
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAminocyclopropenium as a novel hydrogen bonding organocatalyst for cycloaddition of carbon disulfide and epoxide to prepare cyclic dithiocarbonate†
Xinru Dua,
Ziqi Liub,
Zhenjiang Li *b,
Xin Yuanb,
Chunyu Lib,
Min Zhangb,
Zhihao Zhangb,
Xin Hu*a and
Kai Guob
*b,
Xin Yuanb,
Chunyu Lib,
Min Zhangb,
Zhihao Zhangb,
Xin Hu*a and
Kai Guob
aCollege of Materials Science and Engineering, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, China. E-mail: xinhu@njtech.edu.cn
bState Key Laboratory Materials-Oriented Chemical Engineering, College of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Road South, Nanjing 211816, China. E-mail: zjli@njtech.edu.cn
First published on 2nd April 2024
Abstract
The smallest Hückel aromatic ring cyclopropenium substituted by electron-donating C-amino groups produced a aminocyclopropenium electron-rich cation. A bifunctional aminocyclopropenium halide catalyst installed with bis-(hydroxyethyl) functions on the amino group was then designed. A typical (diethanolamino)cyclopropenium halide catalyst C5·I was screened optimally for the cycloaddition of carbon disulfide into an epoxide to produce cyclic dithiocarbonate with an excellent conversion (95%) and high selectivity (92%). The electrostatic enhancement of alkyl C–H HBD capability was implemented via vicinal positive charges on the cyclopropenium core, and the acidity of the terminal O–H hydrogen proton increased by intramolecular H-bonding between the two hydroxy groups on the diethanolamino group (O–H⋯O–H). Then, a hybrid H-bond donor comprising enhanced alkyl C–H and hydroxy O–H was formed. The hybrid HBD offered by aminocyclopropenium was vital in activating the epoxide and stabilizing the intermediate, resulting in reduced O/S scrambling. Moreover, weakly coordinated iodide anion served as a nucleophilic reagent to open the ring of the epoxide. The cooperative catalytic mechanism of the HBD cation and halide anion was supported by NMR titrations and control experiments. Eleven epoxides with various substituents were converted into the corresponding cyclic thiocarbonate with high conversion and selectivity under mild conditions (25 °C, 6 h) without a solvent. The cycloaddition of carbon disulfide with epoxides catalyzed by aminocyclopropenium developed a new working model for hydrogen bonding organocatalysis.
1 Introduction
Epoxides are not only easily prepared but also react with numerous reagents and are versatile intermediates for organic synthesis.1 The cycloaddition reaction of epoxide and heterocumulene is a convenient, efficient, and atom-economical approach to synthesizing high-value-added five-membered heterocyclic compounds.2–5 In recent years, the cycloaddition of epoxide with carbon dioxide (CO2) to produce cyclic carbonate has been well studied and is quite promising for industrial applications.6–12 Cyclic dithiocarbonates are sulfur-containing analogs of cyclic carbonates, which can also be produced by coupling epoxides with carbon disulfide (CS2).13 Despite potential applications in monomers for sulfur-containing polymers14–18 and radiation protection material,19 relatively few studies have been published. CS2 is an isoelectronic analogue of CO2,20 which occurs naturally in gases released from the surface and produced by microbial metabolism;21 this bulk chemical can also be considered an inexpensive C1 building block.13 Studies on the cycloaddition of carbon disulfide with epoxides were first published in 1960.22,23 In contrast to carbon dioxide, the cycloaddition of carbon disulfide and epoxide often yielded complex by-products at high temperatures or long reaction times (Scheme 1). This is primarily due to the frequent scrambling of oxygen and sulfur atoms, leading to a decrease in the selectivity of the reaction, which was first documented by Endo et al.24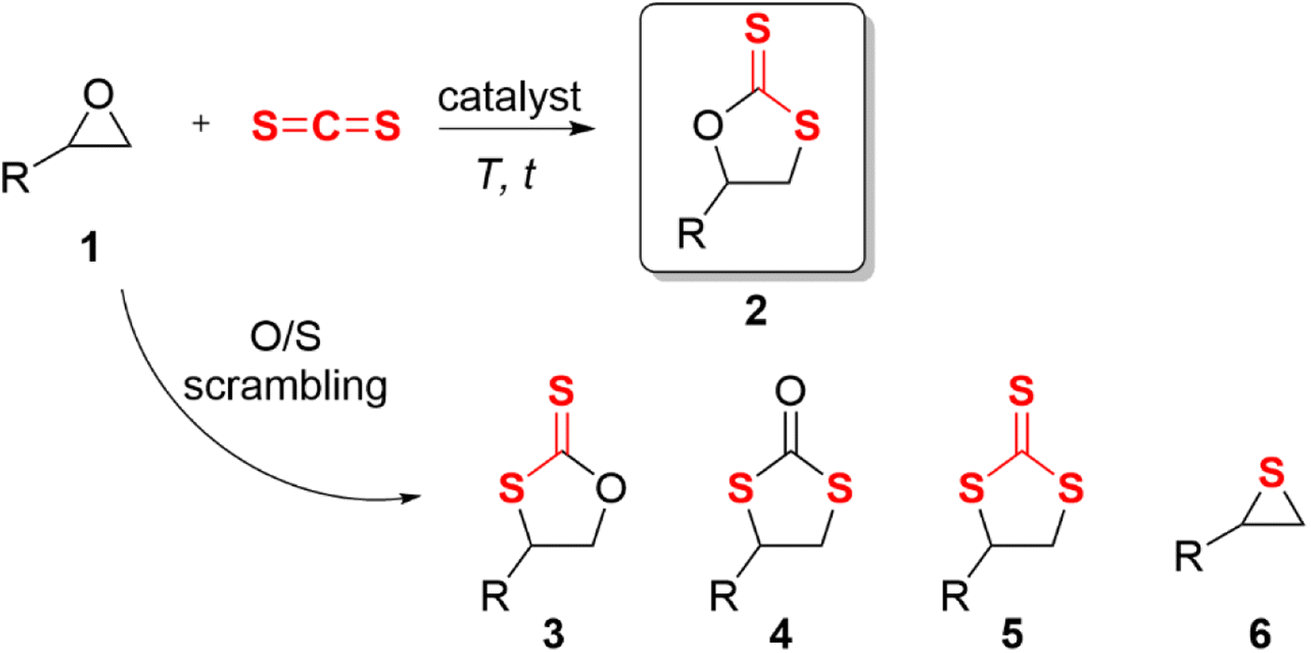 | ||
| Scheme 1 Cycloaddition of epoxides with carbon disulfide and other side reactions. | ||
For the insertion of CS2 in epoxides, base catalysts were most frequently used. The nucleophilic base-activated carbon disulfide forms an adduct intermediate, and then the sulfur anion attacks the epoxide causing the ring-opening20,23,25–31 (Scheme 2a). The monofunctional metal halide offered a nucleophilic halide anion to open the three-membered ring of the epoxide24,32–34 (Scheme 2b). Another catalytic mode was a metal complex (MLn) and a halogen anion binary co-catalysis system.2,5,35–38 The metal complex served as Lewis acid to activate the epoxide, and the halogen anion nucleophilically attacked the methylene carbon of the epoxide to open the ring (Scheme 2c). Most of them require co-catalysts, high catalyst loading, toxic solvents, or harsh reaction conditions. The prolonged time and high temperature sharply increased the risk of generating unwanted by-products, such as cyclic trithiocarbonate 5 or sulfur-containing polycarbonate.38 Moreover, the base may lead to ring opening or self-polymerization of the epoxide.39 There is little information available in the literature about bifunctional hydrogen bonding catalysis for this reaction.33,40,41 Noncovalent H-bonding interactions between charge-deficient hydrogen protons and electrophilic species can activate substrates and/or stabilize intermediates,42 which has emerged as a key strategy for organic transformations.43 We predicted that the O/S scrambling during the coupling of CS2 with epoxide can be well controlled through H-bonding. Herein, a neutral H-bonding bifunctional organocatalyst was proposed to facilitate its gentle conversion.
