
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring acetaminophen prodrugs and hybrids: a review
Vladimir V. Kouznetsov *
*
Laboratorio de Química Orgánica y Biomolecular, Escuela de Química, Universidad Industrial de Santander, Cl. 9 # Cra 27, A.A. 680006, Bucaramanga, Colombia. E-mail: kouznet@uis.edu.co; vkuznechnik@gmail.com
First published on 22nd March 2024
Abstract
This critical review highlights the advances in developing new molecules for treating pain syndrome, an important issue for human health. Acetaminophen (APAP, known as paracetamol) and nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used in clinical practice despite their adverse effects. Research is being conducted to develop innovative drugs with improved pharmaceutical properties to mitigate these effects. A more practical way to achieve that is to study well-known and time-tested drugs in their molecular combinations. Accordingly, the present work explores APAP and their combined chemical entities, i.e., prodrugs (soft drugs), codrugs (mutual prodrugs), and hybrids. Due to their molecular structure, APAP prodrugs or codrugs could be considered merged or conjugated hybrids; all these names are very fluid terms. This article proposed a structural classification of these entities to better analyze their advances. So, the following: carrier-linked O-modified APAP, -linked N-modified APAP derivatives (prodrugs), and direct- and spacer-N,O-linked APAP hybrids (codrugs) are the central parts of this review and are examined, especially ester and amide NSAID–APAP molecules. The C-linked APAP and nitric oxide (NO)-releasing APAP hybrids were also briefly discussed. Prime examples of APAP-based drugs such as propacetamol, benorylate, acetaminosalol, nitroparacetamol, and agent JNJ-10450232 weave well into this classification. The proposed classification is the first and original, giving a better understanding of the SAR studies for new pain relievers research and the design development for the analgesic APAP-(or NSAID)-based compounds.
Introduction
Pain considerably impacts the patient's biological, psychological, sociological, and economic welfare, which cannot be ignored. Being a significant clinical problem, pharmacologic therapy for pain has received increasing attention in recent years.1–3 Nevertheless, its adequate treatment has yet to be attained poorly, with only around one in four patients obtaining significant pain assistance.4 This treatment, usually centered on pharmacological therapy, is supplied by dispensing familiar analgesic agents such as acetaminophen (APAP), nonsteroidal anti-inflammatory drugs (NSAIDs), and opioid analgesics.5 Single doses of acetaminophen display analgesic activity in various acute pain syndromes; thus, its combination with other analgesics helps ease the pain.6Acetaminophen (N-acetyl-P-aminophenol), widely known in the USA (also branded in Europe as paracetamol, from the shortened form of para-acetyl-amino-phenol), is one of the most used drugs both over-the-counter and on prescription for pain (analgesic) and fever (antipyretic) in adults and children.7 WHO has included APAP in the list of essential medicines that are the most effective, safe, and economical for priority conditions. The antipyretic properties of APAP are also highly valued, and the versatility of the mechanism of action and therapeutic effects determine a wide range of applications for various diseases. Due to its hepatic effects and moderate chemical stability, aqueous solubility, etc., research is underway to develop innovative APAP-based drugs for treating pain syndrome, and possibly, new molecules will be synthesized in the next decade. However, the design and discovery of novel drug candidates represent the initial and, probably, the most vital step in the small molecular drug development process. It takes over 10–15 years with an average cost of over $1–3 billion for each new drug to be approved for clinical use.8–10 Thus, this identification step of new chemical leads, i.e., lead optimization through molecular modification strategy, are very risky, complex, and expensive processes. Noteworthy is the fact that the concepts of structural hybridization, prodrugs (soft drugs), and codrugs (mutual prodrugs) contribute a lot to the molecular optimization strategy.11–15 These promising designs possess several advantages, such as reduced adverse reactions and toxicity, improved patient compliance, and a simpler pharmacokinetic profile, which reduces drug–drug interactions and should assist the drug development process.
On the other hand, searching for new drugs commonly suggests the interactions of a single selective molecule, a pharmacophore unit, and a single target for a single mechanism.16 That is the current paradigm of drug discovery, based on the classic approach from Emil Fischer's “lock-key” model (1884) or Paul Ehrlich's “magic bullet” concept (1900). Therefore, a molecular drug (“one-molecule” or active pharmaceutical ingredient, API) goes straight to its intended cell-structural target (“one-target”).17 However, growing evidence that numerous small molecular drugs exert their effects through interactions with multiple targets improves drug research development that confronts the data reductionism approach.18–20 Indeed, pain syndromes like neurodegenerative, cancer, lipid, and psychiatric disorders, metabolic/cardiovascular diseases, infectious diseases, and multifactorial natures cannot be efficiently cured by single-target drug treatment. In this case, agents that can modulate multiple targets simultaneously (polypharmacology) are needed.
Born and developed on the pharmacology basis of the interaction of receptors and selective ligands, i.e., the bivalent ligand approach described by Portoghese,21 the multifunctional ligands concept has been intensively exploited since the 2000s, when Morphy et al. and others highlighted and analyzed the small molecules a single chemical entity that acts at multiple targets. Such an entity often delivers superior efficacy against complex diseases compared to compounds with high specificity for a single target.22–25 This strategy (multimodal APIs, one pill) is a working model for the rational design and development of novel drugs. Having the same idea and goal, different terms for the constructed bioactive molecules are being used. Thus, in current literature, the following terms are found for such experimental therapeutics: “dual-acting compounds”, “designed multiple ligands”, “multitarget-directed compounds”, “single molecule multiple targets”, or “hybrid ligands”, and others. There are so many names, but the molecular essence is the same, i.e., a single chemical entity with the combined two or more pharmacophore units (so-called multiple magic bullet) to modulate multiple targets simultaneously.26 In this work, molecular hybrid terms are used to describe such experimental therapeutics.
Therefore, due to the increasing interest in developing multiple-target approaches for rational drug discovery and the APAP drug role in medicine (pain relief and Parkinson's disease control), APAP is a spotlight of discussion in this work focused on exploring its derivatives and their combinations. Therefore, diverse APAP derivatives and their combined chemical entities (APAP prodrugs, codrugs, and hybrids) are surveyed, trying to structurally classify and examine them. Additionally, the NO-releasing APAP hybrids were also discussed. The present work did not include metal complexes with APAP and APAP co-crystals.
Acetaminophen drug: historical background and mechanisms of actions
Its active ingredient, N-acetyl-p-aminophenol 3 (Fig. 1), a functionalized benzenoid aromatic compound, was synthesized from p-nitrophenol 1 using tin in glacial acetic acid in 1878 by H. N. Morse, an American chemist. Then, it was rediscovered in 1888 and later recognized in 1949 as the main and active metabolite of phenacetin 2. This is the first non-opioid analgesic without anti-inflammatory properties. N-Acetyl-p-aminophenol (APAP or paracetamol) was introduced as an analgesic and fever-reducing drug only in the 1950s under the trade name Tylenol (USA) and Padanol (Britain). They are still used around the world. Interestingly, the first observation (1893) of APAP activity was that acetaminophen was inferior to phenacetin. It later turned out to be a mistake, possibly mediated by impurities, but this contributed to widespread use of phenacetin in the first half of the XX century until the 1970s when it was withdrawn from the market in most countries due to adverse effects on the kidneys (nephrotoxicity and carcinogenesis).27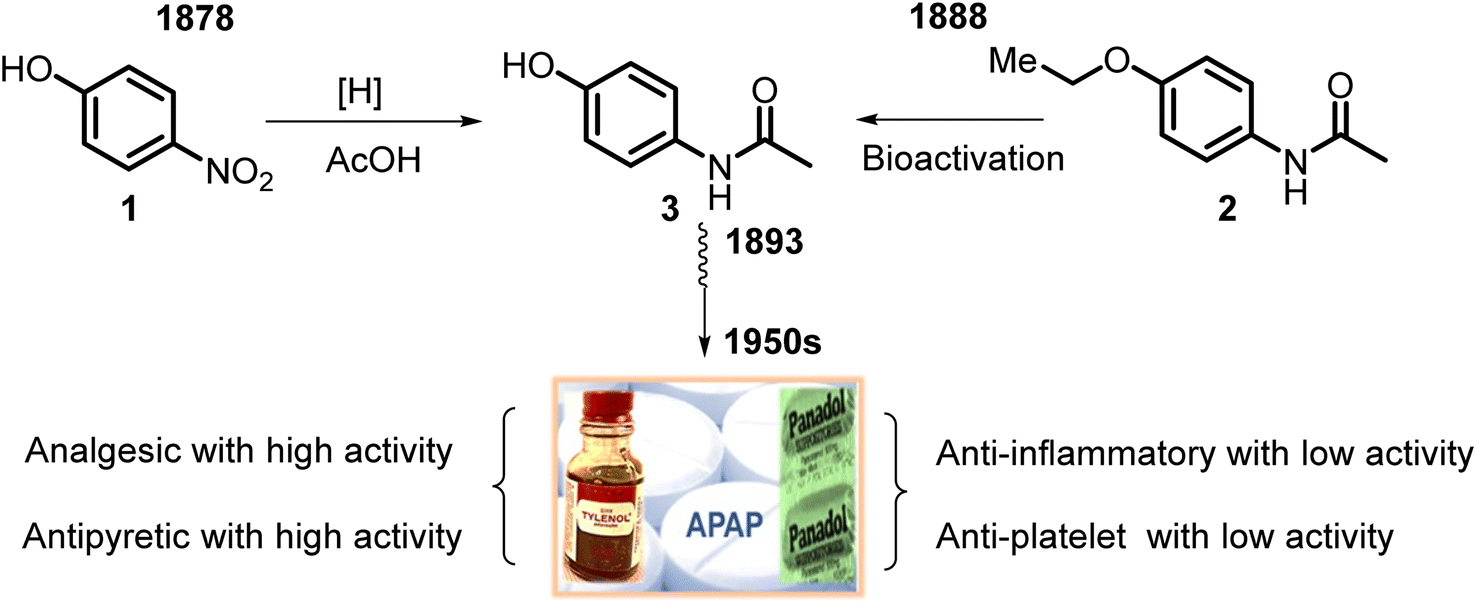 | ||
| Fig. 1 Graphical representation of APAP discovery and its main clinical pharmacological activities. APAP was synthesized in 1878 by Morse, and in 1887, von Mering used it clinically but discarded it because of the false assumption of its toxicity. In 1948, Brodie and Axelrod indicated the practical usefulness of the APAP (“APAP rediscovery”) that led to marketing in the 1950s in the United States. | ||
Thus, APAP is a member of the aniline family of analgesics, having a phenol natural. It is better tolerated as an analgesic than the non-steroidal anti-inflammatory drugs (NSAIDs), although it may be somewhat less efficacious. It is normally employed to release mild pain, such as headache and toothache, and is a preferred alternative to aspirin, particularly for patients who cannot tolerate aspirin.28,29 Its clinical pharmacological activities include potent analgesic and antipyretic effects, minimal anti-inflammatory activity, and minor antiplatelet effects. Also, APAP shows anti-nociceptive effects7 (Fig. 1). APAP is safe to take, without stomach and small intestine irritant effects.7,30,31
The mechanisms of the basic pharmacological effects of acetaminophen are not completely known and could encompass peripheral and central nervous system sites of action.30,32 However, the role of APAP as an inhibitor of the prostanoid (prostaglandins-PGs, prostacyclin, and thromboxane) synthesis from the arachidonic acid (ACA) was studied more. APAP has an apparent selectivity for the cyclooxygenase (COX-1 and COX-2) enzymes, which have two sites: a COX site at the hydrophobic channel in the core of the enzyme and peroxidase (POX) site at the heme-containing active site at the protein surface28,33–36 (Fig. 2). Noteworthy that COX exists in two isoforms, constitutive COX-1, responsible for physiological functions and inducible COX-2, which is involved in the inflammation process. Unlike other non-selective (ibuprofen, ketoprofen, naproxen, etc.) and selective (“coxibs”) NSAIDs that block both COX-1/COX-2 and only COX-2, respectively, APAP appears to inhibit reversibly endoperoxide H2 synthase (PGHS) activity through its capacity to serve as a reducing co-substrate for the POX active site.
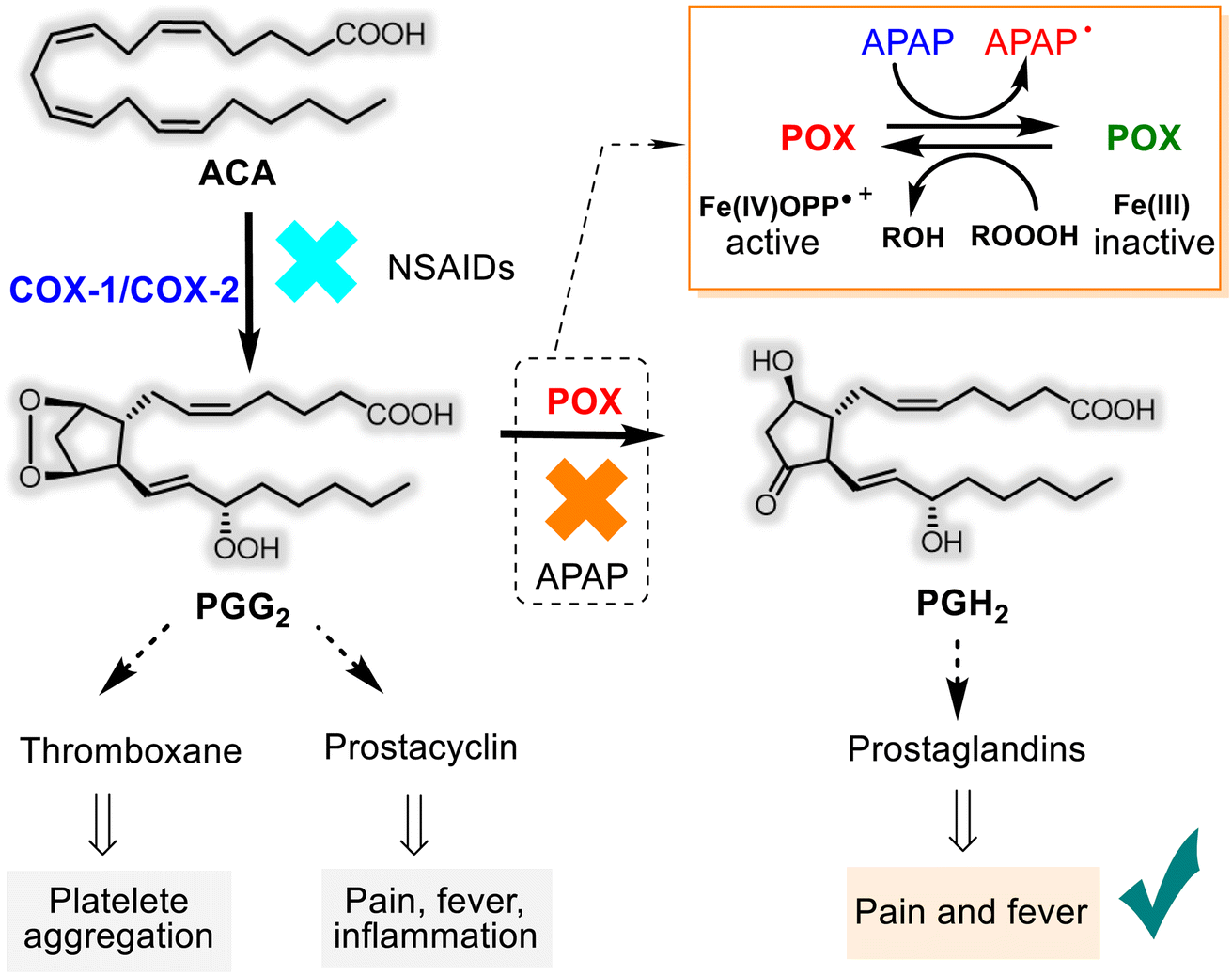 | ||
| Fig. 2 The predominant hypothesis on the mechanism of APAP action mode is the so-called “COX-associated Peroxidase Hypothesis”. The biosynthesis of PGs from ACA involves a key bifunctional enzyme containing two separate catalytic domains that are responsible for converting arachidonic acid to PGH2: (i) a cyclooxygenase domain that engenders an unstable peroxide intermediate (PGG2) that is blocked by non-selective and selective NSAIDs and (ii) a peroxidase (POX) domain containing a heme group that converts the unstable intermediate to PGH2 where APAP inhibits the POX catalytic step by converting its heme group (Fe(IV)OPP˙+) to an inactive reduced state, – Fe(III). | ||
Consequently, APAP has little or no anti-platelet or anti-inflammatory effects. In contrast, it can exert antipyretic and analgesic effects by blocking the production of prostaglandins (Fig. 2). It's easier to say that the first step of the ACA transformation to PGG2 depends on the cyclooxygenase enzymes. But, their generation, in turn, relies on the availability of ferryl protoporphyrin IX radical cation (Fe(IV)OPP˙+) belonging to the POX site. As it works at the second step of PG bioformation, both sites cooperate in the PGH2 production, and APAP obstructs this cooperation. This hypothesis on the mechanism of APAP action mode is called the “COX-associated Peroxidase Hypothesis” and is predominant.
Another theory, the so-called “Cannabinoid Hypothesis”, is that APAP affects CNS, where the COX-3 isoenzyme is present and inhibited by the APAP metabolite (N-arachidonoyl-4-aminophenol, AM404). The latter interacts with the endocannabinoid system through both direct and indirect activation of cannabinoid receptor type 1 (CB1R)37 and transient receptor potential vanilloid-1 receptor (TRPV1), being a weak agonist.38,39 Both hypotheses could be true and complementary. But that's not all. The anti-nociceptive effects of APAP can be related to the endogenous neuronal pain-inhibitory systems, principally through descending serotonin and noradrenaline neurotransmitters, as well as endogenous cannabinoids and opioids, whose effects are inhibited by APAP. The activation of the descending systems significantly modifies the release of glutamate and the discharge of GABA and glycine.7,40
APAP is a desired drug for treating pain and fever in patients allergic to aspirin because salicylates are poorly tolerated. It is also preferred to aspirin for the treatment of pain/fever in children with viral infections. However, its use is among the most common causes of worldwide poisoning. According to the U.S. Acute Liver Failure Study Group, APAP overdose is the leading cause of calls to Poison Control Centers (>100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per year).41
000 per year).41
In addition, it is a drug whose use is counted in thousands of tons, and its environmental presence and hazardous impact on non-target organisms could rapidly increase other pharmaceutical agents.42
APAP metabolism and roles of its metabolites
Being a low-molecular-mass compound and a weak acid (pKa 9.7), APAP is unionized at human physiological pH values (7.35–7.45). With an ideal partition coefficient between octanol and water (3.2), APAP, a moderately lipid-soluble molecule, is rapidly absorbed from the gastrointestinal tract and quickly distributed throughout the body, going through cell membranes by passive diffusion mode, and its binding to plasma proteins is insignificant. Thus, it is well absorbed and reaches the peak blood concentrations within 90 minutes after ingestion. On the other hand, APAP crosses easily the blood–brain barrier and is distributed homogeneously throughout the central nervous system (CNS).APAP metabolism mainly occurs within liver microsomes at the microscopic level. Three main hepatic pathways are glucuronidation, sulfation, and P450 oxidation.39,43,44 After a therapeutic dose (for adults is 2 tablets of 500 mg, each taken orally every 4 hours up to a maximum of 8 tablets for any 24 hours), APAP has generally transformed into two pharmacologically inactive forms: harmless glucuronide (APAP–Glu) and sulfate (APAP–SO3H) conjugates. That occurs with UDP-glucuronosyl transferases (UGT) and sulfotransferases (SULT), corresponding to almost 80–90% of the absorbed APAP. A small fraction (<5%) of unchanged APAP reaches the urine. The remaining fraction (5–15%) is oxidized by the P450 (CYP 450) mixed-function oxidase system (primarily CYP2E1) to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) (Fig. 3). However, NAPQI is also generated in the brain by the CYP450 oxidation.
