
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational studies and synthesis of 131iodine-labeled nocardiotide A analogs as a peptide-based theragnostic radiopharmaceutical ligand for cancer targeting SSTR2†
Rizky Juwita Sugihartiab,
Rani Maharani *c,
Fransiska Kurniawana,
Rahmana Emran Kartasasmitaa and
Daryono Hadi Tjahjono*a
*c,
Fransiska Kurniawana,
Rahmana Emran Kartasasmitaa and
Daryono Hadi Tjahjono*a
aSchool of Pharmacy, Bandung Institute of Technology, Bandung, Indonesia. E-mail: daryonohadi@itb.ac.id
bResearch Center for Radioisotope, Radiopharmaceutical, and Biodosimetry Technology, National Research and Innovation Agency, Indonesia
cDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor, Indonesia. E-mail: r.maharani@unpad.ac.id
First published on 4th April 2024
Abstract
Radiolabeled peptides belong to a highly specific group of radiotracers used in oncology, particularly for diagnostics and cancer therapy. With the notable advantages of high binding affinity and selectivity to cancer cells, they have proven to be very useful in nuclear medicine. As a result, efforts have been focused on discovering new peptide sequences for radiopeptide preparation. Nocardiotide A, a cyclic hexapeptide comprising the amino acids cyclo-Trp-Ile-Trp-Leu-Val-Ala (cWIWLVA) isolated from Nocardiopsis sp., has shown significant cytotoxicity against cancer cells, rendering it a suitable candidate for the process. Therefore, the present study aimed to design a stable and effective radiopeptide by labeling nocardiotide A with iodine-131 (131I), ensuring that its affinity to SSTR2 is not compromised. In silico study showed that structural modification of nocardiotide A labeled with 131iodine exhibited good affinity value, forming hydrogen bonds with key residues, such as Q.102 and T.194, which are essential in SSTR2. Based on the results, cyclic hexapeptides of cWIWLYA were selected for further synthesis, and its peptide product was confirmed by the presence of an ionic molecule peak m/z [M + Na]+ 855.4332 (yield, 25.60%). In vitro tests conducted on cWIWLYA showed that cWIWLYA can bind to HeLa cancer cells. Radiopeptide synthesis was initiated with radiolabeling of cWIWLYA by 131I using the chloramine-T method that showed a radiochemical yield of 93.37%. Non-radioactive iodine labeling reaction showed that iodination was successful, which detected the presence of di-iodinated peptide (I2-cWIWLYA) with m/z [M + Na]+ 1107.1138. In summary, a radiopeptide derived from nocardiotide A showed great potential for further development as a diagnostic and therapeutic agent in cancer treatment.
1 Introduction
Radiolabeled peptides, also known as radiopeptides, serve as a carrier to transport radioisotopes to the cancer site for its diagnosis and therapy.1 They specifically target overexpressed peptide receptors in cancer tissues, exhibiting selectivity towards tumor-specific cells. This unique property, stemming from their high-affinity characteristics, enables the radiopeptide to be highly effective in early cancer detection and treatment initiation. With these advantages, the search and development of novel peptide compounds are on the rise.2 One of the promising candidates is a radiopeptide designed to target the peptide receptor of somatostatin receptor 2 (SSTR2),3,4 offering enhanced visualization for cancer imaging and targeted therapy.5Naturally occurring peptides with cytotoxic properties were predominantly developed as anticancer peptides (ACPs) due to their ability to traverse cell membranes and penetrate the cancer cell.6 To enhance penetration, it is crucial to utilize peptides with low molecular weight (Mw < 10 kDa). It is also required to resist proteolytic degradation through possible modification by cyclization, resulting in increased affinity.7
Nocardiotide A, a cyclic hexapeptide composed of cyclo-Trp-Ile-Trp-Leu-Val-Ala (cWIWLVA), is derived from marine sponges of Callyspongia sp. This compound exhibits significant cytotoxicity against human MM.1S multiple myeloma, human HeLa cervix carcinoma, and murine cells CT26 colon carcinoma with IC50 8, 11, and 12 μM mL−1, respectively.8 The distinctive biological activity demonstrated by nocardiotide A makes it an attractive candidate for the development of an ACP. Furthermore, the total chemical synthesis of this compound has been achieved through a combination of solid-phase peptide synthesis (SPPS) and liquid-phase peptide synthesis (LPPS), resulting in a yield of 20%.9 Given present evidence on its selective anticancer activity and the ease of synthesis, nocardiotide A holds a great potential as a lead compound that can be modified, synthesized, and developed into a radiopeptide for cancer detection and therapy.
The choice of a radioisotope that can be easily incorporated into the substrate with minimal structural modification is crucial for the production process. In the previous computational study, nocardiotide A labeled by technetium-99m showed radiolabeling had an impact with decreasing the binding free energies.10 Among the available options for radiolabeling, 131I stands out as one of the most favorable. It possesses a relatively long half-life of 8.05 days as well as emits gamma radiation (82%, 0.364 MeV) and beta particles (86%, 0.607 MeV). This combination of characteristics makes 131I an ideal candidate for the development of theragnostic agents, which can be utilized in diagnostic procedures, therapy, and monitoring the efficacy of therapy in nuclear imaging.11 A variety of small-molecule polypeptides have radiolabeled by iodine-131.12–14 In addition, the iodine radioisotope also presents as 123I for diagnostic application with SPECT and 124I for PET imaging.
The labelling process using 131I involves its incorporation into tyrosine or histidine residues within the peptide. However, the original structure of nocardiotide A does not provide accessibility to these residues. To circumvent this limitation, a modification of nocardiotide A can be conducted by incorporating histidine and tyrosine into the original structure. Therefore, the present study aims to design an analog of nocardiotide A and synthesize the selected peptide sequence based on docking results.15–17 Additionally, cytotoxicity was assessed and a preliminary study on radiolabelling of the newly developed peptide by 131I for radiopeptide candidates was conducted.
