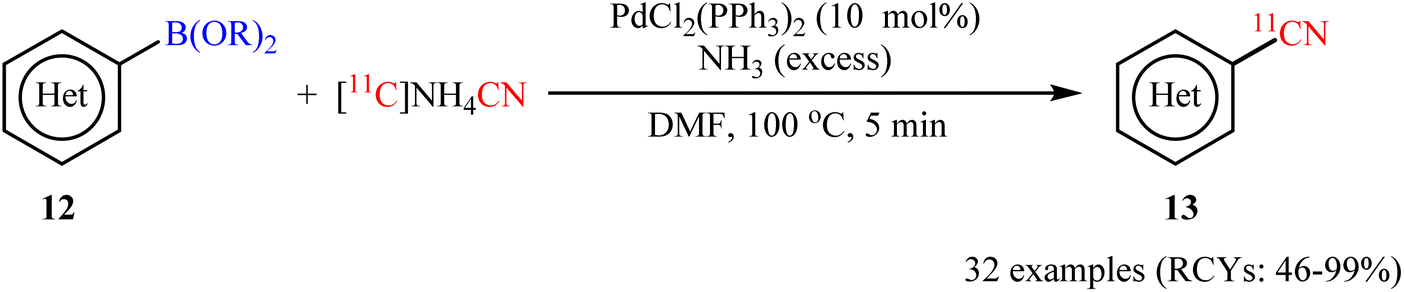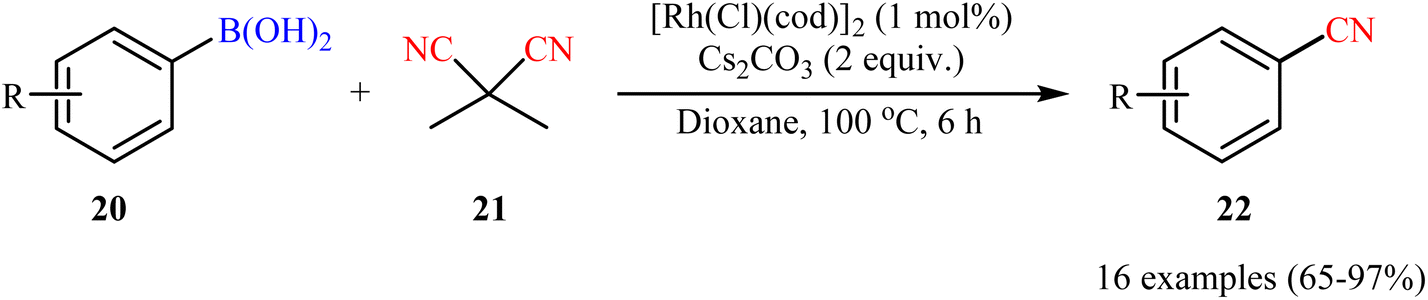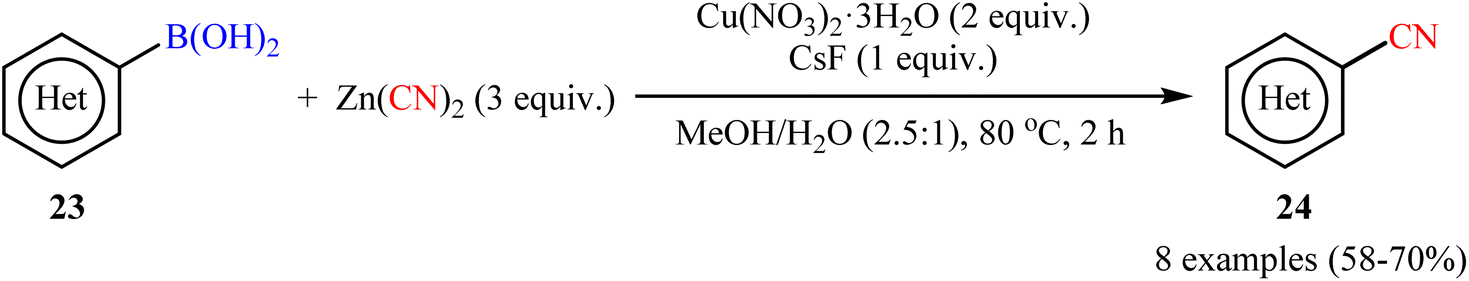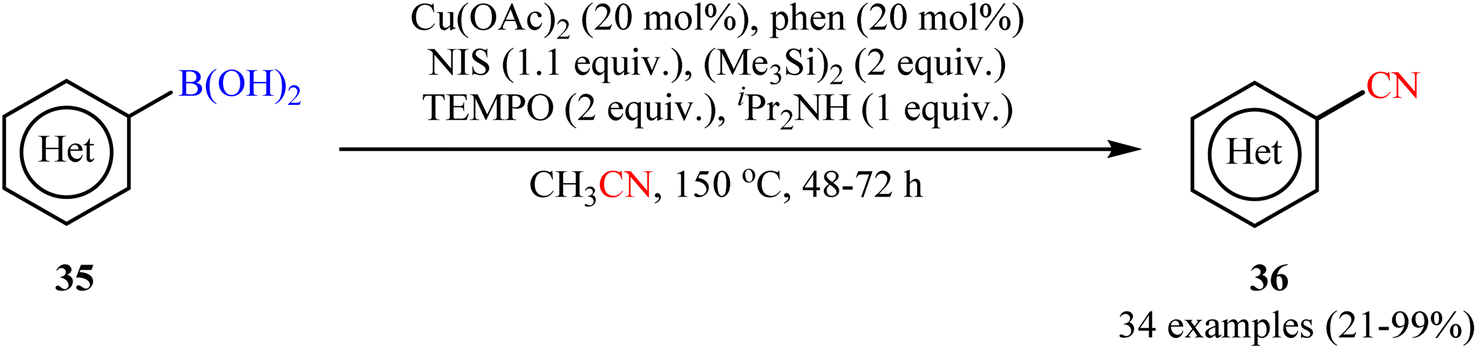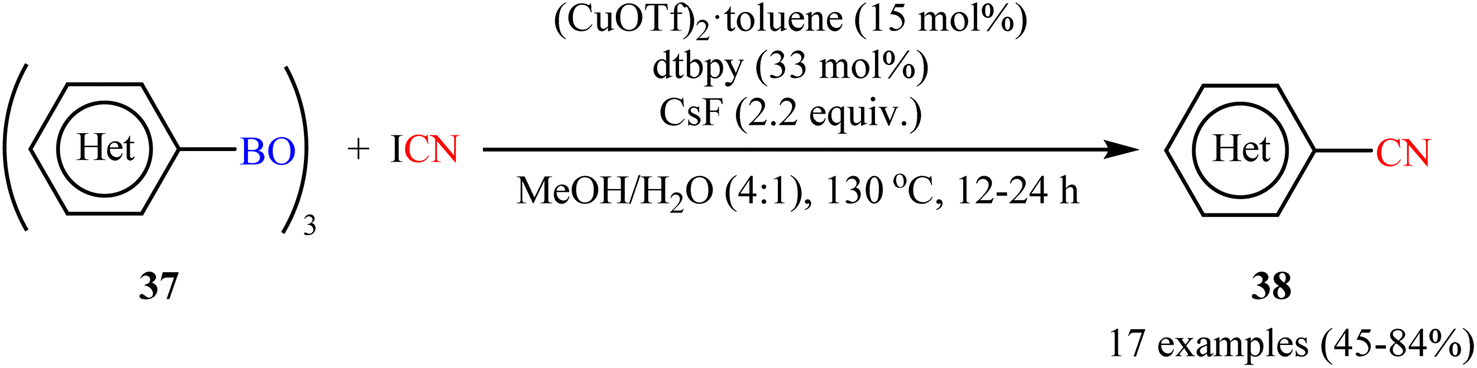DOI:
10.1039/D4RA00487F
(Review Article)
RSC Adv., 2024,
14, 9184-9199
Recent investigations into deborylative (thio-/seleno-) cyanation of aryl boronic acids
Received
18th January 2024
, Accepted 11th March 2024
First published on 19th March 2024
Abstract
In this review, we intend to summarize the most important discoveries in the deborylative (thio-/seleno-) cyanation of aryl boronic acids from 2006 to the end of 2023. Thus, the review is divided into three parts. The first section focuses exclusively on cyanation of aryl boronic acids into aryl nitriles. The second section covers the available literature on the synthesis of aryl thiocyanates through thiocyanation of respective aryl boronic acids. The third will discuss selenocyanation of aryl boronic acids into aryl selenocyanates.
1. Introduction
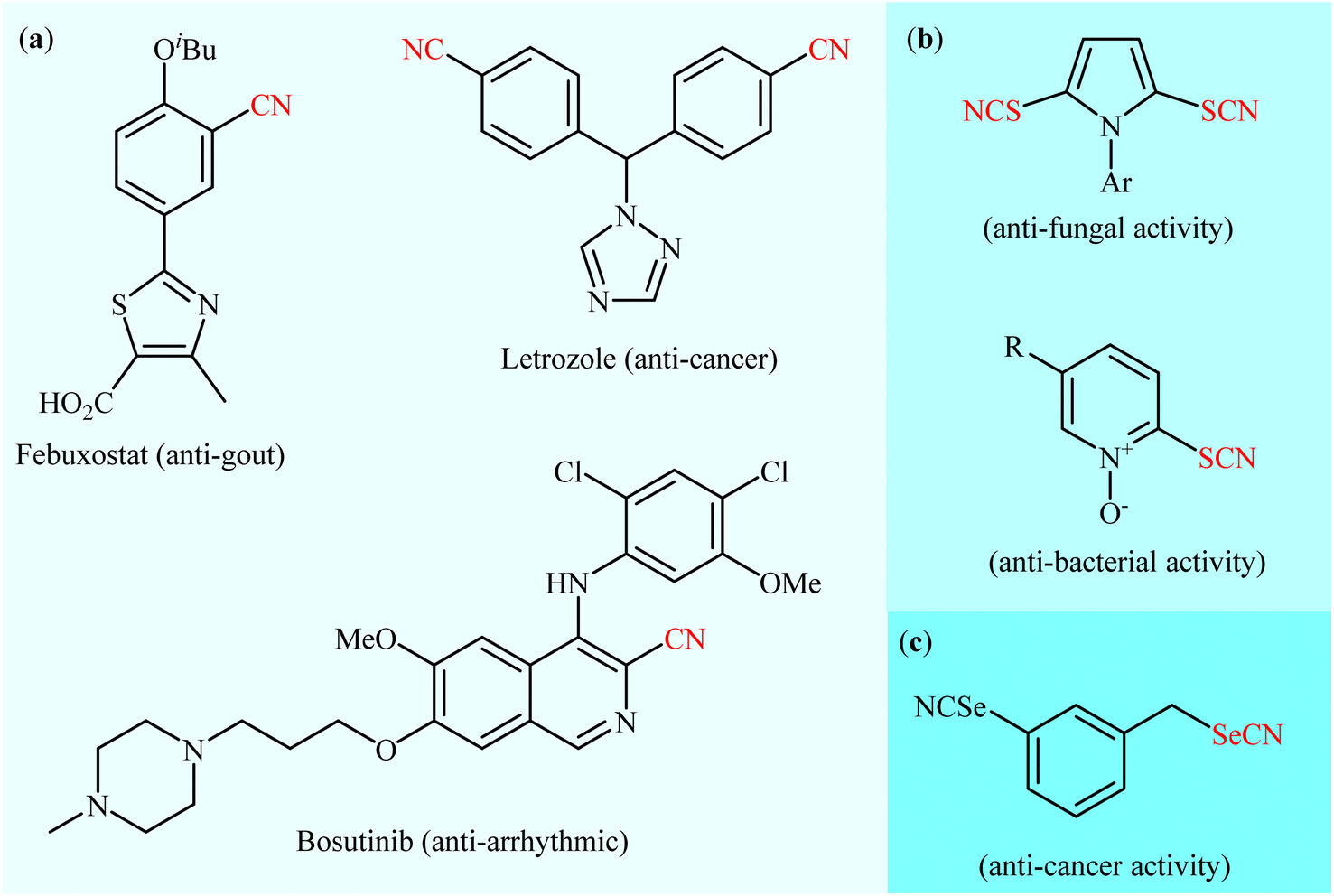
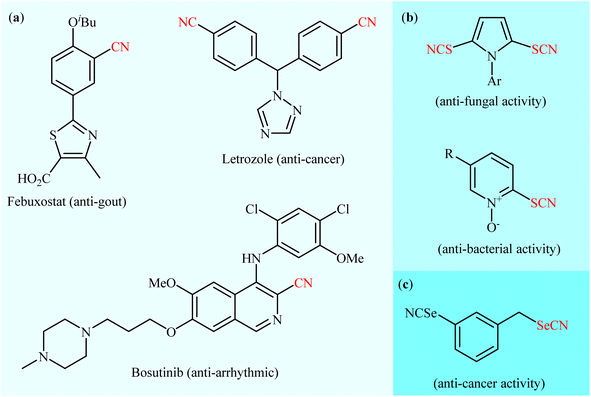
As one of the most important and versatile functional groups, the nitrile group (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) plays a vital role in organic chemistry and serves as a precursor for various functional groups, such as amine, amide, aldehyde, ketone, carboxylic acid, triazole, and tetrazole groups.1 More significantly, aryl nitrile skeletal structures are widely found in pharmaceuticals (Scheme 1a),2 agrochemicals,3 herbicides,4 dyes5 and natural products.6 In a similar way, thiocyanate (–SCN) is a structurally related moiety to nitrile, widely encountered in medicinal chemistry (Scheme 1b)7 and natural products.8 Aryl thiocyanates are particularly relevant because they can be easily converted into a number of other useful organic compounds, such as thioethers,9 disulfides,10 thiophenols,11 sulfonyl cyanides,12 and trifluoromethyl thioethers.13 Likewise, the selenium analogue of thiocyanates, selenocyanates (R–SeCN) are an important class of seleno-organic compounds due to their significant biological activity14 and numerous applications in organic synthesis.15 In this class of compounds, aryl selenocyanates have recently attracted strong interest in medicinal chemistry owing to their notable biological activities especially as anti-cancer and chemopreventive agents (Scheme 1c).16,17
N) plays a vital role in organic chemistry and serves as a precursor for various functional groups, such as amine, amide, aldehyde, ketone, carboxylic acid, triazole, and tetrazole groups.1 More significantly, aryl nitrile skeletal structures are widely found in pharmaceuticals (Scheme 1a),2 agrochemicals,3 herbicides,4 dyes5 and natural products.6 In a similar way, thiocyanate (–SCN) is a structurally related moiety to nitrile, widely encountered in medicinal chemistry (Scheme 1b)7 and natural products.8 Aryl thiocyanates are particularly relevant because they can be easily converted into a number of other useful organic compounds, such as thioethers,9 disulfides,10 thiophenols,11 sulfonyl cyanides,12 and trifluoromethyl thioethers.13 Likewise, the selenium analogue of thiocyanates, selenocyanates (R–SeCN) are an important class of seleno-organic compounds due to their significant biological activity14 and numerous applications in organic synthesis.15 In this class of compounds, aryl selenocyanates have recently attracted strong interest in medicinal chemistry owing to their notable biological activities especially as anti-cancer and chemopreventive agents (Scheme 1c).16,17
 |
| | Scheme 1 Selected examples of biologically active aryl (thio-/seleno-)cyanates. | |
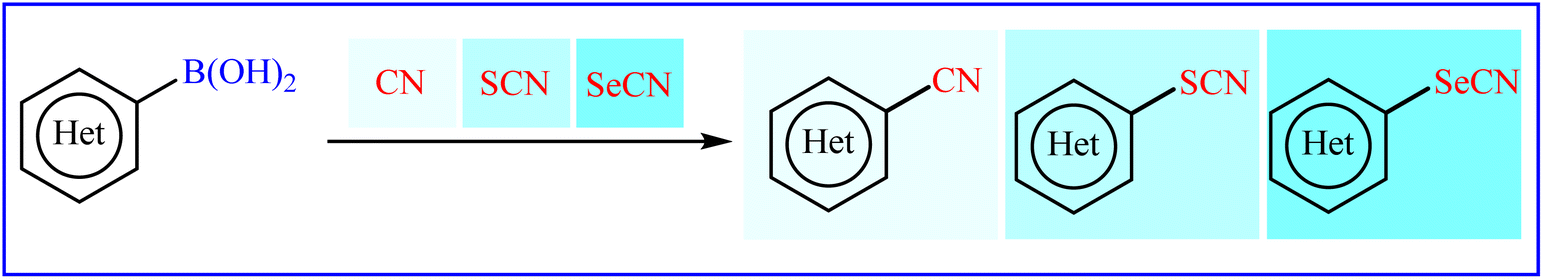
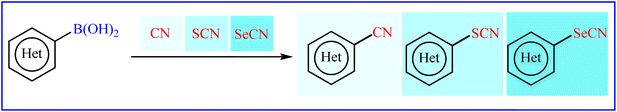
Traditionally, aryl (thio-/seleno-)cyanates are prepared by reaction of (thio-/seleno-)cyanate sources with active aryl species such as aryl halides18–20 and aryl diazonium salts.21–23 However, the mutagenicity and toxicity of some aryl halides24 and high reactivity, explosive nature and instability of aryl diazonium salts25 can be considered as the main drawbacks of these protocols. Recent endeavor in this domain has allowed the use of aryl boronic acids as easy accessible, non-toxic, air- and moisture-stable aryl species. In 2017, Yan, Zhang, and Wang published an interesting review paper entitled “Recent advances in the synthesis of aryl nitrile compounds” that highlights some of the important investigations into the deborylative cyanation of aryl boronic acid derivatives.26 Since significant progress and improvements in this fast-growing research area has been made during the past few years, it seems an appropriate time to summarize those developments as well as the available literature on thio-/seleno-cyanation of aryl boronic acids in an updated and comprehensive review. Therefore, herein, we try to provide a comprehensive overview of the synthesis of aryl (thio-/seleno-)cyanates through deborylative (thio-/seleno-)cyanation of respective aryl boronic acids (Fig. 1), with an aim to encourage scientists to make further progress in this appealing research area.
