
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the potential of MB2 MBene family as promising anodes for Li-ion batteries†
Ying Hana,
Lianli Wang *a,
Bin Zhenga,
Jinlei Wanga,
Li Zhanga and
Beibei Xiaob
*a,
Bin Zhenga,
Jinlei Wanga,
Li Zhanga and
Beibei Xiaob
aSchool of Materials Science and Engineering, Xi'an University of Science and Technology, Xi'an 710054, PR China. E-mail: wanglianli@xust.edu.cn
bSchool of Energy and Power Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu 212003, PR China
First published on 8th April 2024
Abstract
In recent years, finding high-performance energy storage materials has become a major challenge for Li-ion batteries. B-based two-dimensional materials have become the focus of attention because of their abundant reserves and non-toxic characteristics. A series of two-dimensional transition metal borides (MBenes) are reported and their electrochemical properties as anode materials for Li-ion batteries are investigated by density functional theory (DFT) calculations. The surface of MB2 possesses medium adsorption strength and diffusion energy barrier for Li atoms, which are conducive to the insertion and extraction of Li-ions during the charge/discharge process of Li-ion batteries. Herein, we explore the potential of MB2 (M = Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Cu and Zn) as the anode material for LIBs. Excitingly, the Li atom can be stably adsorbed on the surface of MB2 (M = Sc, Ti, V, Nb, Mo, W) monolayers, and the theoretical capacity of the MB2 monolayer is high (521.77–1610.20 mA h g−1). The average open circuit voltage range is within 0.10–1.00 V (vs. Li/Li+). The relationship between the p-band center of the B atom and the adsorption energy of Li on the surface of MB2 is also investigated. Furthermore, it is found that the charge transfer of Li atom and metallic center in the most stable position is strongly related to the corresponding value of diffusion energy barrier. These results confirm that MB2 monolayers are promising 2D anode materials for Li-ion batteries, demonstrating the application prospects of B-based 2D materials.
1. Introduction
The rapidly increasing global demand for advanced energy storage and conversion technologies has made lithium-ion batteries (LIBs) an important electrochemical energy storage candidate in the field of energy storage.1–4 The performance of LIBs depends largely on the properties of the electrode materials, especially the anode materials, which mainly limit the performance of LIBs.5–7 Consequently, the pursuit of high-performance anode materials has become a key challenge for the development of next-generation LIBs. Compared with three-dimensional materials, e.g., graphite, two-dimensional materials are widely used as anode materials for LIBs owing to their large surface area, excellent physical and chemical properties, and high charge carrier mobility.8 The successful extraction of 2D graphene has garnered significant research attention; unfortunately, high diffusion barrier and lithium nucleation over graphene limit its application in terms of durability.9–11Apart from graphene, 2D materials with novel compositions and properties are being actively pursued for advanced applications. One of the latest members of the 2D material family is MXene, which is produced from the topochemical deintercalation of the A layer from a laminate MAX phase with a general formula of Mn+1AXn, where M refers to an early transition metal, A represents an A-group element (Al or Si), and X denotes C and/or N.12,13 The specific 2D structure coupled with the metal sites endows MXenes with attractive performance for energy storage and catalytic applications.14–16 So far, MXenes are limited to carbides, nitrides, or carbonitrides due to the constraints of MAX phase precursors. Recently, analogous 2D MBenes (transitional metal borides) have been predicted by theoretical calculations as excellent alternatives for a wide range of applications, such as metal-ion batteries17 and catalysts.18 In general, MBenes can refer to all 2D transition borides, such as FeB2,19 MoB2 (ref. 20) and TiB12,21 showing a wide variety of structures. Among currently reported MBenes, MoB, and FeB monolayers have been theoretically verified to possess an omnidirectional small diffusion energy barrier and high storage capacity for Li,17 whereas VB, CrB and MnB monolayers have been demonstrated to possess high Young's modulus and unique in-plane anisotropy, low diffusion potential and low open-circuit voltage as anode materials for LIBs.22 Moreover, Mo2B2 and Fe2B2 possess metal-like electronic conductivity, small diffusion energy barrier and high Li storage capacity.17
Furthermore, monolayer MB2 (M = Mn, Be, Mg, Mo, Fe and Ti) have been explored as promising anode candidates for high-energy-density LIBs due to their excellent electrical conductivity during the lithiation process.23–27 Therefore, the proposal of MB2 monolayers may offer a novel strategy for the rational design of 2D anode materials for LIBs. Hence, Zhang et al. have examined 34 MB2 monolayers with different M elements, ranging from group IIA to IVA, to screen out stable materials for promising applications using particle swarm optimization (PSO) and density functional theory (DFT) computations. The screening process identified eight stable MB2 monolayers with M = Be, Mg, Ti, Hf, V, Nb, Ta and Fe.28 Therefore, the systematic research of MB2 for secondary-ion batteries is still scarce, which leads to sluggish development of MB2. Hence, more fundamental studies of vast MB2 are required. Specifically, the covalent network of borides can facilitate the formation of monolayer structures, which are tempting for the large specific surface and high utilization.
Herein, we explore the potential of MB2 in energy storage by investigating the potential of a class of MB2 monolayers as anodes in LIBs using first-principle calculations. We mainly use 2D MB2 as an electrode of LIBs to predict its Li-ion storage performance. Then, the kinetic stability and electrochemical properties of these single-layer materials as anode materials for LIBs are calculated. In order to understand the Li storage interaction mechanism of MB2, we have also investigated the projective state density, p-band center and charge density difference. The current work aims to explore potential high-performance anode materials, providing a reference for the subsequent research on the application of 2D borides in Li-ion batteries.
