
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of large-sized spherical Co–C alloys with soft magnetic properties though a high-pressure solid-state metathesis reaction
Xu Jia a,
Leilei Zhang*b,
Yi Tiana,
Binbin Wua,
Yu Taoa,
Duanwei Hea,
Baocheng Yangb,
Filippo S. Boic and
Li Lei*a
a,
Leilei Zhang*b,
Yi Tiana,
Binbin Wua,
Yu Taoa,
Duanwei Hea,
Baocheng Yangb,
Filippo S. Boic and
Li Lei*a
aInstitute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China. E-mail: lei@scu.edu.cn
bInstitute of Nano-Structured Functional Materials, Huanghe Science and Technology College, Zhengzhou 450063, China. E-mail: luanzhanglei123@163.com
cCollege of Physics, Sichuan University, Chengdu 610065, China
First published on 4th March 2024
Abstract
In this work, we report a novel high-pressure solid-state metathesis (HSM) reaction to produce spherical bulk (diameters 2–4 mm) Co–C alloys (Co3C and Co1−xCx). At 2–5 GPa and 1300 °C, C atoms preferentially occupy the interstitial sites of the face-centered cubic (fcc) Co lattice, leading to the formation of metastable Pnma Co3C. The Co3C decomposes above 1400 °C at 2–5 GPa, C atoms infiltrate the interstitial sites of the fcc Co lattice, saturating the C content in Co, forming an fcc Co1−xCx solid solution while the C atoms in excess are found to precipitate in the form of graphite. The Vickers hardness of the Co–C alloys is approximately 6.1 GPa, representing a 19.6% increase compared to hexagonal close-packed (hcp) Co. First-principles calculations indicate that the presence of C atoms in the Pnma Co3C structure leads to a relative decrease in the magnetic moments of the two distinct Co atom occupancies. The Co–C alloys exhibited a soft magnetic behavior with saturation magnetization up to 93.71 emu g−1 and coercivity of 74.8 Oe; coercivity increased as the synthesis pressure rises.
Introduction
Iron-group metal carbides refer to compounds composed of elements from the iron-group, namely iron (Fe), cobalt (Co), and nickel (Ni), in combination with carbon atoms. These alloy compounds can exhibit a diverse range of crystal structures and properties, which are contingent upon the composition,1 proportions of elements,2–5 and the arrangement of the crystal lattice.6–8 A noteworthy observation lies in the dependence of the magnetic moment on the unit cell volume.9 This imbues them with substantial potential applications in the realm of materials science.1,4,10 High pressure is of utmost significance in determining the phase stability of iron-group metal carbides.4,11,12Carbon atoms readily integrate into the crystal lattice of iron-group metals. Within the realm of iron-group metal carbides, the formation enthalpy (Hform.) of Ni–C or Co–C systems is higher than that of the Fe–C system. This observation also indicates relatively lower stability and greater synthetic challenges for the carbide phases in the Ni–C and Co–C systems compared to the Fe–C.3,13 The solubility of C (in these systems) may not be the same. Iron carbides can conventionally be synthesized by direct reaction between Fe and C at relatively low temperatures. However, the formation of Co–C alloy is restricted under such conditions, requiring the presence of elevated temperatures along with the application of pressure. Fe, Co, and Ni share similarities in their chemical properties. The chemical bonding within carbides is predominantly attributed to metallic bonds; however, due to the hybridization of C and metallic states, contributions from covalent bonds are also present.13 The unique characteristics of these chemically distinctive bonds formed between metals and C atoms confer distinctive attributes upon iron-group metal carbides.
Co3C has recently attracted extensive attention in areas such as magnetism, hydrogen storage, and catalysis.5,14–22 Experimental and theoretical studies have confirmed its notable magnetization strength and coercivity, suggesting that it could serve as an ideal rare-earth-free permanent magnet material with promising developmental prospects.5,14 Co3C nanoparticles have been synthesized using methods like wet chemical synthesis,5,23,24 mechanical alloying,3,6,25 ion irradiation,26 and the one-pot template-free solvothermal approach.27 P. V. Marshall et al. utilized a DAC (diamond anvil cell) device with a size of 300 μm to synthesize microscale Co3C under conditions of 5–15 GPa and 1200 K.28 However, stable carbides are not present in the Co–C system. Instead, metastable cobalt carbides are commonly found in mixed forms. The computational analysis indicates that the Hform. of Co3C is lower than that of the competing phase, Co2C, under high pressure. Consequently, the production of single-phase (Co3C) samples will be kinetically favorable in high pressure environments.28
Under experimental conditions of 7 GPa and 1400–1500 °C, S. Naka et al. reported absence of stoichiometric cobalt carbide when cobalt was used as a catalyst for diamond synthesis.29 Absence of cobalt carbides was shown also by W. Utsumi et al. in their in situ X-ray diffraction study of graphite-to-diamond transformation using various solvent-catalysts under high pressure and high-temperature conditions.30 When measuring the solubility of diamond in metallic cobalt under conditions of 5 GPa and 1100–1300 °C, Y. Tian et al. did not observe the presence of cobalt carbide products.31 Despite the important contributions reported in literature, extended investigations are needed in order to gain new insights on the stabilization of Co3C phases through high pressure methods. Despite the important progress in this research area, little has been reported on the high pressure behavior of Co3C.
The HSM reaction has been proven to be an effective approach for synthesizing various metal nitrides,32,33 including GaN,34 Fe3N,35 RexN,36–38 Fe2.322Co0.678N0.888 (ref. 39) and CoNx.40 This method allows to reduce the reaction enthalpy, to enhance the activation energy, and prevent the thermal decomposition and volatilization of the precursors and products, forming the bulk samples with excellent crystallinity.
In this work, we proposed a novel high pressure chemical reaction method to synthesize bulk Co–C alloys and its high pressure decomposition behavior was investigated under a variable range of pressure and temperature (P–T) conditions. The hardness and magnetic properties of the Co–C alloys were characterized using a Vickers hardness tester, Vibrating Sample Magnetometer (VSM), and the Density Functional Theory (DFT). The aim of the present study is to provide novel insights on the formation and decomposition mechanism of Co–C alloys under high pressure.
Experimental and theoretical calculations
High pressure synthesis experiments were conducted using a large-volume press facility (DS 6 × 14 MN, China). The temperature of the sample was directly measured by a WRe5/26 thermocouple inserted through the sample chamber. The temperature gradient within the sample chamber was approximately 10 °C. The chamber pressure was determined by considering the relationship between the melting temperature of silver and pressure,41 with an estimated pressure error of approximately ±0.1 GPa.Co–C alloys were synthesized through the high-pressure solid-state metathesis (HSM) reaction using optimized molar ratios of precursor materials Li2CO3, BN, and Co2O3 (e.g., Li2CO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BN:Co2O3 = 1:1:2). The mixed powders were firstly ground until a uniform color distribution was achieved and then pre-compressed into cylindrical shapes (8 mm diameter, 8 mm height) using a mold. The pre-compressed cylindrical samples were sealed within a graphite heating device, as shown in Fig. 1a.
:BN:Co2O3 = 1:1:2). The mixed powders were firstly ground until a uniform color distribution was achieved and then pre-compressed into cylindrical shapes (8 mm diameter, 8 mm height) using a mold. The pre-compressed cylindrical samples were sealed within a graphite heating device, as shown in Fig. 1a.