2 Experimental
2.1 Docking studies
A complete 3D structural model of SSTR2 was obtained from RSCB with PDB ID 7XAT. In order to introduce a radiolabeling target into the original nocardiotide A, its analogs were designed by incorporating histidine and tyrosine. The molecular interaction assessment of the 131I labeled nocardiotide A analogs with SSTR2 were conducted for both mono-iodinated [131I-peptide] and di-iodinated peptide [131I2-peptide] structures as shown in Fig. 1. The obtained structure from geometry optimization was subsequently utilized for molecular docking simulation, where the interactions study of selected analogs to the SSTR2 was performed by AutoDock Vina 1.1.6.18,19 To determine hydrogen and hydrophobic bonds, MGLTools 1.5.6 and Discovery Studio 2020 were employed. Finally, molecular dynamics simulations were performed using AMBER18, running for 200 ns for each peptide and labeled iodine peptide with the best value of free binding energy in SSTR2.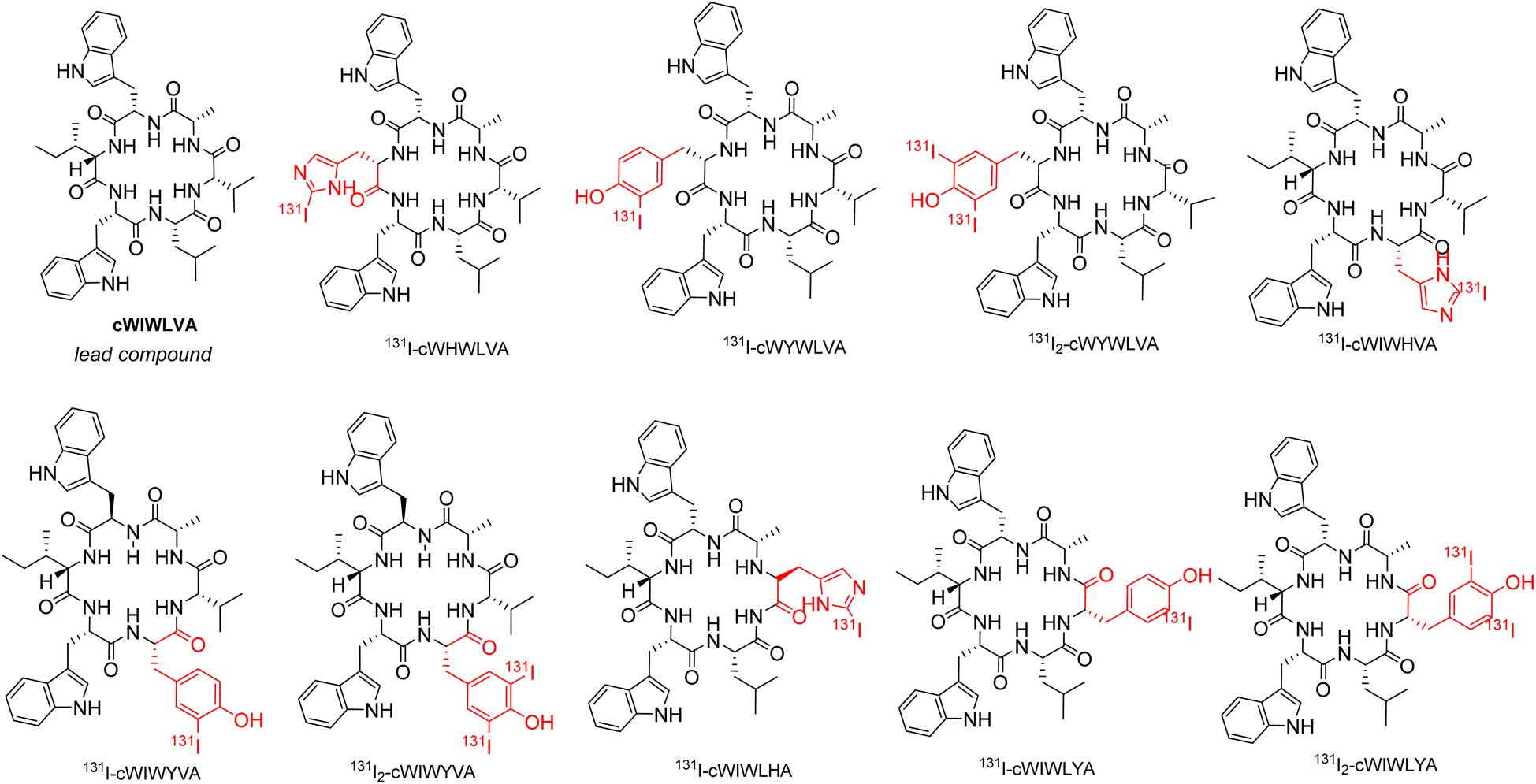 | ||
| Fig. 1 Chemical structure of nocardiotide-A (cWIWLVA) and its analogs labeled by 131I. | ||
2.2 Synthesis of the selected peptide
The SPPS method was employed to synthesize linear hexapeptides using 2-chlorotrityl chloride resin and the Fmoc strategy. A mixture of dichloromethane (DCM) and N,N-dimethylformamide (DMF) served as the solvent, while N,N′-diisopropylcarbodiimide (DIC) and oxyma were utilized as coupling reagents. The Fmoc group was removed with 20% piperidine solution in DMF, and the completion of the coupling protocol was monitored by the chloranil test. In the synthesis, all amino acids used were Fmoc-protected and all side-chain functionalities were protected with TFA labile groups. To release the linear peptides from the resin, a mixture of acetic acid (AcOH), trifluoroethanol (TFE), and dichloromethane (DCM) in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:6 was used as the cleavage solution for a duration of 2 hours. This resulted in linear hexapeptides with protected side chains characterized by TOF-ESI-MS (Waters Q-Tof MS Xevo). Following this, the protected precursor was used for the cyclization reaction without purification.