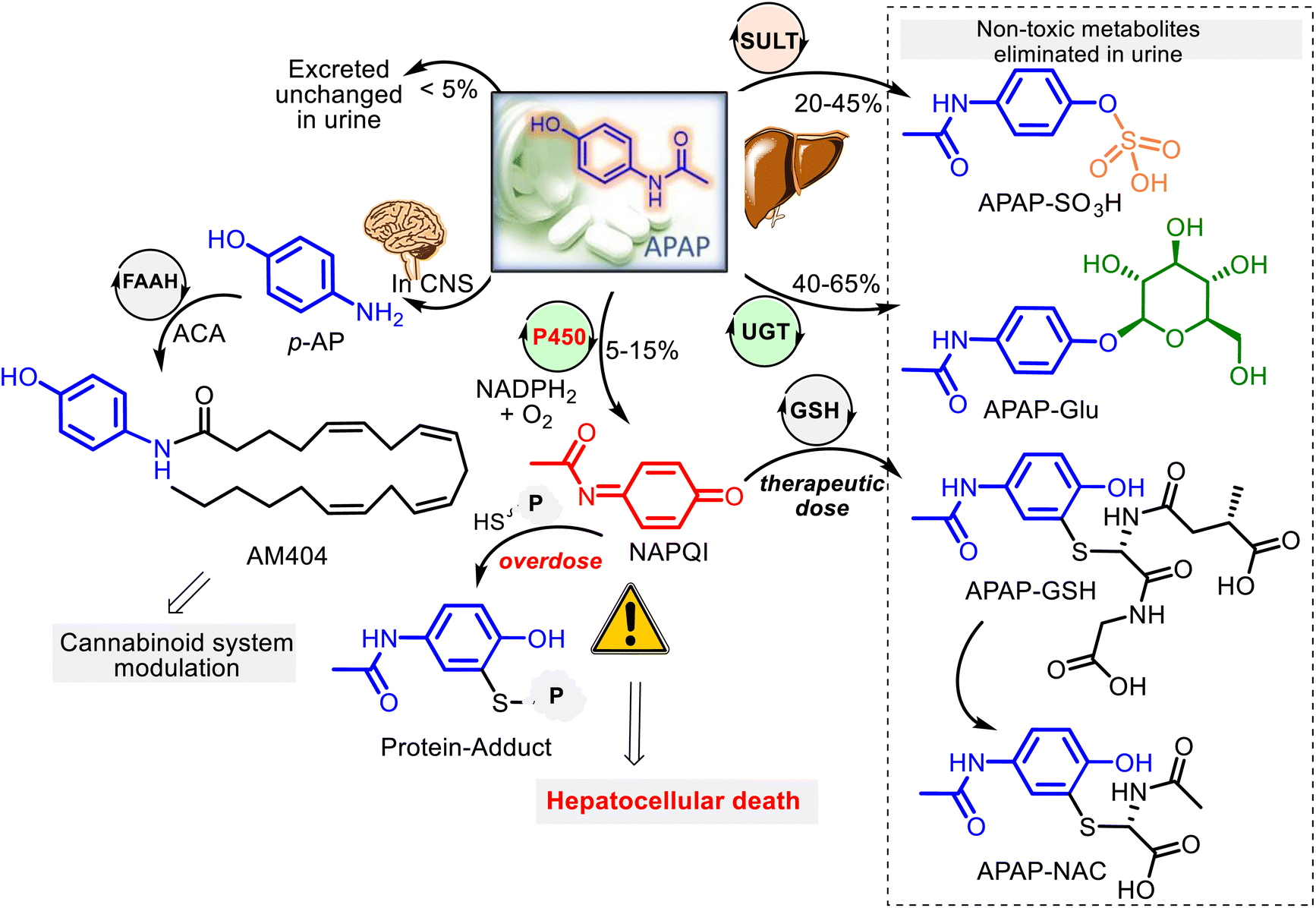 | ||
| Fig. 3 Schematic representation of the predominant theories on the APAP metabolism in humans. Liver APAP metabolism. Main metabolic pathways of APAP in the liver after administration of therapeutic or toxic doses. Glucuronidation (non-toxic APAP–Glu formation) is the main pathway of acetaminophen metabolism, followed by sulfation (non-toxic APAP–SO3H formation) and a minor contribution from the oxidation route (toxic NAPQI formation). Rapid conjugation of NAPQI to glutathione (GSH) allows detoxification of NAPQI through non-toxic glutathione or cysteine conjugates, which are eliminated in urine. CNS (brain) APAP metabolism. APAP deacetylation process to produce para-aminophenol (p-AP). Its conjugation with arachidonic acid by fatty acid amide hydrolase (FAAH) enzymes forms N-arachidinoyl-phenolamine (AM404), which exhibited TRPV1 agonism with an EC50 value of 26 nm. | ||
In addition to the described pathways of acetaminophen metabolism, acetaminophen might undergo a deacetylation process. Animal studies revealed that the deacetylation reaction of APAP by the liver enzyme N-deacetylase could produce a minor metabolite p-aminophenol (p-AP), which was found to cause nephrotoxicity in rodent models.45–47 Still, in humans, it was shown that when this metabolite forms in the liver, it enters the brain. In the brain and the cerebrospinal fluid, p-AP is conjugated with arachidonic acid (ACA) by the intracellular enzyme fatty acid amide hydrolase (FAAH) to form an active metabolite AM404, capable of triggering the CB1R and the TRPV-1 channels. Both channels are involved in the modulation of pain signaling and are proposed to mediate the central analgesic action of APAP (Fig. 3).32,48–50 Thus, despite being in clinical use for over a century, the precise mechanism of action of the familiar drug APAP remains a mystery. Additionally, APAP and its metabolite, NAPQI, were found to extend the therapeutic window of L-DOPA treatment for Parkinson's disease (PD) through selective inhibition of key enzymes monoamine oxidase (MAO), catechol-o-methyltransferase (COMT) and aldehyde dehydrogenase (ALDH), involved in the dopamine pathway and PD.51
Fixed-dose APAP combinations with other drugs in one tablet
A combination of APAP with other drug groups is often used in clinical practice. It helps ease the pain. These drug combinations, i.e., drug cocktails (Fig. 4A), may enhance the therapeutic effect of individual drugs, but there are unfavorable APAP–drug interactions. This APAP-based treatment is supplied by administering familiar analgesic agents such as APAP, NSAID group, and opioid analgesics.52 The improved form of these combinations is a drug combination into one pill (or other formulation). It includes two or more APIs in a single dosage form at a fixed dose, which needs to consider the diverse pharmacokinetics, physiological variations, and drug dose regimes. The so-called multiple-targeting fixed-dose drug combinations (FDC) (Fig. 4B) are cost-effective processes that can assist in reducing pill load and, hence, managerial and manufacturing costs.53,54 FDCs are designed to treat more effectively diverse pathologies.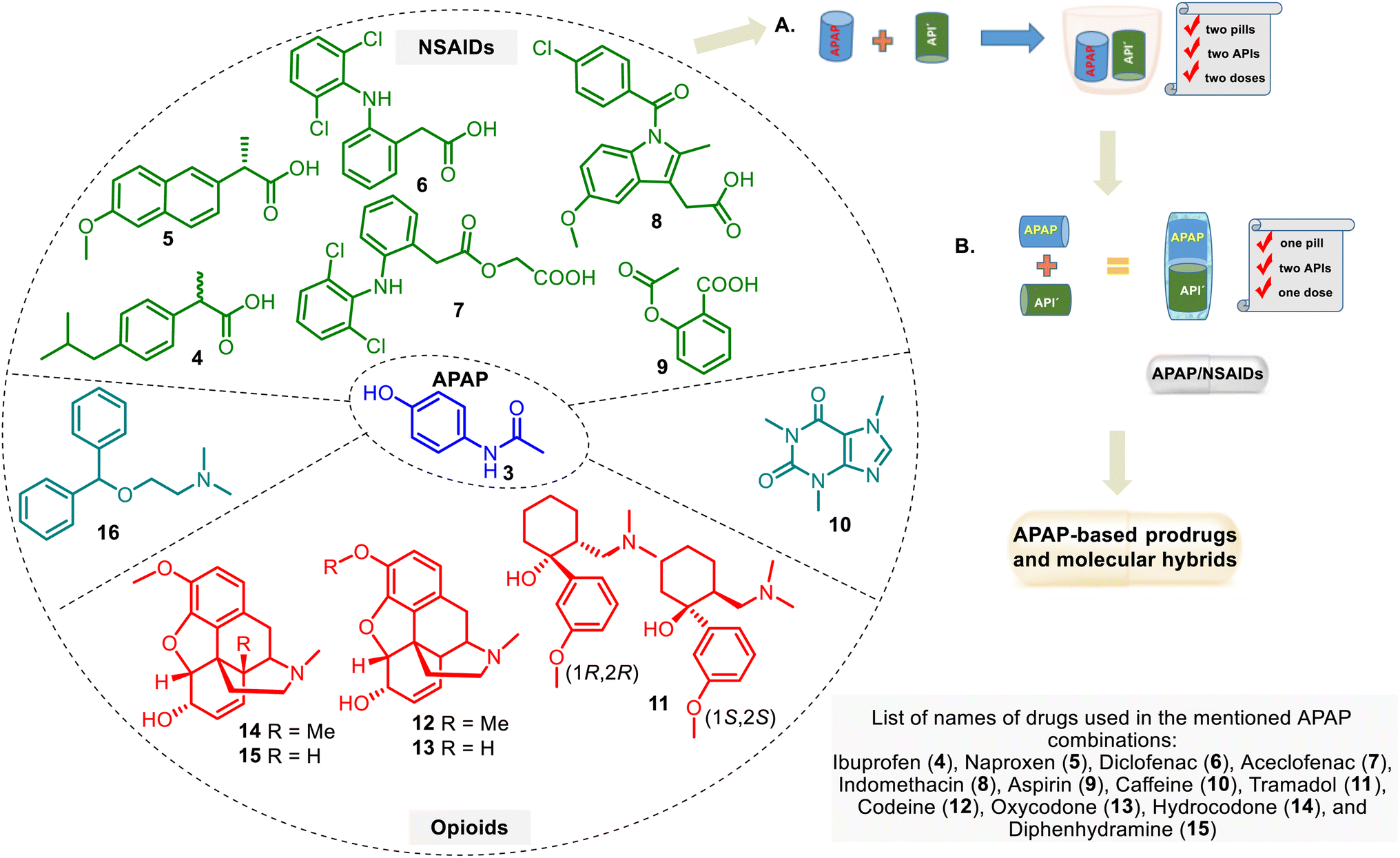 | ||
| Fig. 4 Structures of APIs, which form fixed-dose combinations with APAP and non-opioid analgesics (NSAIDs) or opioid (narcotic) pain relievers, and pictorial representation of their combination: (A) drug cocktails combine more than one drug (API) simultaneously. It can be more effective than single drugs. Drug combinations may enhance the therapeutic effect of individual drugs, while drug–drug interactions are generally unfavorable. (B) The multiple-targeting double-fixed-dose drug combination approach with the participation of APAP and other APIs (API′). | ||
This approach has the following potential advantages: rapid achievement of the desired effect, reduction of toxicity, simple administration, etc.54 However, there are also shortcomings, including stability issues, solubility differences, and incompatibility between the parent drugs.55 Regarding APAP combinations, their therapeutic superiority over either drug remains controversial. Although some APAP formulations are still suspicious by physicians in pain relief, it has been acknowledged that fixed-dose in drug combination with APAP is a natural trend in improving pain treatment.6 In any case, this approach serves as a stimulating and rational basis for developing APAP-based hybrid drugs. The design of these hybrids, perhaps, could help avoid the abovementioned problems of the APAP–FDC approach in managing pain and finding solutions for other diseases.
Prodrugs and molecular hybrids: general aspects
Studying some first drugs, e.g., prontosil, aspirin, or the mentioned phenacetin, scientists recognized that these were transformed in vivo into metabolites more active than themselves; thus, this phenomenon stimulated more drug investigation. When exploring and revising drug behavior at the end of the 1950s, Albert and Harper introduced their respective terms of prodrugs and drug latentiation, which coincide with the release of the API from prodrug form under enzymatic and/or chemical transformation in vivo.56,57 In general, the rationale behind using prodrugs (also known as mutual drugs) is to optimize the ADMET properties of the parent drugs and improve drug targeting.According to the IUPAC definition, a prodrug is a compound that undergoes biotransformation before exhibiting pharmacological effects.16 Depending on the molecular structure of this compound (prodrug), we can find two main groups, i.e., the bioprecursor prodrugs and the carrier-linked prodrugs (Fig. 5).
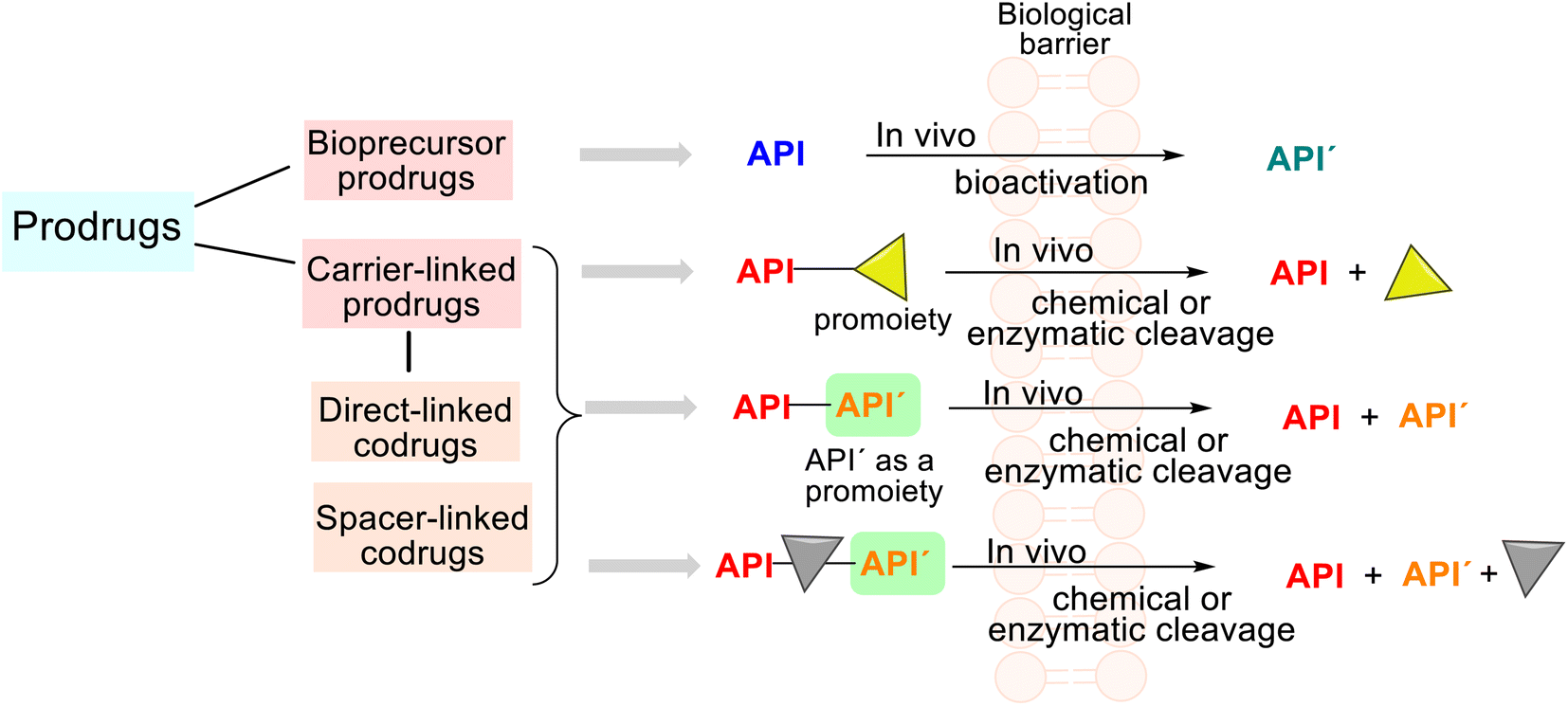 | ||
| Fig. 5 General classification of prodrugs using chemical criteria and a simplified illustration of the prodrug concept. Historically, Albert and Harper introduced the terms prodrugs and latentiated drugs to describe biologically active compounds that undergo biotransformation before revealing their pharmacological effects. Currently, according to their chemical structure, mechanism of activation, and modified functional groups, prodrugs are sorted into a bioprecursor group and carrier-linked prodrugs, also known as conventional prodrugs. | ||
The third group is drug delivery systems not discussed in this analysis. The carrier-linked prodrugs are based on an API temporarily connected to a carrier (a promoiety) through a bioreversible covalent chemical linkage. Such prodrugs undergo biotransformation, releasing the parent API and the carrier (Fig. 5). Certain functional groups (amino, hydroxyl, carbonyl, or carboxylic functions) must be present on molecule drugs, and the carrier moiety that promotes absorption must be present to form that linkage. In contrast, the bioprecursor prodrug (API) does not contain any carriers and provides a new, more active API′ through a molecular metabolic or chemical modification.58,59
Thus, prodrugs are simple chemical derivatives that need only one to two chemical or enzymatic conversion steps to produce the active parent drug. In some cases, a prodrug may comprise two and more APIs bonded via a covalent chemical linkage in a single molecule so that each API acts as a promoiety for the other. Such derivatives are called codrugs and can be classified generally as spacer-linked and direct-linked. Both APIs are usually conjugated directly using an ester/amide bond in the latter.15 These molecular combinations could also be considered conjugated hybrid molecular entities.
Another tactic for discovering novel drug candidates and their improvement is the mentioned molecular hybridization approach based on the multi-pharmacophore hybrid model. According to this model, molecular hybrids are divided into four distinct types, corresponding to the degree of overlapping pharmacophores, i.e., conjugated hybrids, fused hybrids, and merged hybrids12–14 (Fig. 6). The first type of hybrids with increasing molecular size and structural complexity can be connected by non-cleavable (rigid, non-hydrolyzable, or enzymatically stable) spacers or cleavable, flexible linkers. The following spacers esters, amides, carbamates, or disulfide groups are the most used because plasma proteins or others can cleave them to release two separate bioactive ingredients with independent actions.
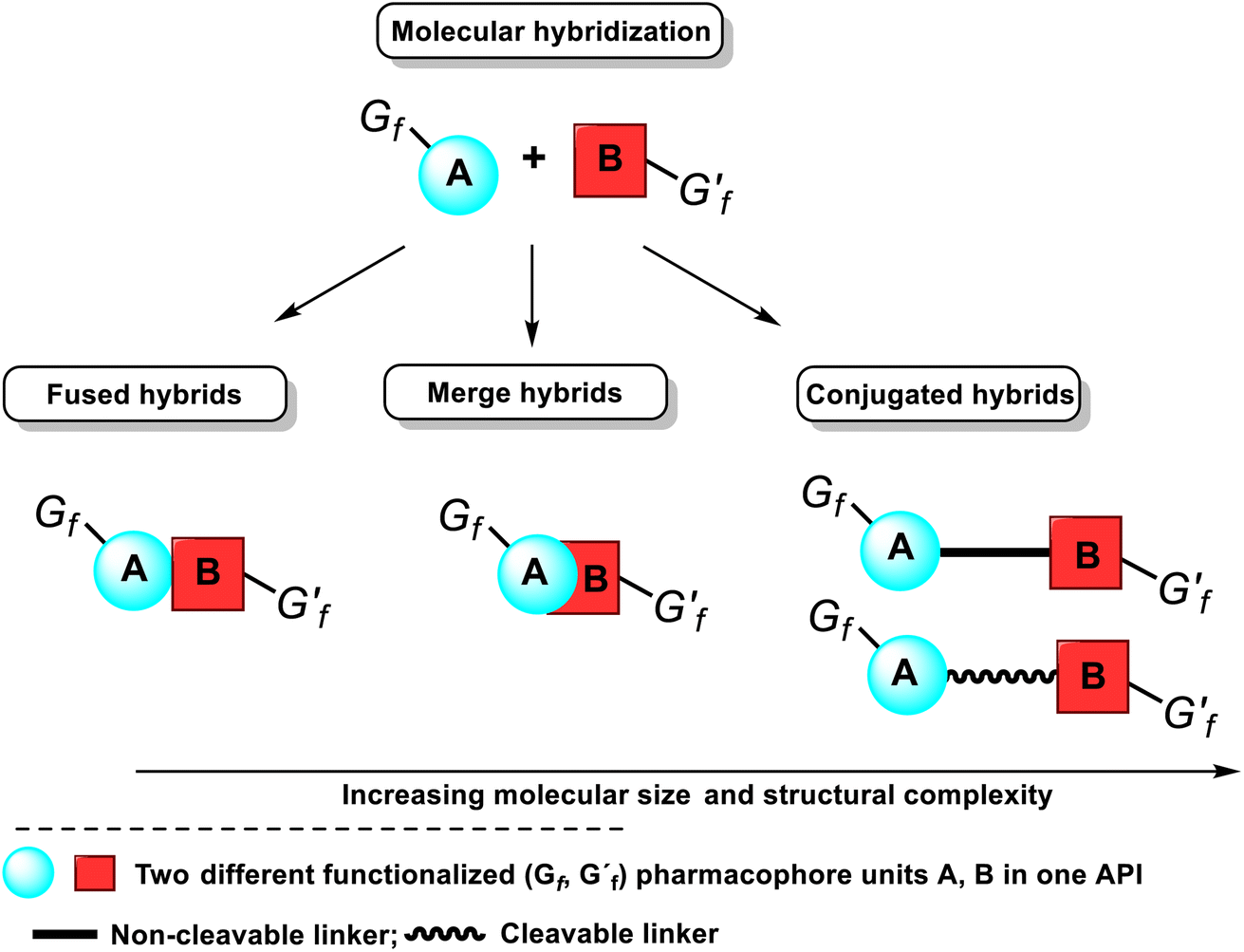 | ||
| Fig. 6 Classification of molecular hybrids and their structural representation and the integration of their pharmacophores in a bioactive single hybrid molecule, i.e., API. | ||
The cleavable hybrid construction aims to increase poor pharmacokinetic properties, i.e., improve the desired activity and slowly deliver the two active units to the respective targets. That enhances the designed hybrid's selectivity. The non-cleavable spacer group can be a single bond, a polymeric chain (generally a methylenic chain), or, in some cases, an aromatic, heteroaromatic, or nonaromatic cycle. The main rationale for exploiting a linker is to provide a bridge to connect two pharmacophore units and modulate the release of individual active units in vivo. That retains their biological activity, specificity, and affinity for their targets. However, using these linkers often leads to hybrid molecules with unfavorable drug-likeness profiles of Lipinski and Veber rules. Fused hybrids are those that contain underlying two or more pharmacophore units without using any spacers, i.e., each molecule A or B provides a functional group to build a link, usually resulting in an enzymatically hydrolyzable ester, carbamate, or amide (Fig. 6). When there is an overlapping structural motif of both pharmacophores of the participating molecules in the hybridization process, molecules are called merged hybrids. This overlapping approach looks more attractive from a biopharmaceutical standpoint.
The mentioned APAP derivatives, so-called molecular hybrids and/or prodrugs with two or more “heads”, in which APAP structure in a “head”, are discussed in the next section.