2. Computational details
Fig. 1 shows top and side views of the relaxed crystal structure of MB2 (M = Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Cu and Zn) monolayer (2 × 2 × 1 supercell). Each single cell of MB2 contains one M atom and two B atoms. The basic building block of MB2 is composed of M and B atomic layers, where the arrangement of the upper B atomic layer is similar to the honeycomb distribution of C atoms in graphene and M atoms are placed directly below the center of the B–B honeycomb. The layer thickness of MB2 are shown in Table S1.†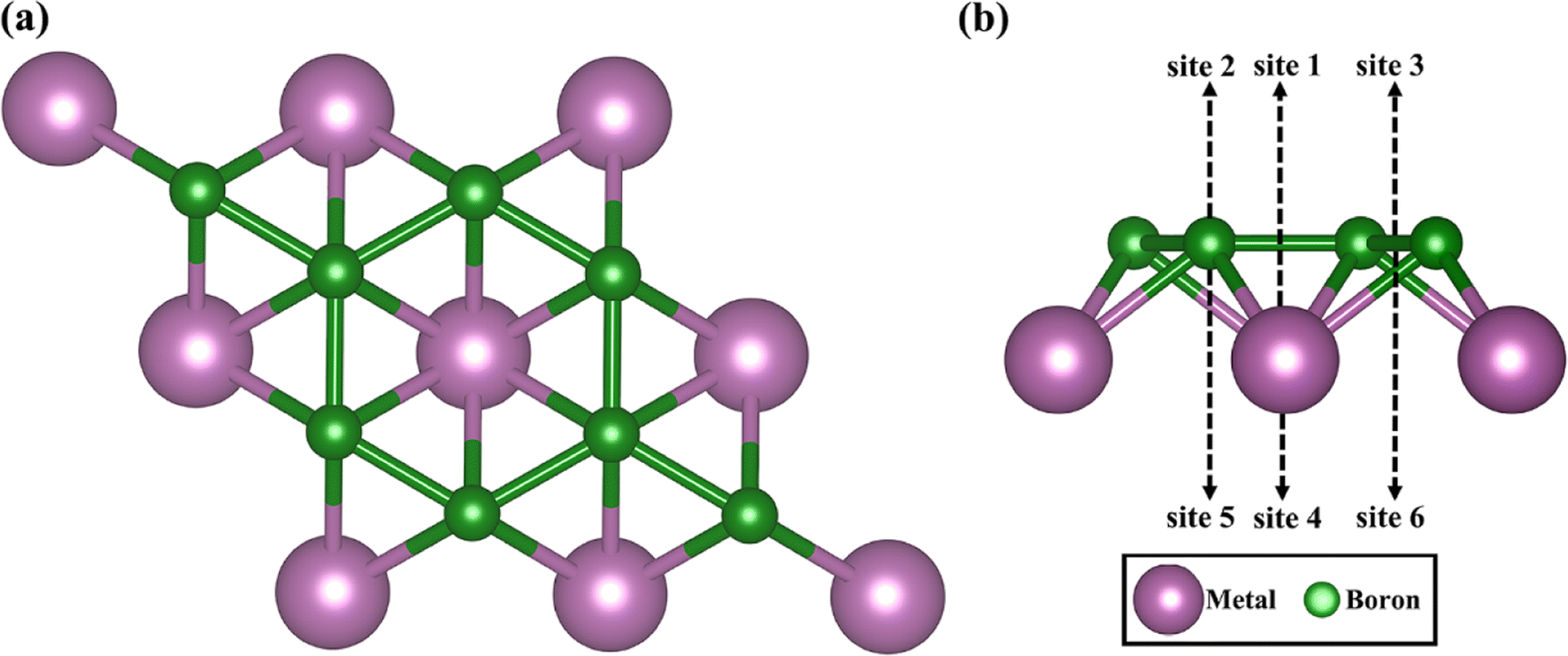 | ||
| Fig. 1 (a) Top and (b) side views of MB2 monolayers (M = Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Cu and Zn), site 1 to 6 are different adsorption sites, where purple and green colors denote the transition metal and boron atoms, respectively. | ||
After an extensive structural search using the CALYPSO structure prediction method,29,30 two-dimensional monolayer MB2 were obtained. All calculations, such as structural optimization, electronic properties and energy, within the DFT framework, were implemented in the Vienna ab initio Simulation Package (VASP).31 The exchange–correlation energy of the electrons was described within the generalized gradient approximation (GGA) framework using the Perdew–Burke–Ernzerhof (PBE) functional.32 The interactions between nuclear electrons and valence electrons were described using the projector-enhanced wave (PAW) method.33 The cut-off energy was set to 520.00 eV. All structures were fully relaxed until the convergence tolerance for the energy and force on each atom were less than 1.0 × 10−7 eV and 0.01 eV Å−1, respectively. All atomic structures and charge density distributions were visualized using the VESTA package.34 The 5 × 5 × 1 k-point lattices were used in structural optimization calculations, and the 11 × 11 × 1 k-point lattices were applied in electronic structure analysis. Thermodynamic stability is assessed through ab initio molecular dynamics (AIMD) simulations conducted within the canonical ensemble (NVT) at 300 K, employing a Nose–Hoover thermostat with a 6 ps period, and utilizing a time step of 2 fs. The DFT-D3 method was employed to correct van der Waals interactions between the adsorption atom and layers since it can correctly handle remote dispersion interactions.35 An inter-layer vacuum space of 20 Å was used to avoid the interactions between adjacent layers. To confirm the dynamic stability of MB2 monolayers, the phonon dispersion curve was calculated using a finite displacement method approach as implemented in the PHONOPY code with convergence accuracy consistent with the electronic calculation.36 With the help of complete Linear Synchronous Transit (LST)/Quadratic Synchronous Transit (QST) method and nudged elastic band (NEB) tools in the Dmol3 code,37 the transition state and diffusion pathways were predicted.38 The double numerical atomic orbital augmented by a polarization function (DNP) is chosen as the basis set.39 A smearing of 0.005 Ha (1 Ha = 27.21 eV) to the orbital occupation is applied to achieve accurate electronic convergence. In transition state search calculations, the convergence tolerances of energy, maximum force and displacement are 1.0 × 10−5 Ha, 0.002 Ha Å−1 and 0.005 Å, respectively. Incorporating spin polarization was necessary to achieve precise determination of the ground state energy. The Bader charge analysis serves to measure and evaluate the transfer of charges.
The adsorption energy equation of a single Li atom on the surface of MB2 can be given as follows:
| Ead = EMB2+Li − EMB2 − ELi | (1) |
The charge density difference can be given as follows:
| Δρ = ρLiMB2 − ρMB2 − ρLi | (2) |
The charge/discharge process follows the usual half-cell reaction in an aqueous solution, as follows:40
| MB2 + xLi+ + xe− ↔ LixMB2 (M = Sc, Ti, V, Nb, Mo, W) |
The average adsorption energy, which is defined as the difference between the total energy of the system before and after the Li atom adsorption process, can be expressed as:
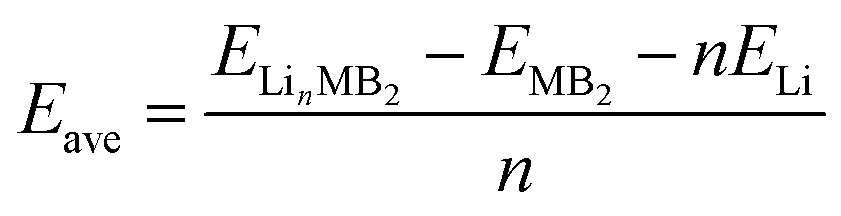 | (3) |
On the premise of ignoring the influence of volume, pressure and entropy, the open circuit voltage (OCV) can be given as follows:41
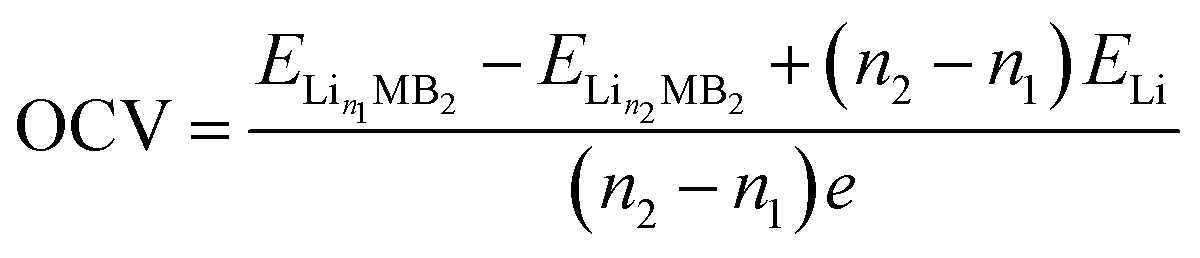 | (4) |
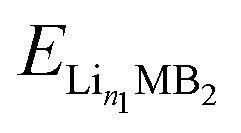 ,
, 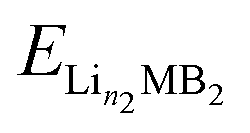 and ELi represent the energy of Lin1MB2, Lin2MB2 and per atom for bulk Li metal, respectively, and n1 and n2 denote the number of adsorbed Li atoms.
and ELi represent the energy of Lin1MB2, Lin2MB2 and per atom for bulk Li metal, respectively, and n1 and n2 denote the number of adsorbed Li atoms.
