
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective extraction and determination of 24 quinolones in water and egg samples using a novel magnetic covalent organic framework combined with UPLC-MS/MS†
Na Lia,
Mengnan Lianga,
Hao Zhanga,
Zhongxia Huabc,
Ling Ma*bc,
Yanyu Qi *a and
Ke Wang*abc
*a and
Ke Wang*abc
aCollege of Chemistry and Materials Science, Hebei Normal University, Shijiazhuang 050023, China. E-mail: wkecdc@163.com; hbsdqyy@hebtu.edu.cn
bShijiazhuang Center for Disease Control and Prevention, Shijiazhuang 050011, China. E-mail: mamalin001@163.com
cShijiazhuang Technology Innovation Center for Chemical Poison Detection and Risk Early Warning, Shijiazhuang 050011, China
First published on 14th March 2024
Abstract
The excessive use of quinolones (QNs) has seriously threatened human health. In this study, a novel functionalized magnetic covalent organic framework Fe3O4@SiO2@Ah-COF was fabricated with biphenyl-3,3′,5,5′-tetracarbaldehyde and hydrazine hydrate (85%) as monomers and was used as a magnetic solid-phase extraction (MSPE) absorbent for the determination of 24 QNs in water and egg samples through ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The extraction parameters of MSPE were optimized, including pH, adsorbent dosage, adsorption time, and eluent type. An effective and rapid detection method was then established, which showed good linearity (R2 ≥ 0.9990), low limits of detection (0.003–0.036 μg L−1) and low limits of quantitation (0.008–0.110 μg L−1) for QNs. The good recoveries of 24 QNs in water and egg samples were in the range of 70.3–106.1% and 70.4–119.7%, respectively, with relative standard deviations lower than 10% (n = 5). As a result, Fe3O4@SiO2@Ah-COF is a promising magnetic adsorbent, and the established method was successfully applied for the determination of 24 QNs in water and egg samples.
1. Introduction
Quinolones (QNs) are an important and successful group of synthetic antibiotics with a 4-quinolone structure, which have been developed to the fourth generation.1 The fourth generation of quinolones have better medicinal properties and stronger antibacterial ability; thus, they are widely used in human health care and animal disease treatment.2 Their mechanism of action is that the presence of an F atom and piperazinyl group prevents the synthesis of bacterial DNA.3 On the one hand, as the consumption of QNs increases, their residues in foods of animal origin can have a variety of toxic side effects on the consumers through the migration of the food chain, seriously threatening human health.4 On the other hand, due to the low solubility, bioavailability and biodegradation rates,5 QNs are difficult to be fully absorbed by humans and animals. Most of them are discharged into the environment in their original form or as active metabolites through feces and urine, resulting in water and soil pollution. In recent years, some reports have indicated that QNs exist at high levels in aquatic environments, interfering with microbial driven nitrogen cycling processes and becoming an important factor affecting water eutrophication and greenhouse gas emissions.6 Given the significant residual hazards of QNs, many countries and organizations (including the United States, the European Union and China) have set corresponding maximum residue limits (MRLs) for QNs. However, MRLs for QNs in drinking water have not been specified in GB 5749-2022, and a unified standard detection method for QNs in water quality has not yet been established. Therefore, it is of great significance to develop sensitive, reliable and high-throughput analytical methods to detect the concentration of QNs in food and water.To date, a variety of detection methods have been applied for the analysis of QNs, such as immunoassays,7 fluorescence-based methods,8 capillary electrophoresis (CE),9 high performance liquid chromatography (HPLC),10 and liquid chromatography-tandem mass spectrometry (LC-MS/MS).11,12 Among them, ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS)12 has been gradually developed owing to its advantages of high sensitivity, specificity and simultaneous qualitative and quantitative analysis.
Nevertheless, the complicated matrix and extremely low concentration of QNs in food and water samples can influence detection results; therefore, an effective sample pretreatment is indispensable. Common sample pretreatment methods include liquid–liquid extraction(LLE),13 dispersive liquid–liquid micro-extraction (DLLME),14 solid phase extraction (SPE),15–17 dispersive solid-phase extraction (DSPE)18,19 and magnetic solid phase extraction (MSPE).20–22 Different from conventional sample pretreatment methods, MSPE is a dispersed solid-phase extraction technique that uses magnetic materials as adsorbents to achieve the separation and enrichment of analytes by an external magnetic field. It avoids the disadvantages of cumbersome operation and high consumption of organic solvents and has been applied in the pretreatment of environmental, biological and food samples.23,24
The absorbent is clearly the most important influential factor on MSPE. Commonly used adsorbents include nanotubes, graphene,25,26 molecularly imprinted polymers,27 and metal–organic frameworks.28 Although the above adsorbents have their characteristics, the defects of large amounts of materials, relatively lower adsorption capacity and instability limited their extensive applications as MSPE adsorbents. Relatively speaking, covalent organic frameworks (COFs) are a new type of porous polymer, which have a unique topological structure and functionalized modification characteristics. COFs have high-affinity interactions with the target compounds, high specific surface area and pore volume, and have been used in adsorption, separation, catalysis, sensing and other fields.29–32 Notably, COFs have shown great potential as adsorbents for the extraction and removal of various substances such as pharmaceutical contaminants,33 glycopeptides,34 and environmental contaminants.35 At present, according to the types of covalent bonds formed, COFs are mainly divided into boron-based COFs, imine-based COFs, and triazine-based COFs.36 Among them, the stability of boron-based COFs in water and moisture is poor. Furthermore, the preparation conditions of triazine-based COFs are harsh, the synthesis is difficult, and the crystallinity of the products is poor. Only imine-linked COFs have simple synthesis conditions, diverse synthesis methods and good chemical stability, and can be stably present in common organic solvents, water, and even acid and alkali solutions.37 However, it is difficult to precipitate COFs with low density from the complex matrix, and the adsorption efficiency is low.38 Fascinatingly, the combination of COFs and Fe3O4 perfectly makes up for this shortcoming well, making magnetic covalent organic frameworks (MCOFs) that not only have rich functional groups but also the merits of easy separation, improving the enrichment capacity and adsorption capacity of the target analyte.39,40
To the best of our knowledge, there are few reports on the application of MCOF materials in the field of adsorbing QNs. In the reports on the application of the MSPE method to determine QNs,23,41–44 there are few reports on methods that can simultaneously detect QNs in different matrices, and the number of QNs is no more than 14. Therefore, developing a unified method capable of simultaneously detecting large amounts of QNs residues in different matrices remains a challenge. Moreover, the reusability and narrow spectrum adsorption of magnetic materials remains to be explored and improved in further development.