APAP-prodrugs and -hybrid molecules
APAP prodrugs, simple derivatives of N-(4-hydroxyphenyl)acetamide (4-hydroxy-acetanilide, 3), are old, classic but interesting models in the research for the improvement of ADME characteristics of the parent drug (APAP) modifying undesirable drug properties. Generally, new APAP prodrugs are achieved by modifying the phenolic hydroxyl group (O-modified APAP prodrugs) and the acetamide group (N-modified APAP prodrugs); the first variant is more used.Carrier-linked O/N-modified APAP prodrugs
According to the chemical classification of prodrugs, the structurally diverse carrier-linked O/N-modified APAP prodrugs were found in the literature. Interestingly, note that APAP itself is a metabolic product of the first synthetic analgesic drugs, acetanilide (Antifebrin®) and phenacetin (acetophenetidin), i.e., bioprecursor-type prodrugs (Fig. 7).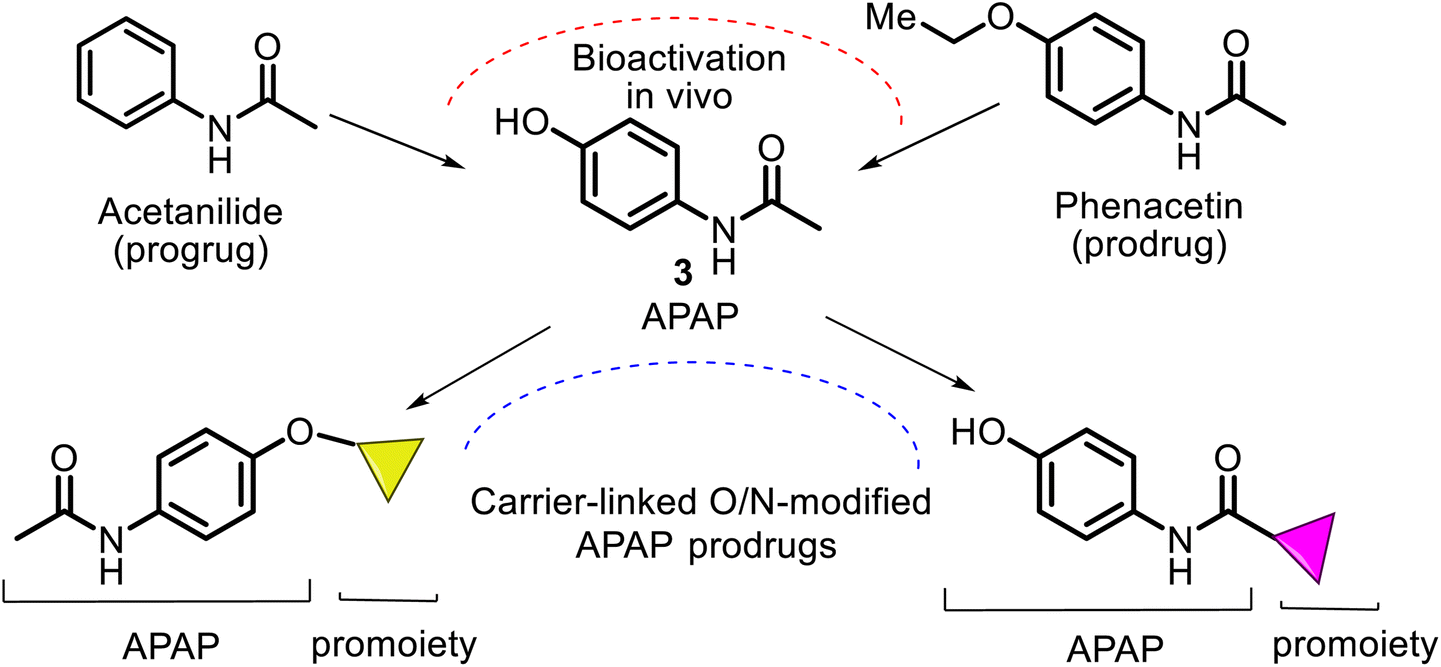 | ||
| Fig. 7 Structures of the first synthetic analgesic drugs, acetanilide and phenacetin, resulted in precursor prodrugs of acetaminophen. Nevertheless, both were not originally designed as prodrugs, so their prodrug nature was determined in hindsight. Both are more toxic (renal toxicity) and less active (antipyretic and analgesic activities) than their metabolite, APAP, which is formed through aromatic hydroxylation and O-de-ethylating processes from the respective acetanilide and phenacetin prodrugs. They and their analogs comprise an APAP ring linked to single carrier moiety and are also known as bipartite prodrugs. | ||
Most of the described works on the APAP modification via a prodrug approach have primarily focused on the corresponding ether or ester, amino acid, or peptide ester carriers. In contrast, sulfate or phosphate ester promoieties are less common. In the design of new carrier-linked O-modified APAP prodrugs, the following types of functional groups or fragments: alkyl- or aryl-carbonyl (AC), alkyl-oxycarbonyl (AOC), alkyl-carbonyl-oxymethyl (ACOM), amino-alkyl-carbonyl (AAC), N,N′-dialkyl-amino-alkyl-carbonyl (DAAC), alkyl-oxycarbonyl-oxymethyl (AOCOM), and N-alkyl-N-alkyl-oxycarbonyl-aminomethyl (NANAOCAM) were employed to improving the poor bioavailability and the low water solubility of APAP (about 1.3 g per 100 mL). That is achieved through balancing aqueous and lipid solubilities (i.e., hydrophilic–lipophilic balance). Structurally, the AOCOM promoiety is formed by inserting a –OCH2– group between the carbonyl carbon and the phenol oxygen, while the NANAOCAM fragment is designed by introducing a –NR′CH2– fragment instead of the –OCH2– group into the AOC promoiety. Generally, these O-modified APAPs comprise an acyl-enabling functional group which, upon hydrolysis, results in an unstable intermediate that spontaneously decomposes into the active form drug.
The use of AC and AOC carriers represents an almost universal strategy of masking negative effects and improving the pharmaceutical properties of drugs. So, in 1968, Dittert et al.60,61 realized the first systematic studies on the relationship between physicochemical properties (lipid and water solubilities, partition coefficients, and in vitro hydrolysis rates) and in vivo biological activity (toxicity and analgesic activity studied in mice and rats) of the carboxylate esters and carbonate esters of APAP 17 and 18 (i.e., 4-AC-APAP and 4-AOC-APAP prodrugs, respectively) (Fig. 8).
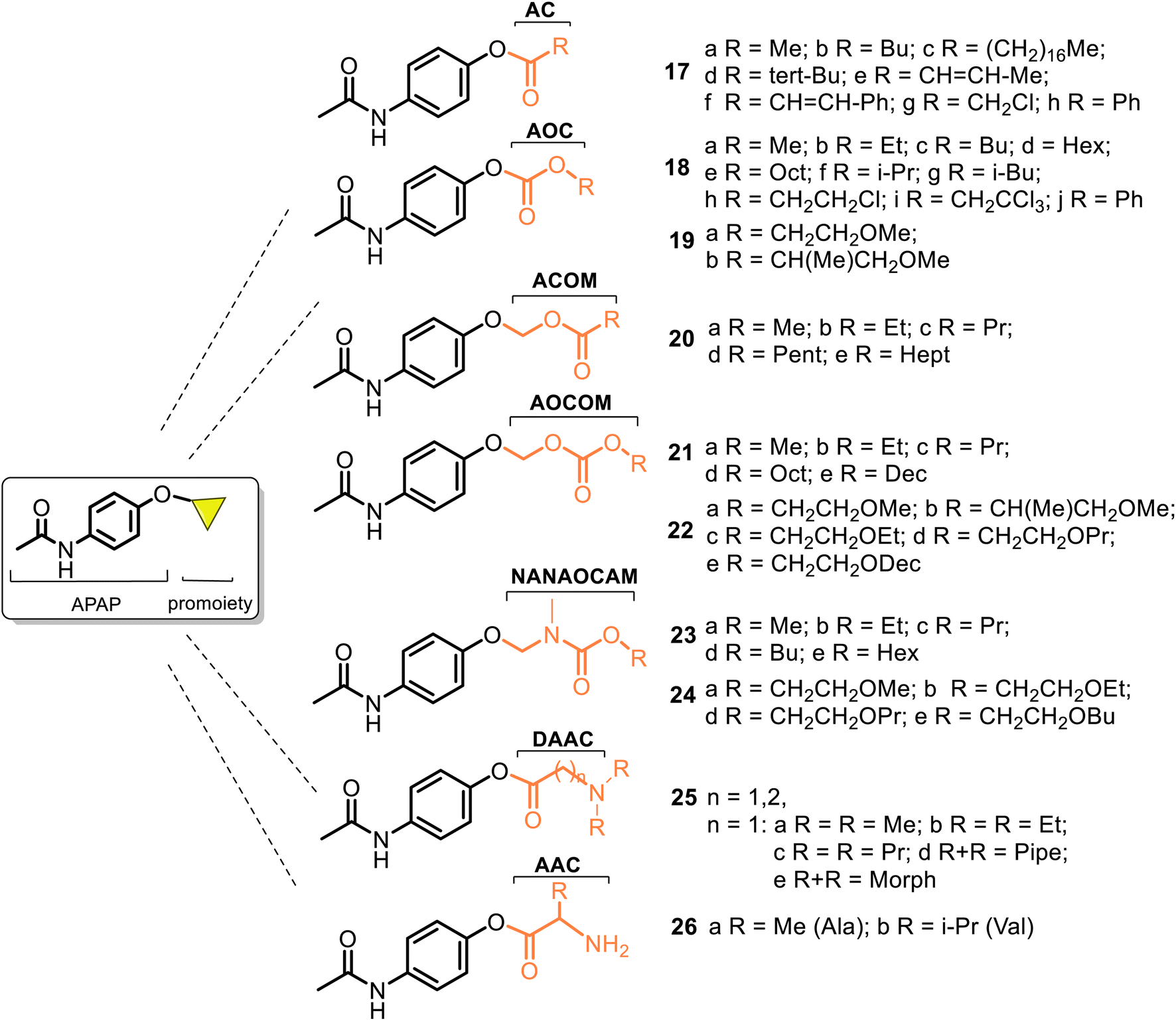 | ||
| Fig. 8 Structures of the reported carrier-linked O-modified APAP prodrugs. The 4-AC-APAP (17) and 4-AOC-APAP (18) series were first examined to be administered orally, while the prodrugs 19–26 were developed and studied as topical analgesics. In general, such phenols, when administered orally, exhibit poor bioavailability. This major problem can be avoided with dermal delivery. Starting the 4-AOC-APAP prodrug 18 studies as topical analgesic agents, the AOC promoiety has been modified to enhance the topical delivery of APAP. Thus, numerous and different ACOM, AAC, DAAC, AOCOM, and NANAOCAM functional groups/fragments in the APAP core. | ||
Their main points and observations were: (1) according to the analgesic and toxicity activity results of the prepared APAP derivatives 17–18, which act as prodrugs, the lipid and water solubilities of these compounds, rather than their enzyme-catalyzed hydrolysis rates, feasibly control the availability of APAP following oral administration and (2) the 4-AOC-APAP prodrugs 18 showed significant analgesic activity, indicating the usefulness of the carbonate linkage for creating new APAP-based drugs. Some important findings of these O-modified APAP prodrug series are given in Table 1.
| APAP prodrug | Promoiety type | Characteristic pharmacologic actions and main remarks |
|---|---|---|
| a In units of μmol cm−2 h−1.b Experimental maximum flux. | ||
| 17 | AC | • Tested for oral delivery60,61 |
| • Prodrugs 17, 18 were much less soluble in water and more soluble in the nonpolar solvent (cyclohexane) than APAP, and thus, their cyclohexane/water partition coefficients were higher than the parent drug. These tendencies in 18 were more pronounced | ||
| 18 | AOC | • They all hydrolyzed slowly in pH 7.4 phosphate buffer; most exhibited half-lives over 24 h. The 4-AOC-APAP prodrugs 18 are susceptible to enzymatic hydrolysis to release free APAP in the tissues of humans and animals |
| • The oral LD50 (toxicity) values for 4-AOC-APAP 18a–c, f, g, and j were 1759, 1450, 1614, 3500, 2150, and 340 mg kg−1, respectively. They were less toxic than APAP (oral LD50 = 1080 mg kg−1). The series 17 was not tested | ||
| • Only carbonates 18i and 18j could show significant analgesic activity | ||
| 18–19 | AOC | • Tested for topical delivery63 |
| • logSIPM (mM): 0.28 (APAP), 1.08 (18a), 0.97 (18b), 1.37 (18, R = Pr), 1.14 (18c), 1.12 (18d), 1.01 (19a), 0.53 (19b) |
||
| • logSAQ (mM): 0.45 (APAP), 1.31 (18a), 0.58 (18b), 0.43 (18, R = Pr), −0.37 (18c), −1.32 (18d), 1.54 (19a),0.52 (19b) |
||
| • logJMIPMa: −0.57 (APAP), −1.08 (18a), −1.73 (18b), −1.82 (18, R = Pr), −2.15 (18c), −2.71 (28d), −1.12 (19a), −1.59 (19b) |
||
| 20 | ACOM | • Tested for topical delivery.64 Showed a considerable improvement in the delivery of APAP compared with the 4-AOC-APAP prodrugs 18–19 |
| • SIPM (mM): 1.9 (APAP), 8.4 (20a), 62.0 (20b), 73.5 (20c), 109 (20d), 98.7 (20e) | ||
| • SAQ (mM): 100 (APAP), 15.2 (20a), 24.7 (20b), 7.1 (20c), 0.5 (20d), 0.06 (20e) | ||
| • JIPMa: 0.51 (APAP), 0.73 (29a), 1.86 (20b), 0.77 (20c), 0.34 (20d), 0.11 (20e) | ||
| • 4-ACOC-APAP prodrugs 20 are more hydrolytically stable than derivatives 18–19 since the leaving group during hydrolysis is an aryl hemiacetal (pKa ∼ 11) rather than a phenol (pKa ∼ 8–10) | ||
| 21–22 | AOCOM | • Tested for topical delivery. The ACOM → AOCOM conversion causes a reduction in lipid and water solubilities with little enhancement65–67 |
| • SIPM (mM): 1.9 (APAP), 7.9 (21a), 20.7 (21b), 45.8 (21c), 66.4 (21d), 130 (21e) | ||
| • SAQ (mM): 100 (APAP), 7.2 (21a), 7.7 (21b), 2.0 (21c), 0.004 (21d), 0.0005 (21e) | ||
| • JMMIPMa,b: 0.51 (APAP), 0.44 (21a), 0.66 (21b), 0.28 (21c), 0.02 (21d), 0.007 (21e) | ||
| 23–24 | NANAOCAM | • Tested for topical delivery.67–69 The pair of series 23 and 24 showed similar properties to those of the respective series 21 and 22 |
| • Only prodrugs 22a and 24a with a 2-methoxyethyleneoxy group showed better maximum flux values than APAP, albeit marginally: 1.41 and 1.36 times, respectively. JMMIPMa,b: 0.51 (APAP), 0.72 (22a), 0.67 (24b) | ||
| 25 | DAAC | • Tested for topical delivery.70 The incorporation of a basic amine functional group into an acyl prodrug is one more step in developing new prodrugs, which appears promising |
| 26 | ACC˙ | • JMMIPMa,b: 0.51 (APAP), 1.05 (25e), 0.54 (25e·HCl) |
Despite the complex relationships between the structure, physicochemical properties, and activity found in these series, the analysis of the 4-AOC-APAPs series 18a–e was very useful. They resulted in good starting points in the research for the topical (dermal and transdermal) delivery of APAP-based analgesics. Sloan and co-workers have contributed much to studying and developing this type of prodrugs.62–70 Starting their research, they examined the 4-AOC-APAPs prodrugs 18 with the C1–C6 alkyl groups and similar derivatives 19 measuring their delivery (i.e., the fluxes, JMIPM) through hairless mouse skin from suspensions of each prodrug in isopropyl myristate (IPM) (Fig. 8).63 The studied compounds 18a–d and 19a–b were more soluble in IPM (SIPM) and water (SAQ) than APAP, while their SIPM and SAQ values decreased as the number of carbons in the alkyl chain increased. Although the paradigm of “the more water-soluble members of this more lipophilic series are the most effective at enhancing the delivery of total APAP species” remained broad, only comp. 18a and 19a were more effective than APAP, albeit marginally, <2 times (Table 1).
With the same objective of improving the topical delivery of APAP, new 4-ACOM-APAPs 20 and 4-AOCOM-APAP derivatives 21 and 22 (Fig. 8) were designed, prepared, and studied using the same experimental methods.64–67 Compounds 20 are distinguished from the respective series 18 with the presence of a methylene spacer, which separates the carbonyl moiety. This is the cause of the change in their physicochemical properties, and thus, 4-ACOM-APAPs 20 are more soluble in IPM (SIPM) and water (SAQ) than the corresponding members of the AOC series 18. However, compounds 20a–e resulted in 4–60 times more lipid soluble than APAP but were much less water soluble than the parent drug (Table 1). Their SIPM values are growing as the alkyl chain increases (20a–d) in contrast to series 18a–e and 19a–b, whereas the increasing SAQ values are only observed going from 20a to 20b. The SAQ values in the rest of the series 20c–e decreased as the alkyl chain length increased. Regarding flux (JIPM) or permeation of these compounds, the series 20b–c were 2–11 times more effective at delivery of APAP than the corresponding members of 18b–c. Notably, the flux of the most permeable derivative, 20b was 3.6 times greater than that of APAP.64
Other series, 4-AOCOM-APAPs 21 and 22, were obtained and analyzed in the same manner, comparing the above-mentioned physicochemical properties to those of 18 and 20.65–67 It was observed that as a whole, the group of the 4-AOCOM-APAP prodrugs 21 were less effective at delivering APAP than derivatives 18 and 20. However, the trend in fluxes followed almost exactly the trend in water solubility, i.e., the more water-soluble derivative usually gave a higher flux value in this comparison of derivatives 18, 20, and 21 (Table 1). The inefficacy in delivering the APAP of the latter was explained due to their low water solubility.66 Being compounds with the highest molecular weight, 4-AOCOM-APAP derivatives 22, incorporated by ethylene oxy groups into the promoiety, work worse, displaying an unsuitable balance of lipid (SIPM) and water (SAQ) phases that is important for optimizing delivery. The two series, 23–24, with other soft alkyl promoieties, NANAOCAM, showed similar behavior67–69 (Fig. 8). To improve the skin permeation properties through achieving the best SIPM/SAQ equilibrium of these compounds, the incorporation of a basic amine group into the promoiety of an acyl prodrug (DAAC or AAC) was also made, and the prepared series 4-DAAC-APAPs 25 and 4-AAC-APAPs 26 were evaluated70 (Fig. 8). Generally, the delivery of a topically applied comp. 25–26 and improvement in their fluxes was not reached. However, the flux value of prodrug 25e was two times higher compared to APAP, and its hydrochloride flux was equal to that of APAP (Table 1).
Therefore, optimizing these O-modified APAP prodrugs was insufficient for clinically effective delivery. Still, the accumulated information would be helpful in the research to improve APAP characteristics because APAP drugs are delivered not only via oral and topical routes but also through intravenous or rectal routes.