The estimate of theoretical specific capacity (TSC) is obtained by the following equation:
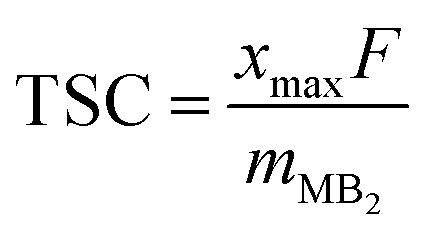 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801 mA h mol−1, and mMB2 stands for the molecular mass of MB2.
801 mA h mol−1, and mMB2 stands for the molecular mass of MB2.
3. Results and discussion
3.1 Adsorption of Li atom on MB2 monolayer
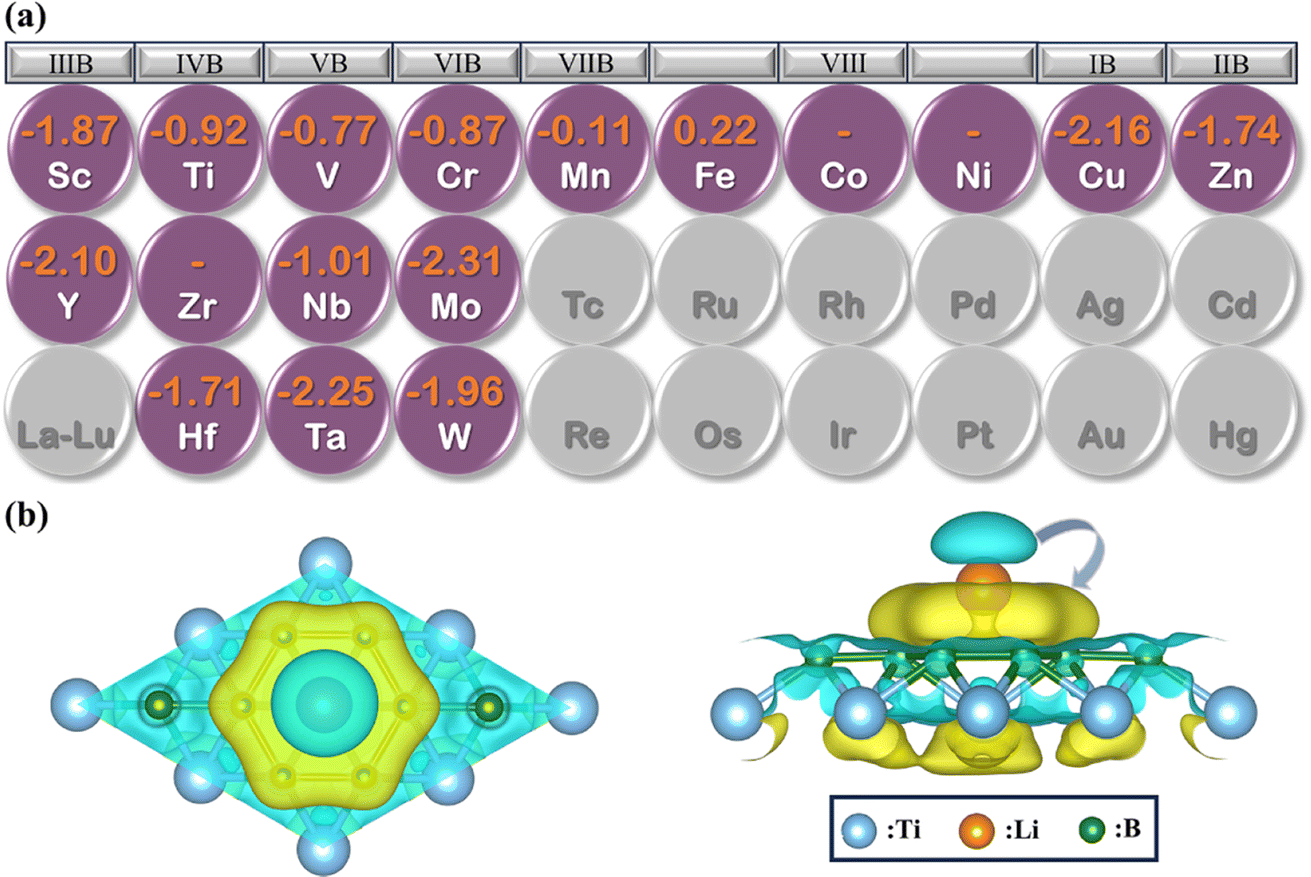
To explore the potential of MB2 (M = Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Cu and Zn) as an anode material, we have investigated the adsorption of Li atom on the surface of MB2. According to the symmetry of MB2 structure, there are six adsorption sites for Li atom (sites 1 to 6 in Fig. 1), which can be divided into two parts based on the location of Li atom, whether it is closer to the B atomic side or the metal side. The adsorption energy of a single Li atom on MB2 was calculated using eqn (1). The calculation results are shown in Table S2.† For MB2 (M = Sc, Ti, V, Cr, Cu, Zn, Y, Nb, Mo, Hf, Ta and W), Li tends to be close to B atom of MB2, especially the site 1 in the center of the B-layer six-membered ring is the most favorable adsorption position for Li atom. However, in the case of FeB2 and MnB2, Li atoms tend to be close to metal atoms of MB2, and the best adsorption site is site 5. In addition, it is found that CoB2, NiB2 and ZrB2 systems adsorb a single Li atom and exhibit obvious structural distortion, indicating that these systems are not suitable anode candidates.Based on the preferential adsorption site, the adsorption energy (Ead) of Li atom the B atomic side in fourteen MB2 structures was computed (Fig. 2a). Our computed values are in the range of −0.11 to −2.31 eV, which are close to the reported boride results (−0.36 to −2.70 eV).42–46 Furthermore, the results reveal that the strongest adsorption of Li atom occurs on MoB2 surface (−2.31 eV), while MnB2 exhibits relatively weak binding with Li atom (−0.11 eV). At the same time, the Li atom is difficult to attach to the B atom side of FeB2. As shown in Fig. S1,† the adsorption energy of Li atom on the metal side of TiB2 and NbB2 becomes positive, indicating that Li also is difficult to adsorb on the metal side. For FeB2 and MnB2, the adsorption strength of Li atom on the metal side is stronger than the B atomic side. However, the adsorption strength on the metal side for other structures is weaker than the adsorption strength on the B atomic side.
 | ||
| Fig. 2 (a) The adsorption energy of Li atom on the B atomic side of MB2 monolayers; (b) top and side views of differential charge density diagram of Li atom adsorbed on TiB2 monolayer, where the green region indicates charge depletion and yellow region indicates charge accumulation and blue, orange, and green colors denote the Ti, Li and B atoms, respectively. | ||
To gain a deeper understanding of the adsorption mechanism between Li atom and MB2 surface, we have calculated the charge density difference (Δρ) between Li and MB2 using eqn (2), where the result of TiB2 is shown in Fig. 2b. It is worth noting that the charge transfers from Li to B. Subsequently, the charges deplete around the Li atom and accumulate near the B atomic layer, respectively. This can be attributed to the electronegativity of B atoms. In addition, Bader charge analysis was performed to quantify the charge transfer effect.47 About 0.85 charges were observed to transfer from the Li atom to the B atomic layer. Similarly, the charge density difference and Bader charge of the most stable adsorption sites on the surface of other structures are shown in Fig. S6, Tables S3 and S4.† The amount of charge transfer from the Li atom to the nearest B atom of MB2 varies slightly within the range of 0.837 to 0.874, indicating that the charge transfer of the Li atom is almost complete, which is consistent with other borophenes48 and borides.25 Moreover, the strong attractive electrostatic interactions between Li atom and MB2 monolayer can avoid the formation of Li clusters, thereby ensuring better electronic conductivity throughout the charge/discharge process and improving the safety of LIBs.
