHz, CDCl3) δ 8.19 (t, J = 3.1 Hz, 1H), 3.34–3.28 (m, 2H), 3.15–3.09 (m, 2H), 1.45 (s, 9H), 1.20 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 165.6, 59.4, 57.2, 41.2, 27.5, 23.6 (3C), 22.5 (3C); HRMS (TOF-ESI) m/z: calcd for C11H24NO3S2 [M + H]+ 282.1192, found 282.1205.3-(tert-Butylsulfonyl)-1-(4-chlorophenyl)propan-1-one (3ha)
3ha was prepared following the general procedure by reacting 1h (33 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 5 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3) afforded the title compound 3ha as a white solid (16 mg, 28%). M.p.: 146–147 °C; IR (neat) νmax/cm−1 2982, 1674, 1588, 1301, 1265, 1116; 1H NMR (300 MHz, CDCl3) δ 7.94 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 8.7 Hz, 2H), 3.55 (dd, J = 8.5, 6.4 Hz, 2H), 3.38 (dd, J = 8.5, 6.5 Hz, 2H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 195.3, 140.5, 134.4, 129.7 (2C), 129.3 (2C), 59.3, 40.5, 29.8, 23.5 (3C); HRMS (TOF-ESI) m/z: calcd for C13H17O3NaSCl [M + Na]+ 311.0479, found 311.0476.3-(tert-Butylsulfonyl)-1-(4-fluorophenyl)propan-1-one (3ia)
3ia was prepared following the general procedure by reacting 1i (30 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 5 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3 to 6/4) afforded the title compound 3ia as a white solid (16 mg, 29%). M.p.: 103–104 °C; IR (neat) νmax/cm−1 2925, 1684, 1596, 1267, 1108, 979, 736; 1H NMR (300 MHz, CDCl3) δ 8.03 (m, 2H), 7.16 (br t, J = 8.7 Hz, 2H), 3.56 (dd, J = 8.4, 6.3 Hz, 1H), 3.38 (dd, J = 8.5, 6.4 Hz, 1H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 194.9, 166.3 (d, J = 254.4 Hz), 131.0 (d, J = 9.3 Hz, 2C), 116.1 (d, J = 21.9 Hz, 2C), 59.4, 40.7, 29.8, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C13H17O3NaSF [M + Na]+ 295.0775, found 295.0779.3-(tert-Butylsulfonyl)-1-phenylpropan-1-one (3ja)
3ja was prepared following the general procedure by reacting 1 h (26.4 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 5 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3 to 6/4) afforded the title compound 3ja as a white solid (31 mg, 61%). M.p.: 120–121 °C; IR (neat) νmax/cm−1 2935, 2920, 1687, 1262, 1108, 1002, 740, 688; 1H NMR (300 MHz, CDCl3) δ 8.00 (br d, J = 7.3 Hz, 2H), 7.60 (br t, J = 7.6 Hz, 1H), 7.49 (br t, J = 7.7 Hz, 2H), 3.60 (dd, J = 8.5, 6.6 Hz, 1H), 3.39 (dd, J = 8.6, 6.7 Hz, 1H), 1.47 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 196.5, 136.1, 133.9, 128.9, 128.3, 59.3, 40.6, 29.8, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C13H18O3NaS [M + Na]+ 277.0869, found 277.0887.3-(tert-Butylsulfonyl)-1-(4-methoxyphenyl)propan-1-one (3ka)
3ka was prepared following the general procedure by reacting 1k (32 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 4 hours. Flash chromatography (cyclohexane/ethyl acetate: 5/5) afforded the title compound 3ka as a white solid (33 mg, 58%). M.p.: 75–76 °C; IR (neat) νmax/cm−1 2926, 1674, 1601, 1573, 1251, 1112, 977; 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 8.9 Hz, 2H), 6.95 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 3.54 (dd, J = 8.5, 6.8 Hz, 2H), 3.37 (dd, J = 8.5, 6.6 Hz, 1H), 1.46 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 194.9, 164.1, 130.6 (2C), 129.2, 114.1 (2C), 59.3, 55.7, 40.7, 29.3, 23.5 (3C); HRMS (TOF-ESI) m/z: calcd for C14H20O4NaS [M + Na]+ 307.0975, found 307.0982.3-(tert-Butylsulfonyl)-1,3-diphenyl propan-1-one (3la)