A representative example is propacetamol 27a, which is a parenteral prodrug based on APAP and N,N′-diethylglycine ester, i.e., is one of the 4-AAC-APAP derivatives (Fig. 9). Being more water-soluble than APAP, propacetamol hydrochloride, devoid of the major contraindications. It is used in intensive and postoperative care and delivered via the intravenous (IV) route.71 As a result, its onset of analgesia is more rapid than that from APAP ordered orally or rectally.72–74 Thus, propacetamol cannot be administered orally due to its greater water solubility. Interestingly, 4-AAC-APAP derivatives were proposed by Kovach et al. in 1981 as potentially useful prodrugs.75 Although 4-AAC-APAP derivatives such as the Ala-APAP 27b,76 Pro-APAP 27c,77 and Gly-APAP 27d78 prodrugs (Fig. 9) seem to be not stable enough to be used in a transdermal formulation. Currently, they are being investigated to be orally formulated as chewable dosage forms by masking the bitter taste of APAP (27c) or to prevent a decrease in hepatic glutathione (GSH) levels, i.e., to avoid the toxic effects of APAP (27b,d). It was reported that dipeptide ester prodrugs of acetaminophen (28a–c) did not disturb hepatic glutathione levels (in mice). They showed better stability than these 4-AAC-APAP derivatives.79 Like these O-modified APAP prodrugs, D-α-galactopyranose-APAP showed low hepatotoxicity (human liver cells) and prolonged analgesic effects (animal pain model) that indicate the viability of the galactose ring as a suitable carrier in the development of prodrugs for painkilling cure.80
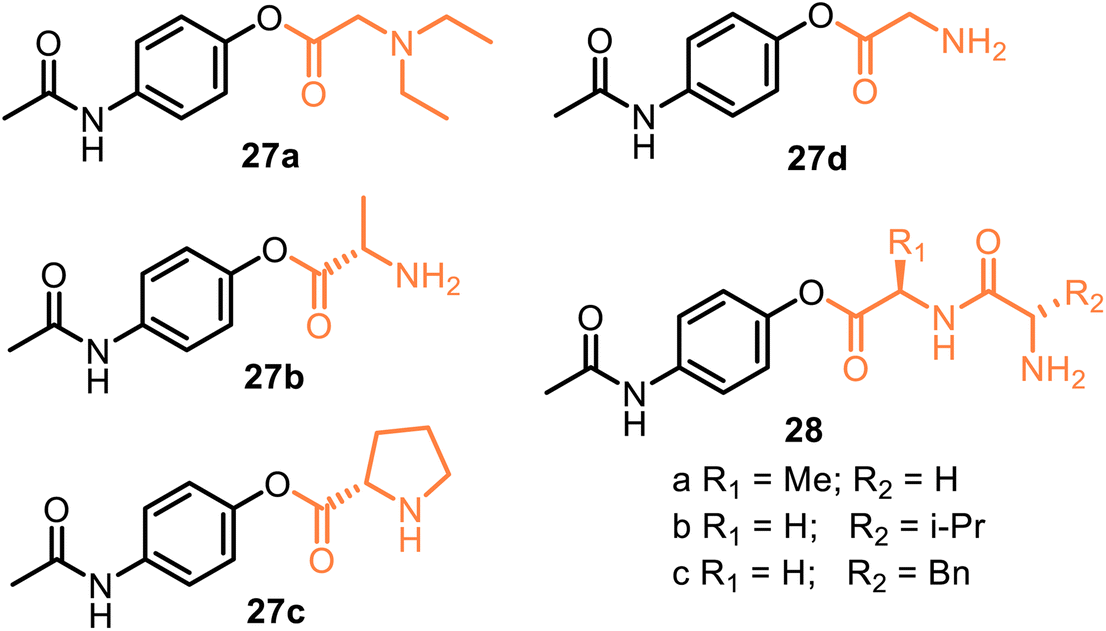 | ||
| Fig. 9 Structures of the O-modified APAP prodrugs with amino acid (4-AAC-APAP prodrugs) or dipeptide carriers. Among them, parenteral APAP prodrug 27a (propacetamol) is an illustrative example. Nonspecific plasma esterases completely and rapidly hydrolyze propacetamol to APAP and diethylglycine in a 1:1 ratio. Thus, the IV administration of 1 g of propacetamol produces 0.5 g of APAP. Noteworthy that after the IV injection of propacetamol, APAP easily crosses the blood–brain barrier, guaranteeing a central analgesic effect. However, there is evidence that a single dose of IV propacetamol provides around four hours of effective analgesia for about 36% of patients with acute postoperative pain. | ||
All these discussed promoieties are created on the usually unionizable functional fragments based on the carboxyl group to improve water solubility and oral, topical, and/or transdermal delivery. However, incorporating an ionizable group in the promoieties can increase water solubility. For example, sulfate or phosphate ester promoieties were studied in the 80 s, reporting that APAP phosphate (Pho-APAP, 29) (Fig. 10) as a sodium salt was a freely water-soluble, practically tasteless and hygroscopic substance. But it is stable in aqueous solution at neutral and acidic pH, in vivo hydrolyzable to APAP by alkaline phosphatase. It may be administered orally as an aqueous solution.81 In contrast, potassium APAP sulfate 30, a natural APAP metabolite, is also rather stable in solution but is highly resistant to enzymatic hydrolysis in vivo and very toxic during intravenous administration.82
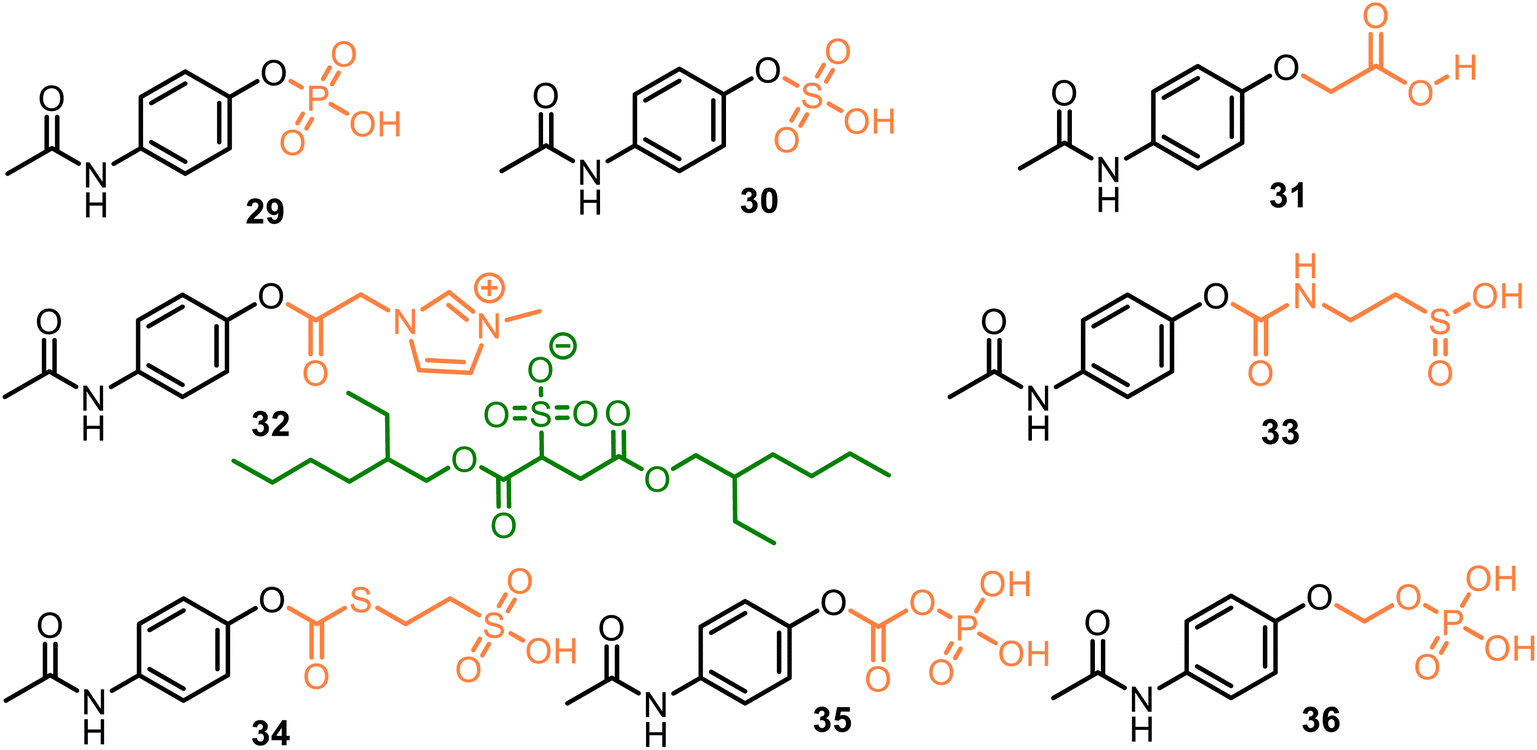 | ||
| Fig. 10 Structures of APAP drugs based on polar, ionizable phosphate, sulfonate, or carboxylate moieties. Among them, sodium APAP phosphate 29 is in vivo hydrolyzable to APAP by alkaline phosphatase, abundant in the lumen of the small intestine. Prodrug 31 was also examined in vivo analgesic, antipyretic, and anti-inflammatory tests. Its anti-inflammatory action was better than APAP: its % inhibition of edema was 37.8% compared with that of APAP (31.7%). | ||
As a result, phosphate-APAP 29 in the form of its salts (sodium, glycine, lysine, and adenosine salts) was patented as a water-soluble analgesic and antipyretic medicine.83 Still, its physiochemical characteristics were not reported, nor the (4-acetylaminophenoxy) acetic acid 31 (Fig. 10). The latter displayed analgesic (with mice, tail flick method) and antipyretic (with rats) activity similar to that of APAP. It also exhibited anti-inflammatory activity (with mice), which was more appreciable than the parent drug.84 Thus, the aqueous solubility, permeability, and absorption of the APAP derivatives could be increased via the attachment of polar or ionizable phosphate, sulfonate, or carboxylate moieties, which were termed pharmaceutically acceptable carriers.
We can also include some ionic liquids in this group of ionizable moieties. Ionic liquid 32, an APAP derivative with an ionic imidazolium/docusate pair, demonstrated a fast release rate of APAP in the acidic medium (pH = 1.2) despite its low water solubility. This fact reveals that high water solubility may not be necessary to properly deliver the prodrug into the body.85 However, it must be proven.
Meanwhile, from 2014 to 2023, the new APAP derivatives 33–36, which comprise the following carbonyl-amino-ethane methanesulfinic acid (33) or carbonyl-thio-ethane sulfonic acid (34) and carbonic phosphoric (35) anhydride, or methyl phosphate (36) carriers and their K, Na salts, were patented.86–92 APAP derivatives 33 and 34, based on the APAP core and the respective promoieties of 2-aminoethanesulfinic acid and 2-sulfanylethane sulfonic acid, provided a more rapid onset of APAP action as compared to the administration of acetaminophen. They can confer hepatoprotection from acetaminophen due to the in vivo-generated carriers. The latter act as neuroprotective agents against acetaminophen-induced hepatorenal oxidative damage.86
(4-Acetamidophenyl carbonic) phosphoric anhydride 35, a carbonic analog of 29, related to the 4-AOC-APAPs (see comp. 20, Fig. 8), showed increased water solubility relative to its parent drug. It was 10 times more soluble in water than APAP (152 mg mL−1 and 15 mg mL−1, respectively). As a result, its conversion to APAP in human plasma was more efficient. Still, meanwhile, it was not capable of transforming water into the parent drug, and its toxic dosage was higher than the parent drug.87–90 (4-Acetamidophenoxy) methyl phosphate 36 was proposed as a suitable agent for the rapid treatment of a disease that is responsive to APAP, such as pain, fever, inflammation, or ischemic injury (such as myocardial and/or cerebral). Prodrug 36 and its sodium salt are soluble in water at room temperature, showing 145 mg mL−1 and 160 mg mL−1 values, respectively. Moreover, they are relatively stable under some conditions (e.g., during storage and/or preparation in a saline solution) while being easily converted to their parent drug in human plasma.91,92 Research and development of new O-modified APAP prodrugs with these pharmaceutically acceptable carriers is an actual task.
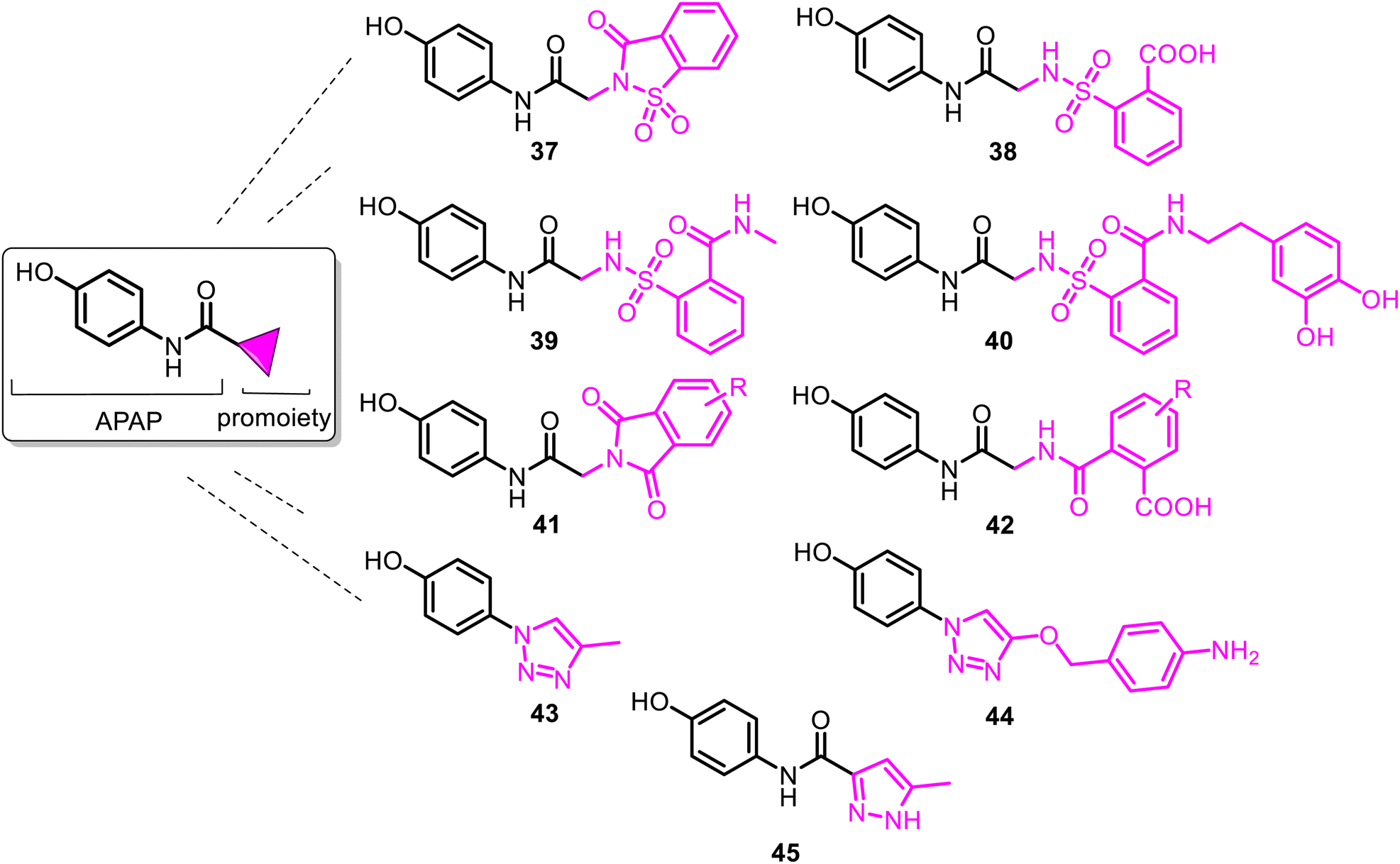 | ||
| Fig. 11 Structures of the reported carrier-linked N-modified APAP prodrugs with reduced hepatotoxicity. SCP-1 (37) was the first developed N-modified APAP lead. It contains the saccharin skeleton. Accidently discovered in 1878 by a Russian chemist Constantin Fahlberg, saccharin was the first commercially recognized as a sweet-tasting agent. N-Sulpharyl-APAP prodrugs 39 and 40 displayed similar analgesic properties (ED50 = 45.2 μmol kg−1 and 14.7 μmol kg−1, respectively) to APAP (ED50 = 68.6 μmol kg−1) as well as antipyretic activity (ED50 = 197.5 μmol kg−1 and 176.6 μmol kg−1, respectively), compared with APAP (ED50 = 245.1 μmol kg−1). JNJ-10450232 (NTM-006) agent (45) is a novel, promising analgesic and antipyretic APAP prodrug synthesized by Janssen Pharmaceutical Research & Development, LLC (JPRD, Spring House, PA). It was neurologically well-tolerated following single oral administration at up to the highest dose of 750 mg kg−1 tested in Sprague-Dawley rats (modified Irwin Test). | ||
Among them, the APAP derivative with the saccharin (1,2-benzisothiazol-3(2H)-ona-1,1-dioxide-2-yl) moiety 37 (named SCP-1) was the first developed N-modified APAP lead. It showed an in vivo analgesic profile similar to APAP and significantly diminished hepatotoxicity, thus contributing to its safety profile. Notably, when compared with APAP on a molar basis, 37 was a more potent analgesic (2- to 7-fold). However, compared to APAP, SCP-1 does not possess an antipyretic activity.93–95 Additionally, SCP-1 is quickly metabolized in vivo, generating the main metabolite 38 (SCP-123), which is quite hydrosoluble and easily eliminated by urine due to the formed carboxylic group.96,97 SCP-123 (38) and its corresponding sodium salt (SCP-123 ss) are equipotent on a molar basis with APAP in analgesic models (tail-flick and hot plate tests with mice). Their close analogs exhibited good activity against Escherichia coli and Bacillus megaterium.98
However, the most important detail is that SCP-1 (37) and its metabolite 38 served as another biomodel in the search for improvement in the pharmacological profile of the N-modified APAP prodrug. Thus, in 2020, N-sulpharyl-APAP prodrugs 39–40 (Fig. 11) were developed.99 They are one of the most promising analgesics among the novel 2-(benzenesulfonamide)-N-(4-hydroxyphenyl) acetamide class. With modifying a benzenesulfonic acid moiety in 38, APAP derivatives 39–40 exhibited increased stability and elevated lipophilicity. Their hydrolysis of the amide group is slower than that of a metabolically unstable lipophilic 37. Moreover, they retain both analgesia and antipyresis. They are not hepatotoxic because they do not generate toxic metabolite NAPQI, even in concentrations equal to a toxic dose of APAP (600 mg kg−1, mice). These safer agents with a marked improvement in the hepatotoxicity profile could be an important new tool to combat acute and chronic pain as analgesics and kidney and liver disease associated with the COVID-19 infection as antipyretics. Phthalimide-APAP derivatives 41–42, structurally related to 37–38, were prepared and proposed as potential analgesic and antipyretic agents but were not studied.100,101
To avoid or moderate APAP-induced hepatotoxicity, the acetamide, –NH(CO)CH3 fragment of APAP, was bioisosterically replaced with a 1,2,3-triazole scaffold constructing pharmacologically potent APAP-triazole derivatives such as 43–44 (Fig. 11).102,103 The bioisosterism approach (1,2,3-triazole ring as an amide function)104 is an elegant solution to the APAP hepatotoxicity problem. Sahu and co-workers showed that triazole incorporation into the APAP nucleus allowed obtaining new, structurally diverse N-modified APAP derivatives 43–44. These exhibit increased efficacy (anti-inflammatory, analgesic, and antipyretic activity) and decreased toxicity of the parent APAP drug. Compare the respective LD50 > 1000 mg kg−1 and 2000 mg kg−1 (43 and 44) with the APAP toxicity (LD50 < 1000 mg kg−1).103 Thus, displaying efficacy superior to that of APAP, especially in anti-inflammatory potency, and negating hepatotoxic manifestation of the parent APAP drug, compounds 43 and 44 are new, effective and safe leads for clinical studies. Another heterocyclic ARAP derivative 45 called JNJ-10450232 (NTM-006) (Fig. 11) exhibited similar antinociceptive and antipyretic effects to that of APAP (in rodent pharmacology models). It showed the potential to have a longer duration of action and, at the same time, markedly reduced the risk for hepatotoxicity on overdose.105
The metabolism and disposition of comp. 45 following oral administration to rats, dogs, monkeys, and humans were studied. It was reported that its primary metabolic pathway in monkeys and humans was the direct O- and N-glucuronidation reactions. At the same time, amide hydrolysis was another primary metabolic pathway in rats and dogs.106 Noteworthy that a minor bioactivation pathway to quinone-imine like NAPQI from APAP was detected only in monkeys and humans. Agent JNJ-10450232 is now proposed for first-in-human clinical studies107 and a Phase 2 dental pain study.108 Comparing the hepatotoxicity of pairs of APAP derivatives 37–38, 43–44 with that of 45 (Fig. 12). The authors concluded that their potential mechanisms are minimally hepatotoxic through the deactivation of the formation of the quinone-imine system.
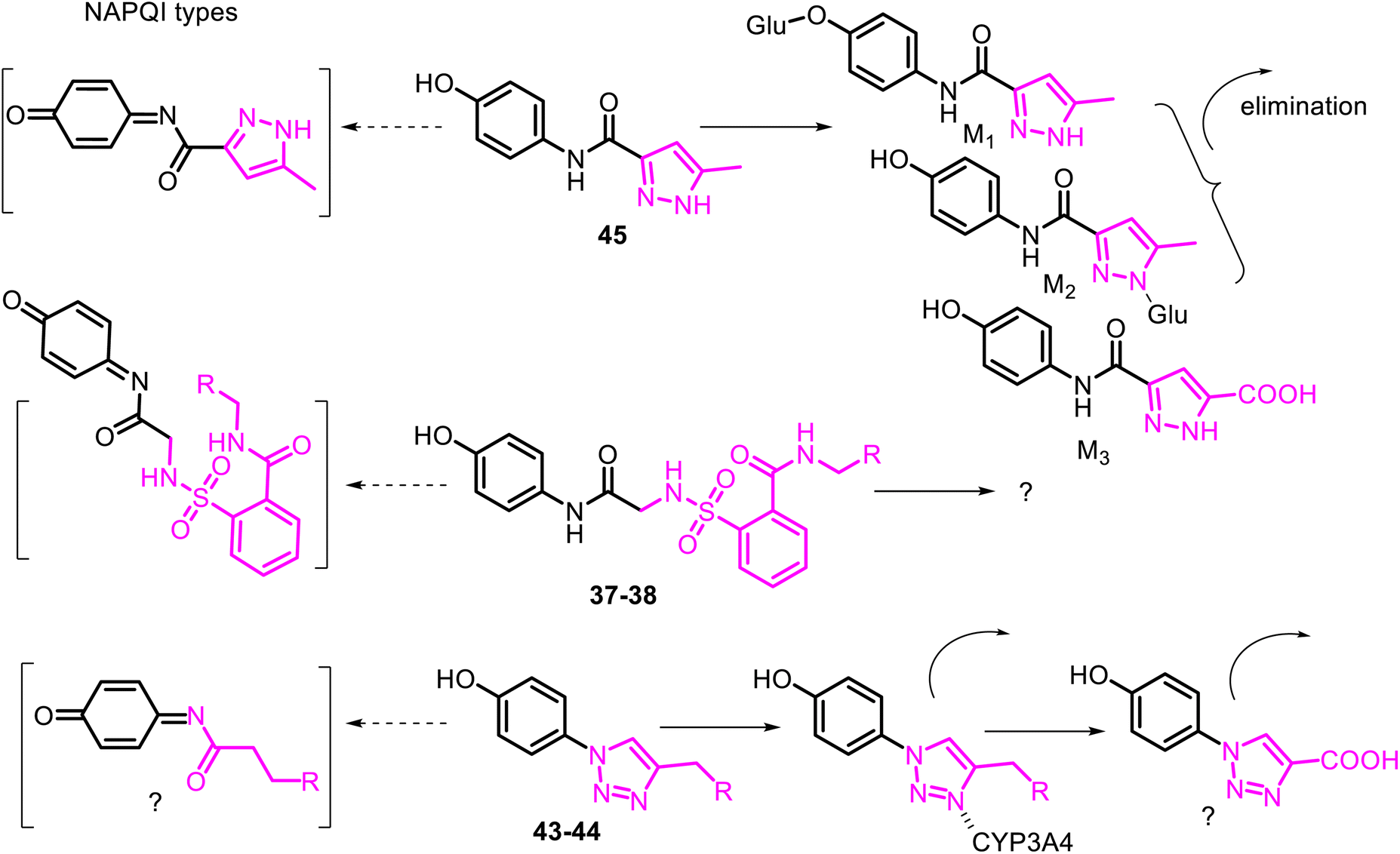 | ||
| Fig. 12 Proposed biotransformation pathways of non-hepatotoxic APAP derivatives 37–38, 43–44 and 45. The lack of hepatotoxicity was due to a decrease or lack of formation of the toxic quinone-imine type NAPQI. The elimination of JNJ-10450232 (45) and its major metabolites M1–M3 was mainly via urinary excretion. Derivatives 37–38 showed favorable cytochrome P450 isozyme profiles, and NAPQI generation by P450 isozymes was not detected in the serum of a mouse. Optical microscopy and confocal scanning light microscopy techniques indicated no considerable change in the hepatic tissues (liver of Wistar albino rats) after acute toxicity with treatments of 43–44, in contrast to the APAP treatment, in which distinct necrotic lesions were found that can be concluded that treatment of 43–44 is devoid of any toxic manifestation, i.e., NAPQI formation did not occur. | ||
Thus, the deactivating role of the aminooxoethyl-sulfamoyl (37–38), triazolyl (43–44), and aminoacylpyrazolyl (45) fragments against the enzyme CYP450 complex can be realized by metabolic switching from oxidation of the p-aminophenol core to another potential oxidizable moiety. For example, activated methyl group linked to the π-amphoteric triazole and pyrazole rings, or pyrazolyl NH function in the respective comp. 43–45. Another metabolic switching could be possible from direct O- or/and N-glucuronidation reactions of the –OH or/and –NH function of the APAP derivative 45 (ref. 106) and even by incorporating into P450 isozymes (CYP3A4) through the heme/iron–nitrogen interactions, and then efficiently metabolize in the comp. 43–44.109,110 It should be noted that the 1,2,3-triazole ring, like triazolylphenoles 43–44, is not protonated at physiological pH because of their poor basicity. It thus would be metabolically stable and able to participate as a hydrogen-bond acceptor, which could be favorable in binding biomolecular targets. From all these speculations, the metabolism profile of the APAP derivative 45 has been correctly studied (Fig. 12).
