
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Capture of mechanically interlocked molecules by rhodium-mediated terminal alkyne dimerisation†
Thomas M. Hood *,
Samantha Lau and
Adrian B. Chaplin*
*,
Samantha Lau and
Adrian B. Chaplin*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: tom.m.hood@warwick.ac.uk; a.b.chaplin@warwick.ac.uk
First published on 5th March 2024
Abstract
The transition metal-mediated dimerisation of terminal alkynes is an attractive and atom-economic method for preparing conjugated 1,3-enynes. Using a phosphine-based macrocyclic pincer ligand, we demonstrate how this transformation can be extended to the synthesis of novel, hydrocarbon-based interlocked molecules: a rotaxane by ‘active’ metal template synthesis and a catenane by sequential ‘active’ and ‘passive’ metal template procedures.
Introduction
Coordination chemistry is a prominent feature of contemporary methods for constructing mechanically interlocked molecules, such as rotaxanes and catenanes.1 Active metal template synthesis has emerged as a particularly effective strategy, exploiting a metal to position and covalently fuse the precursor fragments together in an entangled arrangement (Fig. 1A).2 Whilst this strategy has been successfully implemented using a variety of metal-mediated transformations, the overwhelming majority of examples are based on copper-mediated alkyne–azide cycloaddition reactions (I) or Glaser couplings (II) in combination with bidentate nitrogen-based macrocycles.2,3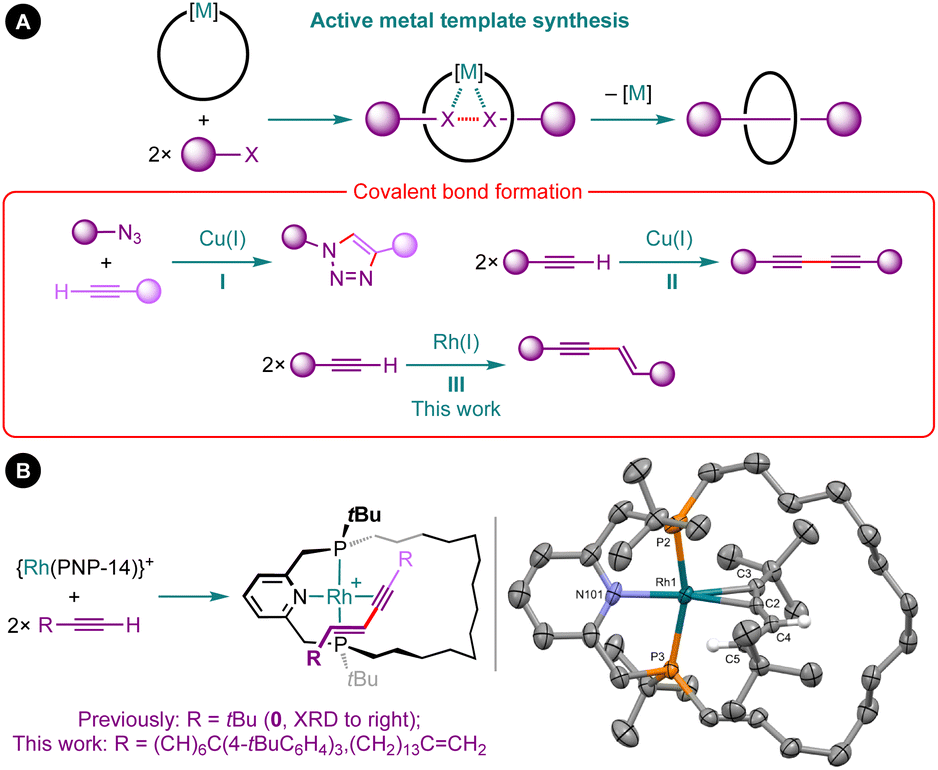 | ||
| Fig. 1 (A) Active metal template synthesis of interlocked molecules and (B) terminal alkyne dimerisation reactions promoted by macrocyclic rhodium(I) PNP pincer complexes. Solid-state structure of complex 0 shown with thermal ellipsoids drawn at 30% probability and most H-atoms omitted. | ||
As part of our work exploring the organometallic chemistry of terminal alkyne dimerisation reactions4 promoted by macrocyclic pincer complexes (Fig. 1B),5,6 we speculated that this transformation could be adapted into an active metal template procedure for the preparation of mechanically interlocked 1,3-enynes (III). We herein describe the preparation of hydrocarbon-based rotaxane 1 and catenane 2 derived from the phosphine-based macrocyclic pincer ligand PNP-14 (Fig. 2).7 Despite the widespread use of phosphine ligands in organotransition metal chemistry and homogenous catalysis,8 this constitutes the first application in active metal template synthesis of mechanically interlocked molecules.