:2:6 was used as the cleavage solution for a duration of 2 hours. This resulted in linear hexapeptides with protected side chains characterized by TOF-ESI-MS (Waters Q-Tof MS Xevo). Following this, the protected precursor was used for the cyclization reaction without purification.
The cyclization of peptide was performed in the solution phase using 1.14 equivalents of PyBOP under a diluted concentration of 2.0 mM DMF in the presence of basic DIPEA (1.65 eq.) and NaCl in H2O (5 eq.). The reaction mixture was stirred for 24 hours at room temperature and monitored by thin-layer chromatography using iso-propanol:n-hexane (8:2) as the eluent. Subsequently, it was extracted using ethyl acetate and saturated NaCl solution. The ethyl acetate phase was collected and evaporated to remove the solvent. The crude cyclic product from the extraction was subjected to deprotection by 95% TFA in water.20 The peptide synthesis procedures are shown in S.1,† with the resulting product being purified, characterized by TOF-ESI-MS and RP-HPLC (Agilent 1260). Analytical RP-HPLC was performed with C18 column (ZORBAX Eclipse XBD C18 4,6 × 150 mm) at a flow rate of 1.0 mL min−1 (with wave length detection of 254 nm) with 100% H2O (solvent A) and 100% acetonitrile (solvent B). The analytical HPLC starts from initial settings 100% A for 10 min and is followed by a linear gradient of 100% A and 0% B to 20% A and 80% B over 40 min. The gradient remained at this position for 25 min before switching back to initial settings of 100% A for another 5 min.
2.3 In vitro study
To evaluate the cytotoxic activity of the selected peptide, an MTT test was performed on HeLa cancer cells, with synthesized cWIWLVA serving as the lead compound for comparison. The cells were cultured in RPMI medium supplemented with FBS and penicillin–streptomycin, and then incubated in a 5% CO2 incubator at 37 °C. After 48 h, the cultures were transferred into 96 well plates with a certain density per well and incubated for an additional 48 h. They were treated with peptides at various concentration range and incubated for another 48 h. Following the treatment period, 100 μL of culture medium containing MTT at a concentration of 5 mg mL−1 was added to each well, and the plates were incubated for 3 hours at 37 °C. The purple formazan crystals that formed were dissolved by adding 10% sodium dodecyl sulphate (SDS) stopper reagent and left in the dark overnight. Furthermore, the quantity of formazan, which was presumed to be directly proportional to the number of viable cells, was measured using an ELISA reader by calculating the absorbance at 595 nm.21,22 The IC50 values of the selected peptide were estimated and compared to those of the lead compound using Prism GraphPad 8.4.3 software.2.4 Radiolabeling of peptide
The process began by dissolving 3 mg of the peptide in a small amount of DMSO, then H2O was added to up 1.5 mL. The reagents including chloramine-T (8 mg mL−1) and sodium metabisulfite (7 mg mL−1) were individually dissolved in 1 mL of 0.05 mol L−1 PBS (pH 7.4). Radioisotope 131I (3 mCi) was added to the 450 μL peptide solution followed by 100 μL chloramine-T. The mixture was vortexed at room temperature for 2 minutes and 100 μL sodium metabisulfite was then added to stop the reaction. The radiolabelling efficiency was assessed by thin layer chromatography (Whatmann 3MM) using 0.9% saline as the mobile phase. The stability of radiolabeled peptide at room temperature was then evaluated for several days, while its stability in human plasma was observed for 72 hours. Radiochemical yield was estimated by TLC scanner (TLC 204, Comecer) and γ-counter (The Capintec™ CAPRAC-t Wipe-Test Counter).Subsequently, an additional synthesis of the non-radioactive iodine labeling reaction was conducted, and its successful iodination was confirmed by TOF-ESI-MS. The experiment was performed using the same method that was started by replacing 131I with 0.6 mg KI dissolved in 1 mL of 0.05 mol L−1 PBS (E. Merck). The experiment was conducted in a small glass vial, with a molar ratio of peptide:KI:chloramine-T set at 1:1:1.
3 Results and discussion
In this study, analogs of nocardiotide A were designed as a radiopeptide labelled with 131I, specifically targeting SSTR2. Their binding affinity is essential in cancer imaging as it directly affects the quality of visualizing cancer cells within the body.6,23 Therefore, interactions between the designed analogs labelled with 131I and SSTR2 were explored to determine the effect of geometry changes upon the radiolabelling process.To develop a radiopeptide based on 131I, it is necessary to incorporate either histidine or tyrosine into the peptide structure. Consequently, modification to the original structure of nocardiotide A is required.
According to a previous synthetic study conducted by Muhajir et al., the site between alanine and tryptophan at the C terminus and N-terminus, respectively, was selected for the cyclization of the linear precursor, rendering the two residues irreplaceable. Previous studies highlighted the significant role of tryptophan as amino acid residues in the effects of ACP on cancer cells, particularly in terms of their toxicity. Considering this, the modification of nocardiotide A analogs conducted, allowing for the presence of two tryptophan in the peptide analogs' structure.24 To achieve this, amino acids histidine and tyrosine were incorporated individually to replace valine, leucine, and isoleucine residues in the original structure. As a result, 6 nocardiotide A analogs, namely cWHWLVA, cWIWHVA, cWIWLHA, cWIWLYA, cWIWYVA, and cWYWLVA were designed and subjected to a docking study. The addition of 131I to tyrosine and histidine by radioiodination through direct labeling resulted in the chemical structure shown in Fig. 1.
Molecular docking was conducted to analyze and identify the important residues responsible for the receptor–ligand interaction between the radiopeptide derived from nocardiotide A and SSTR2 (PDB ID 7XAT) within the proposed binding site of SSTR2 which defines as D89, F92, Q102, A104, M119, D122, N125, Q126, R190, C193, T194, Y205, I209, T212, G216, F265, W269, F272, Y273, N276, K291, V298, T301, Y302, N304 and S305.