Herein, we first designed and fabricated a novel MCOFs (Fe3O4@SiO2@Ah-COF) based on the properties of QNs via a simple reaction of biphenyl-3,3′,5,5′-tetracarbaldehyde and hydrazine hydrate (85%). Considering the biphenyl skeleton and unsaturated bonds of Fe3O4@SiO2@Ah-COF, and that QNs contain the carboxyl groups and amino group on the piperazine ring side chain,45 the great adsorption effect was obtained through the synergistic effects of p–π interaction, π–π interaction and hydrogen bonding. Then, Fe3O4@SiO2@Ah-COF was characterized and the optimal parameters affecting the extraction efficiency were investigated. Finally, the developed MSPE-UPLC-MS/MS method was applied for the determination of 24 QNs in water and egg samples.
2. Experimental
2.1 Reagents and materials
Ferric chloride hexahydrate (FeCl3·6H2O), ethylene glycol (EG), sodium acetate trihydrate (CH3COONa·3H2O), 1,4-dioxane, sodium chloride (NaCl) and anhydrous ethanol were purchased from Tianjin Yongda Chemical Reagent Co., Ltd (Tianjin, China). Biphenyl-3,3′,5,5′-tetracarbaldehyde was purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Hydrazine hydrate (85%) was purchased from National Pharmaceutical Group Chemical Reagent Co., Ltd (Shanghai, China). Tetraethoxysilane (TEOS) was obtained from Aladdin Chemistry Co., Ltd (Shanghai, China). Polyethylene glycol (PEG-4000) was supplied by Xiya Chemical Technology Co., Ltd (Shandong, China). 25% (w/w) ammonia solution and 37% (w/w) hydrochloric acid were acquired from Tianjin Kernel Chemical Reagent Co., Ltd (Tianjin, China). 99.9% (w/w) formic acid was acquired from Dikma (USA). Acetonitrile was obtained from Merck (Darmstadt, Germany), and methanol was purchased from Thermo Fisher Co., Ltd (USA). Methanol and acetonitrile were of HPLC grade, and other reagents were of analytical grade. Ultra-pure water was prepared using a Millipore Milli-Q Gradient Water Purification System (USA).Lomefloxacin (LOM) was purchased from Dr Ehrenstorfer, GmbH (Augsburg, Germany), and all other reagents were purchased from Tianjin Alta Technology Co., Ltd. The purity of Tosufloxacin (TOS) is 70.6%, and those of other standards are all above 95.0%. The properties of QNs are listed in Table S1.†
2.2 Instrumental and analytical conditions
The AB SCIEX Exion-TRIPLE QUAD 5500 ultra-high performance liquid chromatography-tandem mass spectrometry with electrospray ionization source (ESI) was used for the detection.A Waters Acquity UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 μm) was used for the separation of QNs at the constant column temperature of 40 °C. The mobile phases were composed of acetonitrile (mobile phase B) and 0.1% formic acid aqueous solution (mobile phase A). The gradient elution procedure was as follows: (1) 0–10 min, 13% B (v/v); (2) 10–10.1 min, 13–18% B (v/v); (3) 10.1–16 min, 18% B (v/v) (4) 16–16.1 min, 18–85% B (v/v); (5) 16.1–18 min, 85% B (v/v); (6) 18–18.1 min, 85–13% B (v/v); (7) 18.1–20 min, 13% B (v/v). The flow rate and injection volume were 0.2 mL min−1 and 3 μL, respectively. Chromatograms of 24 QNs are shown in Fig. S3.†
Electrospray ionization (ESI) in positive ion mode was used, and data were acquired in the multiple reaction monitoring (MRM) mode. The flow rate of the drying gas was 10 L min−1 at the temperature of 550 °C. The capillary voltage was 5500 V. The MRM parameters comprising the precursor ion, production ion, retention time, collision energy (CE), declustering potential (DP) of 24 QNs are optimized, and the detailed information is listed in Table S2.†
Fourier transform infrared spectroscopy (FT-IR) (Thermo Nicolet Co., Ltd, USA), Brunauer–Emmett–Teller (BET) (Quantachrome Instruments, USA), D8A X-ray diffractometer (XRD) (Bruker, Germany), SQUID XL-7 vibrating sample magnetometer (Quantum Design, USA), scanning electron microscopy (SEM) with a Regulus 8100 instrument (Hitachi, Japan), transmission electron microscopy (TEM) with a FEI Tecnai G2 F20 (FEI, USA) and energy-dispersive X-ray spectroscopy (EDS) (EDAX, USA) were used for the characterization of the prepared materials.
2.3 Synthesis of Fe3O4@SiO2@Ah-COF
The magnetic Fe3O4 nanoparticles were synthesized by the solvent thermal method.46 Typically, 1.62 g FeCl3·6H2O was dissolved in 60 mL of ethylene glycol (EG) under vigorous stirring at room temperature. Then, 7.2 g of CH3COONa·3H2O and 2.0 g of PEG-4000 were added to the solution with stirring for 30 min. Subsequently, the obtained homogeneous yellow solution was transferred into a Teflon-lined autoclave and heated to 200 °C for 8 h. The resultant nanoparticles were washed with ethanol three times, and then dried at 60 °C for 4 h in a vacuum oven.Fe3O4@SiO2 nanoparticles were synthesized by the sol–gel method.47,48 The Fe3O4 nanoparticles (200 mg) were dispersed in a mixed solution of 160 mL of anhydrous ethanol and 40 mL of ultrapure water. The mixture was sonicated for 30 min to ensure that it was evenly dispersed. After that, 3 mL of 25% (w/w) ammonia was added to the mixture and sonicated for 5 min. Finally, 2 mL of tetraethoxysilane (TEOS) was slowly added dropwise under magnetic stirring. The mixture was allowed to react for 24 h at 30 °C to get Fe3O4@SiO2, washed three times with anhydrous ethanol and ultrapure water, and dried under vacuum at 60 °C for 3 h.