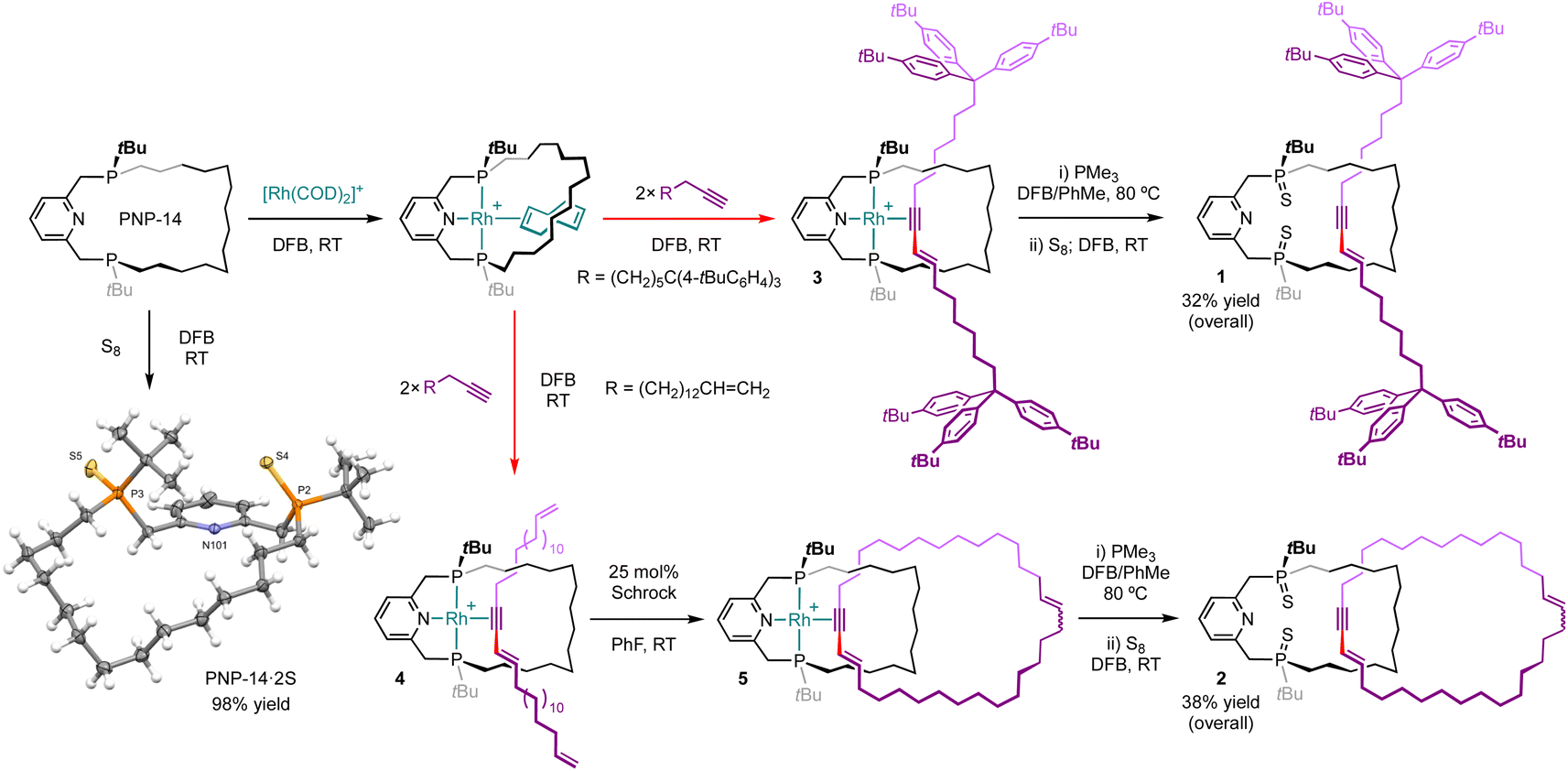 | ||
| Fig. 2 Synthesis of rotaxane 1 and catenane 2. [B(3,5-(CF3)2C6H3)4]− counterions omitted for clarity and solid-state structure of PNP-14·2S shown with thermal ellipsoids drawn at 50% probability. | ||
Results and discussion
Using a protocol developed previously for rhodium(I) E-enyne complex 0 (Fig. 1B),6,9 rotaxanate 3 and pseudo-rotaxanate 4 were obtained in quantitative spectroscopic yield upon treatment of [Rh(PNP-14)(η2-COD)]+ (COD = cyclooctadiene; δ31P 57.4/45.9, 2JPP = 312 Hz, 1JRhP ∼135 Hz) with the novel bulky aryl stoppered terminal alkyne HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)6C(4-tBuC6H4)3 and methylene-spaced ene–yne HCC(CH2)13CH
C(CH2)6C(4-tBuC6H4)3 and methylene-spaced ene–yne HCC(CH2)13CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, respectively, in the weakly coordinating solvent 1,2-difluorobenzene (DFB) at room temperature (Fig. 2).10 The new rhodium derivatives present 1H NMR resonances at δ 6.95/5.94 (3) and 7.01/5.98 (4) with 3JHH coupling constants of ∼15 Hz that are diagnostic for coordinated E-enynes, whilst the C1 symmetry of the molecules is manifested in the observation of inequivalent 31P NMR resonances at δ 58.6/54.9 (3) and 56.9/53.1 (4) that are coupled to 103Rh (1JRhP = 128 Hz) and display trans 2JPP coupling constants of ∼393 Hz.11 Subsequent treatment of 4 with 25 mol% Schrock's catalyst (CAS: 139220-25-0) at room temperature enabled capture of catenate 5 by ring closing olefin metathesis, with complete conversion confirmed after monitoring the reaction in situ for 5 days at room temperature using 1H and 31P{1H} NMR spectroscopy and (tandem) ESI-MS.