Table 1 provides a summary of the SSTR2-peptide interactions observed in the docking studies. The results of a molecular docking showed that both peptides and their labelled peptides were successfully accommodated in the binding site. These results presented the depiction of hydrogen bonds and hydrophobic interactions in the complex system, which played a crucial role in protein–ligand interactions and were related to binding affinity.25 Hydrogen bonds identified in the designed and labeled peptides were following the previous in silico study. They corresponded to critical residues in the active site of SSTR2, which included Q.102 and T.194,26 and this led to the high binding energy of the designed radiopeptide. The binding (affinity) energies ranged from −10.3 to −11.7 kcal mol−1, indicating minor structural alteration from the original substrate in iodine radiolabelling.
| No. | Peptide | ΔG kcal mol−1 | Hydrogen bond | Hydrophobic interaction |
|---|---|---|---|---|
| 1 | cWHWLVA | −9.7 | Q102, L290, V103, K291 | R184, S192, F294, V298 |
| 131I-cWHWLVA | −9.6 | T194 | L99, I177, I195, F208, I209, F275, S279, F294, L290, V298 | |
| 2 | cWIWHVA | −10.5 | Q102, T194, L290 | F294, V298 |
| 131I-cWIWHVA | −10.4 | Q102, L290 | V103, F294, V298 | |
| 3 | cWIWLHA | −10.1 | Q102, T194 | V298, L99, F294, L290 |
| 131I-cWIWLHA | −9.6 | Q102, T194 | L99, R190, L290, F294, V298 | |
| 4 | cWIWLYA | −10.1 | Q102, N186, L290 | L99, R184, R190, T194, F294, V298 |
| 131I-cWIWLYA | −9.9 | N196 | L99, I195, N196, L290, F294, V298 | |
| 131I2-cWIWLYA | −9.7 | I195, N196 | L99, F208, L290, F294, V298 | |
| 5 | cWIWYVA | −10.2 | Q102 | L290, K291, F294, V298 |
| 131I-cWIWYVA | −10.3 | Q102, L290 | K291, F294, V298 | |
| 131I2-cWIWYVA | 10.1 | Q102 | V103, L290, F294, V298 | |
| 6 | cWYWLVA | −9.9 | T194, F275 | F272, L290, F294 |
| 131I-cWYWLVA | −9.9 | T194, I195, F275 | F208, F294 | |
| 131I2-cWYWLVA | −9.9 | R184, T194, F275 | I195, F208, F294 |
Although all designed radiopeptides showed different interactions with changed binding residue in the binding pocket of SSTR2, the computational study revealed that the designed radiopeptide showed no significant change in the affinity value for SSTR2 compared to the original peptide. Thus, for synthesis purposes, analogs cWIWLHA and cWIWLYA were selected as target peptides with the consideration of small residue leucine and alanine between histidine or tyrosine, which facilitated the process. Meanwhile, other analogs such as cWYWLVA, WHWLVA, cWIWHVA, and cWIWYVA contain two adjacent tryptophan residues, as well as two adjacent tryptophan and valine residues. They were initially considered challenging to synthesize due to steric hindrance.
The visualization of the computational study selected peptide cWIWLYA is shown below. Fig. 2 shows a superimposed image of cWIWLYA with its radiolabelled iodine. Fig. 3 shows the interactions of cWIWLYA labeled by 131I in the active site of SSTR2, highlighting the significant amino acids that contributed to the ligand–protein binding.
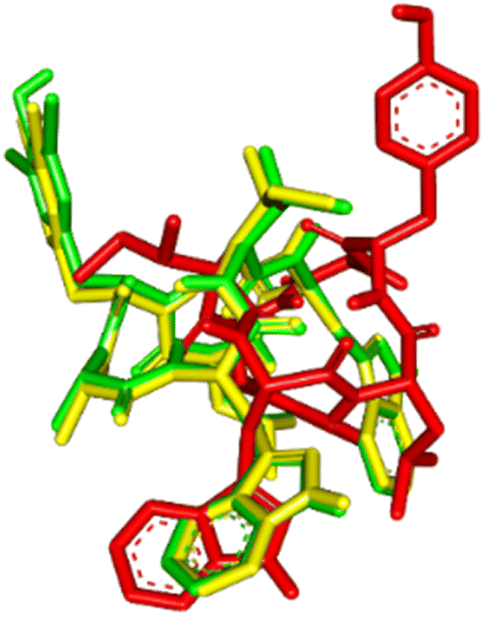 | ||
| Fig. 2 Superimpose of cWIWLYA (red), 131I-cWIWLYA (yellow) and 131I2-cWIWLYA (blue) in the active sites of SSTR2. | ||
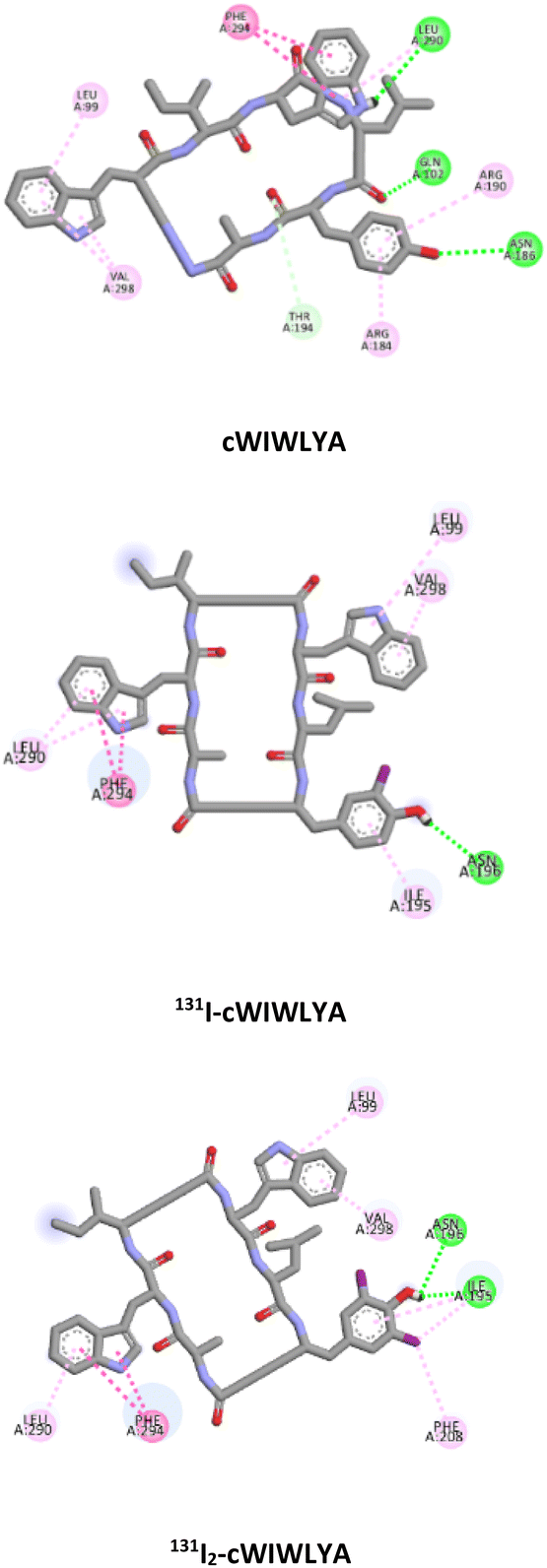 | ||
| Fig. 3 Interaction of cWIWLYA, 131I-cWIWLYA and 131I2-cWIWLYA in the binding sites of SSTR2. | ||
