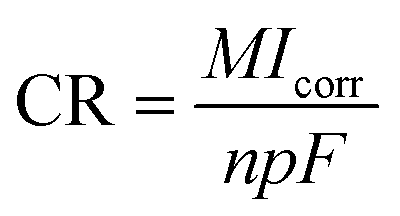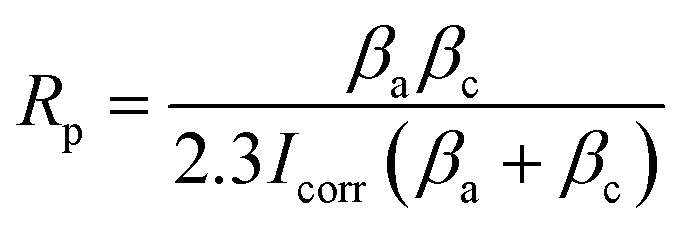DOI:
10.1039/D4RA00707G
(Paper)
RSC Adv., 2024,
14, 11616-11631
Preparation of HA-MAO coatings on β-type alloys and its corrosion resistance in high glucose environments
Received
28th January 2024
, Accepted 31st March 2024
First published on 10th April 2024
Abstract
Aim to provide practical clinical guidance for the treatment of implants in diabetic patients, this study investigated the corrosion mechanism of bionic coatings containing different Ca/P ratios in diabetic environments. The bionic coatings were prepared in β-titanium alloys using micro-arc oxidation (MAO) technology and evaluated for corrosion mechanism, biocompatibility, and safety by cytotoxicity, electrochemical corrosion, and coating bonding force experiments. Ca and P from the electrolyte were integrated into the coating during MAO discharge process to form hydroxyapatite. The coating Ca/P ratio initially increased and then decreased with the electrolyte Ca/P ratio. In vitro cellular experiments demonstrated that increasing the porosity of HA-containing coatings would be beneficial to the growth of cells adhering to their surfaces. Corrosion tests revealed that the corrosion tendency of the coating at higher sugar content was more severe, and a proper elevation of the Ca/P ratio was better for the corrosion resistance of the coating. The bonding analysis of the coatings before and after corrosion showed that an increase in the Ca/P ratio would improve the bonding of the MAO coatings in higher glucose content environments, thus improving the safety of the implants in diabetic patients.
1 Introduction
β titanium alloys exhibit desirable characteristics in the field of biomedical engineering, including a low elastic modulus, high elastic strain allowance, and favorable biocompatibility.1–3 These attributes make them attractive for various applications such as dental implants, cranial bone repair implants, heart valves, and spinal fixation devices.4–6 However, the mismatch in elastic modulus between the alloy and bone tissue,7–9 as well as its biologically inert surface properties,10,11 impose limitations on its wider usage. To overcome these limitations and further optimize the application of β titanium alloys in the medical field, suitable surface treatments have become imperative. Presently, several surface technologies are commonly employed for titanium alloys, including plasma spraying, physical vapor deposition, and chemical vapor deposition.12,13 Nevertheless, these methods entail intricate preparation procedures, relatively high costs, and inadequate adhesion between the coating and the substrate.14 As a novel surface treatment technology, micro-arc oxidation (MAO) offers notable advantages in terms of simplicity, high efficiency, and environmental friendliness. In particular, the technology allows surface treatment of workpieces of different shapes, providing a proven treatment method for personalized medicine.
The mechanism underlying MAO technology suggests that the composition and morphology of the coating are significantly influenced by the electrolyte composition. Researchers have successfully fabricated MAO coatings with diverse functional additives on titanium alloys. These additives had demonstrated enhancements in the mechanical properties and biocompatibility of the coatings. Notably, hydroxyapatite (HA, Ca10(PO4)2(OH)6), which represents the principal inorganic component of human bones, plays a vital role in providing mechanical support as the bone scaffold. HA possesses multiple surface cell-binding sites, facilitating its tight integration with tissue cells. Consequently, HA has emerged as a widely employed biomaterial component in current clinical applications for hard tissue repair.15 However, due to inherent mechanical limitations, such as low toughness, coating techniques are commonly employed to overlay HA onto the surface of metallic implants. MAO technology could combine the excellent mechanical properties of metals with the favorable bioactivity of HA.16 Extensive research has been conducted on MAO coatings incorporating calcium (Ca) and phosphorus (P) in titanium alloys. For instance, Kim et al.17 introduced HA particles into the electrolyte to fabricate MAO TiO2 ceramic coatings containing HA. These coatings exhibited strong adhesion to the substrate and effectively improved the surface biocompatibility of titanium alloys. Zhang et al.18 utilized MAO to produce TiO2/HA composite coatings and investigated the structural variations of the coatings with oxidation time. They observed that prolonged oxidation time led to an increased content of the Ca–P phase within the composite coating, resulting in enhanced corrosion resistance and biocompatibility. Muhammad Qadir et al.19 delved into the ramifications of modulating the mass concentrations of Ca(CH3COO)2 and EDTA-2Na within the electrolyte on the calcium and phosphorus content, as well as their atomic ratio, in the coating engendered by MAO. The study elucidated that as the Ca(CH3COO)2 concentration ascended, there was a noticeable augmentation in the coating calcium content, a decline in phosphorus content, and an expanded calcium to phosphorus atomic ratio. Conversely, with an upsurge in the EDTA-2Na concentration, the coating manifested a significant uptick in calcium content. Yet, the multifaceted nature of the electrolytes and their concentrations, such as the intricacies of Ca/P ratio preparation, currently lack a holistic and methodical discourse.
Implants not only need to promote wound healing, but are also challenged by corrosive damage to the patient's physiologic environment. In diabetic patients, in addition to corrosion due to exposure to body fluids, implants are exposed to the formation of a localized microenvironment, in particular the inhomogeneous accumulation of advanced glycation end products and inflammatory factors (proteins) on the surface of the implant, which accelerates the electro-chemical corrosion reaction.20,21 In the context of the corrosive effects of hyperglycemia on materials, studies conducted by R.-C. Zeng et al.22 have revealed that the presence of glucose expedites the corrosion rate of pure Mg materials in conventional physiological saline, whereas the corrosion rate is constrained when exposed to Hank's balanced salt solution. The researchers propose that glucose can undergo conversion to gluconic acid within physiological saline, thereby reducing the solution pH and promoting Cl− attack on the Mg surface, consequently accelerating the corrosion rate. Furthermore, L.-Y. Cui et al.23 reported that glucose has the capability to dissolve the Mg(OH)2 coating, thereby hastening the corrosion rate of Mg–1.35Ca alloy in a salt solution. Subsequent investigations by L.-Y. Li et al.24 demonstrated that Mg2+ ions can form complexes at lower glucose concentrations (1 g L−1), effectively impeding the contact between Cl− ions and the metal surface, consequently inhibiting the corrosion behavior of AZ31 magnesium alloy. However, at higher glucose concentrations (2, 3 g L−1), the conversion to gluconic acid lowers the pH of the solution, promoting the absorption of Cl− ions onto the surface of the magnesium alloy and subsequently increasing its corrosion rate. Nevertheless, limited research exists on the corrosion behavior of MAO coatings with varying Ca/P ratios on titanium alloy surfaces exposed to a high glucose environment, as well as the influence of corrosion on the coating bonding.
In this study, we would manipulate the Ca/P ratio in the electrolyte solution to MAO coatings with diverse morphologies and compositions. The effects of these coatings on cytotoxicity and cell proliferation were investigated. The corrosion properties and bond strength of these coatings in diabetic environments were also scrutinized to elucidate their durability. This study would provide valuable insights into the fabrication of biocompatible coatings on titanium alloys and their potential application in diabetic patients.
2 Experimental materials and methods
2.1 Experimental materials and coating preparation
The substrate used in this study was a β titanium alloy (Guangxi Medical University Biotechnology Laboratory Self-made) with the following elemental compositions: Ti 69.2%, Nb 24.9%, Zr 4.3%, and Sn 1.6%. The samples were cut into circular discs (with a diameter of 10 mm and a thickness of 2 mm) and were then ground using sandpapers of varying grit sizes ranging from 80# to 1000#. After cleaning with distilled water and ethanol in an ultrasonic cleaner, the samples were air-dried for later use.
The coating process employed a bespoke MAO power supply: employing a stainless-steel electrolytic tank as the cathode and the titanium substrate as the anode. The electrical parameters were set as follows: a duty cycle of 15%, a frequency of 400 Hz, a voltage of 450 V, and an oxidation time of 30 minutes. The electrolyte used consisted of 0.12 mol L−1 of NaH2PO4·12H2O with added concentrations of 0.2, 0.26, 0.32 mol L−1 of Ca(CH3COO)2·H2O, formed the electrolyte with Ca/P ratios of 1.67, 2.17, and 2.67. For ease of description, the coatings made by different Ca/P ratios electrolyte were referred to as 1.67 coating, 2.17 coating, and 2.67 coating.
2.2 Analysis of coating structure
2.2.1 Physical analysis of coatings. The coating morphology, pre- and post-corrosion, was scrutinized utilizing a scanning electron microscope (SEM; SN-3400, Hitachi, Japan). Concurrently, elemental analyses were executed via the integrated energy-dispersive spectrometer (EDS, Hitachi PV8200). EDS data were collected from 10 different regions of each sample and subsequently averaged to obtain the calcium–phosphorus ratio, according to relevant studies, the uncertainty of determining the ratio of calcium to phosphorus by this method was 7%.25 The phase composition of the samples was analyzed by X-ray diffraction (XRD, MiniFlex600-C, Japan) using Kα radiation from a Cu target (λ = 1.5406 Å) at a speed of 10° min−1. XRD spectra were obtained at 100 mA, 40 kV, and 2θ ranging from 10° to 85° (in steps of 0.02°), the phase composition was also analyzed using HighScore Plus. To insight into the elemental composition and chemical bonding, coatings were subjected to X-ray photoelectron spectroscopy (XPS; Rigaku D/MAX 2500V) before and after experiencing corrosion. For calibrating the XPS outcomes, the C 1s binding energy (284.8 eV) was harnessed as a benchmark. In discerning the coating thickness, an eddy current thickness gauge (TT260B, Shenzhen Excellent Instrumentation Co.) was employed, with readings harvested from five disparate regions on each specimen, subsequently averaged to obtain a representative value.
2.2.2 Coating wettability analysis. Hydrophobicity analysis of the coatings was conducted at room temperature (∼28 °C) using the DSA100E (KYUSS, Germany). A droplet volume of 4 μL was employed, and the contact angle results of the water droplet were subsequently interpreted using the Young–Laplace fitting method. For each sample, five parallel experiments were carried out, and the outcomes were averaged, with standard deviations noted.
2.2.3 Coating bonding analysis. The adhesion strength of the coating was assessed using a WS-2005 type automatic scratch tester (Shanghai Wujiu Automation Equipment Co.) for coating adhesion. A diamond indenter, with a cone angle of 120° and a curvature radius of 0.2 mm, was employed. The maximum loading force was 40 N and the scratch speed was set at 40 N min−1, while the worktable movement speed ranged from 2 to 10 mm min−1. Outcomes were determined based on conductivity, and the morphology of the scratches was subsequently observed using SEM.
2.3 Coating biocompatibility analysis
2.3.1 Cell proliferation. Cell viability was quantitatively assessed using the CCK-8 assay. Samples were placed in a 24-well plate, and human venous endothelial cells (HVECs) were seeded onto the sample surface with a density of 1 × 104 cells per cm3, followed by incubation in a cell culture chamber for 48 h. After rinsing with PBS, 500 μL of complete culture medium containing 10% CCK-8 solution was added to each well. The 24-well plate was then returned to the cell incubator for an additional 4 h of cultivation. Once the solution was fully stabilized, 200 μL of the reaction mixture was transferred to a 96-well plate. Absorbance values at 450 nm were subsequently measured using a microplate reader.
2.3.2 Cytoskeleton observation. Samples were placed in a 24-well plate, and human venous endothelial cells were seeded onto the sample surface at an initial density of 1 × 104 cells per cm2. The samples were subsequently incubated in a cell culture chamber for 48 hours. After a PBS wash, cells were fixed using 4% paraformaldehyde. Permeabilization of the cells was achieved by treating them with 0.2% Triton X-100 in PBS for 5 minutes. The cells were then incubated in the dark for 12 hours at 4 °C with an appropriate volume of rhodamine-labeled phalloidin in PBS at a concentration of 5 units per mL. Following another PBS rinse, the cells were stained with Hoechst 33258 for 10 minutes at 4 °C. After a final PBS wash, cells on the sample surface were visualized and imaged using a confocal laser scanning microscope (CLSM).
2.4 Coating corrosion resistance analysis
Corrosion tests were conducted in a simulated body fluid (SBF) comprised of NaCl (8.0 g L−1), KCl (0.4 g L−1), NaHCO3 (0.35 g L−1), MgSO4·7H2O (0.06 g L−1), CaCl2 (0.14 g L−1), Na2HPO4 (0.06 g mL−1), and KH2PO4 (0.06 g L−1). These tests were carried out using both electrochemical methods and immersion techniques. Additionally, glucose concentrations were varied in the SBF at 0 g L−1, 0.99 g L−1, 5 g L−1, and 8 g L−1 to further study the corrosion behavior.