Fe3O4@SiO2@Ah-COF was prepared based on the modified literature method.49 A total quantity of 150 mg of Fe3O4@SiO2 was added to a 100 mL round-bottom flask, along with 35 mL of 1,4-dioxane solvent. The mixture was ultrasonicated for about 10 min, and then placed into an oil bath agitator. Next, biphenyl-3,3′,5,5′-tetracarbaldehyde (80 mg, 0.30 mmol) was dissolved in 5 mL of dimethyl sulfoxide solution, and dispersed evenly by ultrasonication. Then, hydrazine hydrate (85%, 0.80 mmol) and pure glacial acetic acid solution (1.10 mL) were separately added. The mixture was stirred for 1 h at room temperature, and then heated at 70 °C for 24 h. After cooling to room temperature, the blue-gray solid was isolated with a magnet, and washed thoroughly with 1,4-dioxane solvent and anhydrous ethanol alternately three times to remove unreacted chemicals. The final product was dried under vacuum at 60 °C for 2.5 h to get Fe3O4@SiO2@Ah-COF.
2.4 Sample collection and pretreatment
Twenty water samples were collected from relevant rivers and filtered by 0.45 μm filter membrane. The pH of the water sample was adjusted to 4.0 with hydrochloric acid and stored in a refrigerator at 4 °C for the MSPE procedure.Fifteen egg samples were obtained from the market. Then, 0.50 g of the homogenized sample was transferred to a 50 mL polypropylene tube and diluted with 10 mL ultra-pure water. The mixture was then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. All supernatants were collected, and the pH of sample solution was adjusted to 4.0 by adding hydrochloric acid for MSPE.
000 rpm for 10 min. All supernatants were collected, and the pH of sample solution was adjusted to 4.0 by adding hydrochloric acid for MSPE.
2.5 Procedure of MSPE
The MSPE program is shown in Scheme 1. Firstly, 10 mL of the sample solution and 10 mg of Fe3O4@SiO2@Ah-COF were added to a 50 mL centrifuge tube. Sample adsorption was performed by using the vortex method for 20 min. After that, the magnetic material was collected under the action of the external magnetic field, and the supernatant was discarded. QNs were eluted from Fe3O4@SiO2@Ah-COF using 8 mL of 3% (v/v) acidified methanol under vibration for 8 min. The eluent was separated with a magnet and then dried at 40 °C under N2. Finally, the residue was redissolved using 1 mL of mixed solution of acetonitrile and 0.1% formic acid solution (2:8, v/v), filtered through a 0.20 μm membrane, and introduced into UPLC-MS/MS for analysis. In addition, the Fe3O4@SiO2@Ah-COF was washed three times with 3% acidified methanol and recycled after vacuum drying at 60 °C.
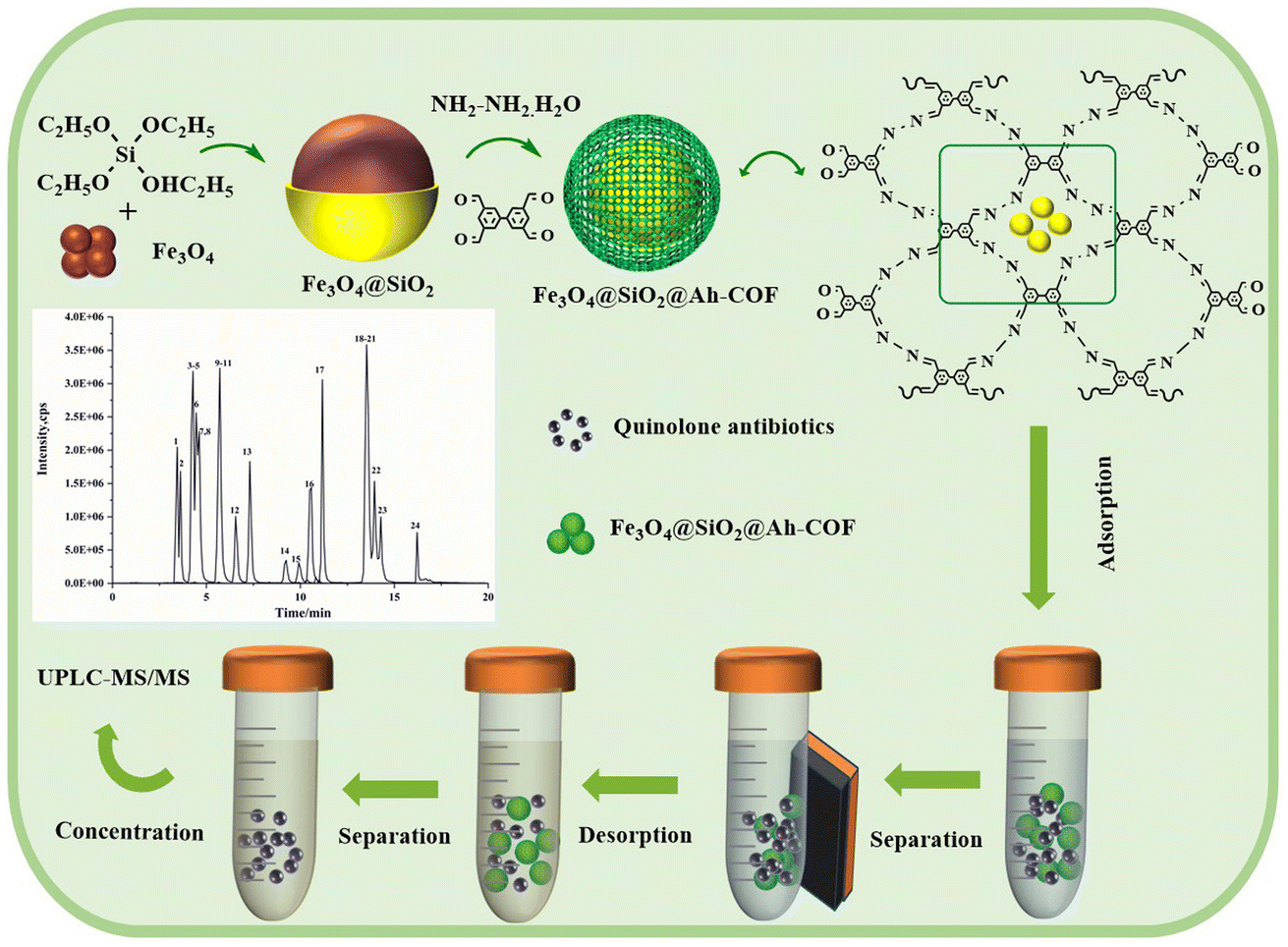 | ||
| Scheme 1 The synthetic route for the Fe3O4@SiO2@Ah-COF and the procedure of MSPE. | ||
3. Result and discussion
3.1 Characterization of the Fe3O4@SiO2@Ah-COF
The SEM and TEM images show the structural characteristics of the Fe3O4 nanoparticles and Fe3O4@SiO2@Ah-COF nanoparticles. As shown in Fig. 1A and B, it can be easily found that Fe3O4 had a smooth and well-dispersed surface, while Fe3O4@SiO2@Ah-COF had a rough surface. The typical Fe3O4 nanoparticle nuclei and the shell structure of the gray COF layer can be further clearly seen in Fig. 1C and D (TEM images). | ||
| Fig. 1 The SEM of (A) Fe3O4 and (B) Fe3O4@SiO2@Ah-COF; the TEM of (C) Fe3O4 and (D) Fe3O4@SiO2@Ah-COF. | ||
Fig. 2 shows the energy-dispersive X-ray spectrum (EDS) of Fe3O4@SiO2@Ah-COF. As shown in the figure, the five elements (C, N, O, Si, and Fe) are distributed on Fe3O4@SiO2@Ah-COF, with contents of 59.17 wt%, 6.86 wt%, 17.66 wt%, 2.97 wt% and 13.34 wt%, respectively. These obvious differences all indicated the successful synthesis of the COF layer on the surface of Fe3O4.
