
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of length of branched alkyl side chains on thiazolothiazole-based small molecular acceptors in non-fullerene polymer solar cells†
Wenhong Peng *ac,
Jiyu Xionga,
Tao Chena,
Dong Zhaoa,
Jinran Liua,
Ning Zhanga,
Yefang Tenga,
Junting Yu*b and
Weiguo Zhu*b
*ac,
Jiyu Xionga,
Tao Chena,
Dong Zhaoa,
Jinran Liua,
Ning Zhanga,
Yefang Tenga,
Junting Yu*b and
Weiguo Zhu*b
aSchool of Materials Engineering, Changzhou Vocational Institute of Industry Technology, Changzhou, 213164, China. E-mail: pengwh88@126.com
bSchool of Materials Science and Engineering, Jiangsu Engineering Research Center of Light-Electricity-Heat Energy-Converting Materials and Applications, Jiangsu Collaborative Innovation Center of Photovoltaic Science and Engineering, Jiangsu Key Laboratories of Environment-Friendly Polymers, National Experimental Demonstration Center for Materials Science and Engineering, Changzhou University, Changzhou, 213164, China. E-mail: zhuwg18@126.com
cHunan Provincial Key Laboratory of Environmental Catalysis & Waste Recycling, School of Materials and Chemical Engineering, Hunan Institute of Engineering, Xiangtan, 411104, China
First published on 8th March 2024
Abstract
It has been reported that the length of branched alkyl side chains on fused-ring electron acceptors confers different impacts on properties versus solubility of BJH blends. However, because this impact on a non-fused acceptor backbone has rarely been studied, we examined the impact of molecular optimization from alkyl chain tuning based on non-fused thiazolothiazole small-molecule acceptors. The length of the side chain on the thiophene bridge was modified from 2-butyloctyl to 2-ethylhexyl, which corresponds to small molecules TTz3(C4C6) and TTz3(C2C4), respectively. Compared with the reported TTz3(C6C8) with long alkyl side chains, TTz3(C4C6) and TTz3(C2C4) exhibited stronger molecular aggregation, higher absorption coefficients, and greater redshifted UV absorption. Unexpectedly, after the alkyl chain was slightly shortened in this type of acceptor system, devices were successfully fabricated, but it was necessary to reduce the blending concentration at low rotation speeds due to the sharp decrease in the solubility of corresponding acceptor materials. Thus, the obtained unfavorable thickness and morphology of the active layer caused a decrease in Jsc and FF. As a consequence, TTz3(C4C6)- and TTz3(C2C4)-based devices showed an unsatisfactory power conversion efficiency of 6.02% and 2.71%, respectively, when donors were paired with the wide bandgap donor J71, which is inferior to that of TTz3(C6C8)-based devices (8.76%). These results indicate that it is challenging to determine the limit of the adjustable range of side chains to modify non-fused thiazolothiazole small-molecule acceptors for high-performance non-fullerene solar cells.
Introduction
In the development of organic photovoltaic cells, there has been a great deal of research in recent years on non-fullerene organic solar cells (OSCs) using a non-fullerene derivative as the acceptor component.1–4 Non-fullerene acceptors including polymeric and small-molecule acceptors have emerged as promising alternatives to replace fullerene derivatives. The power conversion efficiency (PCE) of non-fullerene OSCs based on small-molecule acceptors has increased by over 18% through versatile molecular engineering and chemical modification of acceptors.5–12 In particular, the structural modification of fused-ring electron acceptors of acceptor–donor–acceptor (A–D–A)-type planar molecules and A–DA′D–A-type Y-series molecules can be summarized as core engineering, side-chain engineering, π-bridge and terminal engineering, as well as asymmetric strategy.13–16Side-chain engineering is a simple and effective method to manipulate the performance of electron acceptors, including optical and electrical properties, molecular arrangement, and crystalline and aggregation properties.1,17 For instance, by replacing the alkoxy side chains of a naphtho[1,2-b:5,6-b′]dithiophene core with alkyl side chains, the red-shifted absorption, stronger π–π stacking and higher electron mobility of the resulting A–D–A type molecule named IOIC3 yielded a higher PCE of 13.1%.18 Zheng and co-workers reported an M-series acceptor M3 by tailoring the bulkiness of neighboring side chains, and the molecular orientation of the resulting molecule was altered from edge-on (M32) to face-on (M3), which improved the vertical charge transport so that a PCE of 16.66% was achieved with a largely enhanced FF.19
Moreover, in recent years, there has been a systematic study of side chains, including N-alkyl chains and those on the lateral TT unit of Y6.20 Zou and co-workers found that alkyl chains with a 3-position branch of the Y6 core will endow the corresponding acceptor, N3, with increased solubility and more optimal BJH morphology, resulting in a higher PCE in binary and ternary devices and an efficiency of 16.42%.21 Another efficient Y-series acceptor reported by Hou and co-workers, namely BTP-eC9, in which a set of alkyl chain optimizations was produced by elongating the N-alkyl from 2-ethylhexyl (EH) to 2-butyloctyl (BO) and shortening the lateral undecyl into nonyl on the TT unit, provided a high PCE of 17.8% along with an exceptionally high FF of 81.1%.22 Notably, there are numerous reports of side-chain engineering that incorporate IDT-series, M-series and Y-series fused-ring electron acceptors; however, studies on non-fused electron acceptors using this strategy are rare.
The thiazolothiazole (TTz) unit is a simple, rigid and coplanar building block that is widely applied for wide bandgap polymer donors. Additionally, given weak electron-deficient characteristics and highly extended π-electron system, polymer donors based on the TTz unit can be well-matched with non-fullerene acceptors so that devices acquire high efficiencies greater than 18%.23–28 In 2018, a series of efficient TTz-based electron acceptors (TTz1, TTz2, TTz3(C6C8)(TTz3) and TTz4) with simple structures was first reported by our group and binary devices based on TTz3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :J71 delivered a PCE of nearly 9% with a high Jsc of 16.12 mA cm−2 and a bandgap of 1.42 eV.29,30
:J71 delivered a PCE of nearly 9% with a high Jsc of 16.12 mA cm−2 and a bandgap of 1.42 eV.29,30
Further narrowing of the bandgap for absorption in the NIR region would further increase the efficiency. Herein, to understand the effect of side chain engineering in non-fused electron acceptors, fine tuning of the alkyl chains of thiazolothiazole-based electron acceptors was performed to enable full exploration of the photovoltaic potential. In this work, we employed a non-fused acceptor TTz3(C6C8) as the model material to construct acceptors TTz3(C4C6) and TTz3(C2C4) bearing different lengths of branched alkyl side chains, and effects on solubility, thermal stability, crystallinity, and photovoltaic performance were investigated. The chemical structures of three acceptors that possess the same backbone and essentially a similar molecular structure are shown in Chart 1.