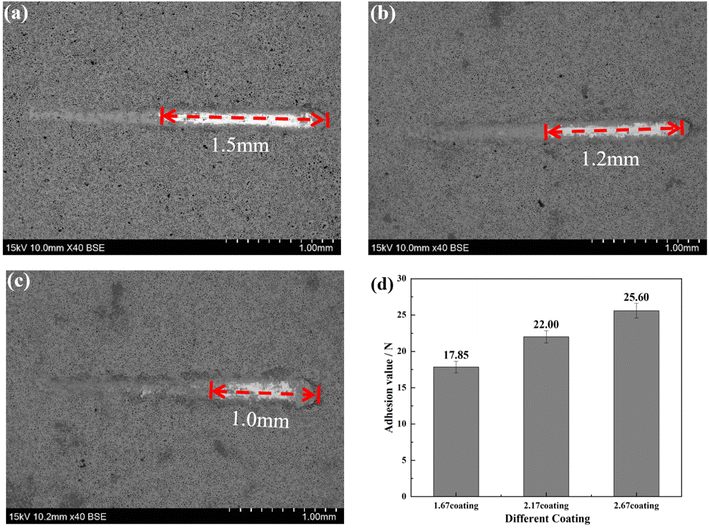
2.4.1 Electrochemical corrosion. To investigate the corrosion behavior of the samples, electrochemical impedance and potentiodynamic polarization tests were carried out using a four-channel electrochemical workstation (CS350, China). The open circuit potential (OCP) was initially determined, followed by measurements of the electrochemical impedance spectroscopy (EIS) and potentiodynamic polarization curves (PDP) after a 10 minute interval. A three-electrode system was employed for the electrochemical tests, with a saturated calomel electrode as the reference, platinum as the counter electrode, and the sample serving as the working electrode. The sample surface was sealed with paraffin, exposing only 1 cm2 for testing. The corrosion solution used was the previously prepared Hank's solution. The frequency range for testing the electrochemical impedance was 105 to 10−2 Hz, utilizing a sinusoidal wave with an amplitude of 10 mV. Experimental data were fitted using ZView software, with equivalent circuit models plotted. Potentiodynamic polarization tests were conducted at a scanning rate of 2 mV s−1. The corrosion potential Ecorr (V) and corrosion current density Icorr (A cm−2) were analyzed using the Tafel extrapolation method with the built-in software. Based on the Stern–Geary equation, the corrosion rate CR (mm per annum) and polarization resistance Rp (Ω cm2) were calculated as follows:26,27| |
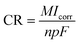 | (2.1) |
| |
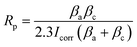 | (2.2) |
In this equation, M is the relative atomic mass of the metal (g), n is the ionic valence of the metal; ρ is the density of the metal (g cm−3), F is Faraday constant; and βa and βc are the anodic and cathodic Tafel slopes (mV dec−1), respectively.
2.4.2 Immersion corrosion. The samples were subjected to in vitro degradation experiments in hanks physiological solution at 37 ± 1 °C for 30 days. In order to quantitatively assess the corrosion process and to calculate the corrosion rate, the mass changes were recorded daily during submergence by means of a high-precision electronic balance, which facilitated the calculation of the corrosion rate. In addition, the evolution of the microstructure was characterized by SEM, EDS, and XRD to record the changes in phase and morphology.
3 Results and discussion
3.1 Coating structure analysis
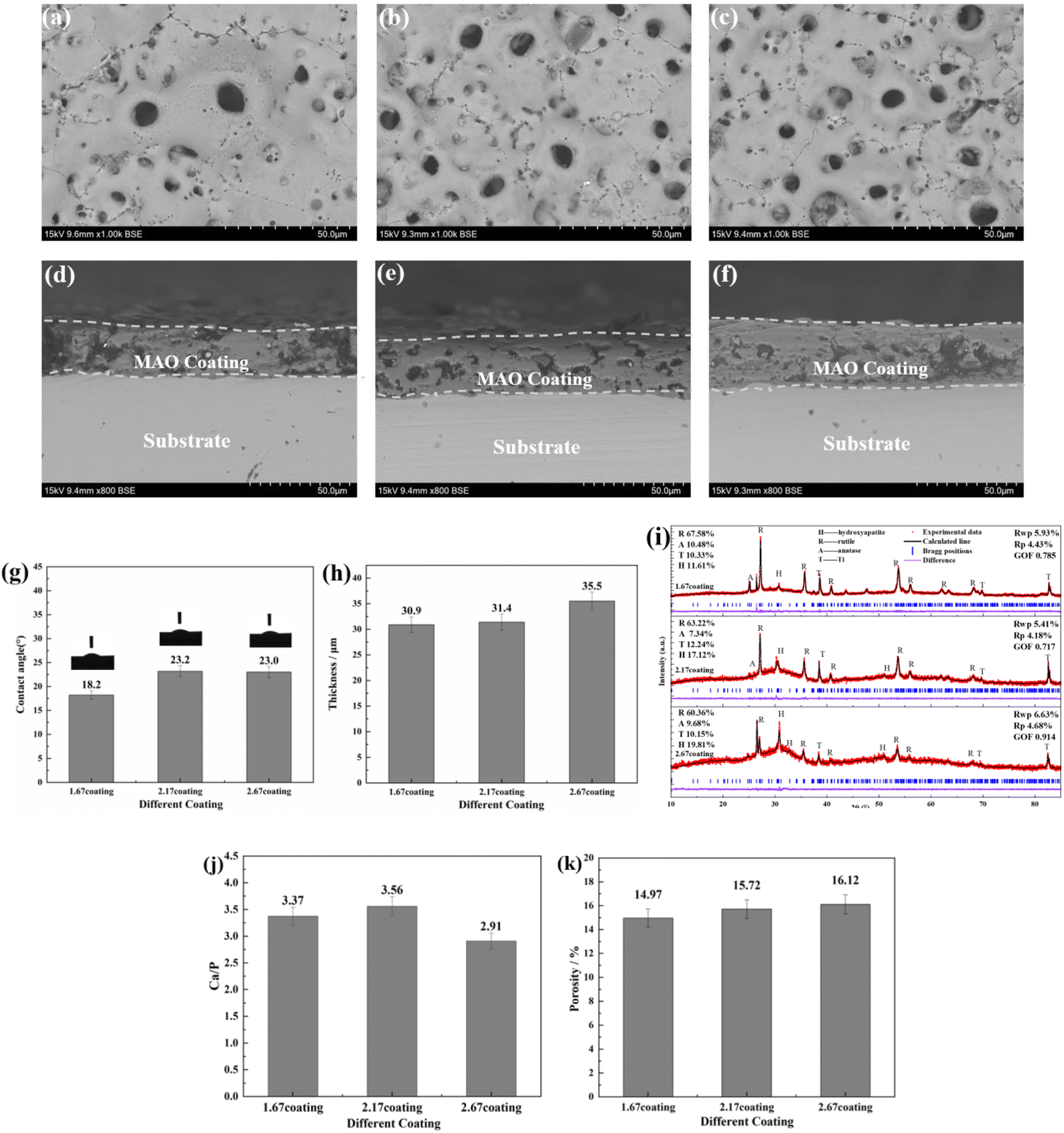
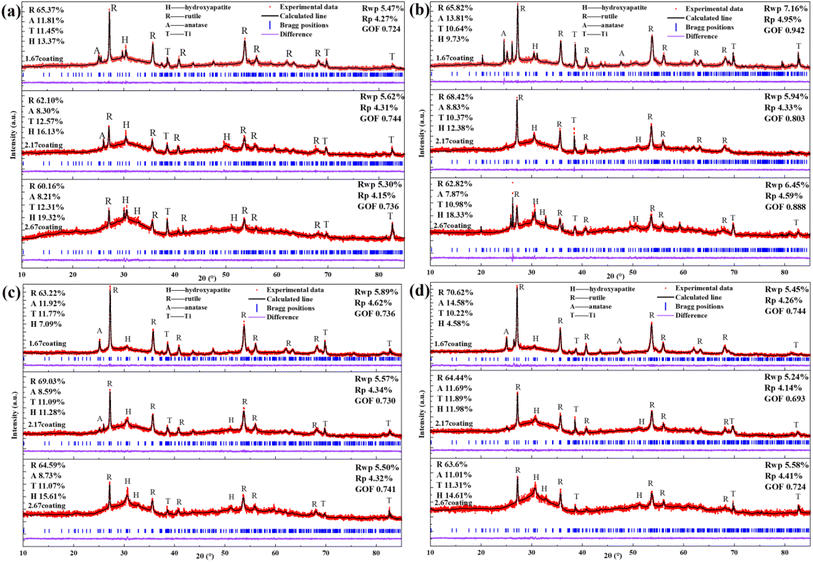
The composition of the electrolyte determines its density and electrical conductivity, which influences the MAO discharging process and thus the coating morphology. The morphologies of the coatings obtained under different Ca/P electrolytes were depicted in Fig. 1. The coatings surface were uniformly distributed with pores of varying diameters, exhibiting the loose and porous characteristic (showed as Fig. 1a–c). These micro-pores serve as discharge channels for plasma during the MAO process. The electrolyte concentration increased with the increase of Ca/P ratio, which enhanced the MAO discharge strength and ultimately lead to the increase of coating discharge pores. According to calculations using Image J, the porosities of the coatings were 14.97% at 1.67 coating, 15.72% at 2.17 coating, and 16.12% at 2.67 coating (Fig. 1k). Electrolyte components could be involved in the formation of coatings through the MAO discharge process. EDS analysis (Table 1) of different coatings showed that, the Ca and P contents in the coatings were enhanced from 10.49 at%, 3.20 at% of 1.67 coating to 13.98 at%, 4.31 at% of 2.67 coating, respectively. After counting the Ca, P elemental content, the Ca/P ratios of different coatings were respectively 3.37, 3.56, and 2.91(Fig. 1). Considering the EDS test method with 7% uncertainty, the difference in Ca/P for 2.17 coating versus 1.67 coating and 2.67 coating was ∼±0.186, ∼±0.676, respectively. These results were significant. From Fig. 1d–f, the cross-sectional topographies showed that the internal structures of the different coatings were also loose and porous. These are left over from the discharge process and are related to the electrolyte concentration. The outline of the coating reveals that, as the Ca/P ratio of the electrolyte continually increased, there was a slight rise in coating thickness. Measurements obtained from the thickness gauge revealed that the thickness of the 1.67 coating was a minimum of 30.9 μm, while the thickness of the 2.17 coating and the 2.67 coating were 31.4 μm and 35.5 μm.
 |
| | Fig. 1 Surface morphology of different coatings: (a) 1.67 coating, (b) 2.16 coating, (c) 2.67 coating. Sections morphology of different coatings: (d) 1.67 coating, (e) 2.16 coating, (f) 2.67 coating. (g) Contact angle, (h) thickness, (i) XRD diffraction spectra, (j) EDS Ca/P ratio, (k) porosity. | |
Table 1 Content of elements in different MAO coatings (at%)
| Sample |
Element |
| Ti |
Nb |
O |
Ca |
P |
Sn |
Zr |
Na |
| 1.67 coating |
21.96 |
7.17 |
55.42 |
10.49 |
3.20 |
0.03 |
1.55 |
0.18 |
| 2.17 coating |
20.41 |
6.98 |
53.41 |
13.46 |
3.91 |
0.1 |
1.34 |
0.38 |
| 2.67 coating |
18.68 |
6.58 |
54.85 |
13.98 |
4.31 |
0.04 |
1.09 |
0.47 |
XRD analysis (Fig. 1i) of different MAO coatings shows that, in addition to the TiO2-dominated crystal structure (both anatase and plagioclase morphologies), the diffraction peaks of the HA crystals appeared at ∼26° and ∼32°. With the elevation of the Ca/P ratio in the electrolyte, the content of anatase on the coating surface relatively decreased, while the more stable rutile content saw a relative increase. This, in turn, resulted in an increase in the solution temperature near the anode, prompting the transformation of anatase to the more thermally stable rutile phase of TiO2 under elevated temperatures.28 Based on the analysis of the phase composition, it was also clear that the HA content increased from 11.61% of 1.67 coating to 19.81% of 2.67 coating. Additionally, the peak intensity of HA was enhanced with the rise in the electrolyte Ca/P ratio, suggesting a relative increase in the HA content within the coating. This was due to the fact that during MAO discharge breakdown, more Ca2+ and HPO42− in the electrolyte mix with the melt ejected from the discharge channel to form the MAO coating.
A reduced contact angle indicated enhanced hydrophilicity, which subsequently leads to better cell spreading on the surface and inherently improved cell affinity. Contact angle measurements revealed that the contact angles for all samples were consistently less than 90° (Fig. 1g). The results of the contact angle experiments showed that the contact angle of the 1.67 coating was a minimum of 18.2°, while the contact angle of the 2.17 coating and the 2.67 coating were 23.2° and 23°, respectively. Combined with SEM and XRD analyses, it was observed that as the Ca/P ratio increased, the increase in the porosity of the material surface improved the wettability of the material surface.