 |
| | Fig. 1 Deborylative (thio-/seleno-)cyanation of aryl boronic acids. | |
2. Cyanation of aryl boronic acids
2.1. Palladium-catalyzed reactions
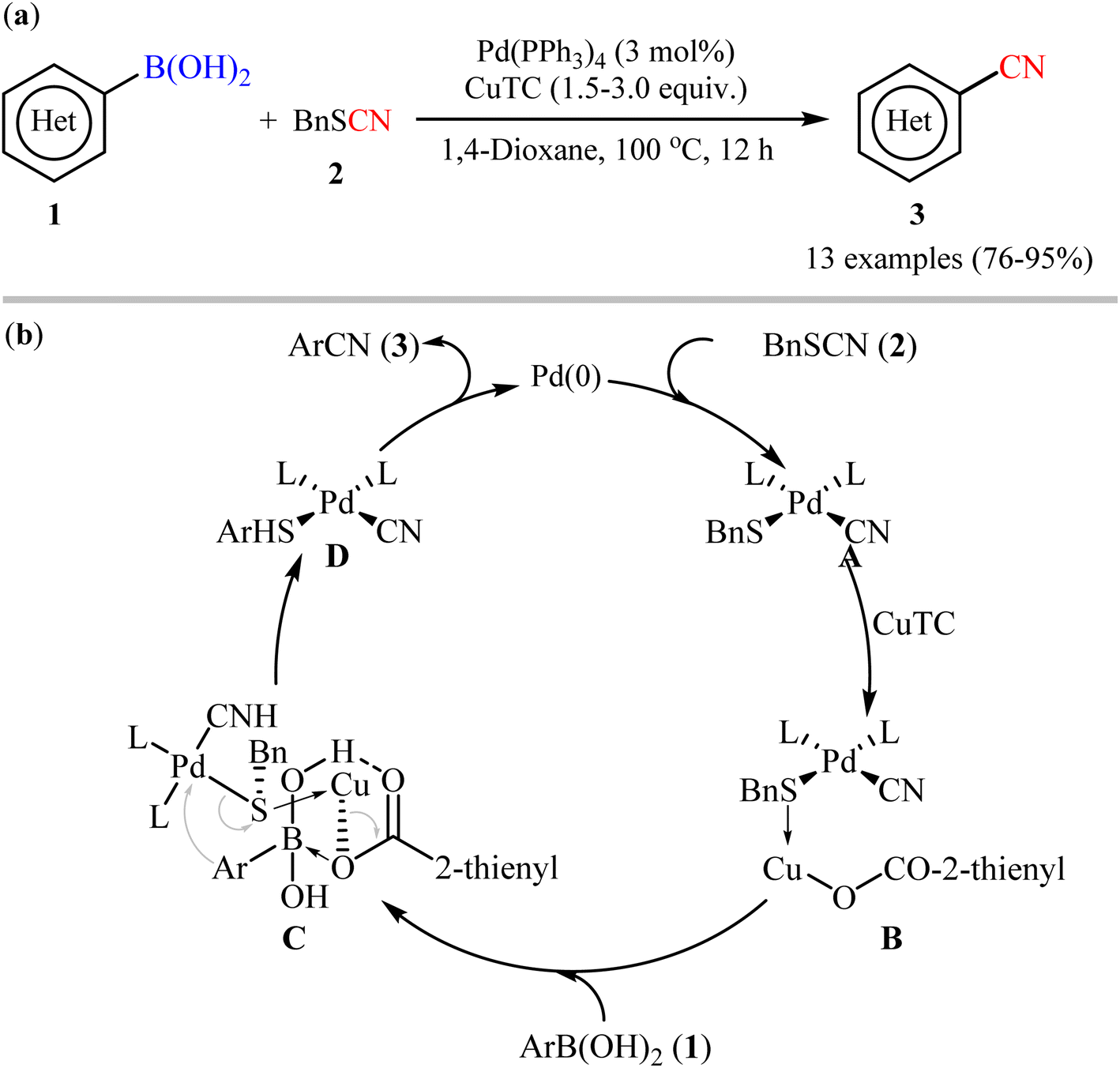
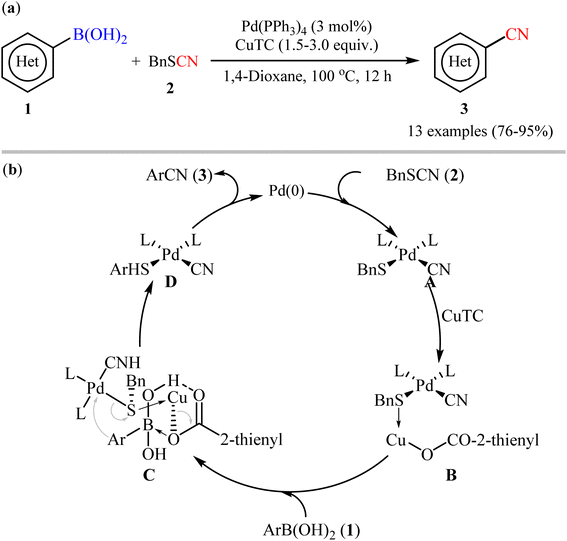
One of the first pioneering work of palladium-catalyzed cyanation of aryl boronic acids has been successfully demonstrated by Zhang and Liebeskind in 2006,27 who disclosed that the treatment of (hetero)aryl boronic acids 1 with commercially available low-cost benzyl thiocyanate 2 as cyanating agent in the presence of a catalytic amount of Pd(PPh3)4 and superstoichiometric amounts of copper(I) thiophene-2-carboxylate (CuTC), resulted in the formation of the corresponding (hetero)aryl nitriles 3 in good to excellent yields. As shown in Scheme 2a, both electron-rich and electron-poor aryl boronic acids (with the exception of nitro-substituted substrates) were compatible with this reaction. Additionally, the authors also carried out the cyanation of two alkenylboronic acid derivatives, affording the corresponding products in high yields. However, neither boronate esters nor boroxines were effective substrates in this transformation. According to author's anticipated mechanism (Scheme 2b), the reaction starts with an oxidative addition of the thiocyanate to Pd(0) to form complex A. Next, coordination of the sulfur atom of the SCN group to CuTC gives intermediate B that, after transmetalation with boronic acid 2 affords complex D through a chair transition state C. Finally, reductive elimination of this intermediate provides the desired nitrile 3 and regenerates the catalytically active Pd(0).
 |
| | Scheme 2 (a) Pd-catalyzed cyanation of (hetero)aryl boronic acids 1 with benzyl thiocyanate; (b) plausible mechanism for the formation of(hetero)aryl nitriles 3. | |
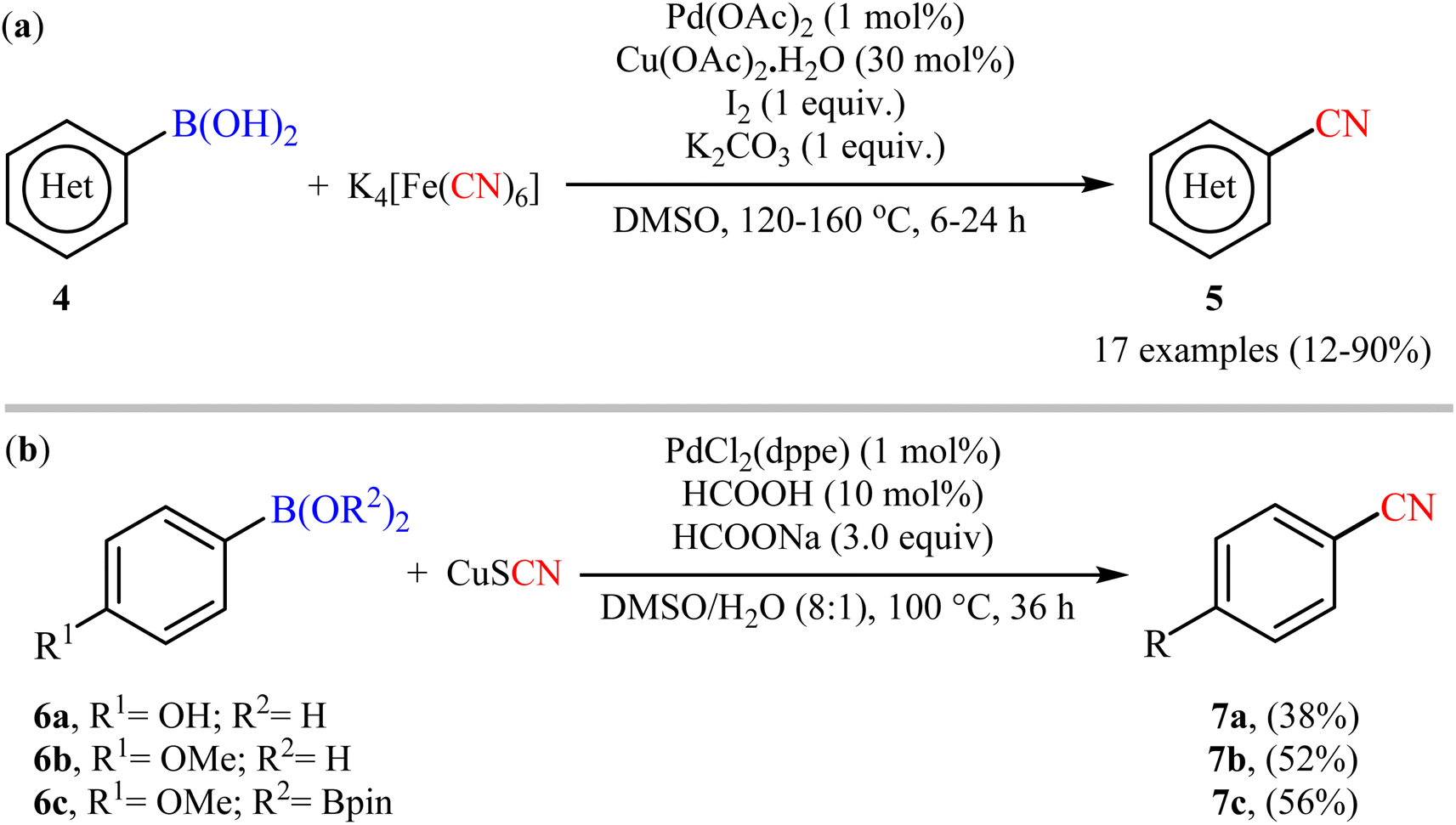
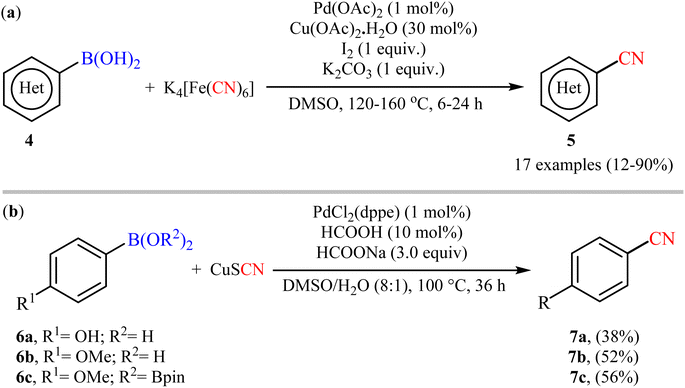
Six years later, Tian and co-workers unravelled an elegant route for the synthesis of diverse (hetero)aryl nitriles 5 from the corresponding (hetero)aryl boronic acids 4 using nontoxic and inexpensive K4[Fe(CN)6] as a cyanating agent under the action of a catalytic system comprised of Pd(OAc)2, Cu(OAc)2·H2O, I2, and K2CO3 (Scheme 3a).28 The current methodology exhibited a relatively broad substrate scope, good functional group tolerance and satisfactory yields. However, the drastic conditions of temperature (120–160 °C) limited its range of applications. On the basis of mechanistic studies, the author demonstrated that this protocol proceeded via two-step pathway initiated with the iodination of aryl boronic acids with I2, followed by a subsequent cyanation of in situ generated aryl iodides with K4[Fe(CN)6]. Soon after, Cheng and colleagues explored the reactivity of cuprous thiocyanate (CuSCN) as a suitable and safe cyanating agent under the Pd-catalyzed coupling reaction with aryl boronic acids/esters 6 (Scheme 3b).29 Although only three examples were disclosed, this work represents the first example of cyanation of organoboron compounds with CuSCN. Notably, the method was found to be equally efficient for both boronic acids and pinacol boronic esters. Therefore, the compatibility of this method with boronic esters can be considered as one of its main advantages over the previous Pd-catalyzed protocols that were limited to boronic acids.