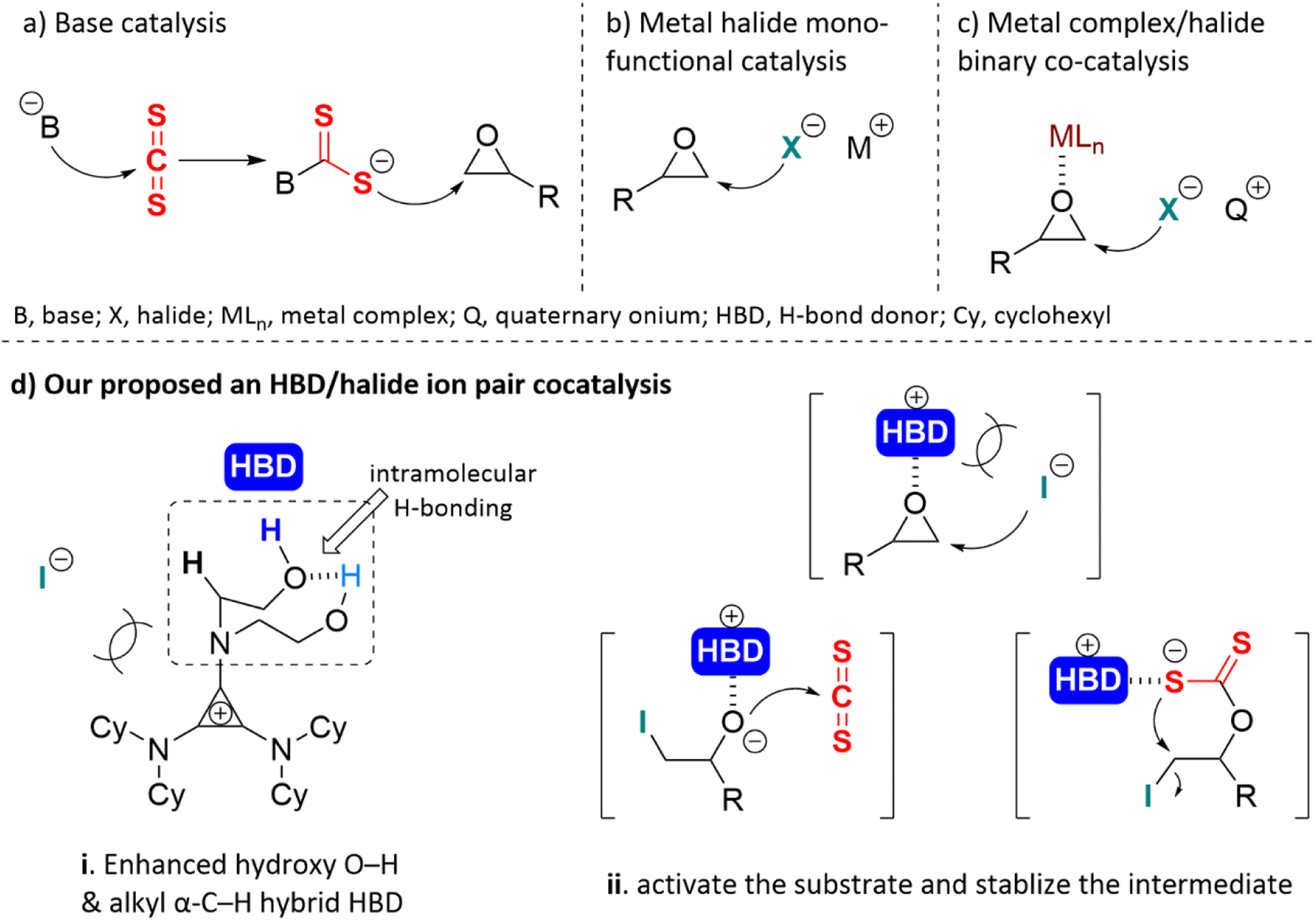 | ||
| Scheme 2 (a) Base activated carbon disulfide first, then the sulfur anion opened the ring of the epoxide; (b) the monofunctional metal halide offered a halide anion to attack the methylene carbon of the epoxide; (c) the metal complex (MLn) activated the epoxide, and the halide anion opened the ring of the epoxide; (d) a novel H-bonding catalyst was proposed to facilitate the cycloaddition of the epoxide and carbon disulfide. (i) Intramolecular H-bonding between the hydroxy group enhanced O–H, formed a hybrid HBD system with α-C–H. (ii) The HBD system could activate the epoxide and stabilize the anion intermediate. | ||
Cyclopropenium is the smallest aromatic compound that satisfies the Hückel's rule and was first synthesized in 1957 by Breslow et al.44 When it was substituted with amino groups, the aromatic cation electrostatically enhanced the acidity of the side chain α-C–H,45 which can be regarded as a nonclassical H-bond donor. The orbital repulsion between the electron-excess ion pair would offset some of the coulombic force.46,47 This phenomenon gave rise to what is known as “ion pair strain”, characterized by the weakened coordination of the halide anion with cyclopropenium, and it was predicted that the catalytic activity of these anions was very high.48 Aminocyclopropenium as an H-bonding catalyst has been utilized in many organic reactions,49–52 such as ring-opening polymerization,53 cycloaddition of CO2 into epoxide,54 pyranylation of alcohols,55 and Payne-type rearrangement of glycidol.56 Based on previous studies, a novel HBD aminocyclopropenium ion pair catalyst was designed to efficiently convert epoxide and carbon disulfide to cyclic carbonate under solvent-free and mild conditions. The intramolecular H-bonding between two hydroxyl groups improved the acidity of the hydrogen proton of the terminal hydroxyl group (O–H⋯O–H*).57 The enhanced O–H and α-C–H formed a hybrid H-bond donor system (Scheme 2d(i)). The hybrid HBD polarized the epoxide and stabilized the anion intermediate via H-bonding (Scheme 2d(ii)).
2 Experimental section
2.1 General procedure for the cycloaddition reaction of carbon sulfide and epoxides
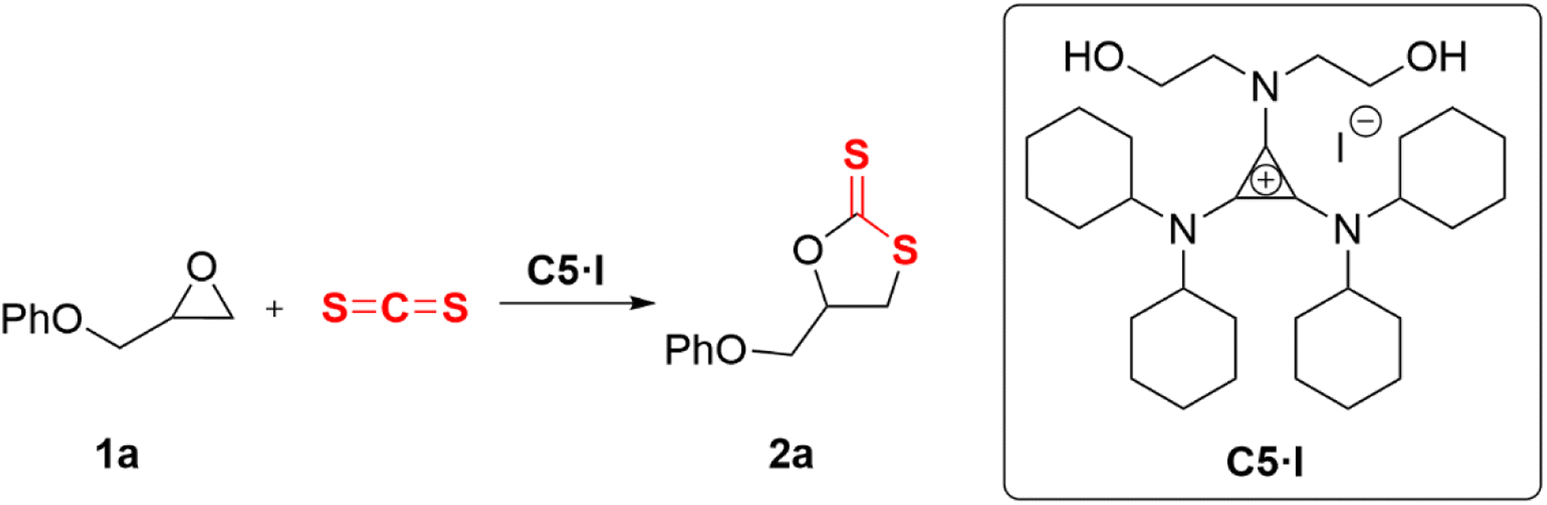
All operations were performed using standard Schlenk techniques with a nitrogen atmosphere to reduce exposure to water and oxygen. A flame-dried 10 mL Schlenk tube containing a magnetic stirring bar was charged with carbon sulfide (0.14 mL, 2.4 mmol, 1.2 equiv.), epoxide 1 (2 mmol, 1 equiv.), and 1-diethanolamino-2,3-bis(dicyclohexylamino)cyclopropenium iodide (C5·I) (63 mg, 0.1 mmol, 0.05 equiv.) under an argon atmosphere. The reaction mixture was stirred at 25 °C for 6 h. After that, the mixture was purified by silica gel flash column chromatography (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA = 5:1) to give the corresponding products 2a–2k.
:EA = 5:1) to give the corresponding products 2a–2k.