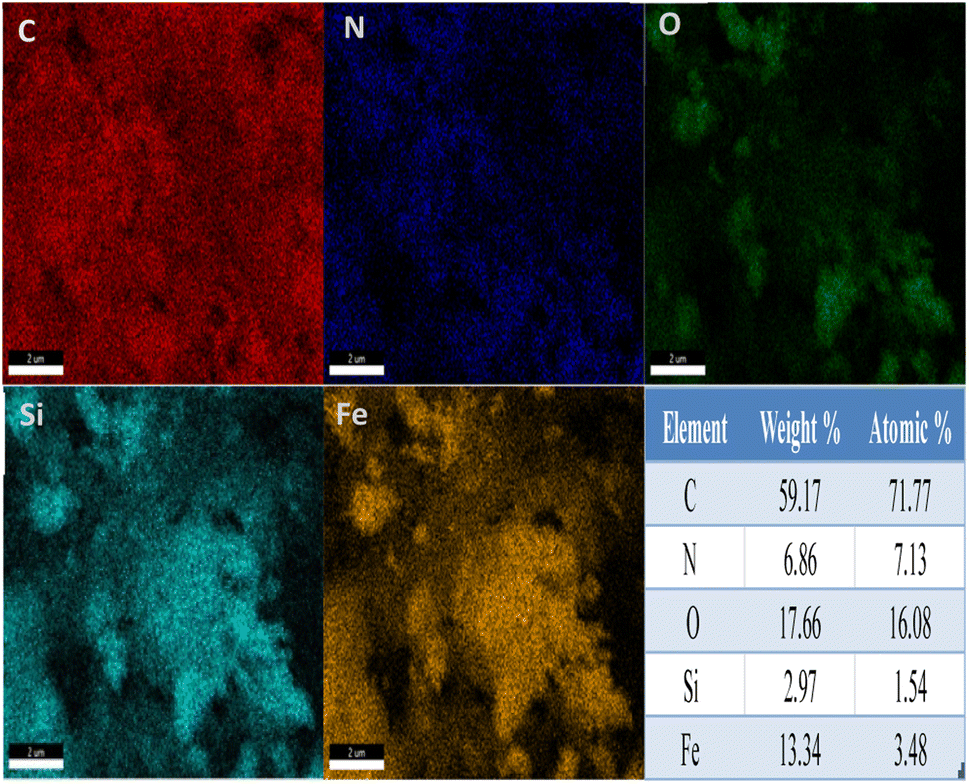 | ||
| Fig. 2 EDS elemental mapping of Fe3O4@SiO2@Ah-COF. | ||
To further prove the successful synthesis of Fe3O4@SiO2@Ah-COF, the crystal morphology and characteristic spectra of the Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@Ah-COF samples were determined through XRD and FT-IR.
As seen in Fig. 3A, the XRD diagram of Fe3O4 showed six characteristic diffraction peaks at 2θ = 30.1°, 35.48°, 43.15°, 53.44°, 57.01°, and 62.61°, which corresponded to the (220), (311), (400), (422), (511) and (440) planes, respectively, and were in good agreement with the standard magnetite XRD patterns (JCPDS No. 75-1610).50 The XRD results validated that the crystalline phase of Fe3O4 was not destroyed in all samples during the preparation of Fe3O4@SiO2 and Fe3O4@SiO2@Ah-COF.
 | ||
| Fig. 3 (A) XRD analysis of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@Ah-COF. (B) FT-IR spectra of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@Ah-COF. (C) VSM magnetization curves of Fe3O4 and Fe3O4@SiO2@Ah-COF. (D) N2 adsorption–desorption isotherms of Fe3O4 and Fe3O4@SiO2@Ah-COF. | ||
The synthesis process of Fe3O4@SiO2@Ah-COF was evaluated by FT-IR. As depicted in Fig. 3B, the typical band at 587 cm−1 was assigned to the Fe–O–Fe vibration. The broad bands of –OH at 3441 cm−1 and 1631 cm−1 might be assigned to the stretching and bending vibrations of water molecules on the surface of Fe3O4, respectively. The peaks around 1091 cm−1 could be related to the Si–O bending vibration. This verified that the silica shells were successfully encapsulated onto the surface of Fe3O4. Compared with the spectra of Fe3O4 and Fe3O4@SiO2, the almost identical characteristic absorption peaks described above were also observed in the spectrum of Fe3O4@SiO2@Ah-COF. Furthermore, the emerging stretching features at 1438 cm−1 and 1623 cm−1 were caused by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode, demonstrating that the COFs successfully covered the surface of Fe3O4@SiO2 via Schiff-base condensation reaction. The FT-IR results confirmed the successful fabrication of Fe3O4@SiO2@Ah-COF.
N stretching mode, demonstrating that the COFs successfully covered the surface of Fe3O4@SiO2 via Schiff-base condensation reaction. The FT-IR results confirmed the successful fabrication of Fe3O4@SiO2@Ah-COF.