The result of the molecular dynamic study showed that each peptide and labeled peptide were stable in the binding pocket for 200 ns, without significant conformational changes as seen on Fig. 4.
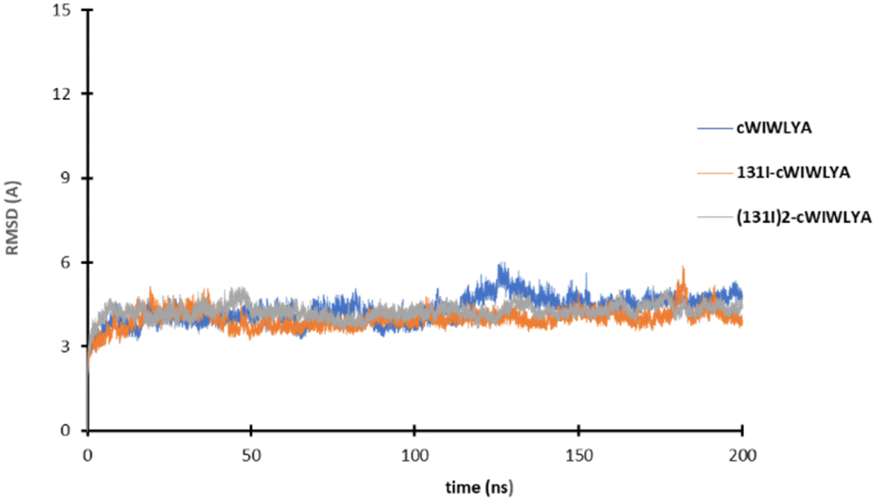 | ||
| Fig. 4 The plot of the RMSD value of cWIWLYA, 131I-cWIWLYA and 131I2-cWIWLYA during the molecular dynamic simulation. | ||
All amino acids used in the synthesis of analogs cWIWLHA and cWIWLYA were Fmoc-protected, with the indole group, histidine side chain, and tyrosine side chain being protected by tert butyloxycarbonyl (Boc) group, tritly (Trt) group, and tert-butyl (tBu), respectively. The procedure for synthesizing nocardiotide A analogs is based on its total synthesis by Muhajir et al., where alanine and tyrosine were selected as C termini and N termini for cyclization due to a combination of small and large residues. The protected linear peptides cWIWLHA and cWIWLYA were prepared by attaching the first amino acid Fmoc-L-Ala-OH onto resin. This was followed by the attachment of the second amino acid Fmoc-L-His(Trt)-OH or Fmoc-L-Tyr (tBu)-OH, which was then continued to the sixth amino acid Fmoc-L-Trp(Boc)OH (Fig. S.1).† Furthermore, the crude of protected linear WIWLHA and WIWLYA were characterized by ESI-MS showing confirmed molecular ion peaks at m/z [M + H]+ 1267.6594 (calculated m/z 1267.6556) with a yield of 9.20% and [M + H]+ 1107.629 (calculated m/z 1107.6130) with a yield of 52.90% (Fig. S.2).†
During the synthesis of linear WIWLHA precursors, an issue arose with the yield of the synthesized product. It was suspected that a coupling failure had occurred during histidine as the second amino acid. This was attributed to the bulky structure of the Trt as a protective group, which could have interfered with the amide bond formation reaction due to aggregation, thereby making the coupling process difficult.
The results of the mass spectrum of the protected linear WIWLHA compounds showed a trace of the product at the baseline. It indicated that the synthetic product is minor and not prospective to proceed to the next stage. As a linear of analog WIWLYA was obtained in good yield, the linear precursor was then characterized directly without further purification of the precursor using mass spectrometry and 1H-NMR to confirm the successfulness of the synthesis (Fig. S.3).† The precursor was further cyclized using PyBOP and subsequently Boc- and t-Bu deprotected, giving the desired crude cyclic product of cWIWLYA. The fractions containing cWIWLYA from the open column chromatography using C-18 silica gel were collected and evaporated giving the desired cyclic peptide. The purified product was then characterized by mass spectrometry. Furthermore, the synthetic cWIWLYA was analysed by analytical RP-HPLC showing the peak in the chromatogram with a purity of 80%. The profiles of analytical RP-HPLC chromatogram of the cWIWLYA was compared to the those of the protected linear WIWLYA and cWIWLYA, indicating the peaks of the main products in the cyclization (Fig. 5). The molecular mass determined experimentally by ESI-MS corresponded with the theoretically calculated monoisotopic mass for cyclic peptide cWIWLYA m/z [M + Na]+ 855.4332 (calculated m/z 855.4350) (Fig. S.4).† The 1H-NMR and 13C-NMR data of product showed the number of protons and carbons that corresponded to the structure of the target compound (Fig. S.5 and S.6).† Finally, the purified white solid product of crude peptides had an overall yield of 25.6%.