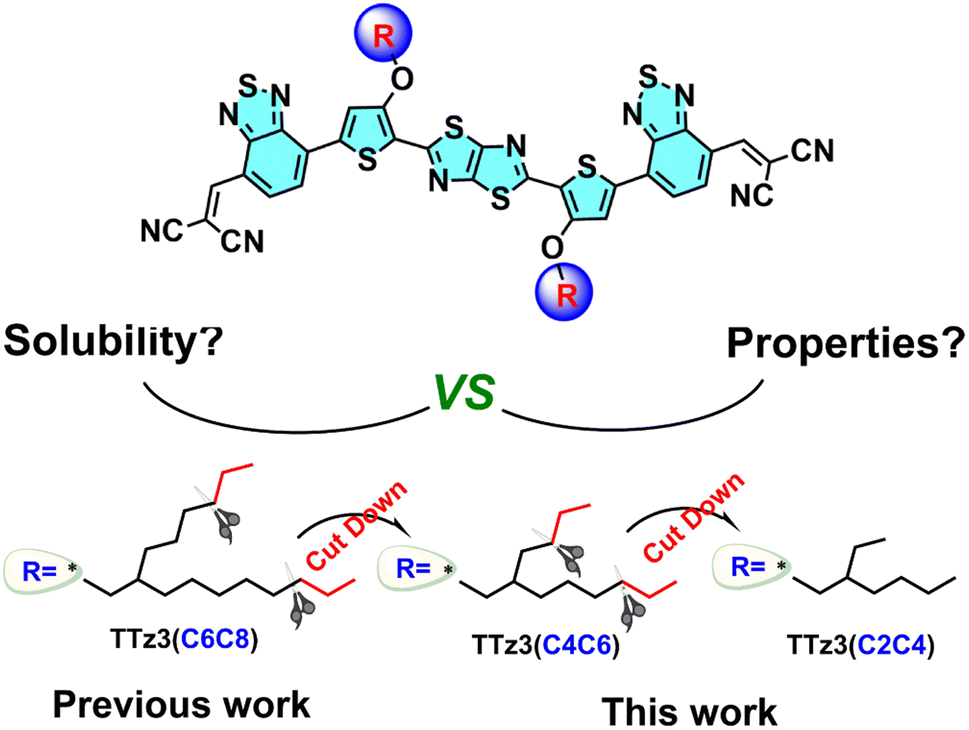 | ||
| Chart 1 Chemical structures of acceptors TTz3(R) bearing different branched alkyl side chains. | ||
The acceptor TTz3(C2C4) with the shortest alkyl side chain showed a more red-shifted absorption with a narrow bandgap of 1.36 eV. Compared with TTz3(C6C8), TTz3(C4C6) and TTz3(C2C4) exhibited a lower optical bandgap, a down-shifted lowest unoccupied molecular orbital (LUMO) level, a higher absorption coefficient and stronger molecular aggregation. However, the TTz3(C4C6)- and TTz3(C2C4)-based devices blended with J71 as the polymer donor displayed decreasing PCEs of 6.02% and 2.71% (8.76% for TTz3(C6C8)), respectively, due to the greatly deteriorated solubility. This work indicates that the selection of lateral side chain lengths of thiazolothiazole-based non-fused acceptors is delicate and sensitive to the solubility, miscibility, and the formation of phase separation. Therefore, poor choices may cause unfavorable blend morphology and poor solution processability.
Results and discussion
Synthesis, characterization and thermal stability
The synthesis route of TTz3(C4C6) and TTz3(C2C4) are depicted in Scheme 1. The synthesis method refers to the synthesis of TTz3(C6C8) described in our previous work,30 and corresponding reaction details are shown in the ESI Experimental section.† The results indicate that the length of the branched alkyl side chains has little effect on the synthesis of the intermediate. However, their difference in solubility caused by the length of alkyl chain is gradually reflected with the increasing conjugated degree of the molecules, in that the shorter the alkyl chain, the lower the solubility of monomer 7-R. Therefore, a greater amount of reaction solvent is required for the synthesis of corresponding small molecule TTz3(R), and moreover, the final yield will be lower. Moreover, the length of the alkyl chain has a great influence on the purification and structural characterization of small molecules. The purification of TTz3(C4C6) and TTz3(C2C4) is mainly based on deposition (chloroform/acetone), Soxhlet extraction (acetone) and recrystallization (chloroform/acetone), while the column chromatography separation process was difficult to implement. Finally, these compounds were confirmed by 1H NMR and time-of-flight mass spectrometry, as shown in Fig. S1–S14.† Relatively low NMR spectroscopy peaks of the acceptor with short alkyl chain were obtained. The experimental result indicated that the solubility of thiazolothiazole-based small molecules is poor, and they are strongly dependent on the alkyl chain. TTz3(C4C6) and TTz3(C2C4) were difficult to process from common solvents, such as chloroform (CF), chlorobenzene (CB), which would lead to an inability by these acceptors to exhibit intrinsic photovoltaic properties.31,32 | ||
| Scheme 1 Synthetic routes for TTz3(C4C6) and TTz3(C2C4). | ||
Fourier transform infrared (FT-IR) spectroscopy was employed for the analysis of the acceptor structure, and its characteristic absorption peaks are shown in Fig. S15 and S16.† The characteristic FT-IR peaks of two small molecules are basically the same due to their same molecular skeleton and characteristic groups. Herein, in the IR spectra, the characteristic peaks are located at 3000–2800 cm−1, which is the stretching vibration of the C–H at the alkyl chain. The characteristic peak at approximately 2223 cm−1 is generated by the stretching vibration of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N in the end group. Moreover, the characteristic peak located at 1900–1200 cm−1 represents the stretching vibration of C
N in the end group. Moreover, the characteristic peak located at 1900–1200 cm−1 represents the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN in the molecule and skeletal vibration of the aromatic ring. The complex peak at <1650 cm−1 is the C–O stretching vibration and C–H deformation vibration. These results are helpful to confirm that the two acceptors were successfully synthesized.
C and CN in the molecule and skeletal vibration of the aromatic ring. The complex peak at <1650 cm−1 is the C–O stretching vibration and C–H deformation vibration. These results are helpful to confirm that the two acceptors were successfully synthesized.