Vibration magnetometer (VSM) was used to measure the magnetic values of Fe3O4 and Fe3O4@SiO2@Ah-COF, and the results are shown in Fig. 3C. The saturation magnetization value of the bare Fe3O4 and Fe3O4@SiO2@Ah-COF was 79.12 and 37.02 emu g−1, respectively. Compared with Fe3O4, the saturated magnetization of Fe3O4@SiO2@Ah-COF was somewhat reduced, which was attributed to the formation of a core–shell structure. Despite the decreased magnetization, the magnetic strength of Fe3O4@SiO2@Ah-COF was still adequate for magnetic separation. Fe3O4@SiO2@Ah-COF was well dispersed in water, and Fe3O4@SiO2@Ah-COF can be collected in 60 s in an external magnetic field, indicating that the obtained Fe3O4@SiO2@Ah-COF exhibited excellent magnetism ability and could be used for the rapid magnetic separation.
To evaluate the porosity and surface area, N2 adsorption–desorption analysis at 77 K was carried out. As displayed in Fig. 3D, there is a well-defined type IV isotherm, indicating the presence of a mesoporous structure, and the average pore size of the magnetic material is 4.51 nm. After calculation, the BET surface area of Fe3O4@SiO2@Ah-COF was 71.82 m2 g−1, which was higher than that of Fe3O4 with a surface area of 7.89 m2 g−1. This suggests that Fe3O4@SiO2@Ah-COF can provide abundant active sites, increase the opportunity for magnetic materials to come into contact with QNs and improve the adsorption efficiency. These results confirmed the successful preparation of Fe3O4@SiO2@Ah-COF.
3.2 Condition of MSPE
To obtain a high extraction recovery, parameters that influence the extraction performance (including the extraction dosage, sample solution pH, NaCl concentration, extraction time, elution solvent type, volume and elution time) were optimized in detail. | ||
| Fig. 4 Effect of the adsorbent dosage. | ||
 | ||
| Fig. 5 Effect of the extraction time (A), elution solvent type (B), the volume of eluent (C) and elution time (D) of 24 QNs. | ||
The eluent is an influential factor to obtain the best elution efficiency. It has been reported that acidity is conducive to the elution of QNs,54 so we first investigated the effects of methanol, acetonitrile, 0.5% (v/v) acidified methanol and 0.5% (v/v) acidified acetonitrile on the recovery. Results showed that the elution efficiency of methanol is dramatically higher than that of acetonitrile. The highest recovery rate of methanol was 75.2%, while that of acetonitrile was only 15.8%. Compared with acidified acetonitrile, acidified methanol led to a higher elution efficiency. The possible reason can be explained as follows: methanol has a polarity that is more similar to that of the QNs, so it has a relatively high dissolution capacity for analytes. The concentration of formic acid in methanol was further investigated. As shown in Fig. 5B, the elution efficiencies of 24 QNs continuously increased with the increase of the formic acid concentration in methanol, and then declined at a formic acid concentration of 5% (v/v). Therefore, 3% (v/v) formic acid in methanol was selected for elution.
Fig. 5C shows that QNs can achieve good recoveries when the eluent volume increased to 8 mL. When the eluent was fixed at 8 mL, the effect of varied elution time was further studied from 3 to 11 min. As shown in Fig. 5D, the recoveries increased gradually with the increasing vortex time from 3 to 8 min, and then slowly decreased over 8 min. The results indicated that reversible adsorption reaches equilibrium after 8 min.55 Therefore, 8 min of elution time was selected in this study.
3.3 Adsorption mechanism
Fe3O4@SiO2@Ah-COF contains an imine structure, abundant carbonyl groups and a large number of benzene ring structures. Among them, the lone pair electrons on the N atom and the benzene ring structure are prone to form p–π and π–π interactions with the piperazine ring of QNs; and the carbonyl groups contained in it form hydrogen bonds with the carbonyl or carboxyl groups of QNs, which makes the magnetic materials have good adsorption effects.In addition, the large specific surface area of the magnetic materials can provide more adsorption sites for the target analytes, improving the adsorption capacity. Moreover, the presence of mesoporous structures is beneficial for reducing the mass transfer resistance, which allows the target substance to quickly pass through the material.
3.4 Reusability of the adsorbent
Reusability is a significant parameter to evaluate the tolerance performance of a certain adsorbent. After the analytes were desorbed from the magnetic material, the magnetic material was washed and dried, and then the adsorption–desorption cycle was tested under optimal condition with spiked blank samples. As shown in Fig. S2,† the results proved that Fe3O4@SiO2@Ah-COF could be reused 5 times for the recoveries of 24 QNs without an obvious decrease, which greatly reduces the experimental cost.3.5 Matrix effect
Matrix effect (ME) refers to matrix interferences with the target analytes, causing the mass spectrometry signal to be suppressed or enhanced during analysis and detection.56 In this method, the ME of 24 QNs in water and egg samples was evaluated by the ratio of slopes between the matrix-matched calibration curves and the solvent calibration curves. According to the calculation results (Table S3†), the ME values of QNs in water and egg samples were 85.4–113.7% and 80.3–116.4%, respectively, suggesting that the QNs had no obvious matrix effect.57 Therefore, we chose to use the external standard method using a solvent standard curve for quantitative analysis of the experiment.3.6 Method validation
The analytical performance of the developed MSPE-UPLC-MS/MS method under the optimal conditions is listed in Table 1. The linear range of 24 QNs was 0.2–200 μg kg−1, with R2 ranging from 0.9990 to 0.9999. For water samples, the limits of detection (LOD) [signal-to-noise ratio (S/N) of 3] was 0.003–0.036 μg L−1 and the limits of quantitation (LOQ) (S/N of 10) was 0.008–0.110 μg L−1. For egg samples, the LOD was 0.009–0.272 μg kg−1 and the LOQ was 0.029–0.908 μg kg−1. The results proved that the method had satisfactory linearity and high sensitivity.| Analytes | Linear range (ng mL−1) | Calibration curve | R2 | LOD | LOQ | ||