According to the previous reports,49–52 there is a certain correlation between the p-band center of non-metal atoms and the adsorption energy (Ead) of the investigated system. The p-band center is of particular interest for application in energy storage and conversion. As an illustration, they can be used as electrode materials in LIBs,51 offering the potential for a dependable and high-energy-density cathode or anode. Consequently, we have explored the correlation between the p-band center of B atoms and the adsorption energy of Li atom on the surface of MB2 (Fig. 3). Fig. 3 illustrates an approximate trend where, as the p-band center shifts toward the Fermi level, the values of Ead gradually increase. Moreover, the location change of p-band center is the result of atomic orbital hybridization. Herein, it is worth noting that the p-band center is affected by the hybridization of the B atom p-orbitals in the MB2. When B atom p-orbitals are hybridized for the MB2 with different M elements, they become more diffused and less strongly bound to the atomic nucleus. We have compared the correlation between the p-band center and the diffusion energy barrier of the B-side Li atom and the adsorption energy and diffusion energy barrier of the metal-side Li atom, respectively, and found that the relationship between them is not obvious (Fig. S2†). According to the report of Zheng et al.,53 there is a correlation between the d-band center of transition metal atoms and the adsorption energy, and we have also analyzed this, and the results are shown in Fig. S3,† which shows that the correlation is not strong enough.
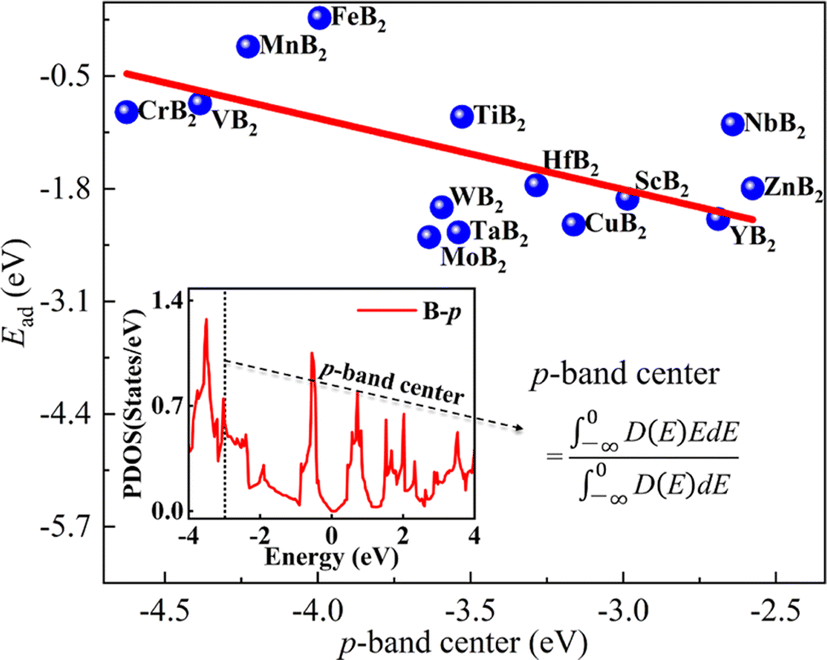 | ||
| Fig. 3 The relationship between the p-band center and the adsorption energy of Li atom on the B atomic side of MB2 monolayers and p-orbital projected density of states (PDOS). The curve in the inset diagram is the PDOS curve of the B atom in TiB2. | ||
3.2 Diffusion of Li atom on MB2 monolayers
Besides the adsorption, the diffusion of Li atoms needs to be considered for the charge and discharge efficiency.54 There are four possible diffusion paths for the Li atom on the B atomic and metal surfaces of the MB2 monolayer (Fig. S4a and b†). On the B atomic side, a direct path through two neighboring sites 1 (path I) and an indirect path through two neighboring sites 1 (through site 2, path II) (Fig. S4a†). On the metal side, a direct path through two adjacent sites 5 (path III) and a indirect path through two adjacent sites 5 (through site 4, path IV) (Fig. S4b†). The diffusion barrier of Li atom along paths I–IV on the MB2 monolayer is calculated (Tables S5 and S6†). We compare the diffusion energy barriers of the four diffusion paths of Li atom on the B atomic side and metal side. It can be seen that the Li atom is more inclined to diffuse through paths II and IV on the B atomic side and the metal side. This is consistent with the previously reported diffusion path of Li atom on the surface of BeB2 and MgB2.24Fig. 4a and S5† present the diffusion barrier profiles for the Li atom and the diffusion path of Li atom on the B atomic side and metal side of MB2 monolayer, respectively. According to the previous reports, the diffusion energy barrier of most 2D materials is in the range of 0.03 to 0.78 eV.55–60 Herein, the computed values are comparable to those of previously studied electrode materials. The computed diffusion energy barrier for Li atom on the B atomic side of MoB2 is 0.63 eV, which is comparable to the previously reported value of 0.52 eV,25 showing the reliability of current results. Furthermore, to explore the law of diffusion energy barrier of different metal elements on the surface of MB2, the relevant discussions were carried out. For Group 1 in Fig. 4b, it can be clearly observed that the smaller adsorption energy leads to a higher diffusion energy barrier. However, for Group 2, the relationship between adsorption energy and diffusion energy barrier is not obvious.