3 Results and discussion
3.1 Design of aminocyclopropenium catalysts and evaluation of the catalytic performances
Aminocyclopropenium acted as a hydrogen bond donor (HBD) by rationally designing the amino substituents. Typical HBDs were O–H and N–H, and C–H was too weak to serve as an H-bond donor from a common perception. However, the electron-donating π-conjugative effect of amino group was stronger than the electron-withdrawing σ-inductive effect in the cyclopropenium system. The α-C–H of the substituted alkylamino group on the cyclopropenium cation was weakly acidic and exhibited a capacity of HBD.54,56 On the basis of the above theoretical foundations and previous research work, three types of HBD cyclopropenium cations were designed and synthesized, in which alkylaminocyclopropenium C1–C3 was nonclassical C–H HBD. Anilino-cyclopropenium C4 synthesized according to the literature procedure58 had a bidentate N–H HBD, and the diethanolamino on C5 provided two hydroxyl groups. To investigate the effect of the amount of O–H on the catalytic behavior, morpholino- and N-aniline-N-ethanolamino-substituted cyclopropenium C6 and C7 were synthesized. With the purpose of verifying the promotion of the cyclopropenium moiety, the catalyst C8 was prepared through Hofmann alkylation of N-methyl-N-diethanolamine.59 Carbon disulfide (CS2, 1 mL) and phenyl glycidyl ether 2a (PGE, 2 mmol) were reacted at 25 °C for 24 h as a template reaction to evaluate the catalytic effect of the catalysts mentioned above (Table 1).
|
|||
|---|---|---|---|
| Entry | Catalyst | Conv.b (%) | Selec.b (%) |
| a All reactions were performed with 2 mmol of 2-(phenoxymethyl)oxirane (1a) and carbon disulfide (1 mL) catalyzed by C5·I (5 mol%) for 24 hours at 25 °C under Ar; n.d. = not determined; Conv. = conversion; Selec. = selectivity; TBAC in entry 13 was tetrabutylammonium chloride.b Conversions (of 1a) and selectivity (to 2a) were determined by 1H NMR with dodecane as an internal standard. | |||
| 1 | C1 | 22 | 83 |
| 2 | C2·Cl | 23 | 85 |
| 3 | C3 | 33 | 90 |
| 4 | TBAC | 12 | n.d. |
| 5 | C2·I | 46 | 87 |
| 6 | C4 | n.d. | n.d. |
| 7 | C5·Cl | 51 | 90 |
| 8 | C5·Br | 76 | 91 |
| 9 | C5·I | 88 | 92 |
| 10 | C5·BF4 | 8 | n.d. |
| 11 | C6 | 33 | 90 |
| 12 | C7 | 25 | 85 |
| 13 | C8 | 19 | n.d. |
| 14 | NaI | n.d. | n.d. |
| 15 | — | n.d. | n.d. |
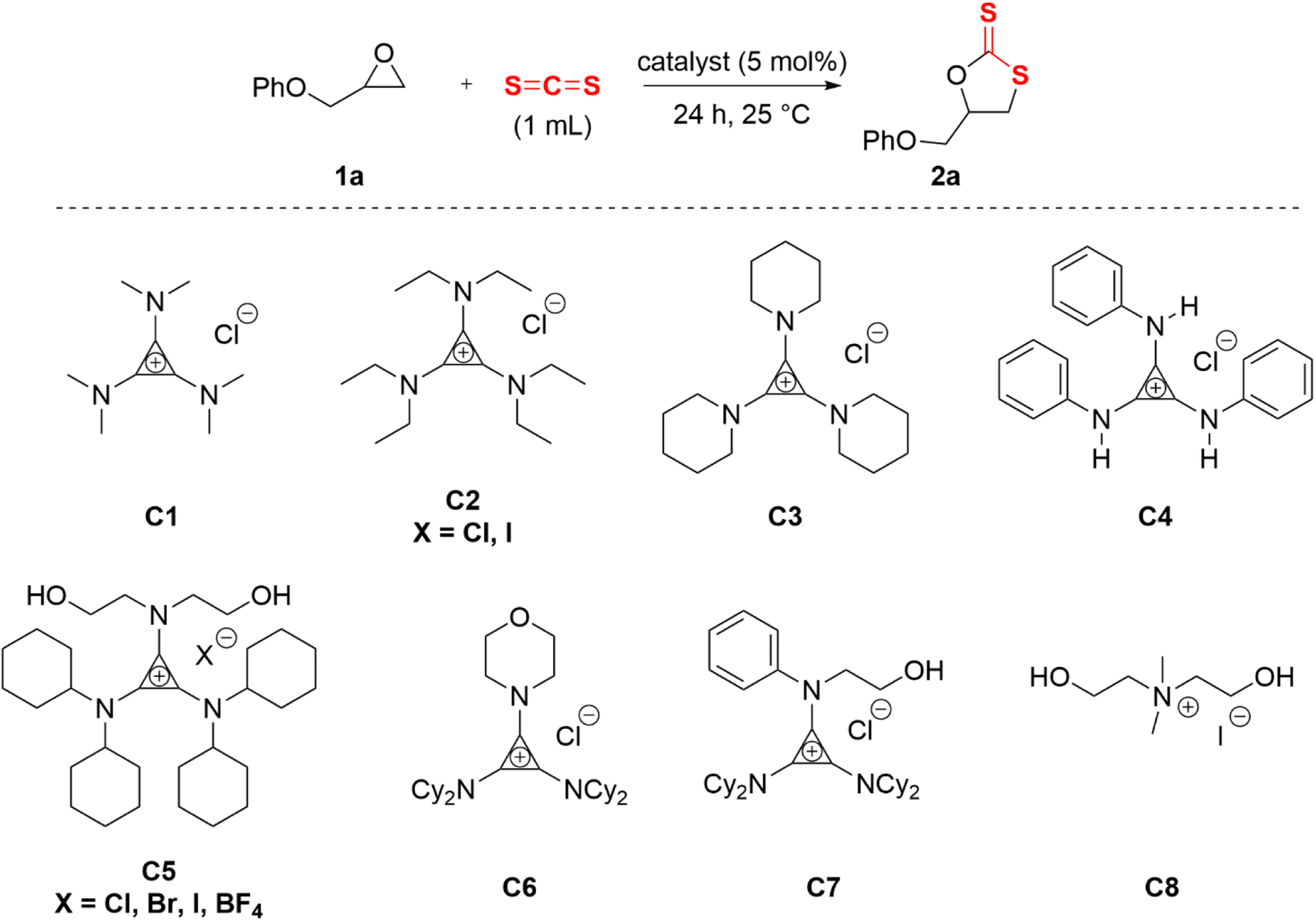
Initially, 2-(phenoxymethyl)oxirane (1a) was selected as a model substrate for the screening of the optimal catalysts. The evaluation results of aminocyclopropenium catalysts are shown in Table 1. With the increase of the alkylamino chain length on the cyclopropenium, the conversion and selectivity of the substrate improved (Table 1, entries 1–3, the detailed analytical method of conversion and selectivity, see ESI†). Tetrabutylammonium chloride (TBAC) was used as a control catalyst without the H-bond donor and exhibited a weak catalytic ability (Table 1, entry 4), which indicated that the aromatic cyclopropenium cation did enhance the acidity of C–H by electrostatic interaction. While the anion was switched to iodide, the conversion improved significantly but was still unsatisfactory at 46% (Table 1, entry 5). The reaction could not proceed when the N–H hydrogen bond donor catalyst C4 was applied (Table 1, entry 6). Diethanolamino-cyclopropenium chloride (C5·CI) catalyzed the cycloaddition of the epoxide and carbon disulfide with 51% conversion and 90% selectivity (Table 1, entry 7). Surprisingly, the anions of C5 were replaced with bromine and iodine with improved conversion and the same selectivity (Table 1, entries 8 and 9, C5·Br: Conv. 76%, Selec. 91%; C5·I: Conv. 88%, Selec. 92%). A similar experimental result has been reported in a previous work.33 The morpholino-substituted catalyst C6 showed comparable activity to C3 (Table 1, entry 11), probably due to both playing the role of C–H HBD. Catalysts with only a single hydroxyl group were less effective and less selective (Table 1, entry 12). The cycloaddition reaction did not occur if the catalyst was omitted (Table 1, entry 15). In fact, N–H HBD could not convert the epoxide and C–H HBD catalysts were poor to fair in the catalytic performance. However, the O–H HBD-containing cyclopropenium iodide was able to promote the cycloaddition of epoxide and carbon disulfide, and the number of hydroxyl groups was also important to the success of the reaction. Moreover, we believed that the anion influenced the conversion of the epoxide and the steric hindrance of the cyclopropenium substituent affected the selectivity of cyclic dithiocarbonate.