The thermal stability of the acceptors TTz3(R) was evaluated by thermogravimetric analysis (TGA) in N2 with a heating ramp of 10 °C min−1, and corresponding TGA curves are shown in Fig. 1a. For visual comparison, the relevant performance curves of small molecule TTz3(C6C8) were also drawn in this study. The decomposition temperature (Td) at 5% loss of initial weight was recorded at 344 °C for TTz3(C4C6) and 364 °C for TTz3(C2C4), which is 2 and 22 °C higher than that of TTz3(C6C8), respectively. When heated to 600 °C, the mass residue also increased with the shortening of the alkyl chain. In particular, TTz3(C2C4) exhibited significantly enhanced thermal stability, revealing that there is a significant impact of alkyl chain length on the thermal stability of small molecules.33–35
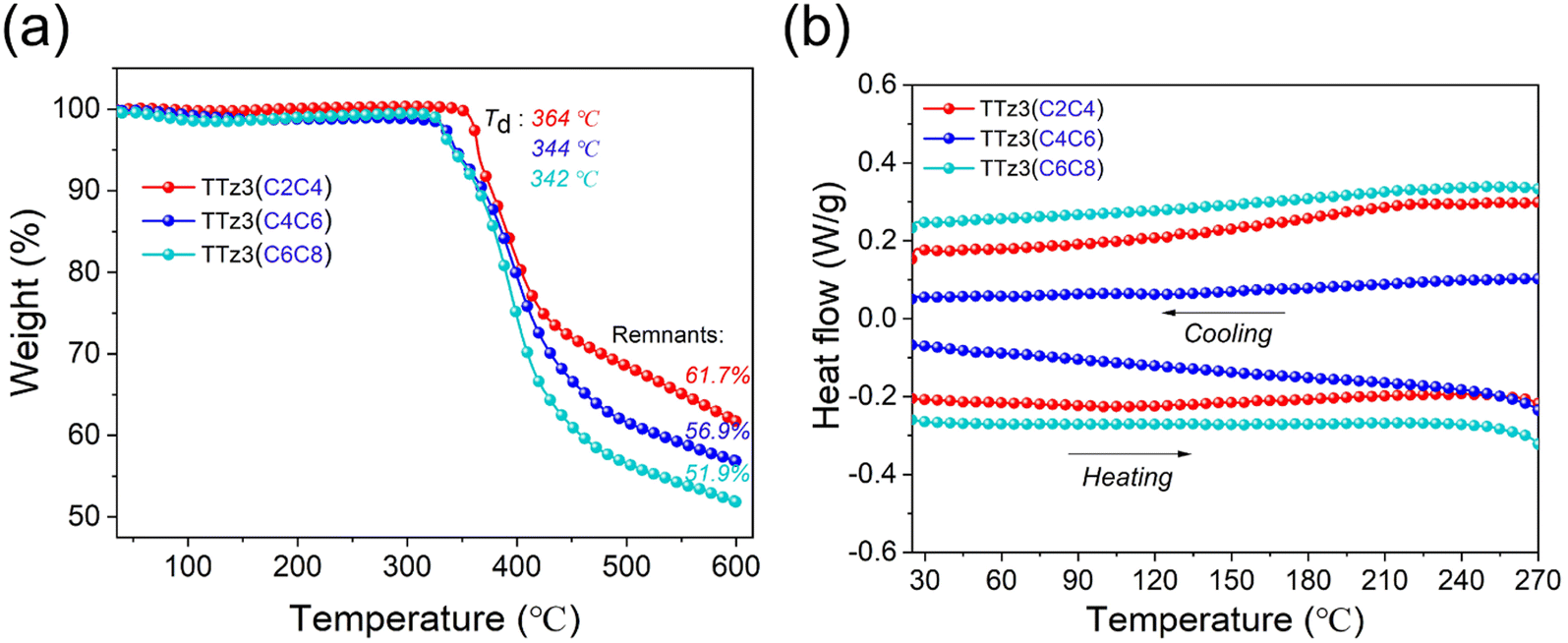 | ||
| Fig. 1 (a) Thermogravimetric analysis and (b) DSC curves of acceptors TTz3(R) in nitrogen atmosphere. | ||
In the differential scanning calorimetry (DSC) measurement, no endotherm or exotherm transitions were observed for TTz3(C4C6) and TTz3(C2C4) from the entire scanning range of repeated heating–cooling DSC cycles between room temperature and 270 °C, and these results were similar to those of TTz3(C6C8) (Fig. 1b). Generally, the crystallinity of compounds depends on the length of the alkyl-substituted chain, and the molecular crystallinity increases with the shortening of the alkyl chain.36,37 However, in this TTz3(R) system based on the thiazolothiazole unit, no glass transition process was observed when the alkyl chain of small molecules was shortened, which resulted in difficult dissolution in conventional solvents.
Electrochemical and optical properties
Electrochemical cyclic voltammetry (CV) measurements were performed to evaluate the energy levels of TTz3(C4C6) and TTz3(C2C4) (Fig. 2a), and corresponding data are listed in Table 1. The Eox/Ered of TTz3(C4C6) and TTz3(C2C4) were determined to be 0.65/−0.89 V and 0.64/−0.85 V, respectively. Correspondingly, their HOMO/LUMO levels were −5.45/−3.91 eV and −5.44/−3.95 eV. Clearly, compared with TTz3(C6C8), the HOMO levels of TTz3(C4C6) and TTz3(C2C4) were all upregulated, while the LUMO levels were all downregulated (Fig. 2b). Therefore, electrochemical bandgaps were all reduced, which is beneficial to the redshifted absorption spectrum. However, a relatively high-lying HOMO level indicates a lower Voc, suggesting that the orbital energy levels of non-fused small molecules are easily affected by the length and steric hindrance of side chains.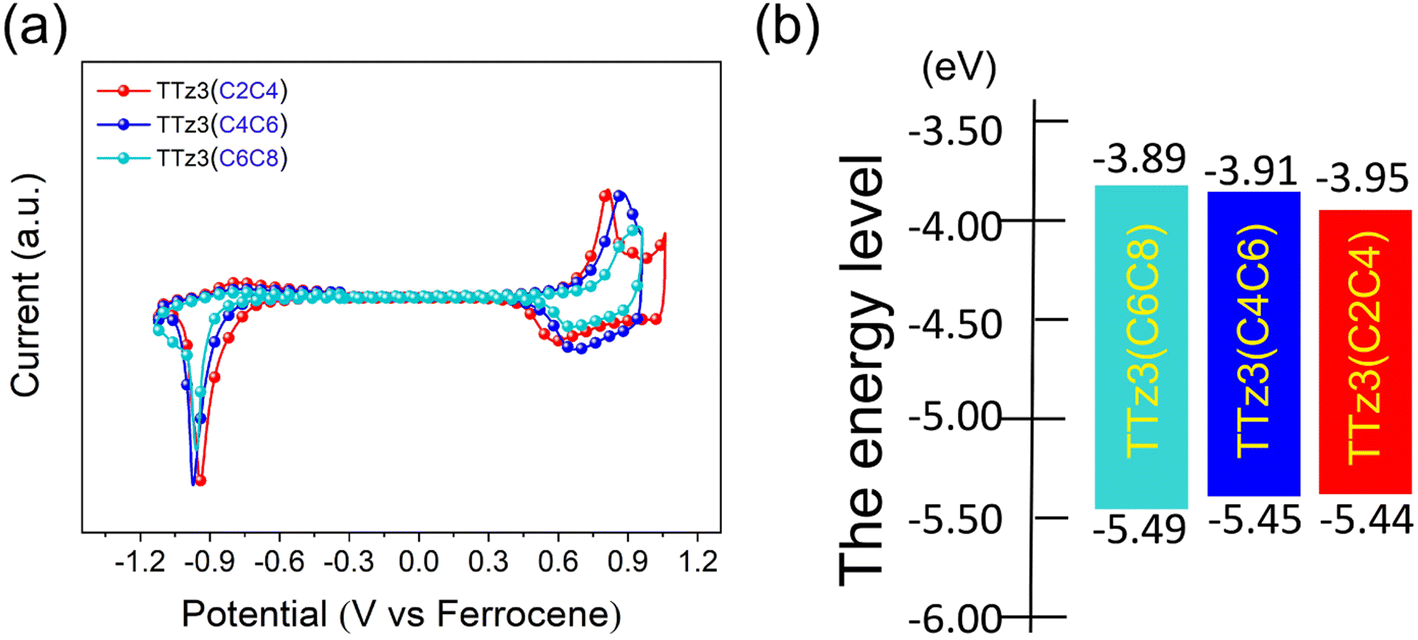 | ||
| Fig. 2 (a) Cyclic voltammetry of acceptors TTz3(R) in chloroform solution, with ferrocene as the standard. (b) Energy levels of acceptors TTz3(R). | ||
| Acceptors | λmax (nm) | λonsetc (nm) | Eoptgd (eV) | Eox/Erede (V) | EHOMO/ELUMOf (eV) | Eecgg (eV) | εmax (M−1 cm−1) | |