|---|---|---|---|---|---|---|---|
| Water (μg L−1) | Egg (μg kg−1) | Water (μg L−1) | Egg (μg kg−1) | ||||
| LuFu | 0.2–200 | Y = 24230.5X + 2717.2 |
0.9993 | 0.011 | 0.272 | 0.037 | 0.908 |
| ENO | 0.2–200 | Y = 45259X + 4032.3 |
0.9996 | 0.007 | 0.083 | 0.023 | 0.276 |
| NOR | 0.2–200 | Y = 26159.1X + 3118.5 |
0.9999 | 0.004 | 0.188 | 0.014 | 0.625 |
| FuLuo | 0.2–200 | Y = 161000X + 10735.5 |
0.9999 | 0.009 | 0.083 | 0.030 | 0.277 |
| OFL | 0.2–200 | Y = 177862X + 62971.9 |
0.9999 | 0.011 | 0.034 | 0.037 | 0.112 |
| PEF | 0.2–200 | Y = 126828X + 1784.1 |
0.9998 | 0.003 | 0.026 | 0.010 | 0.087 |
| ANT | 0.2–200 | Y = 153922X + 62455.4 |
0.9999 | 0.007 | 0.009 | 0.022 | 0.029 |
| CIP | 0.2–200 | Y = 34507.3X + 5276.8 |
0.9998 | 0.024 | 0.152 | 0.074 | 0.507 |
| LOM | 0.2–200 | Y = 78931.8X + 65585.7 |
0.9992 | 0.010 | 0.072 | 0.030 | 0.238 |
| Luosuo | 0.2–200 | Y = 149193X + 10463.8 |
0.9996 | 0.021 | 0.088 | 0.064 | 0.293 |
| DaNuo | 0.2–200 | Y = 218201X + 24850.1 |
0.9991 | 0.007 | 0.023 | 0.022 | 0.076 |
| ENR | 0.2–200 | Y = 152947X + 11765.5 |
0.9999 | 0.013 | 0.025 | 0.038 | 0.082 |
| ORB | 0.2–200 | Y = 206573X + 50924.4 |
0.9999 | 0.020 | 0.022 | 0.059 | 0.073 |
| GAT | 0.2–200 | Y = 43867.2X + 18974.3 |
0.9997 | 0.010 | 0.065 | 0.031 | 0.217 |
| SAR | 0.2–200 | Y = 44213.8X + 5397.6 |
0.9998 | 0.013 | 0.067 | 0.039 | 0.224 |
| DIF | 0.2–200 | Y = 132196X + 10242.1 |
0.9997 | 0.009 | 0.027 | 0.029 | 0.091 |
| SPA | 0.2–200 | Y = 218233X + 149197 |
0.9996 | 0.006 | 0.017 | 0.018 | 0.058 |
| MOX | 0.2–200 | Y = 32873.5X + 6457.6 |
0.9997 | 0.011 | 0.146 | 0.033 | 0.486 |
| TOS | 0.2–200 | Y = 124395X + 48280 |
0.9998 | 0.015 | 0.042 | 0.047 | 0.071 |
| GEM | 0.2–200 | Y = 118134X + 16785.2 |
0.9999 | 0.010 | 0.138 | 0.031 | 0.461 |
| BAL | 0.2–200 | Y = 151779X + 3071.7 |
0.9997 | 0.003 | 0.034 | 0.008 | 0.115 |
| TIMA | 0.2–200 | Y = 78438.7X + 3269.3 |
0.9997 | 0.035 | 0.033 | 0.106 | 0.108 |
| MILO | 0.2–200 | Y = 327264X + 25903.5 |
0.9997 | 0.021 | 0.125 | 0.064 | 0.418 |
| JiaLei | 0.2–200 | Y = 40601.6X + 79814.7 |
0.9992 | 0.013 | 0.143 | 0.041 | 0.478 |
As shown in Table S4,† with three different spiked levels, the 24 QNs obtained good recoveries in both water and egg samples, which were respectively 70.3–106.1% and 70.4–119.7% with RSD below 10%, indicating the reliability of the method.
3.7 Analysis of real samples
The method was further applied to the detection of the QNs residues in 15 water and 20 egg samples. As a result, SAR was detected in 4 egg samples and the levels varied from 4.07 to 7.54 μg kg−1.3.8 Comparison with other methods
This method was compared with other reported methods using different adsorbents for the detection of QNs residues.As seen in Table 2, compared to traditional SPE and DSPE, MSPE simplified the extraction procedure and our method can directly extract the target QNs from real samples without protein precipitation treatment,59,60 which decreased the use of organic solvents and shortened the sample preparation time. It is worth noting that previous reports have mostly focused on the detection of milk and meat samples with less detection in egg samples.41–43,58,59 In reported methods, most of them involved the detection of 3–6 QNS. Only one method for 17 QNs detected in aquaculture wastewater was established, and the testing sample matrix was single. In comparison, this study was able to simultaneously determine 24 QNs in water and eggs with low LOD and less extraction time, which showed high throughput and high sensitivity.
| Method | Adsorbent | Sample (numbers of QNs) | Extraction time (min) | Linearity | Recovery (%) | LOD | Ref. |
|---|---|---|---|---|---|---|---|
| a DAD: diode-array detector, FLD: fluorescence detection, RAM-MIPs: restricted access media-MIPs, RPLC: reversed-phase liquid chromatography. | |||||||
| MSPE-HPLC-MS/MS | Fe3O4@COF(TpBD)@Au-MPS | Pork, chicken and bovine (6) | 30 | 0.005–0.500 mg L−1 | 94.23–98.6 | 0.002–0.004 mg kg−1 | 42 |
| MSPE-HPLC-DAD | Fe3O4@COFs | Milk, pork and human plasma (6) | 60 | 2.5–1500 ng g−1 | 78.7–103.5 | 0.25–0.5 ng g−1 | 43 |
| MSPE-LC-FLD | Fe3O4@PLS | Milk (3) | 20 | 0.4–600 μg kg−1 | 96.5–118.6 | 0.41–0.45 μg L−1 | 58 |
| RAM-MIPs-DSPE-HPLC-UV | RAM-MIPs | Milk and river water (5) | 20 and 15 | 5–200 μg L−1 | 80.7–103.5 | 1.02–3.15 μg L−1 | 59 |
| 85.1–105.9 | 0.93–2.87 μg L−1 | ||||||
| SPE-LC-MS/MS | Oasis HLB SPE cartridges | Aquaculture wastewater (17) | >25 | 0.5–100 μg L−1 | 47.8–118.7 | 0.08–0.3 ng L−1 | 60 |
| MSPE-RPLC-MS/MS | PEI-functionalized Fe3O4/ATP | Chicken muscle (3) | 20 | 0.2–100 μg kg−1 | 83.9–98.7 | 0.02–0.08 μg L−1 | 41 |
| MSPE-UPLC-MS/MS | Fe3O4@SiO2@Ah-COF | Water and egg samples (24) | 20 | 0.2–200 μg kg−1 | 70.3–106.1 | 0.003–0.036 μg L−1 | This work |
| 70.4–119.7 | 0.009–0.272 μg kg−1 | ||||||
4. Conclusion
This study reported the design and synthesis of Fe3O4@SiO2@Ah-COF for the first time, which possessed remarkable properties such as good magnetism, large surface area and abundance of carbonyl groups, leading to excellent adsorption efficiency for 24 QNs. Then, a new MSPE-UPLC-MS/MS method was established for the determination of 24 QNs in water and egg samples, showing a simple extraction process, low LOD and LOQ, good accuracy and wide linear range. Real samples that possibly contained residues of QNs were analysed using the developed method, and SAR was found in four egg samples. In summary, the proposed method is suitable for the simultaneous and high-throughput determination of trace QNs in egg and water samples.Author contributions
Na Li: conceptualization, formal analysis, methodology, writing – original draft. Mengnan Liang: data curation, formal analysis. Hao Zhang: methodology, validation. Zhongxia Hua: methodology, supervision. Ling Ma: conceptualization, supervision, funding acquisition, formal analysis. Yanyu Qi: conceptualization, supervision. Ke Wang: conceptualization, funding acquisition, project administration, resources, writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81903322), and the S & T Program of Hebei (223777116D).References
- S. Guan, H. Wu, L. Yang, Z. Wang and J. Wu, J. Sep. Sci., 2020, 43, 3775–3784 CrossRef CAS PubMed.