 |
| | Scheme 3 (a) Pd-catalyzed cyanation of (hetero)aryl boronic acids 4 with K4[Fe(CN)6]; (b) Pd-catalyzed cyanation of (hetero)aryl boronic acids 6 with CuSCN. | |
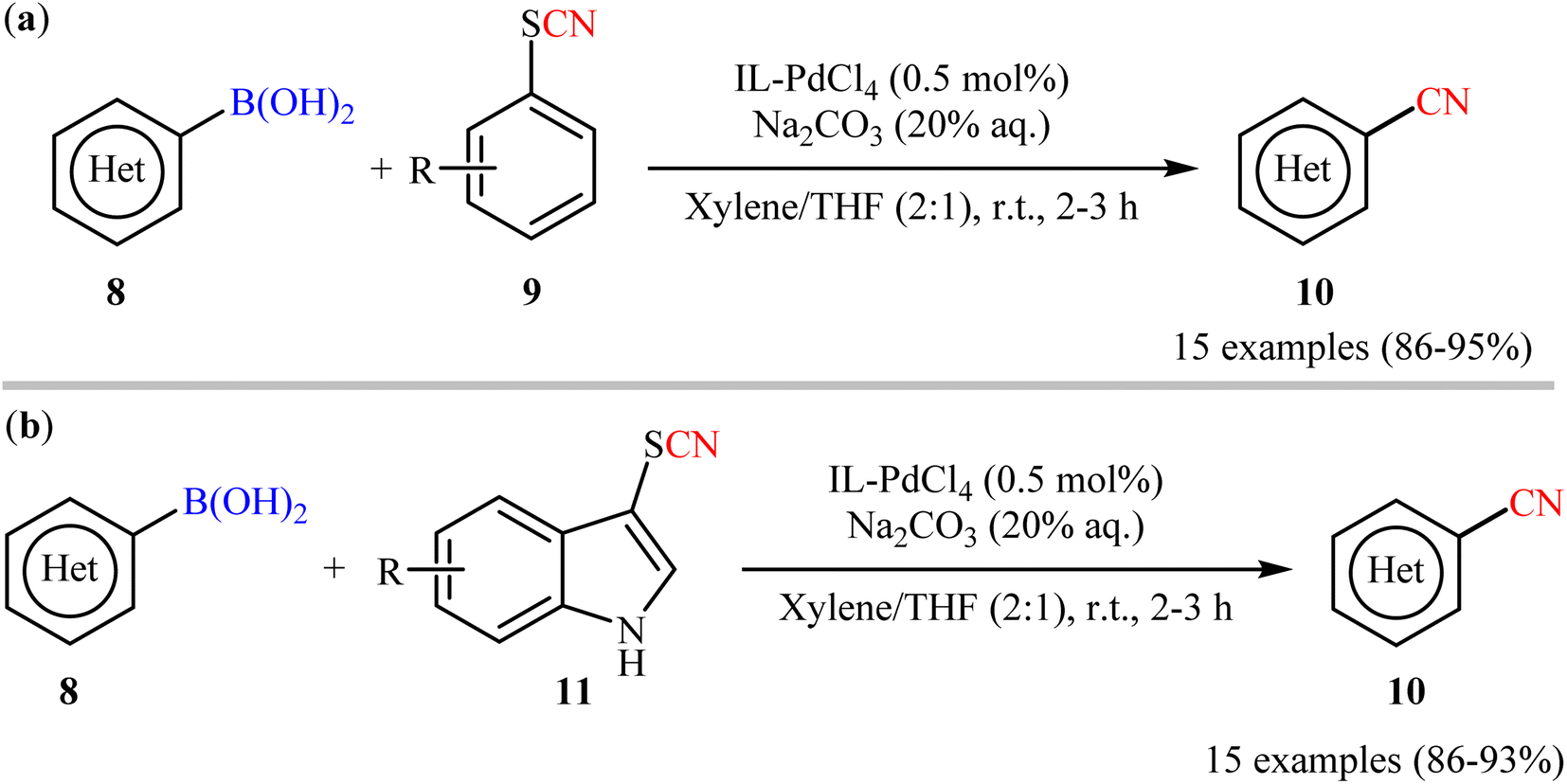
In this realm, Raval's research group disclosed the cyanation reactions of aryl boronic acids with aryl thiocyanates employing N,N,N-trimethyl-2-(sulfooxy)ethanaminium palladium(II) chloride (IL-PdCl4) catalyst.30 Herein, catalytic amounts of IL-PdCl4 in presence of Na2CO3 (20% aq.) enabled deborylative cyanation of various functionalized (hetero)aryl boronic acids 8 with aryl thiocyanate derivatives 9 under mild conditions to dispense the targeted arylnitriles 10 with high functional group compatibility (Scheme 4a). Notably, the same catalytic system was also found very effective for cyanation of the same set of boronic acids 8 with indole thiocyanates 11 (Scheme 4b). Additionally, the catalyst could be reused at least for four times, with only negligible loss of activity. Despite the mild conditions, good yields, and high functional group tolerance; low atom economy and large amounts of waste generation might limit the application profile of these methods.
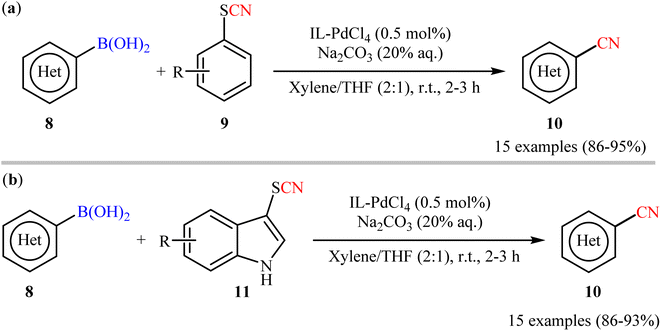 |
| | Scheme 4 Raval's synthesis of aryl nitriles 10. | |
At the same year, an elegant deborylative [11C]cyanation of organoboron compounds using NH411CN as the [11C]cyanide source was developed by Zhang et al.31 The optimal conditions consisted of NH411CN with an excess amount of NH3 and a catalytic amount of bench stable PdCl2(PPh3)2 in DMF at 100 °C. These conditions were successfully used to synthesize 32 (hetero)aryl-[11C]-nitriles 13 in moderate to excellent radiochemical yields (RCYs) from the corresponding aryl boronic acids/esters 12 within minutes (Scheme 5). Using this method, they also reported 11C-labeling of 2-phenylvinylboronic acid pinacol ester (an alkenylboronate) in an excellent RCY of 95 ± 5. It should be mentioned that using superstoichiometric amounts of ammonia as the base in this strategy, eliminates the need to remove the excess NH3 during the production of NH411CN reagent, saving time and resources in the synthesis.
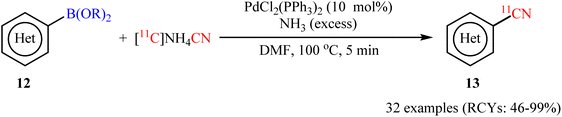 |
| | Scheme 5 Pd-catalyzed [11C]cyanation of aryl boronic acids/esters 12 with [11C]NH4CN. | |
Following these works, Togni and co-workers deeply investigated Pd-catalyzed cyanation reaction of boronic acids with the electrophilic ‘CN’ transfer reagents, N-CN reagents.32 After due screening of six N-CN reagents (N-cyanoheterocyclic reagents, N-cyanoimides, and N-cyanoimines), 90 different ligands, various catalysts (e.g., Pd(dba)2, PEPPSI™-IPr, Pd G3 μ-OMs dimer) and bases (e.g., KOAc, Cs2CO3, and K3PO4), N-cyanosuccinimide 15 was found to be the most promising cyanation agent and the merge of 2.5 mol% of Pd G3 μ-OMs dimer with 5 mol% of xantphos and 20 mol% of K3PO4 was identified as the best catalytic system. Pleasingly, a diverse range of aryl boronic acids 14 bearing various synthetically useful functionalities (e.g., OMe, CN, CHO, CO2Me, CF3, OCF3, NO2, F, Cl, SMe) were amenable under the current Pd catalyzed regime, providing the corresponding aryl nitriles 16 in modest to excellent yields (Scheme 6). However, carboxylic acid, bromo, and polyfluoro-substituted aryl boronic acids as well as N-heteroaromatic boronic acids were found to be unsuitable substrates for this protocol. The authors explained these observations by protonation of K3PO4 with carboxylic acid which is required to activate the precatalyst, homocoupling of bromo-substituted substrate to form 4-bromobiphenyl side product, incompatibility of polyfluoro-substituted substrates with the basic environment, and catalyst poisoning by N-heteroaromatic substrates.
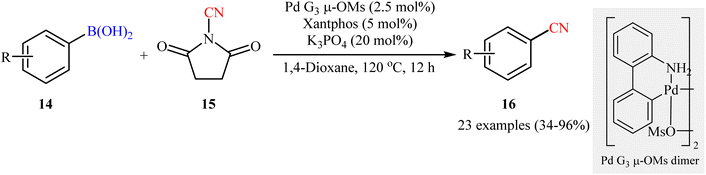 |
| | Scheme 6 Togni's synthesis of aryl nitriles 16. | |
2.2. Rhodium-catalyzed reactions
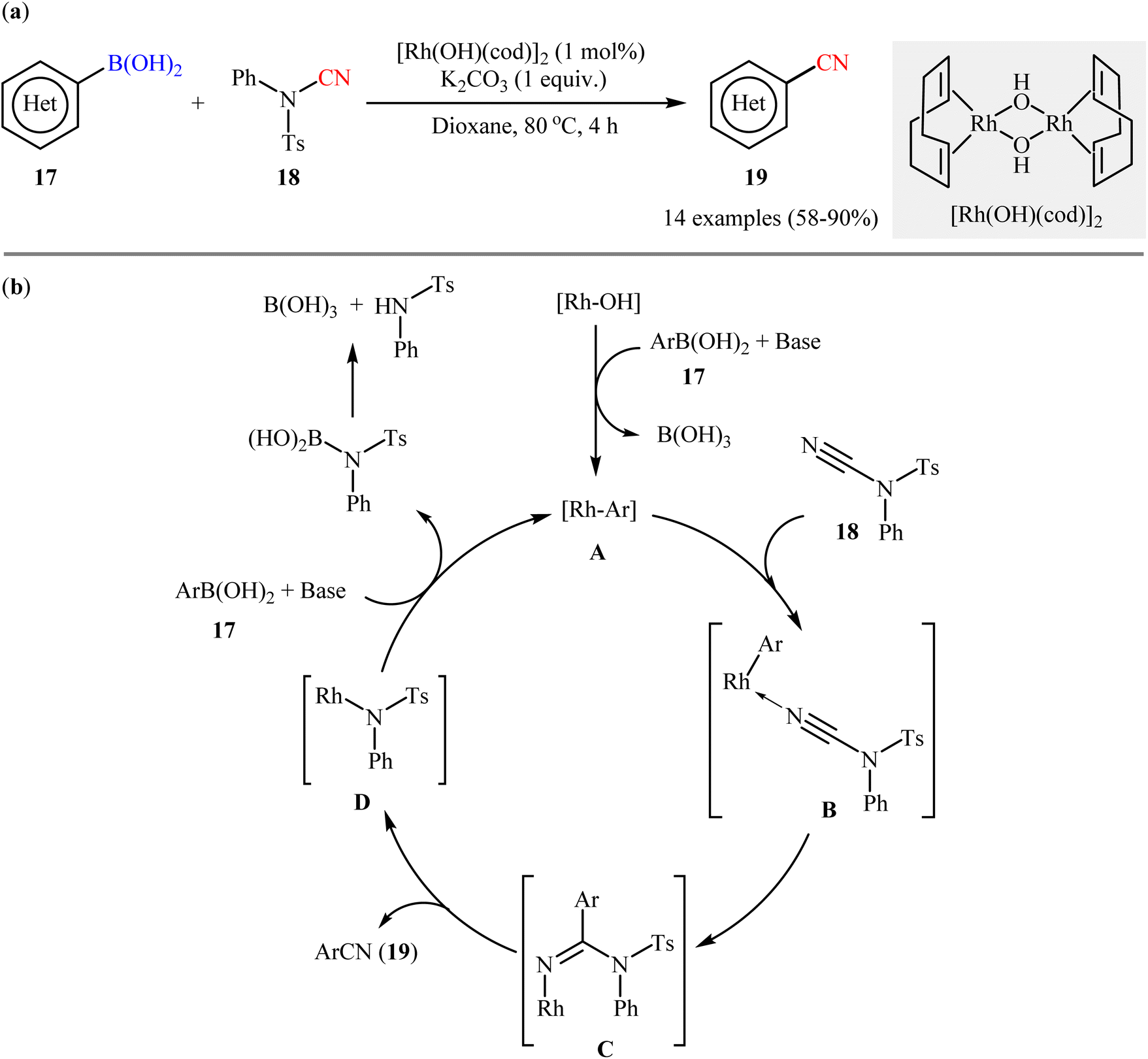
The first accomplishment for the synthesis of organic nitriles via Rh-catalyzed cyanation of respective boronic acids revealed from the laboratory of Prof. Beller in 2011.33 They showed that in the presence of a catalytic amounts of [Rh(OH)(cod)]2 and K2CO3 (1 equiv.) the C–N bond of N-cyano-4-methyl-N-phenylbenzenesulfonamide 18 could be efficiently cleaved and subsequently the CN moiety coupled with various (hetero)aryl boronic acids 17 to afford the corresponding (hetero)aryl nitriles 19 in moderate to high yields, ranging from 58% to 90% (Scheme 7a). It should be mentioned that although the use of boric acid (H3BO3) instead of K2CO3 provided the target products in better yields, K2CO3 was superior to H3BO3 with respect to substrate scope. Successively, the trend of compatibility of an alkenyl boronic acid was also evaluated in the system. The authors also investigated the applicability of their methodology for cyanation of boronic esters and found that the reaction using phenyl boronate neopentylglycol ester afforded the corresponding benzonitrile in moderate yield. However, phenyl boronic acid pinacolate did not undergo cyanation under the identical conditions. Based on literature data, the author proposed a catalytic mechanism for the synthesis of aryl nitriles 19 which encompassed the following steps (Scheme 7b): (i) transmetalation of the aryl boronic acid 17 with an active rhodium(I) species to generate the aryl–rhodium species A; (ii) coordination of the nitrogen atom of the CN group of N-CN reagent 18 to aryl–rhodium species A to give intermediate B; (iii) the insertion of Rh–Ar into the nitrile carbon atom to afford intermediate C; (iv) rearrangement of C to produce the observed aryl nitrile 19 along with the reactive Rh species D; and (v) transmetalation of D with boronic acid 17 to regenerate the aryl rhodium species A and complete the catalytic cycle.
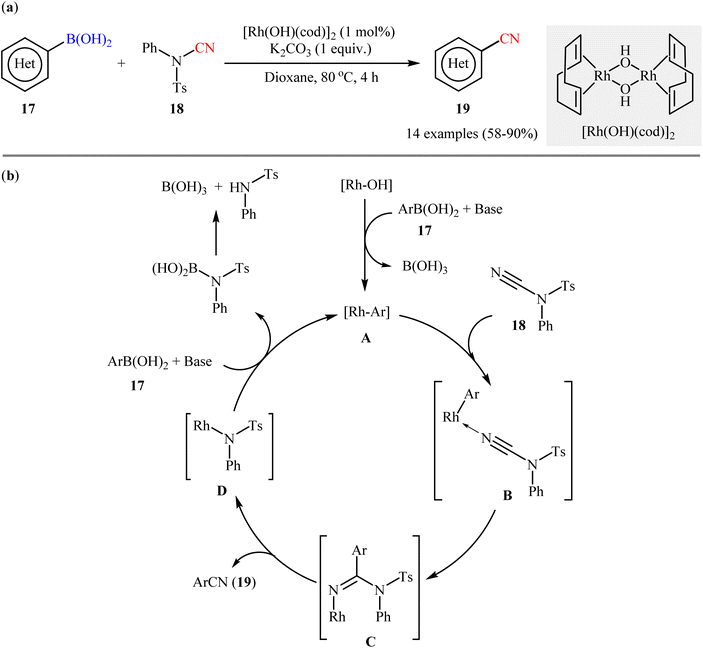 |
| | Scheme 7 (a) Rh-catalyzed cyanation of (hetero)aryl boronic acids 17 with N-cyano-4-methyl-N-phenylbenzenesulfonamide 18; (b) proposed mechanism for the formation of (hetero)aryl nitrile 19. | |
Drawing inspiration from this work, a facile Rh-catalyzed cyanation of organoboron compounds with dimethylmalononitrile (DMMN, 21) as a commercially available, non-toxic and bench-stable cyanating agent was developed by Reeves et al.34 An assembly of [Rh(Cl)(cod)]2 and Cs2CO3 appeared to be the most effective catalytic systems. By this protocol, various aryl boronic acids 20 bearing o/m/p substituents were successfully converted to the corresponding aryl nitriles 22 (Scheme 8). Notably, heteroaryl boronic acids were also well tolerated under the reaction conditions. Besides, both neopentylglycol aryl boronic acid esters and aryl potassium trifluoroborates could also be employed. However, aryl pinacolboronate esters failed to produce any product under the optimized conditions.