CH2, respectively, in the weakly coordinating solvent 1,2-difluorobenzene (DFB) at room temperature (Fig. 2).10 The new rhodium derivatives present 1H NMR resonances at δ 6.95/5.94 (3) and 7.01/5.98 (4) with 3JHH coupling constants of ∼15 Hz that are diagnostic for coordinated E-enynes, whilst the C1 symmetry of the molecules is manifested in the observation of inequivalent 31P NMR resonances at δ 58.6/54.9 (3) and 56.9/53.1 (4) that are coupled to 103Rh (1JRhP = 128 Hz) and display trans 2JPP coupling constants of ∼393 Hz.11 Subsequent treatment of 4 with 25 mol% Schrock's catalyst (CAS: 139220-25-0) at room temperature enabled capture of catenate 5 by ring closing olefin metathesis, with complete conversion confirmed after monitoring the reaction in situ for 5 days at room temperature using 1H and 31P{1H} NMR spectroscopy and (tandem) ESI-MS.
Removal of rhodium from 3 and 5 was achieved by heating with excess PMe3 (20 equiv.) to give the corresponding rotaxane (1′, δ31P 2.42/0.92) and catenane (2′, δ31P 1.46/0.78), alongside [Rh(PMe3)4]+ as the rhodium-containing by-product.12 To facilitate isolation, 1′ and 2′ were converted into the corresponding phosphine sulphides 1 and 2 by treatment with S8 which were thereafter obtained in 32% and 38% overall yield (from PNP-14) following purification on silica. Formation of the new interlocked molecules was corroborated by analysis of isolated materials using NMR spectroscopy and ESI-MS. Threading of the enyne breaks the C2 symmetry of the macrocycle and this desymmetrisation is evident in both the 1H and 31P{1H} NMR spectra of 1 (δ31P 63.71/63.65) and 2 (δ31P 63.6/63.5), alongside perturbation of the macrocycle resonances relative to independently isolated PNP-14·2S (δ31P 64.7, NMR stack plots provided in the ESI†). The interlocked nature of 1 and 2 was also substantiated by high resolution tandem mass spectrometry experiments,13 whereby selective fragmentation of the [M + H]+ ions (1, 1584.1321, calcd 1584.1339 m/z; 2, 982.7566, calcd 982.7549 m/z) gave the [M + H]+ ion of PNP-14·2S (542.3154/542.3159, calcd 542.3167 m/z) as the major species in both cases.
Conclusions
These results serve as proof of principle for the application of transition mediated terminal alkyne dimerisation reactions in the synthesis of mechanically interlocked molecules, whilst also demonstrating how phosphine-based functional groups can be integrated into the structure of these intriguing molecules.Experimental section
All manipulations were performed under an atmosphere of argon using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves were activated by heating at 300 °C in vacuo overnight. Fluorobenzene and 1,2-difluorobenzene (DFB) were pre-dried over Al2O3, distilled from calcium hydride and dried twice over 3 Å molecular sieves.10 CD2Cl2 was freeze–pump–thaw degassed and dried over 3 Å molecular sieves. THF and dioxane were distilled from sodium/benzophenone and stored over 3 Å molecular sieves. DMSO was freeze–pump–thaw degassed and dried up to five times and finally stored over 3 Å molecular sieves. SiMe4 was distilled from liquid Na/K alloy and stored over a potassium mirror. Other anhydrous solvents were purchased from Acros Organics or Sigma-Aldrich, freeze–pump–thaw degassed and stored over 3 Å molecular sieves. PMe3 in toluene and 1,6-dibromohexane were freeze–pump–thawed degassed and dried twice over 3 Å molecular sieves before use. Schrock's catalyst (CAS: 139220-25-0) was recrystallised from SiMe4 at −30 °C before use. nBuLi was titrated before use.14 Br(CH2)6C(4-tBuC6H4)3,15 15-bromo-1-pentadecene,16 [Rh(COD)2][BArF4],17 and PNP-14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 were synthesized according to published procedures. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz. NMR spectra in DFB were recorded using an internal capillary of C6D6. High resolution (HR) ESI-MS and MS/MS were recorded on Bruker Maxis Plus instrument. Microanalysis was performed at the London Metropolitan University by Stephen Boyer.
7 were synthesized according to published procedures. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz. NMR spectra in DFB were recorded using an internal capillary of C6D6. High resolution (HR) ESI-MS and MS/MS were recorded on Bruker Maxis Plus instrument. Microanalysis was performed at the London Metropolitan University by Stephen Boyer.