3la was prepared following the general procedure by reacting 1l (42 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 48 hours. Flash chromatography (cyclohexane/ethyl acetate: 9/1 to 8/2) afforded the title compound 3la as a white solid (38 mg, 57%) M.p.: 136–137 °C; IR (neat) νmax/cm−1 2926, 1687, 1281, 1177, 1108; 1H NMR (300 MHz, CDCl3) δ 7.92 (d, J = 7.3 Hz, 2H), 7.62 (d, J = 7.9 Hz, 2H), 7.55 (t, J = 7.1 Hz, 1H), 7.43 (m, 2H), 7.40–7.29 (m, 3H), 5.19 (dd, J = 9.2 Hz, J = 3.5 Hz, 1H), 4.15 (dd, J = 17.9 Hz, J = 3.5 Hz, 1H), 3.72 (dd, J = 17.9 Hz, J = 9.2 Hz, 1H), 1.24 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 195.4, 136.4, 135.0, 133.7, 129.9 (2C), 129.0 (3C), 128.8 (2C), 128.3 (2C), 62.4, 60.1, 39.6, 24.4 (3C); HRMS (TOF-ESI) m/z: calcd for C19H22O3NaS [M + Na]+ 353.1182, found 353.1189.3-(tert-Butylsulfonyl)-1-(thien-3-yl)propan-1-one (3ma)
3ma was prepared following the general procedure by reacting 1m (28 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 6 hours. Flash chromatography (cyclohexane/ethyl acetate: 6/4) afforded the title compound 3ma as a white solid (29 mg, 55%). M.p.: 133–134 °C; IR (neat) νmax/cm−1 2952, 1669, 1416, 1264, 1106, 771; 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J = 3.4 Hz, 1H), 7.67 (d, J = 4.9 Hz, 1H), 7.15 (t, J = 4.6 Hz, 1H), 3.52 (dd, J = 8.3, 6.2 Hz, 1H), 3.37 (dd, J = 8.5, 6.6 Hz, 1H), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 189.1, 143.0, 134.6, 132.8, 128.5, 59.3, 40.5, 30.2, 23.5 (3C); HRMS (TOF-ESI) m/z: calcd for C11H16O3S2Na [M + Na]+ 283.0433, found 283.0435.3-(tert-Butylsulfonyl)-1-(1-tosyl-1H-indol-3-yl)propan-1-one (3na)
3na was prepared following the general procedure by reacting 1n (65 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3 to 6/4) afforded the title compound 3na as a brown solid (45 mg, 50%). M.p.: 143–144 °C; IR (neat) νmax/cm−1 2971, 1666, 1539, 1266, 1116, 978; 1H NMR (300 MHz, CDCl3) δ 8.34 (s, 1H), 8.25 (dd, J = 7.3, 2.2 Hz, 1H), 7.96 (dd, J = 6.9, 1.3 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.40–7.26 (m, 4H), 3.54–3.50 (m, 2H), 3.44–3.38 (m, 2H), 2.36 (s, 3H, CH3-Ar), 1.47 (s, 9H, tBu); 13C NMR (75 MHz, CDCl3) δ 191.7, 146.2, 135.0, 134.5, 133.2, 132.5, 130.4 (2C), 127.4 (2C), 126.0, 125.1, 122.9, 120.4, 113.3, 59.4, 40.3, 30.7, 23.5 (3C), 21.8; HRMS (TOF-ESI) m/z: calcd for C22H25NO5NaS2 [M + Na]+ 470.1066, found 470.1073.4-(tert-Butylsulfonyl)-1-{[(1R,2S,5R)-2-isopropyl-5-methyl cyclohex-1-yl]oxy} but-3-en-2-one (3oa)
3oa was prepared following the general procedure by reacting 1o (44 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) in a mixture of CH2Cl2/CH3CN (8/2, 1 mL) for 17 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3) afforded the title compound 3oa as a yellow oil (41 mg, 59%). IR (neat) νmax/cm−1 2955, 2920, 1722, 1301, 1116; 1H NMR (300 MHz, CDCl3) δ 4.20 (d, J = 16.8 Hz, 1H), 4.00 (d, J = 16.8 Hz, 1H), 3.26–6.09 (m, 4H), 2.22 (m, 1H), 2.03 (br d, J = 11.3 Hz, 1H), 1.64 (m, 2H), 1.43 (s, 9H), 1.35–1.18 (m, 4H), 0.91 (m, 8H), 0.78 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 206.6, 80.6, 73.9, 59.3, 48.2, 40.0, 39.9, 34.5, 31.6, 30.5, 25.8, 23.5 (3C), 23.4, 22.4, 21.1, 16.4; HRMS (TOF-ESI) m/z: calcd for C18H34O4NaS [M + Na]+ 369.2070, found 369.2080.(11Z,14Z)-1-(tert-butylsulfonyl)icosa-11,14-dien-3-one (3pa)