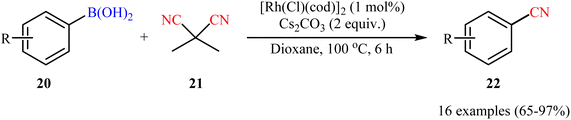 |
| | Scheme 8 Cyanation of aryl boronic acids 20 with DMMN catalyzed by [Rh(Cl)(cod)]2. | |
2.3. Copper-mediated/catalyzed reactions
In 2010, Hartwig and co-workers reported the first general protocol for copper-mediated cyanation of boronic acids/esters using Zn(CN)2 as the cyanide source.35 In this investigation, various copper salts (e.g., Cu(OAc)2, CuCN, CuBr, Cu(NO3)2, CuSO4) and bases (e.g., KOH, K3PO4, K2CO3, KF, CsF) were examined and the Cu(NO3)2/CsF system in MeOH/H2O (2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was found to be optimal for this C–C bond formation reaction. The current methodology tolerated both aromatic and heteroaromatic boronic acids 23 and afforded the corresponding (hetero)aryl nitriles 24 in good yields (Scheme 9). Moreover, alkenyl boronic acids also accomplished the respective products in satisfactory yields. Intriguingly, this procedure could achieve the one-pot meta-cyanation of 1,3-disubstituted arenes by iridium-catalyzed C–H borylation, followed by copper-mediated cyanation of the formed pinacol arylboronate esters. The author nice displayed the applicability of their methodology by high yielding synthesis of etravirine, an anti-HIV medicine. Unfortunately, they did not unravel the plausible mechanistic cycle for the transformation. Interestingly, nowadays, this strategy is widely used as a general, practical, and viable method to build up libraries of nitriles.36–42
:1) was found to be optimal for this C–C bond formation reaction. The current methodology tolerated both aromatic and heteroaromatic boronic acids 23 and afforded the corresponding (hetero)aryl nitriles 24 in good yields (Scheme 9). Moreover, alkenyl boronic acids also accomplished the respective products in satisfactory yields. Intriguingly, this procedure could achieve the one-pot meta-cyanation of 1,3-disubstituted arenes by iridium-catalyzed C–H borylation, followed by copper-mediated cyanation of the formed pinacol arylboronate esters. The author nice displayed the applicability of their methodology by high yielding synthesis of etravirine, an anti-HIV medicine. Unfortunately, they did not unravel the plausible mechanistic cycle for the transformation. Interestingly, nowadays, this strategy is widely used as a general, practical, and viable method to build up libraries of nitriles.36–42
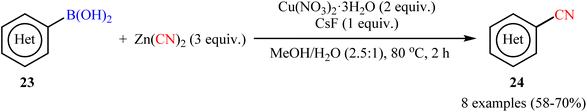 |
| | Scheme 9 Hartwig's synthesis of (hetero)aryl nitriles 24. | |
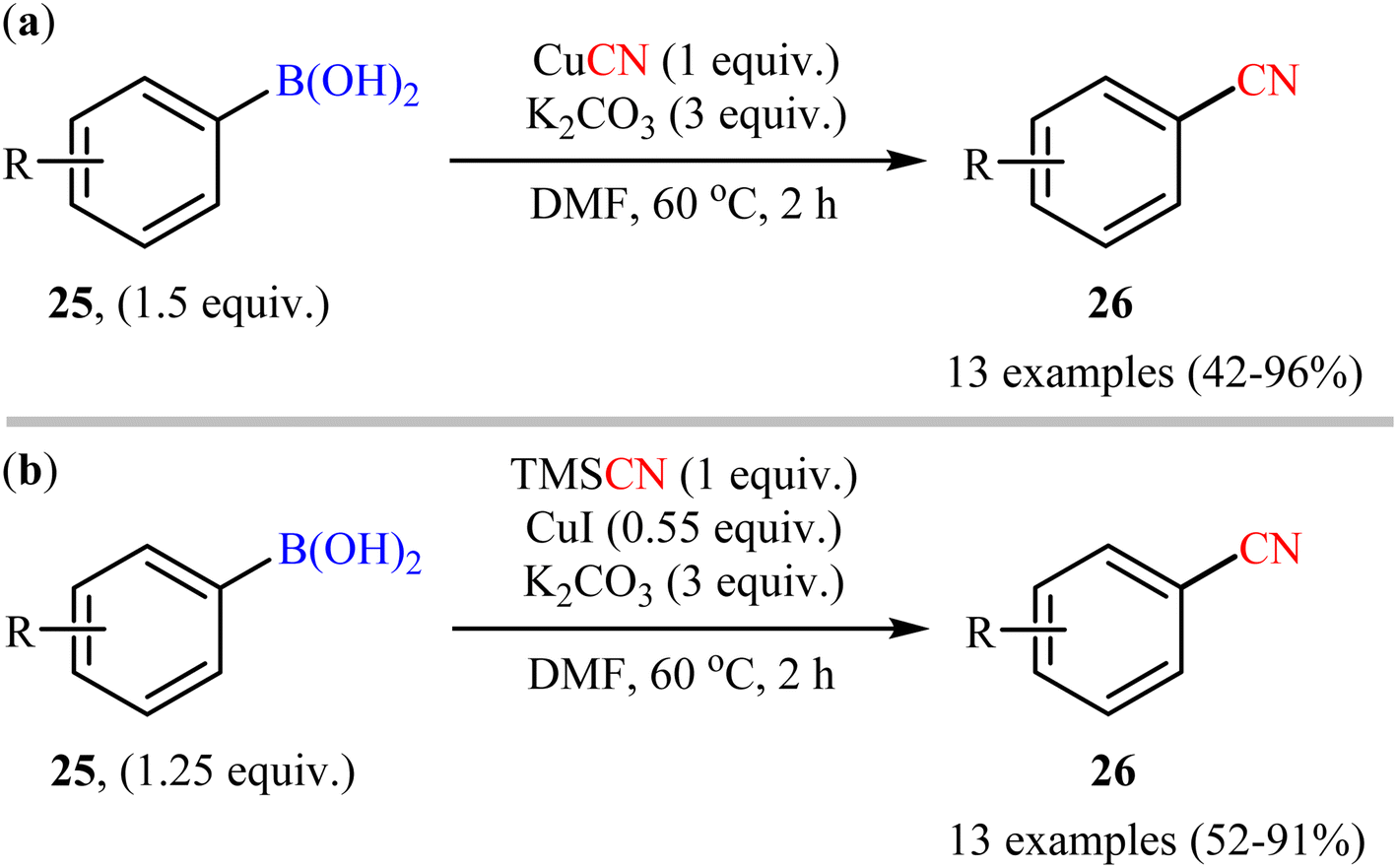
Shortly afterwards, Cheng's research group developed two alternative sets of suitable reaction conditions for the Cu-mediated cyanation of aryl boronic acids: CuCN in the presence of K2CO3, and trimethylsilyl cyanide (TMSCN) in the presence of CuI and K2CO3.43 Thus, a range of functionalized aryl boronic acids 25 were subjected to these complementary protocols, affording the corresponding products 26 in good to moderate yields, irrespective of the nature of substituents (Scheme 10). Notably, both protocols also permitted the usage of arylboroxines and aryl boronic acid pinacol esters under the standard reaction conditions. Unfortunately, no comment was made by the authors regarding the mechanistic aspects of the reactions. A detailed review of the scientific literature revealed that the CuCN-mediated procedure was further successfully applied to the preparation of various (hetero)aryl nitrile derivatives by other groups.44,45 Drawing inspiration from this work, in a closely related investigation, Han and co-workers also reported the synthesis of diverse aryl nitriles by the reaction between corresponding aryl boronic acids/esters with TMSCN using Cu2O as a catalyst and 1,2-dimethylethylenediamine (DMEDA) as a ligand.46 The reactions were carried out at room temperatures under O2 atmosphere and provided moderate to excellent yields of the desired products. The results indicated that the reaction was equally efficient for both aryl boronic acids and esters. However, heteroaryl boronic compounds turned out to be ineffective coupling partners under the standard reaction condition.
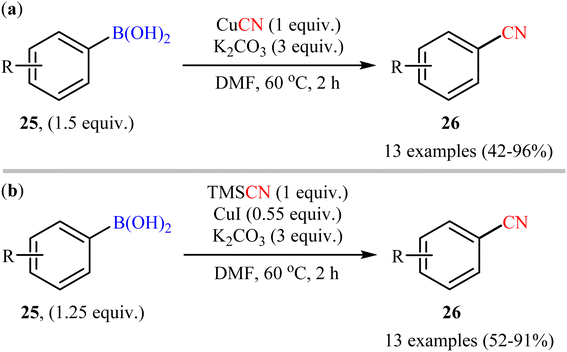 |
| | Scheme 10 Cu(I)-mediated cyanation of aryl boronic acids 25 developed by Cheng. | |
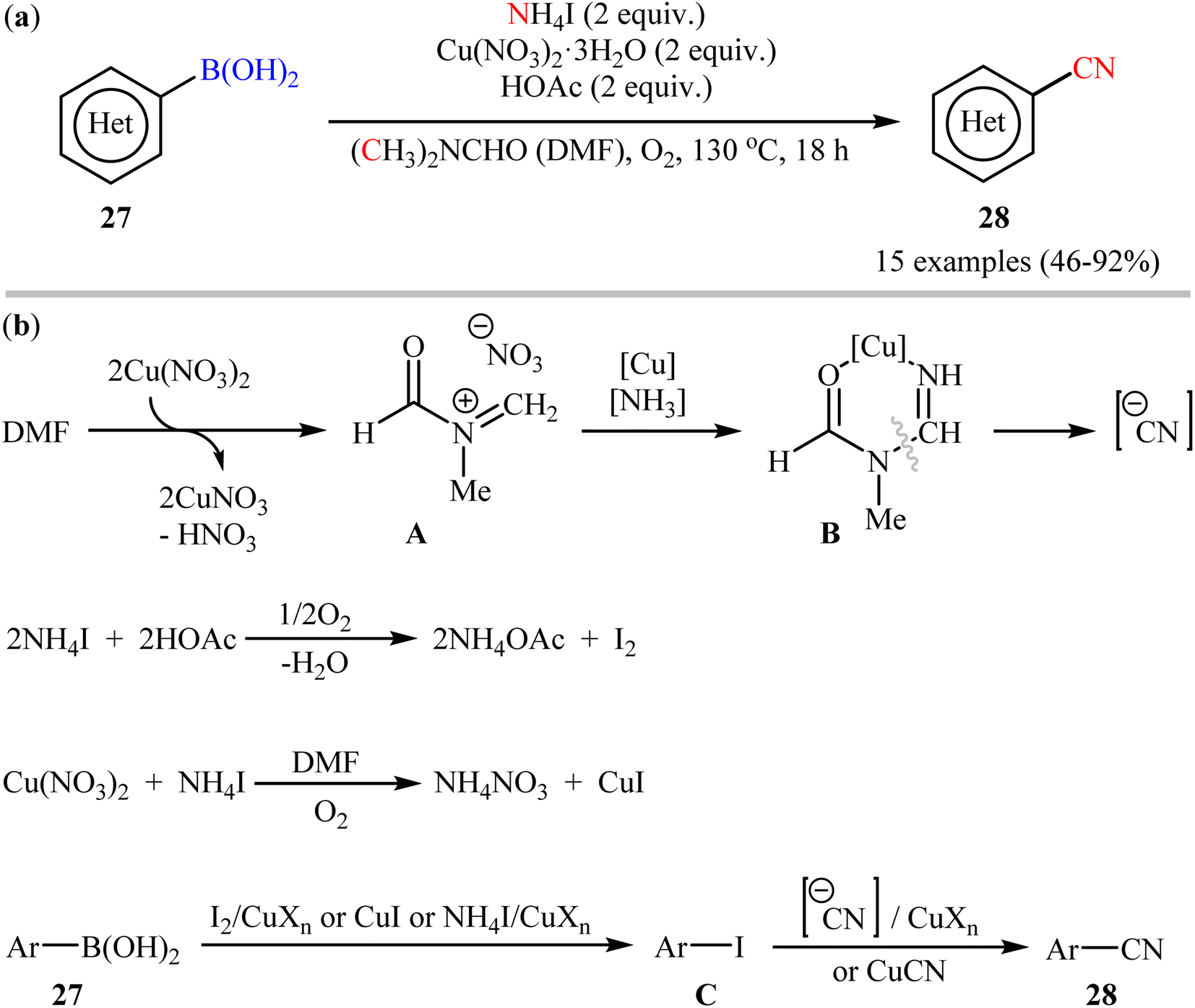
In 2012, Chang and co-workers have made an important contribution to the field by developing a highly efficient cyanation reaction of organoboron compounds using the merge of ammonium iodide (NH4I) with DMF as a safely combined cyano source.47 They disclosed that treatment of various (hetero)aryl boronic acids 27 with NH4I in the presence of Cu(NO3)2·3H2O in DMF under O2 atmosphere afforded the corresponding (hetero)aryl nitriles 28 with yield ranging from 46% to 97% (Scheme 11a). Furthermore, the procedure could also be applied to other substrates including borate salts and electron-rich arenes. In these reactions, NH4I was found to play a dual role: as a donor of the “N” unit of the cyano moiety and as a source of iodide for the formation of iodine. Mechanistically, it was believed the reaction might proceed through an iodination/cyanation sequential process as depicted in Scheme 11b.
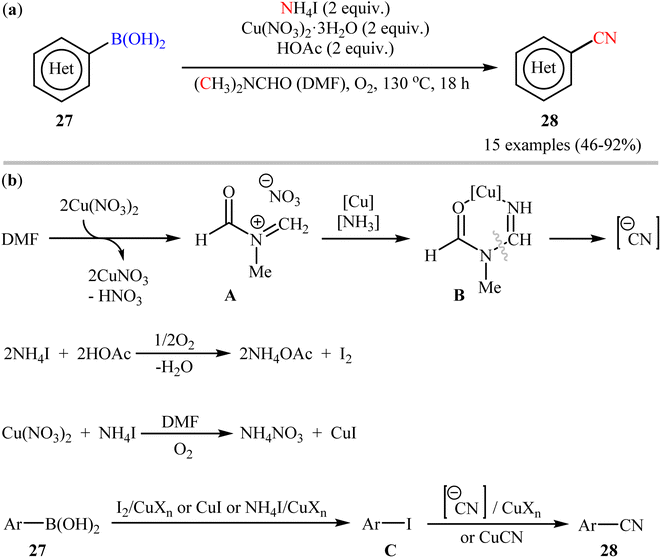 |
| | Scheme 11 (a) Cu-mediated cyanation of (hetero)aryl boronic acids 27 using NH4I and DMF; (b) proposed mechanistic pathway for the formation of (hetero)aryl nitriles 28. | |
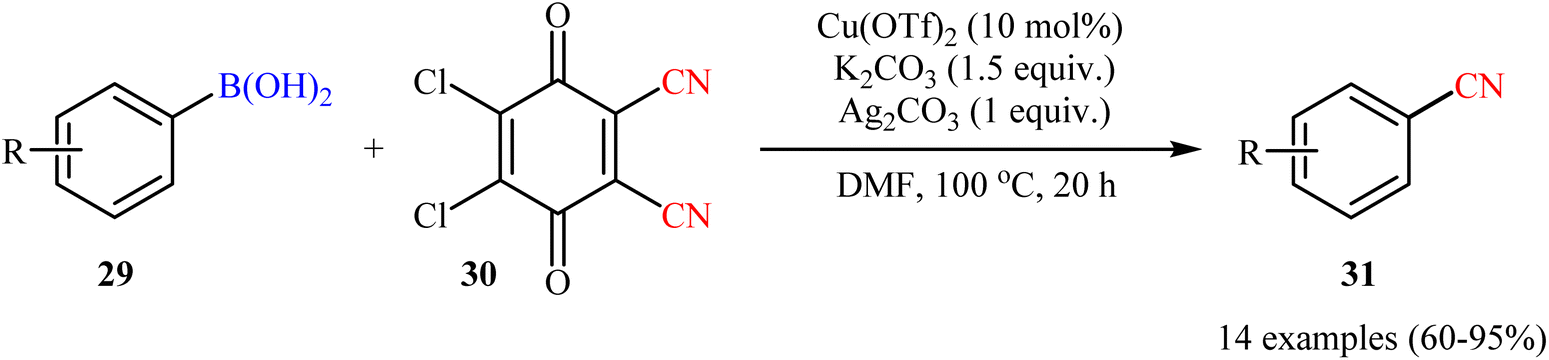
Concurrently, by utilizing 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ; 30) as cyanide source, Cheng and Chen team reported cyanation of a series of aryl boronic acids 29 employing a combination of Cu(OTf)2, K2CO3, and Ag2CO3 as the catalytic system in dry DMF (Scheme 12).48 Various synthetically useful functional groups such as chloro, cyano, nitro, ether, thioether, aldehyde, ketone, and ester functionalities were tolerated on the phenyl ring periphery of boronic acids to produce the nitrile products 31 in moderate to excellent yields. It should be mentioned that the presence of all components of the catalytic system (i.e., Cu(OTf)2, K2CO3, and Ag2CO3) were crucial to the success of this transformation. No target product was detected in the absence of any of them.