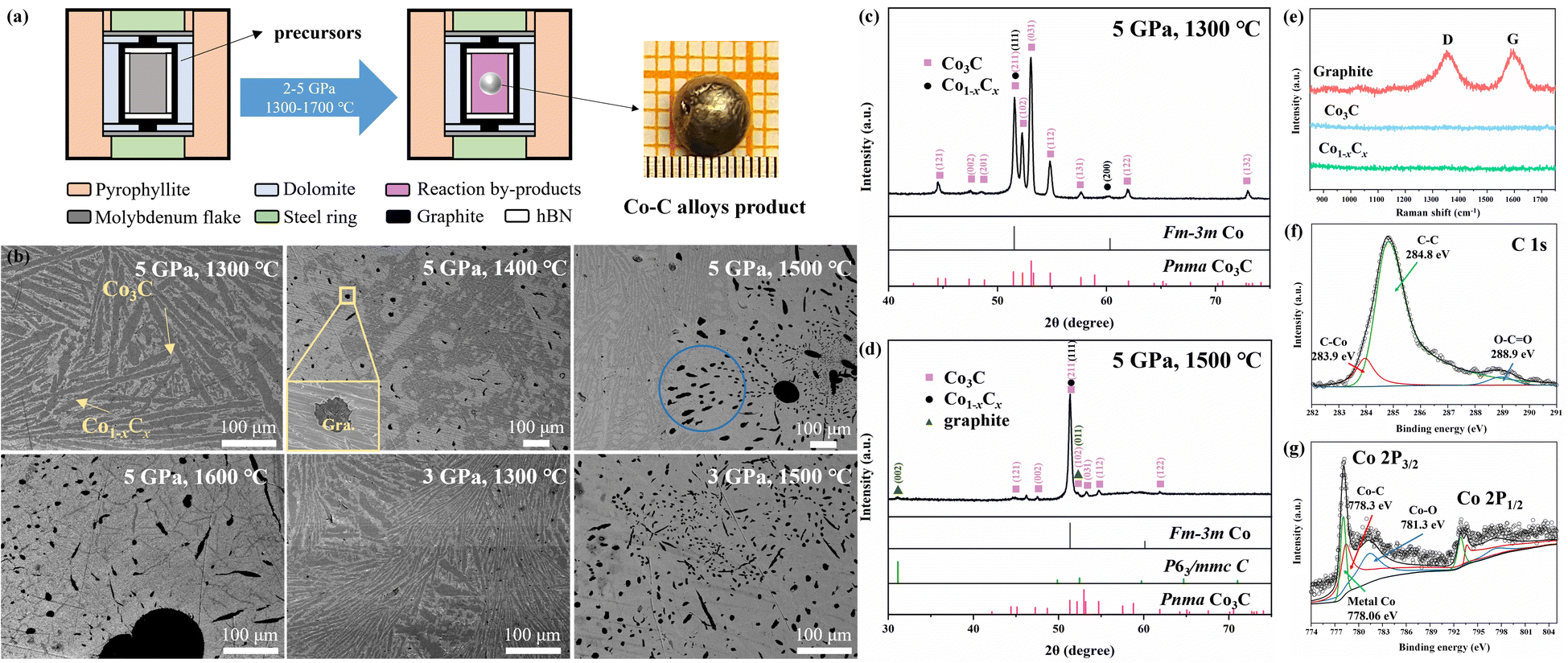 | ||
| Fig. 1 (a) Cell assembly for high pressure experiments before reaction and after reaction, and the inset image is the optical image of the spherical bulk Co–C alloys product. (b) SEM images of spherical bulk Co–C alloys products synthesized at 5 GPa and 1300–1600 °C, 3 GPa and 1300–1500 °C (scale bar 100 μm). The black regions correspond to graphite (Gra.), the light gray regions consist of fcc Co1−xCx solid solution, and the dark gray regions consist of Co3C. The inset is SEM images of the magnified view of the boxed regions. The blue region is measured micro XRD zone. (c) Micro XRD pattern of the cross-section of spherical bulk Co–C alloys product synthesized at 5 GPa and 1300 °C. (d) Micro XRD pattern of the blue region in (b). (e) Raman spectra of the reaction product, the red spectral signal corresponds to graphite, the blue spectral signal corresponds to the Co3C, and the green spectral signal corresponds to the fcc Co1−xCx solid solution. XPS spectra of the spherical bulk Co–C alloys product at 5 GPa and 1500 °C. (f) High resolution spectrum of C 1s. (g) High resolution spectrum of Co 2p. | ||
In the high pressure synthesis experiments, the sample chamber was first loaded to 2–5 GPa, and then heated to the desired temperature (1300–1700 °C) for a 10 minute dwell time before being decompressed to ambient conditions. Typically, after the high pressure experiments, metallic spherical reaction products of 2–4 mm in size and some by-products powders were obtained in the sample chamber. Our synthetic temperature should be lower than the high pressure melting temperature of metal Co.42 Therefore, the formation of the spherical bulk Co–C alloys is unlikely to be the result of conventional melting and recrystallization behaviors; it might be related to the participation of metal borate by-product. Because of their relatively low melting points (<1000 °C), The metal borate would be in the molten state like a kind of solvent of Co–C alloys, and the Co–C alloys products in the LiBO2 melt incline to gather into a spherical body in the effect of the lowest surface energy.35,43 In addition, metal borates are readily washed by water, which makes the purification of alloys products very easy.
The samples were subjected to phase analysis using micro X-ray diffraction (micro XRD, Germany-Bruker D8 Discover Co Kα). The morphology and elemental distribution of the samples were examined using a scanning electron microscope (FE-SEM, JSM-IT500HR, JEOL). The atomic concentration is determined by Energy Dispersive X-ray Spectroscopy (EDS, Aztec Energy X-Max 20, Oxford Instruments). Observation of the morphology and phase distribution of spherical Co–C alloy products was performed using Backscattered Electron Diffraction (BED, Aztec Energy X-Max 20, Oxford Instruments). The chemical states in the samples were analyzed using X-ray photoelectron spectroscopy (XPS, USA-Thermo Scientific ESCALAB Xi+). Raman spectroscopy measurements were performed using a solid-state laser with a wavelength of 532 nm (RGB laser system) and a triple grating monochromator (Andor Shamrock SR-303i-B) equipped with an EMCCD (ANDOR Newton DU970P-BVF). The laser output power was maintained at 50 mW. For each Raman measurement, 20 acquisitions of 3 s were typically performed. The magnetization curves were measured under a field of up to 2 kOe in a vibrating sample magnetometer (VSM, Lakeshore-7404) to analyze the magnetic properties.