3pa was prepared following the general procedure by reacting 1p (58 mg, 0.20 mmol), tert-butylsulfinamide (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 6 hours. Flash chromatography (cyclohexane/ethyl acetate: 7/3) afforded the title compound 3pa as a yellow foam (42 mg, 51%). IR (neat) νmax/cm−1 2982, 1712, 1268, 1102; 1H NMR (300 MHz, CDCl3) δ 5.35–5.27 (m, 4H), 3.21 (t, J = 7.8 Hz, 2H), 2.99 (t, J = 7.2 Hz, 2H), 2.76 (t, J = 5.8 Hz, 2H), 2.49 (t, J = 7.4 Hz, 2H), 2.03 (m, 4H), 1.60 (m, 2H), 1.42 (s, 9H), 1.30 (m, 14H), 0.88 (t, J = 7.0 Hz, 3H, Me); 13C NMR (75 MHz, CDCl3) δ 207.3, 130.3, 130.2, 128.2, 128.0, 59.2, 43.1, 40.2, 33.1, 31.7, 29.7, 29.5, 29.4, 29.2, 28.2, 27.3, 25.8, 24.9, 23.9, 23.5 (3C), 22.7, 14.2; HRMS (TOF-ESI) m/z: calcd for C24H44O3NaS [M + Na]+ 435.2903, found 435.2915.General procedure for the synthesis of hydroxysulfones (4)
The ketosulfone 3 (0.20 mmol) was dissolved in MeOH (c = 0.1 mol L−1) at 0 °C and NaBH4 (0.24 mmol) was then added. After stirring at room temperature for 2 hours, the reaction mixture was quenched by the addition of acetone (2 mL) before evaporation of the solvent. Water (2 mL) was then added, the aqueous layer was extracted with AcOEt (3 × 10 mL) and the combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated. The crude product was purified by flash chromatography with cyclohexane/ethyl acetate to yield the desired hydroxysulfone 4.3-(tert-Butylsulfonyl)cyclopentanol (4ba)
4ba was prepared following the general procedure by reacting 1b (32.8 μL, 0.40 mmol), tert-butylsulfinamide 2a (96 mg, 0.80 mmol), HBF4 (52 μL, 0.40 mmol) for 24 hours. NaBH4 (18 mg, 0.48 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 1/9) afforded the title compound 4ba as a white solid (73 mg, 88%, cis/trans = 10/1). M.p.: 79–80 °C; IR (neat) νmax/cm−1 3499, 2993, 1282, 1143, 688; 1H NMR (300 MHz, CDCl3) δ 4.31 (br s, 1H), 3.72–3.62 (m, 1H), 2.47–2.20 (m, 4H), 2.13–2.06 (m, 1H), 2.02–1.91 (m, 1H), 1.79–1.72 (m, 1H), 1.44 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 72.4, 60.3, 55.7, 37.4, 35.6, 25.8, 24.3 (3C); HRMS (TOF-ESI) m/z: calcd for C9H18O3NaS [M + Na]+ 229.0869, found 229.0868.3-(Benzenesulfonyl)cyclopentanol (4bb)
4bb was prepared following the general procedure by reacting 1b (16.8 μL, 0.20 mmol), benzenesulfinamide 2b (56 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 3/7 to 2/8) afforded the title compound 4bb as a colourless oil (30 mg, 66%, cis/trans = 21/1). IR (neat) νmax/cm−1 3480, 1446, 1284, 1142, 1085; 690; 1H NMR (300 MHz, CDCl3) δ 7.91 (br d, J = 7.4 Hz, 2H), 7.67 (br t, J = 7.8 Hz, 1H), 7.57 (br t, J = 6.9 Hz, 2H), 4.32 (br s, 1H), 3.61 (qt, J = 8.5 Hz, 1H), 2.35–2.10 (m, 4H), 1.96–1.85 (m, 2H), 1.83–1.73 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 138.2, 134.0, 129.5 (2C), 128.7 (2C), 72.7, 63.2, 36.3, 35.6, 24.8; HRMS (TOF-ESI) m/z: calcd for C11H14O3NaS [M + Na]+ 249.0556, found 249.0560.3-(p-Toluensulfonyl)-cyclopentan1-ol (4bc)