|---|---|---|---|---|---|---|---|---|
| Solutiona | Filmb | |||||||
| a Measured in 10−6 M CHCl3 solution.b Measured as a neat film cast from 3 mg per mL CHCl3 solution.c Obtained from the onset wavelength of the film.d Evaluated by Eoptg = 1240/λonset.e Onset potentials of oxidation and reduction as referenced to ferrocene.f EHOMO = −(Eox + 4.8 eV) and ELUMO = −(Ered + 4.8 eV). The formal potential for ferrocene vs. Ag/AgCl is 0.43 V.g Calculated according to ELUMO − EHOMO. | ||||||||
| TTz3(C6C8) | 390, 661 | 421, 691, 740 | 875 | 1.417 | 0.69/−0.91 | −5.49/−3.89 | 1.60 | 6.49 × 104 |
| TTz3(C4C6) | 390, 661 | 422, 710, 752 | 883 | 1.404 | 0.65/−0.89 | −5.45/−3.91 | 1.54 | 6.67 × 104 |
| TTz3(C2C4) | 391, 661 | 423, 718, 755 | 915 | 1.355 | 0.64/−0.85 | −5.44/−3.95 | 1.49 | 6.81 × 104 |
The absorption spectra of the acceptors measured in solution and as thin films are shown in Fig. 3, and the data are summarized in Table 1. As displayed in Fig. 3a, three acceptors exhibited the same absorption in the 300–800 nm region with a maximum absorption peak at 661 nm in chloroform solution. Interestingly, slightly different characteristics are displayed in the film absorption of the acceptors. A slight red-shift of the absorption spectrum occurred with the shortening of the alkyl chain. The absorption edges of two acceptors were separately 883 nm and 915 nm, from which optical bandgap (Eoptg) values were determined to be 1.40 eV and 1.36 eV for TTz3(C4C6) and TTz3(C2C4), respectively. As the alkyl chain shortened, Eoptg gradually decreased, indicating that stronger aggregation and π–π packing interaction could be achieved by modifying the length of the alkyl side chain. Furthermore, the acceptor aggregation caused a slight increase in the absorption coefficients of TTz3(C4C6) and TTz3(C2C4) from 6.67 × 104 to 6.81 × 104 M−1 cm−1, respectively (Fig. 3b).
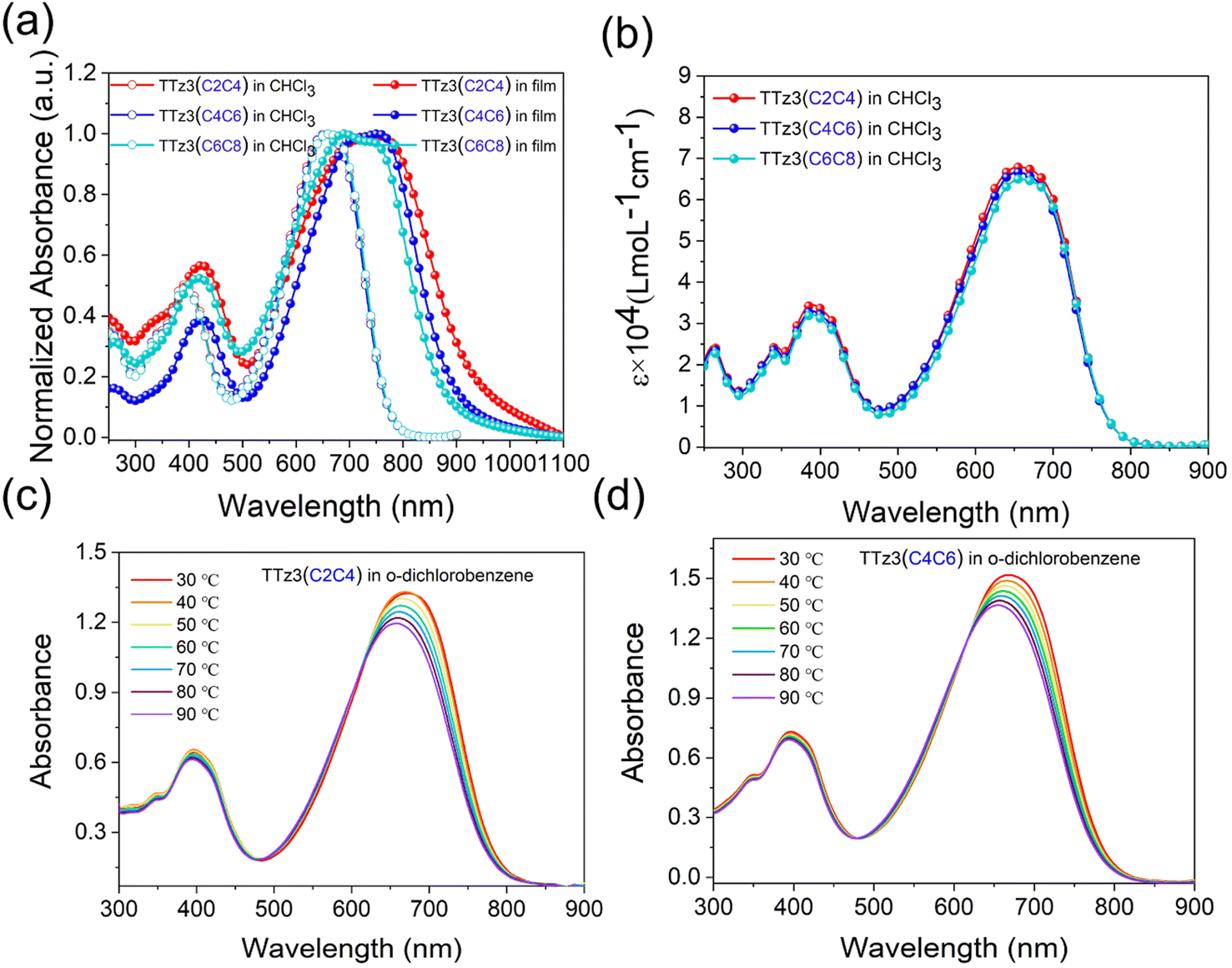 | ||
| Fig. 3 (a) Normalized UV-Vis absorption spectra of acceptors TTz3(R) in CHCl3 and thin films. (b) Molecular absorption coefficients in CHCl3. (c and d) Temperature-dependent UV-Vis absorption spectra of TTz3(C4C6) and TTz3(C2C4) in dilute o-dichlorobenzene solution. | ||
To further investigate the molecular aggregation of these acceptors, temperature-dependent aggregation absorption spectra measurements were performed in dilute o-dichlorobenzene solution, as shown in Fig. 3c and d. When the solution began to cool from 90 to 30 °C with an interval of 10 °C, the maximum absorption peak gradually increased and red-shifted to longer wavelengths for both acceptors, but no temperature-dependent aggregation effect was observed. However, when aggregation is too strong, it is often not conducive to the formation of well-defined heterojunction morphology with nanoscale control.