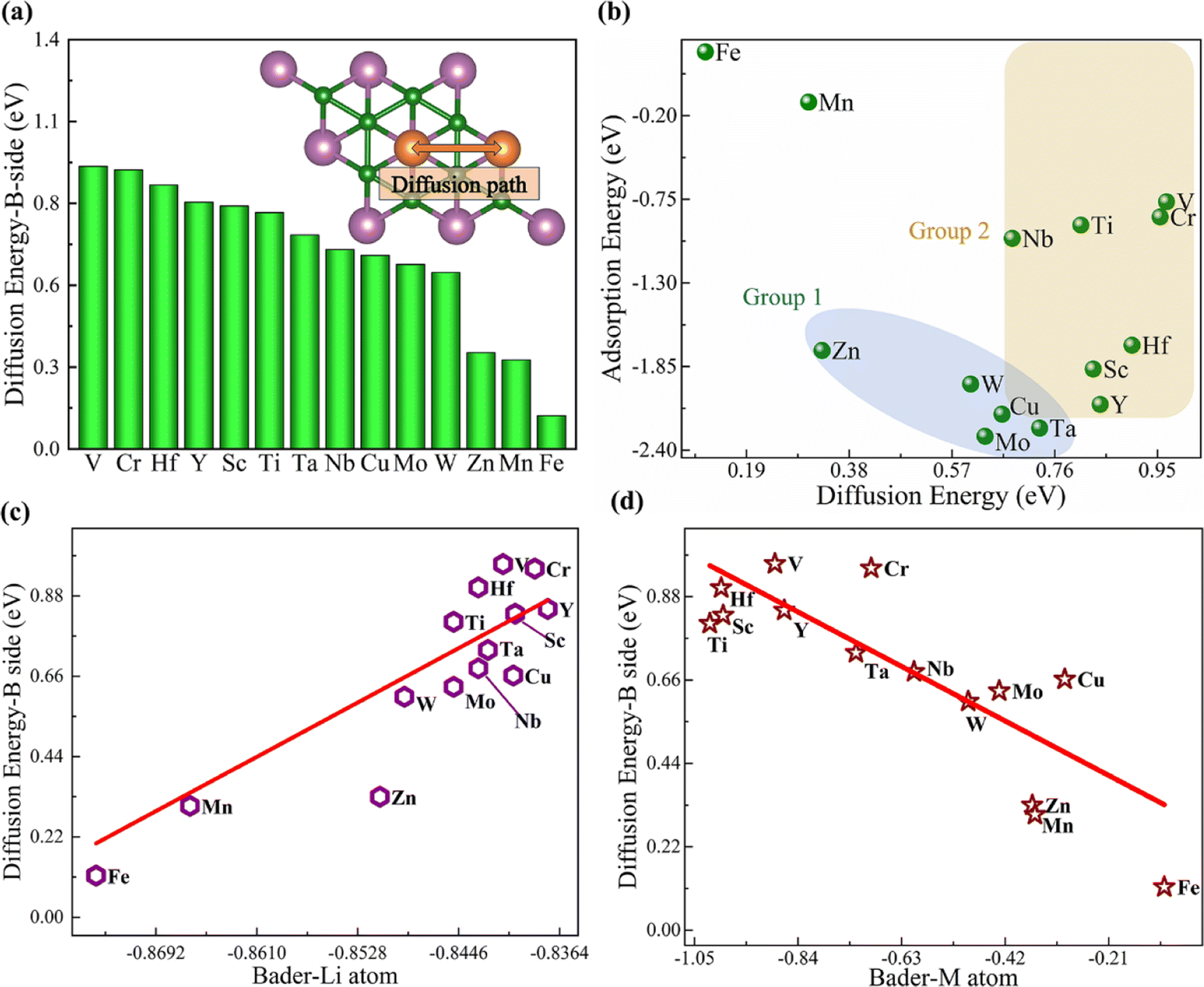 | ||
| Fig. 4 (a) Diffusion energy barrier and diffusion path of Li atom on the B atomic side; (b) adsorption energy and diffusion energy barrier of Li atom on the B atomic side; the diffusion energy barrier of Li atom along path II with the amount of charge transfer of (c) Li atom and (d) metal atom at the most stable position, respectively. | ||
The charge transfer of Li atom and metal atom in the most stable position is strongly related to the corresponding value of diffusion energy barrier (Fig. 4c and d). With the increase of diffusion energy barrier, the charge transfer of Li atom decreases gradually, while the charge transfer of metal atom increases. There is no obvious relationship between the number of transferred charges and adsorption energy, which may be ascribed to the presence of the metal atoms in the structure of MB2.
3.3 Open circuit voltage and theoretical charge storage capacity
From the viewpoint of practical applications and theoretical research, the OCV serves as a crucial parameter for evaluating the performance of LIBs. The Eave and OCV of different MB2 structures for Li atoms can be calculated by eqn (3) and (4), respectively. As a negative electrode material for LIBs, MB2 structure should exhibit a certain stability. Notably, the phonon spectra of ScB2, MoB2, and WB2 monolayers exhibits tiny imaginary frequencies only at the gamma point, which should be consistent with the previous findings of Zhang et al.28 In addition, according to previous reports, the tiny virtual frequency of the gamma point does not affect the stability of the material.20,23,61,62 Based on the above, we think these three materials are also dynamically stable. Herein, MB2 (M = Sc, Ti, V, Nb, Mo, W and Ta) structures are selected via the relevant band structure and phonon spectra owing to their stability and meta metallicity (Fig. S6 and S7†). Referring to Fig. 4b, the diffusion energy barrier and adsorption energy of these six systems are located in the middle region. Hence, appropriate diffusion energy barrier and adsorption strength are also conducive to the insertion and extraction of Li-ions in the anode material.Before calculating OCV, we need to first consider the average adsorption energy of MB2. We strive to systematically build a stable configuration by progressively adsorbing multiple Li atoms on both sides of the MB2 monolayer (Fig. 5a). On the 2 × 2 × 1 supercell of MB2 monolayers, four Li atoms are adsorbed in each layer, and the Li atoms from the first layer to the fourth layer are adsorbed at site 1, site 5, site 2 and site 5, respectively, in which the Li atomic layers of the second and fourth layers are staggered. As the adsorption concentration of each structure increases gradually, the adsorption strength decreases and the rate of adsorption strength reduction becomes smaller (Fig. 5b). Among them, the adsorption strength of ScB2 changes the most when adsorbing the first layer to the second layer.
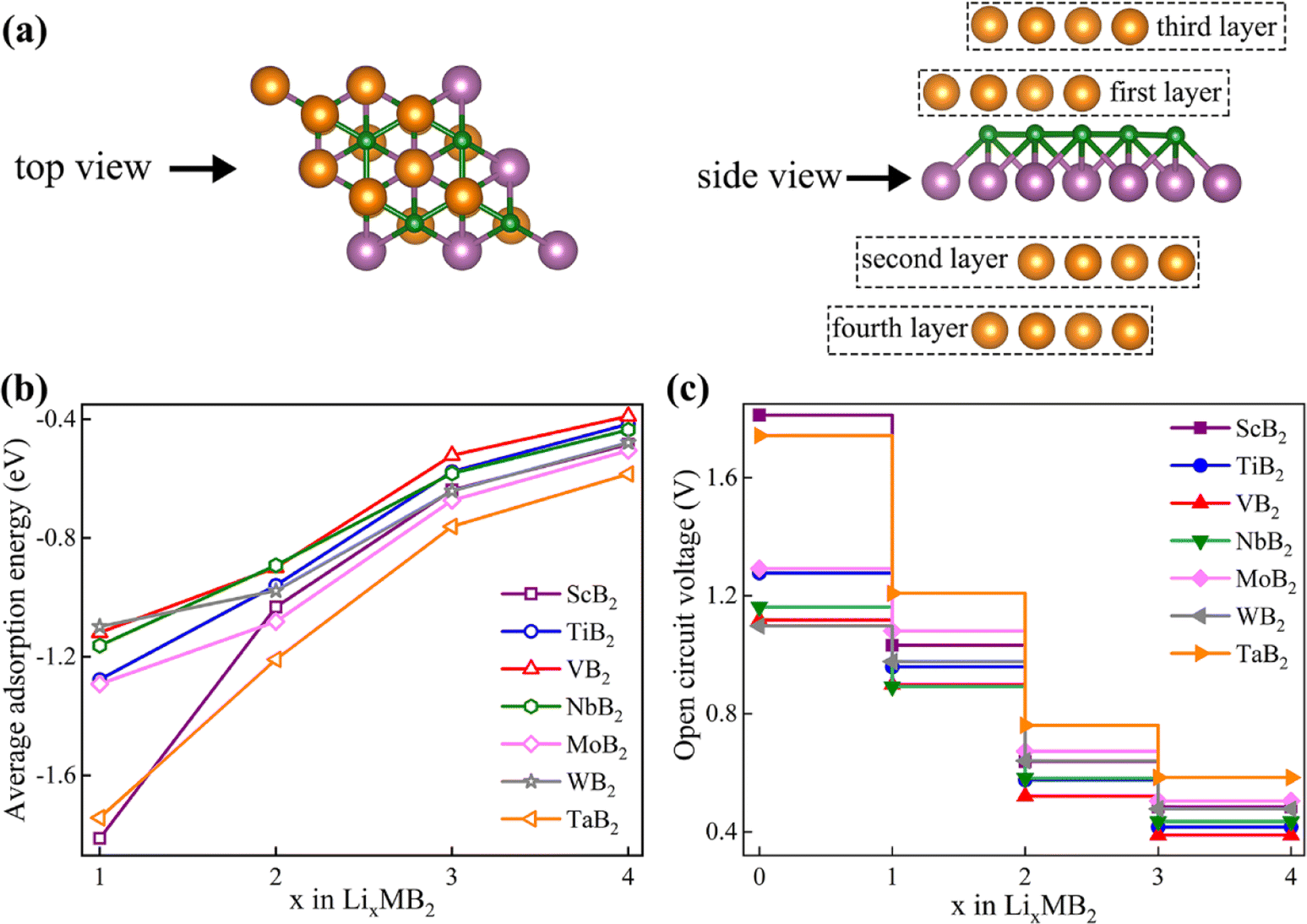 | ||
| Fig. 5 (a) The structural configuration of four layers of Li atoms adsorbed on the surface of MB2; (b) the change in average adsorption energy and (c) the open circuit voltage of LixMB2 (M = Sc, Ti, V, Nb, Mo, W and Ta) as a function of Li concentration. | ||