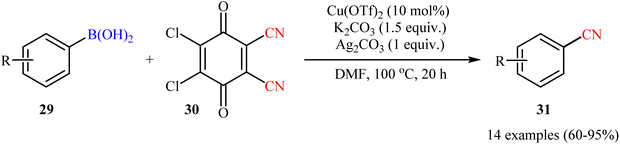 |
| | Scheme 12 Cu-catalyzed cyanation of aryl boronic acids 29 with DDQ. | |
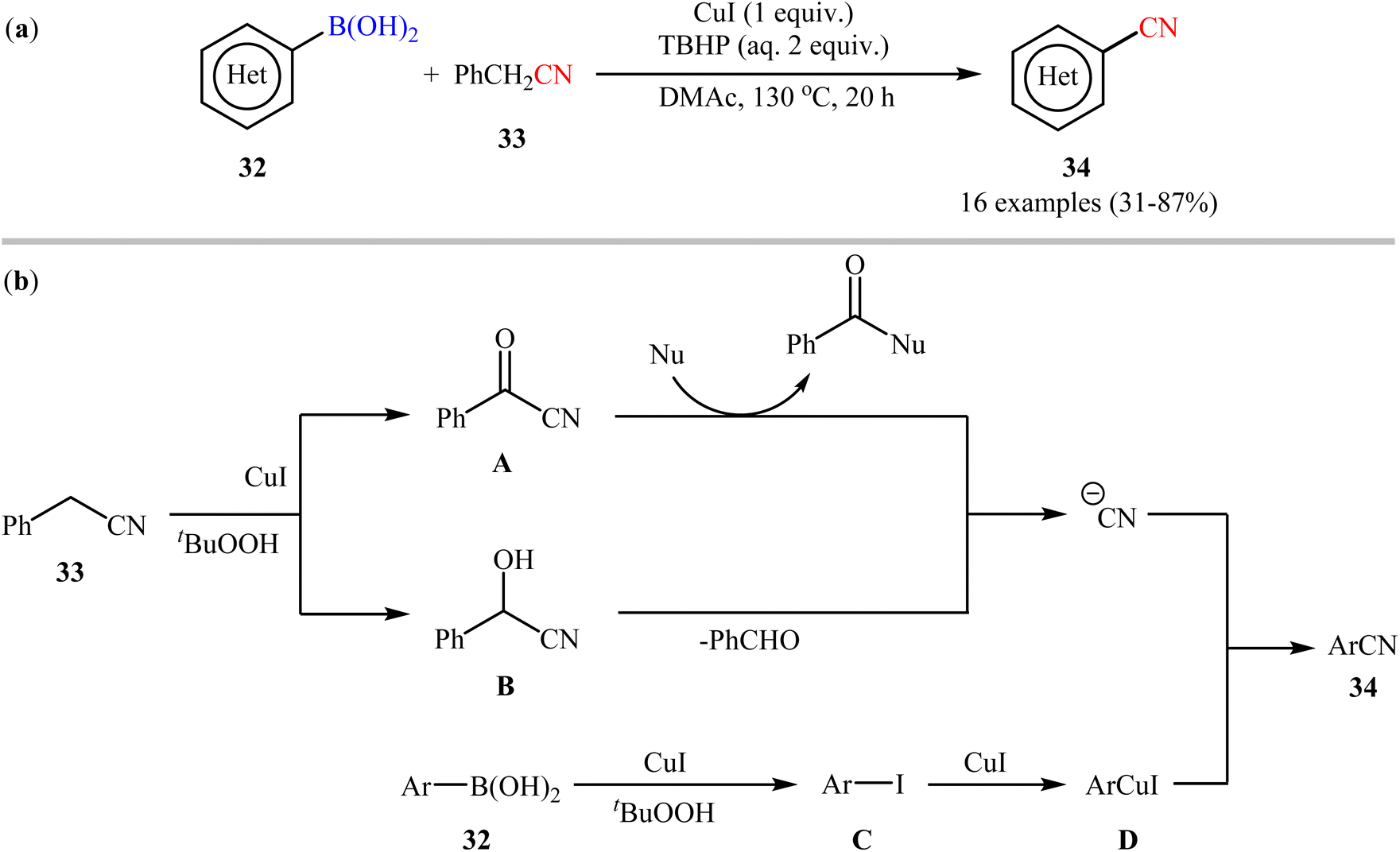
Soon after, Lu and Wang along with their co-workers exploited benzyl cyanide 33 as the CN source for the Cu-mediated cyanation of boronic acids/esters under oxidative conditions.49 Particularly, the reaction was facilitated by CuI (1 equiv.) in N,N-dimethylacetamide (DMAc) using 2 equiv. of tert-butyl hydroperoxide aqueous solution (TBHP) as an oxidant. In this report, 16 (hetero)aryl nitriles 34 were synthesized in fair to good yields from the respective (hetero)aryl boronic acids 32 (Scheme 13a). In addition to aryl boronic acids, the authors investigated the cyanation of aryl boronic esters under their optimal conditions, which generally demonstrated similar reactivity to the parent boronic acids. Despite these good results, it seems that the required elevated temperature (130 °C) may prevent the application of this process in organic synthesis. Moreover, the protocol is not viable for up-scale synthesis due to the drastic reduction in the yield (from 82% in the 0.5 mmol scale, to 56% in the 6.0 mmol scale). The authors also proposed the two possible putative pathways for this transformation which is shown in Scheme 13b. Later, the same catalytic system was found very effective for cyanation of a broad range of aryl boronic acids with ethyl (ethoxymethylene)cyanoacetate.50 In 2017, Wang and Zhang utilized a conceptually similar strategy to construct a library of (hetero)aryl nitriles in acceptable yields.51
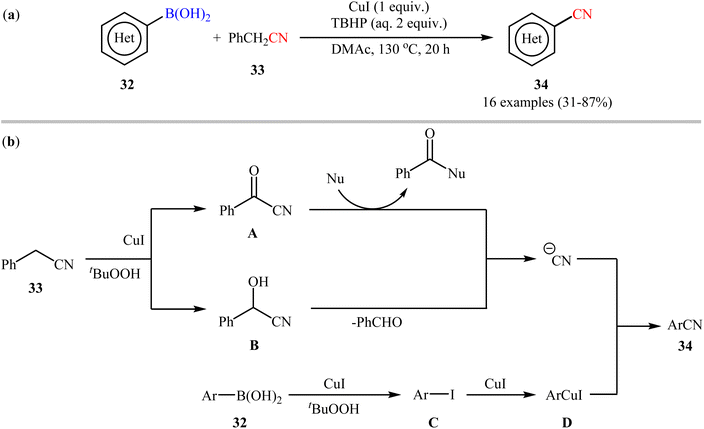 |
| | Scheme 13 (a) Cu-mediated cyanation of (hetero)aryl boronic acids 32 using benzyl cyanide 33; (b) mechanistic proposal for the formation of (hetero)aryl nitriles 34. | |
In 2015, Shen and co-workers reported an elegant way for the cyanation of organoboron compounds with Cu-catalyst by using acetonitrile as the solvent and a slow dosage CN source with Cu(OAc)2/phen/NIS/(Me3Si)2/TEMPO/iPr2NH system.52 In this process, TEMPO played a dual role: firstly, in the generation of the active cyanating agent (TEMPOCH2CN) and secondly, as an oxidant in this catalytic cyanation. As shown in Scheme 14, the system enabled the cyanation of a broad range of (hetero)aryl boronic acids 35 to provide (hetero)aromatic nitriles 36 in fair to quantitative yields. Fortunately, the protocol also functioned smoothly with boronic esters (naphthalene-2-boronic acid pinacol ester) and borate salts (potassium trifluoro(phenyl)borate). However, the method suffers from certain disadvantages, such as prolonged reaction times (2–3 days), harsh conditions (150 °C) and require several catalytic components which may leads to the production of large amounts of chemical waste. According to mechanistic studies, this cyanation reaction proceeds through two sequential steps: initial iodination followed by cyanation.
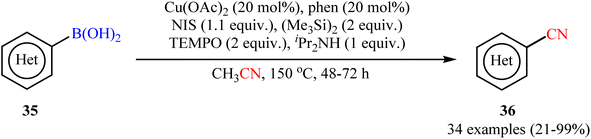 |
| | Scheme 14 Shen's synthesis of (hetero)aromatic nitriles 36. | |
Encouraged by the above results, Okamoto-Ohe's research group developed an efficient method for the cyanation of different (hetero)aryl boroxines 37 on the use of cyanogen iodide (ICN) in the presence of 15 mol% (CuOTf)2·toluene as catalyst, 33 mol% of dtbpy as ligand and 2.2 equiv. CsF as base.53 The reactions were performed in a Schlenk tube in a mixture of MeOH/H2O medium at 130 °C, tolerated a wide range of useful functionalities, and generally provided the desired (hetero)aryl nitriles 38 in moderate to high yields (Scheme 15). The reaction was also compatible with alkenyl boroxines, affording the corresponding alkenyl nitriles in good yields. Double cyanation of a diboronic acid also proceeded successfully by using double the amount of cyanating agent. The author illustrated a reasonable mechanism by taking cues from the control experiments which involved two catalytic cycles, including a Cu(II)-catalyzed iodination of boroxines, and the following CuCN-mediated cyanation of organic iodides.
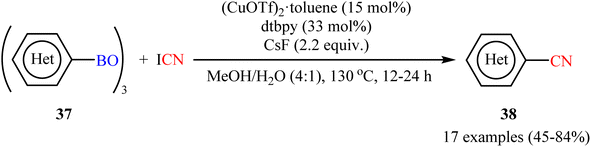 |
| | Scheme 15 Cu-catalyzed cyanation of (hetero)aryl boroxines 37 with ICN. | |
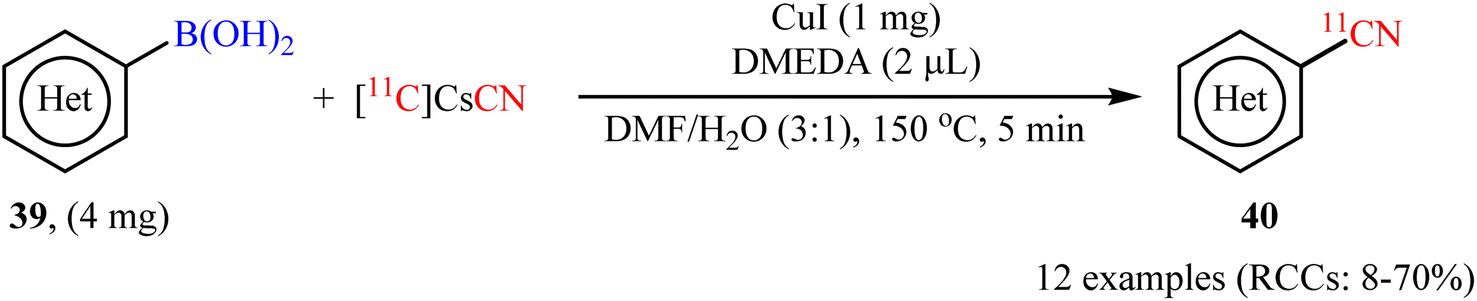
In 2017, Liang and co-workers described the first Cu-catalyzed [11C]cyanation of aryl boronic acids with [11C]CsCN to access 11C-labelled nitriles.54 To evaluate the activity of different catalytic systems, (4-bromo/methoxy-phenyl)boronic acids were chosen as the model reactants. The combination of inexpensive and commercially available CuI with DMEDA was found to be more effective, which gave better yields of 11CN-substituted products. The solvents such as DMF, DMA, DMSO, MeCN, 1,4-dioxane, and DMF/H2O were examined and a good yield of product was obtained when using the binary solvent. Under standard conditions, various aromatic and heteroaromatic boronic acids 39 reacted efficiently to give the desired 11C-labelled products 40 in radiochemical conversions (RCCs) up 70%, determined by radioTLC (Scheme 16). Subsequently, Scott's research team further extended this labeling precursors to other boron compounds (boronic esters, trifluoroborate salts) and arylstannanes by using Cu(OTf)2/pyridine system.55
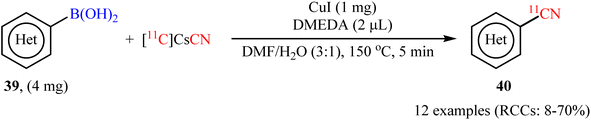 |
| | Scheme 16 Cu-catalyzed [11C]cyanation of aryl boronic acids 39 with [11C]CsCN developed by Liang. | |
2.4. Manganese-mediated reactions
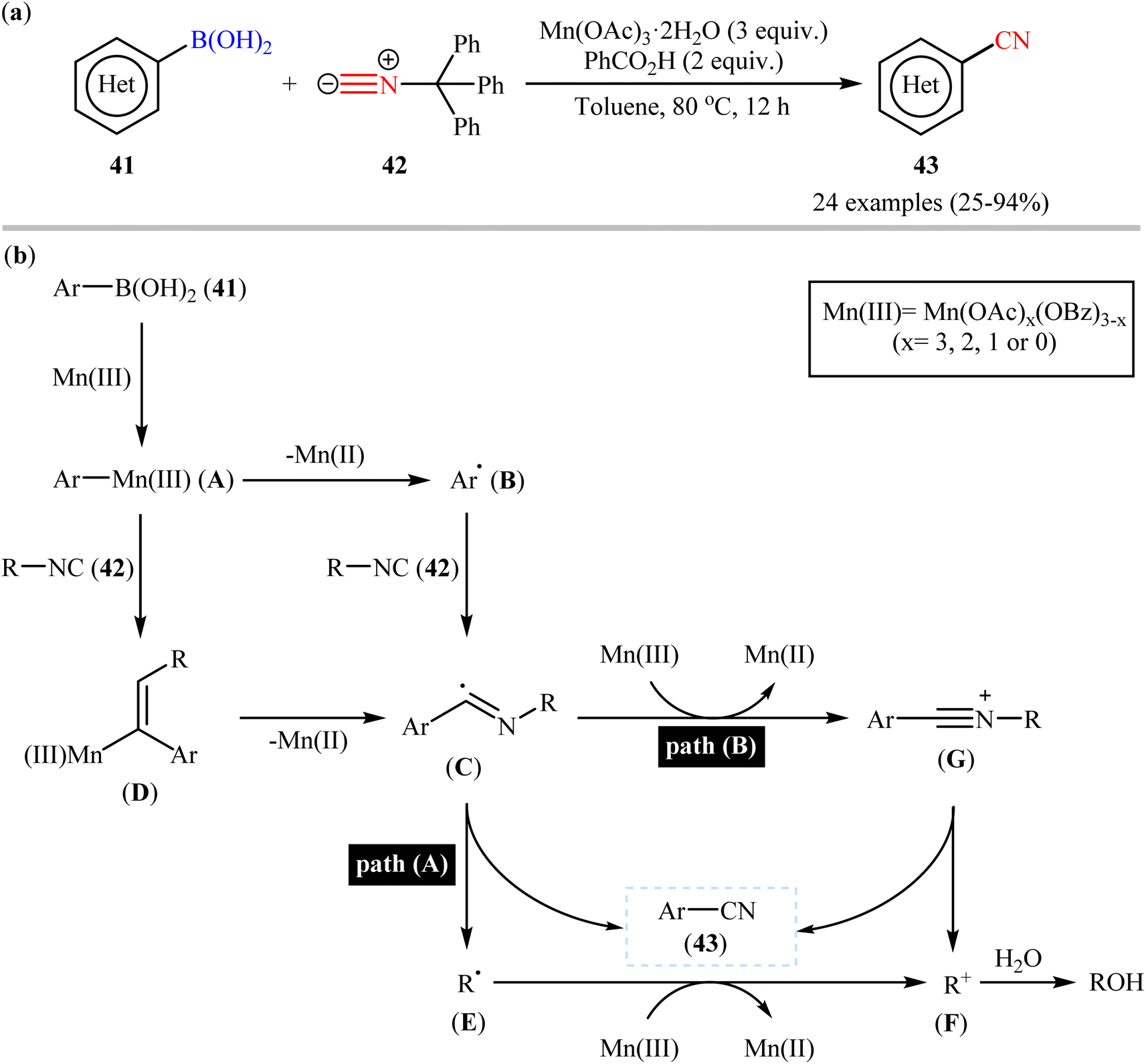
Very recently, a beautiful and efficient manganese-mediated cyanation of (hetero)aryl boronic acids 41 with commercially available trityl isocyanide 42 as cyanating agent using the system comprising of cheap and non-toxic Mn(OAc)3·2H2O as mediator and benzoic acid as an additive was developed by Li and coworkers.56 As shown in Scheme 17a, a wide range of ortho-, meta-, and para-substituted aryl boronic acids with diverse steric and electronic properties were amenable in this reaction, rendering the respective nitriles 43 in modest to excellent yields. However, alkenyl- and alkylboronic acids were incompatible in the reaction and applicability of boronic esters as starting materials was not investigated in this study. Notably, the authors claimed that compared with palladium-, rhodium-, or copper-catalyzed transnitrilation, their methodology showed better functional group compatibility. Based on the previous literature report, they proposed a mechanistic scenario which is depicted in Scheme 17b. At first, aryl boronic acid 41 undergoes transmetalation with Mn(III) to form Mn(III)-aryl species A, which after Mn homolysis leads to aryl radical B and Mn(II). Subsequently, addition of radical B to the isocyanide 42 affords an imidoyl radical C (this radical can be also formed through the intermediate D) that, then loses a R˙ radical E to give the expected aryl nitrile 43 (Scheme 17b, path A). In another possibility, oxidation of the imidoyl radical D leads to the corresponding cation G that, after elimination of the R+ cation F produces the observed aryl nitrile 43 (Scheme 17b, path B).