X-ray diffraction
Powder X-ray diffraction (PXRD) was conducted to obtain the patterns of acceptors and investigate their crystallinity, and corresponding results are presented in Fig. S17 and S18 and Table S1.† Three clear diffraction peaks (100, 200, and 010) were observed for TTz3(C6C8) and TTz3(C4C6). However, TTz3(C2C4) exhibited highly ordered diffraction patterns with four diffraction peaks (100, 200, 300, and 010). The (100) and (010) diffraction peaks of TTz3(C4C6) and TTz3(C2C4) are approximately located at 2θ = 6.59–5.91° and 25.85°, corresponding to the lamellar and π–π stacking distances of 13.41–14.93 Å and 3.44 Å, respectively. In comparison with TTz3(C6C8), the lamellar and π–π stacking distances clearly decreased for TTz3(C4C6) and TTz3(C2C4), indicating that fine-tuning the alkyl chains of thiazolothiazole-based electron acceptors resulted in an increase in the crystallinity, and this may contribute to the closer stacking in the film, which supports the red-shifted absorption.Photovoltaic properties
To demonstrate the potential impact of the length of the branched alkyl side chains on device performance, the devices were fabricated and examined under AM 1.5G solar illumination (100 mW cm−2). The optimal preparation conditions for the devices are listed in Table 2. According to the previous work,30 chloroform was used as the solvent for preparing devices, a J71:acceptor weight ratio of 1:2 was used, and the same proportion of additive (0.25% DIO) was added. However, during the experiment, it was found that the solubility significantly decreased with the shortening of the alkyl side chain of the acceptor, resulting in a donor–acceptor blend concentration for the preparation of the device that could only be reduced.
:TTz3(R) blends
| Active layer | D/A ratio | Solvent | Total concentration | DIO | Rotating speed | Thickness |
|---|---|---|---|---|---|---|
| J71:TTz3(C6C8) |
1:2 |
Chloroform | 15 mg mL−1 | 0.25% | 3000 rpm | 100 nm |
| J71:TTz3(C4C6) |
1:2 |
Chloroform | 12 mg mL−1 | 0.25% | 2500 rpm | 100 nm |
| J71:TTz3(C2C4) |
1:2 |
Chloroform | 7.2 mg mL−1 | 0.25% | 1500 rpm | 65 nm |
J71:TTz3(C2C4) only reached 7.2 mg mL−1, which also increased the difficulty of device preparation. Moreover, the spin-coating rate was reduced to maintain a similar active-layer thickness to prevent the different donor–acceptor blend concentrations from influencing the photovoltaic performance of the device. After optimization, an active-layer thickness of ≈100 nm (prepared by spin-coating a solution of 12 mg mL−1 at 2500 rpm) was created for the TTz3(C4C6) device, while the TTz3(C2C4) device received a layer of ≈65 nm (prepared by spin-coating a solution of 7.2 mg mL−1 at 1500 rpm), which would be inconducive to the intrinsic photovoltaic performance of acceptors TTz3(C4C6) and TTz3(C2C4) due to the poor solution processability.
The current density–voltage (J–V) characteristics of optimized devices are exhibited in Fig. 4a, and photovoltaic parameters are summarized in Table 3. Under optimal conditions, TTz3(C4C6) offered a moderate PCE of 6.02% with a slightly reduced Voc of 0.84 V, a short-circuit current density (Jsc) of 13.31 mA cm−2 and a fill factor (FF) of 53.84%. TTz3(C2C4) offered a poor PCE of 2.71% with a significantly reduced Voc of 0.81 V, Jsc of 9.30 mA cm−2 and FF of 35.98% compared with TTz3(C6C8). The lower Voc afforded by TTz3(C4C6) and TTz3(C2C4) was ascribed to their low-lying LUMO level, and the decreasing Jsc and FF might result from unfavorable active layer morphology and undesirable charge transport ability caused by the poor solubility of the acceptor.