In this study, we have performed 6 types of high pressure experiments, with the high pressure chemical reaction experiment as shown in Table 1. Exp. 1 represents the conventional HSM reaction between Li2CO3 and BN at 5 GPa and 1400 °C, which could result in the formation of graphite, LiBO2, and N2.33 Exp. 2 represents the reaction between Co2O3 and BN at 5 GPa and 1500 °C, which yields reaction products consisting of CoNx, B2O3, and N2.40 Exp. 3 demonstrates the absence of a reaction between Li2CO3 and Co2O3. The combination of Exp. 1 and Exp. 3 yields a novel HSM reaction pathway, as depicted in Exp. 4. This reaction occurs under conditions of 2–5 GPa and 1300–1700 °C, resulting in the formation of Co3C, Co1−xCx, LiBO2, LiBC, and N2 (CoNx cannot be found). This reaction occurs under conditions of 3 GPa at 1300 °C and 5 GPa at 1300 °C, resulting in the formation of Co3C, fcc Co1−xCx solid solution, LiBO2, LiBC, and N2 (CoNx cannot be found). We found that the occurrence of the reaction was not observed at P–T ranging from 1300–1500 °C at 10–15 GPa and below 1200 °C at 3–5 GPa. Exp. 5 signifies the reaction initiating Co3C decomposition at temperatures exceeding 1400 °C under pressure in the range of 2–5 GPa. Exp. 6 signifies the reaction where Co3C has fully decomposed at temperatures exceeding 1600 °C under pressure of 3 GPa.
| Exp. | Reaction precursors | P–T conditions | Reaction products | Ref. |
|---|---|---|---|---|
| 1 | Li2CO3 + BN | 5 GPa, 1400 °C | Graphite + LiBO2 + N2 | 33 |
| 2 | Co2O3 + BN | 5 GPa, 1400 °C | CoNx + B2O3 + N2 | 40 |
| 3 | Li2CO3 + Co2O3 | 5 GPa, 1400 °C | — | This work |
| 4 | Li2CO3 + BN + Co2O3 | 2–5 GPa, 1300 °C | Co3C + Co1−xCx + LiBO2 + LiBC + N2 | This work |
| 5 | Li2CO3 + BN + Co2O3 | 2–5 GPa, 1400–1700 °C | Co3C + Co1−xCx + graphite + LiBO2 + LiBC + N2 | This work |
| 6 | Li2CO3 + BN + Co2O3 | 3 GPa, 1600 °C | Co1−xCx + graphite + LiBO2 + LiBC + N2 | This work |
The DFT calculations were conducted via the commonly used Vienna Ab initio Simulation Package (VASP) program.44–51 The ionic cores and valence electrons were taken into consideration using the projector-augmented wave (PAW) pseudo-potential method and plane-wave basis set.47,52 For the electronic exchange–correlation functional, the generalized-gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) was employed.53 To guarantee the consistency and dependability of the results, the same plane-wave basis set cutoff (750 eV) and k-point mesh (spacing 0.1 Å−1) were used. The 3d74s2 and 2s22p2 electrons were included in the valence space for the PAW pseudo-potentials of Co and C atoms, respectively. All structures under investigation are fully relaxed until the Hellmann–Feynman forces acting on all atoms are less than 0.01 eV Å−1 and the total energy is smaller than 1 × 10−5 eV. For electronic structure and total energy calculation, the electronic self-consistent iteration were finished when it reach 1 × 10−6 eV. Bader charge analysis method54,55 was applied to characterize the charge state and charge transfer.
Results and discussion
Following high pressure high-temperature experiments, the central portion of the high pressure sample chamber reveals a metallic sphere with a diameter of 2–4 mm (Fig. 1a). The observed molar ratio between precursors also exerts an influence on the chemical reaction. Assuming the precursor molar ratio as Li2CO3:BN:Co2O3 = 1:1:m, the high pressure chemical reaction only occurs for values of m between 2 and 5.
The SEM analyses show that the spherical bulk Co–C alloys product synthesized at 5 GPa 1300 °C are mainly divided into dark gray and light gray regions (Fig. 1b), and micro XRD pattern (Fig. 1c) reveals two distinct color regions, corresponding to Co3C and fcc Co. C atoms generated during the chemical reaction infiltrate the fcc Co lattice, resulting in the formation of the fcc Co1−xCx solid solution. However, notice that according to the phase diagram, fcc Co is generally observed at high temperatures and high pressures, and it will transform back to hcp Co at ambient conditions.40 Interstitial C atom tends to stabilize the fcc structure in ambient conditions.
Upon the synthetic temperature above 1400 °C, three distinct color regions (light gray, dark gray and black) can be observed in the SEM image (Fig. 1b). The black regions exhibit a flake-like morphology, always surrounded by light gray regions. Raman measurements (Fig. 1e) indicate that the black region originates from a graphite-like sample, with two distinct fingerprint Raman modes corresponding to the D and G bands of graphite. Co3C could decompose into fcc Co and graphite at higher temperature (above 1400 °C). Notably, the dark gray region exhibits a higher carbon content as compared to the light gray region, because the C atoms in excess are found to precipitate in the form of graphite. Co acts as an good solvent for C, allowing C atoms to readily intercalate into the Co lattice under high temperature and high pressure conditions. Therefore, the light gray region corresponds to the fcc Co1−xCx (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) solid solution, while the dark gray region, with a higher carbon content, corresponds to Co3C.
m) solid solution, while the dark gray region, with a higher carbon content, corresponds to Co3C.
The SEM image and micro XRD pattern acquired from the spherical bulk Co–C alloys product synthesized at 5 GPa and 1500 °C are shown in Fig. 1b and d. The XRD pattern shows a stronger peak-intensity from fcc Co, while weaker contributions arise from Pnma Co3C, graphite (space group P63/mmc), and fcc Co1−xCx solid solution.
The high resolution spectrum of C 1s displayed binding energies at 283.9, 284.8 and 288.9 eV as depicted in Fig. 1f. The high resolution spectrum of Co 2p exhibited binding energies at 778.06, 778.3, and 781.3 eV for Co 2p3/2, with corresponding peaks observed in Co 2p1/2 (Fig. 1g). The Co–C bond binding energies are located at 283.9 and 778.3 eV, which are in good agreement with the reported results.17,56–59 The binding energies at 284.4 and 778.06 eV are attributed to graphite and metallic Co, respectively. The binding energies observed at 288.9 and 781.3 eV are due to the oxidation of the sample surface. Elemental measurements were carried out on by EDS (Energy Dispersive Spectrometer) all samples produced by the high-pressure chemical reaction, which were composed of C, Co and a small amount of O. The ratio of C and Co elements measured in different color areas of the sample is in good agreement with the experimental results.
Experiments were conducted by altering the P–T conditions for spherical bulk Co–C alloys products synthesis, yet pure phase Pnma Co3C could not be successfully obtained. Fig. 1b shows SEM micrographs of spherical bulk Co–C alloys products obtained from high pressure experiments conducted at temperatures ranging from 1300 to 1600 °C at 5 GPa and 1300 to 1500 °C at 3 GPa. An increase in the synthesis pressure effectively alleviated the decomposition of Co3C. SEM micrographs of spherical bulk Co–C alloys products synthesized at 5 GPa and temperatures between 1300 and 1600 °C reveal an increase in the proportional area of black P63/mmc graphite regions and light gray fcc Co1−xCx solid solution region within the products as the temperature rises. It has been reported that metastable Pnma Co3C, decomposes into fcc Co and graphite at certain temperatures.6,22,60–63
The decomposition enthalpy of metastable Co3C is relatively high (−ΔH = 23 kJ mol−1).3,6 In an isobaric environment, the variation in Gibbs free energy (ΔG) can be expressed as ΔG = ΔH − TΔS, where ΔH represents enthalpy change, and ΔS represents entropy change. When Co3C decomposes into Co and C, an increase in internal disorder is observed within the system, leading to a positive ΔS (>0). Consequently, as the ambient temperature rises to a critical threshold (∼1400 °C), ΔG becomes negative, indicating the spontaneous decomposition of the metastable phase, Co3C. At higher temperatures (∼1400 °C), Co3C experiences decomposition, with Co–C bond cleavage leading to release of a substantial amount of free C atoms. Due to the elevated temperature and pressure conditions, during the decomposition of Co3C, Co atoms with broken covalent bonds re-establish metallic bonds with other Co atoms, leading to a transformation of the lattice from Pnma to Fmm.