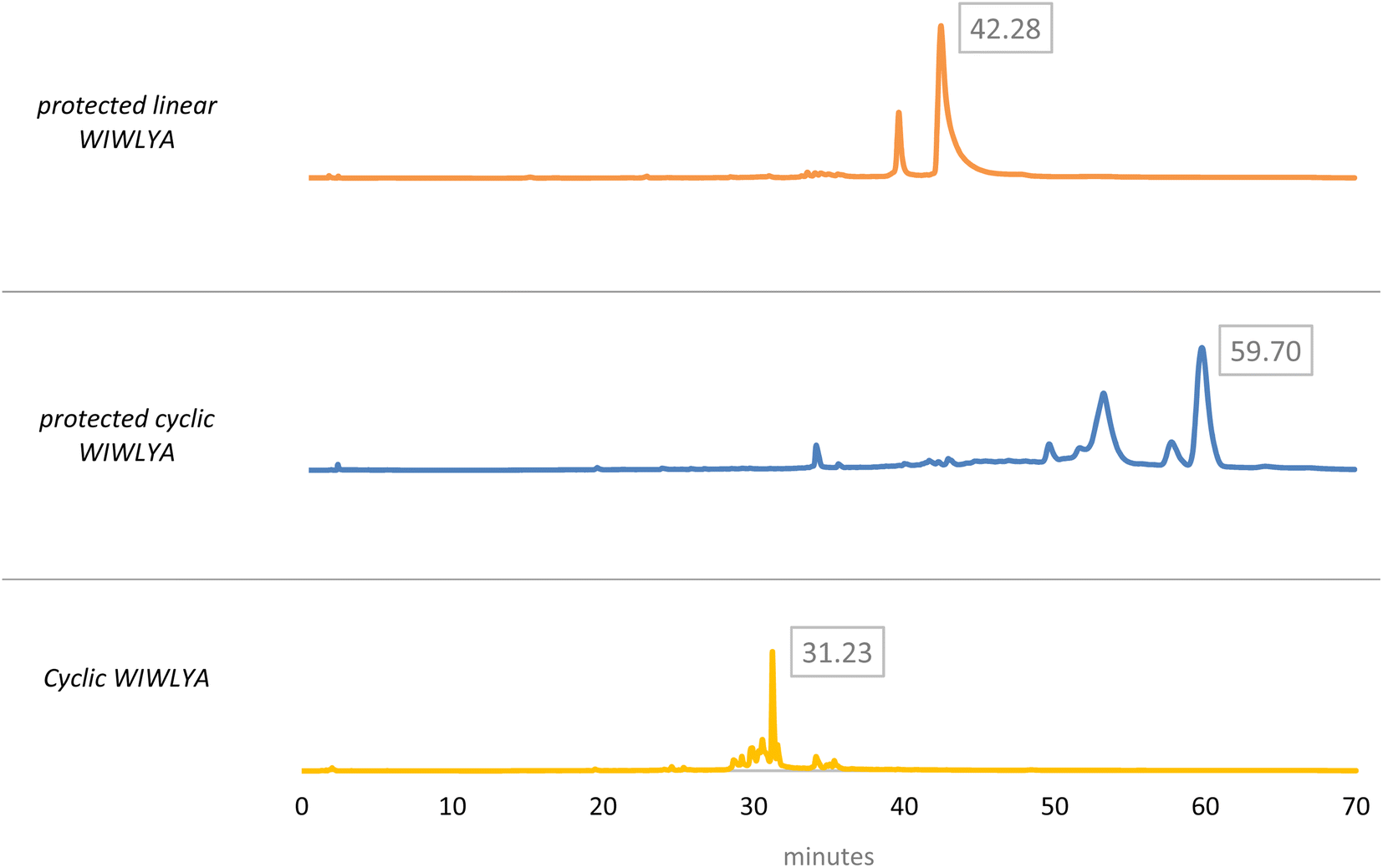 | ||
| Fig. 5 Analytical HPLC chromatogram of the cWIWLYA cyclization synthesis process. | ||
The developed radiopeptide was derived from nocardiotide A which is found as a peptide that has anticancer activity. Therefore, the cytotoxicity test was conducted to observe the change of IC50 value of the analogue peptide where the composition of one of the amino acids in the lead compound was changed (valine replaced by tyrosine). In the in vitro test conducted on HeLa cancer cells, cWIWLYA exhibited an IC50 value of 169.30 μg mL−1, while that of the lead compound cWIWLVA was 52.06 μg mL−1 (Fig. S.7).† Despite exhibiting higher IC50 than the lead compound, the presence of cell death indicated that the active side of the compound could bind to cancer cells. This suggests inhibitory activity and sensitivity of cWIWLYA, making it a potential candidate for a radiopeptide in cancer diagnosis and therapy.
The purified peptide was iodinated using the chloramine-T method (Fig. S.8).† The result of radiolabelling of 131I-cWIWLYA showed that labeling efficiency provide a high radiochemical yield (>90%) (Fig. 6). Further stability study revealed that the product at room temperature was stable for up to three days (Fig. 7). The extent of proteolytic degradation of the 131I-peptide was determined by incubating the radiolabelled peptide with human plasma at 37 °C for up to 72 h. The results showed that the radiolabelled peptide had radioactivity greater than 90% after 5 h of incubation, while on 24 hours remained at 84.9% and on 72 hours 57.5% (Fig. 8). Thus, it is reasonable for further diagnosis and therapy study.