4bc was prepared following the general procedure by reacting 1b (16.8 μL, 0.20 mmol), p-toluenesulfinamide 2c (62 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 24 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 4/6) afforded the title compound 4bc as a colourless oil (36 mg, 75%, cis/trans = 17/1). IR (neat) νmax/cm−1 3492, 1478, 1284, 1185; 1H NMR (300 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 4.31 (br s, 1H), 3.58 (qt, J = 6.7 Hz, 1H), 2.45 (s, 3H), 2.28–2.13 (m, 4H), 1.95–1.84 (m, 2H), 1.79–1.74 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 144.9, 135.0, 130.0 (2C), 128.6 (2C), 72.6, 63.2, 36.2, 35.5, 24.6, 21.6; HRMS (TOF-ESI) m/z: calcd for C12H16O3NaS [M + Na]+ 263.0712, found 263.0710. The data presented above are in agreement with that detailed in the literature.213-(tert-Butylsulfonyl)-1-(4-chlorophenyl)propan-1-ol (4ha)
4ha was prepared following the general procedure by reacting 1h (33 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 5 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 4/6) afforded the title compound 4ha as a colourless oil (11 mg, 18%). IR (neat) νmax/cm−1 3469, 2924, 1282, 1110, 748; 1H NMR (300 MHz, CDCl3) δ 7.32 (m, 4H), 4.95 (dd, J = 8.5, 4.5 Hz, 1H), 3.07 (t, J = 7.3 Hz, 2H), 2.33–2.21 (m, 2H), 1.41 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 142.0, 133.8, 129.0 (2C), 127.2 (2C), 71.8, 59.3, 42.1, 30.2, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C13H19O3NaSCl [M + Na]+ 313.0636, found 313.0638.3-(tert-Butylsulfonyl)-1-(4-fluorophenyl)propan-1-ol (4ia)
4ia was prepared following the general procedure by reacting 1i (30 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 5 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 4/6 to 3/7) afforded the title compound 4ia as a white solid (17 mg, 31%). M.p.: 61–62 °C; IR (neat) νmax/cm−1 3479, 2990, 1509, 1260, 1109; 1H NMR (300 MHz, CDCl3) δ 7.34 (dd, J = 8.7 Hz, J = 5.4 Hz, 2H), 7.04 (t, J = 8.6 Hz, 2H), 4.91 (dd, J = 8.0, 4.6 Hz, 1H), 3.07 (t, J = 7.5 Hz, 2H), 2.52 (br s, 1H), 2.31–2.22 (m, 2H), 1.41 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 162.5 (d, J = 245 Hz), 139.3, 127.5 (d, J = 8.0 Hz, 2C), 115.6 (d, J = 21.3 Hz, 2C), 71.9, 59.3, 42.2, 30.2, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C13H19O3FNaS [M + Na]+ 297.0931, found 297.0937.3-(tert-Butylsulfonyl)-1-(4-methoxyphenyl)propan-1-ol (4ka)
4ka was prepared following the general procedure by reacting 1k (32 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 4 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 4/6) afforded the title compound 4ka as a yellow oil (28 mg, 49%). IR (neat) νmax/cm−1 3490, 2938, 1672, 1512, 1244, 1109, 1030; 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.84 (t, J = 6.5 Hz, 2H), 3.80 (s, 3H), 3.11–2.98 (m, 2H), 2.27 (q, J = 7.5 Hz, 2H), 1.40 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 159.5, 135.6, 127.1 (2C), 114.2 (2C), 72.3, 59.2, 55.4, 42.5, 30.0, 23.6 (3C); HRMS (TOF-ESI) m/z: calcd for C14H22O4NaS [M + Na]+ 309.1131, found 309.1143.3-(tert-Butylsulfonyl)-1-(thien-3-yl)propan-1-ol (4ma)
4ma was prepared following the general procedure by reacting 1m (28 mg, 0.20 mmol), tert-butylsulfinamide 2a (48 mg, 0.40 mmol), HBF4 (26 μL, 0.20 mmol) for 6 hours. NaBH4 (9.1 mg, 0.24 mmol) was next added. Flash chromatography (cyclohexane/ethyl acetate: 4/6) afforded the title compound 4ma as a white solid (24 mg, 47%). M.p.: 82–83 °C; IR (neat) νmax/cm−1 3473, 2977, 1255, 1086, 777; 1H NMR (300 MHz, CDCl3) δ 7.25 (d, J = 4.7 Hz, 1H), 7.00–6.96 (m, 2H), 5.17 (dd, J = 7.3, 5.3 Hz, 1H), 3.11 (t, J = 7.4 Hz, 2H), 2.71 (br s, 1H), 2.45–2.35 (m, 2H), 1.41 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 147.2, 127.0, 125.0, 124.1, 68.6, 59.3, 42.2, 30.3, 25.6 (3C); HRMS (TOF-ESI) m/z: calcd for C11H18O2NaS2 [M + Na]+ 285.0590, found 285.0601.