3.2 Binding force analysis
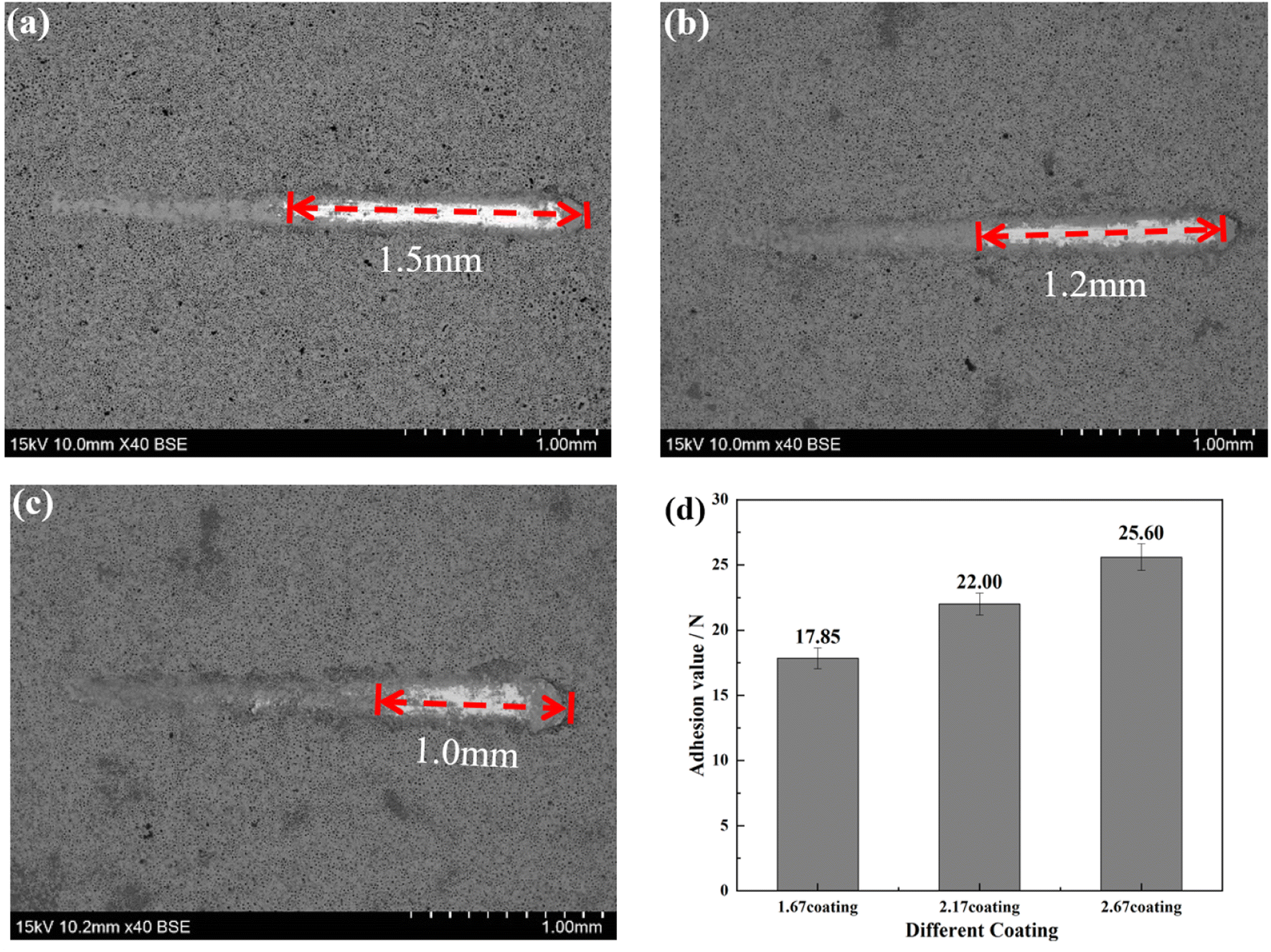
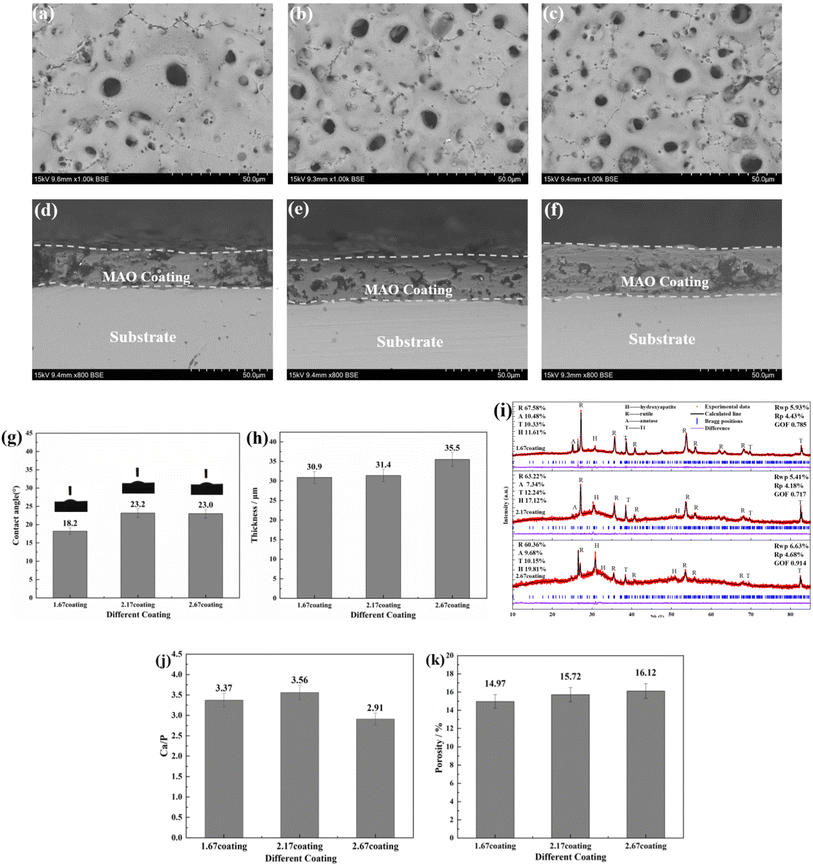
The bonding of different coatings was examined by scratch method and the results were shown in Fig. 2. Scratch assay results, unequivocally demonstrate that an escalation in the Ca/P ratio corresponds with a concomitant trend toward a diminished scratch area on the sample surfaces (Fig. 2a–c). Specifically, the scratch length reduced from 1.5 mm for the 1.67 coating to 1.0 mm for the 2.67 coating, with corresponding bonding forces of 17.85 N and 25.60 N, respectively (Fig. 2d). This indicated an augmentation of the coating hardness and wear resistance with an elevated calcium to phosphorus ratio. The increase of Ca/P ratios enabled increased Ca2+ ion incorporation into the coating matrix, inciting a rise in coating thickness. These crystalline transformations further ameliorate the coating resistance to degradation and mechanical wear while enhancing its biocompatibility. The increase in Ca/P ratio enhances the MAO discharge strength, which increased the coating thickness, and the transformation of the coating crystal structure improved the coating mechanical wearability.29,30
 |
| | Fig. 2 Bonding of different coatings (a) 1.67 coating, (b) 2.17 coating, (c) 2.67 coating, (d) bonding strength. | |
3.3 Biocompatibility analysis
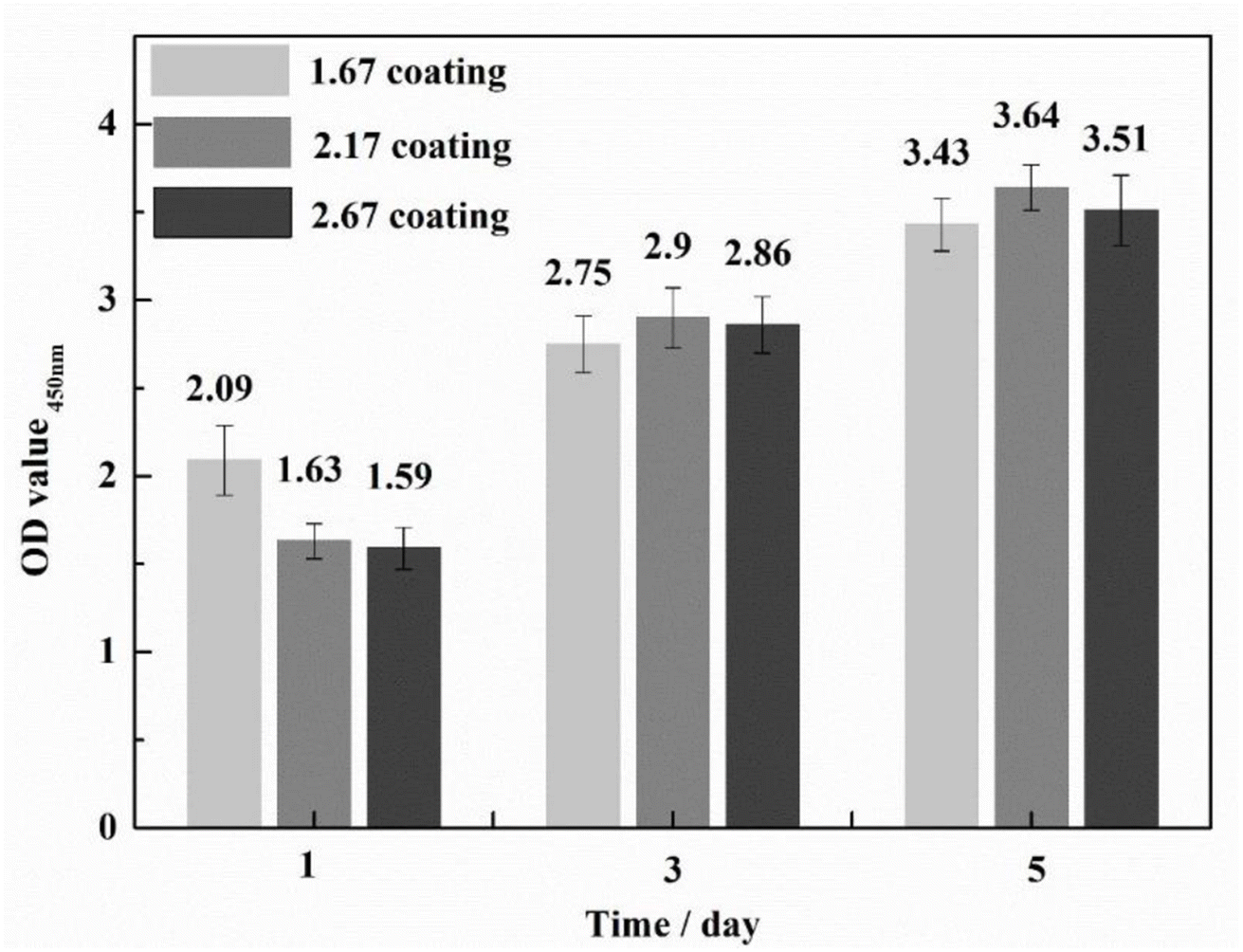
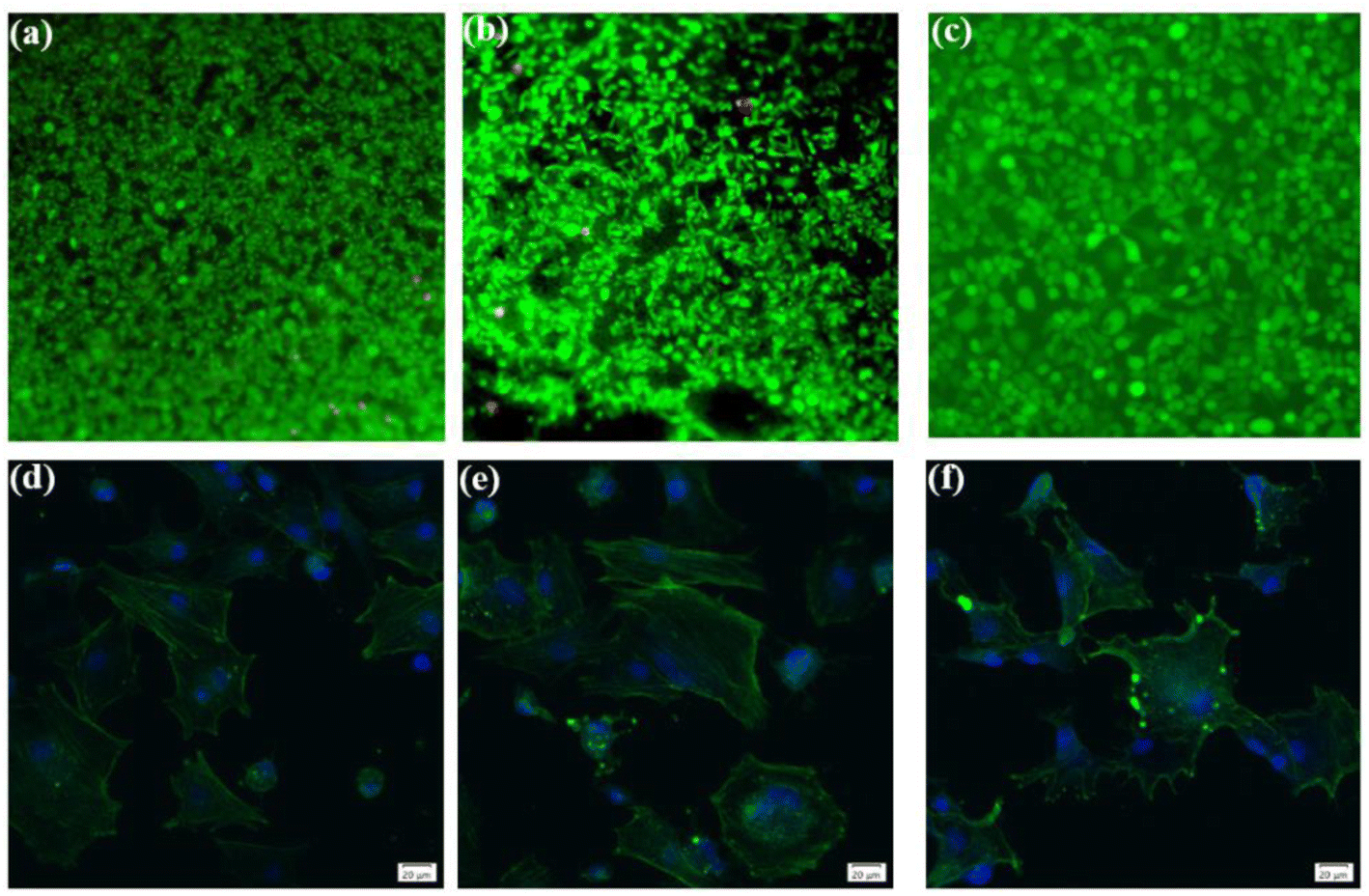
To ascertain the biocompatibility of our coatings as well as their contributory effects on wound healing, it was imperative to monitor not only the normal proliferation of cells on the coated surfaces but also to preclude anomalous cell mortality. To this end, the Cell Counting Kit-8 (CCK-8) assay and live/dead staining techniques were deployed to evaluate the biocompatibility across various samples (Fig. 3). Cytotoxicity assays showed the highest cell viability at 1.67 coating on day 1. From day 3 onwards, it was found that the higher the porosity of the coating, the better the cell proliferation rate, while more live cells were found in 2.67 coating, reflecting its lower cytotoxicity and better biocompatibility. Specific results were depicted in Fig. 4, it is discernible that after a two-day co-culture with various coatings, the cells maintained considerable viability, and their morphologies were clearly observable. As the Ca/P ratio increased from 1.67 to 2.67, there were relatively few dead cells on the coating surface (red color in Fig. 4a–c), indicating that these coatings have low toxicity and good biocompatibility. It could be assumed that the increased HA content in the coating provided a large number of active sites for cell attachment and growth, and its enhanced porosity ensured an adequate supply of nutrients and oxygen for cell proliferation.