Fig. 2b presents the P–T conditions for all experiments in this study, illustrating the delineation of boundaries for the formation of spherical Co–C alloys products, the decomposition of Co3C, and its complete decomposition. It is important to note that the experimental P–T conditions of this study do not fall within the P–T region where graphite transforms into diamond due to catalysis. Instead, they are situated within the stable graphite phase region.
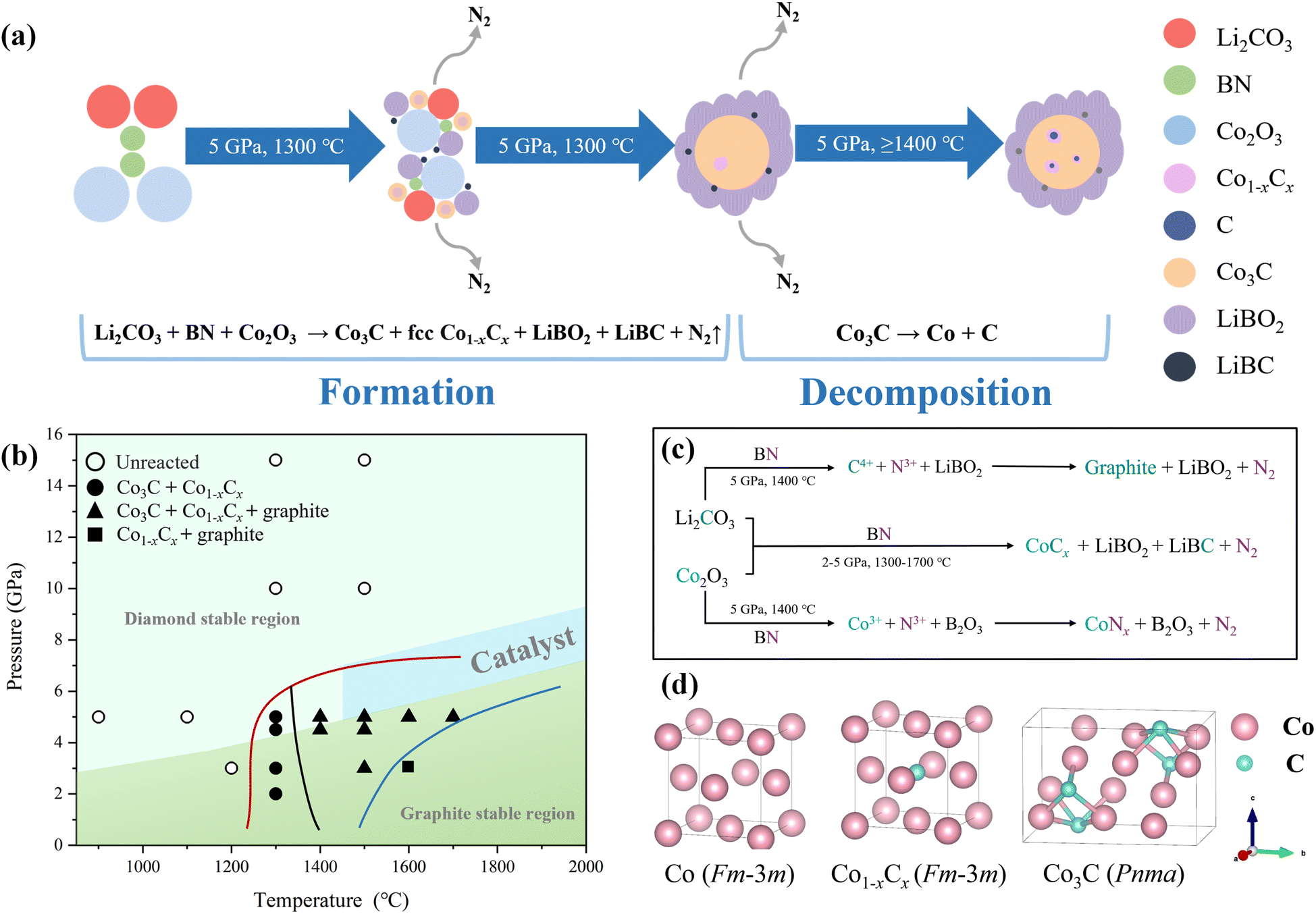 | ||
| Fig. 2 (a) Schematic representation of spherical bulk Co–C alloys formation and decomposition. (b) Plot presenting the experimental conditions employed in this work in the P–T phase diagram of carbon. The regions of different colors, both above and below, correspond to the phase stability domains of diamond and graphite, respectively. The light blue region signifies the P–T range in which graphite transforms into diamond under catalytic conditions, based on data from ref. 64. Black circles denote P–T conditions where no reaction occurred during the experiment. Black points signify the initial formation of Co3C and Co1−xCx at specific P–T conditions. The black rectangle represents the P–T conditions when Co3C completely decomposes. The red line marks the synthesis boundary of spherical Co–C alloys blocks, the black line indicates the decomposition boundary of Co3C, and the blue line signifies the boundary of complete Co3C decomposition. (c) Flowchart of the novel HSM reaction orientation. (d) Crystal structure of fcc Co, Co3C, and fcc Co1−xCx solid solution. Thin solid lines indicate the boundaries of the unit cell. | ||
The novel HSM reaction mechanism could be regarded as a two stage process as shown in Fig. 2c. BN exhibits reducibility, enabling rapid reactions with oxides under high temperature and pressure conditions. In the first-stage of the reaction, B undergoes in distinct ion exchange behavior with both Co and C and at the same time the formation of the LiBO2 and LiBC; the second-stage of the reaction is the combination of Co and C followed by the formation of the Co–C alloys (Co3C and fcc Co1−xCx).
| Li2CO3 + BN + Co2O3 → [C4+ + Co3+] → Co3C + Co1−xCx | (1) |
| Co3C → Co1−xCx + graphite | (2) |
At a synthesis temperature of 1300 °C, then the Co–C alloys could form, comprising a mixture of Co3C and fcc Co1−xCx solid solution, as depicted in eqn (1). When the synthesis temperature exceeds 1400 °C, decomposition of Co3C occurs, resulting in the formation of fcc Co1−xCx solid solution and graphite, as illustrated in eqn (2).
After the experiment, we observed significant voids in the by-products within the sample chamber. XRD analysis of the powder products reveals the presence of LiBO2 and LiBC, with no evidence of nitride products. Drawing on traditional HSM reactions, we infer the generation of nitrogen gas. LiBC could potentially be a product of the ion exchange process. The reaction by-products have also been reported elsewhere.32,33
We have depicted the formation and decomposition processes of bulk Co3C through the novel HSM reaction in a schematic diagram (Fig. 2a). Under conditions of 5 GPa and temperatures ranging from 1300 to 1700 °C, precursor materials Li2CO3, Co2O3, and BN promptly undergo a reaction, resulting in the formation of some smaller spherical bulk Co–C alloys composed of metastable Co3C (space group Pnma) and fcc Co1−xCx solid solution. Fig. 2d illustrates the atomic lattice of fcc Co, Pnma Co3C and fcc Co1−xCx (solid solution). Due to the good solubility (above 7 at%) of C in Co,6 C atoms preferentially occupy octahedral interstitial positions within the fcc Co lattice, displaying a higher electron affinity for metals. In contrast, N atoms face challenges in diffusing within the Co lattice, hindering the formation of metal nitrides. Under high temperature and pressure conditions, a saturated fcc Co1−xCx solid solution transforms into an Pnma Co3C. Obtaining pure metastable Co3C is likely to require even higher pressures. This reaction is accompanied by the generation of N2. Under these experimental conditions (5 GPa, 1300–1700 °C), the metal borate (LiBO2) is in a molten state, facilitating gradual aggregation of these spherical products into larger alloys spheres within this molten environment.