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank K. Leblanc for performing mass analyses. The Université Paris-Saclay, the French Ministry of Superior Education and Research, the CNRS, and the National Research Agency (ANR-17-CE11-0030-03) are gratefully acknowledged for financial support.Notes and references
- For recent examples, see: (a) M. J. Tilby and M. C. Willis, Expert Opin. Drug Discovery, 2021, 16, 1227–1231 CrossRef PubMed; (b) F. L. Cherblanc, R. W. M. Davidson, P. Di Fruscia, N. Srimongkolpithak and M. J. Fuchter, Nat. Prod. Rep., 2013, 30, 605–624 RSC; (c) E. Iwasa, Y. Hamashima and M. Sodeoka, Isr. J. Chem., 2011, 51, 420–433 CrossRef CAS.
- For recent examples, see: (a) T. Neumann, L. Benajiba, S. Göring, K. Stegmaier and B. Schmidt, J. Med. Chem., 2015, 58, 8907–8919 CrossRef CAS PubMed; (b) N. Boyer, K. C. Morrison, J. Kim, P. J. Hergenrother and M. Movassaghi, Chem. Sci., 2013, 4, 1646–1657 RSC; (c) H. Rueeger, R. Lueoend, O. Rogel, J.-M. Rondeau, H. Möbitz, R. Machauer, L. Jacobson, M. Staufenbiel, S. Desrayaud and U. Neumann, J. Med. Chem., 2012, 55, 3364–3386 CrossRef CAS PubMed.
- (a) S. Tamamuku and P. Jannasch, Macromolecules, 2012, 45, 6538–6546 CrossRef; (b) S.-M. Liu, Y. Yang, Z.-J. Jiang, Y.-H. Zhou, J. Zuo and J.-Q. Zhao, J. Appl. Polym. Sci., 2012, 124, 4502–4511 CrossRef CAS.
- For recent reviews, see: (a) Y. Fang, Z. Luo and X. Xua, RSC Adv., 2016, 6, 59661–59676 RSC; (b) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939–1976 CrossRef CAS; (c) For selected examples of sulfones synthesis, see: E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS; (d) Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS PubMed; (e) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404–4407 CrossRef CAS PubMed; (f) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 CrossRef CAS PubMed; (g) M. Jerek, Green Chem., 2012, 14, 3047–3052 RSC; (h) X. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS PubMed.
- (a) S. Trivedi, P. C. Patidar, P. K. Chaurasiya, R. S. Pawar, U. K. Patil and P. K. Singour, Pharma Chem., 2010, 2, 369–377 CAS; (b) J. Xiang, M. Ipek, V. Suri, M. Tam, Y. Xing, N. Huang, Y. Zhang, J. Tobin, T. S. Mansour and J. McKew, Bioorg. Med. Chem., 2007, 15, 4396–4405 CrossRef CAS PubMed.
- For a recent review, see: (a) R. J. Reddy and A. H. Kumari, J. Org. Biomol. Chem., 2021, 19, 3087–3118 RSC; (b) For recent and selected examples, see: B. Dam, A. K. Sahooa and B. K. Patel, Green Chem., 2022, 24, 7122–7130 RSC; (c) Y. Wang, Y. Zhao, C. Cai, L. Wang and H. Gong, Org. Lett., 2021, 23, 8296–8301 CrossRef CAS PubMed; (d) X. Chen, S. Lu, Y. Zheng, J. Wang, L. Yang and P. Sun, Adv. Synth. Catal., 2022, 364, 1305–1312 CrossRef CAS; (e) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205–4208 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Winkler, Y. S. Raupp, L. A. M. Köhl, H. E. Wagner and M. A. R. Meier, Macromolecules, 2014, 47, 2842–2846 CrossRef CAS; (b) W. Chen, Z. Jing, K. F. Chin, B. Qiao, Y. Zhao, L. Yan, C.-H. Tan and Z. Jiang, Adv. Synth. Catal., 2014, 356, 1292–1300 CrossRef CAS.