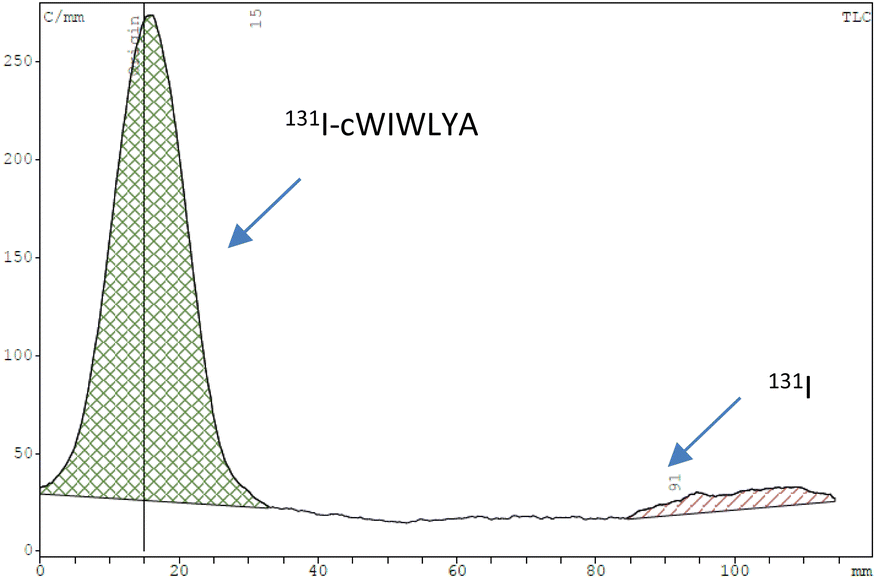 | ||
| Fig. 6 Radiochromatogram profile of the cWIWLYA labeled iodine-131. | ||
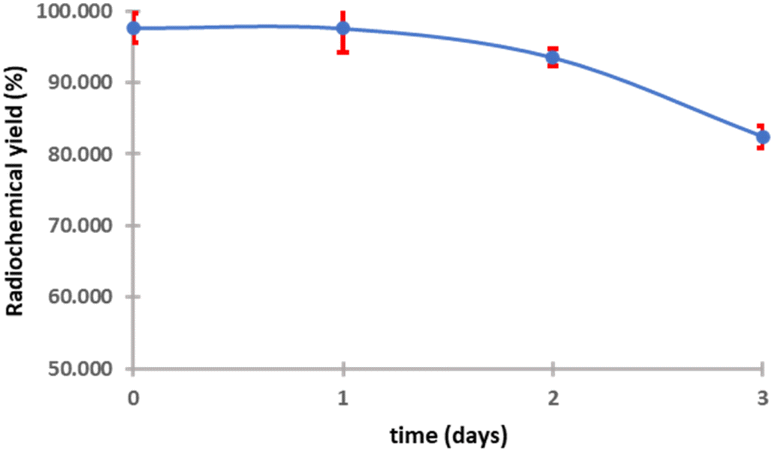 | ||
| Fig. 7 Stability of 131I-cWIWLYA at room temperature. | ||
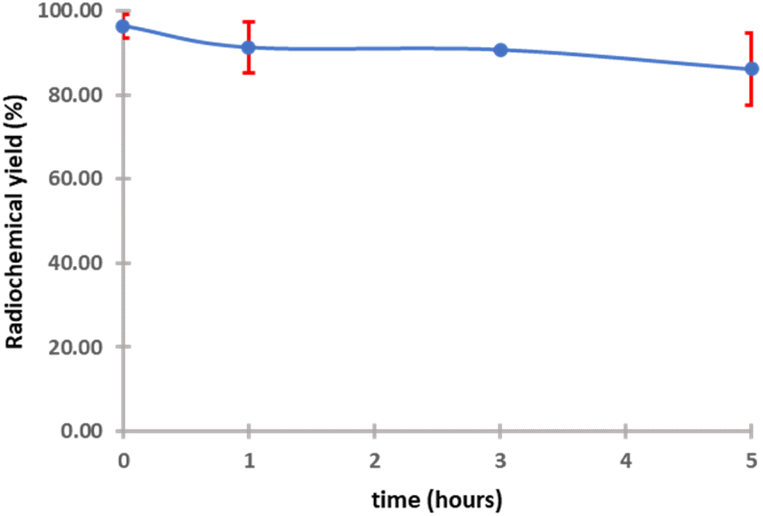 | ||
| Fig. 8 Stability of 131I-cWIWLYA in plasma. | ||
To confirm successful iodination, an additional study was carried out using a non-radioactive method using 1 eq. of peptide. Radioiodination of nocardiotide A analogs by direct labeling will give iodine addition to tyrosine residue at the primary site. The tyrosine moiety can be labeled twice, resulting in a mixture of mono and di-iodinated species. It should be noted that the formation of di-iodinated tyrosine occurs at a faster rate than mono-iodinated.27 The result of iodine-labeled cWIWLYA was characterized using ESI-MS, showing an m/z [M + Na]+ 1107.1138 (calculated m/z 1107.2103) (Fig. S.9).† This confirmed that the product had 2 iodine atoms conjugated to the tyrosine group (di-iodinated). This result supported the notion that a nocardiotide A analog containing a tyrosine residue could be translated into a radioiodine-labeled peptide for the further development of a radiopeptide as a theragnostic agent.
4 Conclusions
The applied computational study showed that all the modified nocardiotide A and designed radiopeptides exhibited binding affinity to SSTR2. The peptide analog that has been successfully synthesized and easily labeled, makes it a prospective candidate for radiopeptide and theragnostic purpose and needs further studies.Author contributions
RJS: conducted computation, completed experiments, collected, and analyzed the data, and wrote the draft manuscript; RM: conducted supervision in peptide synthesis, analyzed data, and corrected the manuscript; FK: conducted supervision in computation, analyzed data, and corrected the manuscript; REK: supervised and corrected the manuscript; DHT: designed the method, conducted supervision, provided resources, and corrected the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the National Research and Innovation Institute Scholarship Program Fiscal Year of 2022–2023 and the Bandung Institute of Technology (28/IT1.C10/SK-DA/2023). The authors acknowledge the facilities and scientific as well as technical support from Advanced Characterization Laboratories Serpong, National Research and Innovation Institute through E-Layanan Sains. The authors also thank the researchers at the G. A. Siwabessy Multi-Purpose Reactor, Directorate of Nuclear Facility Management, and Radioisotope Research Group (PRTRRB)-BRIN for their valuable help in providing iodine-131.References
- M. Fani, H. R. Maecke and S. M. Okarvi, Theranostics, 2012, 2, 481–501 CrossRef CAS PubMed.
- P. Krȩcisz, K. Czarnecka, L. Królicki, E. Mikiciuk-Olasik and P. Szymański, Bioconjugate Chem., 2021, 32, 25–42 CrossRef PubMed.
- R. Mikołajczak and H. R. Maecke, Nuclear Medicine Review, 2016, 19, 126–132 CrossRef PubMed.
- R. Eychenne, C. Bouvry, M. Bourgeois, P. Loyer, E. Benoist and N. Lepareur, Molecules, 2020, 25, 4012 CrossRef CAS PubMed.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Front. Chem., 2020, 8, 1–24 CrossRef PubMed.
- A. Borrelli, A. L. Tornesello, M. L. Tornesello and F. M. Buonaguro, Molecules, 2018, 23(2), 295 CrossRef PubMed.
- P. Scodeller and E. K. Asciutto, Molecules, 2020, 25, 1–24 CrossRef PubMed.
- A. H. Ibrahim, E. Z. Attia, D. Hajjar, M. A. Anany, S. Y. Desoukey, M. A. Fouad, M. S. Kamel, H. Wajant, T. A. M. Gulder and U. R. Abdelmohsen, Mar. Drugs, 2018, 16, 1–13 CrossRef PubMed.
- M. I. Muhajir, A. Hardianto, J. Al-Anshori, D. Sumiarsa, T. Mayanti, Nurlelasari, D. Harneti, A. T. Hidayat, U. Supratman and R. Maharani, ChemistrySelect, 2021, 6, 12941–12946 CrossRef CAS.
- R. J. Sugiharti, R. Maharani, R. E. Kartasasmita and D. H. Tjahjono, J. Math. Fundam. Sci., 2023, 55, 77–91 CrossRef.
- A. Yordanova, E. Eppard, S. Kürpig, R. A. Bundschuh, S. Schönberger, M. Gonzalez-Carmona, G. Feldmann, H. Ahmadzadehfar and M. Essler, OncoTargets Ther., 2017, 10, 4821–4828 CrossRef PubMed.
- T. He, J. Du, K. Zhu, Y. Zhou, Z. Xiao, W. Liu, W. Ren, X. Liu, T. Chen, W. Liu, Z. Chen, G. Ni, X. Liu, T. Wang, J. Quan, P. Zhang and J. Yuan, Biomed. Pharmacother., 2023, 164, 114891 CrossRef CAS PubMed.
- R. Lin, B. Ma, N. Liu, L. Zhang, T. He, X. Liu, T. Chen, W. Liu, Y. Liang, T. Wang, G. Ni, X. Liu, N. Yang, J. Zhang and J. Yuan, Ann. Nucl. Med., 2021, 35, 811–822 CrossRef CAS PubMed.
- Z. Chen, H. Gao, M. Li, S. Fang, G. Li and L. Guo, Anticancer Drugs, 2017, 28, 480–488 CrossRef CAS PubMed.
- L. Pinzi and G. Rastelli, Int. J. Mol. Sci., 2019, 20, 4331 CrossRef CAS PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7(2), 146–157 CrossRef CAS PubMed.
- M. Ciemny, M. Kurcinski, K. Kamel, A. Kolinski, N. Alam, O. Schueler-Furman and S. Kmiecik, Drug Discovery Today, 2018, 23, 1530–1537 CrossRef CAS PubMed.
- M. Ciemny, M. Kurcinski, K. Kamel, A. Kolinski, N. Alam, O. Schueler-Furman and S. Kmiecik, Drug Discovery Today, 2018, 23, 1530–1537 CrossRef CAS PubMed.
- N. London, B. Raveh and O. Schueler-Furman, Curr. Opin. Struct. Biol., 2013, 23, 894–902 CrossRef CAS PubMed.
- M. Liu, Y. C. Tang, K. Q. Fan, X. Jiang, L. H. Lai and Y. H. Ye, J. Pept. Res., 2005, 65, 55–64 CrossRef CAS PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harb. Protoc., 2018, 2018, 469–471 Search PubMed.
- M. Ghasemi, T. Tyron, S. Sebastian and I. Kempson, Int. J. Mol. Sci., 2021, 22, 1–30 Search PubMed.
- A. L. Tornesello, M. L. Tornesello and F. M. Buonaguro, Mini-Rev. Med. Chem., 2017, 17, 758–770 CrossRef CAS PubMed.
- W. Chiangjong, S. Chutipongtanate and S. Hongeng, Int. J. Oncol., 2020, 57, 678–696 CrossRef CAS PubMed.
- D. Chen, N. Oezguen, P. Urvil, C. Ferguson, S. M. Dann and T. C. Savidge, Sci. Adv., 2016, 2, e1501240 CrossRef PubMed.
- K. S. Nagarajan, S. Babu, H. Sohn, P. Devaraju and T. Madhavan, J. Biomol. Struct. Dyn., 2019, 37, 3081–3102 CrossRef PubMed.
- K. Kumar and K. Woolum, Molecules, 2021, 26, 4344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00684d |
| This journal is © The Royal Society of Chemistry 2024 |