Interesting 3-aryl-3-(morpholin-4-yl)-N-(4-hydroxyphenyl)propanamides, prepared using the Mannich reaction of APAP and benzaldehydes, could be classified as carrier-linked N-modified APAP molecules. Unfortunately, these compounds were examined only for antibacterial activity; some of them showed significant activity against Bacillus subtilis and Pseudomonas aeruginosa.111
Direct- and spacer-linked APAP codrugs
This type of APAP codrug could be a particular case of the merged or conjugated APAP hybrids, respectively. The respective literature examples are known. According to their structural aspects, they were grouped like linked O-modified APAP codrugs (i.e., merged ester NSAID–APAP hybrids), linked N-modified APAP codrugs (merged amide NSAID–APAP hybrids), merged ester/amide NSAID–APAP hybrids, and linked C-modified APAP hybrids. All of these will be discussed below.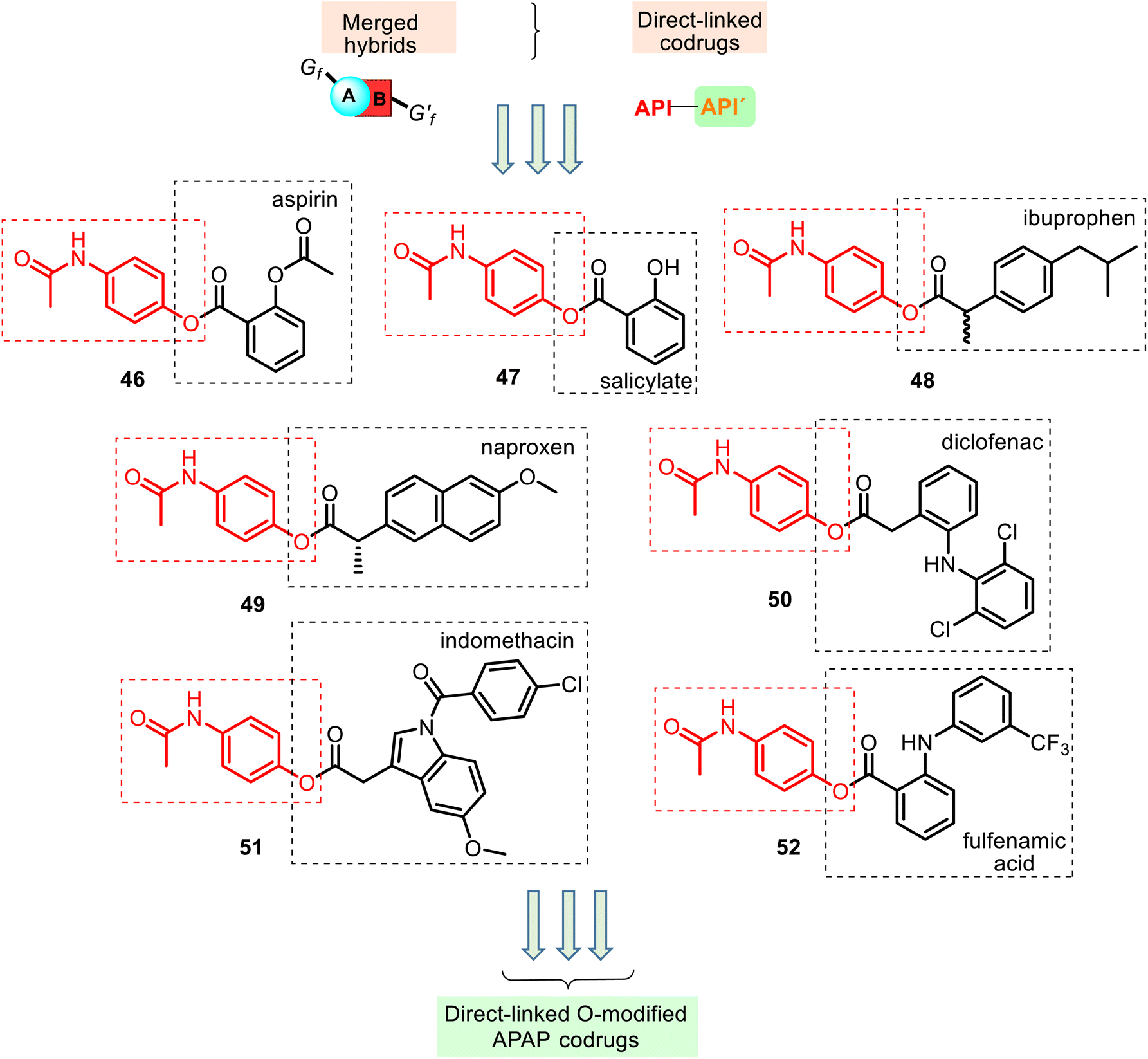 | ||
| Fig. 13 Structures of merged hybrids containing two different APIs connected via a carboxylic ester function, so-called non-identical twin drugs, or mutual bipartite prodrugs, in which both of the drug molecules are directly linked to each other by a covalent bond. Benorylate (46) was first synthesized by Robertson in 1963. It is a white, odorless, tasteless, stable compound, very soluble in lipids, slightly soluble in ethanol, but practically insoluble in water. Acetaminosalol (47) did not hydrolyze in the gastric juice and was more slowly absorbed than acetylsalicylic acid APAP. | ||
Other merged hybrids 48–52 containing two different APIs connected via a carboxylic ester function (Fig. 13) were designed and prepared118–122 using the Steglich esterification reactions of APAP and the NSAIDs mentioned above 4–6, 8 (Fig. 4) and fulfenamic acid, another NSAID. They are used in double-fixed-dose combinations of APAP that generally result in synergistic analgesia and decrease the required dose, which would reduce the adverse effects (renal disturbances and cardiovascular events).6
The association of these acidic NSAIDs and APAP with an overlap mode also aims to improve the therapeutic index by preventing NSAIDs' gastrointestinal toxicity and APAP's hepatotoxicity. However, in this merged hybrid series, APAP's role seems to be a temporary blocker of the free carboxylic group present in the NSAIDs until their systemic absorption. Moreover, the acidic moiety is essential for COX inhibitory activity, which is thought to be mediated by the antipyretic, analgesic, and anti-inflammatory actions of NSAIDs.
Thus, this derivatization of NSAIDs by APAP (comp. 46–52) may avoid their major side effects, and it can spontaneously release the parent drugs or enzymatically in the blood following their absorption. Studying the chemical and enzymatic stability and the analgesic and ulcerogenic activities of ester NSAID–APAP codrugs 46, 48–52, Fadl and Omar, in 1998, reported119 that these codrugs are sufficiently chemically stable in non-enzymatic simulated gastric fluid (hydrochloric acid buffer of pH 1.3 and in phosphate buffer of pH 7.4). However, they are susceptible to enzymatic hydrolysis (human plasma and rat liver homogenate), releasing the corresponding NSAID and APAP. Generally, in vitro and in vivo evaluation of these prodrugs revealed improvement in the therapeutic index of the parent drugs. The following interesting finding is that (a) benorylate 46, with the smallest acyl moiety, was found to be the most labile in both investigated enzyme systems; (b) all these codrugs induced very little irritancy in the gastric mucosa of mice after oral administration for 4 days (ibuprofen–APAP hybrid 48 was less damaging, while diclofenac–APAP hybrid 50 produced more gastric mucosal injury), and (c) their analgesic activity significantly improved over time, i.e., these codrugs per se are lacking analgesic activity, and the observed latent analgesia results from hydrolysis to the parent drugs.
The anti-inflammatory (in vitro COX inhibition and carrageenan paw edema test), anticoagulant (prothrombin time test), and antinociceptive (formalin test in rats) activities of the hybrids 48 and 49 were studied.120,121 The results demonstrated that these hybrids showed significant antinociceptive effects (ED25 = 0.7 ± 0.3 and 0.9 ± 0.3 mg per paw, respectively).120 Their COX-1/COX-2 enzyme inhibition data demonstrated more inhibition activity towards COX-1 and COX-2 enzymes than the parent individual NSAID drugs. Moreover, the low anticoagulant effect of the studied hybrids will probably have fewer bleeding side effects.121 Therefore, NSAID–APAP hybrids 48–49 can improve patient compliance and pharmacokinetics properties.
Another codrug, diclofenac–APAP hybrid 50, was also studied by Visagaperumal et al.122 They showed that its anti-inflammatory activity (male Wistar rats) resulted in better activity than diclofenac (% inhibition of paw volume for 8 h, 84% and 76%, respectively) and the ulcerogenic activity produced by 50 is negligible compared to diclofenac (ulcer index, 1.4 and 6.3, respective).
The combination of phenylamino-benzoic, phenylamino-nicotinic, or phenoxyphenyl acrylic acid scaffolds and APAP allowed for obtaining new O-modified APAP carboxylic esters, such as merged hybrids 53–57 (Fig. 14). APAP hybrids 53–56 were designed, prepared and studied starting with the premise that some NSAIDs and their amides (ibuprofen, fulfenamic or niflumic acids) can act as inhibitors of fatty acid amide hydrolase (FAAH), a key hydrolytic enzyme for the endogenous cannabinoid receptor ligand anandamide, and APAP also modulate FAAH indirectly via AM404 metabolite.123,124
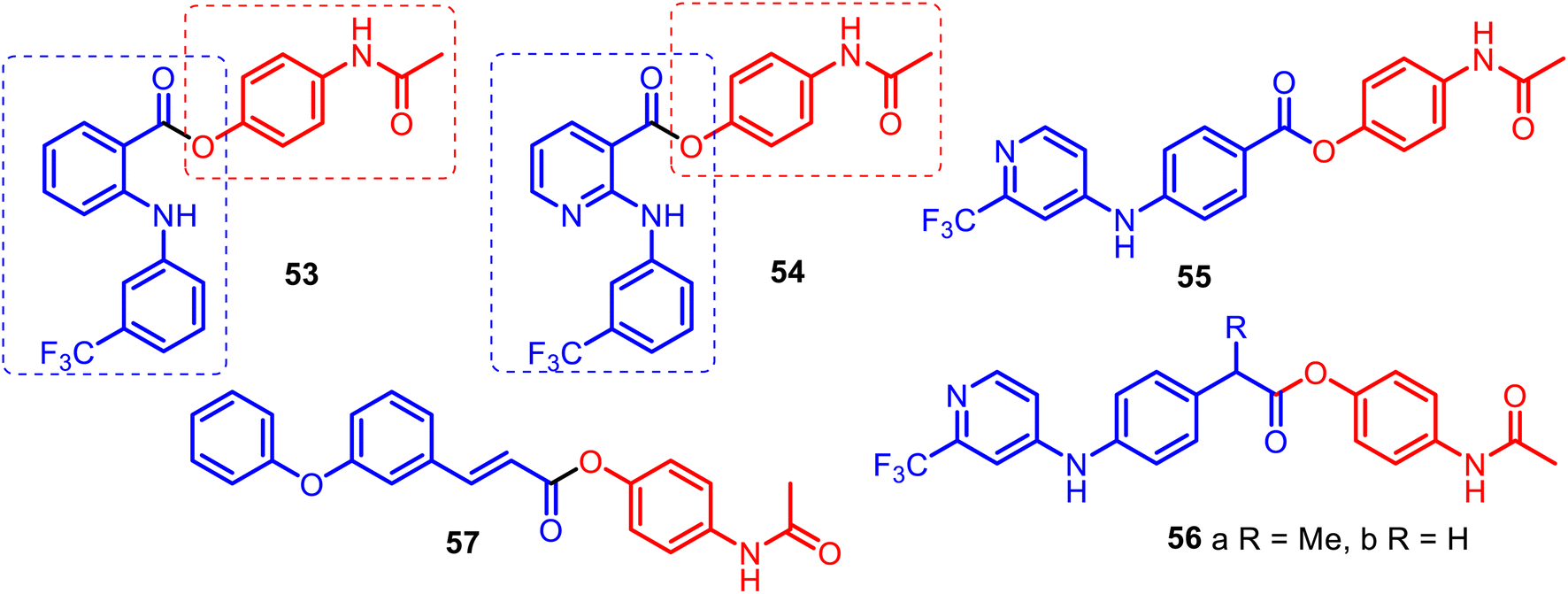 | ||
| Fig. 14 Ester APAP hybrids 53–56 are potent inhibitors of FAAH and APAP hybrid 57 as promising multifunctional agents with high anti-nociception response associated with inflammatory pain. Pharmacological inactivation of FAAH produces analgesic, anti-inflammatory, anxiolytic, and antidepressant phenotypes without showing the undesirable side effects of direct cannabinoid receptor agonists. That indicates that FAAH may be a promising therapeutic target. All hybrids 53–56 contain the CF3 group on the peripheral aryl ring. Thus, it is clear that the trifluoromethyl group is primarily responsible for activity in this series. | ||
Indeed, Onnis and co-workers reported the development of potent APAP hybrids with an extra methylene linker between the phenyl ring and ester moiety 56a–b. These hybrids inhibited rat brain FAAH with IC50 values of 0.1 μM and 0.18 μM, respectively. However, APAP lacks FAAH inhibitory activity (IC50 value > 300 μM).124
This was due to the SAR studies of APAP derivatives based on the ortho-phenylamino-benzoic and phenylamino-nicotinic acid carcasses (fulfenamic and niflumic acids) 53–54, and para-phenylamino benzoic acid skeleton 55, which presented IC50 values of 8.7, >300, and 2.7 μM, respectively. It is noteworthy that APAP hybrid 56a competitively inhibited FAAH activity with a Ki value of 0.16 μM and inactivated monoacylglycerol lipase (MGL), the enzyme responsible for the hydrolysis of the endogenous cannabinoid receptor ligand 2-arachidonoylglycerol, with an IC50 value of 1.9 μM.124 Thus, it can be valuable as a template for designing novel reversible FAAH inhibitors derived through the O-conjugation of the APAP core with para-phenylamino benzoic acid scaffolds. The O-conjugation of the APAP core with meta-phenoxyphenyl acrylate acid gave a new hybrid 57, which showed high analgesic activity (91%), also presenting high anti-lipid peroxidation activity (100%). It was also found that its anti-inflammatory activity (carrageenin rat paw edema test) was low (36.5%) compared to indomethacin (47%) but higher than APAP (21%). According to locomotor function recovery studies, hybrid 57 could be a promising agent for treating peripheral nerve injuries.125
Instead of the NSAID core, diverse heterocyclic rings generally could be incorporated into the APAP structure. For example, alicyclic N-substituted carbamate APAP derivatives 58–60 were specially designed for the releasing parent drugs, based on the heterocyclic rings: muscle relaxant drug (chlorzoxazone 58a, metaxalone 59a, and mephenoxalone 60a) and APAP (Fig. 15).126,127 These O-modified APAP carbamates efficiently operate in aqueous (pH 6–11) and plasma (pH 7.4) media as drug delivery systems through intramolecular cyclization reactions due to a hydroxyl nucleophile.