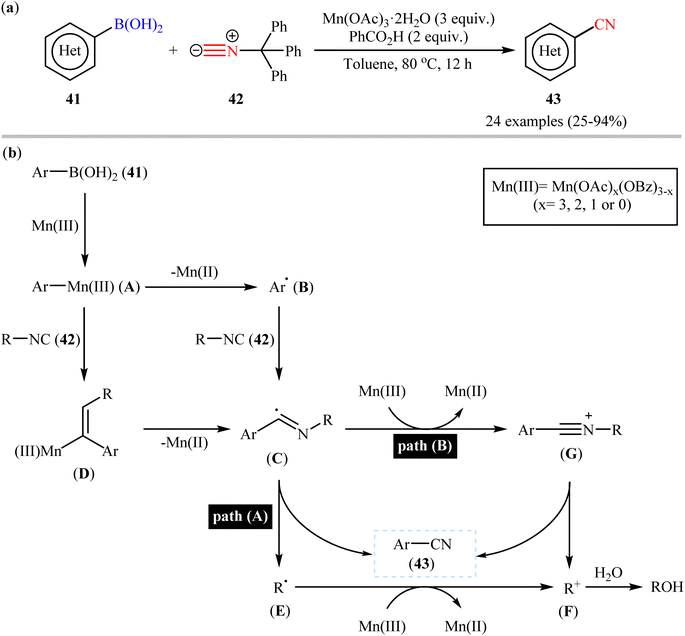 |
| | Scheme 17 (a) Mn-mediated cyanation of (hetero)aryl boronic acids 41 with trityl isocyanide 42; (b) proposed mechanism for the formation of (hetero) aryl nitrile 43. | |
3. Thiocyanation of aryl boronic acids
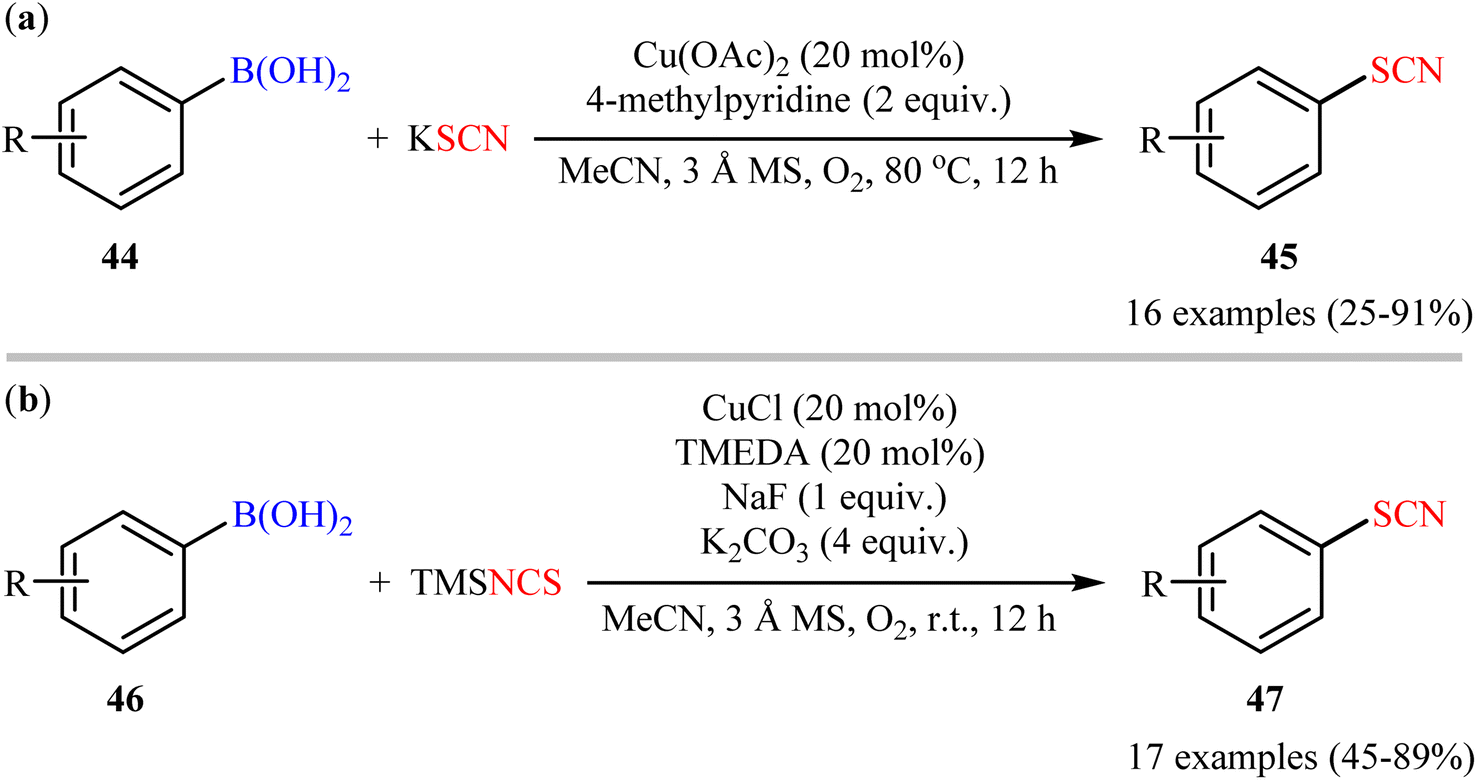
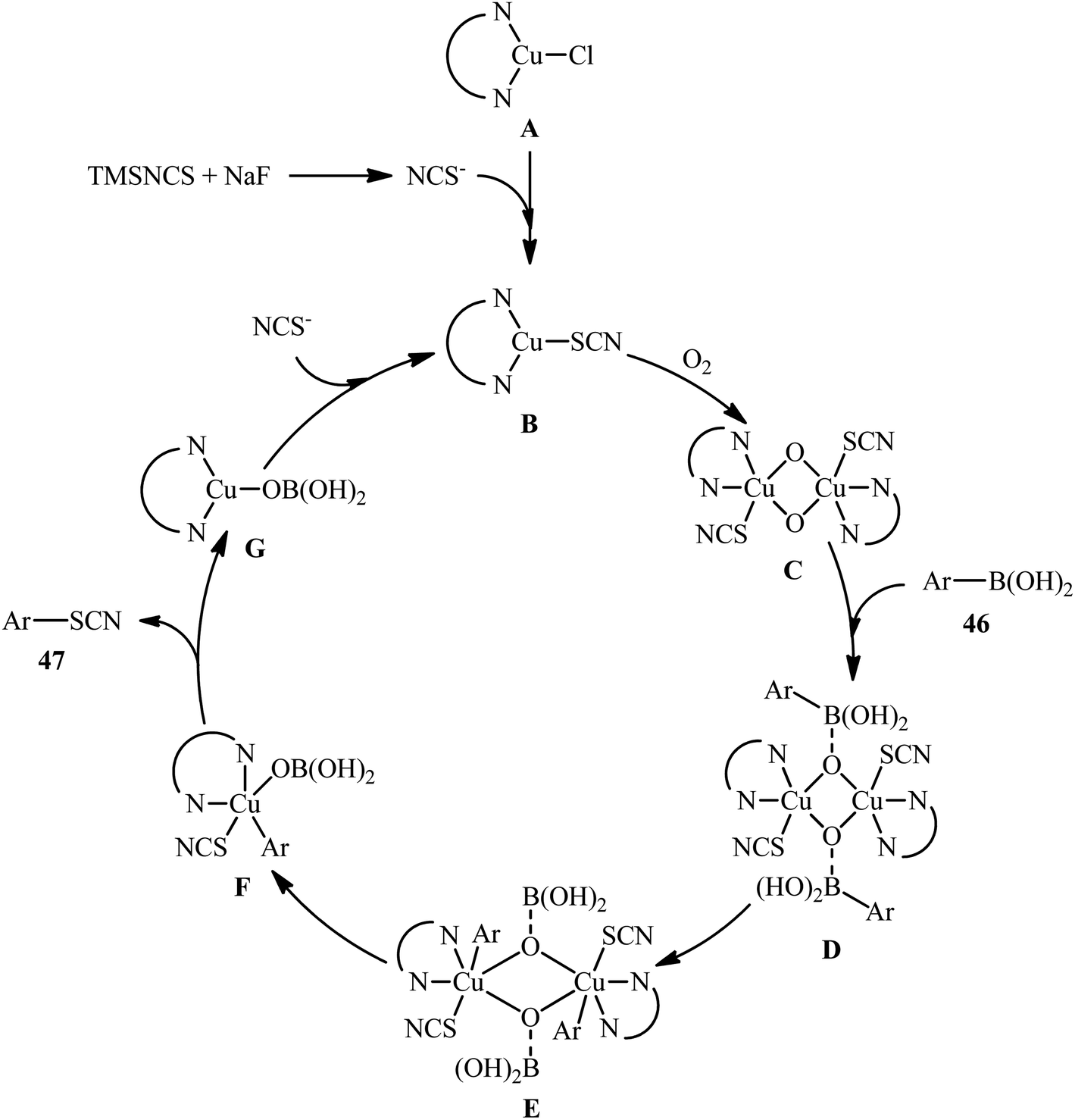
The possibility of synthesizing aryl thiocyanates through deborylative thiocyanation of aryl boronic acids was first demonstrated by Hu and co-workers in 2013, who synthesized a library of 16 aryl thiocyanate derivatives 45 through the reaction of corresponding aryl boronic acids 44 with inexpensive and stable KSCN in the presence of 20 mol% of Cu(OAc)2 as a catalyst and 2.0 equivalents 4-methylpyridine as both a ligand and a base using molecular oxygen as a terminal oxidant.57 This Cu-catalyzed C–S coupling reaction tolerated both electron-rich and electron-poor aryl boronic acids 44 and provided the respective aryl thiocyanates 45 in up to 91% yields (Scheme 18a). However, the protocol was unsuccessful for NO2-substituted substrates. Furthermore, neither alkylboronic acids nor vinylboronic acids were reacted under the conditions employed. Subsequently, the authors improved their methodology in terms of yield and substrate scope using trimethylsilylisothiocyanate (TMSNCS) as SCN source and a combination of CuCl, TMEDA, NaF, and K2CO3 as the catalytic system.58 Irrespective of nature and position of the substituents, both electron-donating (Me, OMe, vinyl) and electron-withdrawing (F, Cl, Br, CF3, CHO, COMe, CO2Me, CN, NO2) functionalities were tolerable under the current protocol, delivering the corresponding aryl thiocyanates 47 in moderate to high outcomes (Scheme 18b). However, as the same results in their previous report, heteroaryl, alkyl, and alkenyl boronic acids were proven not to be adaptable substrates for this reaction. In addition, when boronic esters and borate salts were employed under the optimal reaction conditions, only trace amounts of corresponding aryl thiocyanates were obtained. In Scheme 19, a reasonable mechanistic pathway for this thiocyanation reaction is suggested. Initially, LCu(I)Cl A undergoes exchange with the in situ generated NCS− anion to give reactive LCu(I)SCN B, which after oxidation by oxygen leads to the bis(μ-oxo)dicopper(III) complex C. Next, this complex C reacts with two molecules of aryl boronic acid 46 to form intermediate D which after aryl group transfer to the Cu atom affords E. Subsequent decomposition of complex E generates two monomeric copper(III) complex F. Later, reductive elimination of this complex F affords the desired product 47 and LCu(I)–OB(OH)2 G. Finally, the formed G undergoes further exchange with the NCS− anion to complete the catalytic cycle by reproducing LCu(I)SCN B.
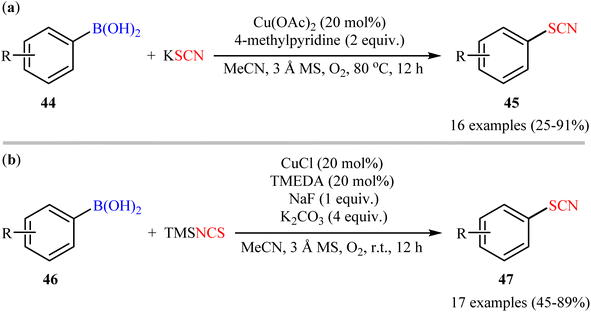 |
| | Scheme 18 (a) Cu-catalyzed oxidative coupling between aryl boronic acids 44 and KSCN; (b) Cu-catalyzed oxidative thiocyanation of aryl boronic acids 46 with TMSNCS. | |
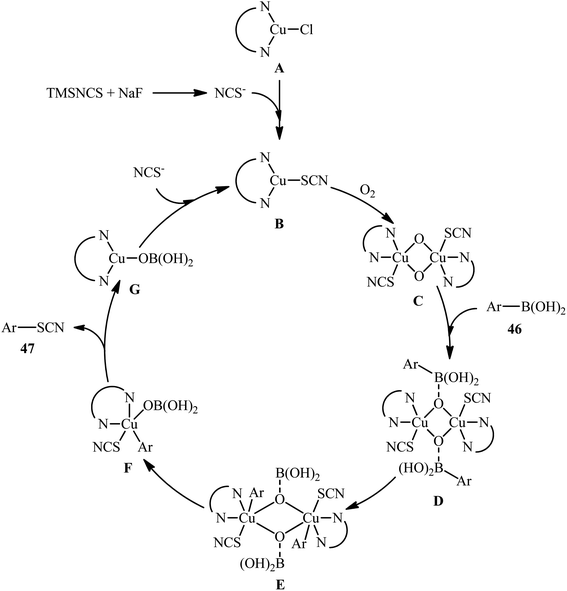 |
| | Scheme 19 Plausible reaction pathway for the formation of aryl thiocyanates 47. | |
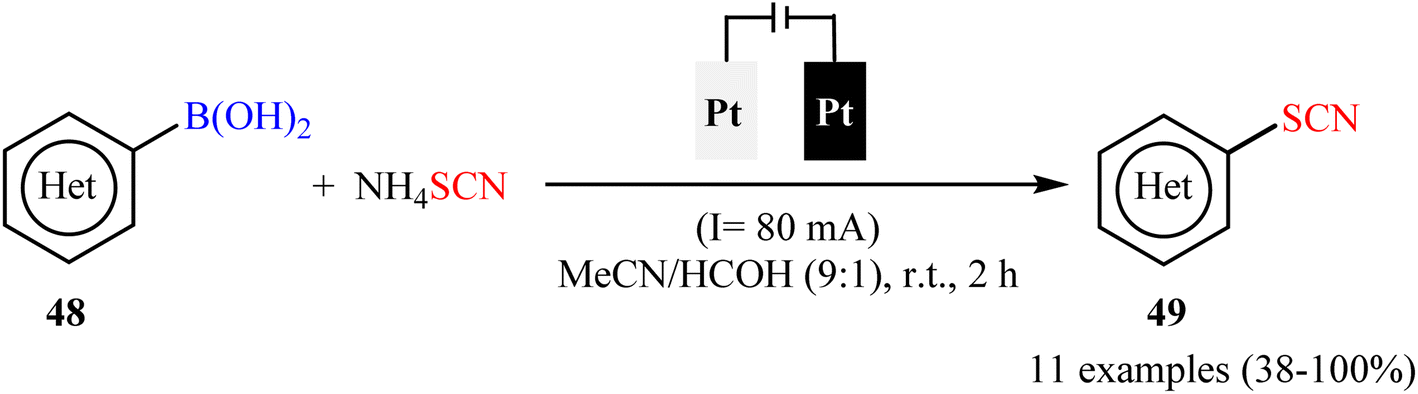
Subsequently, electrochemical version of this thiocyanation reaction was developed by Gooßen and co-workers.59 They showed that the treatment of various (hetero)aryl boronic acids 48 with ammonium thiocyanate (NH4SCN) in an undivided cell assembled with simple Pt sheet electrodes under constant current conditions provided the corresponding (hetero)aryl thiocyanates 49 within 2 h (Scheme 20). Beside short reaction time, mild reaction conditions, simplicity, safety, and no need for metal-catalysts, bases, and additives can be considered as the advantages of this synthetic strategy. In this study, the authors found some limitations in their protocol when they attempted to react electron-neutral phenyl-, 1-naphthyl-, and 2-naphthyl-boronic acids, electron-rich tolyl- and biphenyl-boronic acids, and strongly electro-poor substrates such as 4-cyanophenyl-boronic acid. In these cases, very poor yields or even no desired product was observed at all. Gratifyingly, the authors partly solved this limitation by replacing boronic acids with corresponding trifluoroborates.