Based on the average adsorption energy, we can further calculate the OCV of MB2 monolayer (Fig. 5c). With the increase of adsorption concentration, the value of OCV decreases gradually and the decrease rate becomes smaller and smaller. Specifically, ScB2 exhibits the most significant changes and its OCV decreases by 43.0% from the first to the second layer, 21.7% from the second to the third layer, and 8.5% from the third to the fourth layer. With the increase of adsorbed Li atom content, the average adsorption energy of each structure is positive and the change of OCV is further calculated. It can be found that the OCV of MB2 (M = Sc, Ti, V, Nb, Mo, W and Ta) ranges from 0.48–1.87, 0.42–1.28, 0.39–1.12, 0.44–1.39, 0.51–2.31, 0.48–1.96 and 0.58–2.25 V, respectively. For LIBs systems, the average OCV calculations are 0.87, 0.81, 0.73, 0.77, 0.89, 0.80 and 1.07 V, respectively. Among them, the operating voltage ranges of ScB2, TiB2, VB2, NbB2, MoB2 and WB2 materials are the most suitable for anode materials (0.10–1.00 V). Previous research studies have shown that the excessively low average OCV leads to metallic electroplating, whereas an exorbitantly high average OCV impedes to achieve a high energy density.22,63
Theoretical specific capacity (TSC) is a frequently used index to evaluate the performance of anode materials in LIBs. The TSC of different MB2 structures for Li atoms can be calculated by eqn (5). The correlation between the diffusion energy barrier of the Li atom on B atomic side of MB2 monolayers and theoretical specific capacity is not strong (Fig. 6a). Herein, we divide MB2 structures into three groups. Group 1 exhibits high TSC values with a wide range of diffusion energy barrier for Li atom on MB2 monolayers. In the case of Group 2, the diffusion energy barrier for Li atom on MB2 monolayers is higher than Group 1, but the TSC values are lower. Group 3 exhibits minimum TSC values and a moderate diffusion energy barrier.
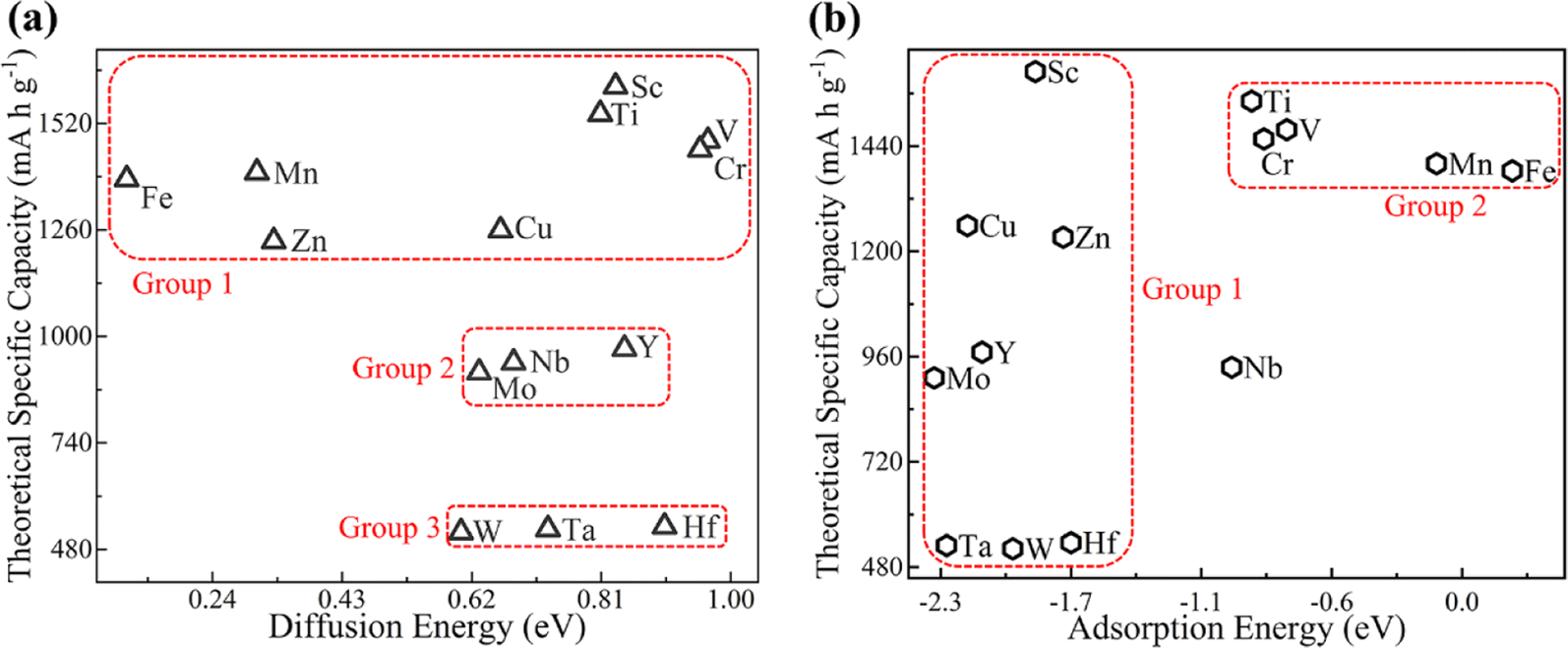 | ||
| Fig. 6 The theoretical specific capacity of different MB2 monolayers with respect to (a) diffusion energy barrier and (b) adsorption energy of Li atoms on B atomic side. | ||
Fig. 6b presents the correlation between the adsorption energy of the Li atom on B atomic side of MB2 monolayers and theoretical specific capacity of the Li–MB2 systems. Overall, the correlation is relatively complicated. The adsorption energy of most MB2 structures is relatively high (Group 1). When the adsorption strength is relatively weak, the theoretical specific capacity is in the upper middle position (Group 2).
According to the maximum adsorption concentration, we find that MB2 (M = Sc, Ti, V, Nb, Mo, and W) adsorbing Li atoms has an ultra-high theoretical specific capacity (521.77–1610.20 mA h g−1). The specific capacity is higher than that of several reported anode materials, such as graphite (372 mA h g−1), pillared MnO2 (588 mA h g−1),64 Ti3C2 (488 mA h g−1),60 and Nb2CS2 (194.36 mA h g−1).5 What's more, the specific theoretical capacity is comparable to that of graphene monolayer (1116 mA h g−1),65 Hf2S (1378 mA h g−1)66 and BN-doped reduced graphene oxide (BN-rGO) (1583 mA h g−1).67 They are expected to become negative electrode materials for LIBs in the future.