In the process of Co3C decomposition, the released C atoms infiltrate into the fcc Co lattice, forming a saturated fcc Co1−xCx solid solution at 5 GPa. Meanwhile, surplus C atoms precipitate out of the fcc Co lattice. At this juncture, the system operates under P–T conditions within the graphite stability region, with excess carbon atoms precipitating in the form of P63/mmc graphite. No transformation into diamond was observed. Variations in the synthesis temperature were also found to impact the carbon content within the samples,31 with higher temperatures accelerating carbon diffusion within cobalt, leading to an increased carbon atom content in samples synthesized at elevated temperatures. Consequently, in the precipitation process, the emergence of P63/mmc graphite becomes more evident in samples synthesized at higher temperatures.
The Vickers hardness of the Co–C alloys synthesized at 5 GPa and 1300 °C and hcp Co was measured with a standard square-pyramidal diamond indenter. At least four indentations were made on each sample. The curves obtained through fitting Vickers hardness measurements under varying loads, are illustrated in the Fig. 3a. The Vickers hardness of the Co–C alloy is approximately 6.1 GPa, representing a 19.6% increase in hardness compared to hcp Co (5.1 GPa). The enhanced hardness in Co–C alloys may stem from two factors: (1) Co3C in Co–C alloys exhibits superior hardness compared to hcp Co,65 and (2) interstitial C stabilizes the fcc Co lattice, resulting in a more denser crystal structure.
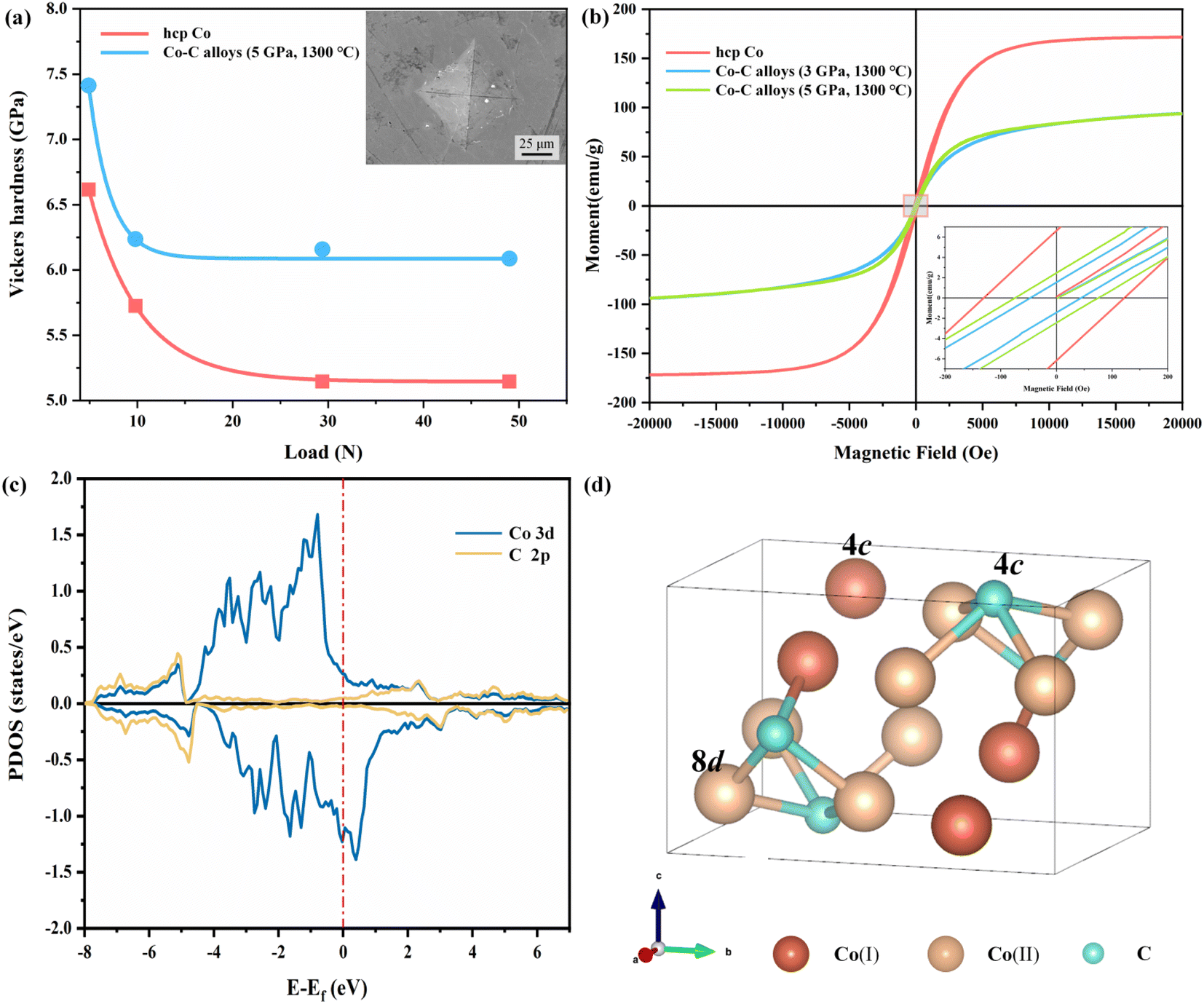 | ||
| Fig. 3 (a) Vickers hardness of sample the Co–C alloys synthesized at 5 GPa and 1300 °C and hcp Co at various applied load and corresponding images of indentations. (b) Hysteresis loops of the Co–C alloys synthesized 5 GPa and 1300 °C, as well as 3 GPa and 1300 °C obtained at room temperature; the inset is the enlarged area. (c) PDOS of Co3C. Co 3d and C 2p states are shown in blue and yellow solid lines, respectively. Vertical red dashed line represents the Fermi level. (d) Crystal structure of Pnma Co3C. Co(I) and Co(II) are two types of atoms. | ||
Fig. 3b shows magnetic hysteresis loops of Co–C alloys synthesized at 3 GPa, 1300 °C, and 5 GPa, 1300 °C, alongside those of hcp Co at room temperature. The Ms of fcc Co is close to that of hcp Co, but the Hc and saturation field of fcc Co are significantly lower than those of hcp Co.66 The Ms for the Co–C alloys synthesized at 3 GPa, 1300 °C, and 5 GPa, 1300 °C are 93.82 and 93.71 emu g−1, respectively. The measured hysteresis loops demonstrate that the magnetization of Co–C alloys increases more slowly than that of hcp Co. The partial density of state (PDOS) of Co3C are presented in Fig. 3c in which the hybridizations between C 2p states and the d orbitals of the Co atoms are clearly observed. There are two nonequivalent Co sites in Co3C, with a small difference in their local magnetic moments. It has been reported that changes in the magnetic moment are associated with narrowing or broadening of the PDOS width.67 Compared to fcc Co and hcp Co,67 the broadening of PDOS width in Co3C leads to weaker spin polarization, resulting in a smaller magnetic moment.