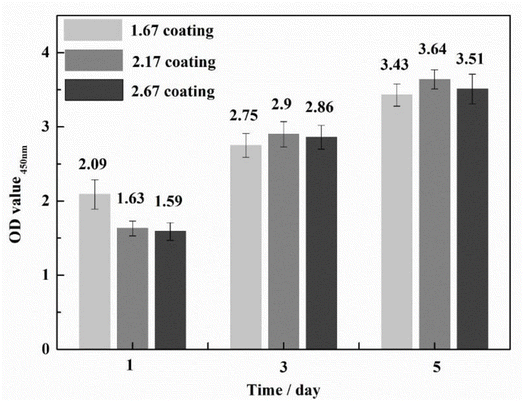 |
| | Fig. 3 CCK-8 analysis of cells co-cultured with different coatings. | |
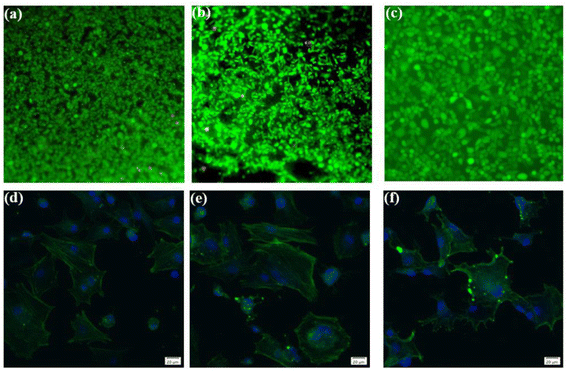 |
| | Fig. 4 Live/dead staining analysis of different coatings in diabetic environment ((a) 1.67 coating, (b) 2.17 coating, (c) 2.67 coating). Fluorescent staining of cells coated with different Ca/P ratios: ((d) 1.67 coating, (e) 2.17 coating, (f) 2.67 coating). | |
In exploring the biocompatibility of MAO coatings prepared in electrolyte environments with varying Ca/P ratios, cytoskeletal observation methods were used to assess the biological activity of the coatings. In the physiological environment of the human body, cells adhere and proliferate extremely well on the implant surface, favoring in vivo bone reconstruction. As shown in Fig. 4d and e (phalloidin staining), the cells exhibited polygonal structures on different coatings, and the tentacles spread out in all directions, indicating that the cells were better adhered to the coating surfaces. Comparisons revealed that the number and expansion capacity of the cells on the coated surface increased as the Ca/P content increased. It can be seen that an increase in the porosity of the coating as well as an increase in Ca/P can provide more growth active sites for cell growth and promote cell growth.31
3.4 Corrosion analysis
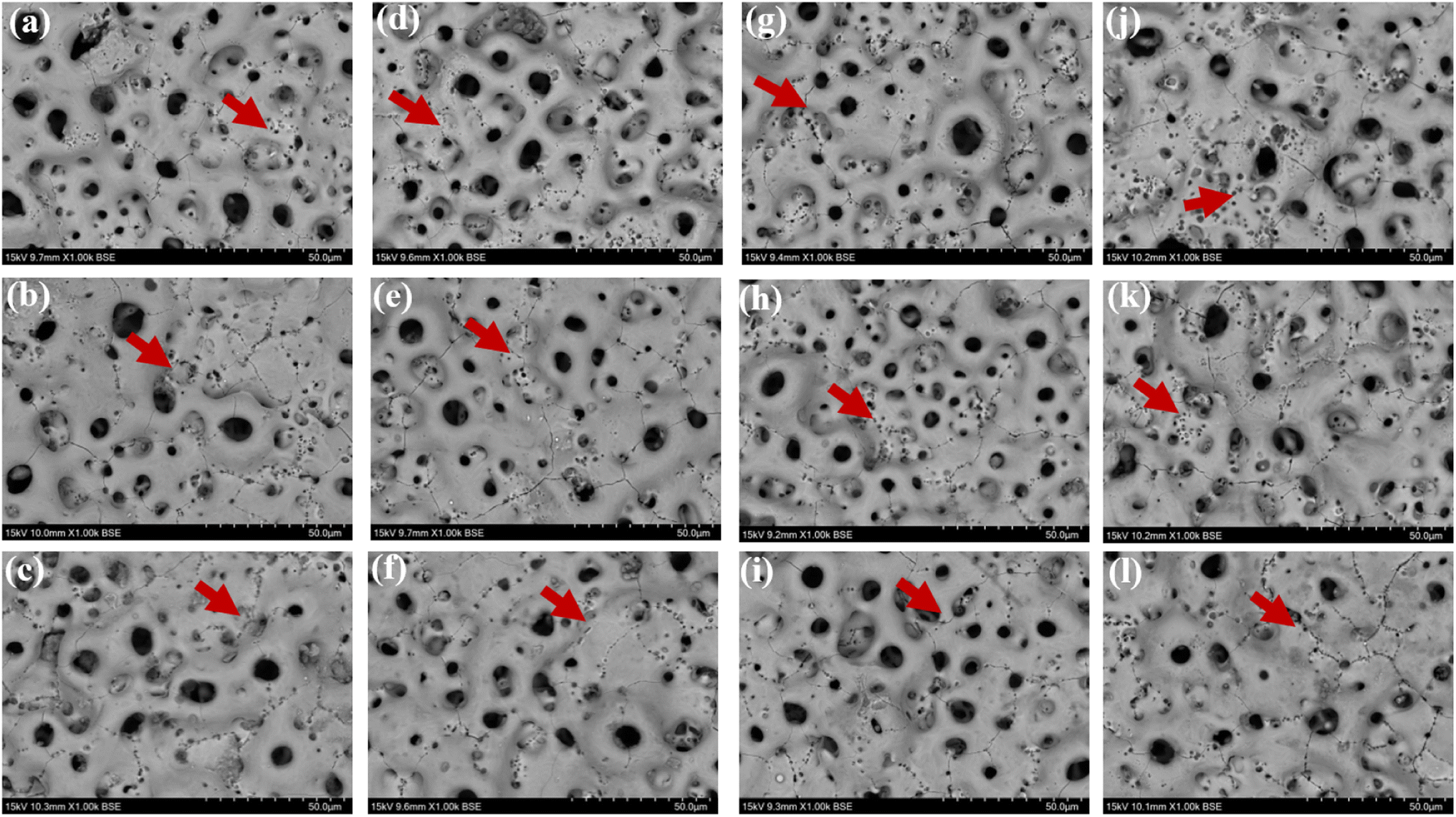
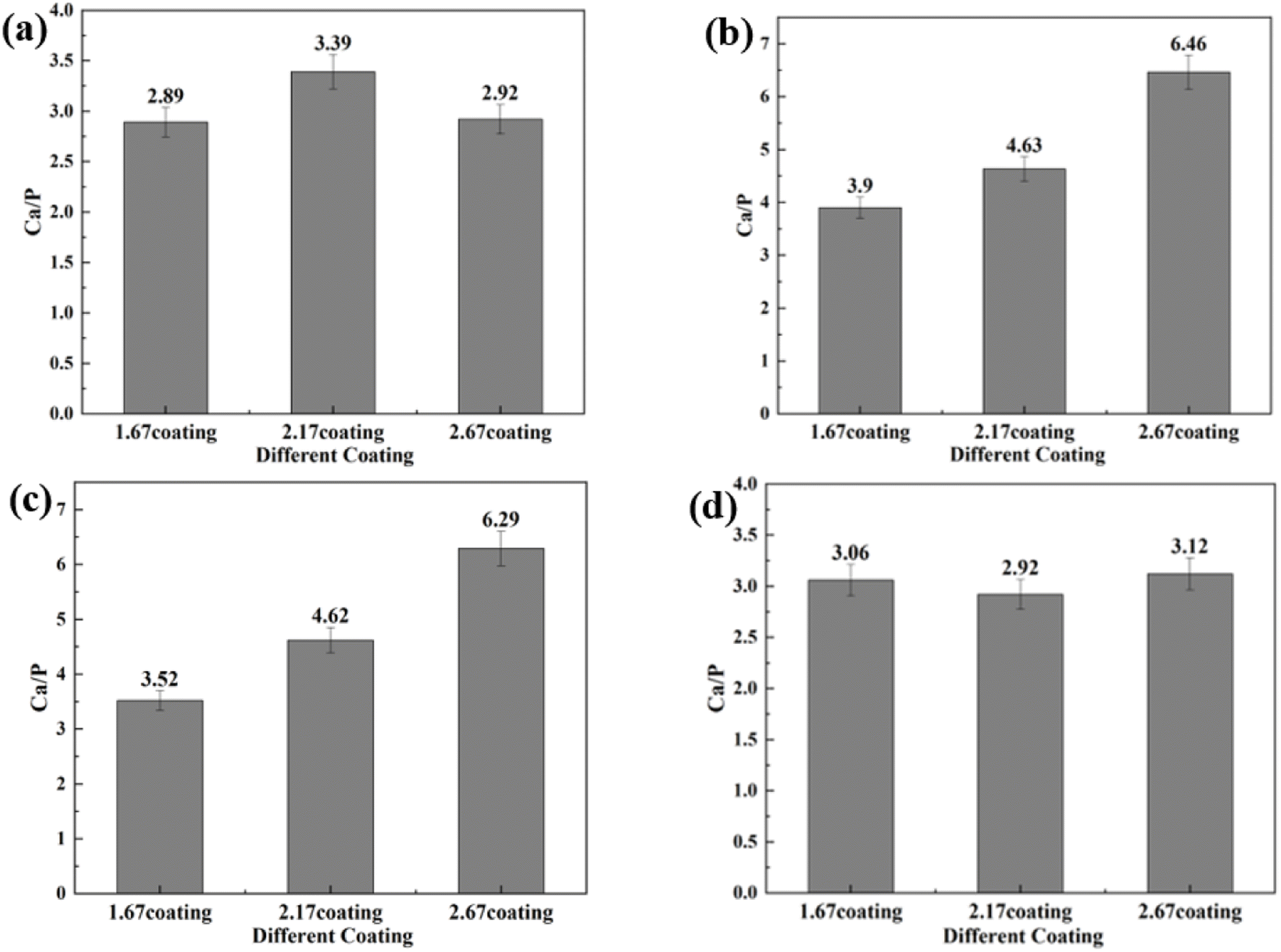
3.4.1 Coating characterization after immersion corrosion. As can be seen from the SEM morphology of Fig. 5, after 30 days of immersion, the different coatings basically maintained their original loose and porous characteristics. However, different degrees of corrosion holes and cracks appeared on the coating surface, as shown by the red arrows in Fig. 5. According to the comparison of the SEM morphology before and after corrosion, it was clearly seen that after 30 days of immersion corrosion, the surface of the coating had small and more intensive corrosion pits. In addition, the cracks on the surface of the coating before corrosion immersion were mainly in the form of relatively uniform irregular polygons. After 30 days of immersion, some small cracks appeared in these polygons, which was also direct evidence of the effect of corrosion immersion on the surface of the coating. When the immersion concentration was 8 g L−1, the crater-like melt on the surface of the coating appeared to be obviously partially missing. Overall, the corrosion condition of the MAO coatings was more serious at the glucose concentration of 8 g L−1, accompanied by an increase in the size of the corrosion holes and an increase in the crack number. The comparison revealed that the 2.67 coating was the most corrosive in different glucose-containing environments, which was attributed to the fact that the elevated Ca/P ratio caused the porosity of the coating to increase, making it easier for the electrolyte to enter the coating and produce corrosion damage. As in Fig. 6, the EDS analysis was performed on the corroded coatings to analyze the pattern of change of the elements. When the glucose concentration was 0 g L−1, the Ca/P ratios on the coating surfaces were 2.89, 3.39, and 2.92 respectively. At a glucose concentration of 0.99 g L−1, the Ca/P ratios were 3.90, 4.63, and 6.46. With a glucose concentration of 5 g L−1, the Ca/P ratios stood at 3.52, 4.62, and 6.29. Lastly, when the glucose concentration reached 8 g L−1, the Ca/P ratios were recorded as 3.06, 2.92, and 3.12, respectively. It indicated that when the glucose content in the solution was ≤5 g L−1, the elevated glucose content accelerated the release of elemental P, so the Ca/P value remaining in the coating was elevated. When the glucose content gone up to 8 g L−1, the Ca was released at a comparable rate to P, so the Ca/P ratio remaining in the coating did not change much. This was mainly due to the increased corrosion rate, which result in the release of different ions from the coating at roughly the same rate.