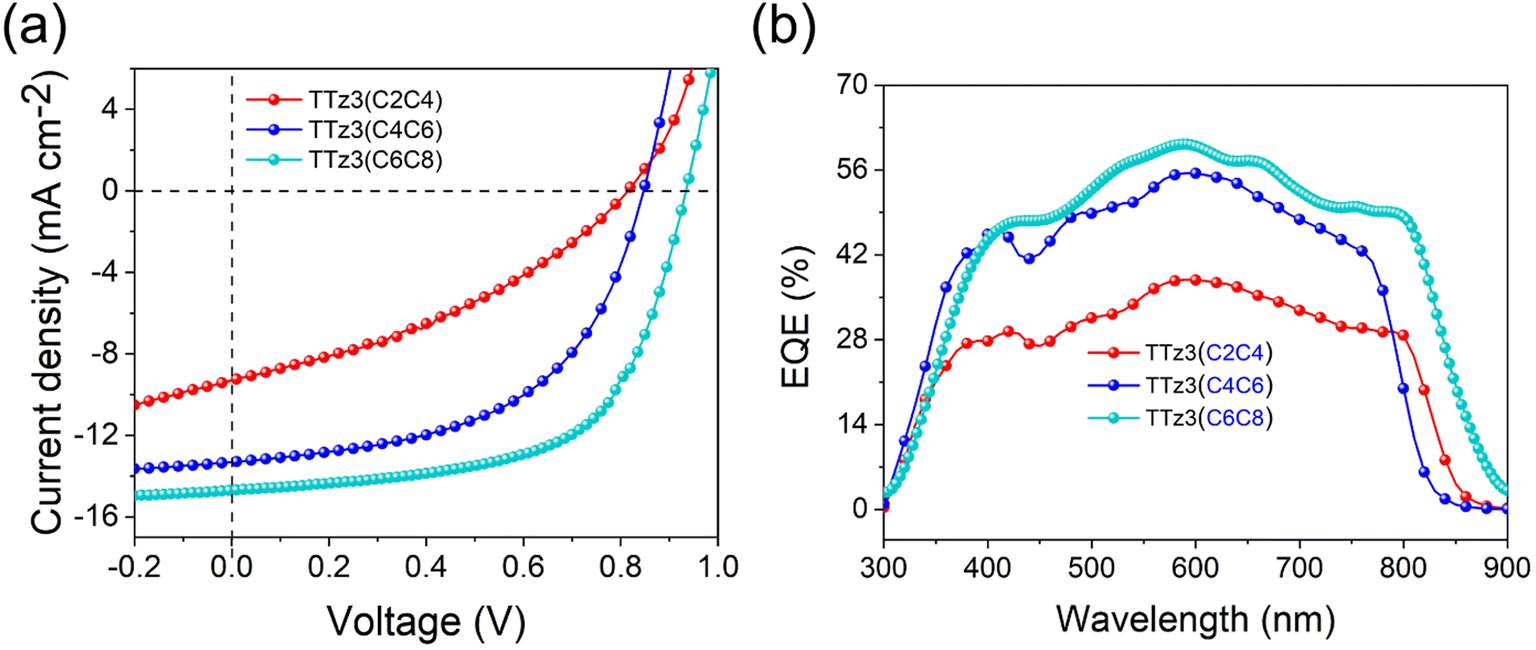 | ||
| Fig. 4 (a) J–V curves and (b) EQE spectrum for acceptor TTz3(R)-based devices. | ||
:J71 (2:1, w/w) devices measured under the illumination of AM 1.5G (100 mW cm−2)
We further note that TTz3(C4C6) and TTz3(C2C4) exhibited relatively higher series resistance (Rs) and lower shunt resistance (Rsh) as compared to TTz3(C6C8) when blended with J71. This implies that there is greater ohmic contact for the J71:TTz3(C6C8) active layer, which allows J71:TTz3(C6C8) devices to achieve a higher FF. The EQE spectra of the optimized devices are shown in Fig. 4b. All the devices covered a broad response ranging from 300 to 900 nm, due to the complementary absorption of the acceptors and J71. In accord with its highest Jsc, the J71:TTz3(C6C8) device showed the widest absorption range and maximum response value. The lower EQEs of TTz3(C4C6) and TTz3(C2C4) indicate the inefficient utilization of incident photons caused by the thickness and morphology of the active layer, which is adverse to the Jsc of the devices. Herein, the photo-to-current response value was not dominated by absorption range or intensity.
Carrier mobility
We also conducted charge carrier mobility measurements by the space charge limited current (SCLC) method, and single carrier devices were prepared at different total concentrations for efficient solution processing. As shown in Fig. 5, the estimated hole mobility (μh) and electron mobility (μe) for the J71:TTz3(C4C6) blend film are 1.61 × 10−4 and 6.18 × 10−5 cm2 V−1 s−1 (μh/μe = 2.61), respectively. The μh and μe values for the J71:TTz3(C2C4) blend film are 1.10 × 10−4 and 3.29 × 10−5 cm2 V−1 s−1 (Table S2†), respectively. In contrast to the J71:TTz3(C6C8) blend film, the acceptors exhibited inefficient charge transport properties due to the knock-on effects of poor solubility, which eventually resulted in the decreased Jsc and FF of the devices.
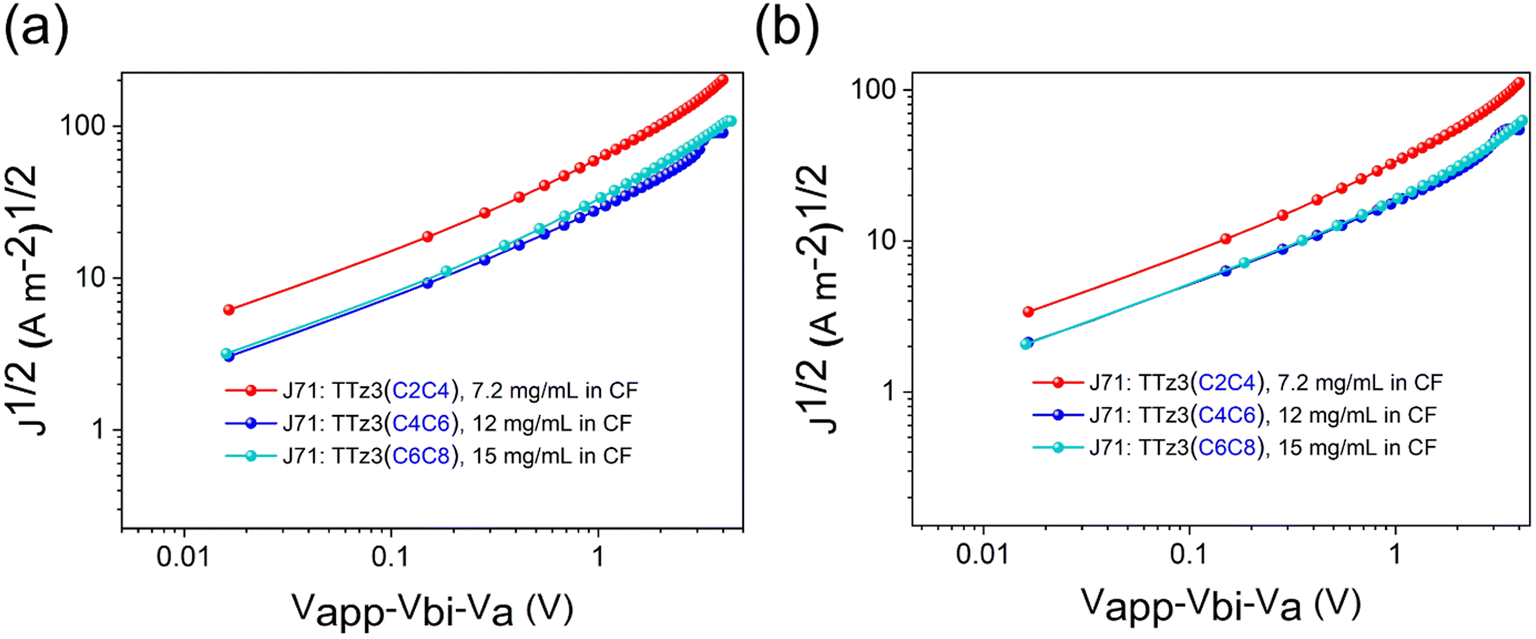 | ||
| Fig. 5 J–V plots of (a) hole mobilities and (b) electron mobilities measured by the SCLC method. | ||
Morphology
Considering that device performance is closely related to blend film morphology, atomic force microscopy (AFM) measurements were carried out to investigate the influences of different side chains of the acceptors on the surface morphology of J71:TTz3(R) blend films. As depicted in Fig. 6, the J71:TTz3(C4C6) and J71:TTz3(C2C4) blend films display a root-mean-square (RMS) roughness of 1.75 and 4.65 nm (Table S2†), respectively, indicating the smoother surface morphology of the blend film based on TTz3(R) with longer alkyl length. The rougher surface and formation of undesirably large aggregates of J71:TTz3(C2C4) blend film indicate enhanced aggregation characteristics and excessive phase separation without the excellent dual continuous interpenetrating network such as that in J71:TTz3(C6C8)-based film, which infers a lack of effective exciton diffusion, charge separation and transport, thus leading to the decrease in the Jsc, FF and PCE.
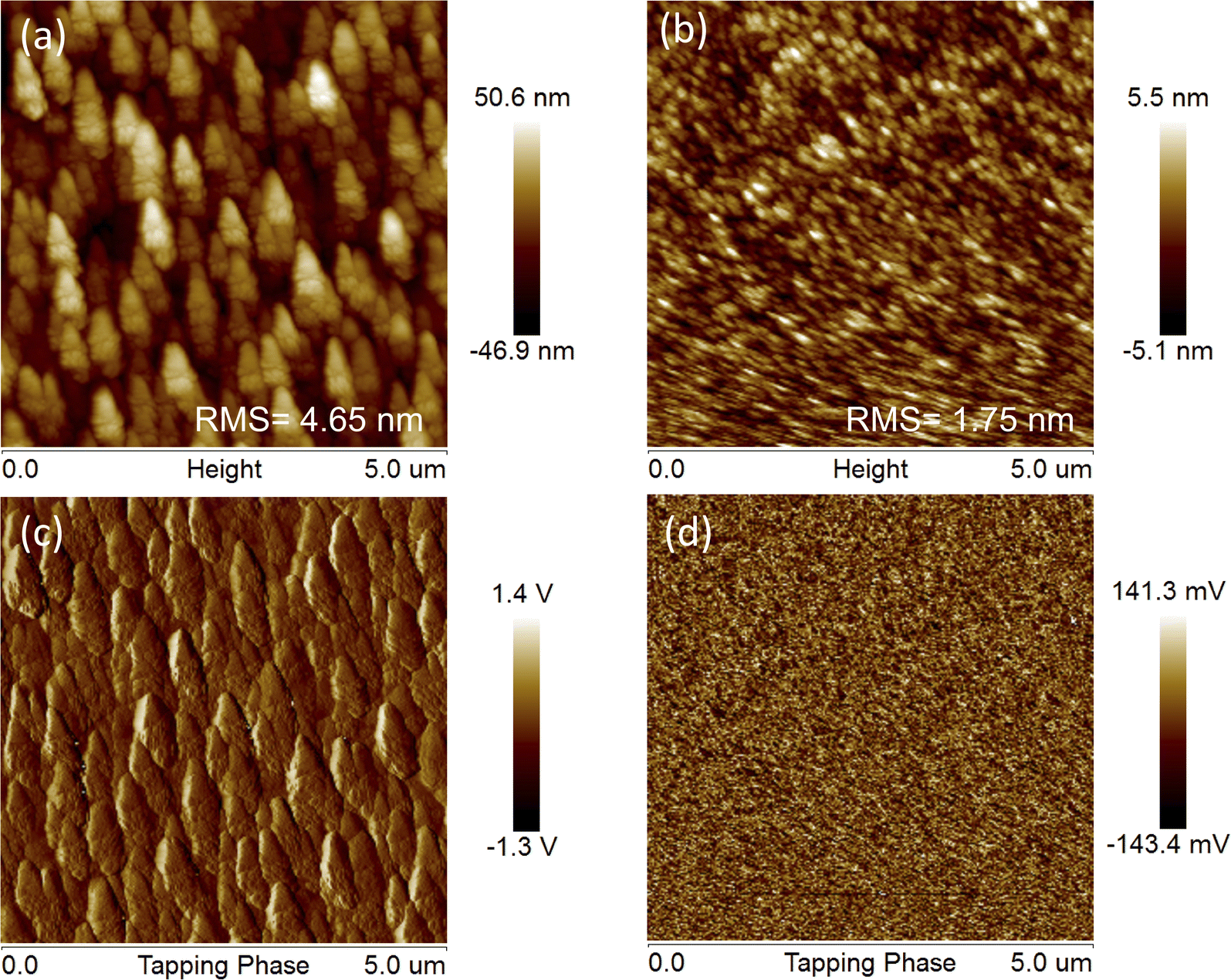 | ||
| Fig. 6 (a) AFM height and (c) phase images of J71:TTz3(C2C4) blends. (b) AFM height and (d) phase images of J71:TTz3(C4C6) blends. | ||
Conclusion
In summary, we designed and synthesized two thiazolothiazole-based non-fused small molecular acceptors, TTz3(C4C6) and TTz3(C2C4), which originated from TTz3(C6C8). The same conjugated skeleton was used, and the alkyl chain length was decreased from 2-butyloctyl to 2-ethylhexyl on the thiophene π-bridge unit, for non-fullerene PSC fabrication. The effects of alkyl chain length on the thermal stability, photophysics, electrochemistry and photovoltaic performance were investigated. We found that the two acceptors exhibited a decreased Eg, increased thermal stability, and obviously enhanced absorption intensity and aggregation in film as the alkyl chain was shortened. However, the modification of the alkyl chain resulted in poor solubility and down-shifted LUMO energy levels, which are unfavourable for a fine morphology by solution processing and would result in a high Voc. As a result, there was decreased PCE for TTz3(C4C6)- and TTz3(C2C4)-based devices blended with the polymer donor J71, from 6.02% to 2.71%, respectively. This study shows that there are significant impacts by the different alkyl chains on the physicochemical properties of small molecules, and further in-depth study of the structure–property relationship of thiazolothiazole-based non-fused small molecular acceptors is required to increase the PCE. If there is a decrease in the alkyl chain length of thiazolothiazole-based small-molecule acceptor materials, satisfactory solution-processing properties can be retained for device preparation, and it is likely that greater improvement in the performance of TTz-based acceptors could be obtained in the future.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We express our thanks to the financial support from National Natural Science Foundation of China (22275024 and 51673031), Natural Science Foundation of Hunan Province of China (2022JJ40119), Scientific Research Fund of Hunan Provincial Education Department (22B0731), Changzhou Leading Innovative Talent Introduction and Cultivation Project (CQ20230070), Natural Science Foundation of the Jiangsu Higher Education Institutions (23KJD430002), Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), The Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Jiangsu Provincial Talents Project of High-Level Innovation and Entrepreneurship.References
- J. Wang and X. Zhan, Acc. Chem. Res., 2021, 54, 132–143 CrossRef CAS PubMed.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- Q. Fan, Y. Li, U. A. Mendez-Romero, X. Guo, E. Wang and M. Zhang, Chem.–Asian J., 2019, 14, 3085–3095 CrossRef CAS PubMed.