From our DFT calculations, the magnetic moments of Co atoms in Co3C, fcc Co, and hcp Co are presented in Table 2. The magnetic moments of Co atoms in fcc Co and hcp Co are comparable, resulting in similar Ms values for both. However, the magnetic moments of the two types of Co atoms (Fig. 3d) in Co3C are only 0.995 and 1.097 μB. Bader charge analysis give the results that the charge states of Co(I), Co(2), and C are +0.29, +0.33, and −0.95, respectively. Due to significant electron transfer from Co's valence electrons towards C atoms in the Co–C system, the magnetic moment of Co atoms decreases in Co3C. The decrease in the magnetic moment of Co atoms in Co3C leads to a reduction in the Ms of the Co–C alloys.
| Site | Pnma Co3C | fcc Co | hcp Co |
|---|---|---|---|
| Co(I) | 0.995 | 1.681 | 1.721 |
| Co(II) | 1.097 | — | — |
Table 3 presents the saturated Ms, and Hc for various Co–C alloys and hcp Co. The Co–C alloys exhibit higher coercivity (45.66 and 74.80 Oe), and Hc increase observed as the synthesis pressure rises. High pressure contributes to the stabilization of Co3C, which has a lower crystal structure symmetry, and increase the internal stress within the bulk material. The internal stress will increase the energy barrier of the domain wall and cause the change of the magnetic crystal anisotropy field, which will affect the Hc.68 Notably, since the decomposition temperature of Co3C is not reached, there is no significant change observed in the Ms. The Hc of Co3C exhibits a strong dependence on particle size.69 The products synthesized through HSM reaction possess larger dimensions and exhibit lower surface anisotropy, resulting in a smaller increase in Hc. Comparatively, the magnetic properties of fcc Co differ from those of Co–C alloys, where the presence of Co3C significantly influences the Hc in the Co–C alloys system. The imposition of higher pressures serves to stabilize the Co3C phase and inhibit its premature decomposition. However, elevated temperatures above 1400 °C cause the decomposition of Co3C, which can lead to a decrease in Hc and a concomitant increase in Ms. Thus, under different P–T synthesis conditions, the magnetic characteristics of the samples vary as a function of the degree of Co3C decomposition.
Conclusions
In this study, spherical bulk Co–C alloys (diameter 2–4 mm) were successfully synthesized using BN, Li2CO3, and Co2O3 as precursor materials through a novel HSM reaction at 2–5 GPa and 1300–1700 °C. The formation and decomposition behaviour of Co3C was investigated at different synthesis P–T. The Vickers hardness of the Co–C alloys is approximately 6.1 GPa, representing a 19.6% increase in hardness compared to hcp Co. The Co–C alloys exhibited a soft magnetic behavior with Ms up to 93.71 emu g−1 and Hc of 74.8 Oe, and Hc increased as the synthesis pressure rises. The DFT calculations results indicate that, compared to hcp Co and fcc Co, the magnetic moment of Co atoms in Co3C decreases with the incorporation of C atoms, leading to a reduction in saturation magnetization. The relatively superior combination of hardness and soft magnetic attributes in bulk Co–C alloys renders them as a promising candidate for electromagnetic applications in environments characterized by high pressure or other demanding conditions.Author contributions
Xu Jia: writing – original draft, investigation. Leilei Zhang: formal analysis, writing – review & editing. Yi Tian: investigation. Binbin Wu: investigation. Yu Tao: investigation. Duanwei He: resources, supervision. Baocheng Yang: software, resources. Filippo S. Boi: writing – review & editing. Li Lei: writing – review & editing, supervision, resources, conceptualization, methodology, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The acknowledgements come at the end of an article after the conclusions and before the notes and references. We appreciate the support of the National Natural Science Foundation of China (NSFC) (Grant No. U2030107) and the Fundamental Research Funds for the Central Universities (Grant No. 2020SCUNL107). The authors thank the High Performance Computing Center of Huanghe Science and Technology College for the computational time provided.References
- W. Yang, S. Rehman, X. Chu, Y. Hou and S. Gao, Transition Metal (Fe, Co and Ni) Carbide and Nitride Nanomaterials: Structure, Chemical Synthesis and Applications, ChemNanoMat, 2015, 1, 376–398 CrossRef CAS.
- H. I. Faraoun, Y. D. Zhang, C. Esling and H. Aourag, Crystalline, electronic, and magnetic structures of θ-Fe3C, χ-Fe5C2, and η-Fe2C from first principle calculation, J. Appl. Phys., 2006, 99, 093508 CrossRef.
- T. Tanaka, K. N. Ishihara and P. H. Shingu, Formation of metastable phases of Ni-C and Co-C systems by mechanical alloying, Metall. Trans. A, 1992, 23, 2431–2435 CrossRef.
- T. Fedotenko, S. Khandarkhaeva, L. Dubrovinsky, K. Glazyrin, P. Sedmak and N. Dubrovinskaia, Synthesis and Compressibility of Novel Nickel Carbide at Pressures of Earth's Outer Core, Minerals, 2021, 11, 516 CrossRef CAS.
- K. J. Carroll, Z. J. Huba, S. R. Spurgeon, M. Qian, S. N. Khanna, D. M. Hudgins, M. L. Taheri and E. E. Carpenter, Magnetic properties of Co2C and Co3C nanoparticles and their assemblies, Appl. Phys. Lett., 2012, 101, 012409 CrossRef.
- V. K. Portnoi and A. V. Leonov, Mechanochemical synthesis of Co-C materials, Inorg. Mater., 2012, 48, 593–600 CrossRef CAS.
- B. X. Liu, J. Wang and Z. Z. Fang, Hexagonal cobalt carbide formed by carbon ion implantation, J. Appl. Phys., 1991, 69, 7342–7344 CrossRef CAS.
- Z. Q. Lv, F. C. Zhang, S. H. Sun, Z. H. Wang, P. Jiang, W. H. Zhang and W. T. Fu, First-principles study on the mechanical, electronic and magnetic properties of Fe3C, Comput. Mater. Sci., 2008, 44, 690–694 CrossRef CAS.
- E. Duman, M. Acet, E. F. Wassermann, J. P. Itié, F. Baudelet, O. Mathon and S. Pascarelli, Magnetic Instabilities in Fe3C Cementite Particles Observed with Fe K-Edge X-Ray Circular Dichroism under Pressure, Phys. Rev. Lett., 2005, 94, 075502 CrossRef CAS.
- A. A. Al-Joubori and C. Suryanarayana, Synthesis and thermal stability of homogeneous nanostructured Fe3C (cementite), J. Mater. Sci., 2018, 53, 7877–7890 CrossRef CAS.
- A. Mussi, P. Cordier, S. Ghosh, N. Garvik, B. C. Nzogang, P. Carrez and S. Garruchet, Transmission electron microscopy of dislocations in cementite deformed at high pressure and high temperature, Philos. Mag., 2016, 96, 1773–1789 CrossRef CAS.
- R. S. Iskhakov, S. V. Stolyar, L. A. Chekanova, E. M. Artem'ev and V. S. Zhigalov, High-pressure phases in nanocrystalline Co(C) films obtained by pulsed plasma vaporization, J. Exp. Theor. Phys. Lett., 2000, 72, 316–319 CrossRef CAS.