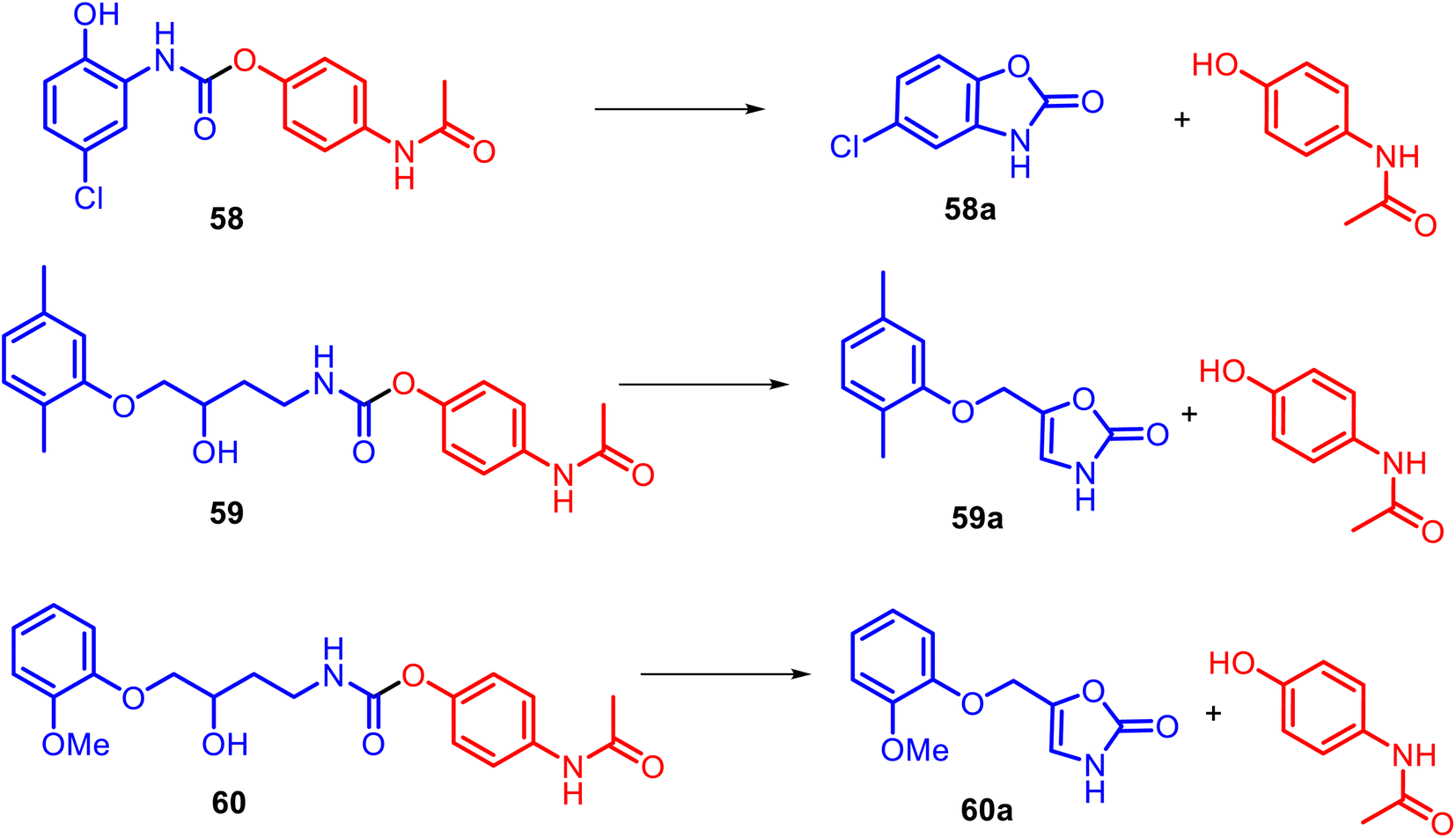 | ||
| Fig. 15 Efficient drug delivery systems based on masked active benzoxazolone and oxazolidinone rings and structures of the respective linear carbamate–APAP hybrids 58 and 59–60, which release corresponding muscle relaxant drugs chlorzoxazone 58a, metaxalone 59a, and mephenoxalone 60a and APAP. Muscle relaxants are usually recommended for neck or back pain caused by muscle spasms. These medications help to reduce muscle spasms and tension. | ||
It was found that oxazolidinone derivatives 59a–60a were released from their mutual prodrugs through a rate-limiting elimination–addition reaction. In contrast, the release of benzoxazolone 58a was followed by a cyclization mechanism involving a change in the rate-limiting step from forming a cyclic tetrahedral intermediate with the expulsion of APAP. Thus, so-called cyclization-activated APAP prodrugs could be a promising design for new efficient muscle relaxants for acute pain.128
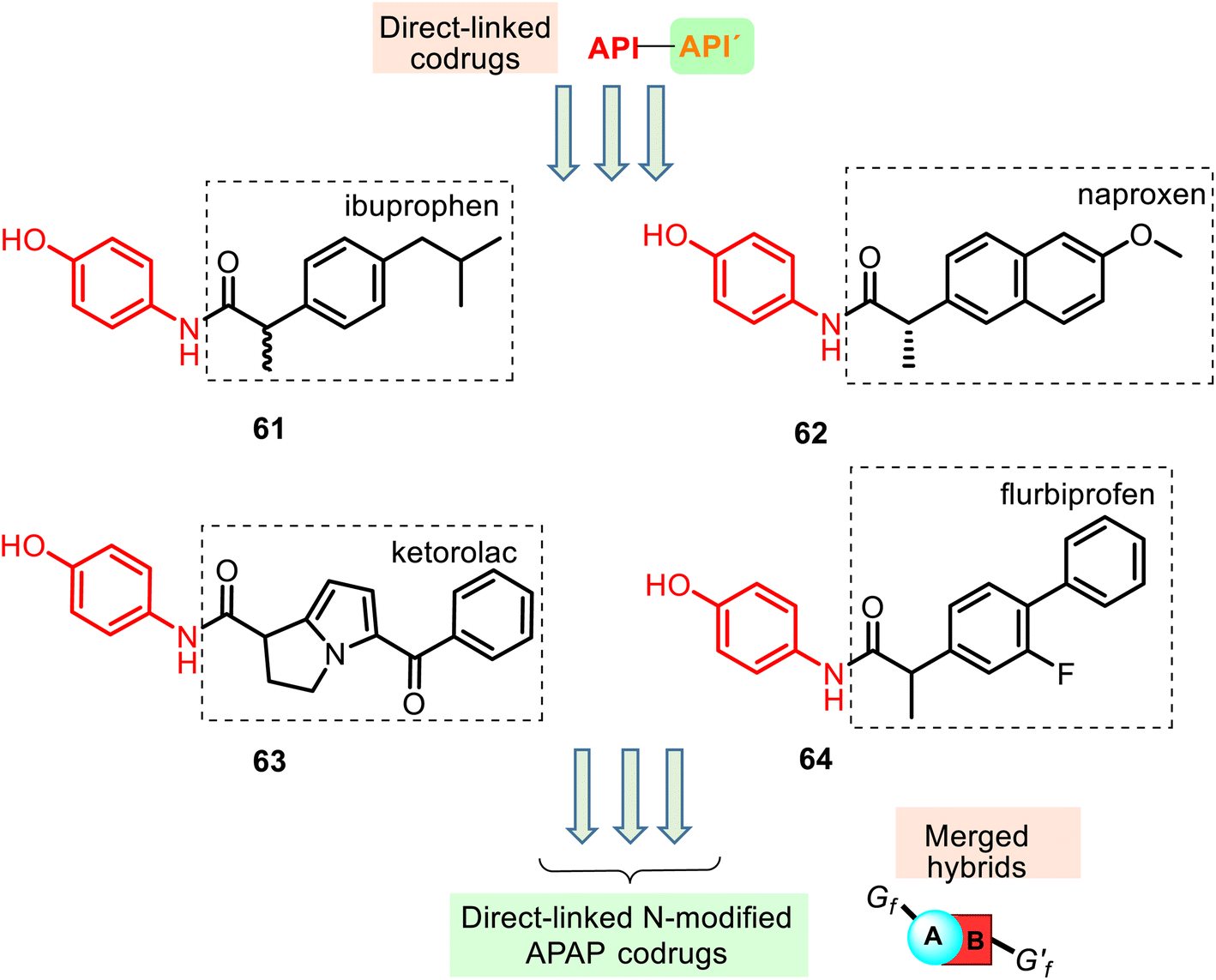 | ||
| Fig. 16 Structures of the amide NSAID–APAP codrugs (or linked N-modified APAP bipartite mutual prodrugs) 61–64 belonging to the merged hybrid group. They were designed and prepared with a view to combine the antipyretic activity component of APAP into the NSAID skeleton with their normal anti-inflammatory activity but without GIT ulceration. They were evaluated for their antipyretic activity in animal models (Male Sprague-Dawley rats). The pyrexia % values at a dose of 25 mg kg−1 for 61–64 prodrugs were 87.4%, 59.5%, 55.8%, and 58.8%, while APAP showed a 63.8% reversal of body temperature. Their anti-inflammatory activity in the in vivo model using carrageenan-induced paw edema provided the following % inhibition values: 34.8% (61), 39.4% (62), 86.8% (63), and 68.9% (64). | ||
Furthermore, its structural amide resemblance with APAP allows it to expect substantial antipyretic effect like the parent drug APAP. Indeed, the prepared hybrids showed much improved antipyretic activity and sufficiently good anti-inflammatory properties with nil ulcerogenic action (rat model).130 Amide ibuprofen–APAP hybrid 61 (Fig. 16) presented the best antipyretic activity. However, it showed a reduced anti-inflammatory activity (carrageenan-induced rat paw edema assay) compared with the parent drug (ibuprofen). Other hybrids 62–64 also presented this tendency. Thus, the conversion of the free carboxylic group in the NSAIDs to the amide linkage caused a reduction in the anti-inflammatory activity of the parent drugs.
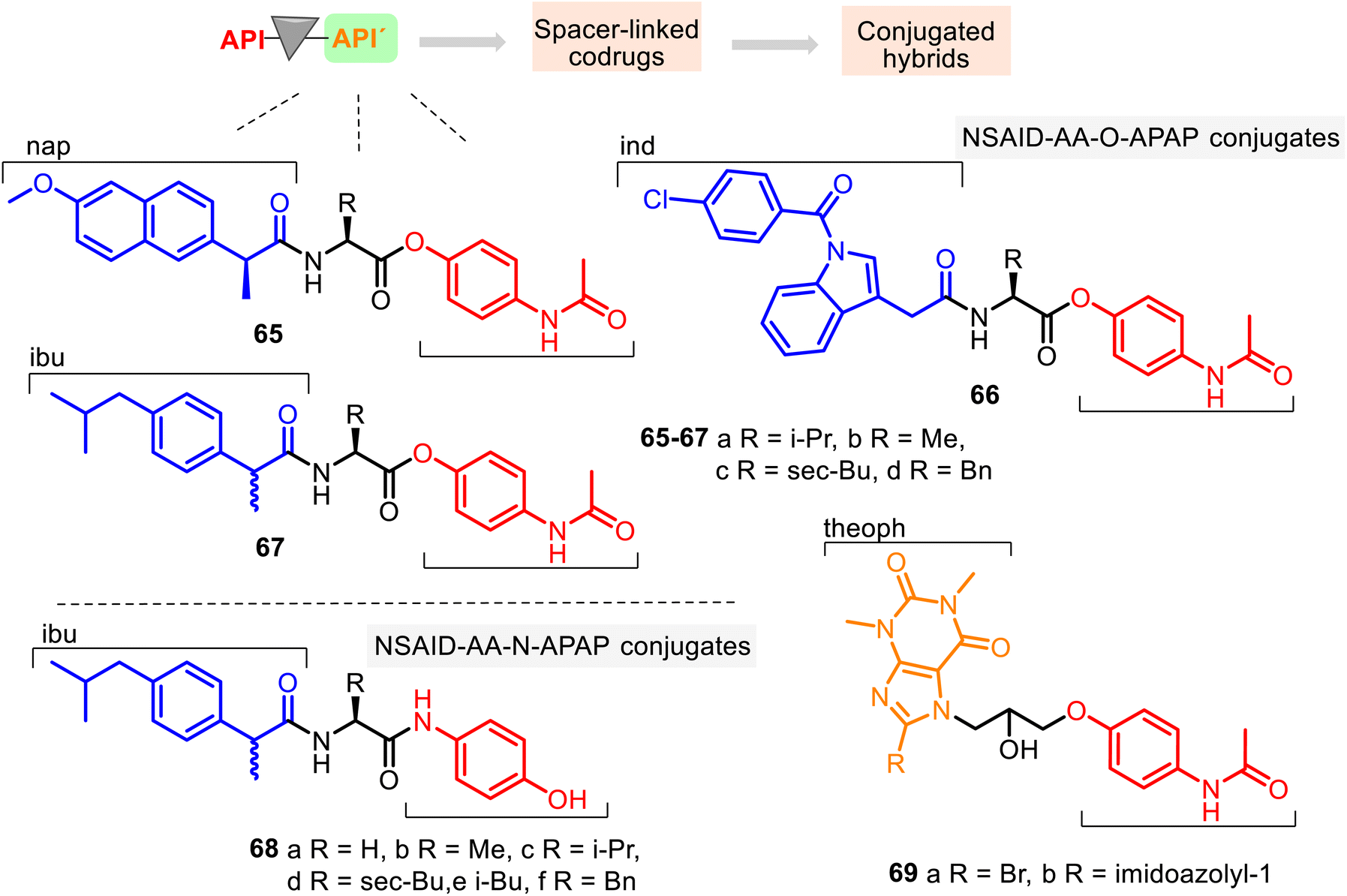 | ||
| Fig. 17 Structures of new conjugated hybrids containing APAP core: NSAID–AA–O–APAP hybrids 65–67, NSAID(Ibu)–AA–N–APAP hybrids 68, and Theoph–CH2CH(OH)CH2–O–APAP hybrids 69. They are also known as tripartite mutual prodrugs. | ||
Since amino acids (AA) have been used as carriers for drugs because of their ability to transport into mammalian tissue (see comp. 28, Fig. 9), a series of NSAID–AA–O–APAP conjugates 65–67, where the APAP core is linked with its phenolic hydroxyl group, was studied for their anti-inflammatory, ulcerogenic, and toxicological properties. Their anti-inflammatory activity was determined in vivo by the acute carrageenan-induced paw edema standard method in rats using the potency magnitude (P), which is the % inhibition of edema for the tested compounds relative to the % inhibition of edema for indomethacin (Ind) as a reference standard.
The following hybrids, L-Nap–L-Val–APAP 65a, L-Nap–L-Phe–APAP 65d, Ind–L-Ile–APAP 66c, and DL-Ibu–L-Phe–APAP 67d, were the most promising anti-inflammatory active agents among 12 conjugates (potency, % inhibition relative to indomethacin at a dose of 10 mg kg−1, P = 81.9, 83.8, 116.3, and 48.5, respectively), being more potent than the corresponding standard drugs (Nap, Ind, and Ibu: P = 75.0, 100, and 48.5, respectively). At the same time, they (except hybrid 66c) showed no visible stomach lesions and no toxicity in the animals tested.131 In contrast, ulcer index values of ibuprofen, naproxen, and indomethacin were 5.50, 8.34, and 6.00, respectively. Through the SAR study, it was found that the R function of the amino acid residue controls the anti-inflammatory potency of the NSAID–AA–O–APAP conjugates. In general, the benzyl group of the amino acid function offers more potent conjugates than alkyl functions (methyl, isopropyl, isobutyl).
Conjugates 68 possess similar NSAID (Ibu), AA, and APAP sequences to the NSAID–AA–APAP derivatives 65–67. Still, the APAP core is linked with its amide group (Fig. 17). Thus, this series may be called NSAID–AA–N–APAP derivatives, where the conjugate 68f with the DL-Ibu–L-Phe–APAP sequence was found to be more effective and selective (COX-2/COX-1, SI = 5.3). It resulted in a safer anti-inflammatory agent than the parent ibuprofen, with suitable peripheral (P = 134) and central analgesic (P = 225) properties.132
APAP hybrids 69 contain theophylline (theoph) skeleton and, thus, also represent interesting combined entities. Theoph is an efficient bronchodilator drug that controls the renal blood flow and exhibits anti-inflammatory activities. Still, it causes toxic side effects such as central nervous stimulation, cardiopathy, and excessive secretion of gastric acid. Therefore, to minimize their side effects, APAP hybrids 69 were prepared.133 Their potential bronchodilator effects were studied using the precontracted Wistar rat tracheal strip method. It was observed that the hybrids 69a and 69b were 6.5 times and 2.5 times, respectively, more active than theophylline at 10−3 M concentration.134 Their toxic effects (LD50 = 423 mg kg−1 and 412 mg kg−1) were less pronounced than theophylline (LD50 = 200 mg kg−1). However, their analgesia, pyrexia, and inflammation properties were not evaluated.
An interesting example of bis-direct-N,O-linked APAP codrugs 70–71 was described by Alwash and co-workers in 2018.135 These codrugs are acetanilide chalcone derivatives linked to aspirin and ibuprofen (Fig. 18) and prepared in a two-step procedure using APAP as starting material (the Claisen–Schmidt condensation and then the mixed anhydride method with ethyl chloroformate). They were screened against bacterial (Escherichia coli) and fungal (Staphylococcus aureus and Candida albicans) strains, but appealing antibacterial and antifungal activity was not observed. Nevertheless, codrugs 70–71 could be an interesting model in researching new APAP-based painkillers. Noteworthy that similar N,O-disubstituted APAP derivatives with the thiazolidine-2,4-dione ring have been developed as promising lead compounds for treating diabetes.136
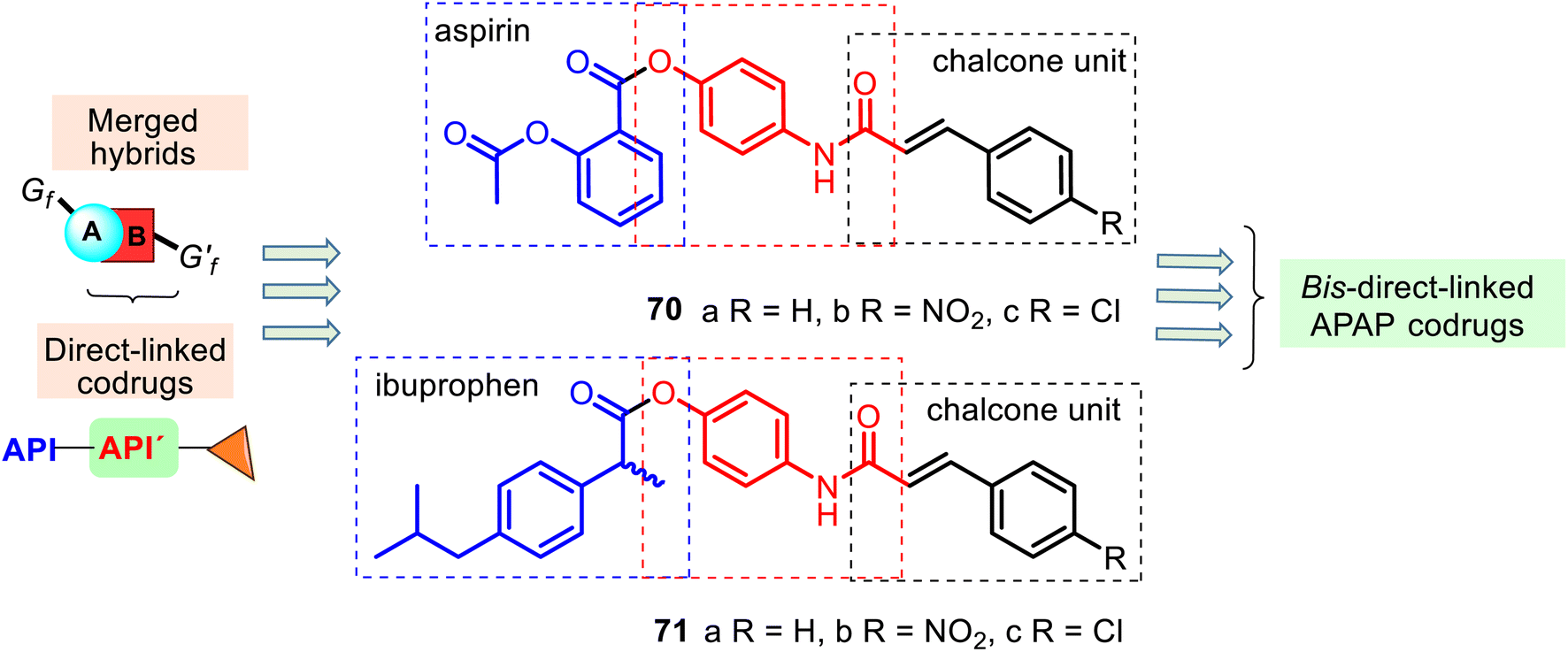 | ||
| Fig. 18 Structures of the bis-direct-N,O-linked APAP codrugs 70–71 are attractive models for researching new APAP-based analgesics. | ||
Another heterocyclic skeleton, the 1,2,3-triazole (TA) ring, can act as a promising pharmacophore (see comp. 43–44, Fig. 11) and as a good linker and valuable structural platform. Triazole molecules possess various pharmacological activities (anti-inflammatory, anticancer, antioxidant, or anticonvulsant activity). As seen above, APAP is an analgesic, antipyretic drug, and potential antioxidant, cardio-, and neuroprotective agent. Thus, 1,2,3-TA-based APAP hybrids 72–74 (Fig. 19) could be interesting models for new pharmacological nontoxic agents endowed with anti-inflammatory, analgesic, or/and anticancer properties.137–139 A good docking score for the Bn–TA–APAP derivatives 72 when docked into the Human Estrogen Receptor Alpha Ligand Binding Domain (PDB Code: 1XP6) has been a reason for studying their inhibition of breast cancer cells. The results of the in vitro cytotoxic studies (human cancer cell line MCF-7) indicated that among the tested compounds, N-benzyl-TA-APAP 72b and 72c exhibited promising cytotoxicity with IC50 values of 19.83 and 20.57 μg mL−1, respectively.137
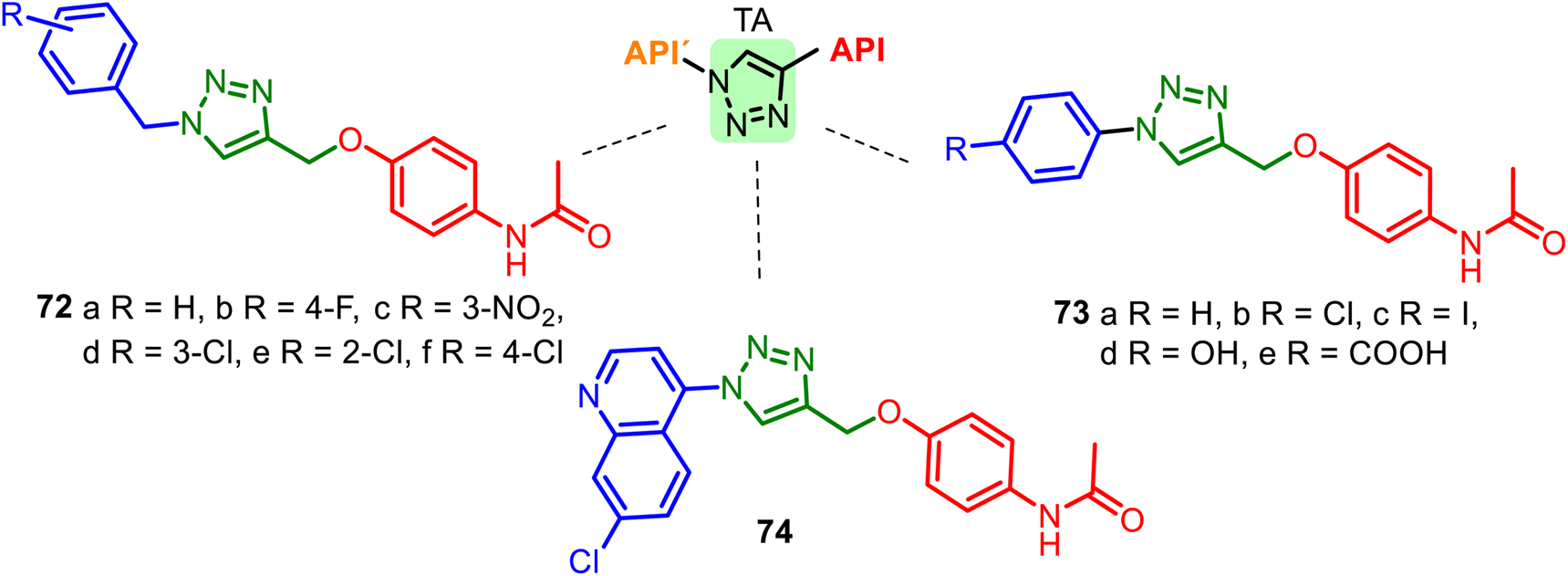 | ||
| Fig. 19 Series of Bn–TA–APAP derivatives 72 and Ar–TA–APAP derivatives 73–74, interesting models for drug research. The 1,2,3-triazole ring plays the role of a linker unit. | ||
Hybrid 72d showed the most potent antioxidant activity (DPPH method) with an IC50 value of 0.4 μg mL−1, more than the well-known antioxidant ascorbic acid. Other Ar–TA–APAP hybrids 73–74 series were not practically studied in biological systems.138,139 Interestingly, 1,2,3-TA-based AMAP (N-acetyl-meta-aminophenol) hybrids were also designed, prepared, and studied, but they resulted in less active as cytotoxic and antioxidant agents than Bn–TA–APAP derivatives 72.140
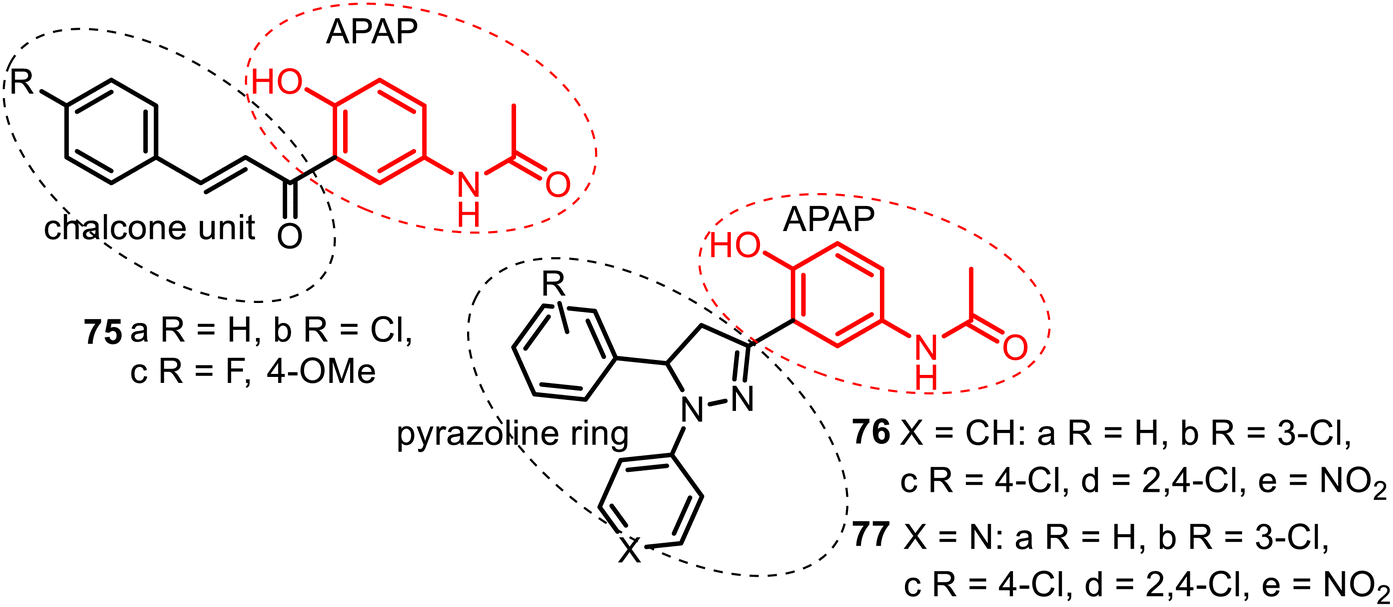 | ||
| Fig. 20 Series of the C-linked APAP compounds: chalcone–APAP (75), N-arylpyrazoline–APAP (76), and N-(pyridin-4-yl)pyrazoline–APAP (77) hybrids. | ||
The antifungal and antibacterial screening of chalcone–APAP hybrids 75 showed that among them, comp. 75c exhibited maximum activity against Candida albicans, but none of these hybrids was active in antimicrobial tests.141 Biological screening of hybrids 76–77 resulted in more promising results. A hybrid 76d from the N-arylpyrazoline–APAP series demonstrated good antibacterial (Pseudomonas aeruginosa and Staphylococcus aureus), antifungal (Aspergillus niger), and antitubercular (Mycobacterium tuberculosis H37Rv) properties. It inhibited these microorganisms with MIC = 6.25 μg mL−1. A hybrid 77e of the N-(pyridine-4-yl)pyrazoline–APAP series resulted in being a potent agent with a broad spectrum of antimicrobial (P. aeruginosa and S. aureus), antifungal (C. albicans) and antitubercular actions presenting a value of MIC = 3.12 μg mL−1.142
Nitric oxide-releasing APAP hybrids
Nitric oxide, the nitrogen monoxide radical (NO•), has been recognized as a ubiquitous gaseous transmitter, and its therapeutic potential has received increasing attention. Still, NO molecule cannot be easily directly administered due to its high reactivity in air and high concentration-dependent physiological roles. Thus, administering NO by applying its donors is considered therapeutically beneficial. Several NO donor compounds are currently in clinical studies as agents with antihypertensive, analgesic, anti-inflammatory, anti-cancer, and antiviral activities.143–147 Small heterocyclic molecules are suitable platforms for developing hybrid NO-releasing molecules (NO-donor hybrid APIs), which form two classic types. Nitrates and furoxans (1,2,5-oxadiazole 2-oxides) hybrids have been more developed. The NO-donor hybrid APIs containing a nitro-oxy moiety (O–NO2) (Fig. 21A) are more studied and, thus, progressed, especially NO–NSAID products.148 The second type, furoxan-based NO-donor prodrugs (Fig. 21B), can exogenous NO release in the presence of thiol cofactors and constitute an important class of perspective NO-donors.149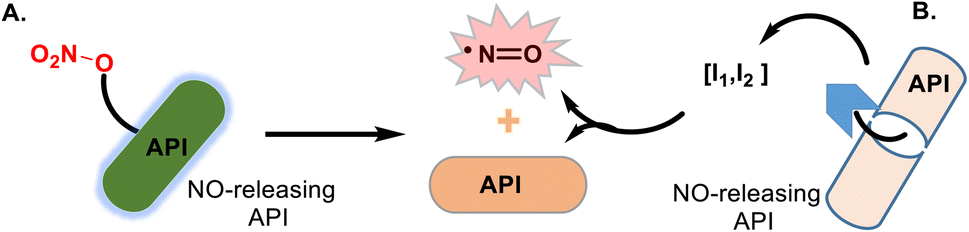 | ||
| Fig. 21 Pictographic representation. (A) NO-donor hybrid APIs is a broad grouping that covers a range of established drugs that have been structurally modified to incorporate NO-containing molecules. The release of nitric oxide from NO-donor hybrid API must be balanced to provide sufficient activity within the concentration range of the parent API. (B) NO-donor hybrid APIs with a nitrate group, such as hybrid NO–NSAIDs, show interesting anti-inflammatory/analgesic properties and attractive effects in several cardiovascular conditions, especially in vascular injury, atherosclerosis, and anticancer therapy. These hybrids are examples of multitarget drugs, the single chemical entities that can modulate more than one target. | ||