Considering that the change of MB2 geometry after the adsorption of Li atom will lead to the change of electronic structure, the band structure and PDOS of Li4MB2 are further analyzed (Fig. 7a–f). It can be seen that the contribution of the states around the Fermi level is mainly from the atoms in the MB2 monolayer, while the contribution of the Li atoms is very small. The above results show that the metal properties of MB2 monolayer can be well maintained before and after the insertion of Li atom into MB2, so that the electronic conductivity of MB2 monolayer can meet the requirements of LIBs during the whole charge and discharge process. Therefore, we believe that MB2 monolayer has very excellent storage capacity and electronic properties, and is a very ideal anode material. In addition, we evaluated the structural thermal stability of the MB2 monolayer at 300 K before and after adsorption of Li atoms using AIMD simulation method (Fig. S8 and 9†). The mean values of potential energy and temperature remained almost constant at room temperature of 300 K throughout the AIMD simulation, and MB2 held the initial structural framework well, indicating that they have good thermal stability. These results indicate that MB2 monolayers have high synthetic potential under certain experimental conditions. In particular, maintaining stability at this temperature is sufficient to ensure the safe operation of LIBs.
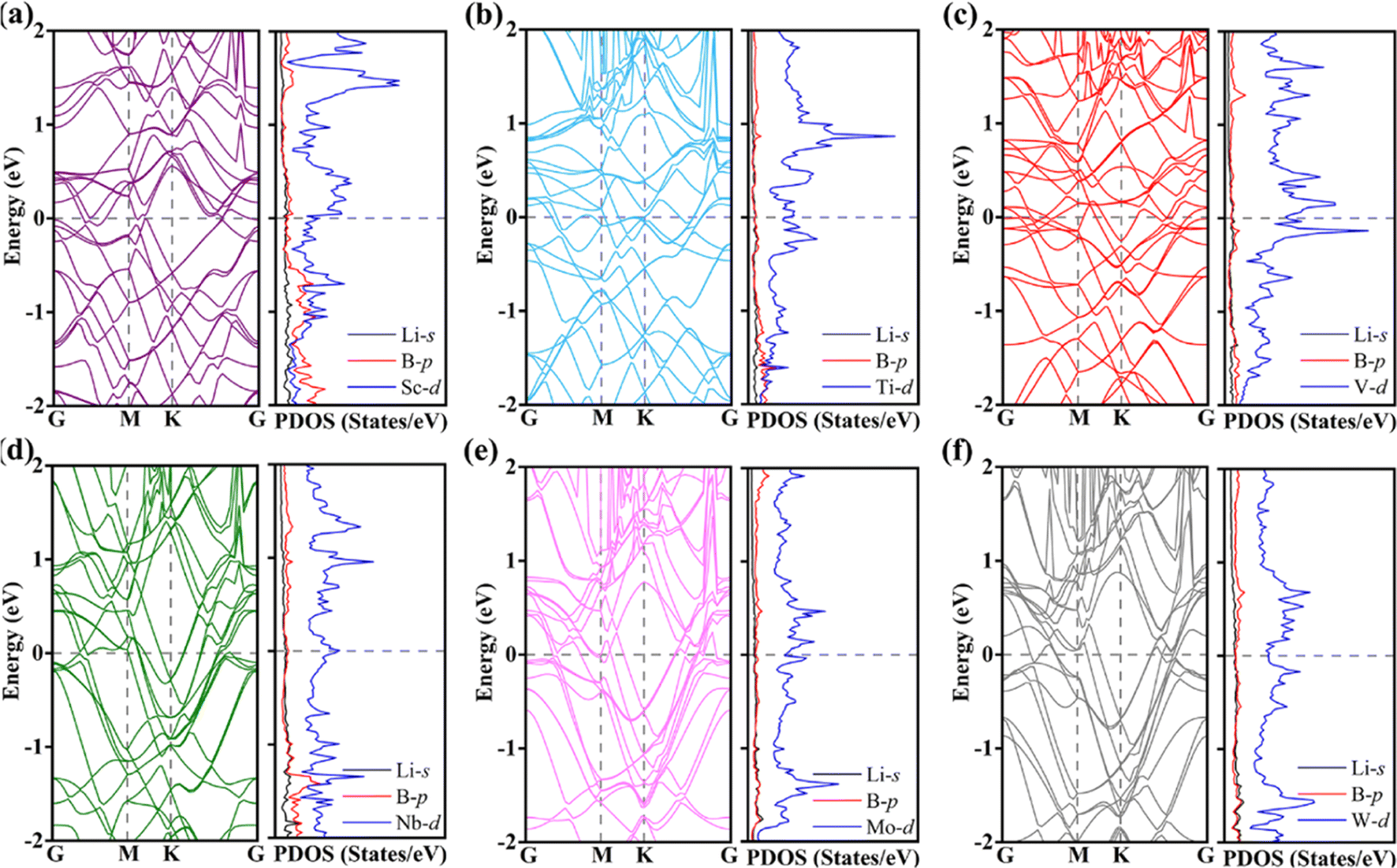 | ||
| Fig. 7 (a–f) Energy band structures and PDOS of the Li4MB2 (M = Sc, Ti, V, Nb, Mo, and W) monolayers. The horizontal black dashed lines represent Fermi levels. | ||
4. Conclusions
Based on the first-principle calculations, we have investigated the potential of MB2 (M = Sc, Ti, V, Nb, Mo and W) as anode materials for LIBs. The surface of MB2 exhibited medium adsorption strength and diffusion energy barrier for Li atom, which is conducive to the insertion and extraction of Li-ions during the charge/discharge process. For different adsorption structures, the mean adsorption energy of MB2 is negative. The correlation between the p-band center of the B atomic atom and the adsorption energy of Li atom on the surface of MB2 is quite obvious. As the p-band center undergoes an upward shift towards the Fermi level, a diminution in the adsorption energy is observed. Finally, MB2 can spontaneously adsorb Li atoms, forming Li4MB2. The theoretical specific capacity range of Li atoms adsorbed on MB2 monolayers (M = Ti, V, Nb, Mo and W) ranges from 521.77 to 1610.20 mA h g−1. These values surpass those of Ti3C2 (488 mA h g−1), V2CS2 (301.12 mA h g−1) and other conventional anode materials. The average voltage range of MB2 falls within the ideal energy range for anode materials (0.10–1.00 V). The results confirm that MB2 monolayer is a promising LIBs electrode material. Additionally, these studies can provide some clues for the design of other high-capacity metal diboride anodes, and inject new impetus for further experimental and theoretical research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51701152, 22002116), the Natural Science Foundation of Shaanxi Province (2018JQ5181).References
- J. Liu, J.-G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. Li, Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS.
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. Proietti Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- E. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng, A. van der Ven, V. Srinivasan, R. Kostecki and G. Ceder, J. Phys. Chem. Lett., 2010, 1, 1176–1180 CrossRef CAS.
- X. Fan, W. T. Zheng, J.-L. Kuo and D. J. Singh, ACS Appl. Mater. Interfaces, 2013, 5, 7793–7797 CrossRef CAS PubMed.