- I. R. Shein, N. I. Medvedeva and A. L. Ivanovskii, Electronic and structural properties of cementite-type M3X (M=Fe, Co, Ni; X=C or B) by first principles calculations, Phys. B, 2006, 371, 126–132 CrossRef CAS.
- M. Zamanpour, S. Bennett, P. Taheri, Y. Chen and V. G. Harris, Magnetic properties and scale-up of nanostructured cobalt carbide permanent magnetic powders, J. Appl. Phys., 2014, 115, 17A747 CrossRef.
- V. G. Harris, Y. Chen, A. Yang, S. Yoon, Z. Chen, A. L. Geiler, J. Gao, C. N. Chinnasamy, L. H. Lewis, C. Vittoria, E. E. Carpenter, K. J. Carroll, R. Goswami, M. A. Willard, L. Kurihara, M. Gjoka and O. Kalogirou, High coercivity cobalt carbide nanoparticles processed via polyol reaction: a new permanent magnet material, J. Phys. D: Appl. Phys., 2010, 43, 165003 CrossRef.
- A. V. Syugaev, N. V. Lyalina, S. F. Lomayeva and A. N. Maratkanova, Electrochemical behavior of Co3C carbide, J. Solid State Electrochem., 2015, 19, 2933–2941 CrossRef CAS.
- R. M. Irfan, M. H. Tahir, S. Iqbal, M. Nadeem, T. Bashir, M. Maqsood, J. Zhao and L. Gao, Co3C as a promising cocatalyst for superior photocatalytic H2 production based on swift electron transfer processes, J. Mater. Chem. C, 2021, 9, 3145–3154 RSC.
- P. Gao, Y. Wang, S. Yang, Y. Chen, Z. Xue, L. Wang, G. Li and Y. Sun, Mechanical alloying preparation of fullerene-like Co3C nanoparticles with high hydrogen storage ability, Int. J. Hydrogen Energy, 2012, 37, 17126–17130 CrossRef CAS.
- H. Yoon, A. Xu, G. E. Sterbinsky, D. A. Arena, Z. Wang, P. W. Stephens, Y. S. Meng and K. J. Carroll, In situ non-aqueous nucleation and growth of next generation rare-earth-free permanent magnets, Phys. Chem. Chem. Phys., 2015, 17, 1070–1076 RSC.
- A. A. El-Gendy, M. Qian, Z. J. Huba, S. N. Khanna and E. E. Carpenter, Enhanced magnetic anisotropy in cobalt-carbide nanoparticles, Appl. Phys. Lett., 2014, 104, 023111 CrossRef.
- M. Sarr, N. Bahlawane, D. Arl, M. Dossot, E. McRae and D. Lenoble, Atomic layer deposition of cobalt carbide films and their magnetic properties using propanol as a reducing agent, Appl. Surf. Sci., 2016, 379, 523–529 CrossRef CAS.
- Z. Turgut, M. S. Lucas, S. Leontsev, S. L. Semiatin and J. Horwath, Metastable Co3C nanocrystalline powder produced via reactive ball milling: synthesis and magnetic properties, J. Alloys Compd., 2016, 676, 187–192 CrossRef CAS.
- K. Kawashima, K. Shin, B. R. Wygant, J.-H. Kim, C. L. Cao, J. Lin, Y. J. Son, Y. Liu, G. Henkelman and C. B. Mullins, Cobalt Metal–Cobalt Carbide Composite Microspheres for Water Reduction Electrocatalysis, ACS Appl. Energy Mater., 2020, 3, 3909–3918 CrossRef CAS.
- J.-H. Kim, K. Kawashima, B. R. Wygant, O. Mabayoje, Y. Liu, J. H. Wang and C. B. Mullins, Transformation of a Cobalt Carbide (Co3C) Oxygen Evolution Precatalyst, ACS Appl. Energy Mater., 2018, 1, 5145–5150 CAS.
- O. I. Nakonechna, M. M. Dashevski, A. M. Kurylyuk and N. M. Bilyavyna, Mechanochemical synthesis of Co3C carbide with carbon nanotubes, Phys. Chem. Solid State, 2019, 20, 13–17 CrossRef CAS.
- Z. Wang, Z. Yusop, P. Ghosh, Y. Hayashi and M. Tanemura, Formation of carbon nanostructures containing single-crystalline cobalt carbides by ion irradiation method, Appl. Surf. Sci., 2011, 257, 3168–3173 CrossRef CAS.
- Z. Chen, L. Liu and Q. Chen, One-pot template-free synthesis of urchin-like Co2C/Co3C hybrid nanoparticles, Mater. Lett., 2016, 164, 554–557 CrossRef CAS.
- P. V. Marshall, Z. Alptekin, S. D. Thiel, D. Smith, Y. Meng and J. P. S. Walsh, High-Pressure Synthesis of Bulk Cobalt Cementite, Co3C, Chem. Mater., 2021, 33, 9601–9607 CrossRef CAS.
- S. Naka, A. Tsuzuki and S.-I. Ihirano, Diamond formation and behaviour of carbides in several 3d-transition metal-graphite systems, J. Mater. Sci., 1984, 19, 259–262 CrossRef CAS.
- W. Utsumi, T. Okada, T. Taniguchi, K. Funakoshi, T. Kikegawa, N. Hamaya and O. Shimomura, In situ x-ray diffraction of graphite–diamond transformation using various catalysts under high pressures and high temperatures, J. Phys.: Condens.Matter, 2004, 16, S1017–S1026 CrossRef CAS.
- Y. Tian, J. Wang, J. Zhang, S. Guan, L. Zhang, B. Wu, Y. Su, M. Huang, L. Zhou and D. He, Solubility and stability of diamond in cobalt under 5 GPa, Diamond Relat. Mater., 2020, 110, 108158 CrossRef CAS.
- L. Lei and L. Zhang, Recent advance in high-pressure solid-state metathesis reactions, Matter Radiat. Extremes, 2018, 3, 95–103 CrossRef.
- L. Lei, W. Yin, X. Jiang, S. Lin and D. He, Synthetic Route to Metal Nitrides: High-Pressure Solid-State Metathesis Reaction, Inorg. Chem., 2013, 52, 13356–13362 CrossRef CAS.
- L. Lei and D. He, Synthesis of GaN Crystals Through Solid-State Metathesis Reaction Under High Pressure, Cryst. Growth Des., 2009, 9, 1264–1266 CrossRef CAS.
- W. Yin, L. Lei, X. Jiang, P. Liu, F. Liu, Y. Li, F. Peng and D. He, High pressure synthesis and properties studies on spherical bulk ε-Fe3N, High Pres. Res., 2014, 34, 317–326 CrossRef CAS.
- L. Qi, L. Lei, Q. Hu, L. Zhang, L. Feng, M. Pu, H. Ohfuji and T. Irifune, Strengthening effects of interstitial nitrogen on rhenium, J. Appl. Phys., 2018, 123, 055901 CrossRef.
- H. Zhang, B. Wu, J. Liu, Z. Liu, F. S. Boi, D. He, T. Irifune and L. Lei, High-Pressure Coupling Reactions to Produce a Spherical Bulk RexN/Fe3N Composite, Inorg. Chem., 2023, 62, 6263–6273 CrossRef CAS.
- X. Jiang, L. Lei, Q. Hu, Z. C. Feng and D. He, High-pressure Raman spectroscopy of Re3N crystals, Solid State Commun., 2015, 201, 107–110 CrossRef CAS.