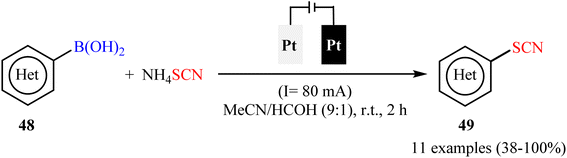 |
| | Scheme 20 Electrochemical ipso-thiocyanation of (hetero)aryl boronic acids 48 with NH4SCN. | |
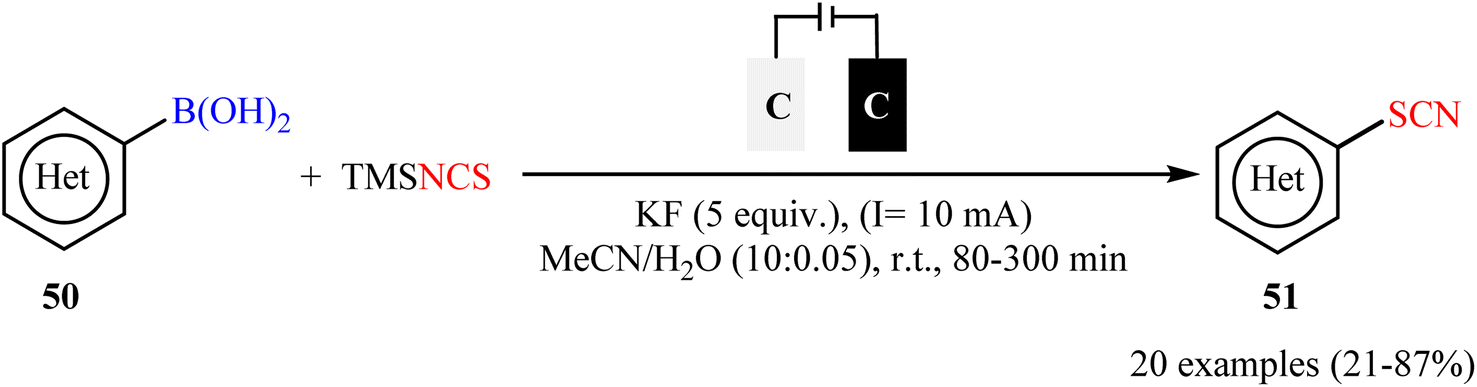
Along this line, Fu, Cai, and their colleagues recently achieved the electrochemical thiocyanation of (hetero)aryl boronic acids 50 with TMSNCS under catalyst- and oxidant-free conditions.60 The reaction was conducted in an undivided cell with graphite electrodes under constant-current electrolysis at 10 mA in anhydrous MeCN/H2O mixed solvents at room temperature and afforded the target (hetero)aryl thiocyanates 51 in relatively poor to high yields (Scheme 21). Although this method showed better functional group tolerance than Gooβen's strategy, but, unfortunately, electron-neutral and electron-poor substrates were sluggish to participate in this protocol. Following these works, Han-Xu's research group successfully synthesized two aryl thiocyanate derivatives in high yields by simple heating of the corresponding aryl trifluoroborates in the presence of NaSCN (4 equiv.) in DMSO/HOAc (1:1) at 80 °C.61
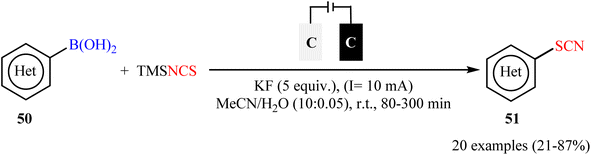 |
| | Scheme 21 Metal-free electrochemical thiocyanation of (hetero)aryl boronic acids 50 with TMSNCS. | |
4. Selenocyanation of aryl boronic acids
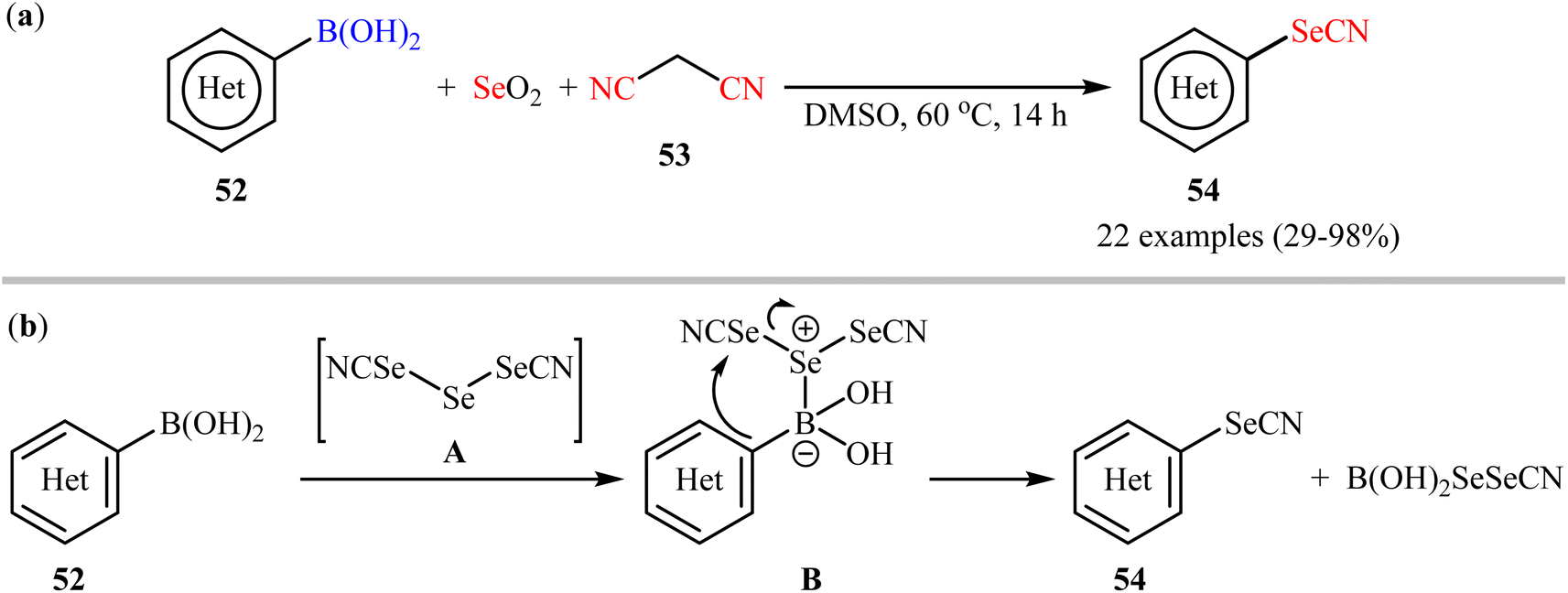
Synthesis of aryl selenocyanates through the deborylative selenocyanation of corresponding boronic acids has been scarcely studied; in fact, only three examples were reported on such reactions till date. In 2019, Redon, Vanelle, and colleagues revealed that the treatment of (hetero)aryl boronic acids 52 with selenium dioxide and malononitrile 53 under catalyst- and additive-free conditions afforded the respective (hetero)aryl selenocyanates 54 in up to 98% yields (Scheme 22a).62 The results indicated that substrates bearing an electron-donating group (e.g., Me, OMe) afforded better yields than those with an electron-withdrawing group (e.g., Cl). Notably, substrates bearing a strongly deactivating (CF3, NO2) group in para-position, failed to participate in this reaction. A possible reaction mechanism for this selenocyanation involves the formation of triselenodicyanide intermediate A through the reaction of selenium dioxide with malononitrile in DMSO. Then, aryl boronic acid 52 reacts with this intermediate A to form complex B, which after intramolecular nucleophilic addition of aryl ring of the aryl boronic acid on to the electrophilic SeCN group affords the desired arylselenocyanate compound 54 (Scheme 22b).
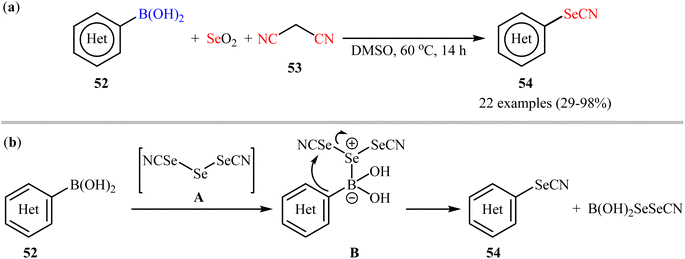 |
| | Scheme 22 (a) Redon-Vanelle's synthesis of (hetero)aryl selenocyanates 54; (b) plausible mechanism for the formation of (hetero)aryl selenocyanates 54. | |
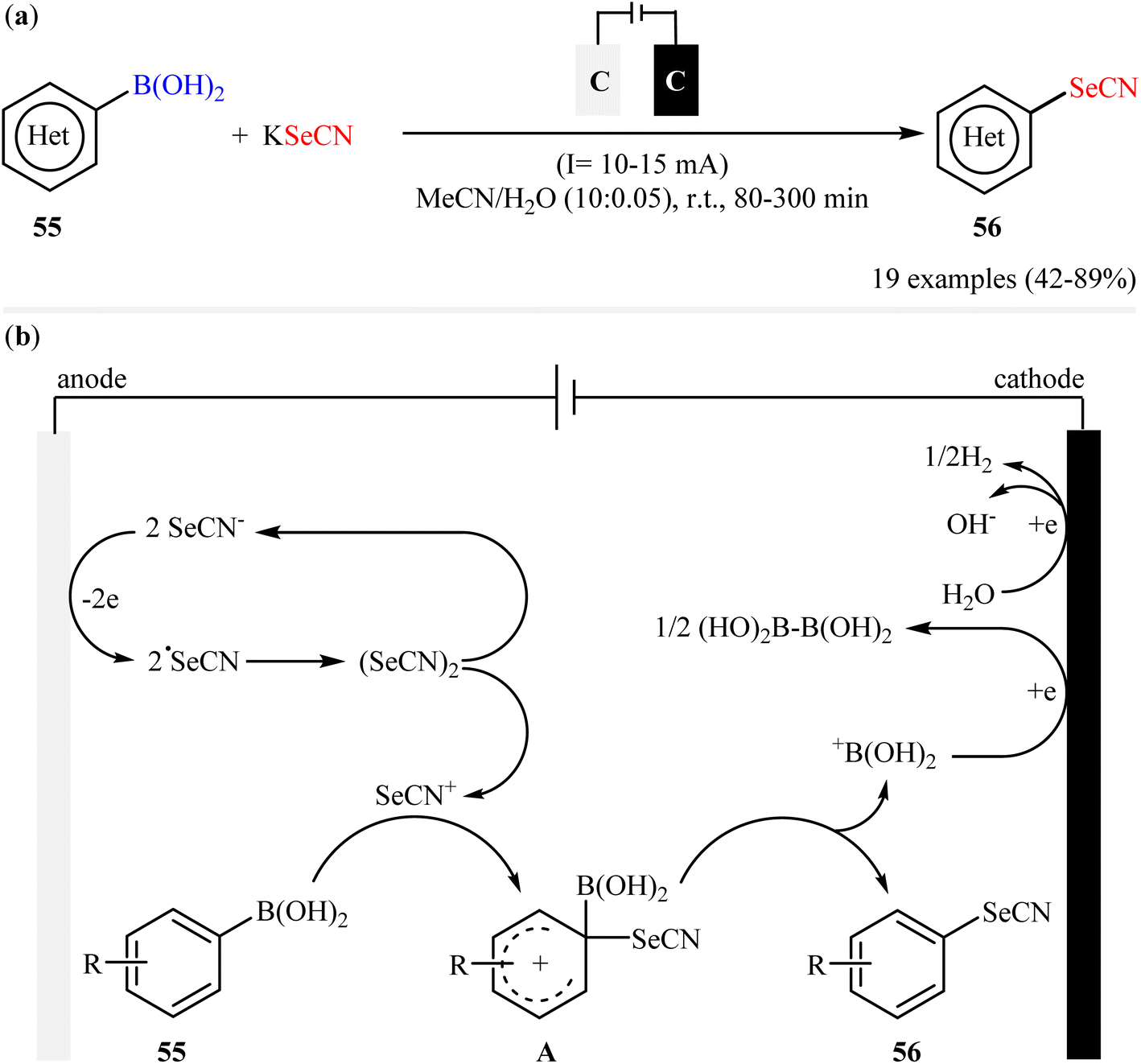
In 2020, in the same paper describing the electrochemical deborylative thiocyanation of aryl boronic acids to aryl thiocyanates using TMSNCS as a thiocyanation reagent in the presence of KF, Cai and co-workers reported the successful electrocatalytic selenocyanation of aryl boronic acids to produce corresponding aryl selenocyanates under metal-free conditions.60 Thus, in a simple undivided cell equipped with two graphite felts under constant current conditions (10–15 mA) in aqueous solution of MeCN, the reaction of various electron-rich aryl boronic acids 55 with KSeCN at room temperature furnished the desired aryl selenocyanates 56 in moderate to high yields (Scheme 23a). Unfortunately, aryl boronic acids bearing electron-withdrawing groups (e.g., Cl, Br, CN, CO2Me) was proven not to be adaptable substrates for this electrosynthesis. In the plausible mechanistic pathway, the authors speculated that this reaction proceeds via generation of selenocyanate radical (˙SeCN) intermediate through a single-electron-transfer process at the anodic. Next, radical coupling of this intermediate forms selenocyanogen, (SeCN)2, which after heterolytic bond-cleavage affords the electrophile SeCN+. Subsequently, electrophilic attack of this cation to aryl boronic acid 55 produces active adduct B. Finally, the elimination of +B(OH)2 from intermediate B leads to the formation of the expected aryl selenocyanate 56 (Scheme 23b).