Preparation of HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C(CH2)6C(4-tBuC6H4)3
C(CH2)6C(4-tBuC6H4)3
A suspension of Br(CH2)6C(4-tBuC6H4)3 (290 mg, 504 μmol) in DMSO (5 mL) was treated dropwise with THF until homogeneous. A suspension of HCCLi·H2N(CH2)2NH2 (51.0 mg, 554 μmol) in THF (5 mL) was then added and the resulting suspension heated at 130 °C for 48 h. The reaction was quenched by addition of H2O (2 mL) and the product extracted into CH2Cl2 (3 × 5 mL). The combined organic extracts were washed with brine (2 × 10 mL), dried over MgSO4 and then concentrated in vacuo to give an oily white solid. Purification using a silica plug (hexane → 1:1 hexane:CH2Cl2) afforded the product as a white solid. Yield: 220 mg (422 μmol, 84%; mp. 142–143 °C).
1H NMR (500 MHz, CDCl3): δ 7.24 (d, 3JHH = 8.4, 6H, m-Ar), 7.14 (d, 3JHH = 8.4, 6H, o-Ar), 2.48–2.52 (m, 2H, Ar3CC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.13 (td, 3JHH = 7.1, 4JHH = 2.6, 2H, C2CCH), 1.91 (t, 4JHH = 2.6, 1H, CCH), 1.44 (pent, 3JHH = 7.1, 2H, C2CH2CCH), 1.25–1.36 (m, 4H, 2×CH2), 1.30 (s, 27H, tBu), 1.05–1.12 (m, 2H, CH2).
2), 2.13 (td, 3JHH = 7.1, 4JHH = 2.6, 2H, C2CCH), 1.91 (t, 4JHH = 2.6, 1H, CCH), 1.44 (pent, 3JHH = 7.1, 2H, C2CH2CCH), 1.25–1.36 (m, 4H, 2×CH2), 1.30 (s, 27H, tBu), 1.05–1.12 (m, 2H, CH2).
13C{1H} NMR (126 MHz, CDCl3): δ 148.2 (s, tBu![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ), 145.0 (s, i-Ar), 129.0 (s, o-Ar), 124.5 (s, m-Ar), 84.9 (s, CH), 68.2 (s, CH), 55.5, (s, Ar3), 40.7 (s, Ar3CH2), 34.4 (s, tBu{C}), 31.5 (s, tBu{CH3}), 30.1 (s, CH2), 28.8 (s, CH2), 28.7 (s, H2CH2CCH), 25.7 (s, CH2), 18.5 (s, H2CCH).
), 145.0 (s, i-Ar), 129.0 (s, o-Ar), 124.5 (s, m-Ar), 84.9 (s, CH), 68.2 (s, CH), 55.5, (s, Ar3), 40.7 (s, Ar3CH2), 34.4 (s, tBu{C}), 31.5 (s, tBu{CH3}), 30.1 (s, CH2), 28.8 (s, CH2), 28.7 (s, H2CH2CCH), 25.7 (s, CH2), 18.5 (s, H2CCH).
HR ESI-MS (positive ion 4 kV): 559.3684 ([M + K]+, calcd 559.3701) m/z.
Preparation of HCC(CH2)13CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH2
CH2
A suspension of 15-bromo-1-pentadecene (1.22 g, 4.24 mmol) and HCCLi·H2N(CH2)2NH2 (0.41 g, 4.45 mmol) in 1,4-dioxane-DMSO (10:5 mL) was stirred at 120 °C for 16 h. The reaction was quenched by addition of H2O (15 mL) and product extracted into hexane (3 × 15 mL). The combined organic extracts were washed with brine (2 × 10 mL), dried over MgSO4 and then concentrated in vacuo to give an off-white oily wax. The analytically pure compound was obtained as a colourless wax by repeated column chromatography (silica, hexane; Rf = 0.37). Yield: 244 mg (1.04 mmol, 25%; mp. 43–48 °C).
1H NMR (500 MHz, CDCl3): δ 5.81 (ddt, 3JHH = 16.9, 10.2, 6.7, 1H, H2CC), 4.99 (ddt, 3JHH = 16.9, 2JHH = 2.2, 4JHH = 1.6, 1H, 2CCH), 4.93 (ddt, 3JHH = 10.2, 2JHH = 2.2, 4JHH = 1.3, 1H, 2CCH), 2.18 (td, 3JHH = 7.1, 4JHH = 2.6, 2H, C2CCH), 2.01–2.07 (m, 2H, H2CCHC2), 1.94 (t, 4JHH = 2.6, 1H, CC), 1.52 (pent, 3JHH = 7.6, 2H, C2CH2CCH), 1.33–1.43 (m, 4H, 2×CH2), 1.22–1.33 (m, 16H, 8×CH2).
13C{1H} NMR (126 MHz, CDCl3): δ 139.4 (s, H2CH), 114.2 (s, H2CH), 85.0 (s, CH), 68.2 (s, CH), 34.0 (s, CH2CHH2), 29.80 (s, 2×CH2), 29.76 (s, 2×CH2), 29.7 (s, 2×CH2), 29.31 (s, CH2), 29.27 (s, CH2), 29.1 (s, CH2), 28.9 (s, CH2), 28.7 (s, CH2), 18.6 (s, H2CCH).