3.2 Optimization of the cycloaddition reaction conditions
Reaction conditions were then optimized with C5·I as the catalyst. The initial experiment was performed with 1a and CS2 in the presence of C5·I. Catalyst loading, solvent, reaction temperature, reaction time, and the equivalent amount of carbon disulfide were the five factors related to optimization.With increase in catalyst loading, the conversion improved and selectivity deteriorated (Table 2, entries 1–5). The conversion of 2a only showed a minor increase when the reaction temperature was raised from 25 °C to 80 °C (Table 2, entries 3, 6 and 7). Elevated temperatures harmed the formation of cyclic dithiocarbonates, resulting in increased occurrence of oxygen/sulfur scrambling in the reaction intermediates, leading to a reduction in selectivity.24 By extending the reaction time to 24 h, a significant improvement in the conversion was observed (Table 2, entries 3 and 8–10). Considering that the catalyst was soluble in chloroform and methanol only, the experiment was conducted utilizing chloroform as the solvent to avoid interference caused by methanol protons. It was observed that the addition of the solvent did not contribute to an increase in the reaction rate (Table 2, entry 11). The desired conversion rate was not obtained even when the reaction time was prolonged to 24 h (Table 2, entry 11, parentheses). Finally, the equivalent of carbon disulfide was also screened. Carbon disulfide served as both the solvent and reactant in the initial experiments. Surprisingly, as the amount of carbon disulfide was gradually reduced to 1.2 equivalents, compound 2a could be rapidly obtained at room temperature with excellent conversion and selectivity (Table 2, entries 12 and 13). Prolonging the reaction time to 24 h did not significantly improve the conversion and selectivity (Table 2, entry 12, parentheses). To summarize, the optimal conditions were established as catalyst C5·I (5 mol%), CS2 (1.2 equiv.), solvent-free, 25 °C, and 6 h.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst loading (mol%) | Solvent | Temperature (°C) | Time (h) | Conv.b | Selec.b |
| a All reactions were performed with 2 mmol of 2-(phenoxymethyl)oxirane (1a) and carbon disulfide (1 mL) catalyzed by C5·I (5 mol%) under Ar; n.d. = not determined; Conv. = conversion; Selec. = selectivity.b Conversions (of 1a) and selectivity (to 2a) were determined by 1H NMR with dodecane as an internal standard.c 1.5 mL solvent was added; prolonging the reaction time to 24 h, the conversion was 57%.d CS2 (0.14 mL, 2.4 mmol, 1.2 equiv.); prolonging the reaction time to 24 h, the conversion was 95%.e CS2 (0.24 mL, 4 mmol, 2 equiv.). | ||||||
| 1 | 1 | Neat | 80 | 24 | 6% | n.d. |
| 2 | 2 | Neat | 25 | 24 | 37% | 88% |
| 3 | 5 | Neat | 25 | 24 | 88% | 92% |
| 4 | 8 | Neat | 25 | 24 | 95% | 86% |
| 5 | 10 | Neat | 25 | 24 | 96% | 65% |
| 6 | 5 | Neat | 40 | 24 | 90% | 88% |
| 7 | 5 | Neat | 80 | 24 | 92% | 86% |
| 8 | 5 | Neat | 25 | 4 | 35% | 90% |
| 9 | 5 | Neat | 25 | 8 | 56% | 91% |
| 10 | 5 | Neat | 25 | 16 | 70% | 91% |
| 11c | 5 | Chloroform | 25 | 6 (24) | 31% (57%) | 90% |
| 12d | 5 | Neat | 25 | 6 (24) | 84% (95%) | 89% |
| 13e | 5 | Neat | 25 | 6 | 75% | 90% |
3.3 Substrate scope studies for cycloaddition of epoxides into CS2 catalyzed by C5·I
With the advent of this efficient cooperative catalytic system, a variety of substituted epoxides were tested to probe the versatility of this catalytic system (Table 3). Aryl glycidyl ethers were generally suitable for this protocol, with 84% of phenyl glycidyl ethers converted to 2a within 6 h. Prolonging the reaction time to 24 h increased the conversion to 95% with 89% selectivity. The reactivity of o-tolyl glycidyl ether was relatively low and afforded 2b in 88% conversion in 12 h. It is noteworthy that not only cyclic trithiocarbonate 5c but also a mixture of the regional isomers of dithiocarbonate 3c was determined through thin-layer chromatography and 1H NMR (Table 3, entry 3, Conv. 92%; Selec. 82%). The cycloaddition reaction of styrene oxide and carbon disulfide generated preferentially the regioisomer 3c because of the accelerated α-cleavage by the phenyl substituent; this phenomenon was supported by previously reported studies.1,24,26 The trifluoromethyl-substituted epoxide 1d was well tolerated with 94% conversion, indicating that the electron-withdrawing substituted substrate was also suitable for this protocol. The lower reaction selectivity of epichlorohydrin may suffer from the generation of cyclic trithiocarbonate (Table 3, entry 5, Conv. 86%; Selec. 80%). Epoxides with electron-donating substituents were utilized to study the influence of the electronic effect of the substrate on the reaction. The methyl- and t-butyl-substituted glycidyl ethers yielded the corresponding products 2h and 2j in 75% and 82% conversions, respectively. Olefin-containing epoxides were able to facilitate the cycloaddition with carbon disulfide with excellent conversions and selectivity (Conv. 2f: 85%; 2g: 92%; 2i: 92%). Besides, the conversion of bisepoxide BPA 1k was 85%, and the selectivity of 2k was 89%, which was the monomer for the sulfur-containing polymers.60,61 Note that 0.5 mL DMF was added as a solvent to promote solid substrate 1k dissolution. However, internal epoxides such as 1l and 1m were not observed to produce the products even after 48 h of reaction, probably the excessive substituent hindering the ring-opening process.