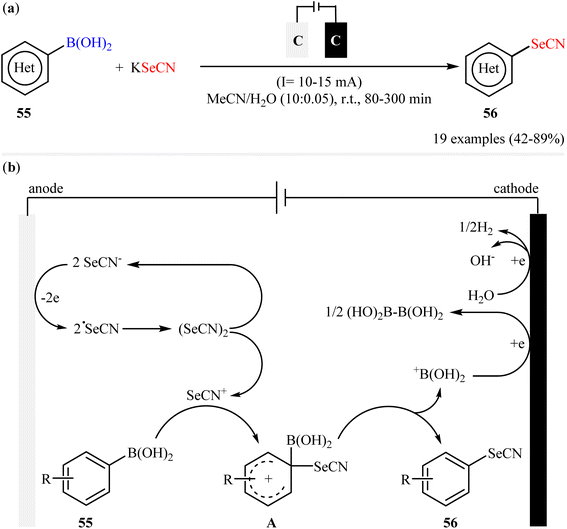 |
| | Scheme 23 (a) Electrochemical deborylative selenocyanation of (hetero)aryl boronic acids 55 with KSeCN; (b) proposed mechanism for the formation of aryl selenocyanates 56. | |
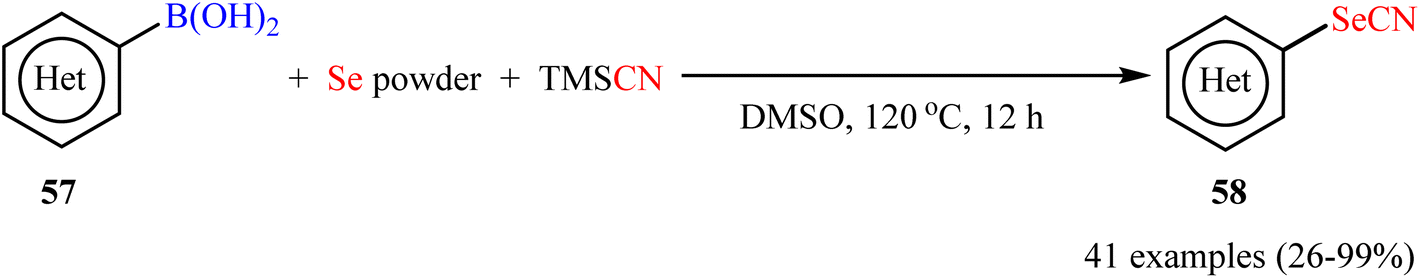
Recently, Zhou-Liu's research team developed a general and straightforward strategy for the synthesis of (hetero)aryl selenocyanates 58 via catalyst- and additive-free selenocyanation of a diverse range of (hetero)aryl boronic acids 57 with TMSCN and elemental selenium (Scheme 24).63 The reactions showed very good tolerance to a number of important and sensitive functional groups such as the trifluoromethyl, trimethylsilyl, trifluoromethoxy, aldehyde, ester, ether, thioether, and halogen groups. Moreover, the authors also successfully extended this protocol to the high yielding synthesis of five- to seven-membered selenaheterocycles.
 |
| | Scheme 24 Catalyst-free three-component reaction of aryl boronic acids 57, Se powder, and TMSCN. | |
5. Conclusion
Aryl nitriles are highly common structural motifs found in a variety of pharmaceuticals, agrochemicals, dyes, and natural products. They also serve as the versatile precursors for synthesis of the diverse functional groups. In a similar way, aryl thiocyanates and selenocyanates are also important structural motifs in pharmaceuticals and biologically active compounds. Additionally, they are used as versatile intermediates in the synthesis of various valuable sulfur- and selenium-containing derivatives, respectively. Conventional methods for aryl (thio-/seleno-)cyanates formation involve (thio-/seleno-)cyanation of active aryl species, mainly aryl halides or aryldiazonium salts. However, these methods suffer from certain disadvantages, such as liberation of hazardous halogen by-products and explode unpredictably of aryl diazonium salts. In order to overpass this limitations, (thio-/seleno-)cyanation of low toxic, high stable, and easily accessible, aryl boronic acids have emerged as viable and general protocol for the preparation of the titled compounds. As shown in this review, a variety of cyanating agents have been utilized for the cyanation of (hetero)aryl boronic acid derivatives. However, almost all of these reactions are limited to the use of palladium and rhodium catalysts or (super)stoichiometric copper salts. Thus the exploration of first row transition metal catalysts on this chemistry will be highly desirable from the standpoint of economy and green chemistry. On the other hand, the number of reported examples on thio- and selenocyanation reactions are narrow and there is further need to study the scope and limitations of these reactions and explore applicability of other thio- and seleno-cyanating agents in these reactions. The literature scan also showed that no reporting guideline exists for the electrochemical cyanation of aryl boronic acids and only a handful of electrochemical methodologies have been reported for thio- and selenocyantion. Considering that the large amounts of wasteful stochiometric oxidants used by many of the methods summarized in this review, it seems that development of external oxidant-free electrochemical strategies can bypass this limitations.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) A. V. Dubrovskiy, T. Kesharwani, N. A. Markina, A. A. Pletnev, C. Raminelli, T. Yao, G. Zeni, L. Zhang, X. Zhang, R. Rozhkov and S. A. Worlikar, Comprehensive Organic Transformations, A Guide to Functional Group Preparations, Wiley-VCH, New York, 2018 Search PubMed;
(b) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(c) A. K. Mahdi, H. Bahir, A. H. Adhab and A. S. Ibrahim, Chem. Rev. Lett., 2023, 6, 213–222 Search PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
-
(a) T. Ohshima, T. Komyoji, S. Mitani, N. Matsuo and T. Nakajima, J. Pest Sci., 2004, 29, 147–152 CrossRef CAS;
(b) J. R. Bertelsen, E. De Neergaard and V. J. P. P. Smedegaard-Petersen, Plant Pathol., 2001, 50, 190–205 CrossRef CAS.
-
(a) K. C. Vaughn, J. C. Hoffman, M. G. Hahn and L. A. Staehelin, Protoplasma, 1996, 194, 117–132 CrossRef CAS;
(b) J. L. Corbett, S. D. Askew, W. E. Thomas and J. W. Wilcut, Weed Technol., 2004, 18, 443–453 CrossRef CAS.
-
(a) C. Astbury, L. K. Conway, C. Gillespie, K. Hodge, E. Innes and A. R. Kennedy, Dyes Pigm., 2013, 97, 100–104 CrossRef CAS;
(b) X. Zhou, P. Zheng, L. Wang and X. Liu, Polym, 2018, 11, 32 CrossRef PubMed.
- F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
-
(a) V. Schulz, Clin. Pharmacokinet., 1984, 9, 239–251 CrossRef CAS PubMed;
(b) E. Elhalem, B. N. Bailey, R. Docampo, I. Ujváry, S. H. Szajnman and J. B. Rodriguez, J. Med. Chem., 2002, 45, 3984–3999 CrossRef CAS PubMed;
(c) V. C. Scoffone, F. Spadaro, C. Udine, V. Makarov, M. Fondi, R. Fani, E. De Rossi, G. Riccardi and S. Buroni, Antimicrob. Agents Chemother., 2014, 58, 2415–2417 CrossRef PubMed.
- T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494–505 RSC.
- G. Segalovich-Gerendash, I. Rozenberg, N. Alassad, N. B. Nechmad, I. Goldberg, S. Kozuch and N. G. Lemcoff, ACS Catal., 2020, 10, 4827–4834 CrossRef CAS.
- L. Yang, Z. Y. Tian and C. P. Zhang, ChemistrySelect, 2019, 4, 311–315 CrossRef CAS.
- E. de Oliveira Lima Filho and I. Malvestiti, ACS Omega, 2020, 5, 33329–33339 CrossRef CAS PubMed.
- J. Chen, T. Wang, T. Wang, A. Lin, H. Yao and J. Xu, Org. Chem. Front., 2017, 4, 130–134 RSC.
- G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Goossen, Chem. Sci., 2014, 5, 1312–1316 RSC.
- Y. T. Ma, M. C. Liu, Y. B. Zhou and H. Y. Wu, Adv. Synth. Catal., 2021, 363, 5386–5406 CrossRef CAS.
-
(a) A. Krief, W. Dumont and C. Delmotte, Angew. Chem., Int. Ed., 2000, 39, 1669–1672 CrossRef CAS;
(b) N. Mukherjee, D. Kundu and B. C. Ranu, Adv. Synth. Catal., 2017, 359, 329–338 CrossRef CAS;
(c) P. Nikolaienko and M. Rueping, Chem.–Eur. J., 2016, 22, 2620–2623 CrossRef CAS PubMed.
- A. Hassanpour, E. Ghavidelaghdam, A. G. Ebadi, M. R. P. Heravi and E. Vessally, RSC Adv., 2021, 11, 22305–22316 RSC.
- W. Ali, M. Álvarez-Pérez, M. A. Marć, N. Salardón-Jiménez, J. Handzlik and E. Domínguez-Álvarez, Curr. Pharmacol. Rep., 2018, 4, 468–481 CrossRef CAS.
-
(a) M. Alterman and A. Hallberg, J. Org. Chem., 2000, 65, 7984–7989 CrossRef CAS;
(b) A. B. Khemnar and B. M. Bhanage, RSC Adv., 2014, 4, 13405–13408 RSC.
-
(a) Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697–6700 CrossRef CAS PubMed;
(b) A. Kamal and G. Chouhan, Tetrahedron Lett., 2005, 46, 1489–1491 CrossRef CAS.
-
(a) C. Paulmier and F. Outurquin, J. Heterocycl. Chem., 1983, 20, 113–119 CrossRef CAS;
(b) A. Frolov, E. Smirnov, O. Kul'bitskaya and A. El'tsov, Zh. Org. Khim., 1980, 16, 2302–2309 CAS;
(c) V. Nefedov, L. Tarygina, L. Kryuchkova and Y. S. Ryabokobylko, Zh. Org. Khim., 1981, 17, 570–584 CAS.
-
(a) G. Petrillo, M. Novi, G. Garbarino and C. Dell'erba, Tetrahedron, 1987, 43, 4625–4634 CrossRef CAS;
(b) I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, J. Organomet. Chem., 2004, 689, 3810–3812 CrossRef CAS.
- G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Goossen, Chem. Sci., 2014, 5, 1312–1316 RSC.
-
(a) H. Rheinboldt and E. Giesbrecht, Chem. Ber., 1956, 89, 631–636 CrossRef CAS;
(b) J. Gosselck and E. Wolters, Chem. Ber., 1962, 95, 1237–1244 CrossRef CAS;
(c) S. Yavuz, A. Dişli, Y. Yıldırır and L. Türker, Molecules, 2005, 10, 1000–1004 CrossRef CAS PubMed.
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS.
-
(a) J. Hofmann and M. R. Heinrich, Tetrahedron Lett., 2016, 57, 4334–4340 CrossRef CAS;
(b) A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed.
- G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- Z. Zhang and L. S. Liebeskind, Org. Lett., 2006, 8, 4331–4333 CrossRef CAS PubMed.
- X. Tian, Y. Sun, C. Dong, K. Zhang, T. Liang, Y. Zhang and C. Hou, Chem. Lett., 2012, 41, 719–721 CrossRef CAS.
- G. Y. Zhang, J. T. Yu, M. L. Hu and J. Cheng, J. Org. Chem., 2013, 78, 2710–2714 CrossRef CAS PubMed.
- B. K. Vaghasiya, S. P. Satasia, R. P. Thummar, R. D. Kamani, J. R. Avalani, N. H. Sapariya and D. K. Raval, J. Sulfur Chem., 2018, 39, 507–515 CrossRef CAS.
- Z. Zhang, T. Niwa, Y. Watanabe and T. Hosoya, Org. Biomol. Chem., 2018, 16, 7711–7716 RSC.
- J. De Jesus Silva, N. Bartalucci, B. Jelier, S. Grosslight, T. Gensch, C. Schünemann, B. Müller, P. C. J. Kamer, C. Copéret, M. S. Sigman and A. Togni, Helv. Chim. Acta, 2021, 104, e2100200 CrossRef CAS.
- P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed. Engl., 2011, 50, 519–522 CrossRef CAS PubMed.
- C. A. Malapit, J. T. Reeves, C. A. Busacca, A. R. Howell and C. H. Senanayake, Angew. Chem., Int. Ed. Engl., 2016, 128, 334–338 CrossRef.
- C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389–11391 CrossRef CAS PubMed.
- W. X. Lv, Z. Li, E. Lin, J. L. Li, D. H. Tan, Y. H. Cai and Q. Li, Chem.–Eur. J., 2019, 25, 4058–4061 CrossRef CAS PubMed.
- F. Xu, O. M. Duke, D. Rojas, H. M. Eichelberger, R. S. Kim, T. B. Clark and D. A. Watson, J. Am. Chem. Soc., 2020, 142, 11988–11992 CrossRef CAS PubMed.
- S. Y. Song, X. Zhou, Z. Ke and S. Xu, Angew. Chem., Int. Ed. Engl., 2023, 62, e202217130 CrossRef CAS PubMed.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS PubMed.
- Y.-J. Niu, G.-H. Sui, H.-X. Zheng, X.-H. Shan, L. Tie, J.-L. Fu, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2019, 84, 10805–10813 CrossRef CAS PubMed.
- H. Zhao, L. Chen, C. Xia and S. Xu, Asian J. Org. Chem., 2023, 12, e202200695 CrossRef CAS.
- K. Jing, L. Chen, P. Zhang and S. Xu, Chin. J. Chem., 2023, 41, 2119–2124 CrossRef CAS.
- G. Zhang, L. Zhang, M. Hu and J. Cheng, Adv. Synth. Catal., 2011, 353, 291–294 CrossRef CAS.
- H. X. Dai and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 134–137 CrossRef CAS PubMed.
- F. Bartoccini, S. Bartolucci, M. Mari and G. Piersanti, Org. Biomol. Chem., 2016, 14, 10095–10100 RSC.
- Y. Ye, Y. Wang, P. Liu and F. Han, Chin. J. Chem., 2013, 31, 27–30 CrossRef CAS.
- K. Jinho, C. Jiho, S. Kwangmin and C. Sukbok, J. Am. Chem. Soc., 2012, 134, 2528–2531 CrossRef PubMed.
- G. Zhang, S. Chen, H. Fei, J. Cheng and F. Chen, Synlett, 2012, 23, 2247–2250 CrossRef CAS.
- Y. Luo, Q. Wen, Z. Wu, J. Jin, P. Lu and Y. Wang, Tetrahedron, 2013, 69, 8400–8404 CrossRef CAS.
- C. Qi, X. Hu and H. He, Synlett, 2016, 27, 1979–1982 CrossRef CAS.
- X. J. Wang and S. L. Zhang, New J. Chem., 2017, 41, 14826–14830 RSC.
- Y. Zhu, L. Li and Z. Shen, Chem.–Eur. J., 2015, 21, 13246–13252 CrossRef CAS PubMed.
- K. Okamoto, N. Sakata and K. Ohe, Org. Lett., 2015, 17, 4670–4673 CrossRef CAS.
- L. Ma, M. S. Placzek, J. M. Hooker, N. Vasdev and S. H. Liang, Chem. Commun., 2017, 53, 6597–6600 RSC.
- K. J. Makaravage, X. Shao, A. F. Brooks, L. Yang, M. S. Sanford and P. J. Scott, Org. Lett., 2018, 20, 1530–1533 CrossRef CAS PubMed.
- Z. Xu, X. Liang and H. Li, Org. Lett., 2022, 24, 9403–9407 CrossRef CAS PubMed.
- N. Sun, H. Zhang, W. Mo, B. Hu, Z. Shen and X. Hu, Synlett, 2013, 24, 1443–1447 CrossRef CAS.
- N. Sun, L. Che, W. Mo, B. Hu, Z. Shen and X. Hu, Org. Biomol. Chem., 2015, 13, 691–696 RSC.
- M. Dyga, D. Hayrapetyan, R. K. Rit and L. J. Gooßen, Adv. Synth. Catal., 2019, 361, 3548–3553 CrossRef CAS.
- D. He, J. Yao, B. Ma, J. Wei, G. Hao, X. Tuo, S. Guo, Z. Fu and H. Cai, Green Chem., 2020, 22, 1559–1564 RSC.
- Y. Yang, X. Gao, X. Zeng, J. Han and B. Xu, Chem.–Eur. J., 2021, 27, 1297–1300 CrossRef CAS.
- S. Redon, A. R. O. Kosso, J. Broggi and P. Vanelle, Synthesis, 2019, 51, 3758–3764 CrossRef CAS.
- X. Zhang, X. B. Huang, Y. B. Zhou, M. C. Liu and H. Y. Wu, Chem.–Eur. J., 2021, 27, 944–948 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence h
h