Anal. calcd for C17H30 (234.43 g mol−1): C, 87.10; H, 12.90; N, 0.00. Found: C, 86.99; H, 13.02; N, 0.00.
Preparation of rotaxane 1
A solution of [Rh(PNP-14)(η2-COD)][BArF4] (8.3 μmol, generated in situ from [Rh(COD)2][BArF4] and PNP-14 as previously described9) in DFB (0.5 mL) was treated with HCC(CH2)6C(4-tBuC6H4)3 (9.5 mg, 18.2 μmol) and stirred at RT for 5 min. Volatiles were removed in vacuo to afford 3 as an orange foam upon removal of volatiles. Crude 3 was then dissolved in DFB (330 μL) and treated with a solution of PMe3 in toluene (170 μL, 1 M, 170 μmol) and the resulting solution heated at 80 °C for 5 days. Volatiles were removed in vacuo and the residue extracted with hexane. The resulting orange oil was treated with S8 (12.6 mg, 49.1 μmol) in DFB (0.5 mL) and stirred at RT for 16 h. Finally, removal of the volatiles in vacuo and purification by preparative TLC (silica; 9:1 CH2Cl2:MeOH; Rf = 0.4–0.5) afforded the product as a white solid. Yield: 4.2 mg (2.7 μmol, 32%; mp 112 °C).
Data for 3
1H NMR (500 MHz, CD2Cl2, selected data): δ 7.76 (t, 3JHH = 7.9, 1H, p-py), 7.33 (d, 3JHH = 7.9, 1H, m-py), 7.31 (d, 3JHH = 7.9, 1H, m-py), 6.95 (dt, 3JHH = 14.6, 3JHH = 6.9, 1H, CCCHC), 5.94 (d, 3JHH = 15.3, 1H, CCHCH), 1.31 (s, 12H, tBuC), 1.30 (s, 38H, tBuC), 0.93 (d, 3JPH = 12.3, 9H, PtBu), 0.89 (d, 3JPH = 12.3, 9H, PtBu).
31P{1H} NMR (162 MHz, CD2Cl2): δ 58.6 (dd, 2JPP = 394, 1JRhP = 129, 1P), 54.9 (dd, 2JPP = 394, 1JRhP = 127, 1P).
1H NMR (400 MHz, DFB, selected data): δ 7.55 (t, 3JHH = 8.0, 1H, p-py), 5.99 (d, 3JHH = 15.0, 1H, CCCCH), 1.12–1.17 (m, 54H, tBuC), 0.82–0.89 (m, 18H, PtBu).
31P{1H} NMR (162 MHz, DFB): δ 56.1 (dd, 2JPP = 393, 1JRhP = 129, 1P), 52.5 (dd, 2JPP = 393, 1JRhP = 127, 1P).
Data for 1′
31P{1H} NMR (162 MHz, PhF, selected data): δ 2.42 (s, 1P), 0.92 (s, 1P).Data for 1
1H NMR (500 MHz, CDCl3): δ 7.45 (d, 3JHH = 7.6, 1H, m-py), 7.41 (t, 3JHH = 7.6, 1H, p-py), 7.24 (d, 3JHH = 8.6, 6H, m-Ar), 7.23 (d, 3JHH = 8.6, 6H, m-Ar), 7.13 (d, 3JHH = 8.3, 12H, 2×o-Ar), 7.12 (obscured, 1H, m-py), 6.17 (dt, 2JHH = 16.0, 3JHH = 6.9, 1H, CCCHC), 5.95 (dt, 2JHH = 16.0, 4JHH = 2.0, 1H, CCCCH), 3.84 (app t, 2JPH = 2JHH = 13.9, 1H, pyC2), 3.60 (dd, 2JHH = 14.1, 2JPH = 11.1, 1H, pyC2), 3.37 (app t, 2JPH = 2JHH = 13.7, 1H, pyC2), 3.35 (dd, 2JHH = 13.8, 2JPH = 11.3, 1H, pyC2), 2.45–2.53 (m, 4H, 2×Ar3CC2), 2.18–2.28 (m, 1H, C2CCCHCH), 2.02–2.16 (m, 4H, CCCHCHC2 [δ 2.11, 2H] + PCH2 [δ 2.08, 1H] + C2CCCHCH [δ 2.07, 1H]), 1.80–1.94 (m, 5H, CH2), 1.11–1.68 (m, 34H, CH2), 1.292 (s, 27H, tBuC), 1.290 (s, 27H, tBuC), 1.24 (d, 3JPH = 15.6, 18H, 2×PtBu), 0.98–1.11 (m, 4H, 2×Ar3CCH2C2).