- L. Lei, L. Zhang, S. Gao, Q. Hu, L. Fang, X. Chen, Y. Xia, X. Wang, H. Ohfuji, Y. Kojima, S. A. T. Redfern, Z. Zeng, B. Chen, D. He and T. Irifune, Neutron diffraction study of the structural and magnetic properties of ε-Fe3N1.098 and ε-Fe2.322Co0.678N0.888, J. Alloys Compd., 2018, 752, 99–105 CrossRef CAS.
- B. Wu, F. Zhang, Q. Hu, Q. Tang, S. Liu, X. Xiang, Y. Xia, L. Fang, H. Ohfuji, T. Irifune and L. Lei, The effect of interstitial-site nitrogen on structural, elastic, and magnetic properties of face-center cubic Co, J. Appl. Phys., 2021, 129, 105901 CrossRef CAS.
- J. Akella and G. C. Kennedy, Melting of gold, silver, and copper—proposal for a new high-pressure calibration scale, J. Geophys. Res., 1971, 76, 4969–4977 CrossRef CAS.
- J. Wang, D. He, X. Li, J. Zhang, Q. Li, Z. Wang, Y. Su, Y. Tian, J. Yang and B. Peng, The melting curve of cobalt under high pressure, Solid State Commun., 2020, 307, 113805 CrossRef CAS.
- L. Lei, D. He, K. He, J. Qin and S. Wang, Pressure-induced coordination changes in LiBO2, J. Solid State Chem., 2009, 182, 3041–3048 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- H. Luo, Q. Gao, H. Liu, Y. Gu, D. Wu, C. Yi, J. Jia, S. Wu, X. Luo, Y. Xu, L. Zhao, Q. Wang, H. Mao, G. Liu, Z. Zhu, Y. Shi, K. Jiang, J. Hu, Z. Xu and X. J. Zhou, Electronic nature of charge density wave and electron-phonon coupling in kagome superconductor KV3Sb5, Nat. Commun., 2022, 13, 273 CrossRef CAS.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- L. Zhang, Q. Wu, S. Li, Y. Sun, X. Yan, Y. Chen and H. Y. Geng, Interplay of Anionic Quasi-Atoms and Interstitial Point Defects in Electrides: Abnormal Interstice Occupation and Colossal Charge State of Point Defects in Dense fcc-Lithium, ACS Appl. Mater. Interfaces, 2021, 13, 6130–6139 CrossRef CAS.
- L. Zhang, H. Y. Geng and Q. Wu, Prediction of anomalous LA-TA splitting in electrides, Matter Radiat. Extremes, 2021, 6, 038403 CrossRef CAS.
- J.-A. Hernandez and R. Caracas, Superionic-Superionic Phase Transitions in Body-Centered Cubic H2O Ice, Phys. Rev. Lett., 2016, 117, 135503 CrossRef PubMed.
- J. Yin, J. Jin, Z. Yin, L. Zhu, X. Du, Y. Peng, P. Xi, C.-H. Yan and S. Sun, The built-in electric field across FeN/Fe3N interface for efficient electrochemical reduction of CO2 to CO, Nat. Commun., 2023, 14, 1724 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011, 134, 064111 CrossRef.
- X. Ma, K. Li, X. Zhang, B. Wei, H. Yang, L. Liu, M. Zhang, X. Zhang and Y. Chen, The surface engineering of cobalt carbide spheres through N, B co-doping achieved by room-temperature in situ anchoring effects for active and durable multifunctional electrocatalysts, J. Mater. Chem. A, 2019, 7, 14904–14915 RSC.
- J. Gautam, T. D. Thanh, K. Maiti, N. H. Kim and J. H. Lee, Highly efficient electrocatalyst of N-doped graphene-encapsulated cobalt-iron carbides towards oxygen reduction reaction, Carbon, 2018, 137, 358–367 CrossRef CAS.
- W. Zhang, Y. Zou, X. Mei, Y. Li, S. Peng and J. Xu, Facile synthesis of Co2(OH) 3Cl/cobalt carbide/reduced graphene oxide composites for enhanced dye-sensitized photocatalytic H2 evolution, Sustain. Energy Fuels, 2020, 4, 6181–6187 RSC.
- J.-H. Kim, K. Kawashima, B. R. Wygant, O. Mabayoje, Y. Liu, J. H. Wang and C. B. Mullins, Transformation of a Cobalt Carbide (Co3C) Oxygen Evolution Precatalyst, ACS Appl. Energy Mater., 2018, 1, 5145–5150 CAS.
- S. Nagakura, Study of Metallic Carbides by Electron Diffraction Part IV. Cobalt Carbides, J. Phys. Soc. Jpn., 1961, 16, 1213–1219 CrossRef CAS.
- L. D. Barriga-Arceo, E. Orozco, V. Garibay-Febles, L. Bucio-Galindo, H. M. León, P. Castillo-Ocampo and A. Montoya, Nanofibre growth from cobalt carbide produced by mechanosynthesis, J. Phys.: Condens.Matter, 2004, 16, S2273–S2277 CrossRef CAS.
- V. G. Harris, Y. Chen, A. Yang, S. Yoon, Z. Chen, A. L. Geiler, J. Gao, C. N. Chinnasamy, L. H. Lewis, C. Vittoria, E. E. Carpenter, K. J. Carroll, R. Goswami, M. A. Willard, L. Kurihara, M. Gjoka and O. Kalogirou, High coercivity cobalt carbide nanoparticles processed via polyol reaction: a new permanent magnet material, J. Phys. D: Appl. Phys., 2010, 43, 165003 CrossRef.
- R. S. Iskhakov, S. V. Stolyar, L. A. Chekanova, E. M. Artem'ev and V. S. Zhigalov, High-pressure phases in nanocrystalline Co(C) films obtained by pulsed plasma vaporization, J. Exp. Theor. Phys. Lett., 2000, 72, 316–319 CrossRef CAS.
- J. Guignard, M. Prakasam and A. Largeteau, A Review of Binderless Polycrystalline Diamonds: Focus on the High-Pressure–High-Temperature Sintering Process, Materials, 2022, 15, 2198 CrossRef CAS.
- Y. Fukumiya, Y. Haga and O. Nittono, Thermal stability and hardness of metastable Co–C composite alloy films, Mater. Sci. Eng., A, 2001, 312, 248–252 CrossRef.
- M. El-Tahawy, L. Péter, L. F. Kiss, J. Gubicza, Zs. Czigány, G. Molnár and I. Bakonyi, Anisotropic magnetoresistance (AMR) of cobalt: hcp-Co vs. fcc-Co, J. Magn. Magn. Mater., 2022, 560, 169660 CrossRef CAS.
- M. Hakamada, F. Hirashima, K. Kajikawa and M. Mabuchi, Magnetism of fcc/fcc, hcp/hcp twin and fcc/hcp twin-like boundaries in cobalt, Appl. Phys. A, 2012, 106, 237–244 CrossRef CAS.
- Y. Zhang, G. S. Chaubey, C. Rong, Y. Ding, N. Poudyal, P. Tsai, Q. Zhang and J. P. Liu, Controlled synthesis and magnetic properties of hard magnetic CoxC (x=2, 3) nanocrystals, J. Magn. Magn. Mater., 2011, 323, 1495–1500 CrossRef CAS.
- Z. Turgut, M. S. Lucas, S. Leontsev, S. L. Semiatin and J. Horwath, Metastable Co3C nanocrystalline powder produced via reactive ball milling: synthesis and magnetic properties, J. Alloys Compd., 2016, 676, 187–192 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |