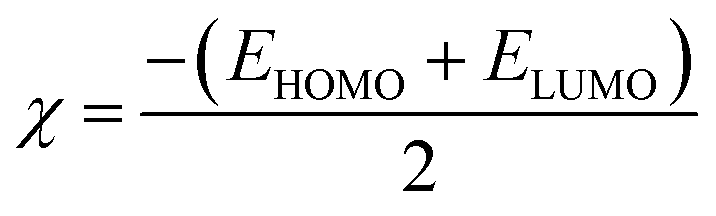DOI:
10.1039/D4RA00602J
(Paper)
RSC Adv., 2024,
14, 10464-10480
Novel pyrazole and imidazolone compounds: synthesis, X-ray crystal structure with theoretical investigation of new pyrazole and imidazolone compounds anticipated insecticide’s activities against targeting Plodia interpunctella and nilaparvata lugens†
Received
23rd January 2024
, Accepted 11th March 2024
First published on 2nd April 2024
Abstract
In this study, we synthesized (2-propoxyphenyl)(3-(p-tolyl)oxiran-2-yl)methanone through oxidizing the double bond of the respective chalcone via the Weitz–Scheffer epoxidation reaction. Additionally, the chalcone with an oxirane ring served as a fundamental building block for the synthesis of various pyrazole and imidazole derivatives, employing diverse nitrogen nucleophiles. All synthesized compounds were confirmed via analytical and spectroscopic analysis, such as FT-IR, 1H NMR, 13C NMR, and mass spectroscopy. Furthermore, all these nitrogen heterocycles were optimized via the DFT/B3LYP/6-31G(d,p) basis set and their physical descriptors were identified. Compound 11 was further confirmed using single-crystal X-ray diffraction with Hirshfeld analysis, and the results were correlated with the optimized structure by comparing their bond length and bond angle, which provided excellent correlation. Additionally, the insecticidal activities of the newly synthesized compounds were tested against P. interpunctella and Nilaparvata lugens. The heterocyclic compounds exhibited remarkable activity compared to the standard reference thiamethoxam. These findings were further confirmed through docking simulation with different proteins, namely PDBID 3aqy and 3wyw. The compounds interacted effectively within the protein pockets, displaying a higher binding energy with amino acids.
1. Introduction
The multifaceted activities of oxiranes and their derivatives in terms of anti-microbial, anti-cancer, and anti-tumor have been investigated by bio-chemists and chemists.1–5 For synthesis of oxiranes from chalcones and alkenes, numerous reagents and catalysts have been utilized.6 These epoxides are flexible chemicals that can easily undergo stereospecific ring-opening reactions to generate multifunctional molecules.7 Alkene epoxidation is one of the most widespread asymmetric transformations where epoxides serve as intermediates for bioactive target molecules that reflect the efficacy of current epoxidation protocols.8 Additionally, they serve as monomers for the construction of polymer materials, including epoxy resin and polyether.9 Through the epoxidation of α,β-unsaturated ketones, the ethylenic bridge was replaced with an oxirane ring.
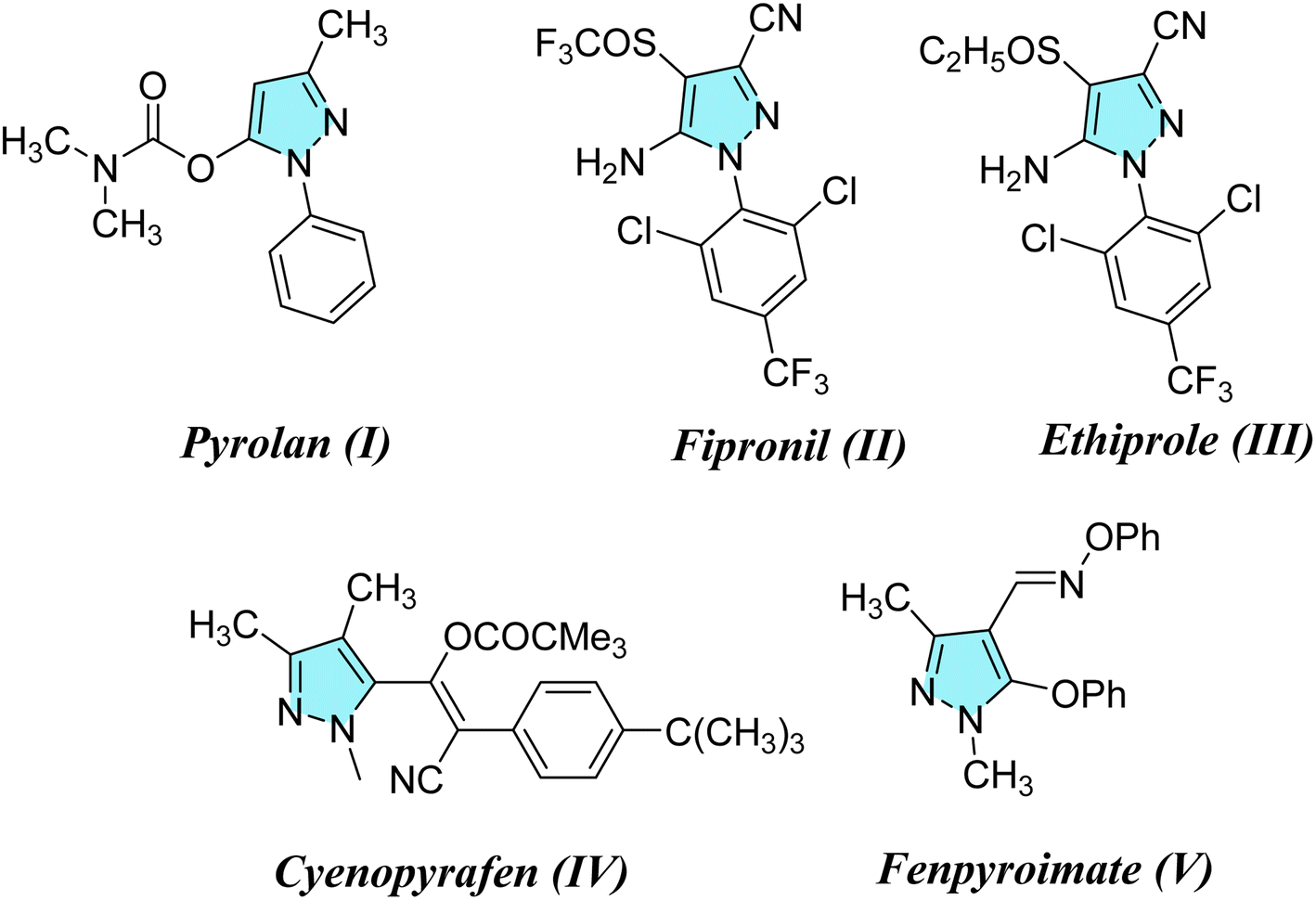
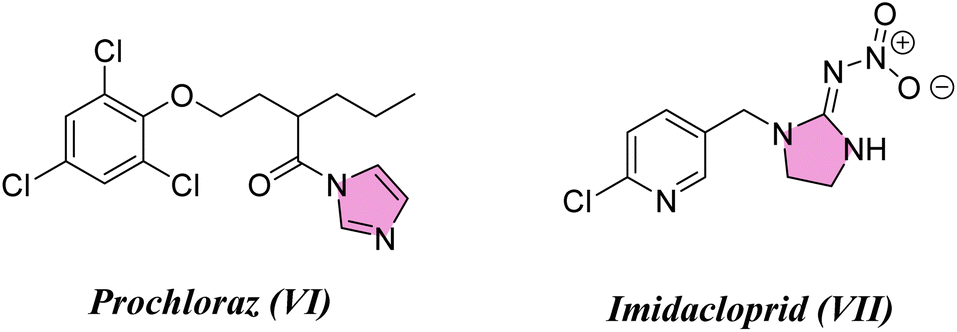
Considering the infestations of pests causing significant financial losses in storage and agricultural environments, crop horticulture and agricultural protection require insecticides that are important, effective, and widely utilized to achieve high quality and improved yields of produce by fighting insect pests.10 Many insect pests attack rice crops throughout the growing season—from seed germination till harvesting. Fighting insects is crucial to reduce food loss from stored items due to insect attacks. P. interpunctella was one of seven insect species whose eggs were found in brown rice. Prenol, isoprenol, dimethyl disulfide, and dimethyl trisulfide were among the volatile substances produced as a result of the P. interpunctella infestation of brown rice. These volatiles have the potential to serve as early biomarkers of insect detection in brown rice.11 Planthoppers or Nilaparvata lugens are the most destructive insect pests because they consume rice plant sap, thus causing plant dehydration and transmitting viral diseases.12 There is a worldwide distribution of these insects. With long-term exposure, insects have developed resistance to a majority of chemical classes used in animal agriculture and public health. Insecticides are essential for the management of insects and pests and are regarded as one of the most significant barriers to applying unique chemical and biological features as well as good safety profiles with relatively low risk for non-target organisms in the environment. Their investigated exposure strategies were flexible and had a low application rate as well as excellent absorption and translocation in plants. Thus, there is a need to find insecticides that combat these insects.13–15 Many pyrazole compounds have been reported to be effective insecticides. Pyrolan (I), fipronil (II), ethiprole (III), cyenopyrafen (IV) and fenpyroximate (V) (Fig. 1) are examples of pyrazole insecticides with high insecticidal activity on the market today.16–19 On the other hand, in agrochemicals, imidazole is widely incorporated as a nitrogen-containing aromatic heterocycle, as shown in Fig. 2.20–22 Prochloraz (VI), for example, is an imidazole fungicide that is widely used in gardening and agriculture. It is applied to wheat, barley, mushrooms, cherries, golf course turf, and flower production. Imidacloprid (IMI) (VII) is a neonicotinoid insecticide that is used for crop protection all over the world. In this study, various pyrazole and imidazole derivatives were successfully synthesized and fully characterized using analytical and spectroscopic techniques. Furthermore, theoretical studies were carried out for nitrogen heterocycles to determine their physical properties. Moreover, this study examines the insecticidal activities of newly synthesized pyrazole and imidazole derivatives against P. interpunctella and Nilaparvata lugens. The results were further supported by docking simulations involving various proteins.
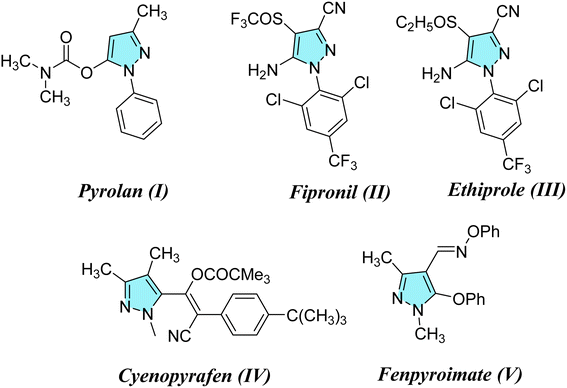 |
| | Fig. 1 Agrochemicals containing pyrazole scaffolds. | |
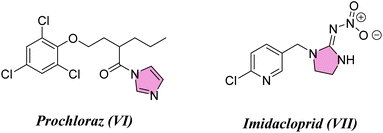 |
| | Fig. 2 Agrochemicals containing imidazole scaffolds. | |
2. Results and discussion
2.1. Chemistry section
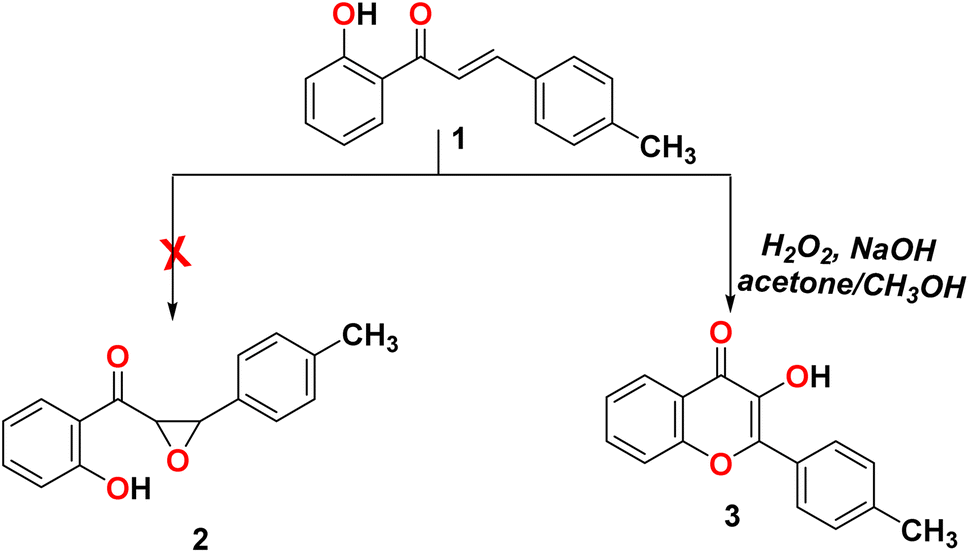
The reaction of 2-(5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (1) with sodium hydroxide/hydrogen peroxide-mediated in acetone and methanol as a solvent did not form the corresponding epoxy compound 2 and formed the C-4′-substituted flavonol 3, as shown in Scheme 1.23
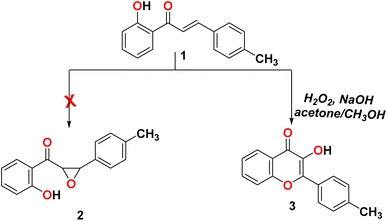 |
| | Scheme 1 Reactivity of 2-(5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenol (1) with H2O2. | |
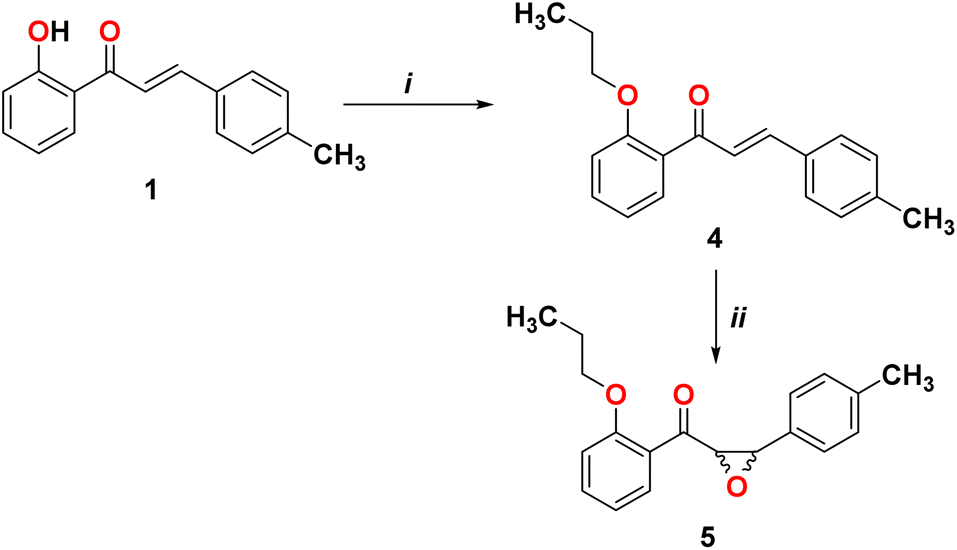
In order to form the epoxide compound, we blocked the OH group in the chalcone compound 1 using alkyl halide, as shown in Scheme 2.
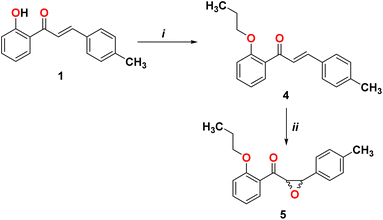 |
| | Scheme 2 Reagents and conditions. (i) Br(CH2)2CH3, K2CO3, and DMF; (ii) H2O2, NaOH, acetone, and CH3OH. | |
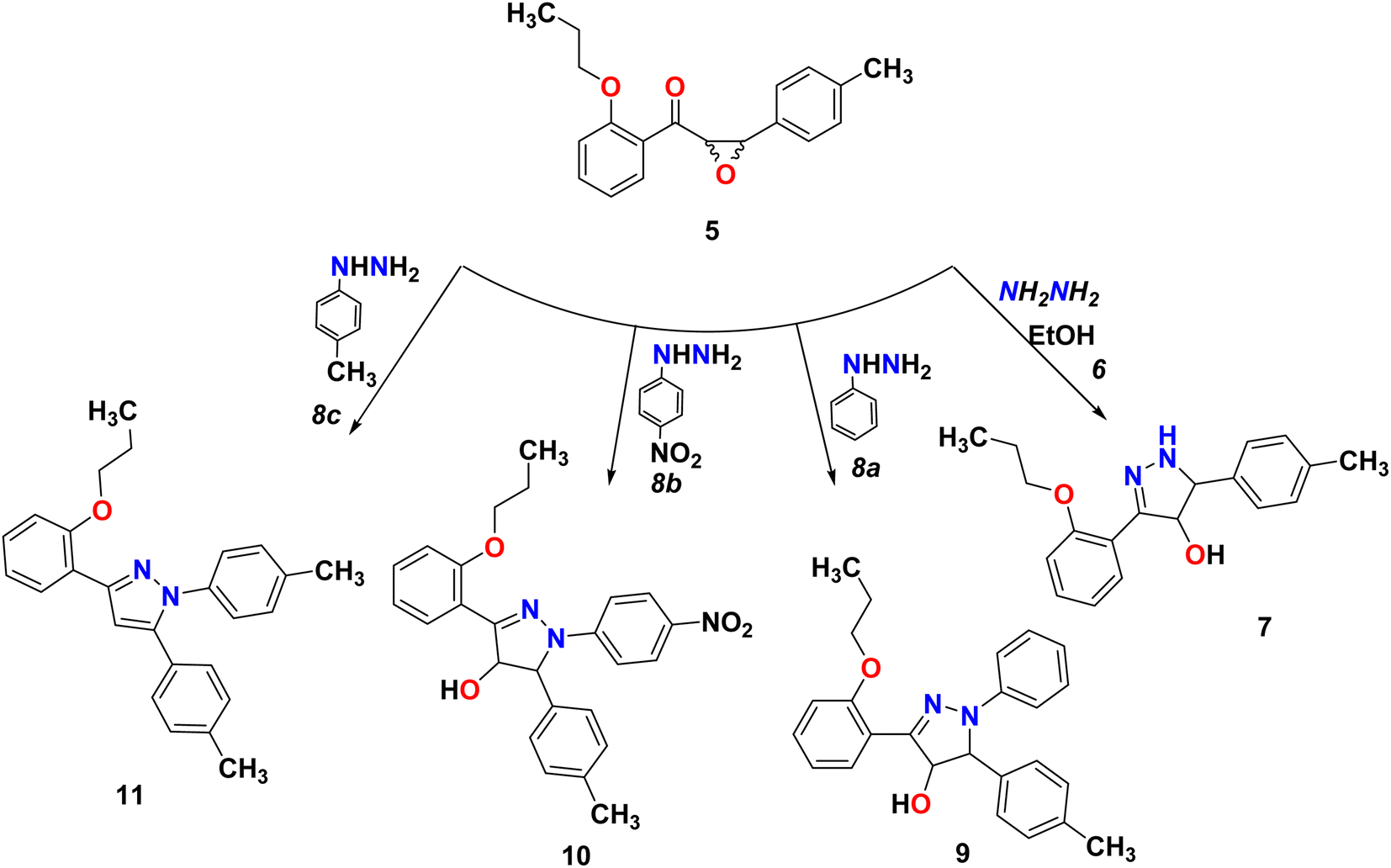
We start converting chalcone 1 to chalcone 4 by the reaction with bromopropane in order to block the OH group. A racemic form of epoxychalcone 5 has been obtained by oxidizing the double bonds of the respective chalcone 4 in a solution of 30% hydrogen peroxide in an alkaline medium via the Weitz–Scheffer epoxidation reaction.24,25 Elemental analysis, IR, NMR, and mass spectroscopy were used to confirm the oxirane product 5. Two doublet signals were detected in the 1HNMR spectrum of compound 5 at 4.00 ppm and at 4.49 ppm, which correspond to the oxirane ring.
Pyrazines can be obtained through the cyclo-condensation of hydrazines with dicarbonyl compounds (Knorr's method)26 and by cycloaddition between alkenes or alkynes and dipolar compounds.27,28 As a nucleophilic reagent, hydrazine hydrate and phenyl hydrazine derivatives were used to perform nucleophilic reaction on epoxychalcone 5. Reacting α,β-epoxyketone 5 with hydrazine hydrate 6 or phenyl hydrazine 8a in boiling ethanol led to the formation of 4,5-dihydro-1H-pyrazol-4-ol derivatives 7 and 9, respectively. However, when epoxychalcones 5 are reacted with phenylhydrazine hydrochloride 8b and 8c under the same conditions, they give the corresponding N-phenyl pyrazolo derivatives 10 and 11 (Scheme 3). In order to monitor the reaction progress, TLC was used. FT-IR and 1H/13C NMR spectra analyses confirmed their chemical structures. The FT-IR spectrum of phenylpyrazol-4-ol 7, 9, and 10 showed broadened stretch bands at 3157, 3538, and 3317 cm−1 for the OH group, respectively, in addition to the stretching band at 3263 cm−1 due to the NH group in compound 7. Also, the IR spectra for these compounds showed bands at about 1595 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group. Based on the 1HNMR spectra of compounds 7–10, the signals corresponding to the hydroxyl group, C4 proton, and C5 proton were detected in the pyrazole ring. In addition, compound 11 was confirmed using single-crystal X-ray diffraction.
N group. Based on the 1HNMR spectra of compounds 7–10, the signals corresponding to the hydroxyl group, C4 proton, and C5 proton were detected in the pyrazole ring. In addition, compound 11 was confirmed using single-crystal X-ray diffraction.
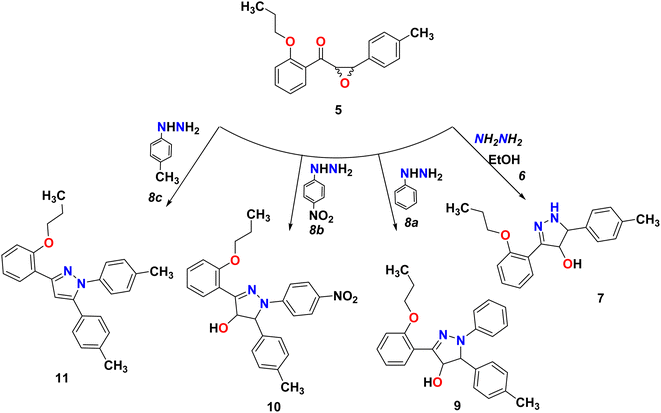 |
| | Scheme 3 Reaction of epoxychalcone 5 with hydrazine hydrate 6 and phenyl hydrazine 8a–c refluxed for 3 hours in ethanol as a solvent. | |
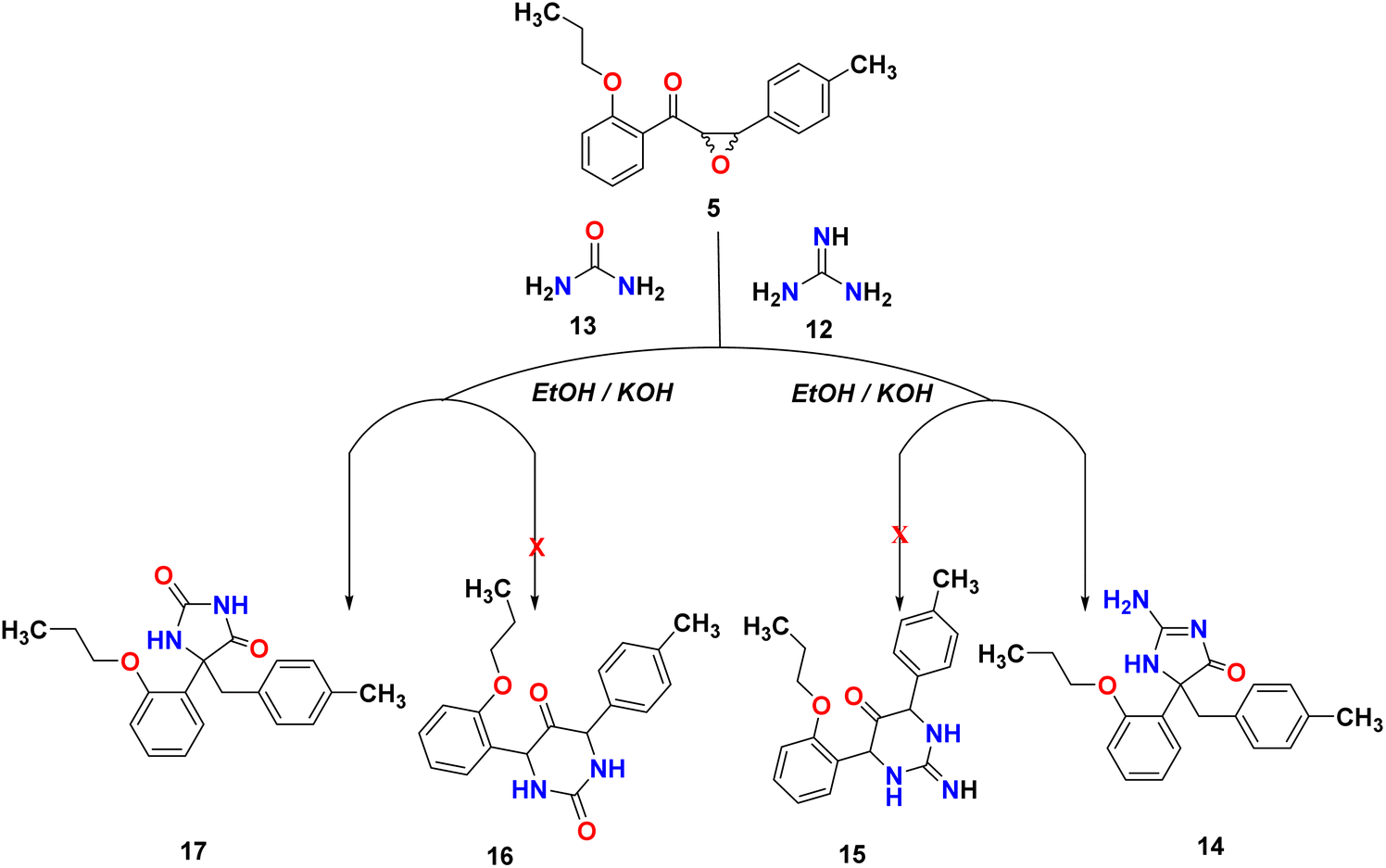
Many natural products and pharmaceutically active compounds contain imidazolone scaffolds. Also, several substituted imidazolones have received considerable attention due to their diverse medicinal applications. On the other hand, hydantoin derivatives are used in polymer chemistry, medicine, and the chemical industry.29–32 In order to generate new substances that have potential pharmacological or medicinal applications, epoxy-chalcone was used as a part of our research program directed toward designing and synthesizing compounds for potentially interesting bioactive compounds. Interestingly, when we treated epoxy chalcone 5 with guanidine hydrochloride in the presence of potassium hydroxide in ethanol (Scheme 4), the imidazolone compound 14 was formed instead of the pyrimidinone 15. Also, the reaction of epoxychalcone 5 with urea in the presence of potassium hydroxide in ethanol (Scheme 4) afforded the imidazolone compound 17 instead of the pyrimidinone 16. The IR spectrum of compound 14 showed characteristic stretching bands at 3265, 3055, 1656, and 1598 cm−1 due to NH, aromatic C–H, CO, and CN groups, respectively, whereas for compound 17, the IR spectrum showed bands at 3356, 3068, and 1763 cm−1 due to aromatic NH, aromatic C–H, and CO groups, respectively. The two compounds 14 and 17 showed two doublets in 1H-NMR at δ 2.92, 3.18 ppm for HA and 3.32, 3.40 for HB of the CH2 group, respectively. Additionally, compound 14 showed a D2O exchangeable singlet peak of NH at 7.37 ppm, while compound 17 showed an exchangeable singlet two peaks at 7.85 and 10.09 ppm, respectively. The chemical shifts of all other aromatic protons were observed in accordance with expectations. Molecular ions from compounds 14 and 17 exhibited peaks at m/z 337 and 338, respectively, in the mass spectrum, as shown in Scheme 4.
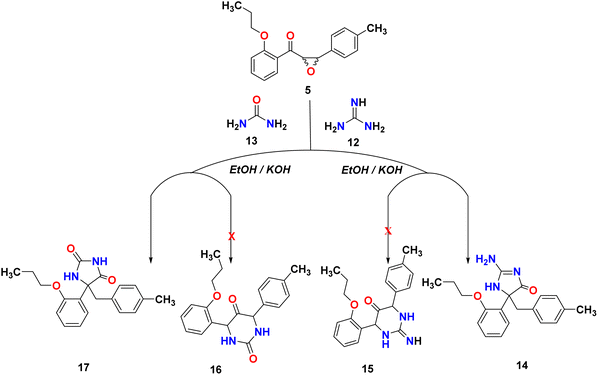 |
| | Scheme 4 Reactivity of epoxy-chalcone 5 with guanidine hydrochloride and urea. | |
Scheme 5 illustrates a possible mechanism for the formation of hydantoin. As in various base-catalyzed reactions of 2,3-epoxydiphenyl ketones,32,33 the first step is the isomerization of the chalcone-epoxide 5 to the corresponding 1,2-dione (A). There are two major pathways that can be envisaged from the intermediate 1,2-dione (A). First, a similar reaction to a benzil-benzilic acid rearrangement occurs when the N-atom of guanidine or urea attacks one of the carbonyl carbons, thereby initiating the migration of an aryl (or aryl–methyl) towards the rearranged products B or C. However, the more stable and final product in the case of urea is hydantoin compound B, while hydantoin compound C is in the case of guanidine. Second, the 1,2-dione may also form 5-membered ring-closed intermediates as the result of condensation products that lead to identical rearranged products B and C via the aryl (or aryl–methyl) migration. Regardless of which groups migrate, both feasible pathways end up producing the same product; therefore, the two isomeric intermediates are possible.
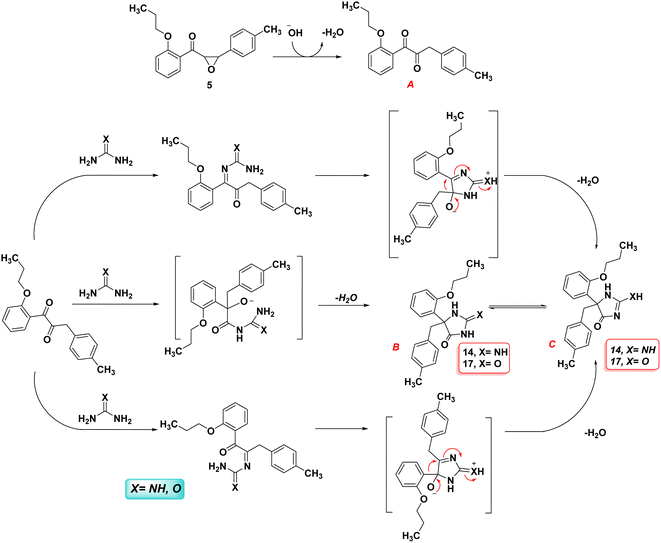 |
| | Scheme 5 Plausible mechanism of the formation of imidazolone compound 14 and 17. | |
2.2. DFT investigation
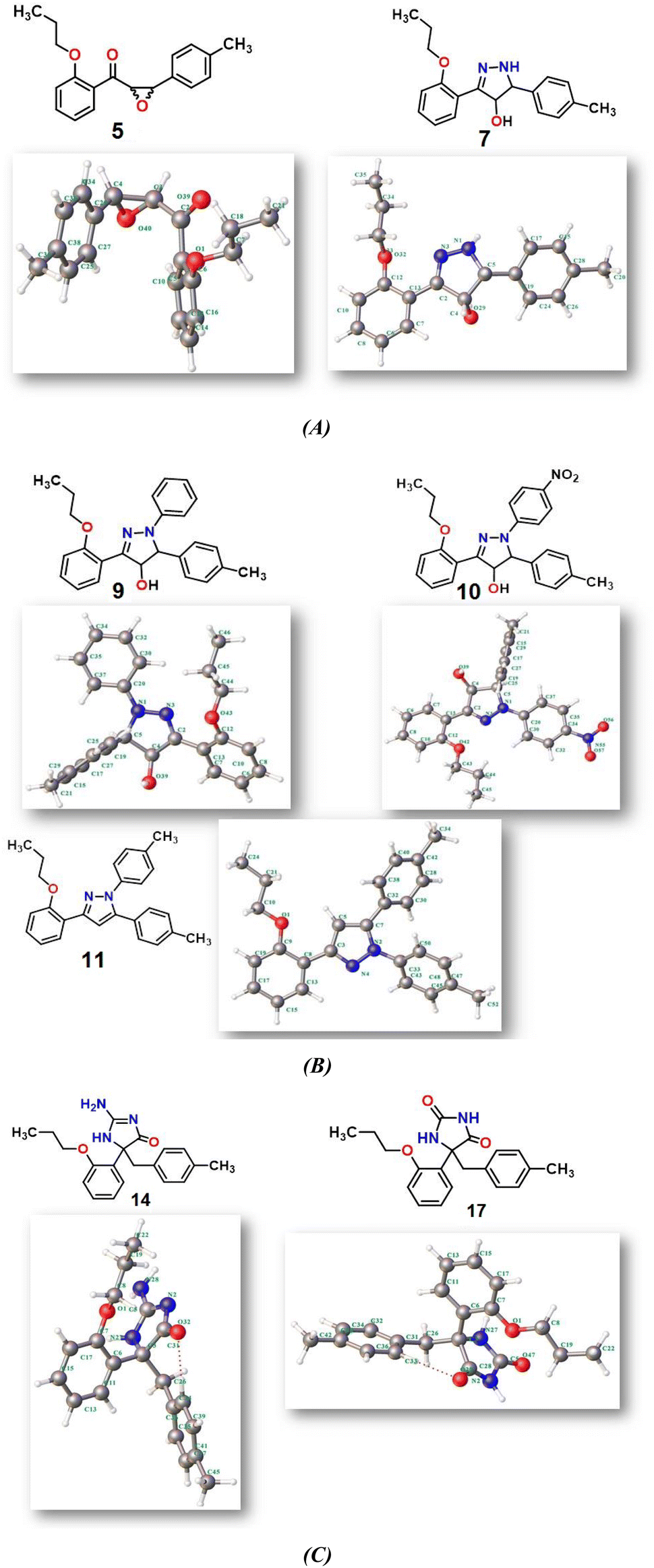
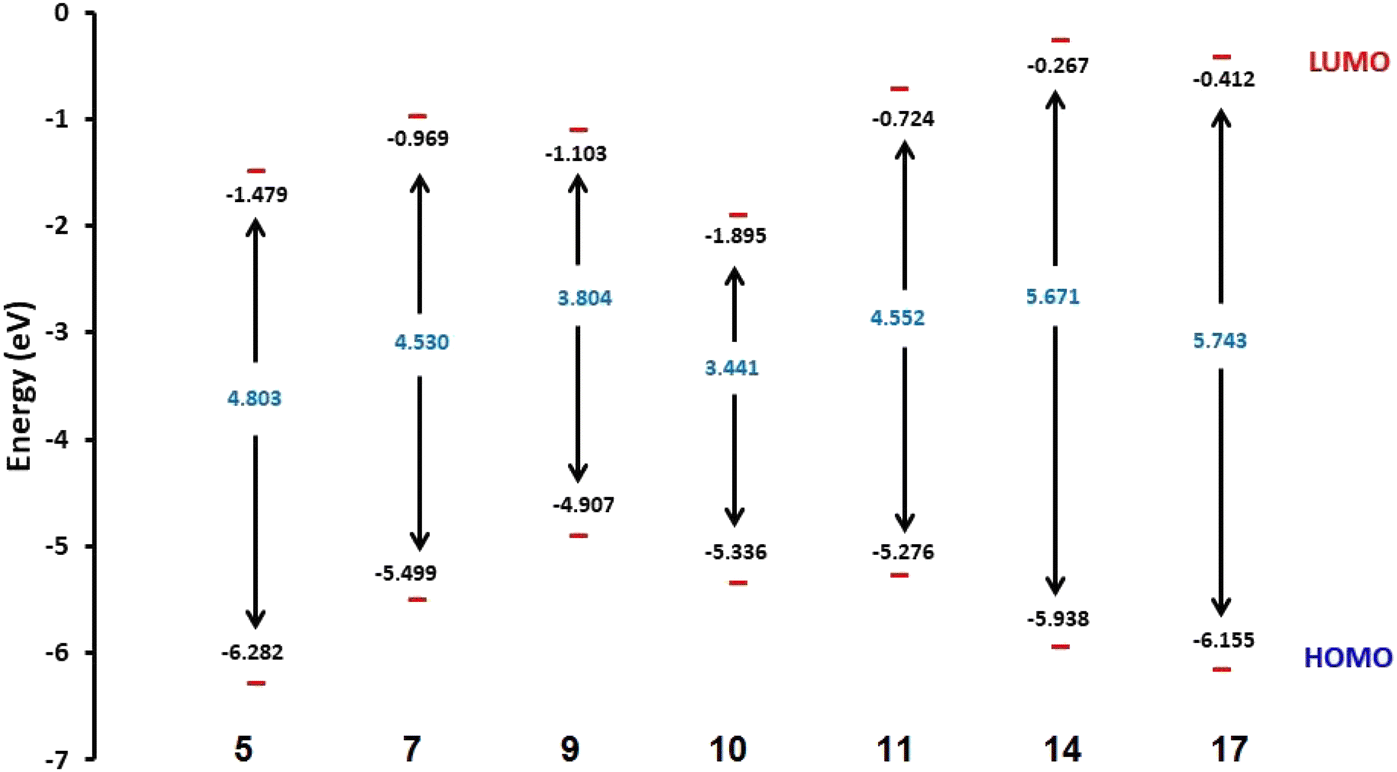
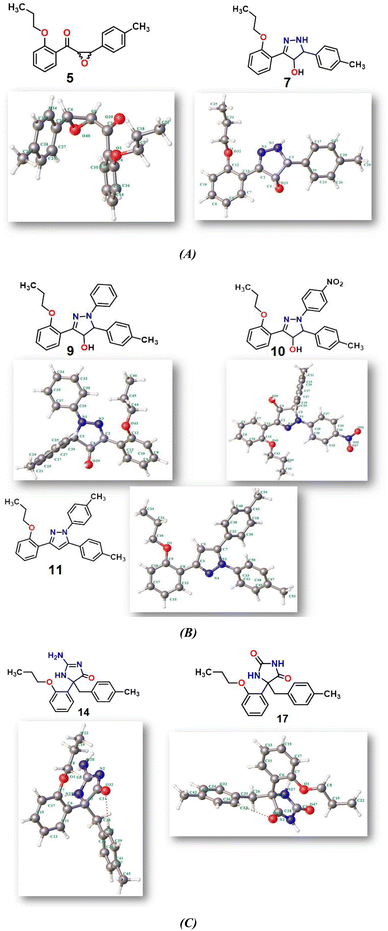
2.2.1. Physical descriptor's. All produced compounds 7, 9, 10, 11, 14 and 17 as well as epoxychalcone 5 were fully optimized using the DFT/B3LYP/6-31G(d,p) basis set,34–36 as shown in Fig. 3. Frequency calculations are performed to ensure that there are no imaginary frequencies. Table 1 shows the physical properties used to optimize the molecular structures of all compounds, which include (σ) absolute softness, (χ) electronegativity, (ΔNmax) electronic charge, (η) absolute hardness, (ω) global electrophilicity, (S) global softness, and (Pi) chemical potential based on eqn (1)–(8) calculated by B3LYP/6-31G(d,p).| |
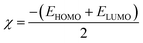 | (2) |
| |
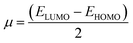 | (3) |
 |
| | Fig. 3 (A–C) Chemical and the optimized structure of compounds 5, 7, 9, 10, 11, 14 and 17 utilizing the DFT/6-31(G) basis set. | |
Table 1 Ground state energies of compound 5, 7, 9, 10, 11, 14 and 17 utilizing DFT/B3LYP/6-31G(d,p) and their physical parameters
| Physical descriptors |
| Compound |
ET (au) |
EHOMO (eV) |
ELUMO (e V) |
Eg (eV) |
μ (D) |
χ (eV) |
η (eV) |
σ (eV) |
Pi (eV) |
S (eV) |
ω (eV) |
ΔNmax |
| 5 |
−961.71 |
−6.282 |
−1.479 |
4.803 |
1.46 |
3.881 |
2.401 |
0.416 |
−3.881 |
0.208 |
3.136 |
1.616 |
| 7 |
−997.20 |
−5.499 |
−0.969 |
4.530 |
1.69 |
3.234 |
2.265 |
0.441 |
−3.234 |
0.220 |
2.308 |
1.427 |
| 9 |
−1228.27 |
−4.907 |
−1.103 |
3.804 |
1.33 |
1.333 |
1.901 |
0.525 |
−3.005 |
0.262 |
2.374 |
1.580 |
| 10 |
−1432.78 |
−5.336 |
−1.895 |
3.441 |
7.92 |
3.616 |
1.720 |
0.581 |
−3.616 |
0.290 |
3.800 |
2.101 |
| 11 |
−1191.19 |
−5.276 |
−0.724 |
4.552 |
2.77 |
3.000 |
2.275 |
0.439 |
−3.000 |
0.219 |
1.977 |
1.318 |
| 14 |
−1090.73 |
−5.938 |
−0.267 |
5.671 |
5.14 |
3.103 |
2.835 |
0.352 |
−3.103 |
0.176 |
1.698 |
1.094 |
| 17 |
−1110.62 |
−6.155 |
−0.412 |
5.743 |
2.76 |
3.284 |
2.871 |
0.348 |
−3.284 |
0.174 |
1.877 |
1.143 |
Fig. 3 shows the results of full optimization using the DFT/B3LYP/6-31G(d,p) basis set for all generated compounds 7–17 including epoxy 5. Table 2 lists the physical properties used in the optimization of molecular structures of all compounds with regard to (σ) absolute softness, (χ) electronegativity, (ΔNmax) electronic charge, (η) absolute hardness, (ω) global electrophilicity, (S) global softness, and (Pi) chemical potential based on eqn (1)–(8) estimated from B3LYP/6-31G(d,p). The total energies of compounds 7–17 were more stable overall than the initial epoxy chalcone 5, indicating their stability. Compound 10 also demonstrated greater stability, with an energy of (−1432.78 au) (−38987.84 eV), which can be attributed to the presence of 4O and 3N in its structure, which increases the compound's electronegativity. Additionally, compared to other compounds, compound 10 has an extremely large dipole moment. Compound 10 and epoxy chalcone 5 have dipole moments that differ by 6.46 D. This is due to the presence of a nitro group, which makes it easier to separate charges and increase reactivity. Table 1 indicates that compound 17 has a higher hardness of 2.87 eV and a low softness of 0.34 eV, suggesting a lower intramolecular charge transfer. Also, compound 17 exhibits the greatest ΔE, indicating its superior kinetic stability when compared to other compounds. On the other hand, compound 10 displays the lowest ΔE, signifying its heightened chemical reactivity. HOMO–LUMO orbitals are illustrated in Fig. 4, and their orbital analyses at the B3LYP/6-31G(d,p) level for compounds 5–17 are shown in Fig. 5. A positive phase was characterized by red colors, while a negative phase was characterized by green colors. Compound 5 has a HOMO with electron clouds mostly over the tolyl ring, epoxy group, and oxygen atom of the ketone group, whereas its LUMO is charged throughout the molecule except for the tolyl group and alkyl group. For the synthesized heterocyclic compounds, the electron clouds in HOMO and LUMO of compound 7 distribute charges over the entire molecule except for the tolyl and alkyl groups. Additionally, the HOMO and LUMO of compound 9 and compound 10 showed a similar charge distribution with the exception of the tolyl group and alkyl group. The HOMO of compound 11 displays electron clouds that are distributed across the entirety of the molecule with the exception of the two methyl and alkyl groups, while the LUMO distributes charges throughout the molecule except for the two methyl and alkoxy groups. On the other hand, compound 14 has a HOMO with electron clouds over the phenyl and tolyl groups in addition to the two nitrogen and oxygen of imidazolone, whereas its LUMO is charged throughout the molecule except for the tolyl group and alkyl group. Furthermore, the HOMO of compound 17 displays electron clouds that are distributed across the entirety of the molecule with the exception of the alkyl group, while the LUMO distributes charges throughout the molecule except for the alkyl and methylbenzyl groups.37
Table 2 Crystal data and refinement parameters of compound 11 (CCDC: 2310940)
| Crystal data of compound 11 |
| Chemical formula |
C26H26N2O |
| Mr |
382.49 |
| Crystal system, space group |
Monoclinic, P21/n |
| Temperature (K) |
296 |
| a, b, c (Å) |
6.7814 (4), 10.6880 (7), 29.8384 (18) |
| β (°) |
94.276 (3) |
| V (Å3) |
2156.7 (2) |
| Z |
4 |
| Radiation type |
Cu Kα |
| μ (mm−1) |
0.56 |
| Crystal size (mm) |
0.21 × 0.16 × 0.11 |
| Diffractometer |
Bruker APEX-II CCD |
| Absorption correction |
Multi-scan SADABS2016/2 – Bruker AXS area detector scaling and absorption correction |
| Tmin, Tmax |
0.89, 0.94 |
| No. of measured, independent & observed [I > 2σ(I)] reflections |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 524, 3725, 3159 524, 3725, 3159 |
| Rint |
0.029 |
| (sinθ/λ)max (Å−1) |
0.595 |
| R[F2 > 2σ(F2)], wR(F2), S |
0.040, 0.125, 1.06 |
| No. of reflections |
3725 |
| No. of parameters |
265 |
| H-atom treatment |
Constrained |
| Δρmax, Δρmin (e Å−3) |
0.14, −0.12 |
 |
| | Fig. 4 Schematic diagrams of HOMO and LUMO energy levels of compounds 5, 7, 9, 10, 11, 14 and 17 obtained from the DFT calculation with the B3LYP/6-31G(d,p) basis set. | |
 |
| | Fig. 5 (A–C) FMO profiles of the synthesized heterocyclic compounds 5, 7, 9, 10, 11, 14 and 17. | |
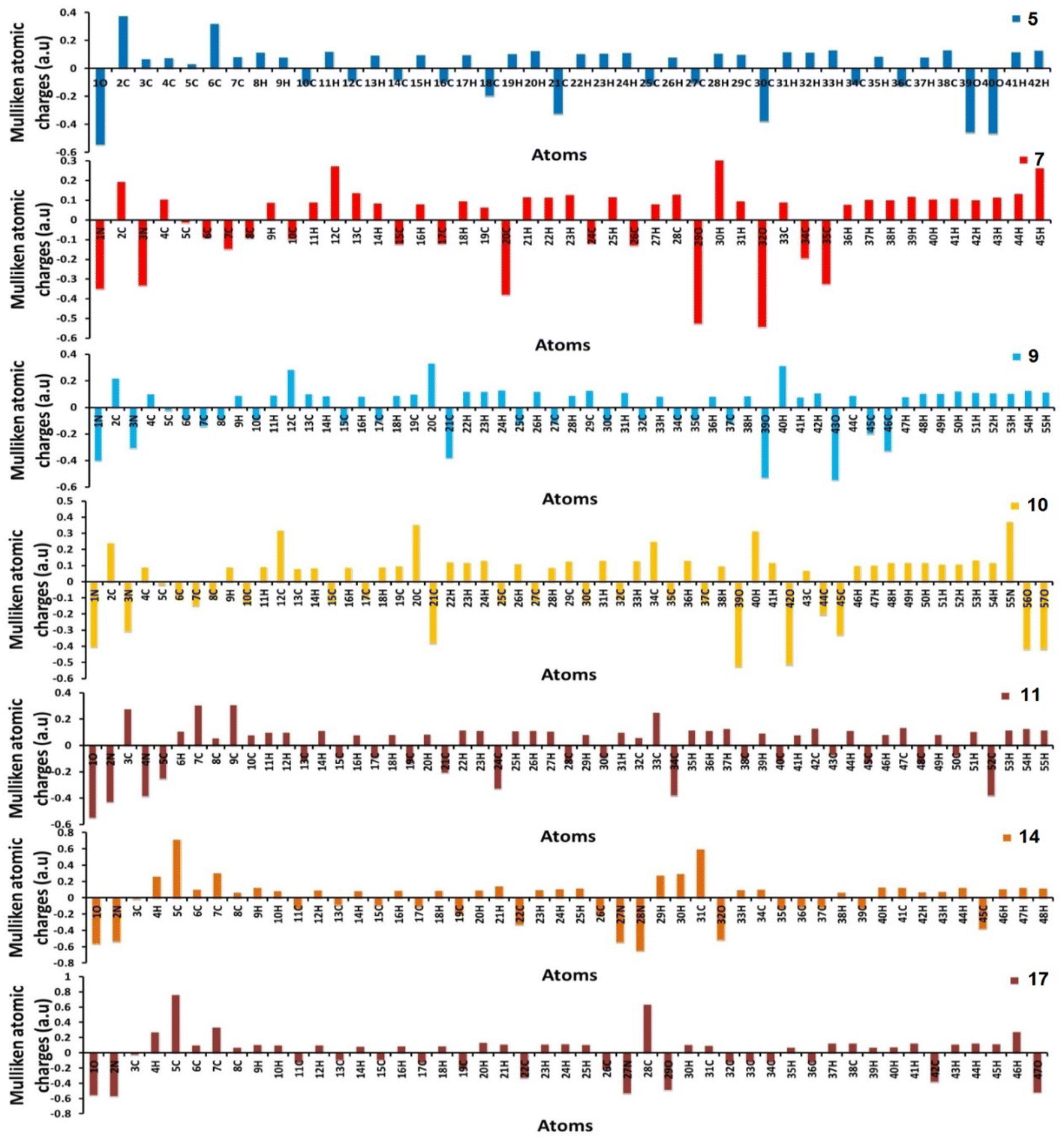
Furthermore, using the B3LYP/6-31G(d,p) basis sets, Mulliken atomic charges and electronic populations were calculated. Electron attraction from atoms with a greater electronegative causes the delocalization of negative charges in atoms with lower electronegative values. Carbon atoms contain both positive and negative charges, as seen in Fig. 6. The heteroatoms (N and O) have a high electron density and a negative charge.
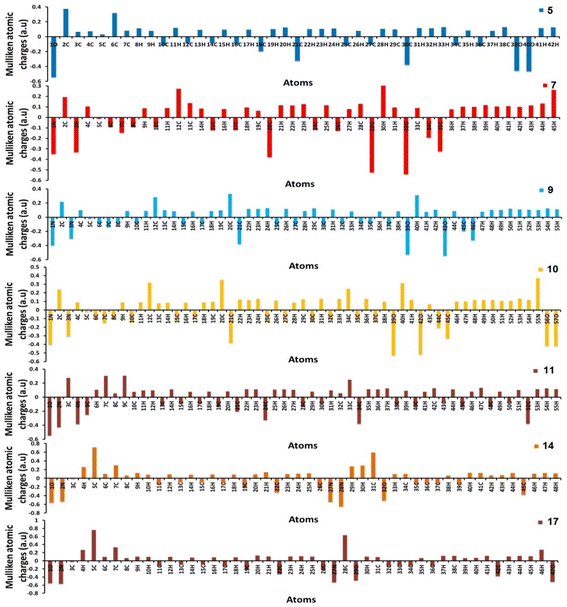 |
| | Fig. 6 Distribution of calculated Mulliken charges for compounds 5, 7, 9, 10, 11, 14, and 17. | |
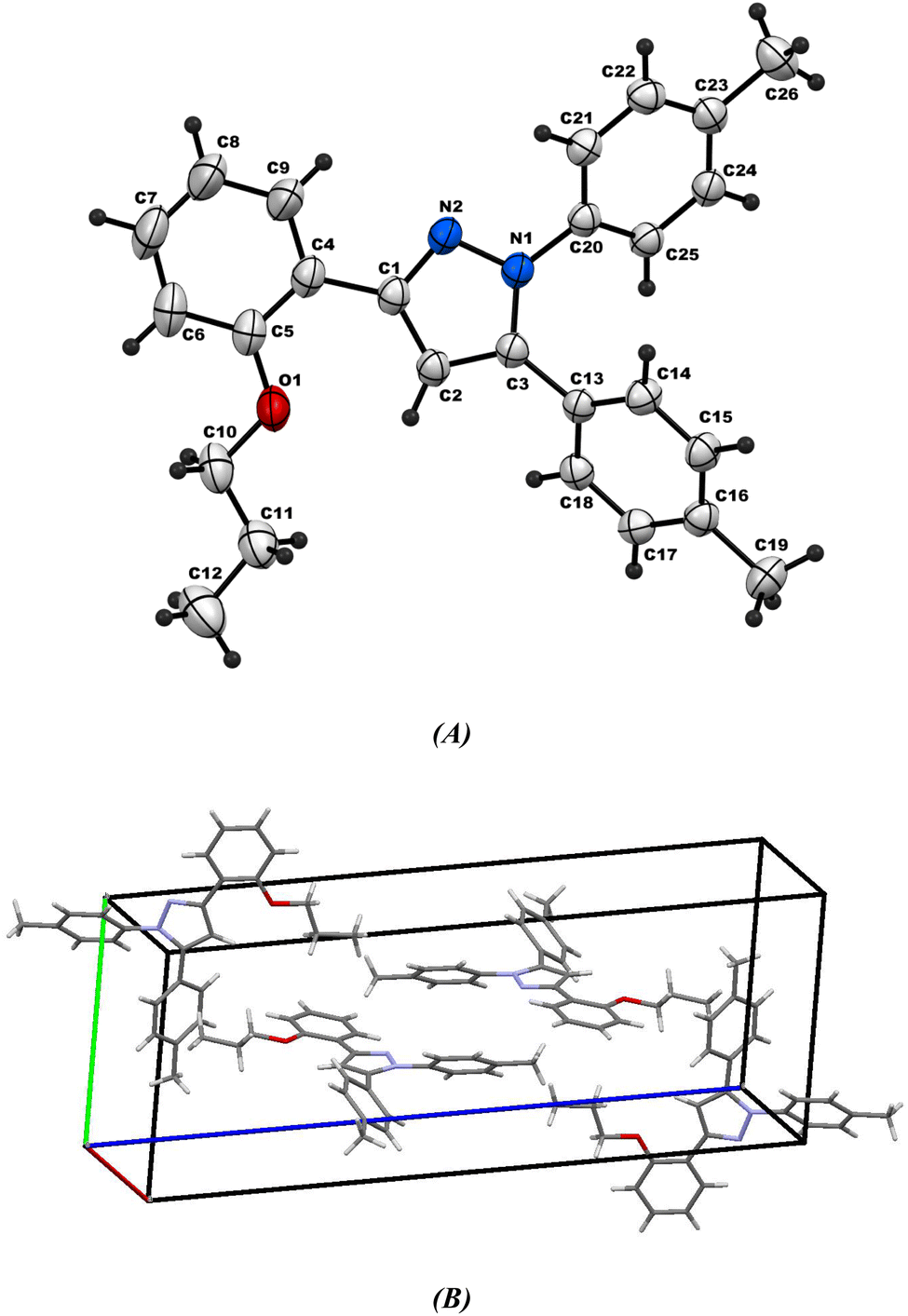
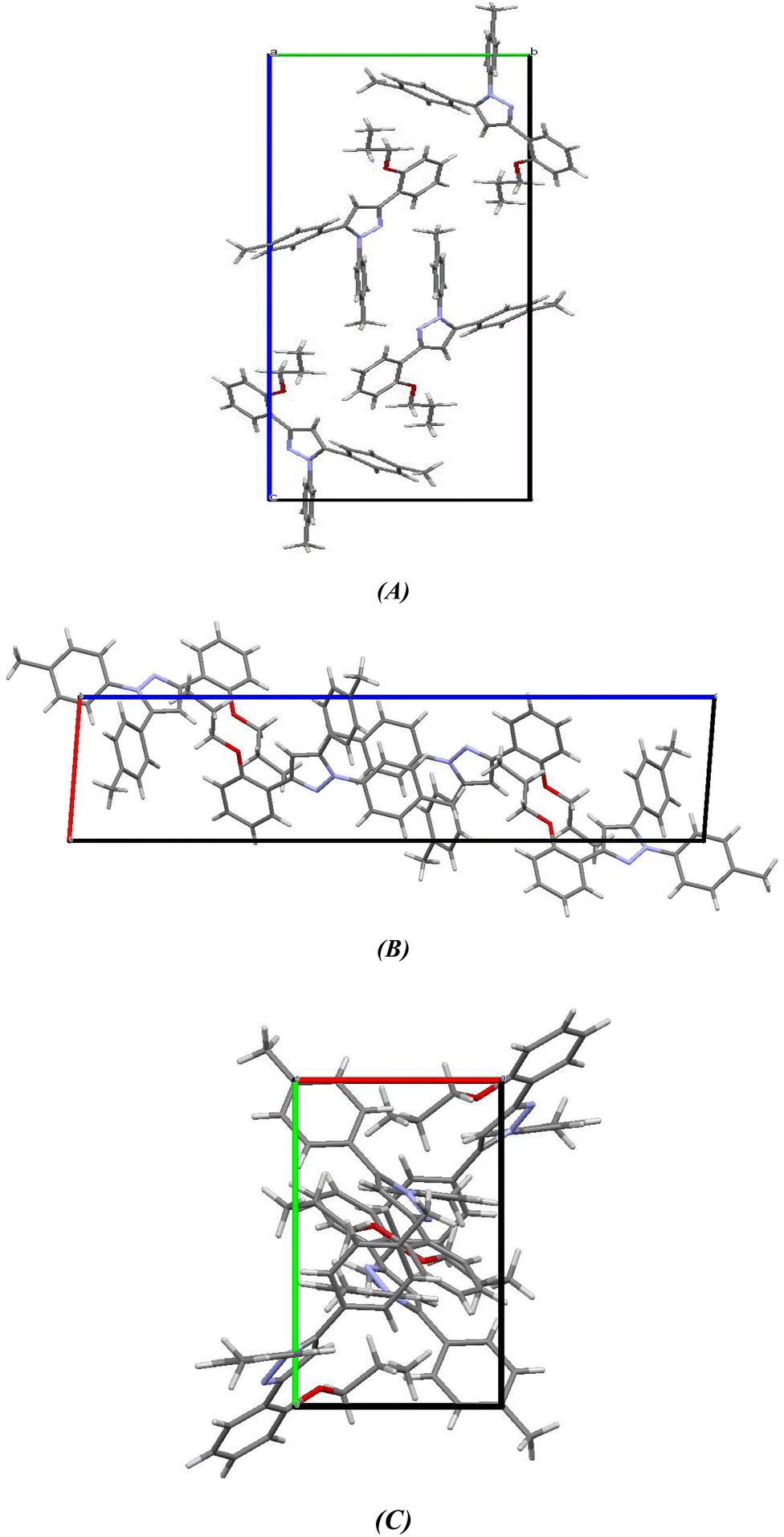
2.2.2. Comparative study between X-ray single crystal and theoretical studies. Table 2 shows the crystallographic and structure refinement data for compound 11, and Fig. 7 shows the ORTEP of the molecule with the thermal ellipsoids drawn with 50% probability (CCDC: 2310940). Table S1 (ESI†) provides a summary of the bond length, bond angles, and torsion angles. The crystallization of compound 11 occurs in the monoclinic space group P21/n. The crystal structure of compound 41 has parameters a = 6.7814 (4) Å, b = 10.6880 (7) Å, and c = 29.8384 (18) Å. Fig. 8 illustrates the three-dimensional packing structure of compound 11, viewed in the a-, b-, and c-directions.
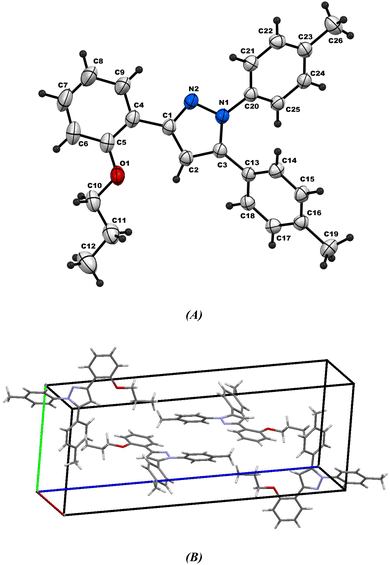 |
| | Fig. 7 (A) Ortep-3 crystal structure diagram of compound 11 and (B) unit cell. | |
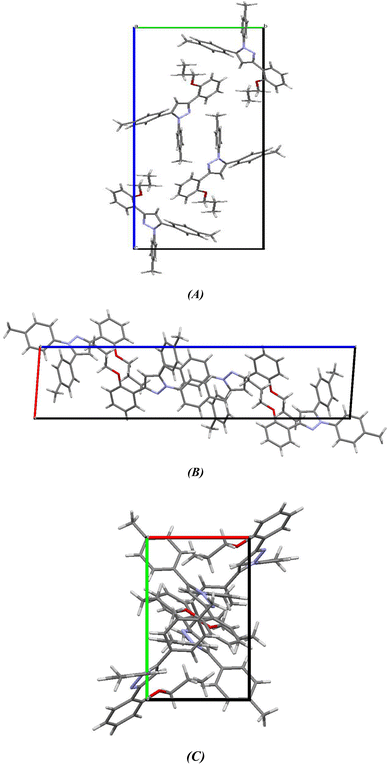 |
| | Fig. 8 Three-dimensional packing model of chromenopyridine 11 along the (A) a-, (B) b- and (C) c-direction. | |
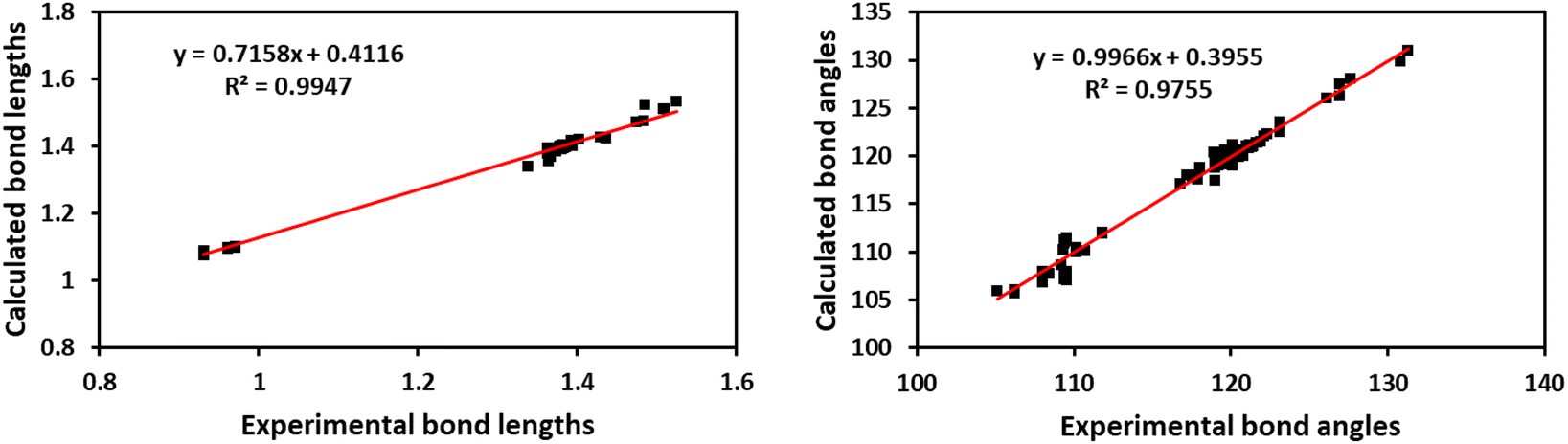
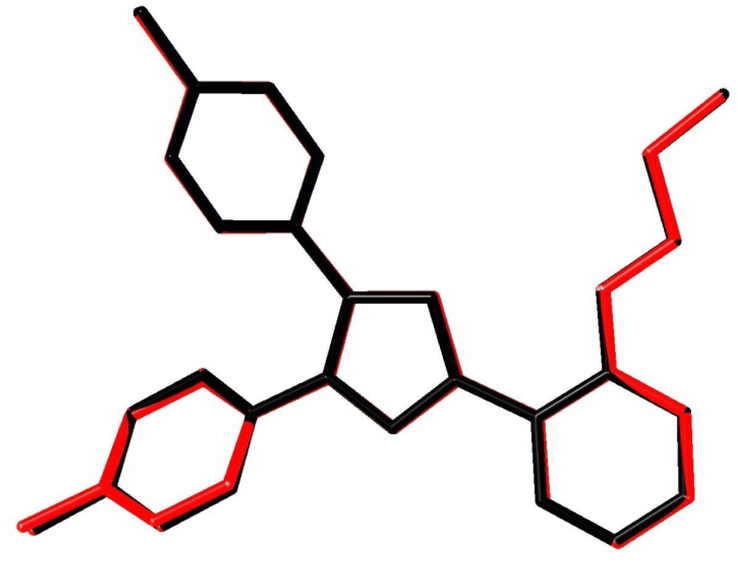
Fig. 7A shows the ORTEP diagram of compound 11 in the solid state, which displays the atomic numbering. The bond lengths and angles can be found in Table S1 (ESI†). It is noteworthy that the computed data is produced from the isolated gaseous phase, while the experimental data is produced from the solid state. Fig. 9 and 10 illustrates a remarkable agreement between theoretical calculations and X-ray analysis of structural molecular geometry.38
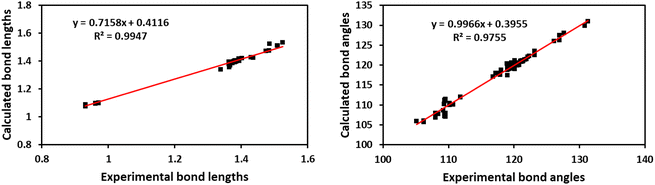 |
| | Fig. 9 Correlation graphic between the experimental and calculated bond lengths and bond angles of compound 11. | |
 |
| | Fig. 10 Atom-by-atom superimposition of compound 11 calculated using DFT/6-31G(d,p) (red) over the X-ray structure (black). Hydrogen atoms are omitted for clarity. | |
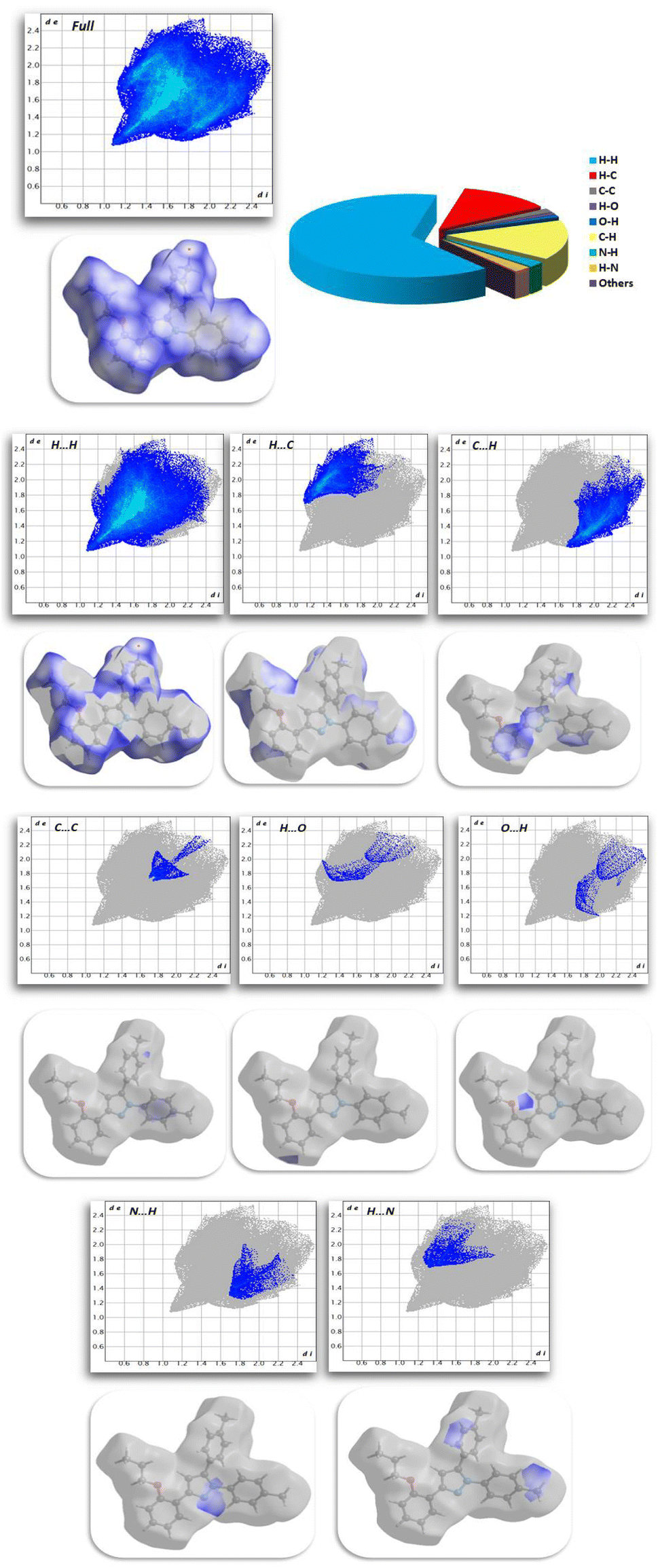
2.2.3. Hirshfeld surface analysis. Fig. 11 shows the Hirshfeld surfaces for compound 11, and Fig. 12 shows both the decomposed dnorm maps and the 2D-fingerprint plot outlining all potential interactions in compound 11. The surface color codes in dnorm can be used to analyze the molecular interactions, with white regions indicating contacts around the van der Waals radii with the red and blue regions representing shorter and longer inter contacts, respectively. The red and blue triangles in the shape index are a clear indication of the π⋯π interactions. Additionally, the presence of the flat green region encircled by a blue edge provided additional proof for the π⋯π interactions.39 Curvedness maps are used to highlight common packing configurations, such as planar stacking arrangements.40 For compound 11, the surface characteristics were mapped across dnorm (0.0130–1.371 Å), shape index (1.0–1.0 Å), and curvedness (4.0–0.4 Å). The distance between the surface and the closest atom is de in the outer zone of the surface and di in the inner zone. The most interactions were H⋯H interactions, which had a narrow point spike at di + de ∼1.1 Å and a contact percentage of 64%. Additionally, the percentage contacts of the C⋯H, H⋯C, N⋯H, C⋯C, O⋯H, and H⋯O interactions were 15.5, 12.4, 2.3, 1.6, 1 and 0.8% with narrow point spikes observed at di + de ∼2.8, 2.8, 3.0, 3.4, 3.0 and 3.0 Å, respectively.
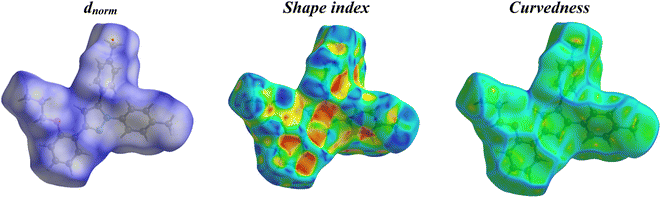 |
| | Fig. 11 Hirshfeld surfaces (dnorm, shape index, and curvedness) of compound 11. | |
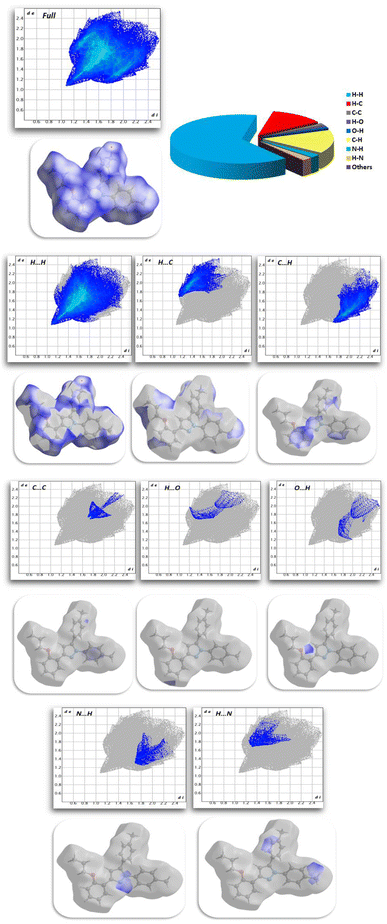 |
| | Fig. 12 Selected fingerprint plots and dnorm surfaces for important interactions in compound 11. | |
2.3. Biological activity
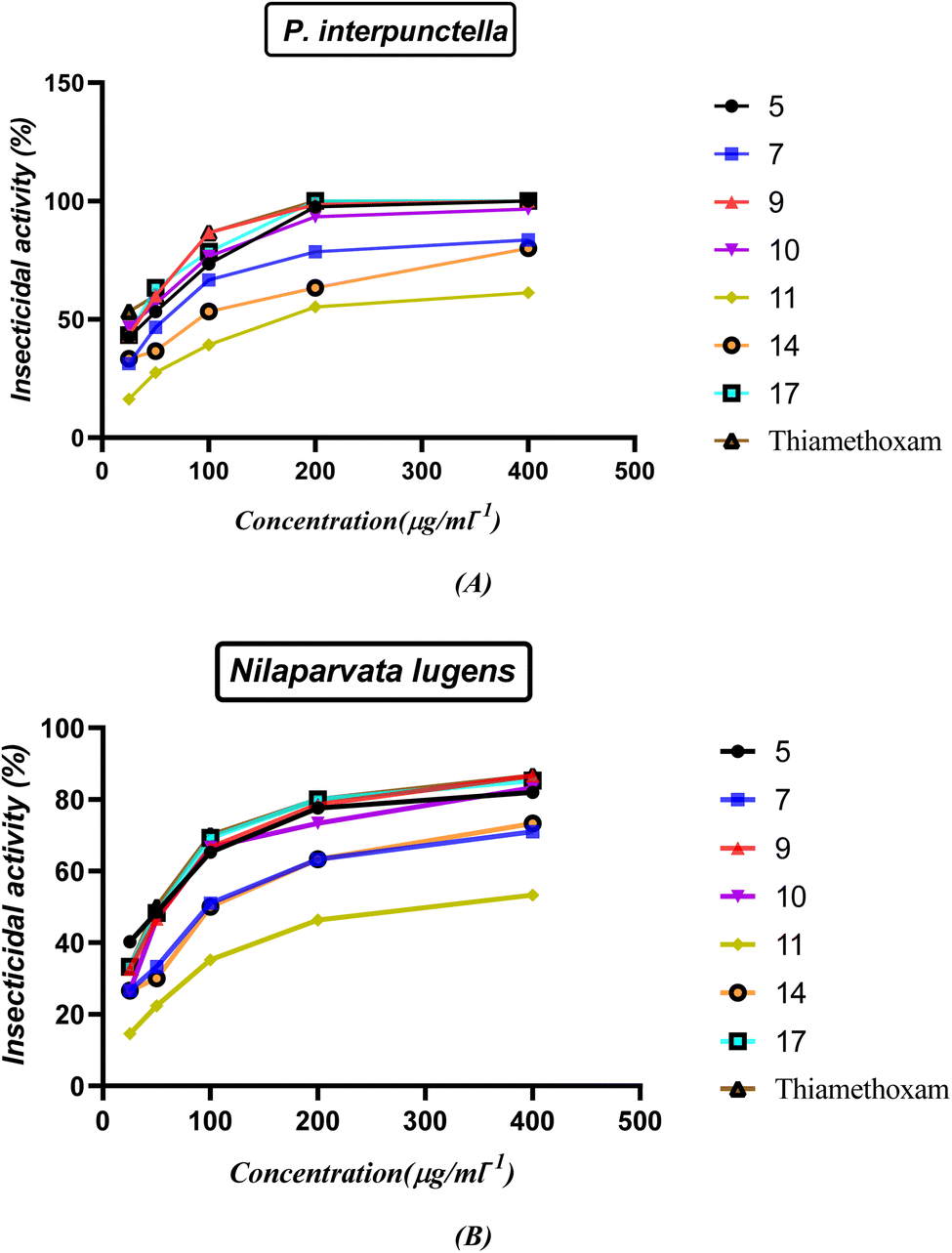
2.3.1. Insecticidal activity. The insecticidal activities of compounds 5, 7, 9, 10, 11, 14 and 17 were measured against healthy late third instar larvae P. interpunctella and Nilaparvata lugens according to the standard test41,42 with a slight modification. The test analogs were dissolved in DMF and serially diluted with water containing Triton X-80 (0.1 mg L−1) to obtain the required concentrations. The insects were reared at 25(±1) °C and groups were transferred to glass Petri dishes. All experiments were carried out in three replicates for statistic requirements. Assessments of mortality were calculated 48 h by the number and size of the live insects relative to those in the negative control. Evaluations were based on a percentage scale of (0 = no activity and 100 = complete eradication). The mortality rates were subjected to probity analysis.43 Thiamethoxam was used as a positive control while water-containing Triton X-80 (0.1 mg L−1) was used as a negative control. The results shown in Fig. 13 and Table S2 (ESI†) indicated that most of the compounds showed good insecticidal activity against the two pests. For example, compounds 5, 9, 10, and 17 were the most effective against both P. interpunctella and Nilaparvata lugens at 400 g mL−1 with nearly 100% activity. The remaining compounds showed moderate insecticidal activity. For example, compounds 5, 9, 10, and 17 had 100%, 100%, 96.6%, and 100% activities at 400 μg mL−1, respectively, against P. interpunctella, whereas thiamethoxam had 100%. Furthermore, compounds 5, 9, 10, and 17 exhibited 82%, 86.6%, 83.3%, and 85.3% activities against Nilaparvata lugens at 400 μg mL−1, respectively, whereas thiamethoxam exhibited 86.6% activity. The authors believed that the trypsin enzymes of P. interpunctella larvae were inhibited by compounds 5, 9, 10, 17 and thiamethoxam control, which also demonstrated the capacity to bind to chitin.
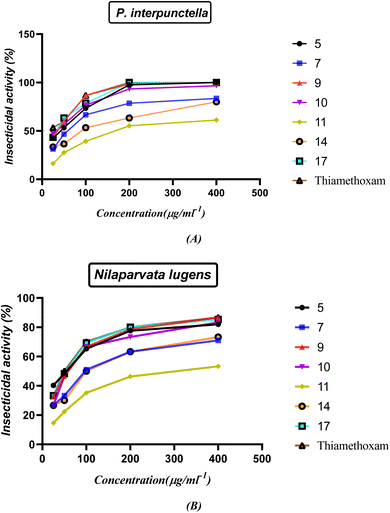 |
| | Fig. 13 (A and B) Insecticidal activity of the synthesized nitrogen heterocycles at different concentrations. | |
The structure–activity relationship (SAR) strategy tries to identify relationships between the biological activity of the examined compounds and their chemical structure. It appears that the presence of an oxirane ring in chalcone 5 contributes significantly to its activity. This is primarily because the ring is easily cleaved, resulting in the formation of an OH group. The OH group plays a pivotal role by readily interacting with OH groups within the biological systems of insects. The confirmation of the interaction is supported by an increase in the activity of compound 1-phenyl-3-(2-propoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-ol (9). The presence of only the OH group and the blocking of NH through the phenyl ring in compound 9 leads to a greater delocalization of electrons and increased their action compared to compound 7. Furthermore, compound 1-(4-nitrophenyl)-3-(2-propoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-ol (10) displays potent activity due to the presence of a withdrawing group, which effectively abstracts electrons, enhancing the attachment of the OH group to the insect cell line through electrostatic hydrogen bond interactions. On the other hand, compounds 11 and 14 exhibit reduced activity compared to the standard drug due to the absence of OH groups in the system, which decrease their action. Finally, compound 5-(4-methylbenzyl)-5-(2-propoxyphenyl)imidazolidine-2,4-dione (17) is noteworthy due to the presence of an imidazoline and dione system. This unique feature allows for the easy conversion to an OH group, consequently increasing its electrostatic hydrogen attraction potential. As a result, this compound exhibits heightened activity against P. interpunctella and Nilaparvata, as displayed in Fig. 13.
The LC50 values of compounds 5, 9, 10, and 17 were subject to further evaluation, and the results are presented in Tables 3 and 4. It is noteworthy that the LC50 values of compounds 9 and 17 for P. interpunctella are notably lower than that of thiamethoxam, suggesting their superior activity compared to the standard reference. In contrast, the LC50 value of compound 5 for Nilaparvata lugens is also lower than that of thiamethoxam, indicating its enhanced efficacy against Nilaparvata lugens when compared to the standard reference.
Table 3 LC50 values of some nitrogen heterocyclic against P. interpunctella
| Compound number |
Y = a + bx |
LC50 (mg L−1) |
95% limits |
Toxic ratio |
| 5 |
Y = 0.1479X + 50.42 |
1.867 |
0.00 |
0.9745 |
| 9 |
Y = 0.1317X + 57.29 |
1.428 |
2.647 |
0.9787 |
| 10 |
Y = 0.1253X + 54.52 |
1.784 |
3.008 |
0.9823 |
| 17 |
Y = 0.1333X + 56.37 |
1.556 |
2.756 |
0.9811 |
| Thiamethoxam |
Y = 0.1191X + 61.33 |
1.686 |
0.00 |
0.9504 |
Table 4 LC50 values of some nitrogen heterocycles against Nilaparvata lugens
| Compound number |
Y = a + bx |
LC50 (mg L−1) |
95% limits |
Toxic ratio |
| 5 |
Y = 0.1575X + 31.66 |
1.567 |
1.324 to 1.811 |
0.9937 |
| 9 |
Y = 0.1762X + 28.57 |
1.720 |
1.613 to 1.830 |
0.9987 |
| 10 |
Y = 0.1714X + 26.86 |
1.727 |
1.482 to 1.991 |
0.9924 |
| 17 |
Y = 0.1720X + 30.29 |
1.670 |
1.500 to 1.845 |
0.9964 |
| Thiamethoxam |
Y = 0.1727X + 30.86 |
1.655 |
1.505 to 1.809 |
0.9972 |
2.3.2. Docking simulation. The docking studies of 5, 7, 9, 10, 11, 14 and 17 using the MOE software44 to classify the biological effect of the most active compounds of nitrogen heterocyclic compounds compatible with the experimental results. The docking of 5, 7, 9, 10, 11, 14 and 17 with different protein receptors, for instance, the crystal structure of Plodia interpunctella beta-GRP/GNBP3 N-terminal domain (PDBID: 3aqy)45 and high resolution structure of Thermus thermophilus enoyl-acyl carrier protein reductase NAD and triclosan-form (PDBID: 3wyw).46 The docking results are elucidated in Fig. 14 and Table 5, which showed the interaction of these nitrogen heterocycles' with PDBID: 3aqy(A). Compound 5 showed high binding energy −8.44 kcal mol−1 and its length 2.75 Å with Arg 81, Lys 54, Arg 48, Thr 59 and showed the attachment of CO of chalcone.47 Additionally, compounds 9 and 10 showed binding tendency with −8.53, −8.87 kcal mol−1 and the presence of OH attract the amino acids with shortage distance between them 1.4, 2.53, and 3.4 Å, while compound 7 displayed a binding energy of −7.32 kcal mol−1 and attached with NH pyrazole and OH groups, resulting in bond lengths of 2.65, 2.72, and 3.03 Å (Arg 61, Arg 48, Arg 63). Moreover, compounds 11 and 14 exhibited moderate binding energies of −7.56 and −8.01 kcal mol−1 and high length range of 3.04–2.43 Å. On the other hand, compound 17 showed enhanced activity inside the pocket of protein with a high binding energy of −9.03 kcal mol−1 (Thr 59, Arg 61, Ile 50) and had a bond length of 1.59 Å, as displayed in Fig. 14(A) and Table 5. These findings align with the biological results and underscore the significance of compound 17 that possess two CO and NH groups, which increase the intermolecular interaction due to the presence of more hydrogen bond interactions. Furthermore, Fig. 14(B) and Table 5 also showed the action of compounds 5, 9, 10 and 17 with a high binding affinity of −8.34, −8.94, −8.62 and −9.23 kcal mol−1, respectively, and attachment with them at the active site position with PDBID: 3wyw(B), which increase its biological evaluation.
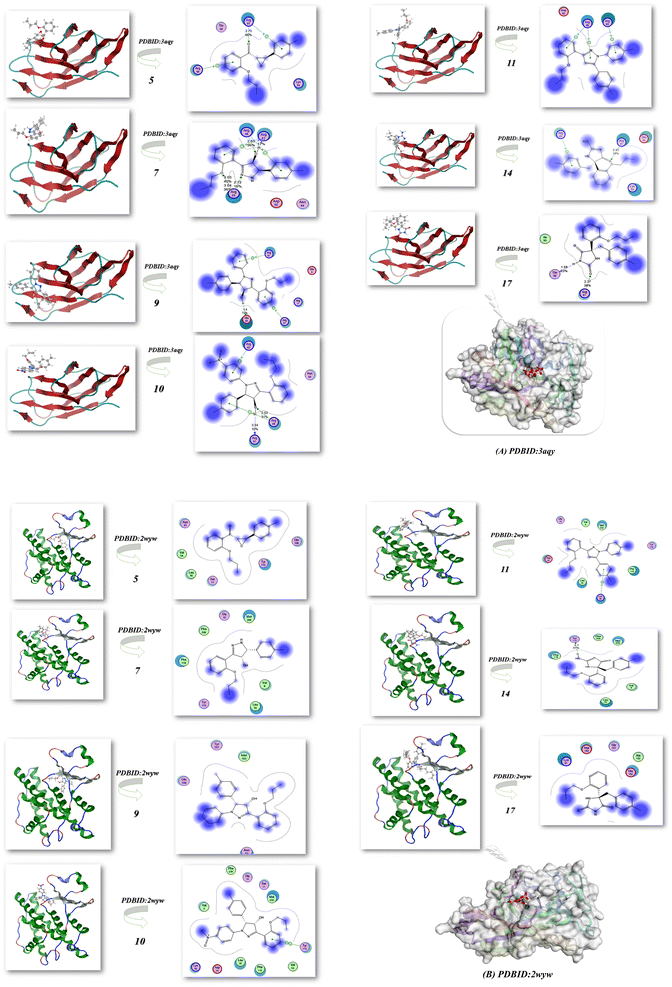 |
| | Fig. 14 (A and B) Docking analysis of the nitrogen nucleophile with PDBID: 3aqy and PDBID: 3wyw. | |
Table 5 Docking simulation heterocycle derivatives with different PDBIDs: 3aqy, 3wyw
| Escherichia coli (PDBID: 3aqy) |
Streptococcus pneumoniae (PDBID: 3wyw) |
| |
Energy affinity (kcal mol−1) |
Distance (Å) |
Amino acids |
|
Energy affinity (kcal mol−1) |
Distance (Å) |
Amino acids |
| 5 |
−8.44 |
2.75 Å |
Arg 81, Lys 54, Arg 48, Thr 59 |
5 |
−8.34 |
−2.01 Å |
Tyr 115, Gln 108, Ser 11, Leu 35, Val 54, Asn 53 |
| 7 |
−7.32 |
2.65, 2.72, 3.03 Å |
Arg 61, Arg 48, Arg 63 |
7 |
−7.11 |
2.54 Å |
Leu 35, Val 8, Tyr 115, Gly 10, Met 209, Phe 206, Phe 119 |
| 9 |
−8.53 |
1.4 Å |
Glu 55, Arg 57, Arg 17, Glu 9, Arg 61 |
9 |
−8.94 |
2.23 Å |
Tyr 107, Met 103, Gln 108, Asn 53 |
| 10 |
−8.87 |
2.53, 3.4 Å |
Arg 63, Asn 64, Arg 48, Arg 61 |
10 |
−8.62 |
2.11 Å |
Tyr 115, Val 54, Phe 11, Leu 35, Ser 11, Met 209, Phe 206, sp 34 |
| 11 |
−7.56 |
3.04 Å |
Arg 48, Arg 63, Asp 62 |
11 |
−6.32 |
−3.04 Å |
Lys 36, Asp 34, Leu 35, Phe 119, Val 54, Tyr 115, Met 209, Ser 11, Gly 10 |
| 14 |
−8.01 |
2.43 Å |
Arg 63, Arg 61, Glu 55, Lys 54 |
14 |
−7.13 |
−2.1 Å |
Tyr 115, Phe 206, Met 209, Val 8, Leu 35, Phe 119 |
| 17 |
−9.032 |
1.59 Å |
Thr 59, Arg 61, Ile 50 |
17 |
−9.23 |
−2.08 Å |
Lys 91, Asp 153, Gly 151, Ala 150, Glu 152 |
3. Experimental section
3.1. General procedure
The Gallenkamp melting point instrument was used to measure the melting points. Thin-layer chromatography (TLC) was conducted on Polygram SIL G/UV254 TLC plates, and the results were visualized with ultraviolet light at 254 nm and 350 nm. A Bruker DPX 400 superconducting NMR spectrometer was used to record the 1H and 13C nuclear magnetic resonance (NMR) spectra, and the IR spectra were measured with a Jasco Fourier transform/IR-6300 FT-IR spectrometer. The Elementar Vario MICRO Cube was used for elemental analysis. Electron impact (EI) was used to determine mass analyses on a Thermo double-focusing sector (DFS) mass spectrometer. A Varian Cary 5 spectrometer and a Shimadzu UV2600 spectrophotometer were used in UV-vis studies. Utilizing X-ray microdiffraction and single-crystal X-ray diffractometers, Rigaku D/MAX Rapid II and Bruker X8 Prospector were used to determine the X-ray crystal structure.
3.2. Materials and reagents
In this study, all solvents and materials were obtained from Aldrich. The synthesis of chalcone 1 was reported in the literature.48,49
3.2.1. Synthesis of (E)-1-(2-propoxyphenyl)-3-(p-tolyl)prop-2-en-1-one (4). A mixture of chalcone 1 (1 g, 4.2 mmol), potassium carbonate (1.16 g, 8.4 mmol, 2 equiv.) and 1-bromopropane (4.6 mmol, 1.1 equiv.) in 20 mL of DMF was stirred and refluxed for 16 h. After the reaction was completed, the reaction mixture was poured into water and then the solution was extracted with EtOAc (2 × 20 mL) and washed with water. The organic layer was dried over MgSO4 and evaporated under reduce pressure to give brown oil. Yield (1.1 g, 94%). FT-IR (νmax, cm−1): 3026 (C–H Ar), 1657 (CO), 1601 (CC), 1568 (ArC–C–). 1H NMR (DMSO-d6, δ ppm): 0.89 (t, 3H, J = 7.2 Hz, H3C), 1.68 (m, 2H, H2C), 2.32 (s, 3H, H3C), 4.03 (t, 2H, J = 6.0 Hz, H2C), 7.02 (t, 1H, J = 7.2 Hz, ArH), 7.15 (d, 1H, J = 8.4 Hz, ArH), 7.23 (d, 2H, J = 8.0 Hz, ArH),7.40–7.53 (m, 5H, ArH, CH),7.59 (d, 1H, J = 8.0 Hz, CH), 13C NMR (DMSO-d6): δ 10.44 (CH3), 20.73 (CH3), 21.10 (CH2), 69.59 (CH2), 112.97 (CH), 120.63 (CH), 126.11 (CH), 128.84 (CH), 129.02 (CH), 129.64 (C), 131.86 (C), 133.05 (CH), 140.42 (C), 142.01 (CH), 157.25 (C), 191.62 (CO). MS (m/z): 280 (M+, 60%), anal. calcd. for C19H20O2 (280.15): C, 81.40; H, 7.19. Found: C, 81.41%; H, 7.12%.
3.2.2. Synthesis of (2-propoxyphenyl)(3-(p-tolyl)oxiran-2-yl)methanone (5). A solution of chalcone 4 (3 g, 10.7 mmol) in acetone (10 mL) and methanol (10 mL) was mixed with 10% sodium hydroxide (10 mL), followed by the dropwise addition of hydrogen peroxide (30%, 10 mL) at 0 °C. The solution was stirred at room temperature for 1 h, then allowed to stand overnight at room temperature; a precipitate of the α-keto epoxide was formed. The residue was filtered, washed with water and recrystallized from EtOH. Yield (2.9 g, 94.5%), Mp 97–99 °C. FT-IR (νmax, cm−1): 3073 (C–H Ar), 1670 (CO) 1597 (ArC–C–). 1H NMR (DMSO-d6, δ ppm): 0.583 (t, 3H, J = 7.2 Hz, H3C), 1.13 (m, 2H, H2C), 2.32 (s, 3H, H3C), 3.82 (m, 2H, H2C), 4.00 (d, 1H, J = 1.6 Hz, CH oxirane), 4.49 (d, 1H, J = 1.6 Hz, CH oxirane), 7.05 (t, 1H, J = 7.6 Hz, ArH), 7.14 (d, 1H, J = 8.4 Hz, ArH), 7.20 (d, 2H, J = 8.0 Hz, ArH), 7.27 (d, 2H, J = 8.0 Hz, ArH), 7.56 (t, 1H, J = 8.0 Hz, ArH), 7.67 (d, 1H, J = 7.6 Hz, ArH), 13C NMR (DMSO-d6): δ 10.29 (CH3), 20.79 (CH3), 21.28 (CH2), 58.88 (CH), 63.19 (CH2), 69.63 (CH), 113.15 (CH), 120.58 (C, CH), 125.66 (CH), 126.11 (CH), 128.89 (CH), 129.84 (C), 133.21 (CH), 135.00 (C), 138.12 (C), 158.72 (CO). MS (m/z): 296 (M+, 56%), anal. calcd for C19H20O3 (296.14): C, 77.00; H, 6.80. Found: C, 77.01%; H, 6.79%.
3.2.3. General synthesis of 7–11. In absolute ethanol (20 mL), oxirane 5 (1 g, 3.37 mmol), and the appropriate hydrazine, namely, hydrazine hydrate or phenyl hydrazine salts (3.37 mmol), were refluxed for 3 hours. The solvent was evaporated under reduced pressure, and the residue was purified by crystallization from ethanol/ether mixture (2:1).
3.2.3.1. Synthesis of 3-(2-propoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-ol (7). White solid. Yield (0.4 g, 40%); Mp 130–131 °C. FT-IR (νmax, cm−1): 3263 (NH), 3157 (OH), 2963 (aliphatic CH), 1597 (CN). 1H NMR (DMSO-d6, δ ppm): 0.79 (t, 3H, J = 7.6 Hz, H3C), 1.57 (m, 2H, H2C), 2.29 (s, 3H, H3C), 3.87 (m, 2H, H2C), 4.42 (dd, 1H, J = 2.8 Hz, OH, D2O exchangeable), 5.16 (t, 1H, J = 6.4 Hz, CH), 5.38 (d, 1H, J = 6.8 Hz, CH), 6.91 (t, 1H, J = 7.2 Hz, ArH), 6.97 (d, 1H, J = 4.4 Hz, ArH), 7.13 (d, 2H, J = 8.0 Hz, ArH), 7.19 (d, 2H, J = 8.0 Hz, ArH), 7.28 (m, 1H, ArH), 7.45 (d, 1H, J = 2.4 Hz, NH, D2O exchangeable), 7.52 (d, 1H, J = 7.6 Hz, ArH), 13C NMR (DMSO-d6): δ 10.91 (CH3), 21.09 (CH3), 22.38 (CH2), 70.04 (CH), 75.61 (CH2), 79.21 (4), 113.26 (CH), 118.20 (CH), 120.76 (CH), 125.39 (CH), 125.89 (CH), 129.79 (CH), 130.89 (C), 133.33 (C), 158.45 (C), 161.33 (C). MS (m/z): 310 (M+, 30%), anal. calcd for C19H22N2O2 (310.17): C, 73.52; H, 7.14; N, 9.03. Found: C, 73.50%; H, 7.13%, N, 9.00%.
3.2.3.2. Synthesis of 1-phenyl-3-(2-propoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-ol (9). Off white solid. Yield (0.6 g, 46%); m.p 93–95 °C. FT-IR (νmax, cm−1): 3538 (OH), 2965 (aliphatic CH), 1595 (CN). 1H NMR (DMSO-d6, δ ppm): 0.74 (t, 3H, J = 7.6 Hz, H3C), 1.55 (m, 2H, H2C), 2.25 (s, 3H, H3C), 3.87 (m, 2H, H2C), 5.05 (d, 1H, J = 2.8 Hz, CH), 5.19 (dd, 1H, J = 6.4 Hz, CH), 5.97 (d, 1H, J = 7.2 Hz, OH, D2O exchangeable), 6.70 (t, 1H, J = 8.4 Hz, ArH), 6.98 (m, 4H, ArH), 7.01 (m, 6H, ArH), 7.31 (t, 1H, J = 6.8 Hz, ArH), 7.73 (dd, 1H, J = 7.6 Hz, ArH), 13C NMR (DMSO-d6): δ 10.37 (CH3), 20.63 (CH3), 21.97 (CH2), 69.47 (CH), 71.34 (CH2), 83.52 (CH), 112.54 (CH), 118.55 (CH), 120.35 (C), 120.81 (CH), 125.76 (CH), 128.90 (CH), 129.41 (CH), 129.58 (CH), 129.88 (CH), 136.15 (C), 136.53 (C), 143.71 (C), 148.51 (C), 156.62 (C). MS (m/z): 386 (M+, 50%), anal. calcd for C25H26N2O2 (386.20): C, 77.69; H, 6.78; N, 7.25. Found: C, 77.63%; H, 6.76%, N, 7.20%.
3.2.3.3. Synthesis of 1-(4-nitrophenyl)-3-(2-propoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-ol (10). Red crystal. Yield (0.4 g, 56%); m.p 162–165 °C. FT-IR (νmax, cm−1): 3395 (OH), 2951 (aliphatic CH), 1594 (CN). 1H NMR (DMSO-d6, δ ppm): 1.04 (t, 3H, H3C), 1.67 (m, 2H, H2C), 2.25 (s, 3H, H3C), 3.95 (m, 2H, H2C), 4.03 (dd, 1H, J = 11.6 Hz, CH), 4.35 (broad, 1H, OH, D2O exchangeable), 5.64 (dd, 1H, J = 4.4 Hz, CH), 7.01–7.15 (m, 8H, ArH), 7.39 (t, 1H, J = 7.6 Hz, ArH), 7.91 (dd, 1H, J = 2 Hz, ArH), 8.04 (d, 2H, J = 9.6 Hz, ArH), 13C NMR (DMSO-d6): δ 10.59 (CH3), 20.64 (CH3), 21.99 (CH2), 46.47 (CH), 61.78 (CH2), 69.67 (CH), 111.80 (CH), 112.96 (CH), 120.16 (C), 120.57 (CH), 125.48 (CH), 125.76 (CH), 128.75 (CH), 129.70 (CH), 131.45 (CH), 136.99 (C), 137.69 (C), 138.20 (C), 148.26 (C), 152.07 (C), 157.17 (C). MS (m/z): 431 (M+, 10%), anal. calcd for C25H25N3O4 (431.18): C, 69.59; H, 5.84; N, 9.74. Found: C, 69.56%; H, 5.81%, N, 9.72%.
3.2.3.4. Synthesis of 3-(2-propoxyphenyl)-1,5-di-p-tolyl-1H-pyrazole (11). White crystal in DMSO and brown oil at room temperature. Yield (0.4 g, 62%). FT-IR (νmax, cm−1): 2963 (aliphatic CH), 1600 (CN). 1H NMR (DMSO-d6, δ ppm): 1.03 (t, 3H, H3C), 1.83 (m, 2H, H2C), 2.26 (s, 3H, H3C), 2.28 (s, 3H, H3C), 4.05 (m, 2H, H2C), 6.94–7.03 (m, 2H, ArH), 7.04 (s, 1H, CH), 7.10–7.22 (m, 6H, ArH), 7.88 (d, 2H, J = 8.4 Hz, ArH), 13C NMR (DMSO-d6): δ 10.53 (CH3), 21.06 (2CH3), 22.22 (CH2), 69.91 (CH2), 108.09 (CH), 113.08 (CH), 120.87 (C), 121.67 (CH), 124.15 (CH), 125.47 (CH), 126.89 (CH), 127.89 (CH), 128.30 (CH), 128.69 (C), 129.59 (CH), 129.93 (C), 137.47 (C), 137.96 (C), 143.33 (C), 148.29 (C), 156.47 (C). MS (m/z): 382 (M+, 70%), anal. calcd for C26H26N2O (382.20): C, 81.64; H, 6.85; N, 7.32. Found: C, 81.60%; H, 6.82%, N, 7.30%.
3.2.4. Synthesis of 2-amino-5-(4-methylbenzyl)-5-(2-propoxyphenyl)-1,5-dihydro-4H-imidazol-4-one (14). Guanidine hydrochloride (0.13 g, 1.35 mmol) and KOH (0.15 g, 2.7 mmol) were added to a solution of epoxychalcone 5 (0.4 g, 1.35 mmol) in ethanol (10 mL). The reaction mixture was refluxed for 3 h. The reaction mixture was filtered, and the solvent was removed using a rotatory evaporator afforded compound 42 (0.24 g, 52%) as a dark yellow solid and recrystallized from EtOH; m.p. 127 °C (decomposed). FT-IR (νmax, cm−1): 3265 (NH), 3055 (aromatic CH), 2966 (aliphatic CH), 1656(CO), 1598 (CN). 1H NMR (DMSO-d6, δ ppm): 1.08 (t, 3H, H3C), 1.94 (m, 2H, H2C), 2.25 (s, 3H, H3C), 2.92 (d, 1H, J = 12.8 Hz, CH2-A), 3.32 (d, 1H, J = 12.8 Hz, CH2–B), 3.99 (m, 2H, H2C), 6.89 (t, 1H, J = 7.6 Hz, ArH), 7.02 (s, 4H, ArH), 7.06 (d, 1H, J = 8.0 Hz, ArH), 7.26 (t, 1H, J = 8.0 Hz, ArH), 7.37 (s, 1H, NH, D2O exchangeable), 7.63 (dd, 1H, J = 7.6 Hz, ArH), 13C NMR (DMSO-d6): δ 10.58 (CH3), 20.58 (CH3), 22.03 (CH2), 41.71 (CH2), 69.14 (CH2), 69.42 (C), 112.25 (C), 119.88 (CH), 126.03 (CH), 126.49 (CH), 128.11 (CH), 128.71 (CH), 130.01 (CH), 132.99 (C), 135.09 (C), 156.43 (C), 170.24 (C), 187.36 (CO). MS (m/z): 337 (M+, 20%), anal. calcd for C20H23N3O2 (337.18): C, 71.19; H, 6.87; N, 12.45. Found: C, 71.15%; H, 6.84%, N, 12.42%.
3.2.5. Synthesis of 5-(4-methylbenzyl)-5-(2-propoxyphenyl)imidazolidine-2,4-dione (17). Urea (0.101 g, 1.68 mmol) and KOH (0.095 g, 1.68 mmol) were added to a solution of epoxychalcone 5 (0.5 g, 1.68 mmol) in ethanol (10 mL). The reaction mixture was refluxed for 3 h. The reaction mixture was filtered, and the solvent was removed using a rotatory evaporator, then rescrystallized with EtOH which afforded compound 17 (0.1 g, 17%) as an off-white solid; m.p. 235–237 °C. FT-IR (νmax, cm−1): 3356, 3195 (NH), 3068 (aromatic CH), 2957 (aliphatic CH), 1763 (C2O), 1717 (C4O). 1H NMR (DMSO-d6, δ ppm): 0.89 (t, 3H, H3C), 1.64 (m, 2H, H2C), 2.28 (s, 3H, H3C), 3.18 (d, 1H, J = 13.2 Hz, CH2-A), 3.40 (d, 1H, J = 13.2 Hz, CH2-B), 3.86 (m, 2H, H2C), 6.96 (t, 1H, J = 8.4 Hz, ArH), 7.04 (d, J = 8 Hz, 1H, ArH), 7.09 (s, 4H, ArH), 7.33 (t, 1H, J = 7.6 Hz, ArH), 7.56 (d, 1H, J = 7.6 Hz, ArH), 7.85 (s, 1H, NH, D2O exchangeable), 10.09 (s, 1H, NH, D2O exchangeable), 13C NMR (DMSO-d6): δ 10.45 (CH3), 20.61 (CH3), 21.58 (CH), 40.00 (CH2), 65.42 (CH2), 69.72 (C), 112.26 (C), 119.81 (CH), 127.06 (CH), 127.31 (CH), 128.30 (CH), 129.54 (CH), 130.52 (CH), 131.46 (C), 135.63 (C), 156.64 (CO), 156.88 (C), 176.25 (CO). MS (m/z): 338 (M+, 8%), anal. calcd for C20H22N2O3 (338.16): C, 70.99; H, 6.55; N, 8.28. Found: C, 70.98%; H, 6.52%, N, 8.24%.
3.3. Hirshfeld surface analysis
The topology analyses were performed using Crystal Explorer 17.5 program.50
3.4. Computational studies
Molecular geometry was directly taken from the experimental outcomes of X-ray diffraction without any constraints. Density functional theory including Becke's three-parameter hybrid functional using the LYP correlation functional (B3LYP) with the 6-31G(d,p) basis set via the Berny method34,35 were proceeded with the Gaussian 09W program.51 The superimposition was performed using Olex2.52 Furthermore, the Mullikan atomic charges of compounds 5, 7, 9, 10, 11, 14 and 17 were calculated as well.
3.5. Insecticidal activity
3.5.1. Evaluation of insecticidal action of heterocycles against larvae of P. interpunctella and Nilaparvata lugens. Laboratory-reared insect colonies of P. interpunctella and Nilaparvata lugens free from insecticides and pathogens obtained from Insect 14 S. A. RIZK ET AL. greenhouse of Pest control Analytical lab, Entom-Aysel Company, Cairo, Egypt were maintained starting from egg rafts. The collected populations of P. interpunctella were reared in plates on an artificial diet comprised of soybean flour, yeast powder, casein, dry maize leaf powder, cholesterol, sugar, agar, ascorbic acid, sorbic acid, methyl p-hydroxybenzoate, vitamin solution, and distilled water, while collected populations of N. lugens were reared on rice (Oryza sativa L.) seedlings (7–10 days after germination) in the laboratory with controlled conditions, such as temperature 37 ± 1 °C and 70–80% relative humidity with a 16–8 h (L/D) photoperiod. The third nymph instars of the second laboratory generation were used for bioassays.53,54
3.5.2. Docking analysis. Utilizing the MOE software, novel compounds were molecularly docked,44 and the Discovery Studio Client (version 4.2) was used to find it.55 The energy of the obtained conformations decreased using the Confirmation Examination module of Auto Dock Vina after systematic conformational research was undertaken out to an RMS gradient of 0.01. The crystal structure of Plodia interpunctella beta-GRP/GNBP3 N-terminal domain (PDBID: 3aqy)45 was determined and the structural characterization of the catalytic site of a Nilaparvata lugens delta-class glutathione transferase (PDBID: 3wyw)56 was done. Ten distributed docking simulations were performed using default parameters, and the confirmations were generated based on the organization of the overall data, the E conformation, and the appropriate use of the pertinent amino acids in each protein's binding pocket.
3.6. Crystal information
CCDC 2310940 is the ESI Publication Number† for the crystallographic data deposited at the Cambridge Crystallographic Data Centre.
4. Conclusion
In conclusion, our research involves the effective synthesis of several pyrazole and imidazole derivatives. A variety of analytical and spectroscopic techniques were used to confirm their identity and structural characterization. Moreover, we utilized DFT calculations and X-ray analysis to optimize and validate the synthesized compounds' structures, affirming their accuracy. In terms of biological activity, compounds 5, 9, 10, and 17 displayed remarkable insecticidal potential, surpassing the performance of the standard reference thiamethoxam. Docking simulations with specific proteins further supported their efficacy, highlighting their strong binding interactions with amino acids.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr Mohammad H. BinSabt gratefully acknowledges the resources of the research sector projects unit (RSPU) general facilities of the Faculty of Science (GFS) (GS03/08) of Kuwait University for X-ray analysis.
References
- B. Kaur and P. Singh, Bioorg. Chem., 2022, 125, 105862 CrossRef CAS PubMed.
- Y. Hu, C. Li, B. A. Kulkarni, G. Strobel, E. Lobkovsky, R. M. Torczynski and J. A. Porco, Org. Lett., 2001, 3, 1649–1652 CrossRef CAS PubMed.
- G. Thirunarayanan and G. Vanangamudi, Spectrochim. Acta, Part A, 2011, 81, 390–396 CrossRef CAS PubMed.
- A. Singh, A. Raza, S. Amin, C. Damodaran and A. K. Sharma, Molecules, 2022, 27, 886 CrossRef CAS PubMed.
- A. M. Fahim, E. H. I. Ismael and H. E. M. Tolan, Polycyclic Aromat. Compd., 2023, 1–42 Search PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed.
- D.-G. Crivoi, A. M. Segarra and F. Medina, J. Catal., 2016, 334, 120–128 CrossRef CAS.
- H. Yi, G. Zou, Q. Li, Q. Chen, J. Tang and M.-y. He, Tetrahedron Lett., 2005, 46, 5665–5668 CrossRef CAS.
- M. Zhang, T. Chen, S. Fang, W. Wu, X. Wang, H. Wu, Y. Xiong, J. Song, C. Li, Z. He and C.-S. Lee, Org. Biomol. Chem., 2021, 19, 2481–2486 RSC.
- J. Liu, F. Li, Z. Hao, Y. Wang, X. Hua, Y. Li and Z. Li, Bioorg. Med. Chem., 2020, 28, 115829 CrossRef CAS PubMed.
- F. Tanaka, Y. Magariyama and A. Miyanoshita, Food Chem., 2020, 303, 125381 CrossRef CAS PubMed.
- M. M. Khan, Q. Huang, T. A. Wagan, H. Hua, W. Cai and J. Zhao, J. Asia-Pac. Entomol., 2020, 23, 269–276 CrossRef.
- M. Calvo-Agudo, J. González-Cabrera, D. Sadutto, Y. Picó, A. Urbaneja, M. Dicke and A. Tena, Environ. Pollut., 2020, 267, 115581 CrossRef CAS PubMed.
- P. Maienfisch, M. Angst, F. Brandl, W. Fischer, D. Hofer, H. Kayser, W. Kobel, A. Rindlisbacher, R. Senn, A. Steinemann and H. Widmer, Pest Manage. Sci., 2001, 57, 906–913 CrossRef CAS PubMed.
- S. A. Rizk, A. Y. Alzahrani and A. M. Abdo, Polycyclic Aromat. Compd., 2023, 1–18, DOI:10.1080/10406638.2023.2227316.
- R. Yang, T. Xu, J. Fan, Q. Zhang, M. Ding, M. Huang, L. Deng, Y. Lu and Y. Guo, Ind. Crops Prod., 2018, 117, 50–57 CrossRef CAS.
- J. Wu, B.-A. Song, D.-Y. Hu, M. Yue and S. Yang, Pest Manage. Sci., 2012, 68, 801–810 CrossRef CAS PubMed.
- S. P. Chandrasekharan, A. Dhami, S. Kumar and K. Mohanan, Org. Biomol. Chem., 2022, 20, 8787–8817 RSC.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. h. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- A. M. Vinggaard, U. Hass, M. Dalgaard, H. R. Andersen, E. Bonefeld-Jørgensen, S. Christiansen, P. Laier and M. E. Poulsen, Int. J. Androl., 2006, 29, 186–192 CrossRef CAS PubMed.
- Y. Gao, D.-C. Huang, C. Liu, Z.-L. Song, J.-R. Liu, S.-K. Guo, J.-Y. Tan, R.-L. Qiu, B. Jin, H. Zhang, N. Mulholland, X. Han, Q. Xia, A. S. Ali, D. Guo, Y. Deng, Y.-C. Gu and M.-Z. Zhang, Bioorg. Med. Chem., 2021, 35, 116073 CrossRef CAS PubMed.
- B. D. Tonietto, A. O. M. Laurentino, M. T. Costa-Valle, L. V. Cestonaro, B. P. Antunes, C. Sates, N. G. dos Santos, E. Dallegrave, S. C. Garcia, M. B. Leal and M. D. Arbo, Environ. Toxicol. Pharmacol., 2022, 94, 103924 CrossRef CAS PubMed.
- M. A. Shalaby, A. M. Fahim and S. A. Rizk, Sci. Rep., 2023, 13, 4999 CrossRef CAS PubMed.
- E. Weitz and A. Scheffer, Ber. Dtsch. Chem. Ges., 1921, 54, 2327–2344 CrossRef.
- Y. Apeloig, M. Karni and Z. Rappoport, J. Am. Chem. Soc., 1983, 105, 2784–2793 CrossRef CAS.
- L. Knorr and A. Blank, Ber. Dtsch. Chem. Ges., 1884, 17, 2049–2052 CrossRef.
- E. C. Cipagauta Esquivel, V. C. Rufino, M. H. Trindade Nogueira, A. C. Carbonaro Souza, J. R. Pliego Júnior and M. S. Valle, J. Mol. Struct., 2020, 1204, 127536 CrossRef CAS.
- D. Nair, P. Pavashe, S. Katiyar and I. N. N. Namboothiri, Tetrahedron Lett., 2016, 57, 3146–3149 CrossRef CAS.
- S. Laufer, G. Wagner and D. Kotschenreuther, Angew. Chem., Int. Ed., 2002, 41, 2290–2293 CrossRef CAS PubMed.
- C. Zhang, S. Sarshar, E. J. Moran, S. Krane, J. C. Rodarte, K. D. Benbatoul, R. Dixon and A. M. M. Mjalli, Bioorg. Med. Chem. Lett., 2000, 10, 2603–2605 CrossRef CAS PubMed.
- K. Faghihi, K. Zamani, A. Mirsamie and M. Reza Sangi, Eur. Polym. J., 2003, 39, 247–254 CrossRef CAS.
- B. A. Bhat, K. L. Dhar, S. C. Puri and M. Spiteller, Synlett, 2006, 2006, 2723–2726 CrossRef.
- L. Varga, T. Nagy, I. Kövesdi, J. Benet-Buchholz, G. Dormán, L. Ürge and F. Darvas, Tetrahedron, 2003, 59, 655–662 CrossRef CAS.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS.
- A. Mohamed, A. M. Fahim and M. A. Ibrahim, J. Mol. Model., 2020, 26, 354 CrossRef CAS PubMed.
- N. A. Kheder, A. M. Fahim, N. S. Mahmoud and K. M. Dawood, J. Mol. Struct., 2024, 1295, 136772 CrossRef CAS.
- M. N. Ahmed, H. Andleeb, A. M. Fahim, M. Madni, S. W. Khan, B. Kaboudin, M. A. A. Ibrahim, P. A. Sidhom and D. M. Gil, J. Mol. Struct., 2024, 1296, 136908 CrossRef CAS.
- H. A. Ghabbour, A. M. Fahim, M. A. Abu El-Enin, S. T. Al-Rashood and H. A. Abdel-Aziz, Mol. Cryst. Liq. Cryst., 2022, 742, 40–55 CrossRef CAS.
- S. Madhankumar, P. Muthuraja and M. Dhandapani, J. Mol. Struct., 2019, 1181, 118–130 CrossRef CAS.
- J. Ma, S. Tong, P. Wang, W. Liao, H. Liu and L. Zhang, J. Econ. Entomol., 2010, 103, 492–496 CrossRef CAS PubMed.
- Z.-P. Che, J.-M. Yang, X.-J. Shan, Y.-E. Tian, S.-M. Liu, X.-M. Lin, J. Jiang, M. Hu and G.-Q. Chen, J. Asian Nat. Prod. Res., 2020, 22, 678–688 CrossRef CAS PubMed.
- X. Zheng and J. Polli, Eur. J. Pharm. Sci., 2010, 41, 43–52 CrossRef CAS PubMed.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- M. Kanagawa, T. Satoh, A. Ikeda, Y. Adachi, N. Ohno and Y. Yamaguchi, J. Biol. Chem., 2011, 286, 29158–29165 CrossRef CAS PubMed.
- J. M. Otero, A.-J. Noel, P. Guardado-Calvo, A. L. Llamas-Saiz, W. Wende, B. Schierling, A. Pingoud and M. J. van Raaij, Acta Crystallogr., Sect. F, 2012, 68, 1139–1148 CrossRef CAS PubMed.
- A. M. Fahim, S. Dacrory and G. H. Elsayed, Sci. Rep., 2023, 13, 14563 CrossRef CAS PubMed.
- B. M. Ivković, K. Nikolic, B. B. Ilić, Ž. S. Žižak, R. B. Novaković, O. A. Čudina and S. M. Vladimirov, Eur. J. Med. Chem., 2013, 63, 239–255 CrossRef PubMed.
- M. A. Shalaby, H. M. Al-Matar, A. M. Fahim and S. A. Rizk, J. Phys. Chem. Solids, 2022, 170, 110933 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. E. R. Gomperts, O. Stratmann, A. Yazyev, J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. Xu, R. Xia, M. Luo, X. Xu, J. Cheng, X. Shao and Z. Li, J. Agric. Food Chem., 2014, 62, 381–390 CrossRef CAS PubMed.
- X. Yu, Y. Liu, Y. Li and Q. Wang, J. Agric. Food Chem., 2015, 63, 9690–9695 CrossRef CAS PubMed.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
- K. Yamamoto, A. Higashiura, M. D. T. Hossain, N. Yamada, T. Shiotsuki and A. Nakagawa, Arch. Biochem. Biophys., 2015, 566, 36–42 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c