13C{1H} NMR (126 MHz, CDCl3): δ 153.6 (dd, 2JPC = 6, 4JPC = 1, o-py), 153.5 (d, 2JPC = 7, o-py), 148.2, (s, tBu), 148.1 (s, tBu), 145.03 (s, i-Ar), 144.97 (s, i-Ar), 143.4 (s, CCCHH), 135.6 (s, p-py), 128.96 (s, o-Ar), 128.95 (s, o-Ar), 124.53 (s, m-Ar), 124.50 (s, m-Ar), 123.4 (br, m-py), 123.3 (br, m-py), 113.3 (s, CCHCH), 92.3 (s, CCHCH), 82.1 (s, CCHCH), 55.43 (s, Ar3), 55.39 (s, Ar3), 40.83 (s, Ar3CH2), 40.80 (s, Ar3CH2), 37.1 (d, 1JPC = 42, pyH2), 36.3 (d, 1JPC = 41, pyH2), 35.3 (d, 1JPC = 47, PtBu{C}), 35.2 (d, 1JPC = 47, PtBu{C}), 34.4 (s, 2×tBu{C}), 33.3 (s, CCCHCHH2), 31.6 (s, 2×tBu{CH3}), 31.2 (d, 2JPC = 15, 2×CH2), 30.8 (s, CH2), 30.6 (s, CH2), 28.8–30.0 (m, 12×CH2), 28.2 (d, 1JPC = 48, PCH2), 27.7 (d, 1JPC = 47, PCH2), 26.1 (s, 2×Ar3CCH2H2), 25.8 (s, 2×PtBu{CH3}), 23.8 (d, 3JPC = 4, CH2), 23.2 (d, 3JPC = 4, CH2), 21.0 (s, H2CCCHCH).
31P{1H} NMR (162 MHz, CDCl3): δ 63.71 (s, 1P), 63.65 (s, 1P).
HR ESI-MS (positive ion, 4 kV): 1584.1321 ([M + H]+, calcd 1584.1339) m/z.
HR ESI-MS/MS (positive ion; 120 eV @ +1584): 542.3154 ([PNP-14·2S + H]+, calcd 542.3167) m/z.
Preparation of catenane 2
A solution of [Rh(PNP-14)(η2-COD)][BArF4] (10.7 μmol, generated in situ from [Rh(COD)2][BArF4] and PNP-14 as previously described9) in DFB (0.5 mL) was treated with HCC(CH2)13CHCH2 (194 μL, 116 mM, 22.5 μmol) and stirred at RT for 5 min. Volatiles were removed in vacuo to afford 4 as an orange oil. Crude 4 was dissolved in fluorobenzene (10 mL) and treated with 25 mol% Schrock's catalyst in 5 mol% portions (0.4 mg, 0.52 μmol) daily over 5 days and periodically assayed by ESI-MS. Volatiles were removed in vacuo to afford 5 as an orange oil. Crude 5 was then dissolved in DFB (300 μL) and treated with a solution of PMe3 in toluene (200 μL, 1 M, 200 μmol) and the resulting solution heated at 80 °C for 5 days. Volatiles were removed in vacuo and the residue extracted with hexane. The resulting orange oil was treated with S8 (12.6 mg, 49.1 μmol) in DFB (0.5 mL) and stirred at RT for 16 h. Finally, removal of the volatiles in vacuo and purification by preparative TLC (silica; 9:1 CH2Cl2:MeOH; Rf = 0.4–0.5) afforded the product as a colourless oil. Yield: 3.7 mg (3.8 μmol, 38%).
Data for 4
1H NMR (500 MHz, CD2Cl2, selected data): δ 7.77 (t, 3JHH = 7.8, 1H, p-py), 7.36 (overlapping d, 3JHH = 7.9, 2H, m-py), 7.01 (dt, 3JHH = 14.6, 3JHH = 6.9, 1H, CCCHC), 5.98 (d, 3JHH = 15.3, 1H, CCCCH), 5.82 (ddt, 3JHH = 16.8, 3JHH = 9.8, 3JHH = 6.7, 2H, CCH2), 4.98 (d, JHH = 17, 2H, CHC2), 4.91 (d, JHH = 10, 2H, CHC2), 3.37–3.51 (m, 4H, pyC2), 0.97 (d, 3JPH = 12.3, 9H, tBu), 0.91 (d, 3JPH = 12.2, 9H, tBu).
31P{1H} NMR (121 MHz, CD2Cl2): δ 56.9 (dd, 2JPP = 394, 1JRhP = 129, 1P), 53.1 (dd, 2JPP = 394, 1JRhP = 127, 1P).
1H NMR (400 MHz, DFB, selected data): δ 8.09–8.15 (m, 8H, ArF), 7.53 (obscured t, 3JHH = 8.2, p-py), 7.49 (br, 4H, ArF), 6.04 (d, 3JHH = 15.4, 1H, CCCCH), 5.82 (dt, 3JHH = 15.4, 3JHH = 8.3, CCH2), 4.80–4.98 (m, 2H, CHC2), 0.87 (app t, JPH = 12.8, 18H, tBu).
31P{1H} NMR (121 MHz, DFB): δ 56.5 (dd, 2JPP = 394, 1JRhP = 128, 1P), 52.1 (dd, 2JPP = 394, 1JRhP = 127, 1P).
HR ESI-MS (positive ion, 4 kV): 1048.7587, ([M]+, calcd 1048.7398) m/z.