- (a) W. Keim, J. Herwig and G. Pelzer, J. Org. Chem., 1997, 62, 422–424 CrossRef CAS PubMed; (b) B. Sreedhar, M. A. Reddy and P. S. Reddy, Synlett, 2008, 1949–1952 CrossRef CAS.
- (a) M. F. Browne and R. L. Shriner, J. Org. Chem., 1957, 22, 1320–1322 CrossRef CAS; (b) I. H. Pitman, E. Shefter and M. Ziser, J. Am. Chem. Soc., 1970, 92, 3413–3417 CrossRef CAS.
- (a) M. J. Tilby, D. F. Dewez, L. R. E. Pantaine, A. Hall, C. Martínez-Lamenca and M. C. Willis, ACS Catal., 2022, 12, 6060–6067 CrossRef CAS PubMed; (b) X. Wang, M. Yang, W. Xie, X. Fan and J. Wu, Chem. Commun., 2019, 55, 6010–6013 RSC; (c) X.-Q. Chu, H. Meng, X.-P. Xu and S.-J. Ji, Chem.–Eur. J., 2015, 21, 11359–11368 CrossRef CAS PubMed; (d) Y. Yang, L. Tang, S. Zhang, X. Guo, Z. Zha and Z. Wang, Green Chem., 2014, 16, 4106–4109 RSC; (e) M. Fernández, U. Uria, L. Orbe, J. L. Vicario, E. Reyes and L. Carrillo, J. Org. Chem., 2014, 79, 441–445 CrossRef PubMed; (f) Z. Jin, J. Xu, S. Yang, B.-A. Song and Y. Robin Chi, Angew. Chem., Int. Ed., 2013, 52, 12354–12358 CrossRef CAS PubMed; (g) A. Bhunia, S. R. Yetra, S. S. Bhojgude and A. T. Biju, Org. Lett., 2012, 14, 2830–2833 CrossRef CAS PubMed.
- (a) E. Drège, J. Oko, P.-E. Venot, N. Gigant and D. Joseph, RSC Adv., 2015, 5, 96720–96724 RSC; (b) Z. Amara, G. Bernadat, P.-E. Venot, P. Retailleau, C. Troufflard, E. Drège, F. Le Bideau and D. Joseph, Org. Biomol. Chem., 2014, 12, 9797–9810 RSC; (c) Z. Amara, E. Drège, C. Troufflard, P. Retailleau, M.-E. Tran Huu-Dau and D. Joseph, Chem.–Eur. J., 2014, 20, 15840–15848 CrossRef CAS PubMed; (d) Z. Amara, E. Drège, C. Troufflard, P. Retailleau and D. Joseph, Org. Biomol. Chem., 2012, 10, 7148–7157 RSC.
- For reviews, see: (a) K. C. Bharadwaj, RSC Adv., 2015, 5, 75923–75946 RSC; (b) P. Xie and Y. Huang, Eur. J. Org Chem., 2013, 6213–6226 CrossRef CAS; (c) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS.
- N. Gigant, E. Drège, P. Retailleau and D. Joseph, Chem.–Eur. J., 2015, 21, 15544–15547 CrossRef CAS PubMed.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- For a review, see: P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed.
- For a review, see: M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed.
- For a review, see: T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- The sulfonated scaffold 3da was isolated in a 56% yield after purification by flash column chromatography.
- N. K. Jana and J. G. Verkade, Org. Lett., 2003, 5, 3787–3790 CrossRef CAS PubMed.
- M. J. Tilby, D. F. Dewez, L. R. E. Pantaine, A. Hall, C. Martínez-Lamenca and M. C. Willis, ACS Catal., 2022, 12, 6060–6067 CrossRef CAS PubMed.
- M. Fernández, U. Uria, L. Orbe, J. L. Vicario, E. Reyes and L. Carrillo, J. Org. Chem., 2014, 79, 441–445 CrossRef PubMed.
- A. Bagno, S. J. Eustace, L. Johansson and G. Scorrano, J. Org. Chem., 1994, 59, 232–233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08675e |
| This journal is © The Royal Society of Chemistry 2024 |