|
||||
|---|---|---|---|---|
| Entry | Epoxide 1 | Product 2 | Conv.b | Selec.b |
| a All reactions were performed with epoxides (2 mmol, 1 equiv.) and carbon disulfide (0.14 mL, 2.4 mmol, 1.2 equiv.) catalyzed by C5·I (5 mol%) for 6 h under Ar; n.d. = not determined; Conv. = conversion; Selec. = selectivity.b Conversions (of substrate 1) and selectivity (to product 2) were determined by 1H NMR with dodecane as an internal standard.c Reaction time was 12 hours.d CS2 (0.48 mL, 8 mmol, 4 equiv.) was used. DMF (0.5 mL) was added as a solvent to dissolve the substrate. | ||||
| 1 | 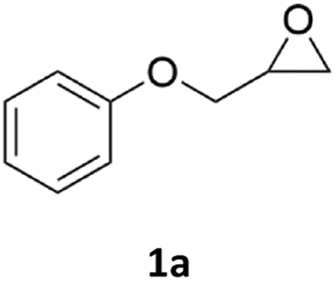 |
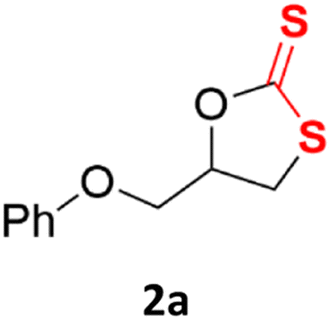 |
84% (95% for 24 h) | 89% |
| 2c | 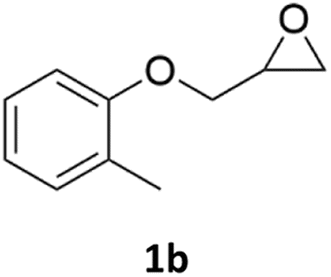 |
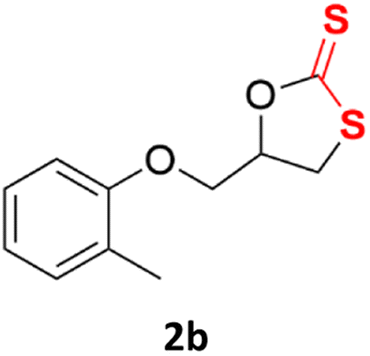 |
88% | 91% |
| 3 | 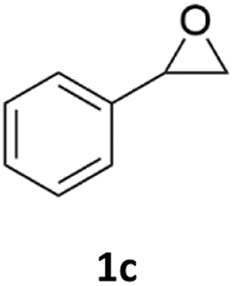 |
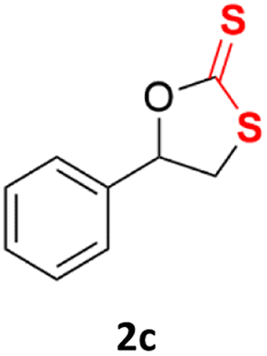 |
92% | 82% |
| 4 | 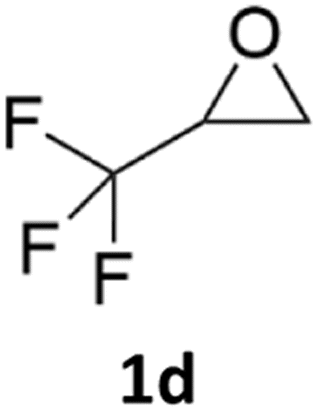 |
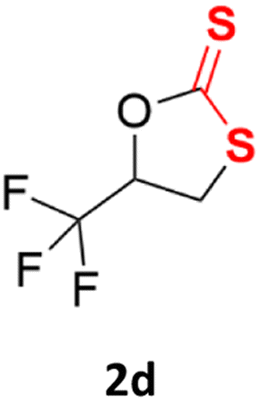 |
94% | 91% |
| 5c | 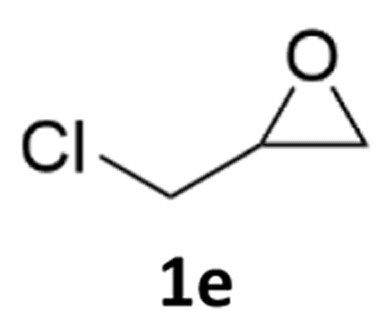 |
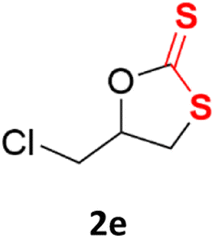 |
86% | 80% |
| 6 | 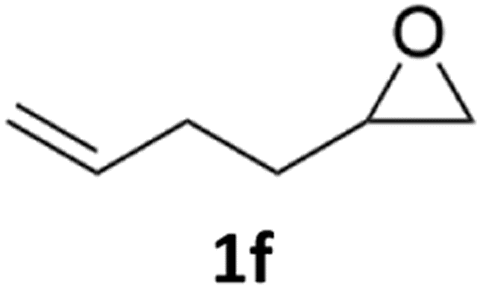 |
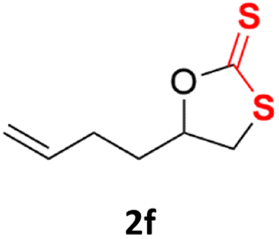 |
85% | 91% |
| 7c | 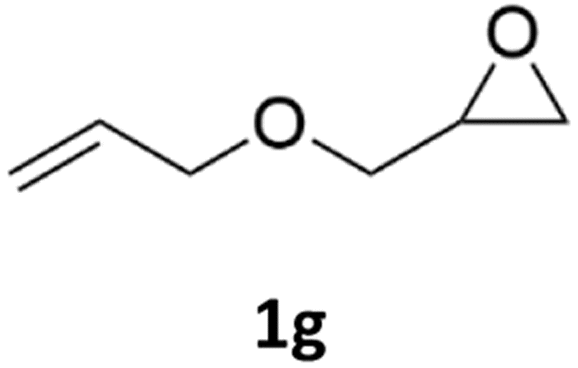 |
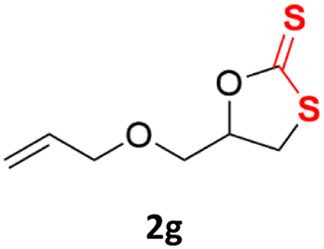 |
92% | 91% |
| 8 | 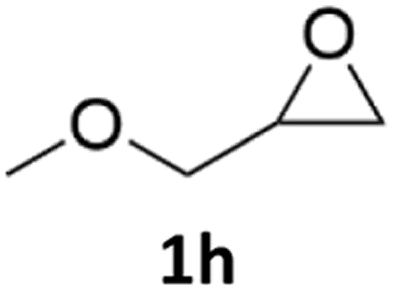 |
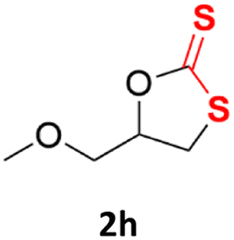 |
75% | 91% |
| 9 | 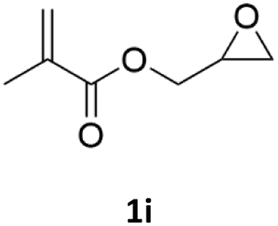 |
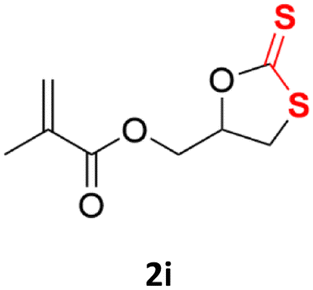 |
92% | 91% |
| 10 | 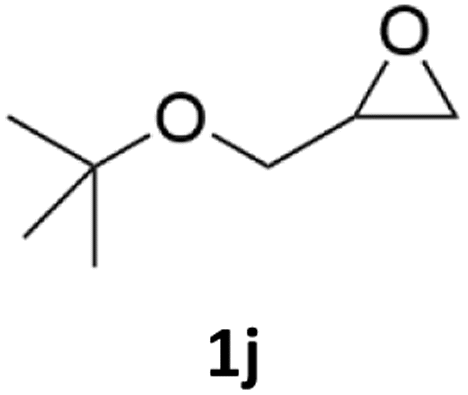 |
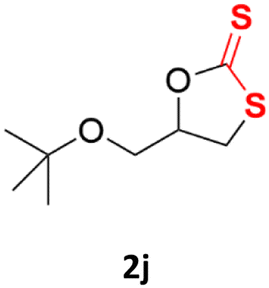 |
82% | 89% |
| 11d | 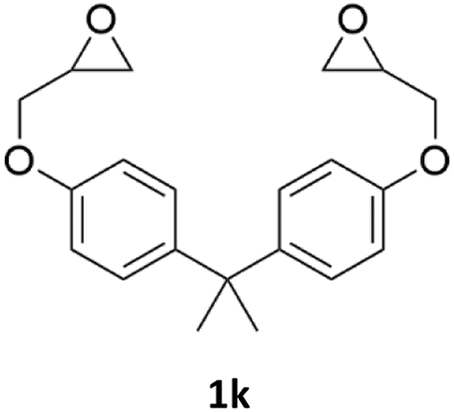 |
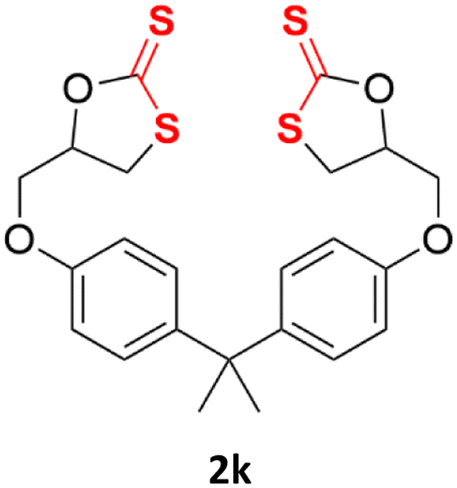 |
85% | 89% |
| 12 | 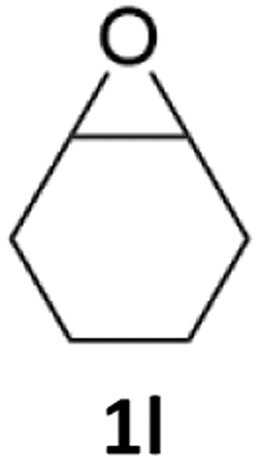 |
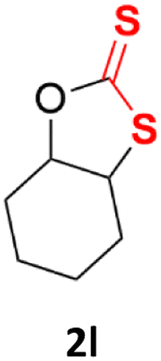 |
n.d. | n.d. |
| 13 | 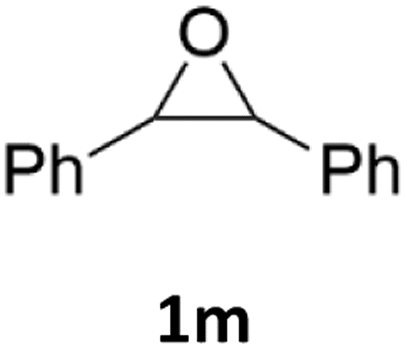 |
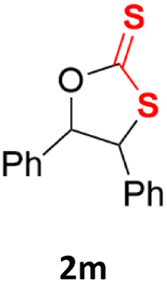 |
n.d. | n.d. |
3.4 Proposition and validation of the mechanism of the cycloaddition reaction catalyzed by C5·I
To validate the role of the catalyst in both its cationic and anionic species in the reaction cycle, analog catalysts C5·BF4 and C8 were synthesized to perform the control experiments. The key step in the cycloaddition reaction was the opening of the epoxide ring by the nucleophilic reagent. Switching the counterion of C5 to non-nucleophilic tetrafluoroborate anion BF4− (C5·BF4) and performing the benchmark reaction under the same conditions. Only a negligible amount of product was generated as per NMR analysis (Table 2, entry 9); the probable explanation for this is that the ion exchange process was unable to reach 100%, and the residual iodide ions led to the formation of the product. Therefore, sodium iodide (Table 1, entry 14) was applied to attempt the benchmark reaction with only a 5% conversion of 1a. The results of the control experiments demonstrated that the halide anion of the strained ion pair played a critical role as the nucleophilic reagent in the catalytic cycle. The catalyst C8 (N,N-dimethyl-dihydroxyethylammonium iodide), which could provide a pair of O–H hydrogen bond donors and an iodide anion, has also been used for the benchmark reaction. Only 19% of 2a was generated via 1H NMR analysis (Table 1, entry 12), which indicated that the H-bond donor aminocyclopropenium was indispensable in catalyzing this reaction.NMR titration experiments were carried out to further support the importance of hydrogen bond donors in activating the substrate (Fig. 1). The phenyl glycidyl ether 1a and catalyst C5·I were mixed in ratios of 1:1, 1:0.8, 1:0.6, 1:0.4, 1:0.2, and 1:0, and CDCl3 and DMSO-d6 were used as solvents for 1H NMR analysis (for the full NMR spectra see ESI†). As the ratio of the substrate to the catalyst increased, the chemical shift of the C–H proton on the methylene carbon of the 1a was observed to downshift from 4.2220 ppm to 4.2363 ppm (Fig. 1a, left, red). Moreover, the chemical shift of α-C–H on C5·I distinctly moved from 3.8529 ppm to 3.8993 ppm (Fig. 1a, right, light blue). Hydrogen protons of the hydroxyl group could not be monitored in CDCl3 due to proton exchange. To verify the activation of O–H HBD, NMR titration experiments were performed in DMSO-d6 at the same ratio (Fig. 1b, deep blue). With a gradually decreasing ratio of 1a and C5·I, the chemical shift of the O–H proton was upshifted from 4.9734 to 4.9688 ppm. These experimental results suggested a strong coordination between the epoxide and the O–H/α-C–H hybrid HBD of the catalyst. Combined with the fact that catalyst C8 had almost no catalytic effect, we suggested that the hybrid H-bond donor system on the cationic part of C5·I played a vital role in activating the substrate.