- S. Li, W. Liu, C.-Z. Li, M. Shi and H. Chen, Small, 2017, 13, 1701120 CrossRef PubMed.
- W. Peng, Y. Lin, S. Y. Jeong, Z. Genene, A. Magomedov, H. Y. Woo, C. Chen, W. Wahyudi, Q. Tao, J. Deng, Y. Han, V. Getautis, W. Zhu, T. D. Anthopoulos and E. Wang, Nano Energy, 2022, 92, 106681 CrossRef CAS.
- T. Zhang, C. An, P. Bi, Q. Lv, J. Qin, L. Hong, Y. Cui, S. Zhang and J. Hou, Adv. Energy Mater., 2021, 11, 2101705 CrossRef CAS.
- C. Guo, Y. Fu, D. Li, L. Wang, B. Zhou, C. Chen, J. Zhou, Y. Sun, Z. Gan, D. Liu, W. Li and T. Wang, Adv. Mater., 2023, 35, 2304921 CrossRef CAS PubMed.
- Y. Cai, Y. Li, R. Wang, H. Wu, Z. Chen, J. Zhang, Z. Ma, X. Hao, Y. Zhao, C. Zhang, F. Huang and Y. Sun, Adv. Mater., 2021, 33, 2101733 CrossRef CAS PubMed.
- Q. Fan, R. Ma, J. Yang, J. Gao, H. Bai, W. Su, Z. Liang, Y. Wu, L. Tang, Y. Li, Q. Wu, K. Wang, L. Yan, R. Zhang, F. Gao, G. Li and W. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308307 CrossRef CAS PubMed.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2022, 6, 171–184 CrossRef CAS.
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, 2109516 CrossRef CAS PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 32, 2102420 CrossRef PubMed.
- C. Li, H. Fu, T. Xia and Y. Sun, Adv. Energy Mater., 2019, 9, 1900999 CrossRef.
- Q. Nie, A. Tang, Q. Guo and E. Zhou, Nano Energy, 2021, 87, 106174 CrossRef CAS.
- X. He, J. Xue and P. Guo, Guangzhou Chem. Ind., 2021, 49, 1–7 Search PubMed.
- A. Markina, K.-H. Lin, W. Liu, C. Poelking, Y. Firdaus, D. R. Villalva, J. I. Khan, S. H. K. Paleti, G. T. Harrison, J. Gorenflot, W. Zhang, S. D. Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, F. Laquai and D. Andrienko, Adv. Energy Mater., 2021, 11, 2102363 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, Y.-J. Pu and T. Park, Chem. Sci., 2021, 12, 14004–14023 RSC.
- J. Zhu, Y. Xiao, J. Wang, K. Liu, H. Jiang, Y. Lin, X. Lu and X. Zhan, Chem. Mater., 2018, 30, 4150–4156 CrossRef CAS.
- Y. Ma, M. Zhang, S. Wan, P. Yin, P. Wang, D. Cai, F. Liu and Q. Zheng, Joule, 2021, 5, 197–209 CrossRef CAS.
- W. Gao, F. R. Lin and A. K.-Y. Jen, Sol. RRL, 2022, 6, 2100868 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- R. Sun, Y. Wu, X. Yang, Y. Gao, Z. Chen, K. Li, J. Qiao, T. Wang, J. Guo, C. Liu, X. Hao, H. Zhu and J. Min, Adv. Mater., 2022, 34, 2110147 CrossRef CAS PubMed.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CrossRef CAS PubMed.
- Z. Cao, J. Chen, S. Liu, X. Jiao, S. Ma, J. Zhao, Q. Li, Y.-P. Cai and F. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 9545–9554 CrossRef CAS PubMed.
- B. Guo, W. Li, X. Guo, X. Meng, W. Ma, M. Zhang and Y. Li, Adv. Mater., 2017, 29, 1702291 CrossRef PubMed.
- G. Reginato, A. Mordini, L. Zani, M. Calamante and A. Dessì, Eur. J. Org Chem., 2016, 2016, 233–251 CrossRef CAS.
- Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087–3106 CrossRef CAS PubMed.
- W. Peng, G. Zhang, L. Shao, C. Ma, B. Zhang, W. Chi, Q. Peng and W. Zhu, J. Mater. Chem. A, 2018, 6, 24267–24276 RSC.
- W. Peng, G. Zhang, M. Zhu, H. Xia, Y. Zhang, H. Tan, Y. Liu, W. Chi, Q. Peng and W. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 48128–48133 CrossRef CAS PubMed.
- J. Liang, M. Pan, Z. Wang, J. Zhang, F. Bai, R. Ma, L. Ding, Y. Chen, X. Li, H. Ade and H. Yan, Chem. Mater., 2022, 34, 2059–2068 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS PubMed.
- H. Feng, X. Song, M. Zhang, J. Yu, Z. Zhang, R. Geng, L. Yang, F. Liu, D. Baran and W. Tang, Mater. Chem. Front., 2019, 3, 702–708 RSC.
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, D. Rodriquez, M. Finn, M. A. Alkhadra, J. M.-H. Wan, J. Ramírez, A. S.-C. Chiang, S. E. Root, S. Savagatrupa and D. J. Lipomi, Polym. Chem., 2018, 9, 4354–4363 RSC.
- J. Liang, M. Pan, Z. Wang, J. Zhang, F. Bai, R. Ma, L. Ding, Y. Chen, X. Li, H. Ade and H. Yan, J. Mater. Chem. A, 2017, 5, 15175–15182 RSC.
- C.-A. Chen, P.-C. Yang, S.-C. Wang, S.-H. Tung and W.-F. Su, Macromolecules, 2018, 51, 7828–7835 CrossRef CAS.
- C. R. Bridges, M. J. Ford, E. M. Thomas, E. M. Thomas, C. Gomez, G. C. Bazan and R. A. Segalman, Macromolecules, 2018, 51, 597–8604 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00572d. |
| This journal is © The Royal Society of Chemistry 2024 |