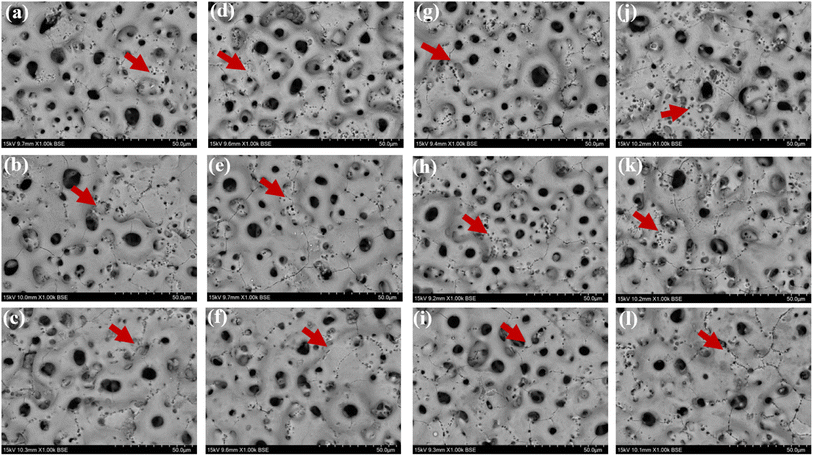 |
| | Fig. 5 Surface morphology of different coatings after immersion corrosion for 30 days: hank's solution: ((a) 1.67 coating, (b) 2.17 coating, (c) 2.67 coating), hank's solution with 0.99 g L−1 glucose: ((d) 1.67 coating, (e) 2.17 coating, (f) 2.67 coating), hank's solution with 5 g L−1 glucose: ((g) 1.67 coating, (h) 2.17 coating, (i) 2.67 coating), hank's solution with 8 g L−1 glucose: ((j) 1.67 coating, (k) 2.17 coating, (l) 2.67 coating). | |
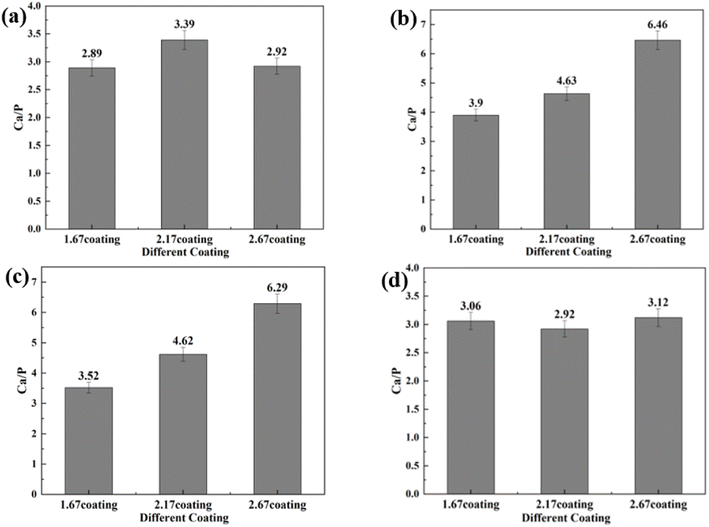 |
| | Fig. 6 EDS of different coatings after immersion corrosion for 30 days: hank's solution (a), hank's solution with 0.99 g L−1 glucose (b), hank's solution with 5 g L−1 glucose (c), hank's solution with 8 g L−1 glucose (d). | |
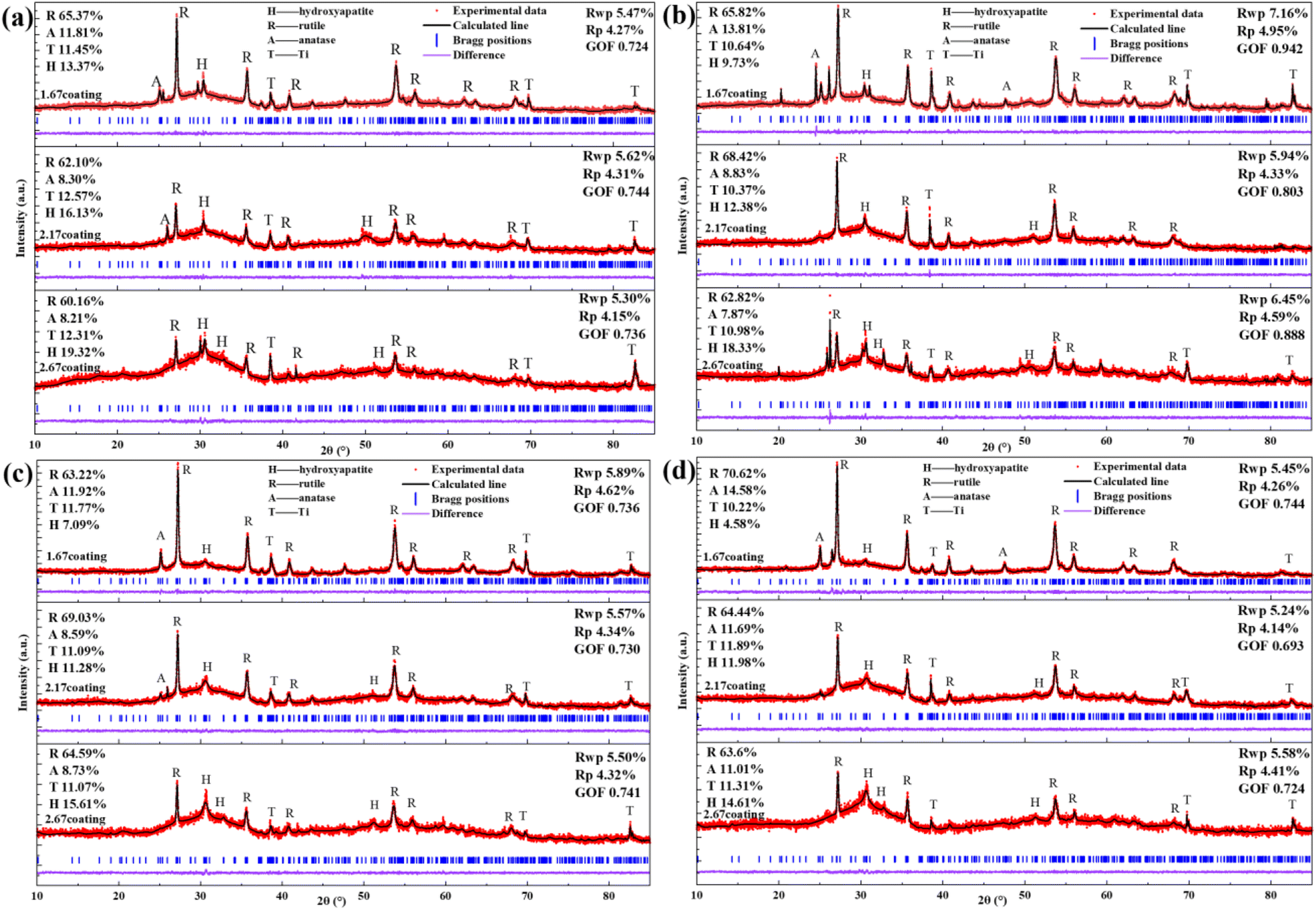
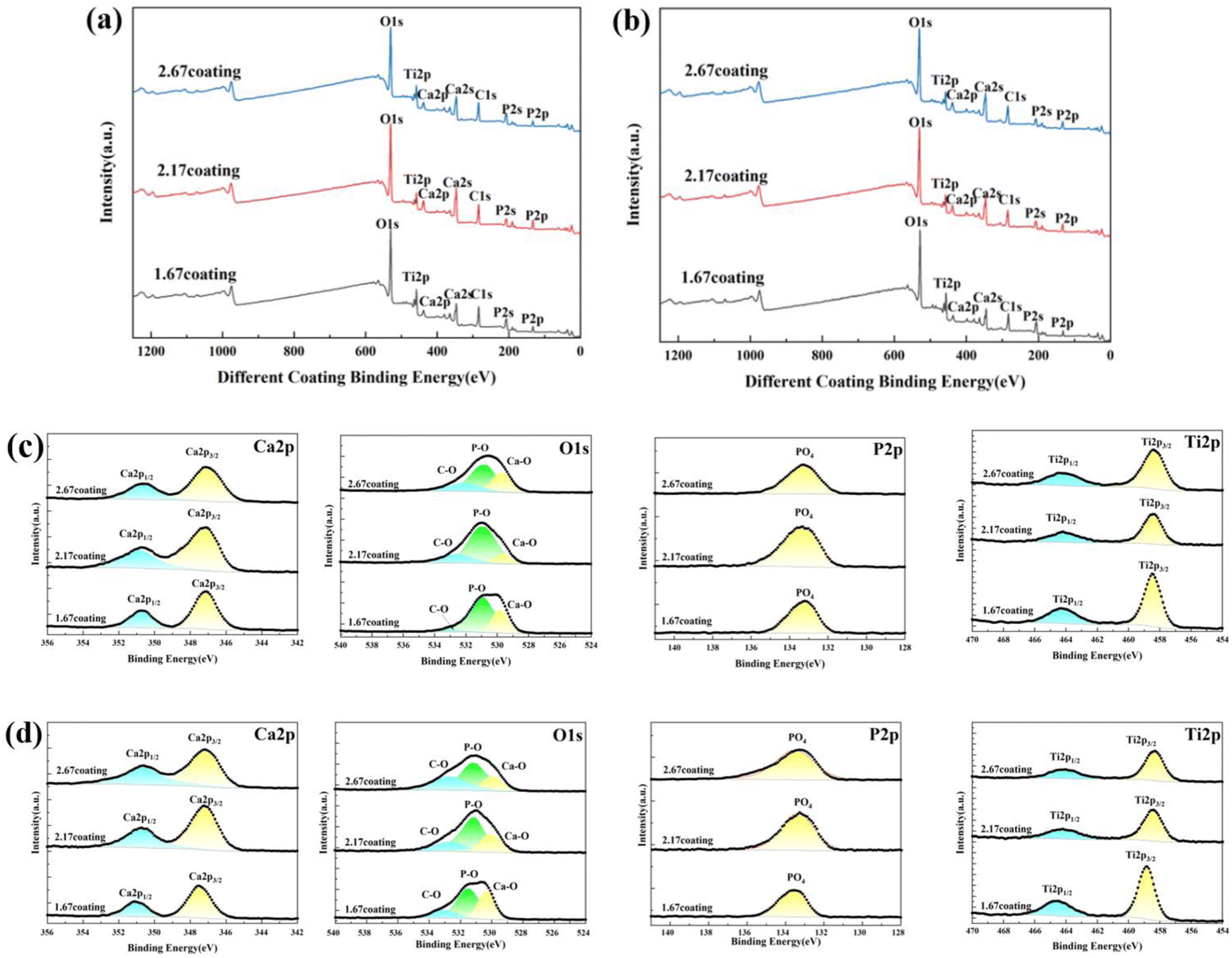
After immersion corrosion, each coating was subjected to XRD and XPS analysis, as illustrated in Fig. 7 and 8. After 30 days of corrosion, all coatings retained the diffraction peaks of TiO2 and HA, suggesting minimal principal structural changes in the coatings. From the XRD spectra, it could be seen that the intensity of the HA diffraction peaks on the coating decreased with the increase of glucose concentration in the soaking solution, while the intensity of the TiO2 peaks remain basically unchanged, which indicated that the HA was relatively easy to dissolve in the high glucose environment. Quantitative XRD analysis further confirmed that the HA content in the coating phase composition decreases with increasing glucose concentration. In particular, the HA content of 2.67 coating had decreased to 14.61% in the solution with 8 g L−1 glucose. As depicted in Fig. 8, XPS analysis was performed on the coatings before and after immersion corrosion with glucose concentrations of 8 g L−1. The XPS spectra indicated the following trends in the elemental peak regions: without immersion, the peak intensities of O 1s, Ca 2p, and P initially increased and then decreased with the rise in the calcium-to-phosphorus ratio, whereas, at diabetic environment, the peak intensities initially rose and then fell with increasing calcium-to-phosphorus ratios. Notably, under diabetic environment, a marked reduction in the peak percentages of P–O bonds and Ca 2p was observed, further confirming the dissolution of calcium and phosphorus elements and the consequent degradation of the hydroxyapatite. This could be due to enhanced reactivity of organic substances in simulated body fluids under diabetic environment, leading to exacerbated erosion of HA.32 This also implies that Ca2+ within HA may undergo ion exchange reactions with amino acids, proteins, and organic acids containing carboxyl groups.
 |
| | Fig. 7 XRD patterns of different coatings after immersion corrosion for 30 days: hank's solution (a), hank's solution with 0.99 g L−1 glucose (b), hank's solution with 5 g L−1 glucose (c), hank's solution with 8 g L−1 glucose (d). | |
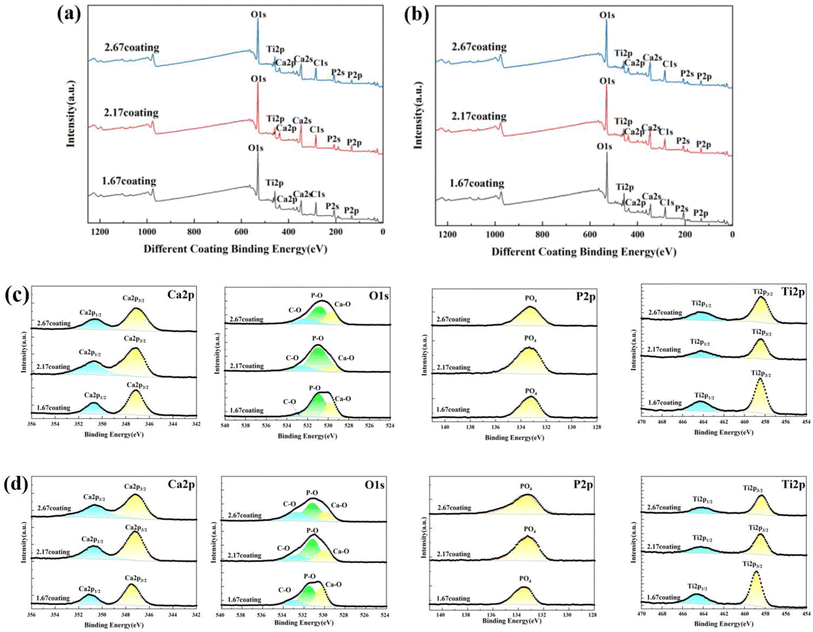 |
| | Fig. 8 XPS spectra of different MAO coatings in hank's solution with different glucose concentrations: (a) un-immersed, (b) 8 g L−1. High-resolution spectra of Ca 2p, Ti 2p, P 2p and O 1s for coating: (c) un-immersed, (d) 8 g L−1. | |
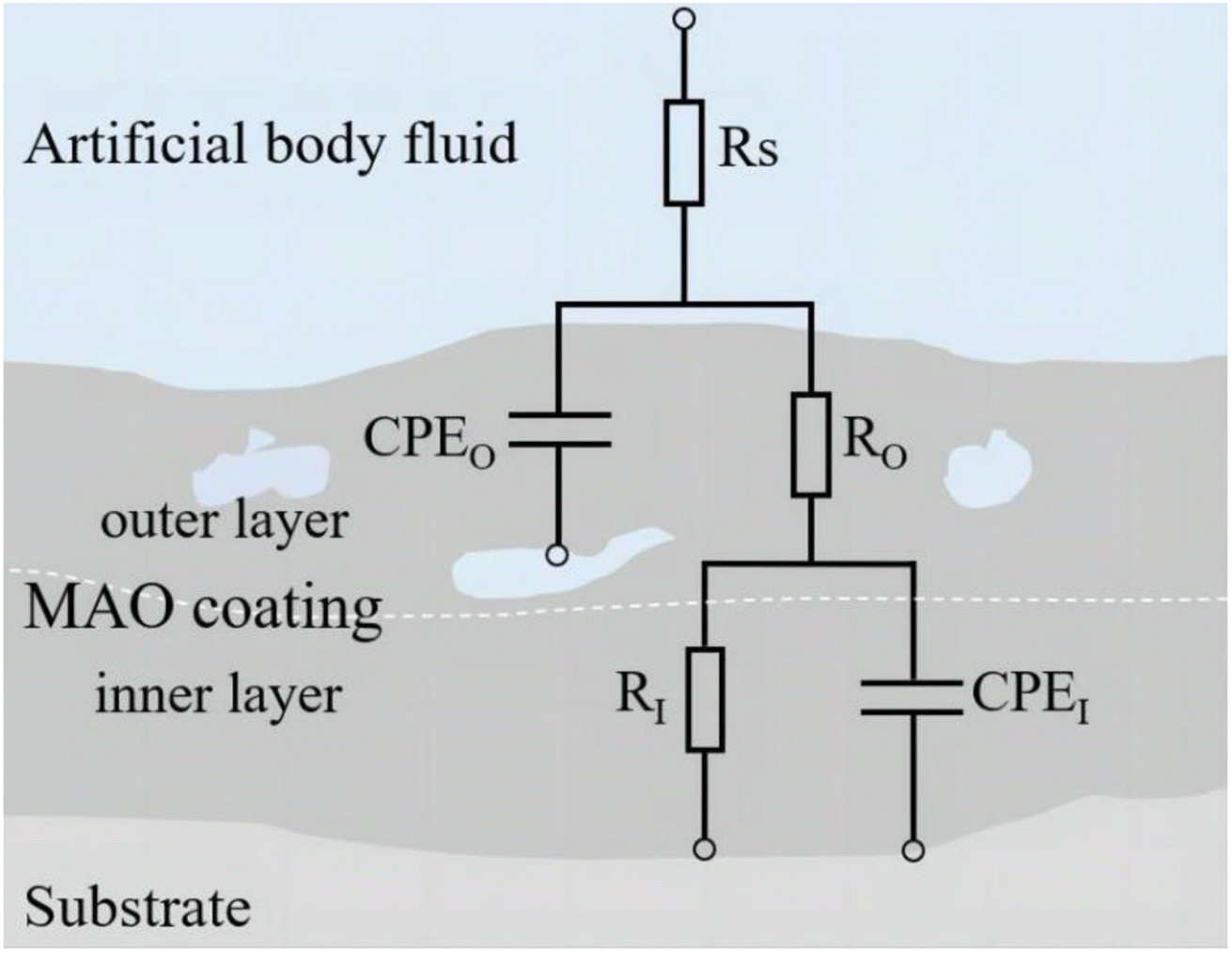
3.4.2 Analysis of electrochemical corrosion and bonding strength. Upon implantation into the human body, implants are subjected to bodily fluid permeation and intricate electrochemical reactions.33 In this study, an electrochemical workstation was employed to analyze the coatings under various simulated body fluid conditions. Fig. 9 presents the Tafel plots of samples under various glucose concentrations, while Table 2 lists the fitting data of these polarization curves. Fig. 10 includes the Electrochemical Impedance Spectroscopy (EIS) of the coatings post-treatment in different glucose concentrations. Fig. 11 depicts the equivalent circuit model featuring elements such as solution resistance (RS), outer layer resistance of the coating (RO), inner layer resistance of the coating (RI), total resistance of the entire coating (RC = RO + RI), outer layer capacitance of the coating (CPEO), and inner layer capacitance of the coating (CPEI), with corresponding fitting data provided in Table 3.