Nitrate group, the oldest and structurally simplest NO donors, still dominates medical research. The combination of the APAP core with the nitro-oxy moiety can allow the generation of diverse hybrids. Still, until the present, there are only known NO-releasing direct-linked O-modified APAP derivatives, such as hybrids 78–81 (Fig. 22). The aim of the APAP coupling with NO via an aliphatic spacer was to increase its anti-inflammatory activity and also to examine the cytoprotective properties of NO to reduce potential liver damage because the activity of NO on cytokine synthesis and regulation of pro-inflammatory mediators are known. Therefore, 4-acetamidophenyl-4-(nitrooxy)butanoate (78) (nitroparacetamol, agent NCX-701) was designed.150–153 Its single-dose phase II study shows it is a better analgesic than APAP in inflammation-induced pain. Moreover, it is safer than its parent compound, showing liver protection properties. This nitrate ester of O-acylated APAP also appears to enhance the effectiveness of other drugs, such as the μ-opioid agonist fentanyl, an opioid with potent and short-lasting antinociceptive effects.152
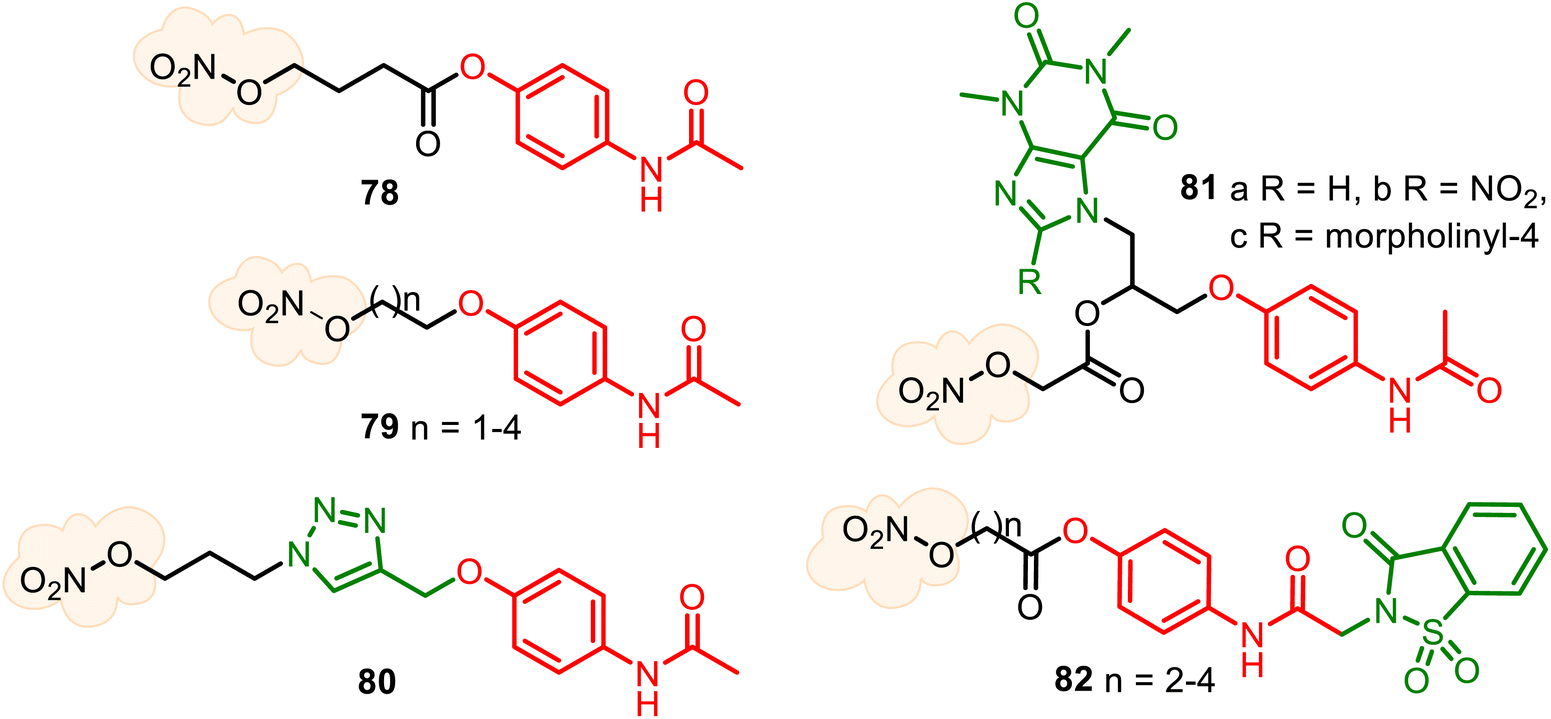 | ||
| Fig. 22 Diverse NO-releasing APAP hybrids. | ||
New nitrate esters of O-alkylated paracetamol 79 were patented as useful analgesic, anti-inflammatory, and lesser toxic agents.154,155 An interesting nitrooxy-TA–APAP hybrid 80, a triazole analog of NCX-701, was recently described but not tested.138 Combining two parent drugs, theophylline and APAP, new conjugated NO-releasing theoph-alkyl-APAP derivatives 81 were synthesized as stable solids, evaluating their toxicity degree (in white Swiss strain male mice) and anti-inflammatory activity (in Wistar adult male rats). The synthesized compounds 81a–c were less toxic (LD50 812, 450, and 433 mg kg−1 body, respectively) than theophylline and APAP (LD50 200 and 338 mg kg−1 body, respectively) and more active, namely with 2.5 times for (81a), 3.3 times (81b), and 2.0 times (81c) than theophylline and of 4.2 times (81a), 5.4 times (81b), and 4.0 times (81c) more active than APAP in the reduction of the subacute inflammatory edema induced by cotton pellet granuloma.156,157
Based on the analgesic and safety profile of the saccharin–APAP hybrid 37 (SCP-1), a series of new nitrate ester derivatives 82 was prepared and studied158 (Fig. 22). Their biological studies confirmed that attachment of a NO-donor moiety to the SCP-1 ring system allowed the preparation of hybrids with enhanced antipyretic activity compared to SCP-1 without increased toxicity. (Benzo)furoxans, as donating NO moieties, are very studied in antiparasitic drug research;149,159 however, are less developed for reducing pain and inflammation agents.160
Conclusion and outlook
Acetaminophen is still among the most popular drugs for relief of acute and chronic pain in recent years. Biochemical research on acetaminophen continues to be relevant and important in pain medicine. Single doses of acetaminophen show analgesic activity in various acute pain syndromes. Thus, its combination with other analgesics helps ease the pain. A combination of APAP with other drug groups (e.g., NSAID group and opioid analgesics). However, the therapeutic advantage of these combinations over either drug alone remains debatable.161 Replacing the combination approach (two or more APIs, one pill) with the multi-target approach (one multimodal API, one pill) seems a better policy.As is usually the case, a simple, small molecule like APAP has many complex biological effects, including toxic and unpleasant properties. To avoid them, the APAP core is modified. Thus, modified APAP derivatives, such as prodrugs and codrugs, are interesting models in the research for enhancing ADME characteristics and modifying undesirable drug properties. Simple chemical modification of the APAP core allows for obtaining new O- and N- or C-modified APAP derivatives designed to improve drug biopharmaceutical profiles. According to their molecular structure, APAP prodrugs or codrugs could be considered merged or conjugated hybrids; all these names are very fluid terms. However, a game of words does not change the objectives of pain treatment research. The search for new effective painkillers is not ending. Still, the worthy alternative to the “good old” acetaminophen could be some of its derivatives or analogs, i.e., combined chemical molecular entities mentioned above.
Thus, the design, synthesis, and study of the O-modified-APAP and N-modified APAP derivatives are constantly developed to seek their improved distribution, transportation, and application via oral and intravenous routes and topical or rectal routes. The O-modified-APAP series were more studied and progressed: propacetamol 27a, benorylate 46, acetaminosalol 47, or nitroparacetamol 78 are prime examples of APAP-based drugs of this series. Normally, these O-modified APAPs include an acyl-enabling functional group in the designed promoiety. Upon hydrolysis, they result in an unstable intermediate that spontaneously decomposes into the active form drug. The promoieties, based on the unionizable functional fragments, can improve water solubility and oral, topical, and/or transdermal delivery. Thus, analgesic and antipyretic action may increase and potentiate. Also, additional pharmacological properties could be generated. For example, O-modified APAP derivatives 29–33 and 33–36, based on the phosphate, sulfonate, or carbonyl-aminoalkyl moieties, can be used not only as analgesic, antipyretic, and anti-inflammatory medicines but also as neuroprotective agents. Unfortunately, the toxicological effects of these derivatives have not yet been studied.
Regarding so-called carrier-linked N-modified APAP prodrugs, their examples are much fewer but no less interesting than O-modified APAP derivatives. This small group of APAP-based analgesics (APAP derivatives 37–45) is characterized by one important property – a significant decrease in hepatotoxicity or its absence. Among them, heterocyclic ARAP derivative 45 (JNJ-10450232) is now under a Phase 2 dental pain study and was suggested for first-in-human clinical studies.
Research on the direct-linked NSAIDs–APAP codrugs in both versions, O-modified NSAIDs–APAP and N-modified NSAIDs–APAP hybrids, arose and was motivated by the useful tactics of the fixed-dose drug combinations (FCD). Early examples are benorylate 46 and acetaminosalol 47, the product's combination of APAP with the respective aspirin and salicylate drugs connected via a carboxylic ester function. Combining the APAP core with ibuprofen, naproxen, diclofenac, indomethacin, ketorolac, flurbiprofen, and fulfenamic, or fulfenamic and niflumic acids allowed to generate promising ester NSAIDs–APAP (49–52) and amide NSAIDs–APAP (61–64) derivatives. APAP's role in this merged hybrid series seems to be a temporary blocker of the free carboxylic group in the NSAIDs until their systemic absorption. Indeed, in laboratory animal experiments, some of these codrugs showed much improved antipyretic activity and good anti-inflammatory properties with nil ulcerogenic action compared to the parent drugs. Unfortunately, comparative studies using the same experimental tests of both series have not yet been carried out.
It would also be interesting to explore more bis-direct-linked APAP codrugs 70–71, 1,2,3-TA-based APAP hybrids 72–74 in which the 1,2,3-triazole ring acts as a linker, and C-linked APAP hybrids 76–77. Its suitable modification may introduce new nontoxic agents endowed with anti-inflammatory, analgesic, and antiparasitic activity, including anticancer properties. This also applies to a promising group of nitric oxide-releasing APAP hybrids (79–82) with its leader, agent NCX-701, under phase II study.
Notably, during the coronavirus disease 2019 (COVID-19) pandemic, APAP remained the drug of choice to control fever and muscle pain in the early stages, considering its potent antipyretic and weak anti-inflammatory activity.162 Thus, it would be advisable to start studying some of the hybrids mentioned above or make new ones, considering experience with new inhibitory medicines against COVID-19. Moreover, novel designs of APA hybrids can raise perspective and make compounds easy to prepare with antioxidant, antimicrobial, antibacterial, and antitumor properties.163,164 A suitable combination of the APAP core with scaffolds from natural products like artemisinin or its derivatives (APAP–dihydroartemisinin molecule, an O-modified APAP prodrug)165 can also allow refilling the arsenal of needed drugs.
Finally, considering that the nature of carries and linkers in each multi-targets drug, such as APAP, and the order of their combinations may vary, so-called combined molecular entities can be organized in an unrepeated way; the proposed classification of APAP derivatives can serve as a suitable guide that helps to understand the interactions of each structural group and makes it easier to do SAR studies.
Author contributions
Vladimir V. Kouznetsov: conceptualization, methodology, review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The author thanks his students and colleagues for motivating and encouraging him, as well as an anonymous reviewer for his valuable comments that improved the main text of this work.References
- R. D. Blondell, M. Azadfard and A. M. Wisniewski, Am. Fam. Physician, 2013, 87, 766 Search PubMed.
- B. R. Beal and M. S. Wallace, Med. Clin., 2016, 100, 65 Search PubMed.
- O. Amaechi, M. M. Human and K. Featherstone, Am. Fam. Physician, 2021, 104, 63 Search PubMed.
- D. C. Turk, H. D. Wilson and A. Cahana, Lancet, 2011, 377, 2226 CrossRef CAS PubMed.
- S. A. Schug and C. Goddard, Ann. Palliat. Med., 2014, 3, 263 Search PubMed.
- C. K. Ong, R. A. Seymour, P. Lirk and A. F. Merry, Anesth. Analg., 2010, 110, 1170 CrossRef CAS PubMed.
- G. G. Graham, M. J. Davies, R. O. Day, A. Mohamudally and K. F. Scott, Inflammopharmacology, 2013, 21, 201 CrossRef CAS PubMed.
- J. P. Hughes, S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239 CrossRef CAS PubMed.
- J. A. DiMasi, H. G. Grabowski and R. W. Hansen, J. Health Econ., 2016, 47, 20 CrossRef PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Acta Pharm. Sin. B, 2022, 12, 3049 CrossRef CAS PubMed.
- H. M. S. Kumar, L. Herrmann and S. B. Tsogoeva, Bioorg. Med. Chem. Lett., 2020, 30, 127514 CrossRef PubMed.
- A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, RSC Adv., 2022, 12, 19470 RSC.
- G. R. Zimmermann, J. Lehar and C. T. Keith, Drug Discovery Today, 2007, 12, 34 CrossRef CAS PubMed.
- J. Zhou, X. Jiang, S. He, H. Jiang, F. Feng, W. Liu, W. Qu and H. Sun, J. Med. Chem., 2019, 62, 8881 CrossRef CAS PubMed.
- N. Das, M. Dhanawat, B. Dash, R. C. Nagarwal and S. K. Shrivastava, Eur. J. Pharm. Sci., 2010, 41, 571 CrossRef CAS PubMed.
- C. G. Wermuth, C. R. Ganellin, P. Lindberg and L. A. Mitscher, Pure Appl. Chem., 1998, 70, 1129 CrossRef CAS.
- J. L. Medina-Franco, M. A. Giulianotti, G. S. Welmaker and R. A. Houghten, Drug Discovery Today, 2013, 18, 495 CrossRef PubMed.
- A. Sadri, J. Med. Chem., 2023, 66, 12651 CrossRef CAS PubMed.
- A. Kabir and A. Muth, Pharmacol. Res., 2021, 76, 106055 Search PubMed.
- E. Proschak, H. Stark and D. Merk, J. Med. Chem., 2018, 62, 420 CrossRef PubMed.
- P. S. Portoghese, Trends Pharmacol. Sci., 1989, 10, 230 CrossRef CAS PubMed.
- R. Morphy, C. Kay and Z. Rankovic, Drug Discovery Today, 2004, 9, 641 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, Curr. Pharm. Des., 2009, 15, 587 CrossRef CAS PubMed.
- M. D. Oliveira Pedrosa, R. M. Duarte da Cruz, J. D. Oliveira Viana, R. O. de Moura, H. M. Ishiki, J. M. Barbosa Filho, M. F. F. M. Diniz, M. T. L. Scotti and F. J. Bezerra Mendonca, Curr. Top. Med. Chem., 2017, 17, 1044 CrossRef PubMed.
- A. Pawełczyk, K. Sowa-Kasprzak, D. Olender and L. Zaprutko, Int. J. Mol. Sci., 2018, 19, 1104 CrossRef PubMed.
- K. Brune, B. Renner and G. Tiegs, Eur. J. Pain, 2015, 19, 953 CrossRef CAS PubMed.
- B. J. Anderson, Paediatr. Anaesth., 2008, 18, 915 CrossRef PubMed.
- U. Klotz, Arzneimittelforschung, 2012, 62, 355 CrossRef CAS PubMed.
- U. Freo, C. Ruocco, A. Valerio, I. Scagnol and E. Nisoli, J. Clin. Med., 2021, 10, 3420 CrossRef CAS PubMed.
- A. Aminoshariae and A. Khan, J. Endod., 2015, 41, 588 CrossRef PubMed.
- S. S. Ayoub, Temperature, 2021, 8, 351 CrossRef PubMed.
- D. M. Aronoff, J. A. Oates and O. Boutaud, Clin. Pharmacol. Ther., 2006, 79, 9 CrossRef CAS PubMed.
- G. W. Przybyła, K. A. Szychowski and J. Gmiński, Clin. Exp. Pharmacol. Physiol., 2021, 48, 3–19 CrossRef PubMed.
- A. Horecka, A. Hordyjewska, T. Blicharski and J. Kurzepa, Bosnian J. Basic Med. Sci., 2022, 22, 488 CAS.