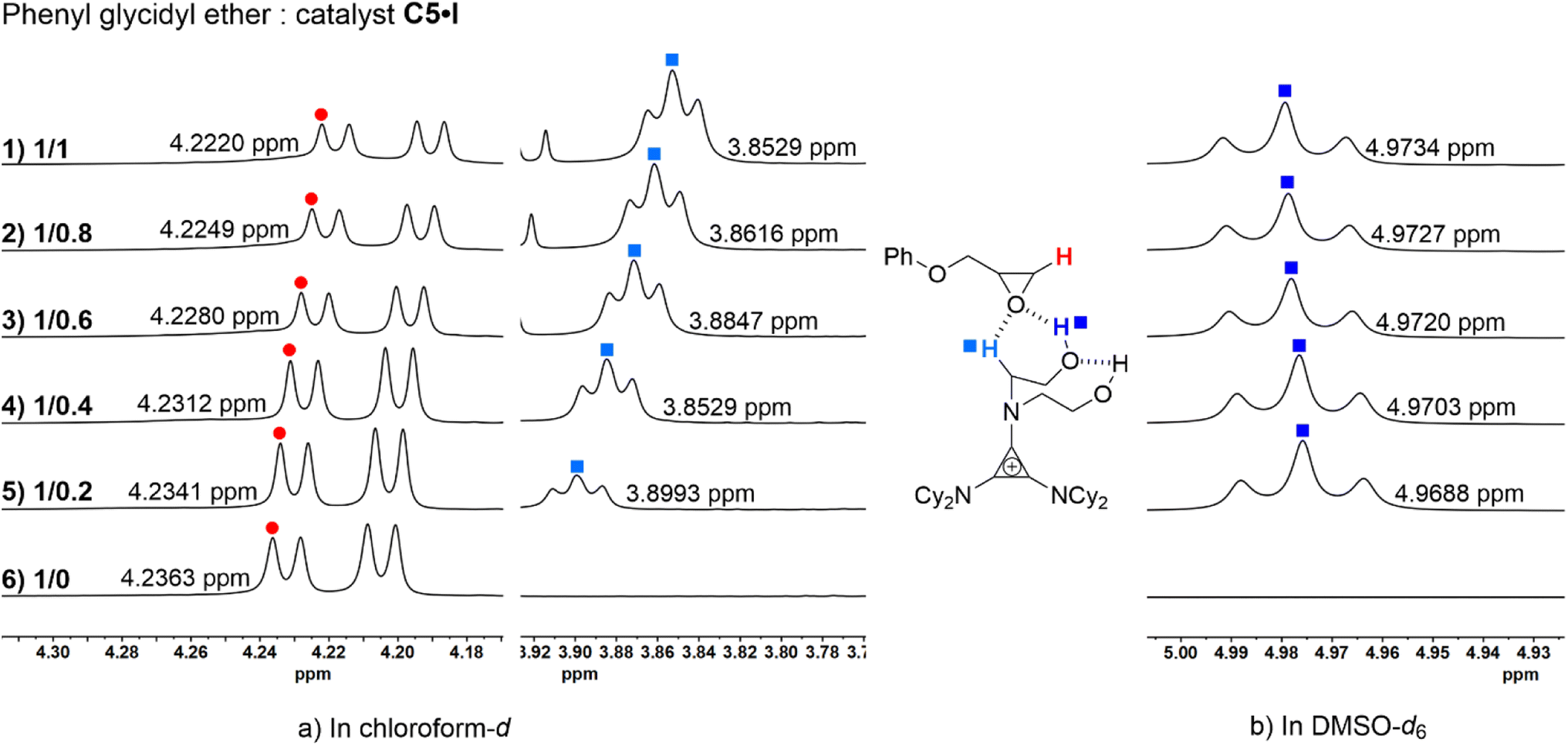 | ||
| Fig. 1 Phenyl glycidyl ether 1a and catalyst C5·I were mixed in the ratio of 1:1, 1:0.8, 1:0.6, 1:0.4, 1:0.2, and 1:0; (a) the chemical shift of the methine proton of 2a (left, red) and α-C–H of C5·I (right, light blue) in the 1H NMR spectra (in CDCl3); and (b) the chemical shift of the hydrogen proton of the O–H (deep blue) of C5·I (DMSO-d6). | ||
Combining previous works5,36 and the results of mechanistic experiments, a plausible H-bond donor/halide anion catalytic cycle was probed (Scheme 3). At the initial stage of the reaction, the intramolecular H-bonding (O–H⋯O–H) between the two hydroxyls enhanced the acidity of the terminal O–H hydrogen proton. The enhanced hydroxyl O–H and alkyl α-C–H formed a hybrid hydrogen bond donor (HBD) system. The iodine anion was in dynamic equilibrium with the cation at a larger distance due to ion pair strain. Upon addition of the epoxide, the HBD coordinated with the oxygen atom of the epoxide to activate it (Scheme 3, step 1); the iodine anion as a nucleophile attacked the methylene carbon of the epoxide for the ring-opening to afford the alkoxide anion intermediate (Scheme 3, step 2); the facile attack of the resulting alkoxide anion attacks at the electrophilic carbon center of the CS2 formed the intermediate alkoxide anion, leading to the incorporation of CS2. The generated dithiocarbonate anion intermediate was subsequently stabilized by the HBD cyclopropenium (Scheme 3, step 3); intramolecular cyclization occurred along with the leaving of iodide to give the target product cyclic dithiocarbonate (Scheme 3, step 4). In this step, the oxygen/sulfur scrambling was reduced by the stabilization of H-bonding, which inhibited the formation of thiirane and other by-products, and improved the selectivity of the reaction.
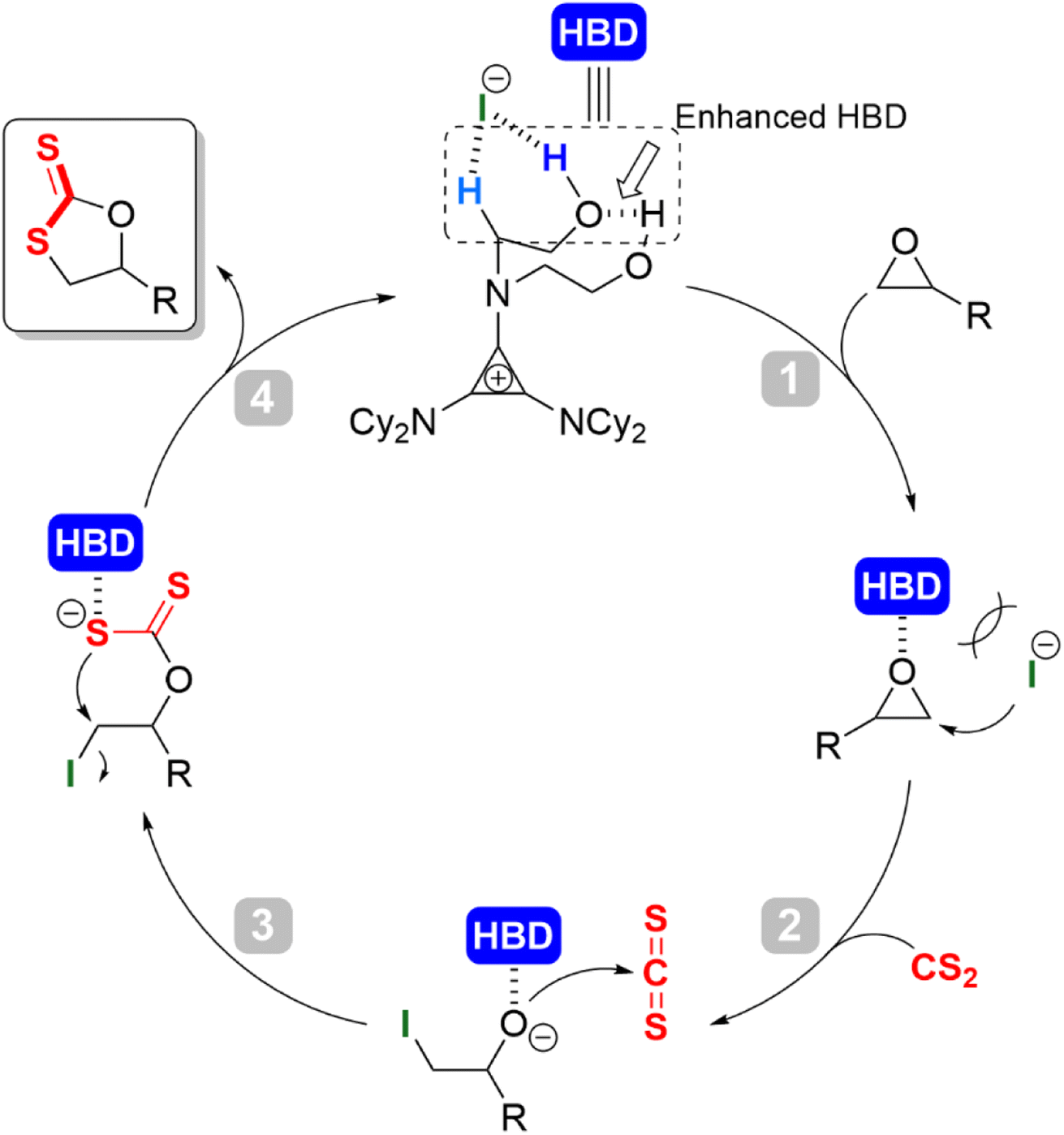 | ||
| Scheme 3 Plausible mechanism for synthesis of cyclic dithiocarbonate. | ||
4 Conclusions
Aminocyclopropenium was first utilized as an H-bonding organocatalyst to facilitate the cycloaddition of epoxide and carbon disulfide under mild conditions for the efficient preparation of cyclic dithiocarbonate. The side chain of cyclopropenium was substituted by the functional amino groups with an H-bond donor (HBD), which could promote activating the substrate and stabilizing the intermediate. The hybrid HBD/nucleophile catalyst C5·I was selected as the optimal catalyst for the cycloaddition reaction. All terminal epoxides were converted to the corresponding cyclic dithiocarbonates at 5 mol% catalyst loading, 25 °C, and solvent-free in 6 h with excellent conversion and selectivity. Based on the NMR titrations and control experiments, a plausible mechanism was proposed. The aromatic cation electronically enhanced the acidity of alkyl α-C–H. Two hydroxy groups of diethanolamino substituted on a cation formed O–H⋯O–H type intramolecular H-bonding. This enhanced alkyl α-C–H and hydroxy O–H constituted a hybrid HBD system that served to activate epoxide substrates and stabilize anion intermediates, suppressing the O/S scrambling and the generation of by-products. The reactivity of the halide anion was increased dramatically due to the repulsion between this type of electron-excess system. The aminocyclopropenium halide cooperative catalysis for the potential application will be further explored.Author contributions
Xinru Du: methodology, investigation. Ziqi Liu: methodology, investigation. Zhenjiang Li: conceptualization, funding acquisition, project administration, resources, supervision, writing – review and editing. Xin Yuan: methodology, investigation. Chunyu Li: methodology, investigation. Min Zhang: methodology, investigation. Zhihao Zhang: investigation, methodology, writing – original draft, writing – review and editing. Xin Hu: funding acquisition, resources, supervision. Kai Guo: conceptualization, funding acquisition, project administration, resources, supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078150), the Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), the project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Jiangsu Synergetic Innovation Center for Advanced Bio-Manufacture (XTB2201), and the Top-Notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP).References
- J. Gorzynski Smith, Synthetically Useful Reactions of Epoxides, Synthesis, 1984, 1984(08), 629–656 CrossRef.