- X. Lei, X. Xu, L. Liu, H. Kuang, L. Xu and C. Xu, J. Dairy Sci., 2022, 105, 1999–2010 CrossRef CAS PubMed.
- C. Fengxian and H. Reti, Ecotoxicol. Environ. Saf., 2017, 136, 111–118 CrossRef PubMed.
- R. Wang, K. Xu and W. Shi, Arch. Pharm., 2019, 352, 1900045 CrossRef PubMed.
- X. Liu, Y. Liu, J.-R. Xu, K.-J. Ren and X.-Z. Meng, Environ. Pollut., 2016, 219, 916–923 CrossRef CAS.
- H. Zhang, X. Wang and Y. Li, Environ. Chem., 2022, 41, 1168–1181 CAS.
- M. Luo, K. Xing, Z. Guo, D. Guo, W. Lai and J. Peng, J. Dairy Sci., 2020, 103, 7791–7800 CrossRef CAS PubMed.
- X. Xu, L. Feng, J. Li, P. Yuan, J. Feng, L. Wei and X. Cheng, Chin. Chem. Lett., 2019, 30, 549–552 CrossRef CAS.
- A. Rusu, G. Hancu, G. Volgyi, G. Toth, B. Noszal and A. Gyeresi, J. Chromatogr. Sci., 2013, 52, 919–925 Search PubMed.
- I. Varenina, N. Bilandžić, Đ. B. Luburić, B. S. Kolanović, I. Varga, M. Sedak and M. Đokić, Food Control, 2023, 148, 109676 CrossRef CAS.
- H. Wang, X. Zhao, J. Xu, Y. Shang, H. Wang, P. Wang, X. He and J. Tan, J. Chromatogr. A, 2021, 1651, 462286 CrossRef CAS PubMed.
- S. Bang Ye, Y. Huang and D.-Y. Lin, Food Chem., 2022, 373, 131466 CrossRef CAS PubMed.
- H. Wang, M. Gao, M. Wang, R. Zhang, W. Wang, R. A. Dahlgren and X. Wang, J. Chromatogr. B, 2015, 985, 62–70 CrossRef CAS PubMed.
- L. Hou, Y. Ji, J. Zhao and L. Zhao, Microchem. J., 2022, 179, 107664 CrossRef CAS.
- Y. Zhu, P. He, H. Hu, M. Qi, T. Li, X. Zhang, Y. Guo, W. Wu, Q. Lan, C. Yang and H. Jin, J. Chromatogr. B, 2022, 1208, 123390 CrossRef CAS PubMed.
- Z. Lu, F. Deng, R. He, L. Tan, X. Luo, X. Pan and Z. Yang, Microchem. J., 2019, 151, 104213 CrossRef CAS.
- W. Huang, P. Wang, P. Jiang, X. Dong and S. Lin, J. Chromatogr. A, 2018, 1573, 59–65 CrossRef CAS.
- H. Yu, Z. Wang, R. Wu, X. Chen and T. W. D. Chan, J. Chromatogr. A, 2019, 1601, 27–34 CrossRef CAS PubMed.
- X. Di, X. Wang, Y. Liu and X. Guo, J. Sep. Sci., 2019, 42, 2187–2298 CrossRef.
- H.-z. Tang, Y.-h. Wang, S. Li, J. Wu, J.-w. Li, H.-y. Zhou and Z.-x. Gao, Anal. Bioanal. Chem., 2019, 411, 7039–7049 CrossRef CAS PubMed.
- A. Wen, G. Li, D. Wu, Y. Yu, Y. Yang, N. Hu, H. Wang, J. Chen and Y. Wu, J. Chromatogr. A, 2020, 1612, 460651 CrossRef CAS PubMed.
- L. Lian, X. Zhang, J. Hao, J. Lv, X. Wang, B. Zhu and D. Lou, J. Chromatogr. A, 2018, 1579, 1–8 CrossRef CAS PubMed.
- H. Yu, Y. Jia, R. Wu, X. Chen and T. W. D. Chan, Anal. Bioanal. Chem., 2019, 411, 2817–2826 CrossRef CAS PubMed.
- Y. Du, X. Yan, Y. Chen, Y. Wu, Q. Qiu, Y. Li and D. Wu, J. Chromatogr. A, 2022, 1675, 463184 CrossRef CAS PubMed.
- L. Fu, H. Zhou, E. Miao, S. Lu, S. Jing, Y. Hu, L. Wei, J. Zhan and M. Wu, Food Chem., 2019, 289, 701–707 CrossRef CAS.
- X. He, G. N. Wang, K. Yang, H. Z. Liu, X. J. Wu and J. P. Wang, Food Chem., 2017, 221, 1226–1231 CrossRef CAS PubMed.
- M. Hernández-Mesa, C. Cruces-Blanco and A. M. García-Campaña, Talanta, 2017, 163, 111–120 CrossRef PubMed.