Data for 5
1H NMR (400 MHz, CD2Cl2, selected data): δ 7.79 (t, 3JHH = 8.3, 1H, p-py), 7.36 (br d, 3JHH = 7.3, 2H, m-py), 7.03 (br, 1H, CCCHC), 6.00 (d, 3JHH = 16, 1H, CCCCH), 5.39 (br, 2H, CHCH), 3.43 (br, 4H, pyC2), 0.98 (d, 3JPH = 11, 9H, PtBu), 0.92 (d, 3JPH = 12, 9H, PtBu).
31P{1H} NMR (121 MHz, CD2Cl2): δ 56.7 (d, 2JPP = 394, 1JRhP = 129, 1P), 53.3 (d, 2JPP = 393, 1JRhP = 127, 1P).
1H NMR (400 MHz, DFB, selected data): δ 8.09–8.15 (m, 8H, ArF), 7.49 (br, 4H, ArF), 6.01 (br d, 3JHH = 14.4, 1H, CCCCH), 5.35 (br, 2H, CHCH), 3.30 (br, 4H, pyC2), 0.88 (br d, 3JPH = 12, 9H, tBu), 0.83 (br d, 3JPH = 10, 9H, tBu).
31P{1H} NMR (121 MHz, DFB): δ 56.6 (d, 2JPP = 394, 1JRhP = 129, 1P), 52.5 (d, 2JPP = 393, 1JRhP = 127, 1P).
HR ESI-MS (positive ion, 4 kV): 1020.7092, ([M]+, calcd 1020.7085) m/z.
HR ESI-MS2 (positive ion, 70 eV @ +1020): 578.2543 ([{Rh(PNP-14)}-H2]+, calcd 578.2546) m/z.
Data for 2′
31P{1H} NMR (162 MHz, DFB, selected data): δ 1.46 (s, 1P), 0.78 (s, 1P).Data for 2
1H NMR (500 MHz, CDCl3): δ 7.45–7.54 (m, 2H, py), 7.20 (br d, 3JHH = 5.2, 1H, py), 6.22 (dt, 3JHH = 16.0, 3JHH = 7.0, 1H, CCCHC), 5.98 (dt, 3JHH = 16.1, 4JHH = 2.0, 1H, CCCCH), 5.36–5.39 (m, 2H, CHCH), 3.88 (app t, JPH = JHH = 14, 1H, pyC2), 3.59–3.69 (m, 1H, CH2), 3.41 (app t, JPH = JHH = 13, 2H, pyC2), 2.04–2.41 (m, 6H, CH2), 1.89–1.99 (m, 9H, CH2), 1.38–1.47 (obscured m, ∼16H, CH2), 1.25–1.35 (m, ∼65H, CH2 + PtBu).
13C{1H} NMR (126 MHz, CDCl3): δ 153.5–153.8 (m, py), 143.3 (s, CCCHH), 135.7 (s, py), 130.54 (s, CHCH), 130.46 (s, CHCH), 123.4 (s, py), 123.3 (s, py), 113.6 (s, CCHCH), 92.5 (s, CCHCH), 82.1 (s, CCHCH), 37.2 (d, 1JPC = 42, pyH2), 36.5 (d, 1JPC = 41, pyH2), 35.5 (d, 1JPC = 23, PtBu{C}), 35.1 (d, 1JPC = 24, PtBu{C}), 32.8 (s, CH2), 32.1 (s, CH2), 32.4 (s, CH2), 31.2 (s, CH2), 29.9 (s, CH2), 29.2–29.8 (m, multiple CH2), 29.2 (s, CH2), 29.12 (s, CH2), 29.09 (s, CH2), 29.07 (s, CH2), 28.88 (s, CH2), 28.86 (s, CH2), 28.7 (s, CH2), 28.3 (s, CH2), 27.9 (s, CH2), 27.6 (s, CH2), 25.77 (s, PtBu{CH3}), 25.75 (s, PtBu{CH3}), 23.8 (d, 3JPC = 4, CH2), 23.3 (d, 3JPC = 4, CH2), 22.9 (s, CH2), 20.8 (s, CH2).
31P{1H} NMR (121 MHz, CDCl3): δ 63.6 (s, 1P), 63.5 (s, 1P).
HR ESI-MS (positive ion, 4 kV): 982.7566, ([M + H]+, calcd 982.7549) m/z.
HR ESI-MS2 (positive ion, 60 eV @ +982): 542.3159 ([PNP-14·2S + H]+, calcd 542.3167) m/z.
Preparation of PNP-14·2S
A solution of PNP-14 (8.5 mg, 17.8 μmol) in DFB (0.5 mL) was treated with S8 (1.2 mg, 4.68 μmol) and stirred at RT for 16 h. Volatiles were removed, and the resulting residue washed with methanol (2 × 0.5 mL) and then dried in vacuo to afford the product as a white microcrystalline solid. Yield: 9.4 mg (17.3 μmol, 97%; mp. 139–140 °C).1H NMR (500 MHz, CDCl3): δ 7.59 (t, 3JHH = 7.8, 1H, p-py), 7.35 (d app t, 3JHH = 7.8, JPH = 2, 2H, m-py), 3.49 (app t, 2JPH = 2JHH = 13, 2H, pyC2), 3.43 (app t, 2JPH = 2JHH = 14, 2H, pyC2), 1.99–2.10 (m, 2H, PCH2), 1.68–1.86 (m, 4H, PCH2 [δ 1.80, 2H] + CH2), 1.24–1.58 (m, 22H, CH2), 1.17 (d, 3JPH = 15.9, 18H, tBu).