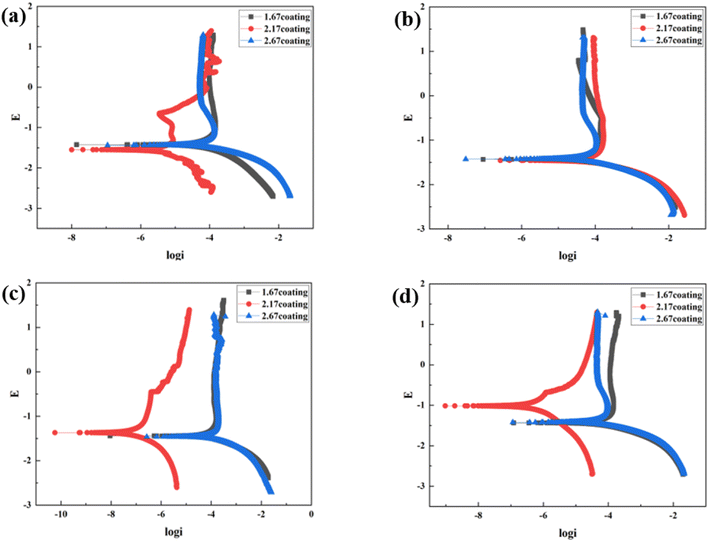 |
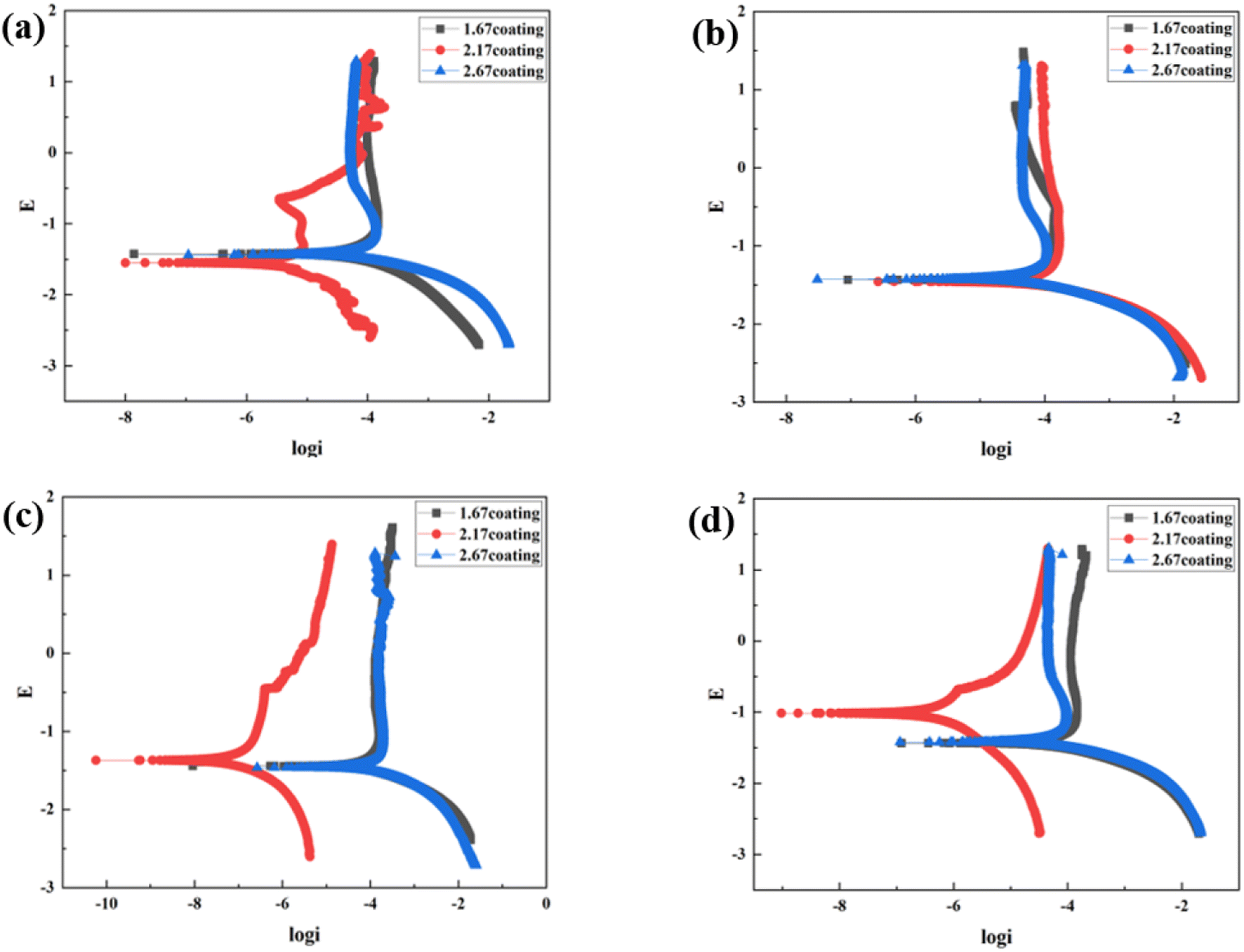
| | Fig. 9 Tafel curves of different coatings after immersion corrosion for 30 days: hank's solution (a), hank's solution with 0.99 g L−1 glucose (b), hank's solution with 5 g L−1 glucose (c), hank's solution with 8 g L−1 glucose (d). | |
Table 2 Fitting results of Tafel polarization curves for coatings in different glucose concentrations
| Glucose concentration |
Sample |
Ecorr (V) |
Icorr (A cm−2) |
CR (mm per annum) |
| 0 g L−1 |
1.67 coating |
−1.42 |
1.44 × 10−4 |
1.30 |
| 2.17 coating |
−1.55 |
3.01 × 10−5 |
0.27 |
| 2.67 coating |
−1.44 |
1.35 × 10−4 |
1.21 |
| 0.99 g L−1 |
1.67 coating |
−1.43 |
9.95 × 10−5 |
0.90 |
| 2.17 coating |
−1.46 |
1.39 × 10−4 |
1.25 |
| 2.67 coating |
−1.43 |
9.07 × 10−5 |
0.82 |
| 5 g L−1 |
1.67 coating |
−1.44 |
1.41 × 10−4 |
1.27 |
| 2.17 coating |
−1.37 |
9.13 × 10−8 |
8.21 × 10−4 |
| 2.67 coating |
−1.46 |
2.60 × 10−4 |
2.3364 |
| 8 g L−1 |
1.67 coating |
−1.44 |
9.72 × 10−5 |
0.87 |
| 2.17 coating |
−1.02 |
1.76 × 10−6 |
1.59 × 10−2 |
| 2.67 coating |
−1.42 |
7.65 × 10−5 |
0.69 |
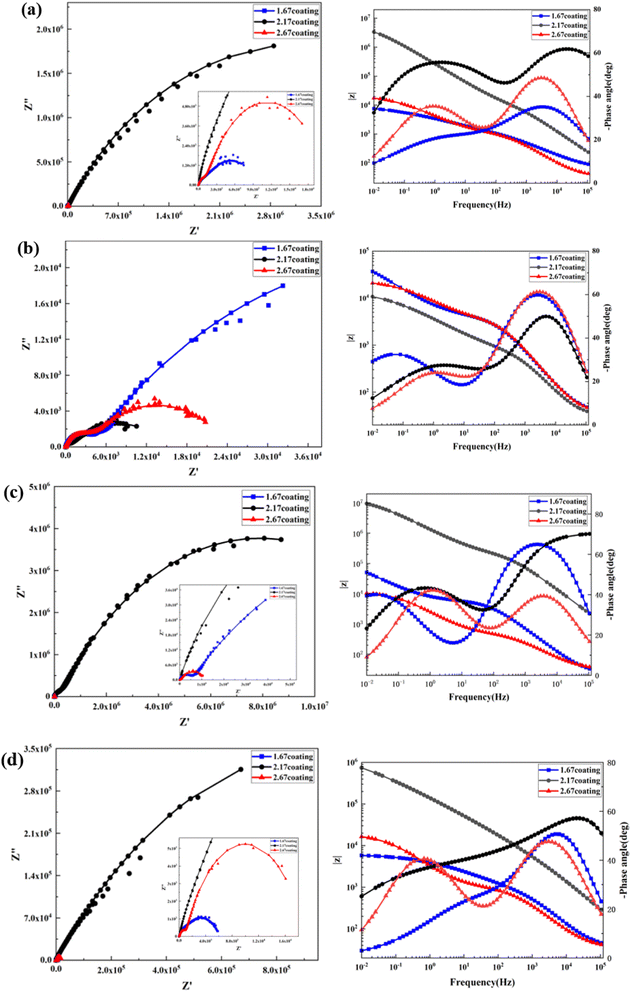 |
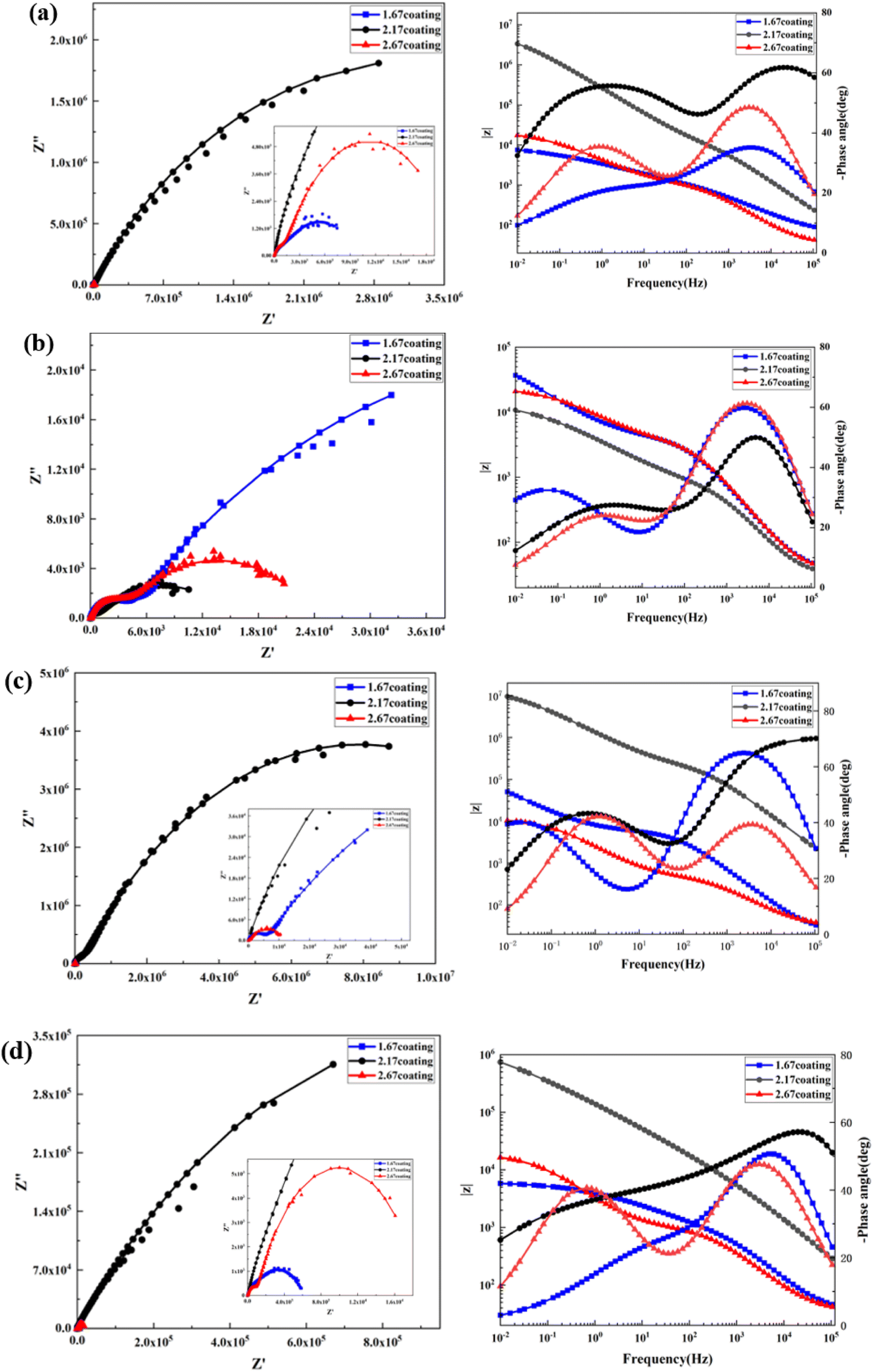
| | Fig. 10 EIS and Bode curves of different coatings after immersion corrosion for 30 days: hank's solution (a), hank's solution with 0.99 g L−1 glucose (b), hank's solution with 5 g L−1 glucose (c), hank's solution with 8 g L−1 glucose (d). | |
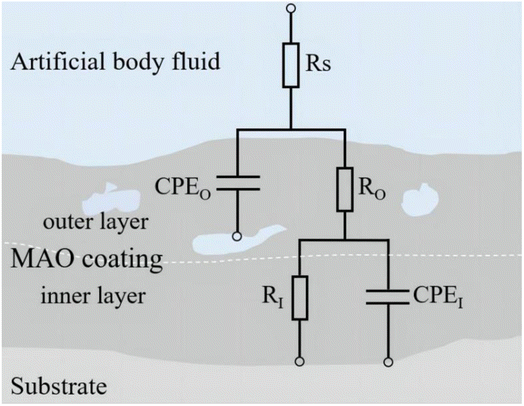 |
| | Fig. 11 Equivalent circuit model diagram. | |
Table 3 Electrochemical parameters obtained by fitting EIS data of coatings in different glucose concentrations
| Glucose concentration |
Sample |
RS (Ω cm2) |
RO (Ω cm2) |
CPEO |
RI (Ω cm2) |
CPEI |
Rc (Ω cm2) |
Chi square |
| YO (Ω−1 sn cm−2) |
nO |
YI (Ω−1 sn cm−2) |
nI |
| 0 g L−1 |
1.67 coating |
57.32 |
1175 |
1.19 × 10−5 |
0.57 |
8809 |
1.40 × 10−4 |
0.39 |
9984 |
1.47 × 10−3 |
| 2.17 coating |
32.81 |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 560 560 |
2.21 × 10−7 |
0.74 |
6.60 × 106 |
9.15 × 10−7 |
0.63 |
6.61 × 106 |
1.82 × 10−3 |
| 2.67 coating |
32.91 |
1139 |
4.28 × 10−6 |
0.72 |
20477 |
8.45 × 10−5 |
0.58 |
21616 |
2.13 × 10−3 |
| 0.99 g L−1 |
1.67 coating |
36.05 |
3936 |
1.45 × 10−6 |
0.77 |
99168 |
9.08 × 10−5 |
0.52 |
1.03 × 105 |
5.09 × 10−3 |
| 2.17 coating |
30.15 |
5422 |
2.33 × 10−5 |
0.76 |
14285 |
1.20 × 10−4 |
0.44 |
15027.2 |
1.51 × 10−3 |
| 2.67 coating |
36.21 |
3477 |
1.09 × 10−6 |
0.80 |
20708 |
5.17 × 10−5 |
0.53 |
24185 |
1.43 × 10−3 |
| 5 g L−1 |
1.67 coating |
22.59 |
5645 |
1.31 × 10−6 |
0.79 |
2.40 × 105 |
9.38 × 10−5 |
0.58 |
2.46 × 105 |
5.76 × 10−3 |
| 2.17 coating |
23.80 |
22300 |
1.21 × 10−8 |
0.78 |
1.54 × 107 |
2.62 × 10−7 |
0.57 |
1.56 × 107 |
6.86 × 10−3 |
| 2.67 coating |
29.74 |
509.9 |
1.05 × 10−5 |
0.66 |
11114 |
1.10 × 10−4 |
0.66 |
11623.9 |
7.98 × 10−3 |
| 8 g L−1 |
1.67 coating |
35.04 |
831.3 |
1.68 × 10−6 |
0.77 |
5327 |
6.44 × 10−5 |
0.45 |
6158.3 |
3.52 × 10−3 |
| 2.17 coating |
92.76 |
1173 |
6.40 × 10−8 |
0.82 |
1.97 × 106 |
2.99 × 10−6 |
0.43 |
1.98 × 106 |
3.57 × 10−3 |
| 2.67 coating |
33.52 |
1019 |
4.59 × 10−6 |
0.72 |
17968 |
9.67 × 10−5 |
0.67 |
18987 |
1.88 × 10−3 |
At the environment with no glucose, the 2.17 coating displayed the lowest self-corrosion current density and corrosion rate (Fig. 9a, Table 2), with 3.01 × 10−5 A cm−2 and 0.27 mm per annum, respectively. The Bode plots indicated that the electrochemical response spectrum of the samples exhibited two time-constants, corresponding to the outer and inner layers of the coating, respectively. The highest low-frequency impedance value |Z| occurred at 2.17 coating, signifying its superior corrosion resistance. In the Nyquist plots, the 2.17 coating exhibited the largest capacitive loop diameter, further validating its corrosion resistance capabilities. However, the worst corrosion resistance is 1.67 coating, its self-corrosion current density and corrosion rate, 1.44 × 10−4 A cm−2 and 1.29 mm per annum, respectively. The main reason was due to its lower coating thickness.