- C. Beattie and M. North, Mechanistic investigation of the reaction of epoxides with heterocumulenes catalysed by a bimetallic aluminium salen complex, Chem.–Eur. J., 2014, 20(26), 8182–8188 CrossRef CAS PubMed.
- K. Chen, R. Yan, Z. Li, W. Huang, L. Gao, T. Duan, H. Tong, Y. Li, J. Sun and K. Guo, Halogen bonding catalysis for the [3+2] cycloaddition reactions of epoxides with CO2, and other heterocumulenes, J. CO2 Util., 2021, 52, 101663 CrossRef CAS.
- Y. Toda, K. Hashimoto, Y. Mori and H. Suga, A Phosphonium Ylide as a Ligand for [3 + 2] Coupling Reactions of Epoxides with Heterocumulenes under Mild Conditions, J. Org. Chem., 2020, 85(16), 10980–10987 CrossRef CAS PubMed.
- C. Beattie and M. North, Titanium(salen)-Catalysed Synthesis of Di- and Trithiocarbonates from Epoxides and Carbon Disulfide, ChemCatChem, 2014, 6, 1252–1259 CrossRef CAS.
- W. D. A. Bezerra, J. L. S. Milani and C. H. D. J. Franco, et al., Bis-benzimidazolium salts as bifunctional organocatalysts for the cycloaddition of CO2 with epoxides, Mol. Catal., 2022, 530, 112632 CrossRef CAS.
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, Recent progress of catalysts for synthesis of cyclic carbonates from CO2 and epoxides, J. CO2 Util., 2023, 68, 102355 CrossRef CAS.
- R. Calmanti, M. Selva and A. Perosa, Tungstate ionic liquids as catalysts for CO2 fixation into epoxides[J], Mol. Catal., 2020, 486, 110854 CrossRef CAS.
- L. Guo, K. J. Lamb and M. North, Recent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates, Green Chem., 2021, 23(1), 77–118 RSC.
- A. Centeno-Pedrazo, J. Perez-Arce and S. Prieto-Fernandez, et al., Phosphonium-based ionic liquids: Economic and efficient catalysts for the solvent-free cycloaddition of CO2 to epoxidized soybean vegetable oil to obtain potential bio-based polymers precursors, J. Mol. Catal., 2021, 515, 111889 CrossRef CAS.
- A. Centeno-Pedrazo, J. Perez-Arce, Z. Freixa, P. Ortiz and E. J. Garcia-Suarez, Catalytic Systems for the Effective Fixation of CO2 into Epoxidized Vegetable Oils and Derivates to Obtain Biobased Cyclic Carbonates as Precursors for Greener Polymers, Ind. Eng. Chem. Res., 2023, 62(8), 3428–3443 CrossRef CAS.
- F. Norouzi and A. Abdolmaleki, Acidic pyridinium ionic liquid: an efficient bifunctional organocatalyst to synthesis carbonate from atmospheric CO2 and epoxide, J. Mol. Catal., 2023, 538, 112988 CrossRef CAS.
- C. Díez-Poza, L. Álvarez-Miguel, M. E. G. Mosquera and C. J. Whiteoak, Synthesis and applications of the sulfur containing analogues of cyclic carbonates, Org. Biomol. Chem., 2023, 21(18), 3733–3755 RSC.
- W. Choi, F. Sanda and T. Endo, A Novel Construction of Living Polymerization by Neighboring Group Participation: Living Cationic Ring-Opening Polymerization of a Five-Membered Cyclic Dithiocarbonate, Macromolecules, 1998, 31(25), 9093–9095 CrossRef CAS.
- S. Krishnamurthy, Y. Yoshida and T. Endo, Cationic ring-opening polymerization of a five membered cyclic dithiocarbonate having a tertiary amine moiety, Polym. Chem., 2022, 13(2), 267–274 RSC.
- C. G. Kim, M. J. Son and J. Y. Do, Cationic living polymerization of cyclic dithiocarbonates involving sulfide-migration, Eur. Polym. J., 2021, 156, 110611 CrossRef CAS.
- G. Coste, C. Negrell and S. Caillol, Cascade (Dithio)carbonate Ring Opening Reactions for Self-Blowing Polyhydroxythiourethane Foams, Macromol. Rapid Commun., 2022, 43(13), 2100833 CrossRef CAS PubMed.
- T. Moriguchi and T. Endo, Polyaddition of Bifunctional Dithiocarbonates Derived from Epoxides and Carbon Disulfide. Synthesis of Novel Poly(thiourethanes), Macromolecules, 1995, 28(15), 5386–5387 CrossRef CAS.
- R. H. Springer, M. B. Scholten, D. E. O'Brien, T. Novinson, J. P. Miller and R. K. Robins, Synthesis and enzymic activity of 6-carbethoxy- and 6-ethoxy-3,7-disubstituted pyrazolo[1,5-a]pyrimidines and related derivatives as adenosine cyclic 3',5'-phosphate phosphodiesterase inhibitors, J. Med. Chem., 1982, 25(3), 235–242 CrossRef CAS PubMed.
- J. Diebler, A. Spannenberg and T. Werner, Regio- and Stereoselective Synthesis of Dithiocarbonates under Ambient and Solvent-Free Conditions, ChemCatChem, 2016, 8(12), 2027–2030 CrossRef CAS.
- F. Warnecke, Die gewerbliche Schwefelkohlenstoffvergiftung, Arch. Gewerbepathol. Gewerbehyg., 1941, 11(2), 198–248 CAS.
- A. M. Creighton and L. N. Owen, 211. Some carbohydrate episulphides, J. Chem. Soc., 1960, 1024–1029 RSC.
- J. A. Durden, H. A. Stansbury and W. H. Catlette, The Preparation of Alkene Trithiocarbonates, J. Am. Chem. Soc., 1960, 82(12), 3082–3084 CrossRef CAS.
- N. Kihara, Y. Nakawaki and T. Endo, Preparation of 1,3-Oxathiolane-2-thiones by the Reaction of Oxirane and Carbon Disulfide, J. Org. Chem., 1995, 60(2), 473–475 CrossRef CAS.
- I. Yavari, M. Ghazanfarpour-Darjani, Z. Hossaini, M. Sabbaghan and N. Hosseini, Methoxide Ion Promoted Efficient Synthesis of 1,3-Oxathiolane-2-thiones by Reaction of Oxiranes and Carbon Disulfide, Synlett, 2008, 2008(6), 889–891 CrossRef.
- A. Z. Halimehjani, F. Ebrahimi, N. Azizi and M. R. Saidi, A simple and novel eco-friendly process for the synthesis of cyclic dithiocarbonates from epoxides and carbon disulfide in water, J. Heterocycl. Chem., 2009, 46(2), 347–350 CrossRef CAS.
- J. Cao, M. Yu, H. Li, L. Wang, X. Zhu, G. Wang, Y. Shi and C. Cao, Synthesis of cyclic di- and trithiocarbonates from epoxides and carbon disulfide catalyzed by N-heterocyclic carbene, Res. Chem. Intermed., 2014, 41(8), 5323–5330 CrossRef.
- N. Aoyagi and T. Endo, Six-Membered Cyclic Amidines as Efficient Catalysts for the Synthesis of Cyclic Dithiocarbonates from Carbon Disulfide and Epoxides under Mild Conditions, Synlett, 2019, 31(01), 92–96 Search PubMed.