13C{1H} NMR (126 MHz, CDCl3): δ 153.7 (dd, 2JPC = 8, 4JPC = 2, o-py), 136.6 (t, 4JPC = 2, p-py), 123.4 (app t, JPC = 3, m-py), 38.9 (d, 1JPC = 39, pyH2), 34.8 (d, 1JPC = 47, tBu{C}), 30.2 (d, 2JPC = 15, CH2), 27.9 (s, CH2), 27.80 (s, CH2), 27.79 (s, CH2), 27.7 (s, CH2), 26.3 (d, 1JPC = 47, PCH2), 25.3 (s, tBu{CH3}), 22.4 (d, 3JPC = 4, CH2).
31P{1H} NMR (162 MHz, CDCl3): δ 64.7 (s).
HR ESI-MS (positive ion, 4 kV): 542.3160 ([M + H]+, calcd 542.3167) m/z.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313) and Royal Society (UF100592, UF150675, A. B. C.) for financial support.Notes and references
- (a) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, 3rd edn, 2022 Search PubMed; (b) J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- M. Denis and S. M. Goldup, Nat. Rev. Chem, 2017, 1, 0061 CrossRef CAS.
- For selected examples see: (a) H. Han, J. S. W. Seale, L. Feng, Y. Qiu and J. F. Stoddart, J. Polym. Sci., 2023, 61, 881–902 CrossRef CAS; (b) A. de Juan, D. Lozano, A. W. Heard, M. A. Jinks, J. M. Suarez, G. J. Tizzard and S. M. Goldup, Nat. Chem., 2022, 14, 179–187 CrossRef CAS PubMed; (c) S. Rashid, Y. Yoshigoe and S. Saito, RSC Adv., 2022, 12, 11318–11344 RSC; (d) A. Acevedo-Jake, A. T. Ball, M. Galli, M. Kukwikila, M. Denis, D. G. Singleton, A. Tavassoli and S. M. Goldup, J. Am. Chem. Soc., 2020, 142, 5985–5990 CrossRef CAS PubMed; (e) J. E. M. Lewis, J. Winn, L. Cera and S. M. Goldup, J. Am. Chem. Soc., 2016, 138, 16329–16336 CrossRef CAS PubMed; (f) L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138(4), 1366–1376 CrossRef CAS PubMed; (g) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC; (h) Z. Baranová, H. Amini, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2014, 33, 6746–6749 CrossRef; (i) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901l RSC; (j) K. D. Hänni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; (k) Y. Sato, R. Yamasaki and S. Saito, Angew. Chem., Int. Ed., 2009, 48, 504–507 CrossRef CAS PubMed; (l) V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed; (m) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- (a) Q. Liang, K. Hayashi and D. Song, ACS Catal., 2020, 10, 4895–4905 CrossRef CAS; (b) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC; (c) Y. Zhou, Y. Zhang and J. Wang, Org. Biomol. Chem., 2016, 14, 6638–6650 RSC.
- (a) T. M. Hood and A. B. Chaplin, Dalton Trans., 2021, 50, 2472–2482 RSC; (b) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Chem.–Eur. J., 2020, 26, 14715–14723 CrossRef CAS PubMed; (c) C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed.
- T. M. Hood and A. B. Chaplin, Dalton Trans., 2020, 49, 16649–16652 RSC.
- T. M. Hood, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2077–2086 RSC.
- (a) Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, 2012 Search PubMed; (b) D. Steinborn, Fundamentals of Organometallic Catalysis, John Wiley & Sons, 2011 Search PubMed.
- M. R. Gyton, T. M. Hood and A. B. Chaplin, Dalton Trans., 2019, 48, 2877–2880 RSC.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- P. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, 2012 Search PubMed.
- J. H. Rivers and R. A. Jones, Chem. Commun., 2010, 46, 4300–4302 RSC.
- C. A. Schalley, P. Ghosh and M. Engeser, Int. J. Mass Spectrom., 2004, 232, 249–258 CrossRef CAS.
- T. R. Hoye, B. M. Eklov and M. Voloshin, Org. Lett., 2004, 6, 2567–2570 CrossRef CAS PubMed.
- G. L. Parker, R. Van Lommel, N. Roig, M. Alonso and A. B. Chaplin, Chem.–Eur. J., 2022, 28, e202202283 CrossRef CAS PubMed.
- R. M. Friedrich, J. Q. Bell, A. Garcia, Z. Shen and G. K. Friestad, J. Org. Chem., 2018, 83, 13650–13669 CrossRef CAS PubMed.
- E. Neumann and A. Pfaltz, Organometallics, 2005, 24, 2008–2011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data and NMR stack plots. CCDC 2063081. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00566j |
| This journal is © The Royal Society of Chemistry 2024 |