- Z. E. Hughes and T. R. Walsh, Nanoscale, 2015, 7, 6883–6908 RSC.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- H. Ding, Y. Li, M. Li, K. Chen, K. Liang, G. Chen, J. Lu, J. Palisaitis, P. O. Å. Persson, P. Eklund, L. Hultman, S. Du, Z. Chai, Y. Gogotsi and Q. Huang, Science, 2023, 379, 1130–1135 CrossRef CAS PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Å. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X.-H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Piñero, P.-L. Taberna, P. Simon and Q. Huang, Nat. Mater., 2020, 19, 894–899 CrossRef CAS PubMed.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- D. Wang, C. Zhou, A. S. Filatov, W. Cho, F. Lagunas, M. Wang, S. Vaikuntanathan, C. Liu, R. F. Klie and D. V. Talapin, Science, 2023, 379, 1242–1247 CrossRef CAS PubMed.
- Z. Guo, J. Zhou and Z. Sun, J. Mater. Chem. A, 2017, 5, 23530–23535 RSC.
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed.
- G. Zhang, X. Li, K. Chen, Y. Guo, D. Ma and K. Chu, Angew. Chem., Int. Ed., 2023, 62, e202300054 CrossRef CAS PubMed.
- Y. An, S. Gong, Y. Hou, J. Li, R. Wu, Z. Jiao, T. Wang and J. Jiao, J. Phys.: Condens. Matter, 2020, 32, 055503 CrossRef CAS PubMed.
- J. Wang, M. Khazaei, M. Arai, N. Umezawa, T. Tada and H. Hosono, Chem. Mater., 2017, 29, 5922–5930 CrossRef CAS.
- J. Jia, B. Li, S. Duan, Z. Cui and H. Gao, Nanoscale, 2019, 11, 20307–20314 RSC.
- B. Zhang, L. Fan, J. Hu, J. Gu, B. Wang and Q. Zhang, Nanoscale, 2019, 11, 7857–7865 RSC.
- M. Wan, S. Zhao, Z. Zhang and N. Zhou, J. Phys. Chem. C, 2022, 126, 9642–9651 CrossRef CAS.
- G. Barik and S. Pal, Phys. Chem. Chem. Phys., 2023, 25, 17667–17679 RSC.
- S. Luo, J. Zhao, Y. Wang, Y. Zhang, Y. Xiong, N. Ma and J. Fan, J. Phys. Chem. C, 2023, 127, 12484–12491 CrossRef CAS.
- A. Varma, R. Badam, A. L. James, K. Higashimine, K. Jasuja and N. Matsumi, ACS Appl. Nano Mater., 2022, 5, 16154–16163 CrossRef CAS.
- C. Zhang, T. He, S. K. Matta, T. Liao, L. Kou, Z. Chen and A. Du, J. Phys. Chem. Lett., 2019, 10, 2567–2573 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- P. G. Klemens and D. F. Pedraza, Carbon, 1994, 32, 735–741 CrossRef CAS.
- J. Gu, S. Magagula, J. Zhao and Z. Chen, Small Methods, 2019, 3, 1800550 CrossRef.
- J. Hu, B. Xu, C. Ouyang, S. A. Yang and Y. Yao, J. Phys. Chem. C, 2014, 118, 24274–24281 CrossRef CAS.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- Y. Li, T. Zhao, L. Li, R. Huang and Y. Wen, Phys. Rev. Mater., 2022, 6, 045801 CrossRef CAS.
- Y. Wu and J. Hou, Phys. Chem. Chem. Phys., 2022, 24, 14953–14963 RSC.
- X. Zhou, X. Chen, C. Shu, Y. Huang, B. Xiao, W. Zhang and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 41169–41181 CrossRef CAS PubMed.
- R. Li, Y. Wang, L.-C. Xu, J. Shen, W. Zhao, Z. Yang, R. Liu, J.-L. Shao, C. Guo and X. Li, Phys. Chem. Chem. Phys., 2020, 22, 22236–22243 RSC.
- M. Xie, J. Pang, W. Jin and X. Kuang, J. Phys. Chem. C, 2022, 126, 17474–17481 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- H. R. Jiang, Z. Lu, M. C. Wu, F. Ciucci and T. S. Zhao, Nano Energy, 2016, 23, 97–104 CrossRef CAS.
- M. Fang, J. Han, S. He, J.-C. Ren, S. Li and W. Liu, J. Am. Chem. Soc., 2023, 145, 12601–12608 CrossRef CAS PubMed.
- J. Gu, Z. Zhao, J. Huang, B. G. Sumpter and Z. Chen, ACS Nano, 2021, 15, 6233–6242 CrossRef CAS PubMed.
- S. Muy, J. Voss, R. Schlem, R. Koerver, S. J. Sedlmaier, F. Maglia, P. Lamp, W. G. Zeier and Y. Shao-Horn, iScience, 2019, 16, 270–282 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Zheng, J. Li, L. Wang, H. Liu, J. Wang, L. Zhang and X. Chen, Int. J. Hydrogen Energy, 2024, 50, 1555–1561 CrossRef CAS.
- K. P. S. S. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, J. Phys. Chem. C, 2015, 119, 15041–15046 CrossRef CAS.
- X. Zhang, Z. Yu, S.-S. Wang, S. Guan, H. Y. Yang, Y. Yao and S. A. Yang, J. Mater. Chem. A, 2016, 4, 15224–15231 RSC.
- S. Zhao, W. Kang and J. Xue, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- M. S. Manju, S. Thomas, S. U. Lee and A. Kulangara Madam, Appl. Surf. Sci., 2021, 541, 148417 CrossRef CAS.
- M. Boroun, S. Abdolhosseini and M. Pourfath, J. Phys. D: Appl. Phys., 2019, 52, 245501 CrossRef CAS.
- G.-C. Guo, D. Wang, X.-L. Wei, Q. Zhang, H. Liu, W.-M. Lau and L.-M. Liu, J. Phys. Chem. Lett., 2015, 6, 5002–5008 CrossRef CAS PubMed.
- K. Belasfar, M. Houmad, M. Boujnah, A. Benyoussef and A. E. L. Kenz, J. Phys. Chem. Solids, 2020, 139, 109319 CrossRef CAS.
- Z. Zhang, X. Liu, B. I. Yakobson and W. Guo, J. Am. Chem. Soc., 2012, 134, 19326–19329 CrossRef CAS PubMed.
- S.-P. Huang, J.-F. Gu, Y.-R. Ren, K.-N. Ding, Y. Li, Y.-F. Zhang, S.-P. Huang, W. Lin and W.-K. Chen, J. Phys. Chem. C, 2022, 126, 5283–5291 CrossRef CAS.
- J. Zhang, L. Xu, C. Yang, X. Zhang, L. Ma, M. Zhang and J. Lu, Appl. Surf. Sci., 2020, 510, 145493 CrossRef CAS.
- J.-H. Li, J. Wu and Y.-X. Yu, J. Phys. Chem. C, 2021, 125, 3725–3732 CrossRef CAS.
- Y.-X. Yu, J. Mater. Chem. A, 2013, 1, 13559–13566 RSC.
- Y. Kadioglu, Phys. Chem. Chem. Phys., 2023, 25, 1114–1122 RSC.
- S. U. D. Shamim, A. A. Piya, M. S. Rahman, S. M. Hasan, M. K. Hossain and F. Ahmed, Phys. Chem. Chem. Phys., 2023, 25, 4047–4061 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00287c |
| This journal is © The Royal Society of Chemistry 2024 |