- H. Duo, X. Lu, X. Nie, L. Wang, S. Wang, X. Liang and Y. Guo, J. Chromatogr. A, 2020, 1626, 461328 CrossRef CAS.
- S. Wang, H. Niu, D. Cao and Y. Cai, Talanta, 2019, 194, 522–527 CrossRef CAS.
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS PubMed.
- W. Huang, J. Byun, I. Rörich, C. Ramanan, P. W. M. Blom, H. Lu, D. Wang, L. Caire da Silva, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8316–8320 CrossRef CAS PubMed.
- D. Zhang, Y. Wang, W. Geng and H. Liu, Sens. Actuators, B, 2019, 285, 546–552 CrossRef CAS.
- M. Ghasemi, M. Khedri, M. Didandeh, M. Taheri, E. Ghasemy, R. Maleki, H. k. Shon and A. Razmjou, Ind. Eng. Chem. Res., 2022, 61, 8809–8820 CrossRef CAS.
- A. Irfan, T. Wang, A. Wang, X. Jing, L. Yang and G. Zhu, Anal. Chim. Acta, 2022, 1209, 339876 CrossRef CAS.
- W. Tan, L. Zhu, L. Tian, H. Zhang, R. Peng, K. Chen, S. Zhao and F. Ye, J. Chromatogr. A, 2022, 1675, 463188 CrossRef CAS PubMed.
- H. Liu, Y. Zhang, D. Zhang, F. Zheng, M. Huang, J. Sun, X. Sun, H. Li, J. Wang and B. Sun, Microchim. Acta, 2019, 186, 182–191 CrossRef PubMed.
- H. Yuan, Z. LU, Y. Li, C. Zhang and G. Li, Chin. J. Chromatogr., 2022, 40, 109–122 CrossRef CAS.
- Y. Li, C.-X. Yang and X.-P. Yan, Chem. Commun., 2017, 53, 2511–2514 RSC.
- J. Xin, X. Wang, N. Li, L. Liu, Y. Lian, M. Wang and R.-S. Zhao, Food Chem., 2020, 330, 127255 CrossRef CAS PubMed.
- N. Li, D. Wu, N. Hu, G. Fan, X. Li, J. Sun, X. Chen, Y. Suo, G. Li and Y. Wu, J. Agric. Food Chem., 2018, 66, 3572–3580 CrossRef CAS.
- X. Li, Y. Chen, S. Chen, C. Hou, R. Xuan, Y. Gao, S. Ren, L. Yao, T. Wang, L. Zhang and Y. Zhang, Anal. Bioanal. Chem., 2021, 413, 3529–3540 CrossRef CAS.
- L. Zhang and W. Jiang, Food Sci. Technol., 2022, 44, 269–277 Search PubMed.
- M. Wang, M. Gao, K. Zhang, L. Wang, W. Wang, Q. Fu, Z. Xia and D. Gao, Microchim. Acta, 2019, 186, 827–850 CrossRef CAS PubMed.
- A. O. Melekhin, V. V. Tolmacheva, N. O. Goncharov, V. V. Apyari, S. G. Dmitrienko, E. G. Shubina and A. I. Grudev, Food Chem., 2022, 387, 132866 CrossRef CAS.
- Y. Li, X. Nie, M. Yang, W. Guo, F. Chen and F. Zhang, Food Sci., 2022, 43, 295–305 Search PubMed.
- Y. Li, H. Zhang, Y. Chen, L. Huang, Z. Lin and Z. Cai, ACS Appl. Mater. Interfaces, 2019, 11, 22492–22500 CrossRef CAS PubMed.
- D. Li, M. He, B. Chen and B. Hu, J. Chromatogr. A, 2019, 1601, 1–8 CrossRef CAS PubMed.
- Y. Chen and Z. Chen, Talanta, 2017, 165, 188–193 CrossRef CAS PubMed.
- W. Zhang, C. Lan, H. Zhang, Y. Zhang, W. Zhang, W. Zhao, C. Johnson, K. Hu, F. Xie and S. Zhang, J. Agric. Food Chem., 2019, 67, 3733–3743 CrossRef CAS PubMed.
- L. Chen, M. Zhang, F. Fu, J. Li and Z. Lin, J. Chromatogr. A, 2018, 1567, 136–146 CrossRef CAS PubMed.
- Y. Liu, H. Wu, X. Du, Y. Feng, Y. Zhao and Y. Chang, J. Anal. Sci., 2018, 34, 196–200 CAS.
- M. K. Abraham, A. S. Madanan, S. Varghese, A. I. Shkhair, G. Indongo, G. Rajeevan, N. S. Vijila and S. George, Analyst, 2023, 149, 231–243 RSC.
- H.-L. Jiang, Q.-B. Fu, M.-L. Wang, J.-M. Lin and R.-S. Zhao, Food Chem., 2021, 345, 128841 CrossRef CAS PubMed.
- C. Vakh, M. Alaboud, S. Lebedinets, D. Korolev, V. Postnov, L. Moskvin, O. Osmolovskaya and A. Bulatov, Anal. Chim. Acta, 2018, 1001, 59–69 CrossRef CAS.
- H.-B. Zheng, J.-Z. Mo, Y. Zhang, Q. Gao, J. Ding, Q.-W. Yu and Y.-Q. Feng, J. Chromatogr. A, 2014, 1329, 17–23 CrossRef CAS PubMed.
- N. Jian, M. Zhao, S. Liang, J. Cao, C. Wang, Q. Xu and J. Li, J. Agric. Food Chem., 2019, 67, 6892–6901 CrossRef CAS PubMed.
- J. Li, X. Xu, W. Guo, Y. Zhang, X. Feng and F. Zhang, Food Chem., 2022, 387, 132821 CrossRef CAS PubMed.
- Z. Yu, B. Liang, Z. Zhao, S. Lin, H. He and S.-X. Liang, J. Food Compos. Anal., 2023, 121, 105352 CrossRef CAS.
- J. Li, Y. Zhou, Z. Sun, T. Cai, X. Wang, S. Zhao, H. Liu and B. Gong, J. Chromatogr. A, 2020, 1626, 461364 CrossRef CAS PubMed.
- Z. Ding, Y. Li and X. Wang, Chin. J. Environ. Eng., 2022, 16, 674–683 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00247d |
| This journal is © The Royal Society of Chemistry 2024 |