With the glucose concentration at 0.99 g L−1, the corrosion resistance of all coatings decreased, with the 2.17 coating presenting the highest self-corrosion current density and corrosion rate, 1.38 × 10−4 A cm−2 and 1.24 mm per annum, respectively. The EIS and Bode plots demonstrated lower low-frequency impedance values |Z| across 2.17 coating, and the smallest capacitive loop diameter in the Nyquist plots. This is due to the fact that glucose reduced the pH of the solution by producing saccharic acid, which promoted the coating corrosion.
At a glucose concentration of 5 g L−1, the 2.17 coating showed the lowest self-corrosion current density and corrosion rate, with the low-frequency impedance value |Z| two orders of magnitude higher than other samples, and the largest capacitive loop diameter in the Nyquist plots. This may be due to the fact that glucose at this concentration had a hindering effect on corrosive ions such as Cl− in solution, reducing their damage to the coating. When the glucose concentration was 8 g L−1, the corrosion resistance of the coatings showed a certain decrease, but the analytical results at this point were similar to those at the glucose concentration of 5 g L−1. Specifically, the 2.17 coating displayed the lowest self-corrosion current density and corrosion rate. In the Nyquist plot, this coating also had the largest capacitive arc diameter, and its low-frequency impedance value |Z| was one to two orders of magnitude higher than the other two samples. This is mainly due to the fact that the 2.17 coating had a relatively high thickness and moderate porosity, and the glucose molecules in the solution could effectively block the damage of corrosive ions to the coating (Fig. 14b).34
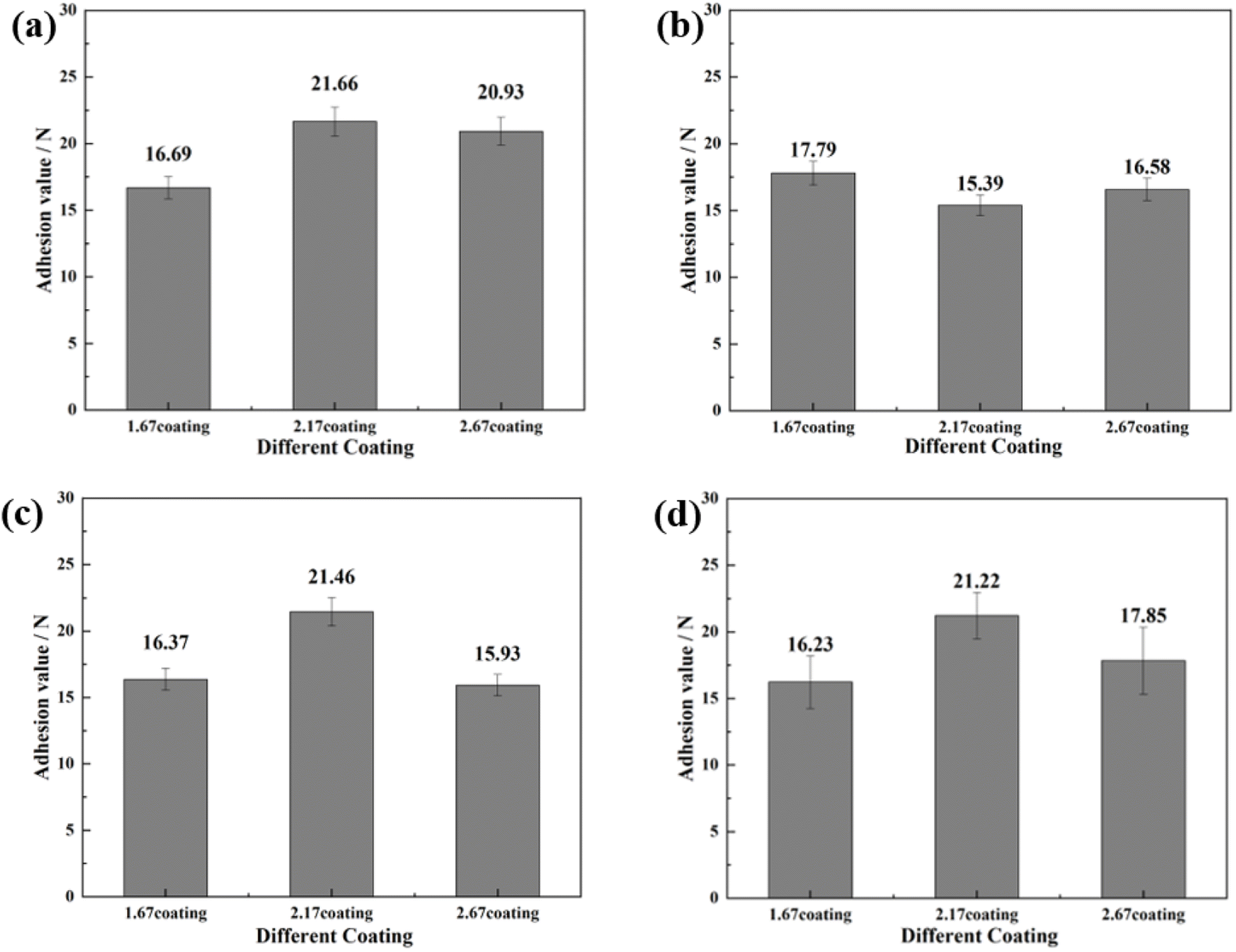
To further investigate the coating safety, scratch test on the coatings after immersion were conducted, as illustrated in Fig. 12 and 13. Observations (Fig. 12) indicated that as the scratches on the MAO coatings became shallower as Ca/P increases. In a glucose-free environment, the scratches for the 2.17 and 2.67 coatings were relatively short, about 1.1–1.3 cm. However, the 2.17 coating corresponded to a higher bonding force of 22.67 N. At the different glucose-content environments, the scratch and bonding strength of the coatings were also different, with the longest scratch and the lowest corresponding bonding strength in the 0.99 g L−1 glucose content environment. Combined with the electrochemical corrosion results, it was possible that the coating was more severely corroded by immersion in this environment, and the internal structure of the coating was corroded and relatively flimsy. After corrosion in environments with ≥5 g L−1 glucose content were 2.17 coatings with shallower scratches and the highest bonding strength. This may be due to the fact that the glucose molecules in the solution adhere to the coating surface, blocking the corrosive ions from damaging the coating and acting as a lubricant during the scratch test, so that the results were comparable to those before corrosion.35 Comparisons revealed that immersion corrosion of the 2.17 coating in electrolyte environments with varying glucose content had little effect on its binding strength, and it possessed the potential for safe use in diabetic patients.
 |
| | Fig. 12 Surface morphology of different coatings after immersion corrosion for 30 days: hank's solution: ((a) 1.67 coating, (b) 2.17 coating, (c) 2.67 coating), hank's solution with 0.99 g L−1 glucose: ((d) 1.67 coating, (e) 2.17 coating, (f) 2.67 coating), hank's solution with 5 g L−1 glucose: ((g) 1.67 coating, (h) 2.17 coating, (i) 2.67 coating), hank's solution with 8 g L−1 glucose: ((j) 1.67 coating, (k) 2.17 coating, (l) 2.67 coating). | |
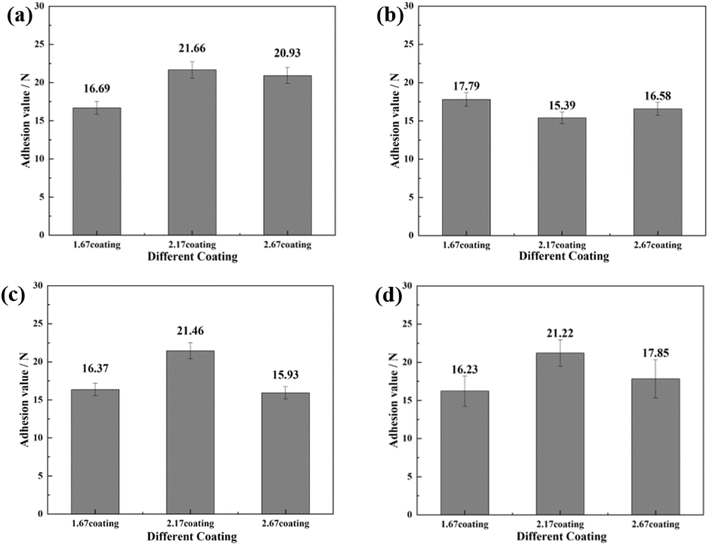 |
| | Fig. 13 Bonding strength of different coatings after immersion corrosion for 30 days: hank's solution (a), hank's solution with 0.99 g L−1 glucose (b), hank's solution with 5 g L−1 glucose (c), hank's solution with 8 g L−1 glucose (d). | |
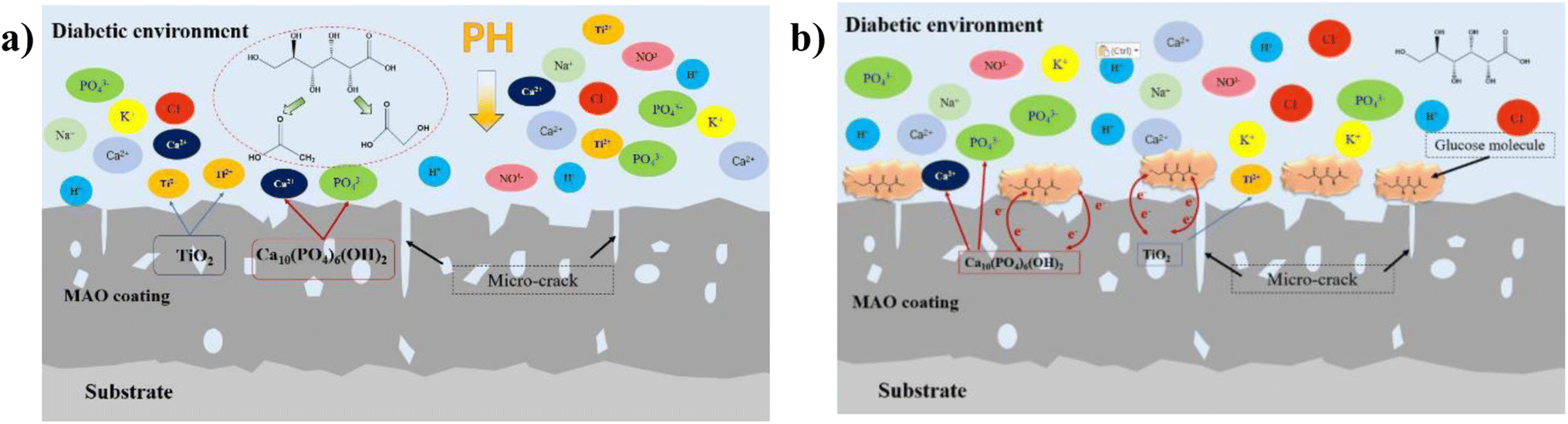
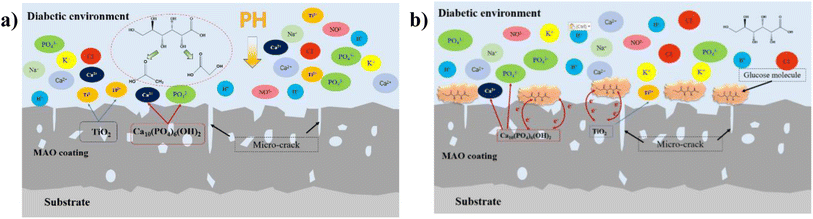 |
| | Fig. 14 Schematic diagram of ion exchange in diabetic environment: (a) low-glucose environment, (b) high glucose environment. | |
In this experiment, the corrosion of HA-containing MAO coatings in electrolyte environments with different glucose contents and its corrosion effect pattern on the coating bonding were investigated. At low concentration (glucose content of 0.99 g L−1), glucose content is low, which is easy to decompose into saccharic acid and reduce the pH of the electrolyte, promoting the corrosive effect of corrosive ions in the solution on the coating, as shown in Fig. 14a. When the concentration of glucose increased, glucose chelated the ions in the electrolyte, so the self-corrosion current and impedance decreased from the electrochemical corrosion results (Fig. 9 and 10). However, in combination with the post-corrosion SEM morphology analysis, it was found that the corrosion phenomenon of the coating became significantly more severe when the glucose content was ≥5 g L−1, which should be attributed to the corrosion reaction due to the glucose directly adhering to the coating surface. At the same time, higher glucose concentration reduced the bonding strength of the coatings after corrosion, which was due to the loose and cracked internal structure of the coating after corrosion. Interestingly, during the corrosion process of the HA-containing coating, Ca and P dissolve into solution while promoting osteogenic differentiation of stem cells, facilitating bone regeneration on the implant surface.
4 Conclusion
In this study, a MAO coating containing HA was successfully fabricated on a β Ti using MAO technology, and its corrosion mechanism, biocompatibility, and safety were comprehensively evaluated. Increasing the Ca/P ratio in MAO coatings enhanced porosity, wettability, thickness, and adhesion, underscoring the role of electrolyte composition in coating properties. HA nucleation and growth within the coating were influenced by the Ca/P ratio, with higher ratios improving corrosion resistance and bioactivity due to enhanced HA crystallinity and optimized chemical composition. Appropriately elevated Ca/P ratios enhanced the bond strength of coatings in diabetic environments. An optimal Ca/P ratio of approximately 2.17 was identified for balancing corrosion resistance and bioactivity in MAO coatings. This study could provide a meaningful theoretical basis for the failure pattern of implants in diabetic patients.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32360245), Open Project of Guangxi Key Laboratory of Regenerative Medicine (202004), and Middle-aged and Young Teachers' Basic Ability Promotion Project of Guangxi (2021KY0120), which are gratefully acknowledged.
References
- Q.-K. Meng, Y.-F. Huo, W. Ma, Y.-W. Sui, J.-Y. Zhang, S. Guo and X.-Q. Zhao, Design and fabrication of a low modulus β-type Ti–Nb–Zr alloy by controlling martensitic transformation, Rare Met., 2018, 37, 789–794, DOI:10.1007/s12598-018-1055-5.
- Y. L. Hao, Z. B. Zhang, S. J. Li and R. Yang, Microstructure and mechanical behavior of a Ti-24Nb-4Zr-8Sn alloy processed by warm swaging and warm rolling, Acta Mater., 2012, 60, 2169–2177, DOI:10.1016/j.actamat.2012.01.003.
- F. He, S. Yang and J. Cao, Effect of Cold Rolling and Aging on the Microstructure and Mechanical Properties of Ti-Nb-Zr Alloy, J. Mater. Eng. Perform., 2020, 29, 3411–3419, DOI:10.1007/s11665-020-04810-0.
- W. Weng, A. Biesiekierski, Y. Li and C. Wen, Effects of selected metallic and interstitial elements on the microstructure and mechanical properties of beta titanium alloys for orthopedic applications, Materialia, 2019, 6, 100323, DOI:10.1016/j.mtla.2019.100323.
- Q. Chen and G. A. Thouas, Metallic implant biomaterials, Mater. Sci. Eng., R, 2015, 87, 1–57, DOI:10.1016/j.mser.2014.10.001.
- S. Tunchel, A. Blay, R. Kolerman, E. Mijiritsky and J. A. Shibli, 3D Printing/Additive Manufacturing Single Titanium Dental Implants: A Prospective Multicenter Study with 3 Years of Follow-Up, Int. J. Dent., 2016, 2016, 8590971, DOI:10.1155/2016/8590971.
- Y. F. Xu, D. Q. Yi, H. Q. Liu, X. Y. Wu, B. Wang and F. L. Yang, Effects of cold deformation on microstructure, texture evolution and mechanical properties of Ti-Nb-Ta-Zr-Fe alloy for biomedical applications, Mater. Sci. Eng., A, 2012, 547, 64–71, DOI:10.1016/j.msea.2012.03.081.
- B. Sun, X. L. Meng, Z. Y. Gao and W. Cai, Martensite structure and mechanical property of Ti-Nb-Ag shape memory alloys for biomedical applications, Vacuum, 2018, 156, 181–186, DOI:10.1016/j.vacuum.2018.07.029.
- S. Shin, C. Zhang and K. S. Vecchio, Phase stability dependence of deformation mode correlated mechanical properties and elastic properties in Ti-Nb gum metal, Mater. Sci. Eng., A, 2017, 702, 173–183, DOI:10.1016/j.msea.2017.06.099.
- Y. Tsutsumi, M. Niinomi, M. Nakai, H. Tsutsumi, H. Doi, N. Nomura and T. Hanawa, Micro-arc oxidation treatment to improve the hard-tissue compatibility of Ti-29Nb-13Ta-4.6Zr alloy, Appl. Surf. Sci., 2012, 262, 34–38, DOI:10.1016/j.apsusc.2012.01.024.
- J. Liu, W. Yang, B. Tao, T. Shen, Y. He, X. Shen and K. Cai, Preparing and immobilizing antimicrobial osteogenic growth peptide on titanium substrate surface, J. Biomed. Mater. Res., Part A, 2018, 106, 3021–3033, DOI:10.1002/jbm.a.36491.
- P. Jiang, Z. Ji, X. Wang and F. Zhou, Surface functionalization – a new functional dimension added to 3D printing, J. Mater. Chem. C, 2020, 8, 12380–12411, 10.1039/D0TC02850A.
- G. Dercz, J. Barczyk, I. Matuła, T. Kubaszek, M. Góral, J. Maszybrocka, D. Bochenek, S. Stach, M. Szklarska, D. Ryszawy and M. Pudełek, Characterization of YSZ Coatings Deposited on cp-Ti Using the PS-PVD Method for Medical Applications, Coatings, 2021, 11, 1348, DOI:10.3390/coatings11111348.
- S. Luo, Q. Wang, R. Ye and C. S. Ramachandran, Effects of electrolyte concentration on the microstructure and properties of plasma electrolytic oxidation coatings on Ti-6Al-4V alloy, Surf. Coat. Technol., 2019, 375, 864–876, DOI:10.1016/j.surfcoat.2019.07.053.
- P. Tabrizian, H. Sun, U. Jargalsaikhan, T. Sui, S. Davis and B. Su, Biomimetic Nacre-like Hydroxyapatite/Polymer Composites for Bone Implants, J. Funct. Biomater., 2023, 14, 393, DOI:10.3390/jfb14080393.
- J. D. Afroze, M. J. Abden and M. A. Islam, An efficient method to prepare magnetic hydroxyapatite-functionalized multi-walled carbon nanotubes nanocomposite for bone defects, Mater. Sci. Eng., C, 2018, 86, 95–102, DOI:10.1016/j.msec.2018.02.002.
- D.-Y. Kim, M. Kim, H.-E. Kim, Y.-H. Koh, H.-W. Kim and J.-H. Jang, Formation of hydroxyapatite within porous TiO2 layer by micro-arc oxidation coupled with electrophoretic deposition, Acta Biomater., 2009, 5, 2196–2205, DOI:10.1016/j.actbio.2009.02.021.
- X. Zhang, Y. Wu, Y. Lv, Y. Yu and Z. Dong, Formation mechanism, corrosion behavior and biological property of hydroxyapatite/TiO2 coatings fabricated by plasma electrolytic oxidation, Surf. Coat. Technol., 2020, 386, 125483, DOI:10.1016/j.surfcoat.2020.125483.
- M. Qadir, Y. Li, K. Munir and C. Wen, Calcium Phosphate-Based Composite Coating by Micro-Arc Oxidation (MAO) for Biomedical Application: A Review, Crit. Rev. Solid State Mater. Sci., 2018, 43, 392–416, DOI:10.1080/10408436.2017.1358148.
- R.-C. Zeng, X.-T. Li, S.-Q. Li, F. Zhang and E.-H. Han, In vitro degradation of pure Mg in response to glucose, Sci. Rep., 2015, 5, 13026, DOI:10.1038/srep13026.
- A. Abdal-hay, T. Amna and J. K. Lim, Biocorrosion and osteoconductivity of PCL/nHAp composite porous film-based coating of magnesium alloy, Solid State Sci., 2013, 18, 131–140, DOI:10.1016/j.solidstatesciences.2012.11.017.
- L.-Y. Li, Z.-Z. Han, R.-C. Zeng, W.-C. Qi, X.-F. Zhai, Y. Yang, Y.-T. Lou, T. Gu, D. Xu and J.-Z. Duan, Microbial ingress and in vitro degradation enhanced by glucose on bioabsorbable Mg–Li–Ca alloy, Bioact. Mater., 2020, 5, 902–916, DOI:10.1016/j.bioactmat.2020.06.014.
- L.-Y. Cui, X.-T. Li, R.-C. Zeng, S.-Q. Li, E.-H. Han and L. Song, In vitro corrosion of Mg–Ca alloy - The influence of glucose content, Front. Mater. Sci., 2017, 11, 284–295, DOI:10.1007/s11706-017-0391-y.
- L.-Y. Li, B. Liu, R.-C. Zeng, S.-Q. Li, F. Zhang, Y.-H. Zou, H. G. Jiang, X.-B. Chen, S.-K. Guan and Q.-Y. Liu, In vitro corrosion of magnesium alloy AZ31- a synergetic influence of glucose and Tris, Front. Mater. Sci., 2018, 12, 184–197, DOI:10.1007/s11706-018-0424-1.
- M. J. Bailey, S. Coe, D. M. Grant, G. W. Grime and C. Jeynes, Accurate determination of the Ca: P ratio in rough hydroxyapatite samples by SEM-EDS, PIXE and RBS- a comparative study, X-Ray Spectrom., 2009, 38, 343–347, DOI:10.1002/xrs.1171.
- M. Kaseem, S. Fatimah, N. Nashrah and Y. G. Ko, Recent progress in surface modification of metals coated by plasma electrolytic oxidation: Principle, structure, and performance, Prog. Mater. Sci., 2021, 117, 100735, DOI:10.1016/j.pmatsci.2020.100735.
- Q. Mo, G. Qin, K. Ling, X. Lv, N. Wang and W. Li, Layer-by-layer self-assembled polyurea layers onto MAO surface for enhancing corrosion protection to aluminum alloy 6063, Surf. Coat. Technol., 2021, 405, 126653, DOI:10.1016/j.surfcoat.2020.126653.
- D. K. Muthee and B. F. Dejene, Effect of annealing temperature on structural, optical, and photocatalytic properties of titanium dioxide nanoparticles, Heliyon, 2021, 7(6), e07269, DOI:10.1016/j.heliyon.2021.e07269.
- F. B. Torstrick, A. S. P. Lin, D. L. Safranski, D. Potter, T. Sulchek, C. S. D. Lee, K. Gall and R. E. Guldberg, Effects of Surface Topography and Chemistry on Polyether-Ether-Ketone (PEEK) and Titanium Osseointegration, Spine, 2020, 45(8), 417–424, DOI:10.1097/BRS.0000000000003303.
- S. Bose, D. Banerjee, A. Shivaram, S. Tarafder and A. Bandyopadhyay, Calcium phosphate coated 3D printed porous titanium with nanoscale surface modification for orthopedic and dental applications, Mater. Des., 2018, 151, 102–112, DOI:10.1016/j.matdes.2018.04.049.
- E. Urbańczyk, A. Krząkała, A. Kazek-Kęsik, J. Michalska, A. Stolarczyk, G. Dercz and W. Simka, Electrochemical modification of Ti–13Nb–13Zr alloy surface in phosphate based solutions, Surf. Coat. Technol., 2016, 291, 79–88, DOI:10.1016/j.surfcoat.2016.02.025.
- A. Arifin, A. B. Sulong, N. Muhamad, J. Syarif and M. I. Ramli, Material processing of hydroxyapatite and titanium alloy (HA/Ti) composite as implant materials using powder metallurgy: A review, Mater. Des., 2014, 55, 165–175, DOI:10.1016/j.matdes.2013.09.045.
- C. Chirico, A. V. Romero, E. Gordo and S. A. Tsipas, Improvement of wear resistance of low-cost powder metallurgy β-titanium alloys for biomedical applications, Surf. Coat. Technol., 2022, 434, 128207, DOI:10.1016/j.surfcoat.2022.128207.
- L.-Y. Li, L.-Y. Cui, B. Liu, R.-C. Zeng, X.-B. Chen, S.-Q. Li, Z.-L. Wang and E.-H. Han, Corrosion resistance of glucose-induced hydrothermal calcium phosphate coating on pure magnesium, Appl. Surf. Sci., 2019, 465, 1066–1077, DOI:10.1016/j.apsusc.2018.09.203.
- W. Yan, Y.-J. Lian, Z.-Y. Zhang, M.-Q. Zeng, Z.-Q. Zhang, Z.-Z. Yin, L.-Y. Cui and R.-C. Zeng, In vitro degradation of pure magnesium- the synergetic influences of glucose and albumin, Bioact. Mater., 2020, 5, 318–333, DOI:10.1016/j.bioactmat.2020.02.015.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b