- J. Diebler, A. Spannenberg and T. Werner, Atom economical synthesis of di- and trithiocarbonates by the lithium tert-butoxide catalyzed addition of carbon disulfide to epoxides and thiiranes, Org. Biomol. Chem., 2016, 14(31), 7480–7489 RSC.
- Y. Taguchi, M. Yasumoto, I. Shibuya and Y. Suhara, The Synthesis of 1,3-Dithiolan-2-ones On the Reaction of Oxiranes with Carbon Disulfide under High Pressure, Bull. Chem. Soc. Jpn., 1989, 62(2), 474–478 CrossRef CAS.
- Y. Taguchi, K. Yanagiya, I. Shibuya and Y. Suhara, The Reaction of Oxiranes with Carbon Disulfide under High Pressure, Bull. Chem. Soc. Jpn., 1988, 61(3), 921–925 CrossRef CAS.
- C. Mei, X. Li, L. Liu, C. Cao, G. Pang and Y. Shi, Selectively synthesis of cyclic di- and trithiocarbonates by N-heterocyclic carbene/LiCl(Br) catalyzed addition of carbon disulfide to epoxides, Tetrahedron, 2017, 73(38), 5706–5714 CrossRef CAS.
- M. Okada, R. Nishiyori, S. Kaneko, K. Igawa and S. Shirakawa, KI-Tetraethylene Glycol Complex as an Effective Catalyst for the Synthesis of Cyclic Thiocarbonates from Epoxides and CS2, Eur. J. Org Chem., 2018, 2018(17), 2022–2027 CrossRef CAS.
- S. Roy, K. Das and S. Halder, Development of Suitable Hydrogen Bond Donor (HBD) Catalysts for the Synthesis of Cyclic Carbonates and Dithiocarbonates from Epoxide, Catal. Lett., 2023 DOI:10.1007/s10562-023-04422-y.
- Y. M. Wang, B. Li, H. Wang, Z. C. Zhang and X. B. Lu, Cooperative catalysis with binary Lewis acid–Lewis base system for the coupling of carbon disulfide and epoxides, Appl. Organomet. Chem., 2012, 26(11), 614–618 CrossRef CAS.
- M. North and P. Villuendas, Aluminium(salen) and Tetrabutylammonium Bromide Catalysed Synthesis of Cyclic Di- and Trithiocarbonates from Epoxides and Carbon Disulfide, Synlett, 2009, 2010(04), 623–627 CrossRef.
- W. Clegg, R. W. Harrington, M. North and P. Villuendas, A bimetallic aluminum(salen) complex for the synthesis of 1,3-oxathiolane-2-thiones and 1,3-dithiolane-2-thiones, J. Org. Chem., 2010, 75(18), 6201–6207 CrossRef CAS PubMed.
- C.-J. Zhang and X.-H. Zhang, Chemoselective Coupling of CS2 and Epoxides for Producing Poly(thioether)s and COS via Oxygen/Sulfur Atom Exchange, Macromolecules, 2019, 53(1), 233–239 CrossRef.
- S. Tanaka, T. Nakashima, T. Maeda, M. Ratanasak, J.-y. Hasegawa, Y. Kon, M. Tamura and K. Sato, Quaternary Alkyl Ammonium Salt-Catalyzed Transformation of Glycidol to Glycidyl Esters by Transesterification of Methyl Esters, ACS Catal., 2018, 8(2), 1097–1103 CrossRef CAS.
- N. Aoyagi, Y. Furusho and T. Endo, Cyclic amidine hydroiodide for the synthesis of cyclic carbonates and cyclic dithiocarbonates from carbon dioxide or carbon disulfide under mild conditions, Tetrahedron, 2019, 75(52), 130781 CrossRef.
- S. Shirakawa, Bifunctional Onium and Potassium Iodides as Nucleophilic Catalysts for the Solvent-Free Syntheses of Carbonates, Thiocarbonates, and Oxazolidinones from Epoxides, Chem. Rec., 2023, 23(10), e202300144 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Definition of the hydrogen bond (IUPAC Recommendations 2011), Pure Appl. Chem., 2011, 83(8), 1637–1641 CrossRef CAS.
- A. G. Doyle and E. N. Jacobsen, Small-Molecule H-Bond Donors in Asymmetric Catalysis, Chem. Rev., 2007, 107(12), 5713–5743 CrossRef CAS PubMed.
- R. Breslow, Synthesis Of The S-Triphenylcyclopropenyl Cation, J. Am. Chem. Soc., 1957, 79(19), 5318 CrossRef CAS.
- Z. Yoshida and Y. Tawara, Aminocyclopropenium ion, J. Am. Chem. Soc., 1971, 93(10), 2573–2574 CrossRef CAS.
- R. Weiss, T. Brenner, F. Hampel and A. Wolski, The Consequences of an Electrostatic“Forced Marriage” between Two Electron-Rich Particles: Strained Ion Pairs, Angew. Chem., Int. Ed., 1995, 34(4), 439–441 CrossRef CAS.
- R. Weiss, M. Rechinger, F. Hampel and A. Wolski, Stable 1 : 1 Adducts from Iodoacetylenes and Iodide Ions: Ion Pair Strain as an Additional Driving Force?, Angew. Chem., Int. Ed., 1995, 34(4), 441–443 CrossRef CAS.
- J. S. Bandar and T. H. Lambert, Aminocyclopropenium Ions: Synthesis, Properties, and Applications, Synthesis, 2013, 45(18), 2485–2498 CrossRef CAS.
- D. J. Lyons, R. D. Crocker, M. Blumel and T. V. Nguyen, Promotion of Organic Reactions by Non-Benzenoid Carbocyclic Aromatic Ions, Angew. Chem., Int. Ed., 2017, 56(6), 1466–1484 CrossRef CAS PubMed.
- R. M. Wilson and T. H. Lambert, Cyclopropenium Ions in Catalysis, Acc. Chem. Res., 2022, 55(20), 3057–3069 CrossRef CAS PubMed.
- P. K. Ranga, F. Ahmad, G. Singh, A. Tyagi and R. Vijaya Anand, Recent advances in the organocatalytic applications of cyclopropene- and cyclopropenium-based small molecules, Org. Biomol. Chem., 2021, 19(44), 9541–9564 RSC.
- P. K. Ranga, F. Ahmad, P. Nager, P. S. Rana and R. Vijaya Anand, Bis(amino)cyclopropenium Ion as a Hydrogen-Bond Donor Catalyst for 1,6-Conjugate Addition Reactions, J. Org. Chem., 2021, 86(7), 4994–5010 CrossRef CAS PubMed.
- J. Xu, J. Liu, Z. Li, S. Xu, H. Wang, T. Guo, Y. Gao, L. Zhang, C. Zhang and K. Guo, Opposite-charge repulsive cation and anion pair cooperative organocatalysis in ring-opening polymerization, Polym. Chem., 2018, 9(16), 2183–2192 RSC.
- J. Xu, A. Xian, Z. Li, J. Liu, Z. Zhang, R. Yan, L. Gao, B. Liu, L. Zhao and K. Guo, A Strained Ion Pair Permits Carbon Dioxide Fixation at Atmospheric Pressure by C–H H-Bonding Organocatalysis, J. Org. Chem., 2021, 86(4), 3422–3432 CrossRef CAS PubMed.
- I. Smajlagic, R. Duran, M. Pilkington and T. Dudding, Cyclopropenium Enhanced Thiourea Catalysis, J. Org. Chem., 2018, 83(22), 13973–13980 CrossRef CAS PubMed.
- Z. Zhang, Y. Ni, Z. Li, J. He, X. Zou, X. Yuan, Z. Liu, S. Cao, C. Ma and K. Guo, Amino-cyclopropenium H-bonding organocatalysis in cycloaddition of glycidol and isocyanate to oxazolidinone, J. Mol. Catal., 2023, 548, 113425 CrossRef CAS.
- J. Bruckhuisen, G. Dhont, A. Roucou, A. Jabri, H. Bayoudh, T. T. Tran, M. Goubet, M. A. Martin-Drumel and A. Cuisset, Intramolecular H-Bond Dynamics of Catechol Investigated by THz High-Resolution Spectroscopy of Its Low-Frequency Modes, Molecules, 2021, 26(12), 3645 CrossRef CAS PubMed.
- R. Weiss and M. Hertel, A nitrogen analogue of deltic acid, J. Chem. Soc., 1980,(5), 223–224 CAS.
- K. D. Green, M. Fridman and S. Garneau-Tsodikova, hChAT: A Tool for the Chemoenzymatic Generation of Potential Acetyl/Butyrylcholinesterase Inhibitors, ChemBioChem, 2009, 10(13), 2191–2194 CrossRef CAS PubMed.
- T. Nakamura, B. Ochiai and T. Endo, Efficient Chemical Recycling System of Networked Polymer: De-Cross-Linking of Cross-Linked Polymer Obtained from Bis(five-membered cyclic dithiocarbonate), Macromolecules, 2005, 38(10), 4065–4066 CrossRef CAS.
- Y. Inoue, K. Matsumoto and T. Endo, Synthesis and properties of poly(thiourethane)s having soft oligoether segments, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(9), 1076–1081 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00937a |
| This journal is © The Royal Society of Chemistry 2024 |