- S. Schildknecht, A. Daiber, S. Ghisla, R. A. Cohen and M. M. Bachschmid, FASEB J., 2008, 22, 215 CrossRef CAS PubMed.
- G. Philippot, S. Hallgren, T. Gordh, A. Fredriksson, R. Fredriksson and H. Viberg, Toxicol. Sci., 2018, 166, 203 CrossRef CAS PubMed.
- P. M. Zygmunt, H. H. Chuang, P. Movahed, D. Julius and E. D. Högestätt, Eur. J. Pharmacol., 2000, 396, 39 CrossRef CAS PubMed.
- A. Bertolini, A. Ferrari, A. Ottani, S. Guerzoni, R. Tacchi and S. Leone, CNS Drug Rev., 2006, 12, 250 CrossRef CAS PubMed.
- Y. Megumu and F. Hidemasa, J. Pharmacol. Sci., 2006, 101, 107 CrossRef PubMed.
- W. M. Lee, Hepatology, 2004, 40, 6 CrossRef CAS PubMed.
- R. A. Moore and N. Moore, Eur. J. Hosp. Pharm., 2016, 23, 187 CrossRef PubMed.
- L. L. Mazaleuskaya, K. Sangkuhl, C. F. Thorn, G. A. FitzGerald, R. B. Altman and T. E. Klein, Pharmacogenet. Genomics, 2015, 25, 416 CrossRef CAS PubMed.
- E. Yoon, A. Babar, M. Choudhary, M. Kutne and N. Pyrsopoulos, J. Clin. Transl. Hepatol., 2016, 4, 131 Search PubMed.
- M. W. Gemborys and G. H. Mudge, Drug Metab. Dispos., 1981, 9, 340 CAS.
- J. M. Davis, K. R. Emslie, R. S. Sweet, L. L. Walker, R. J. Naughton, S. l. Skinner and J. D. Tange, Kidney Int., 1983, 24, 740 CrossRef CAS PubMed.
- B. Costa, D. Siniscalco, A. Trovato, F. Comelli, M. L. Sotgiu, M. Colleoni, S. Maione, F. Rossi and G. Giagnoni, Br. J. Pharmacol., 2006, 148, 1022 CrossRef CAS PubMed.
- E. D. Hogestatt, B. A. Jonsson, A. Ermund, D. A. Andersson, H. Bjork, J. P. Alexander, B. F. Cravatt, A. I. Basbaum and P. M. Zygmunt, J. Biol. Chem., 2005, 280, 31405 CrossRef PubMed.
- C. V. Sharma, J. H. Long, S. Shah, J. Rahman, D. Perrett, S. S. Ayoub and V. Mehta, J. Pain Res., 2017, 10, 2703 CrossRef CAS PubMed.
- C. I. Ghanem, M. J. Pérez, J. E. Manautou and A. D. Mottino, Pharmacol. Res., 2016, 109, 119 CrossRef CAS PubMed.
- J. Harle, C. Slater and M. Cafiero, ACS Omega, 2023, 8, 38053 CrossRef CAS PubMed.
- S. A. Schug and C. Goddard, Ann. Palliat. Med., 2014, 3, 263 Search PubMed.
- T. Herrick and R. Million, Nat. Rev. Drug Discovery, 2007, 6, 513 CrossRef CAS PubMed.
- J. Foucquier and M. Guedj, Pharmacol. Res. Perspect., 2015, 3, e00149 CrossRef PubMed.
- C. S. Gautam and L. Saha, Br. J. Clin. Pharmacol., 2008, 65, 795 CrossRef PubMed.
- A. Albert, Nature, 1958, 182, 421 CrossRef CAS PubMed.
- N. J. Harper, J. Med. Pharm. Chem., 1959, 1, 467 CrossRef CAS PubMed.
- J. B. Zawilska, J. Wojcieszak and A. B. Olejniczak, Pharmacol. Rep., 2013, 65, 1 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255 CrossRef CAS PubMed.
- L. W. Dittert, H. C. Caldwell, H. J. Adams, G. M. Irwin and J. V. Swintosky, J. Pharm. Sci., 1968, 57, 774 CrossRef CAS PubMed.
- L. W. Dittert, G. M. Irwin, C. W. Chong and J. V. Swintosky, J. Pharm. Sci., 1968, 57, 780 CrossRef CAS PubMed.
- K. B. Sloan, Adv. Drug Delivery Rev., 1989, 3, 67 CrossRef CAS.
- S. C. Wasdo and K. B. Sloan, Pharm. Res., 2004, 21, 940 CrossRef CAS PubMed.
- J. D. Thomas and K. B. Sloan, Int. J. Pharm., 2008, 346, 80 CrossRef CAS PubMed.
- J. D. Thomas and K. B. Sloan, Tetrahedron Lett., 2007, 48, 109 CrossRef CAS.
- J. D. Thomas and K. B. Sloan, Int. J. Pharm., 2009, 371, 25 CrossRef CAS PubMed.
- J. D. Thomas, S. Majumdar and K. B. Sloan, Molecules, 2009, 14, 4231 CrossRef CAS PubMed.
- S. Majumdar and K. B. Sloan, Synth. Commun., 2006, 36, 3537 CrossRef CAS.
- S. Majumdar and K. B. Sloan, Int. J. Pharm., 2007, 337, 48 CrossRef CAS PubMed.
- H. Devarajan-Ketha and K. B. Sloan, Bioorg. Med. Chem. Lett., 2011, 21, 4078 CrossRef CAS PubMed.
- M. Depré, A. Van Hecken, R. Verbesselt, T. B. Tjandra-Maga, M. Gerin and P. J. De Schepper, Fundam. Clin. Pharmacol., 1992, 6, 259 CrossRef PubMed.
- E. Viel, A. Langlade, M. Osman, P. Bilbault and J. J. Eledjam, Ann. Fr. Anesth. Reanim., 1999, 18, 332 CrossRef CAS PubMed.
- P. L. Moller, G. I. Juhl, C. Payen-Champenois and L. A. Skoglund, Anesth. Analg., 2005, 101, 90 CrossRef CAS PubMed.
- P. L. Moller, S. Sindet-Pedersen, C. T. Petersen, G. I. Juhl, A. Dillenschneider and L. A. Skoglund, Br. J. Anaesth., 2005, 94, 642 CrossRef CAS PubMed.
- I. M. Kovach, I. H. Pitman and T. Higuchi, J. Pharm. Sci., 1981, 70, 881 CrossRef CAS PubMed.
- A. Parashar, World J. Pharm. Res., 2014, 3, 1111 CAS.
- Z. Wu, A. Patel, R. Dave and X. Yuan, Bioorg. Med. Chem. Lett., 2010, 20, 3851 CrossRef CAS PubMed.
- A. Parashar, Indian J. Pharmacol., 2015, 47, 202 CrossRef CAS PubMed.
- C. Santos, M. L. Mateus, A. P. Santos, R. Moreira, E. De Oliveira and P. Gomes, Bioorg. Med. Chem. Lett., 2005, 15, 1595 CrossRef CAS PubMed.
- F. Sodano, L. Lazzarato, B. Rolando, F. Spyrakis, C. De Caro, S. Magliocca, K. Chegaev, E. Gazzano, C. Riganti, A. Calignano, R. Russo and M. G. Rimoli, Mol. Pharmaceutics, 2019, 16, 4181 CrossRef CAS PubMed.
- M. Taniguchi and M. Nakano, Chem. Pharm. Bull., 1981, 29, 577 CrossRef CAS.
- D. B. Williams, S. A. Varia, V. J. Stella and I. H. Pitman, Int. J. Pharm., 1983, 14, 113 CrossRef CAS.
- M. Stjepanovic, M. Moreau and J. Dugniolle, US Pat., 4322410, 1982 Search PubMed.
- R. Kumar, S. Jain and N. Jain, Pharma Chem., 2013, 5, 73 CAS.
- O. A. Cojocaru, K. Bica, G. Gurau, A. Narita, P. D. McCrary, J. L. Shamshina, P. S. Barber and R. D. Rogers, MedChemComm, 2013, 4, 559 RSC.
- K. R. Bley and B. Jandeleit, US Pat., 9024055, 2015 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 9289501, 2016 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 9683000, 2017 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 10377779, 2019 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 11136342, 2021 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 9981998, 2018 Search PubMed.
- N. Muhammad and K. R. Bley, US Pat., 10442826, 2019 Search PubMed.
- N. G. Bazan and J. Alvarez-Builla, US Pat., 5554636, 1996 Search PubMed.
- N. G. Bazan and J. Alvarez-Builla, US Pat., 5621110, 1997 Search PubMed.
- G. González-Martin, C. Lyndon and C. Sunkel, Eur. J. Pharm. Biopharm., 1998, 46, 293 CrossRef PubMed.
- A. L. Vaccarino, D. Paul, P. K. Mukherjee, E. B. R. de Turco, V. L. Marcheselli, L. Xu, M. L. Trudell, J. M. Minguez, M. P. Matía, C. Sunkel, J. Alvarez-Builla and N. G. Bazan, Bioorg. Med. Chem., 2007, 15, 2206 CrossRef CAS PubMed.
- L. Miao, L. Xu, K. W. Narducy and M. L. Trudell, Org. Process Res. Dev., 2009, 13, 820 CrossRef CAS PubMed.
- Y. D. Reddy, P. P. Kumar, B. R. Devi, P. K. Dubey and Y. B. Kumari, Lett. Drug Des. Discovery, 2013, 10, 226 CAS.
- H. A. Bazan, S. Bhattacharjee, C. Burgos, J. Recio, V. Abet, A. R. Pahng, B. Jun, J. Heap, A. J. Ledet, W. C. Gordon, S. Edwards, D. Paul, J. Alvarez-Builla and N. G. Bazan, Eur. J. Med. Chem., 2020, 202, 112600 CrossRef CAS PubMed.
- Y. D. Reddy, Y. B. Kumari and P. K. Dubey, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2013, 52, 691 Search PubMed.
- Y. D. Reddy, C. V. R. Reddy and P. K. Dubey, Green Chem. Lett. Rev., 2014, 7, 24 CrossRef.
- A. Sahu, D. Das, R. K. Agrawal and A. Gajbhiye, Life Sci., 2019, 228, 176 CrossRef CAS PubMed.
- A. Sahu, D. Das, P. Sahu, S. Mishra, A. Sakthivel, A. Gajbhiye and R. Agrawal, Chem. Res. Toxicol., 2020, 33, 522 Search PubMed.
- D. Lengerli, K. Ibis, Y. Nural and E. Banoglu, Expert Opin. Drug Discovery, 2022, 17, 1209 Search PubMed.
- J. Zhou, S. De Jonghe, E. E. Codd, S. Weiner, D. Gallacher, P. Stahle, M. F. Kelley, E. K. Kuffner, C. M. Flores and G. E. Eichenbaum, Regul. Toxicol. Pharmacol., 2023, 105334 CrossRef PubMed.
- H. K. Lim, J. Chen, W. Lam, Y. Gong, L. Leclercq, J. Silva, R. Salter, J. Berwaerts, C. K. Gelotte, A. M. Vakil, G. E. Eichenbaum, E. K. Kuffner and C. M. Flores, Regul. Toxicol. Pharmacol., 2023, 105379 CrossRef PubMed.
- C. K. Gelotte, A. M. Vakil, J. Berwaerts, B. A. Zimmerman, G. E. Eichenbaum, C. M. Flores and E. K. Kuffner, Regul. Toxicol. Pharmacol., 2022, 134, 105236 CrossRef CAS PubMed.
- C. K. Gelotte, A. M. Vakil, J. Berwaerts, B. A. Zimmerman, P. Zannikos, R. Mishra, C. Eichenbaum, E. K. Kuffner and C. M. Flores, Regul. Toxicol. Pharmacol., 2023, 105480 CrossRef PubMed.
- K. P. Conner, P. Vennam, C. M. Woods, M. D. Krzyaniak, M. K. Bowman and W. M. Atkins, Biochemistry, 2012, 51, 6441 CrossRef CAS PubMed.
- M. N. Podgorski, T. Coleman, P. D. Giang, C. R. Wang, J. B. Bruning, P. V. Bernhardt, J. J. De Voss and S. G. Bell, Inorg. Chem., 2021, 61, 236 CrossRef PubMed.
- K. Srikanth Kumar, A. Lakshmana Rao, M. Bhagya Sri, M. Pravallika, M. Kalyani and K. Seetha Ramudu, Int. J. Pharma Res. Health Sci., 2018, 6, 169 Search PubMed.
- J. E. Murphy, J. Int. Med. Res., 1972, 1, 65 CrossRef CAS.
- V. Wright, Scand. J. Rheumatol., 1976, 4, 5 CrossRef.
- A. Robertson, J. P. Glynn and A. K. Watson, Xenobiotica, 1972, 2, 339 CrossRef CAS PubMed.
- F. M. Williams, U. Moore, R. A. Seymour, E. M. Mutch, E. Nicholson, P. Wright, H. Wynne, P. G. Blain and M. D. Rawlins, Br. J. Clin. Pharmacol., 1989, 28, 703 CrossRef CAS PubMed.
- S. Similä, S. Keinänen and K. Kouvalainen, Eur. J. Pediatr., 1975, 121, 15 CrossRef PubMed.
- R. Q. Brewster, J. Am. Chem. Soc., 1918, 40, 1136 CrossRef CAS.
- A. M. Qandil, Int. J. Mol. Sci., 2012, 13, 17244 CrossRef CAS PubMed.
- T. A. Fadl and F. A. Omar, Inflammopharmacology, 1998, 6, 143 CrossRef CAS PubMed.
- M. E. González-Trujano, G. Uribe-Figueroa, S. Hidalgo-Figueroa, A. L. Martínez, M. Déciga-Campos and G. Navarrete-Vazquez, Biomed. Pharmacother., 2018, 101, 553 CrossRef PubMed.
- M. N. Abualhasan, M. Y. Al-Masri, R. Manasara, L. Yadak and N. S. Abu-Hasan, Scientifica, 2020, 2020, 9817502 CrossRef PubMed.
- D. Visagaperumal, C. Mayuren, N. Anbalagan, P. Raghuveer Varma, B. Srinivas, V. B. Shanker Bontha and G. Ravikumar, Indian Drugs, 2013, 49, 25 Search PubMed.
- C. Sinning, B. Watzer, L. De Petrocellis, V. Di Marzo and P. Imming, ChemMedChem, 2008, 3, 1956 CrossRef CAS PubMed.
- V. Onnis, C. Congiu, E. Björklund, F. Hempel, E. Söderström and C. J. Fowler, J. Med. Chem., 2010, 53, 2286 CrossRef CAS PubMed.
- A. Peperidou, D. Kapoukranidou, C. Kontogiorgis and D. Hadjipavlou-Litina, Molecules, 2014, 19, 20197 CrossRef PubMed.
- A. Vigroux and M. Bergon, Bioorg. Med. Chem. Lett., 1995, 5, 427 CrossRef CAS.
- A. Vigroux, M. Bergon and C. Zedde, J. Med. Chem., 1995, 38, 3983 CrossRef CAS PubMed.
- P. Gomes, N. Vale and R. Moreira, Molecules, 2007, 12, 2484 CrossRef CAS PubMed.
- A. S. Kalgutkar, A. B. Marnett, B. C. Crews, R. P. Remmel and L. J. Marnett, J. Med. Chem., 2000, 43, 2860 CrossRef CAS PubMed.
- M. R. Yadav, D. M. Nimekar, A. Ananthakrishnan, P. S. Brahmkshatriya, S. T. Shirude, R. Giridhar, A. Parmar and R. Balaraman, Bioorg. Med. Chem., 2006, 14, 8701 CrossRef CAS PubMed.
- A. D. Tiwari, S. S. Panda, A. S. Girgis, S. Sahu, R. F. George, A. M. Srour, B. La Starza, A. M. Asiri, C. D. Hall and A. R. Katritzky, Org. Biomol. Chem., 2014, 12, 7238 RSC.
- S. S. Panda, A. S. Girgis, H. H. Honkanadavar, R. F. George and A. M. Srour, Future Med. Chem., 2020, 12, 1369 CrossRef CAS PubMed.
- L. Profire, D. Lupascu, V. Sunel, N. Bibire and C. Vasile, Rev. Chim., 2010, 61, 553 CAS.
- L. Profire, V. Sunel, D. Lupascu, M. C. Baican, N. Bibire and C. Vasile, Farmacia, 2010, 58, 170 CAS.
- A. H. Alwash, A. M. Mahdi and H. J. Al-Karagully, Asian J. Pharm. Clin. Res., 2018, 11, 443 CrossRef CAS.
- G. Navarrete-Vázquez, H. Torres-Gómez, S. Hidalgo-Figueroa, J. J. Ramírez-Espinosa, S. Estrada-Soto, J. L. Medina-Franco, I. León-Rivera, F. J. Alarcón-Aguilar and J. L. Almanza-Pérez, Bioorg. Med. Chem. Lett., 2014, 24, 4575 CrossRef PubMed.
- K. Ramanjaneyulu, J. H. Bindhu, R. R. Babu, Y. R. Prasad, T. U. Naaz, P. H. Ranjani and P. N. S. Gouthami, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2019, 58, 1257 Search PubMed.
- V. V. Kouznetsov, D. Calderón Lamus and C. E. Puerto Galvis, Reactions, 2023, 4, 329 CrossRef CAS.
- P. Coghi, J. P. Ng, A. A. Nasim and V. K. W. Wong, Molbank, 2021, 2021, M1213 CrossRef.
- H. B. Joolakanti, R. Kamepalli, J. Miryala and S. Battu, J. Mol. Struct., 2021, 1242, 130786 CrossRef CAS.
- D. J. Webb and I. L. Megson, Expert Opin. Invest. Drugs, 2002, 1, 587 CrossRef PubMed.
- A. S. Rao, L. Simon, K. K. Srinivasan, S. Moorkoth, Z. A. Haleem, S. S. Jadon, R. M. Matsa and R. Sagiraju, Res. J. Chem. Sci., 2014, 4, 1 Search PubMed.
- A. Ahmad, A. Husain, S. A. Khan, M. Mujeeb and A. Bhandari, J. Saudi Chem. Soc., 2016, 20, 577 CrossRef CAS.
- M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305 CrossRef CAS PubMed.
- Z. Huang, J. Fu and Y. Zhang, J. Med. Chem., 2017, 60, 7617 CrossRef CAS PubMed.
- R. A. Serafim, F. G. Pernichelle and E. I. Ferreira, Expert Opin. Drug Discovery, 2017, 12, 941 CrossRef CAS PubMed.
- S. Kumar, R. K. Singh and T. R. Bhardwaj, Biomed. Pharmacother., 2017, 85, 182 CrossRef CAS PubMed.
- K. R. A. Abdellatif, E. K. A. Abdelall and B. R. Bakr, Curr. Top. Med. Chem., 2017, 17, 941 CrossRef CAS PubMed.
- L. L. Fershtat and N. N. Makhova, ChemMedChem, 2017, 12, 622 CrossRef CAS PubMed.
- O. A. Al-Swayeh, L. E. Futter, R. H. Clifford and P. K. Moore, Br. J. Pharmacol., 2000, 130, 1453 CrossRef CAS PubMed.
- E. A. Romero-Sandoval, J. Mazario, D. Howat and J. F. Herrero, Br. J. Pharmacol., 2002, 135, 1556 CrossRef CAS PubMed.
- P. K. Moore and M. Marshall, Dig. Liver Dis., 2003, 35, S49 CrossRef CAS PubMed.
- E. A. Romero-Sandoval, M. M. Curros-Criado, G. Gaitan, C. Molina and J. F. Herrero, CNS Drug Rev., 2007, 13, 279 CrossRef CAS PubMed.
- T. R. Bhardwaj, M. Kumar, N. Mehta and N. Dhingra, WO 2009/024998, 2009.
- T. R. Bhardwaj, M. Kumar, N. Mehta and N. Dhingra, US Pat., 8207222, 2012 Search PubMed.
- L. Profire, A. Cojocariu, A. M. Oprea, C. E. Lupusoru, C. M. Ghiciuc, C. A. Dehelean and C. Vasile, Rev. Chim., 2010, 61, 1150 CAS.
- L. Profire, M. Apotrosoaei, A. M. Oprea, M. Brebu, F. Lupascu, C. E. Lupusoru and C. Vasile, J. Serb. Chem. Soc., 2014, 79, 389 CrossRef CAS.
- M. Das, S. Bhattacharjee, F. R. Fronczek, N. G. Bazan and M. L. Trudell, Bioorg. Med. Chem. Lett., 2018, 28, 3798 CrossRef CAS PubMed.
- H. Cerecetto and W. Porcal, Mini-Rev. Med. Chem., 2005, 5, 57 CrossRef CAS PubMed.
- S. Bindu, S. Mazumder and U. Bandyopadhyay, Biochem. Pharmacol., 2020, 180, 114147 CrossRef CAS PubMed.
- S. Frantz, Nat. Rev. Drug Discovery, 2006, 5, 881 CrossRef PubMed.
- D. Wojcieszyńska, H. Guzik and U. Guzik, Sci. Total Environ., 2022, 834, 155317 CrossRef PubMed.
- S. Begum and R. Anjum, Int. J. Pharm. Sci. Nanotechnol., 2023, 16, 6309 CAS.
- M. M. Alam, N. I. Alsenani, A. A. Abdelhamid, A. Ahmad, O. A. Baothman, S. A. H. Altayeb, M. S. Nadeem, V. Ahmad, S. Nazreen and A. A. Elhenawy, Arch. Pharm., 2024, 357, 2300340 CrossRef CAS PubMed.
- L. Liu, Y. Ai, J. Ding, B. Li, C. Zeng, X. Zhang, H. Zhong and Z. Zhou, J. Mol. Struct., 2022, 1250, 131758 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |