DOI:
10.1039/D4RA00988F
(Paper)
RSC Adv., 2024,
14, 18373-18384
Regioselective and solvent-dependent photoisomerization induced internal conversion in red fluorescent protein chromophore analogues†
Received
7th February 2024
, Accepted 23rd May 2024
First published on 10th June 2024
Abstract
Photophysical properties of three red fluorescent protein (RFP) chromophore analogues are reported here. The three RFP chromophore analogues differ in the additional conjugation present in the RFP chromophore. The three chromophores do not exhibit any solvent effect in both absorption and fluorescence spectra. The photoirradiation experiments and recording of 1H NMR before and after irradiation on one of the three RFP model chromophores show isomerization of the (Z,E) diastereomer to the (E,E) diastereomer. Calculation of S0 and S1 potential energy curves shows the preference for isomerization through the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with Z-stereochemistry, thus corroborating the experimental results. The computational studies also suggest that torsional motion along the exocyclic CC bond pushes the molecules to a conical intersection, thus paving the pathway for radiationless deactivation.
C bond with Z-stereochemistry, thus corroborating the experimental results. The computational studies also suggest that torsional motion along the exocyclic CC bond pushes the molecules to a conical intersection, thus paving the pathway for radiationless deactivation.
1. Introduction
Fluorescent proteins (FPs) find wide application in cell biology in monitoring live cell processes. In this context, the discovery of the green fluorescent protein (GFP) and its mutant derivatives was a major breakthrough in cell biology for live cell imaging.1 Among all the fluorescent proteins, GFP is perhaps the most versatile protein that has attracted the attention of most cell biologists. When GFP is expressed in a live cell, it does not alter the function of the cell; it is photostable and resistant to alkaline pH, salts, detergents and many proteases.2 Moreover, it does not need a cofactor to fluoresce. These unique properties make GFP useful for a wide range of applications in cell biology. Although the discovery of the mutant derivatives of GFP such as the blue fluorescent protein (BFP) and cyan fluorescent protein (CFP) by Roger Tsien3 widened the visibility inside live cells,4 shifting the imaging light to red was still necessary to reduce background noise and image deep tissues at high resolution.5 The red fluorescent protein cloned from Discosoma coral (DSRed)6 and Kaede7 cloned from stony coral hold great promise of acting as genetically encoded indicators for tracking gene expression deeper inside the cell.6 The red fluorescent protein, DSRed, absorbs at 558 nm and emits at 583 nm,8 whereas wild type GFP absorbs at 395 nm and emits at 508 nm.9 The absorption and emission of DSRed are highly red-shifted compared to those of GFP. The extinction coefficient and fluorescence quantum yield of DSRed at 558 nm excitation are reported to be 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 and 0.7 respectively.10 The red-shifted absorption and emission properties of RFP are due to the additional conjugation present in the benzylidene imidazolinone moiety.11 The red-shifted spectroscopic properties of RFP not only add an additional colour to the spectrum of colours emitted by GFP and its various mutants3 but RFP is also an excellent partner of GFP for fluorescence resonance energy transfer (FRET), which makes RFP an imaging tool in live cells.10 Although the extinction coefficient of DSRed is much higher than that of GFP (extinction coefficient of GFP at 395 nm is 30000 M−1 cm−1), its fluorescence quantum yield is less than that of GFP (quantum yield of GFP is 0.8). Furthermore, the synthetic analogues of the DSRed chromophore are much less fluorescent than those of the wild-type DSRed.12,13
000 M−1 cm−1 and 0.7 respectively.10 The red-shifted absorption and emission properties of RFP are due to the additional conjugation present in the benzylidene imidazolinone moiety.11 The red-shifted spectroscopic properties of RFP not only add an additional colour to the spectrum of colours emitted by GFP and its various mutants3 but RFP is also an excellent partner of GFP for fluorescence resonance energy transfer (FRET), which makes RFP an imaging tool in live cells.10 Although the extinction coefficient of DSRed is much higher than that of GFP (extinction coefficient of GFP at 395 nm is 30000 M−1 cm−1), its fluorescence quantum yield is less than that of GFP (quantum yield of GFP is 0.8). Furthermore, the synthetic analogues of the DSRed chromophore are much less fluorescent than those of the wild-type DSRed.12,13
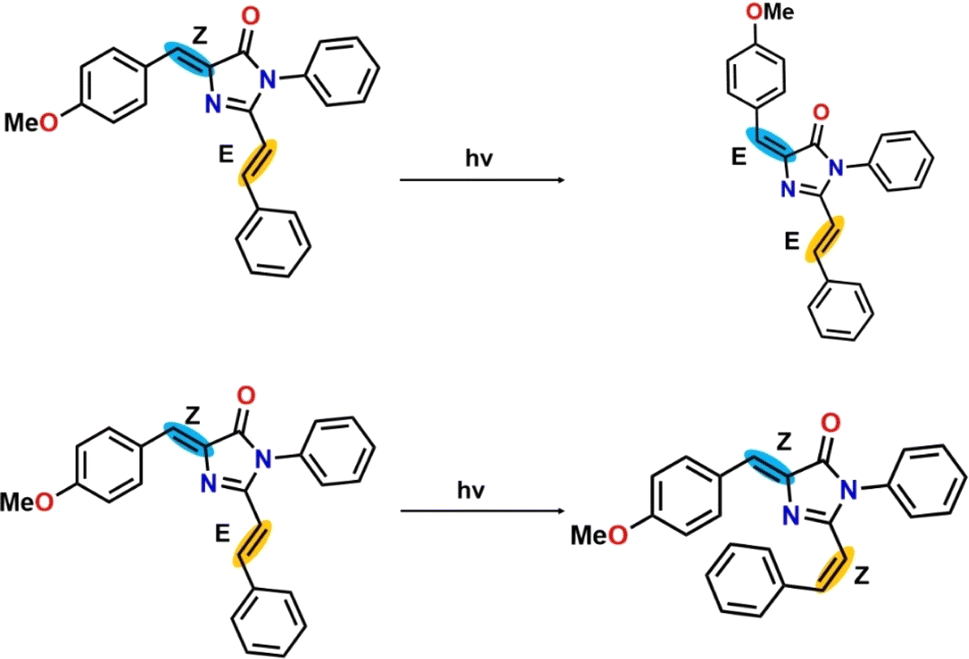
A detailed understanding of the factors that influence the fluorescence emission by fluorescent proteins is important to generate mutant fluorescent proteins for desired applications. Considering this, synthetic chromophore analogues of the fluorescent proteins' chromophores serve as the model system for understanding the fluorescence behaviour of the fluorescent proteins. Studies on the excited state dynamics of synthetic model chromophores of fluorescent proteins facilitate understanding the pathway for the loss of excited state energy of the model chromophores, which in turn facilitates the understanding of factors responsible for fluorescence emission by the fluorescent proteins.14,15 The model RFP chromophores have exocyclic CC and additional CC bonds, which are potential regions for photoisomerization that can trigger internal conversion. Olsen and Smith did computational modelling of the excited state dynamics of a model RFP chromophore and suggested excited state twisted intramolecular charge transfer (TICT) mediated E,Z-photoisomerization about the exocyclic CC bond (C4–C7) to be responsible for weak fluorescence.13 We report here the results of the experimental and computational studies on the photophysical properties of three RFP chromophore analogues (Scheme 1). Solution phase photoirradiation experiments on one of the three analogues revealed regioselective E,Z-isomerization along the exocyclic CC bond (Z-stereochemistry), which has been proposed to be the dominant deactivation channel of the excited state of GFP chromophore analogues16–19 and in RFP chromophore.13 Calculation of the potential energy curves of the ground (S0) and excited state (S1) of the three RFP chromophore analogues shows that the analogues, which exist in the Z,E-diastereomeric forms, prefer to undergo isomerization to form the E,E-diastereomeric forms, thus corroborating the experimental results. The results of these studies suggest that similar to the GFP chromophore analogues, the torsional motion along the exocyclic CC bond acts as the dominant radiationless deactivation channel in the excited state.
 |
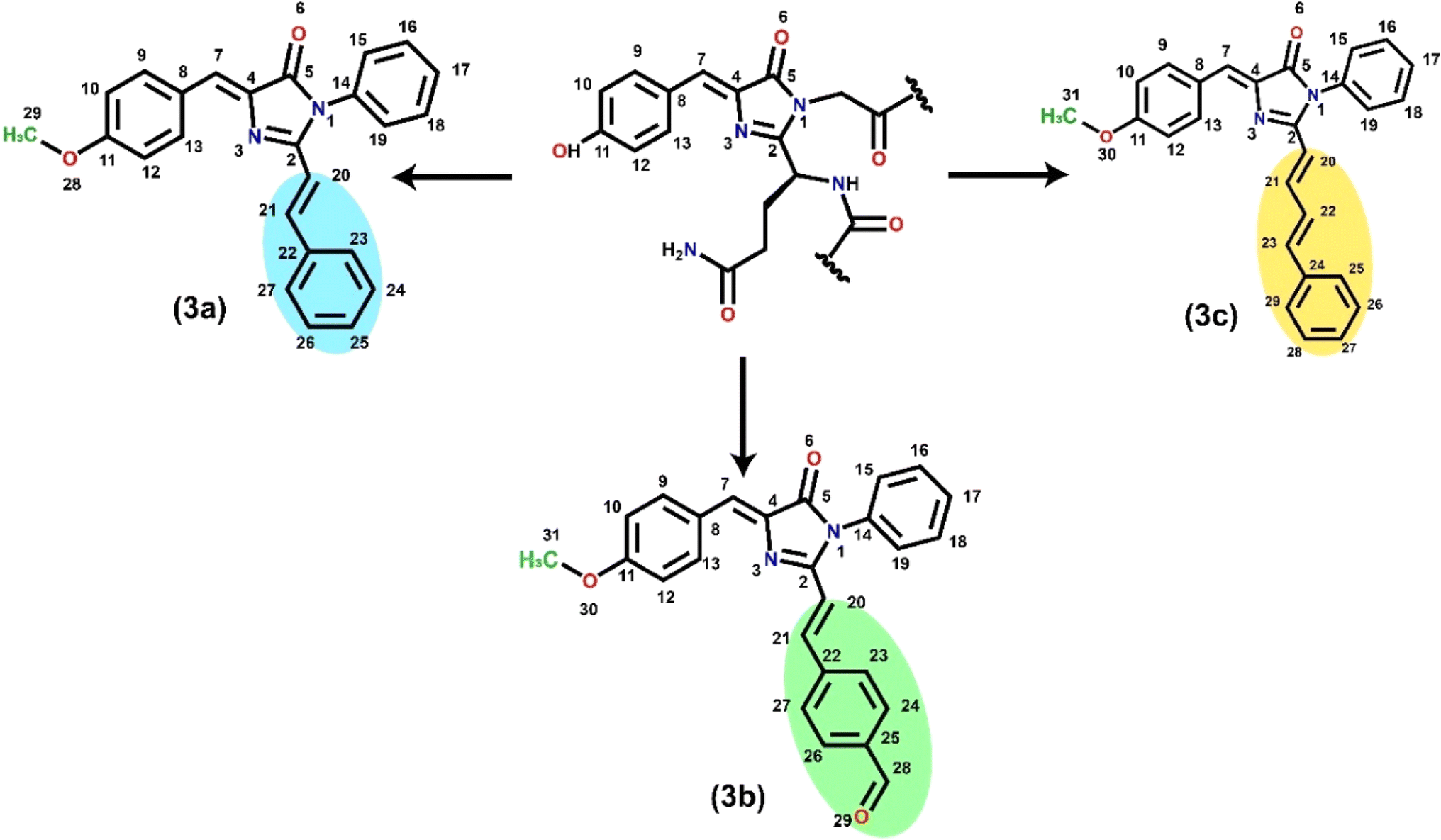
| | Scheme 1 RFP chromophore analogues 3a, 3b, and 3c, which differ in the part that extends the conjugation. The chromophore in the middle is the intrinsic emitter present in the red fluorescent protein. The atom numbering is used to define dihedral angles β (N3–C4–C7–C8) and γ (C2–C20–C21–C22). | |
2. Experimental
2.1 Synthesis and characterization of RFP chromophore analogues
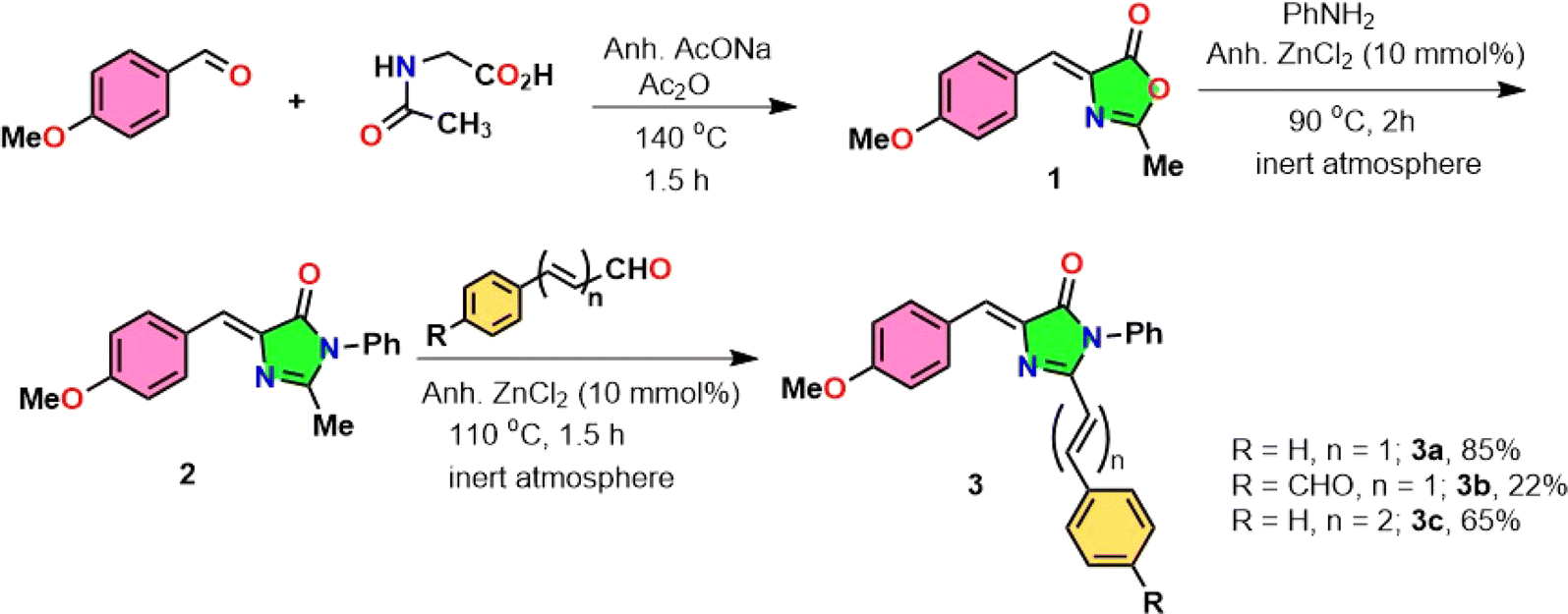
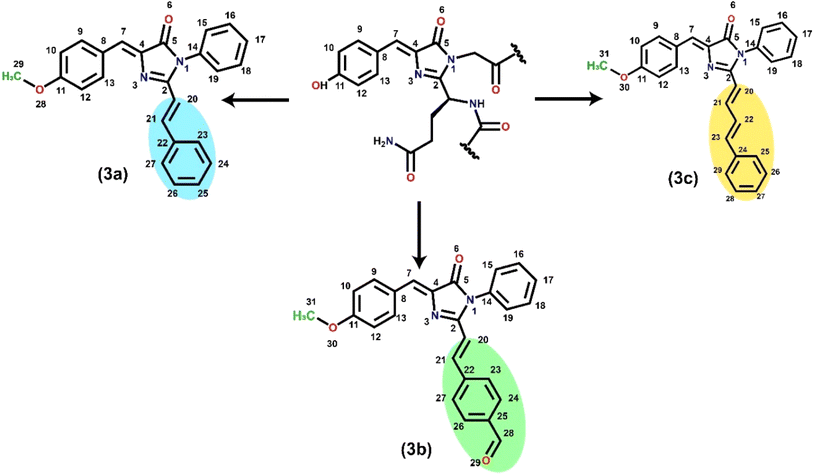
RFP chromophore analogues 3a, 3b and 3c were synthesized in three steps (Scheme 2).
 |
| | Scheme 2 Synthesis of the RFP chromophore analogues 3a–c. | |
The details of the synthesis and characterization of the compounds are given in the ESI (Fig. S1–S6).† Briefly, in the first step, an oxazolone 1 was synthesized by the Erlenmeyer azlactone synthesis method.20 In the second step the oxazolone was converted to imidazolinone 2 by a solvent-free Lewis acid catalyzed reaction.21,22 In the third step, the imidazolinone was further allowed to react with either benzaldehyde, trans-cinnamaldehyde or terephthalaldehyde in a solvent-free Lewis acid catalyzed condensation reaction23–25 to obtain the final compounds 3a, 3b and 3c respectively.
2.2 Measurement of steady-state absorption and fluorescence spectra
The steady state absorbance spectra were recorded on a Jasco V-550 UV-visible spectrophotometer.
The chromophores were dissolved in HPLC-grade solvents for measurement of their absorption and fluorescence spectra. The steady-state fluorescence spectra were recorded on a Fluorolog 3.21 spectrofluorimeter (Horiba Jobin Yvon). The samples were dissolved in cyclohexane, ethyl acetate, acetonitrile and methanol (10 μM) and the solutions were excited at their absorption maxima for comparison of their steady-state fluorescence spectra. Fluorescence quantum yields of the three chromophores were measured in the four solvents taking fluorescein as the reference fluorophore.26 The optical density of the solutions of the chromophores and fluorescein dissolved in 0.1 N NaOH were maintained at 0.1 at 436 nm for 3a and at 450 nm for 3b and 3c and the solutions were excited keeping the bandpass 1, 1 nm. The RFP chromophore analogues 3a, 3b and 3c have additional CC bonds in addition to the exocyclic CC bond attached to the imidazolinone ring. In order to understand which CC bond provides a pathway for non-radiative decay, we irradiated 3a in acetonitrile and recorded absorption spectra at different time intervals. Details of the procedure of the photoirradiation experiment are given in the ESI.†
3. Computational details
The three RFP chromophore analogues were optimized in the ground state followed by the frequency calculation at the B3LYP/TZVP27–29 level of accuracy in the gas and solvent phases. To understand the excited state behaviour, the time-dependent density functional (TDDFT)30–32 method was adopted in conjugation with the long-range corrected Coulomb-attenuated functional (CAM-B3LYP)33 using the TZVP basis set. For solvent phase calculations, the integral equation formalism polarized continuum model (IEFPCM) l34–37 was implemented with solvents of varied dielectric constant (ε) ranging from acetonitrile (ε = 35.688), methanol (ε = 32.7), ethyl acetate (ε = 6.02) and cyclohexane (ε = 2.02).38 However, to understand more details of the optical properties, the absorption spectra of the molecules were calculated at the TDDFT/CAM-B3LYP/TZVP level of theory employing the IEFPCM model.37,39–42 The nature of the electronic excitations and the excited state intramolecular charge transfer were analyzed by implementing the Multifwn43 code through the integral of overlap of hole–electron distribution and transition density.44
In the fluorescence spectra, the calculated electronic vibrations and the force constants of the ground state were used further to compute the emission maxima. The ground state (S0) and excited state (S1) potential energy curves of the RFP chromophore analogue 3a were constructed with respect to torsional motion along the exo-cyclic CC bond and the additional CC bond, which extends conjugation in the chromophore. The corresponding dihedral angles are β (N3–C4–C7–C8) and γ (C2–C20–C21–C22) for 3a. For 3b and 3c, S0 and S1 potential energy curves were constructed with respect to the dihedral angle β (N3–C4–C7–C8). The potential energy curves were constructed at the CAM-B3LYP/TZVP level of accuracy in four different solvents viz. cyclohexane, ethyl acetate, acetonitrile and methanol. The most stable configuration is (Z,E) with the exocyclic CC bond having Z-stereochemistry and additional CC bond having E-stereochemistry and the corresponding dihedral angles are β = 0° and γ = 180°. The potential energy curves were constructed by varying the dihedral angles β from 0° to 180° and γ from 180° and 0° at an interval of 10°. In between 80° to 100°, the potential energy curves were constructed at an interval of 2° to observe the accurate conformation responsible for the occurrence of conical intersection.17,45–47 All the calculations were carried out using the Gaussian 16-suit program.48
4. Results and discussions
4.1 Steady-state absorption and fluorescence spectra
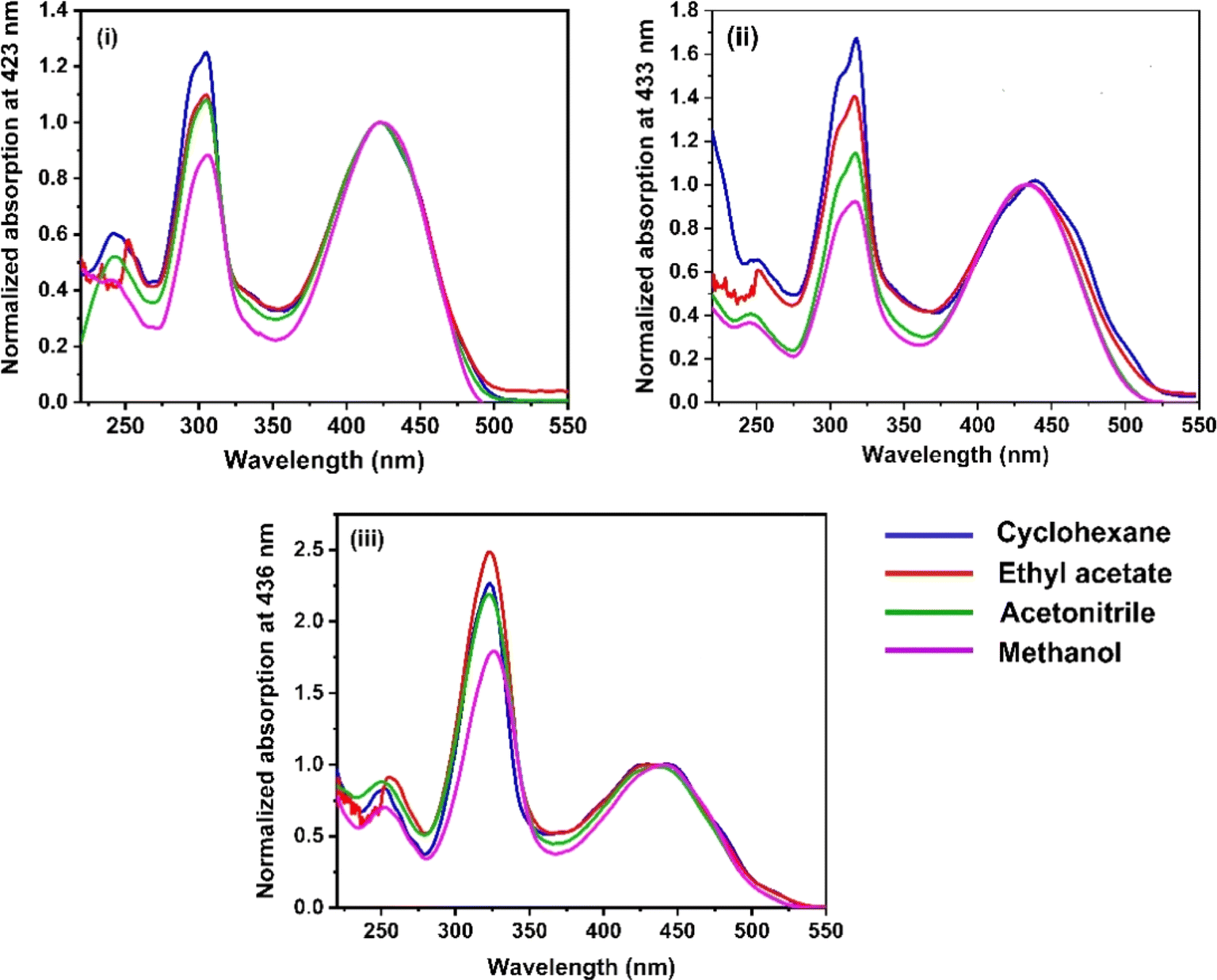
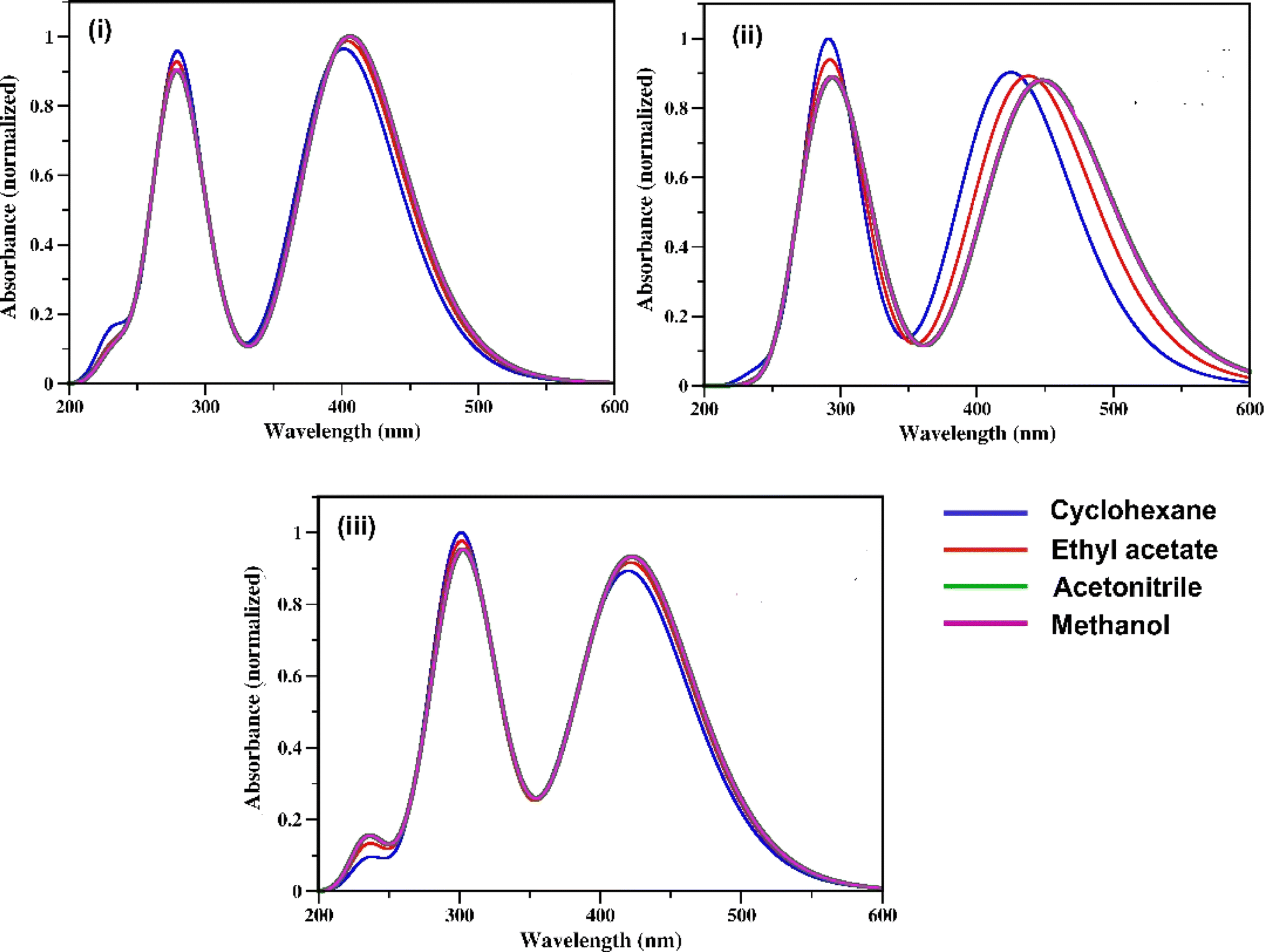
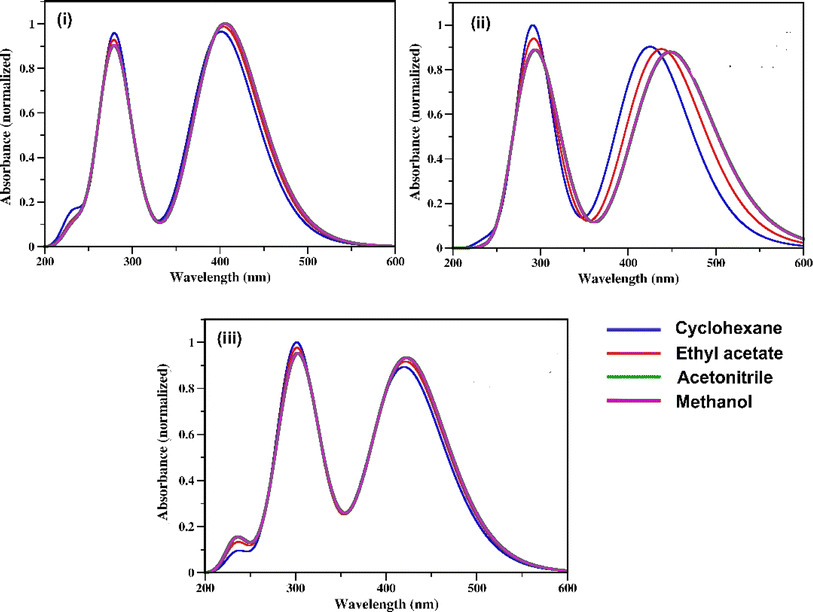
The UV-vis absorption spectra of the three RFP chromophore analogues 3a, 3b and 3c are shown in Fig. 1. The absorption spectra of all these three molecules are characterized by the presence of an absorption band (S2 ← S0) in the near ultra-violet region (305–325 nm) and another absorption band (S1 ← S0) in the visible region (420–440 nm). The absorption spectra of the three RFP chromophore analogues do not show any solvent effect suggesting the absence of strong solute–solvent effect in the ground state. This observation is opposite to that in N(CH3)2 substituted compounds, which exhibit strong solvatochromic red shifts in the absorption spectra with increasing solvent polarity.49 However, on moving from molecule 3a to 3c through 3b, a gradual increase in red shift of the absorption spectra is seen, which is due to increasing π-conjugation length from 3a to 3c through 3b.
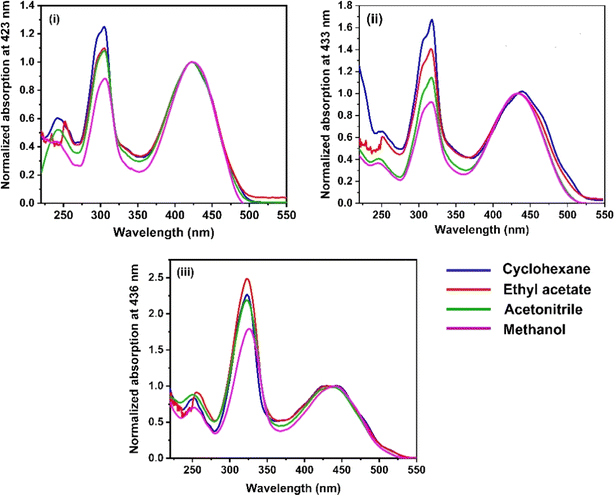 |
| | Fig. 1 UV-vis absorption spectra of RFP chromophores in cyclohexane, ethyl acetate, acetonitrile and methanol. (i) Molecule 3a, (ii) molecule 3b, and (iii) molecule 3c. | |
When 3a was irradiated in acetonitrile for 13 hours and its absorption spectra were recorded at different time intervals, one isosbestic point was observed at 287 nm in the absorption spectra (see Fig. S7, ESI†) suggesting the presence of two isomers in the equilibrium in the solution. With continuous irradiation absorption of the visible absorption band (420 nm) gradually decreases while absorption of the UV absorption band (305 nm) gradually increases. This observation suggests the conversion of the Z-isomer of 3a to the E-isomer and equilibration of the two diastereomers. Further isomerization of the Z-diastereomer to the E-diastereomer was proved by recording 1H-NMR of the photoirradiated sample of 3a. Recording of 1H-NMR spectra of 3a before and after photoirradiation showed that 3a isomerized about the exo-cyclic CC bond (Z-stereochemistry) and not through the other CC bond having E-stereochemistry (Fig. S8–S12, ESI†). Voliani et al. already observed such behavior for a green fluorescent protein (GFP) chromophore analogue.50
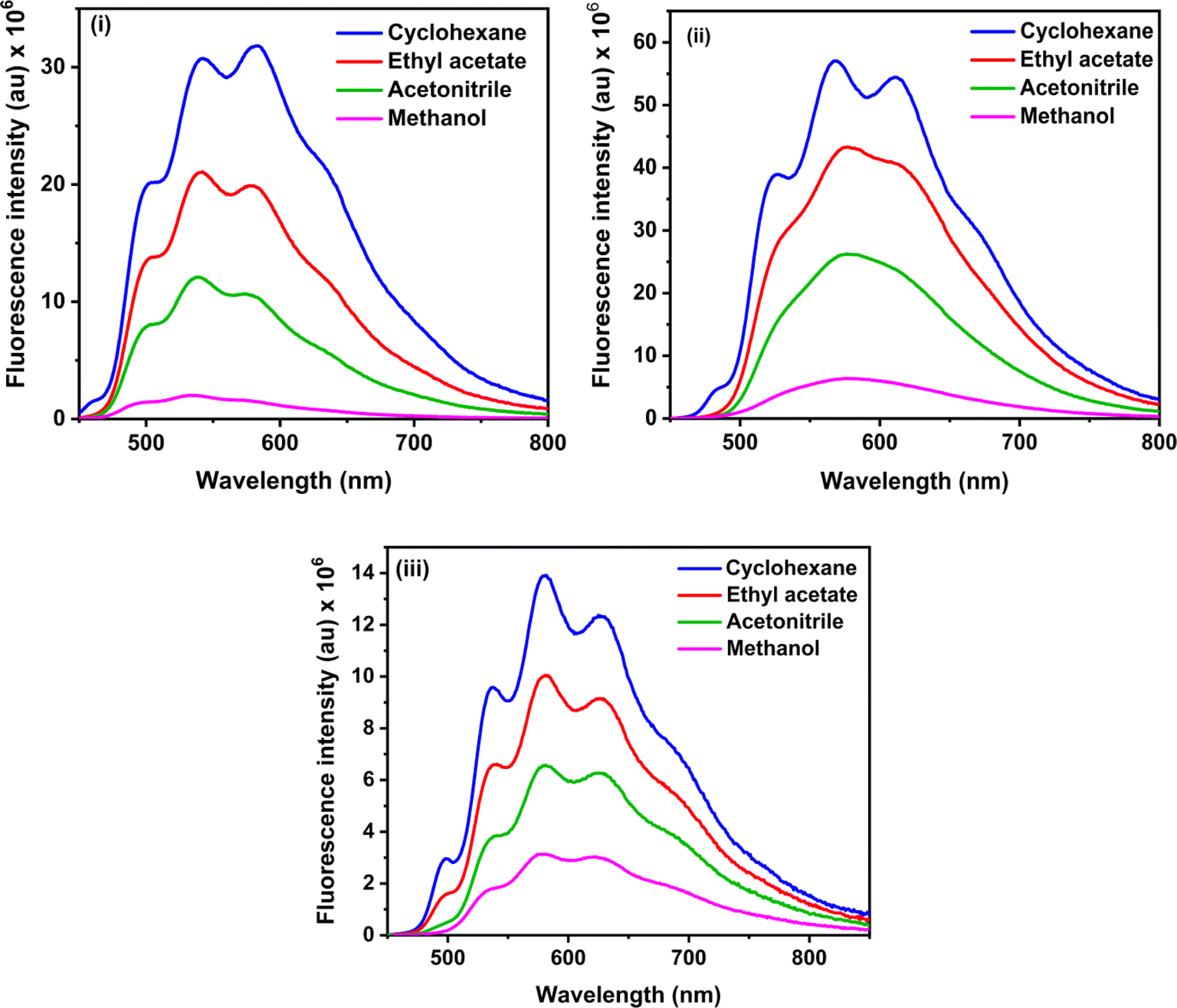
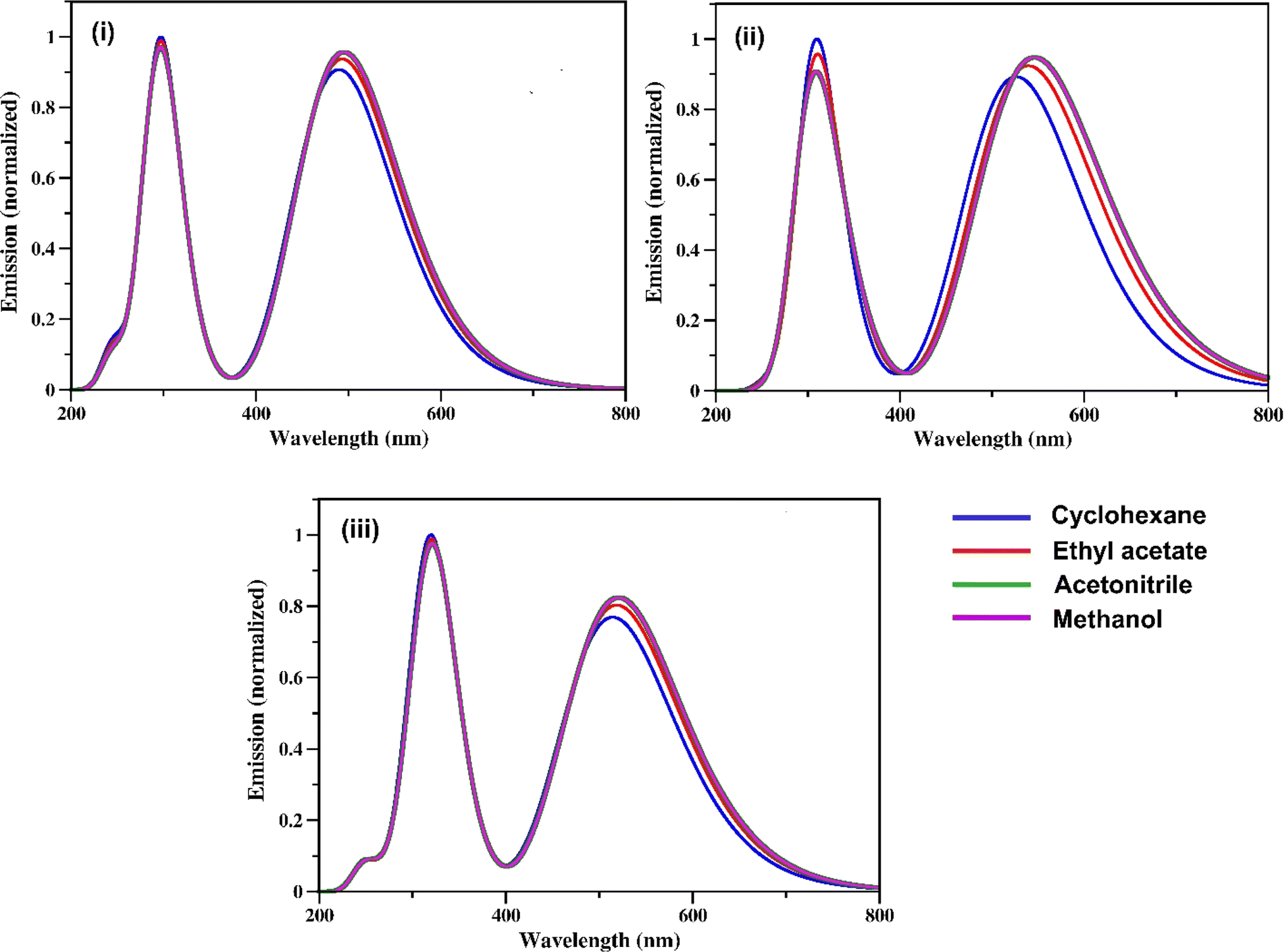
The steady-state fluorescence spectra of the three RFP chromophore analogues 3a, 3b and 3c are shown in Fig. 2. Molecules 3a and 3b show vibrational structure in weakly polar solvents and structureless emission bands in strongly polar solvents, while molecule 3c shows vibrational structure in all solvents, with prominently structured emission in cyclohexane. Among all the three RFP chromophore analogues, 3b is the most fluorescent. The emissions are very broad, no solvent effect is observed and red-shifted from 3a to 3b and 3b to 3c because of the increasing π-conjugation length. While these methoxy-substituted compounds do not exhibit solvent effects in the fluorescence spectra, tertiary amine-substituted compounds exhibit significant solvatochromic effects in fluorescence spectra.49 Excitation at the high energy absorption band gives the same emission spectrum with a higher intensity of emission (see Fig. S13, ESI†). With increasing solvent polarity from cyclohexane to acetonitrile, fluorescence intensity decreases. However, although methanol is less polar than acetonitrile, fluorescence intensity is the least in it.
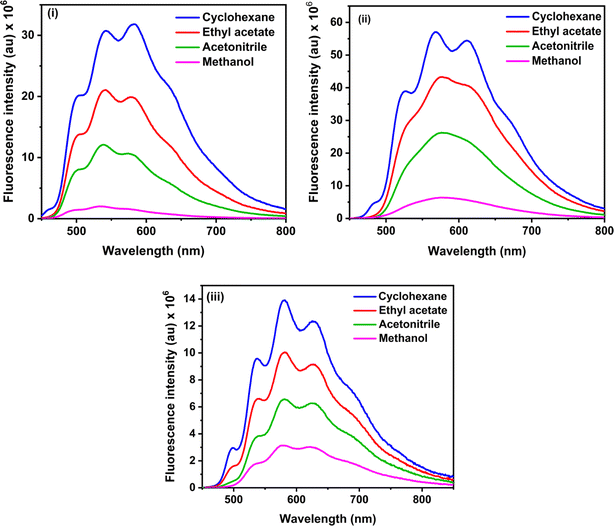 |
| | Fig. 2 Steady state fluorescence spectra of (i) 3a, (ii) 3b and (iii) 3c in cyclohexane, ethyl acetate, acetonitrile and methanol. Excitation wavelengths are 423 nm, 435 nm and 435 nm for 3a, 3b and 3c respectively. Concentration 1 × 10−5 M and bandpass 3, 3 nm are fixed for all fluorescence spectra. | |
Methanol is a hydrogen bond donor solvent and the stretching vibrational modes of the hydrogen bonds formed by methanol with the solute act as a sink for dissipation of the excited state energy of the solute molecules for which fluorescence intensity is least in methanol.16,46
Fluorescence quantum yields of 3a, 3b and 3c are shown in Table 1, and fluorescence quantum yield decreases with increasing solvent polarity and least in methanol due to the hydrogen bond of methanol with the solute molecules. The trend of the measured fluorescence quantum yields accords with the observed decrease of steady-state fluorescence intensity with increasing solvent polarity from cyclohexane to acetonitrile through ethyl acetate and methanol. In general, in a set of similarly rigid fluorophores, fluorescence quantum yield increases with increasing π-conjugation length.51 In the RFP chromophore analogues 3a, 3b and 3c, π-conjugation length increases as 3a < 3b < 3c. Although the π-conjugation length of 3c is the highest among the three RFP chromophore analogues, its fluorescence quantum yield is less than the other two which is due to enhanced flexibility and increased probability of isomerization with the incorporation of one more CC bond in it.
Table 1 Fluorescence quantum yields of RFP chromophore analogues (measurements were performed at temperature 20–25 °C. Φf can vary with maximum ± 0.0008)a
| RFP chromophore |
Solvents |
λex (nm) (reference) |
Φf |
| The λex (nm) of the reference (fluorescein) for 3a is 436 nm and for 3b and 3c it is 450 nm. |
| 3a |
Cyclohexane |
436 (fluorescein) |
0.181 |
| Ethyl acetate |
0.074 |
| Acetonitrile |
0.036 |
| Methanol |
0.006 |
| 3b |
Cyclohexane |
450 (fluorescein) |
0.422 |
| Ethyl acetate |
0.214 |
| Acetonitrile |
0.098 |
| Methanol |
0.026 |
| 3c |
Cyclohexane |
450 (fluorescein) |
0.114 |
| Ethyl acetate |
0.056 |
| Acetonitrile |
0.036 |
| Methanol |
0.025 |
4.2 Absorption spectral characteristics and frontier molecular orbitals analysis
The optimized geometries of the three RFP analogues 3a, 3b and 3c in both the ground state and excited state in acetonitrile solvent are shown in Fig. S14 (see ESI).† The calculated absorption spectra of the three compounds 3a–c in four different solvents acetonitrile (ε = 35.688), methanol (ε = 32.7), ethyl acetate (ε = 6.02) and cyclohexane (ε = 2.02) are shown in Fig. 3. All the detailed information on the vertical excitation energies, oscillator strength (f), and the contributions of the orbitals in the transitions are shown in Table 2. As demonstrated in both Fig. 3 and Table 2, two intense absorption bands are observed in all the four solvents. The positions of the absorption bands in all four solvents were well-matched with those from the experimental results. The calculated vertical excitation peaks for the compound 3a are observed at 305 nm and 441 nm (in acetonitrile), 305 nm and 441 nm (in methanol), 299 nm and 432 nm (in ethyl acetate), and 290 nm and 416 nm (in cyclohexane), which is consistent with the experimental peaks observed at 305 nm and 421 nm (in acetonitrile), 305 nm and 424 nm (in methanol), 305 nm and 422 nm (in ethyl acetate) and 303 nm and 423 nm (in cyclohexane). Similarly, for compounds 3b and 3c, the calculated vertical excitation energies corroborate those from the experimental results. Thus, the results indicate that the absorption bands are not sensitive to the polarity of the solvents.
 |
| | Fig. 3 Calculated electronic absorption spectra of the RFP chromophore analogue (i) 3a (ii) 3b and (iii) 3c in cyclohexane (blue, ε = 2.02), ethyl acetate (red, ε = 6.02) acetonitrile (green, ε = 35.688), and methanol (violet, ε = 32.7). | |
Table 2 Experimental maximum absorption energy Eabexp (nm) calculated vertical excitation energies Eabcalc (nm), oscillator strengths (f), and the % orbital contributions for these transitions (T) of compounds 3a, 3b and 3c in all the four solvents calculated using the TD-DFT/CAM-B3LYP/TZVP level. H = highest occupied molecular orbital and L = lowest unoccupied molecular orbital
| Analogues |
Solvent |
Eabexp (nm) |
Eabcalc (nm) |
f |
Elect. trans. |
% T |
| 3a |
Cyclohexane |
423, 303 |
415.9 |
1.04 |
S0 → S1 |
H → L (98.1%) |
| 289.1 |
0.91 |
S0 → S3 |
H → L + 1 (65.6%) |
| Ethyl acetate |
423, 303 |
431.8 |
1.14 |
S0 → S1 |
H → L (98.1%) |
| 298.7 |
0.91 |
S0 → S3 |
H → L + 1 (80.9%) |
| Acetonitrile |
421, 305 |
441.8 |
1.19 |
S0 → S1 |
H → L (98.1%) |
| 304.7 |
1.06 |
S0 → S3 |
H → L + 1 (81.1%) |
| Methanol |
424, 305 |
440.6 |
1.19 |
S0 → S1 |
H → L (98.1%) |
| 304.6 |
1.07 |
S0 → S3 |
H → L + 1 (65.6%) |
| 3b |
Cyclohexane |
434, 316 |
433.0 |
1.25 |
S0 → S1 |
H → L (98.1%) |
| 303.0 |
1.13 |
S0 → S3 |
H → L + 1 (91.4%) |
| Ethyl acetate |
433, 317 |
447.7 |
1.19 |
S0 → S1 |
H → L (98.1%) |
| 312.7 |
1.05 |
S0 → S3 |
H → L + 1 (99.8%) |
| Acetonitrile |
433, 317 |
455.5 |
1.04 |
S0 → S1 |
H → L (98.1%) |
| 318.9 |
1.12 |
S0 → S3 |
H → L + 1 (99.9%) |
| Methanol |
439, 317 |
455.3 |
1.25 |
S0 → S1 |
H → L (98.3%) |
| 318.8 |
1.13 |
S0 → S3 |
H → L + 1 (99.9%) |
| 3c |
Cyclohexane |
434, 323 |
437.3 |
1.19 |
S0 → S1 |
H → L (97.2%) |
| 315.4 |
0.74 |
S0 → S3 |
H → L + 1 (90.9%) |
| Ethyl acetate |
434, 324 |
455.2 |
1.28 |
S0 → S1 |
H → L (97.1%) |
| 328.9 |
1.38 |
S0 → S3 |
H → L + 1 (91.6%) |
| Acetonitrile |
436, 324 |
465.4 |
1.32 |
S0 → S1 |
H → L (96.9%) |
| 336.8 |
1.44 |
S0 → S3 |
H → L + 1 (92.1%) |
| Methanol |
440, 326 |
465.2 |
1.32 |
S0 → S1 |
H → L (96.9%) |
| 336.7 |
1.44 |
S0 → S3 |
H → L + 1 (92.1%) |
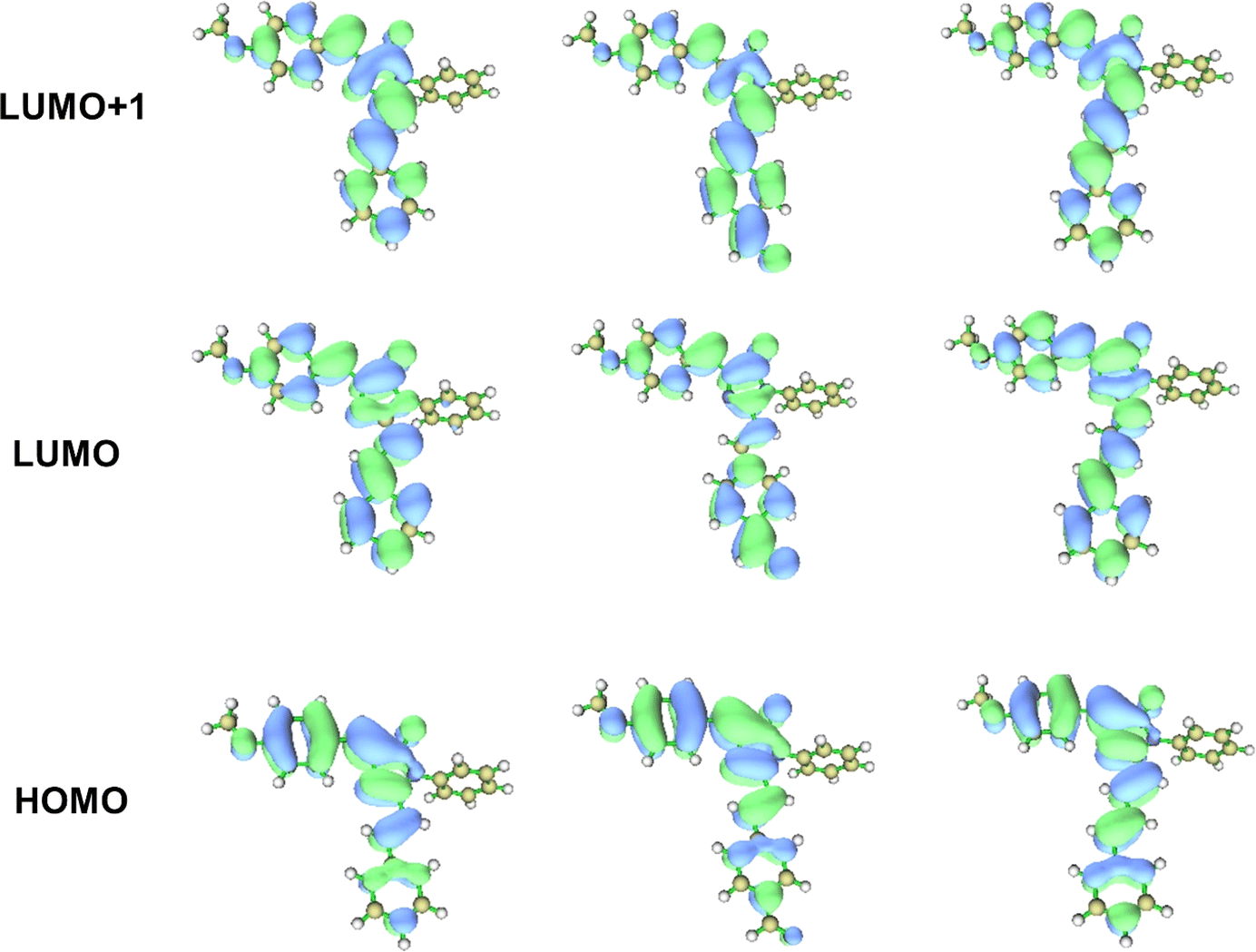
The absorption band appearing around 440 nm and 300 nm for all the analogues 3a, 3b and 3c were obtained due to the two π–π* transitions, and the corresponding components involved during the transitions are HOMO → LUMO and HOMO → LUMO + 1 electronic excitation (Table 2). As shown in the contour plots (Fig. 4) of the frontier MOs of the analogues 3a, 3b and 3c, the HOMO is localized mainly on the benzylidene moiety of the molecule due to the electron-donating nature of the methoxy group. In contrast, because of the increase in the conjugation in the chromophore, the electron density in LUMO and LUMO + 1 is localized on the imidazolinone and the benzene ring through the CC bond(s) present in the extended conjugated part. Thus, these electronic transitions consequently imply that the charge is being transferred from the electron-donating methoxy-substituted benzylidene moiety to the benzene ring present in the extended conjugated part through the imidazolinone moiety and the CC bond(s) of the chromophores.
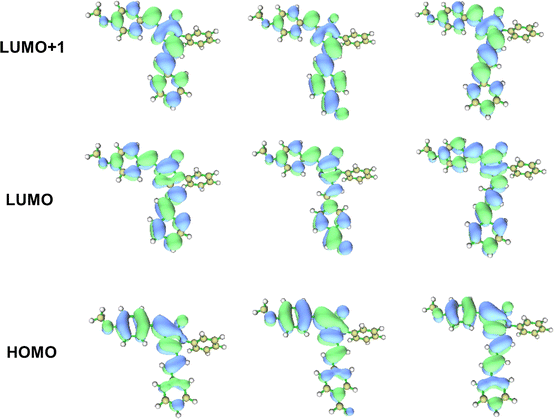 |
| | Fig. 4 Contour plots (isosurface value = 0.0025 au) of HOMO, LUMO and LUMO + 1 orbitals for compound 3a, 3b and 3c calculated at the CAM-B3LYP/TZVP level of the theory in acetonitrile solvent. | |
Another interesting feature observed (see Table 2) during the theoretical quantitative analysis of the electronic transitions is that the orbital contributions for the transition HOMO → LUMO + 1 increase from the chromophore 3a to 3c as the π-conjugations increase. For 3a, the percent of orbital contribution for the transition HOMO → LUMO + 1 in acetonitrile, methanol, ethyl acetate and cyclohexane are 81.1%, 80.9%, 77.8% and 65.6%, respectively. However, compared to 3a, the percentage of orbital contribution for HOMO → LUMO + 1 electronic transition increases for the analogues 3b and 3c and the values are 99.9% and 92.1% (acetonitrile), 99.9% and 92.1% (methanol), 90.4% and 91.6% (ethyl acetate), and 91.4% and 90.9% (cyclohexane). Both the lowest electronic absorption energies of the reference compounds (3a, 3b and 3c) are also slightly red-shifted as the π-conjugations increase. Consequently, this indicates that the intramolecular charge transfer (ICT) character of an electronic transition increases as the π-conjugations increase. To provide a more effective understanding of the ICT phenomenon, the fluorescence spectra of the molecules were also analyzed.
4.3 Fluorescence spectra of the RFP chromophore analogues
The calculated fluorescence spectra of the three synthetic RFP analogues 3a, 3b and 3c are shown in Fig. 5 and the emission maxima are tabulated in Table 3. It is seen that while the experimental fluorescence spectra have structural features in the non-hydroxylic solvents, the calculated fluorescence spectra have two emission bands in all the solvents. The lower energy emission band of 3b shows a red shift with increasing solvent polarity while the fluorescence spectra of 3a and 3c do not show any solvent effect.
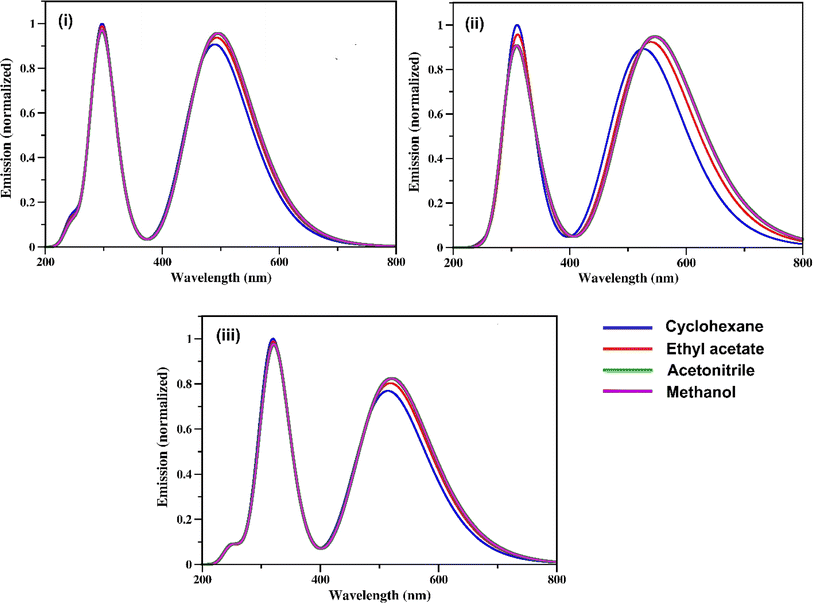 |
| | Fig. 5 Fluorescence spectra of the RFP chromophore analogues (i) 3a (ii) 3b, and (iii) 3c calculated at the CAM-B3LYP/TZVP level of theory. | |
Table 3 Calculated emission maxima (in nm) for the RFP analogues 3a–c in four solvents
| Analogues |
Cyclohexane |
Ethyl acetate |
Acetonitrile |
Methanol |
| 3a |
296, 490 |
296, 494 |
297, 495 |
297, 495 |
| 3b |
307, 527 |
308, 540 |
309, 547 |
309, 547 |
| 3c |
319, 515 |
320, 519 |
320, 521 |
320, 521 |
4.4 Potential energy curves for rotation along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bonds
C bonds
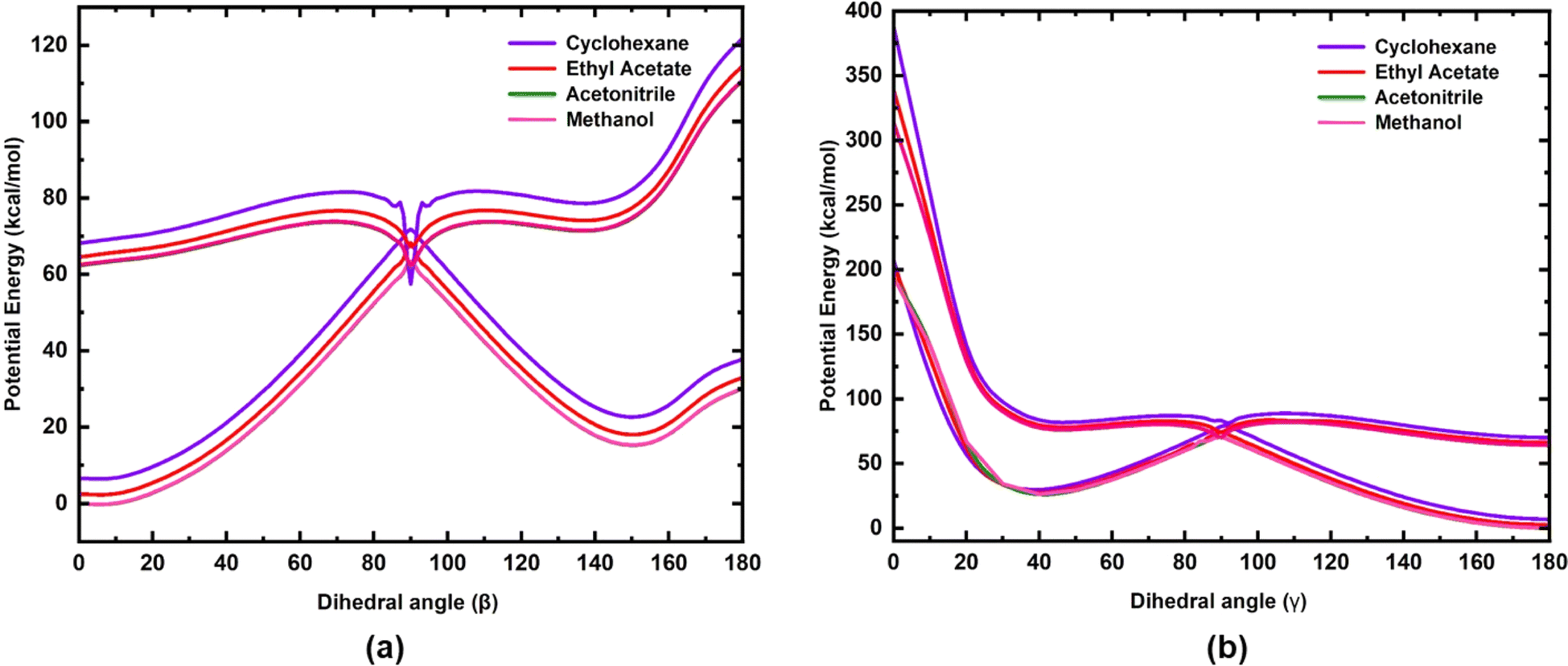
The potential energy curves of S0 and S1 states for rotation along the dihedral angle β (N3–C4–C7–C8) (rotation along the exo-cyclic CC bond, Z stereochemistry) and γ (C2–C20–C21–C22, E stereochemistry) of 3a are shown in Fig. 6. A three-dimensional (3D) potential energy surface scan of S1 for the dihedral angles α (C4–C7–C8–C13) and β (N3–C4–C7–C8) for 3a is shown in the ESI (Fig. S15).† The potential energy curves of the S0 and S1 states of 3b and 3c for the rotation along the β (N3–C4–C7–C8) dihedral angle are shown in the ESI (Fig. S16 and S17).† As seen in Fig. 6(a), for the S1 state, at β = 0°, the chromophore 3a is in the most stable state and it is characterized as the local minima. In the ground state, the potential energy barrier along the minimum energy path (MEP) is ∼63 kcal mol−1. However, as the polarity of the solvent decreases, both S0 and S1 states tend to move away from each other, leading to a slight increase in the energy barrier (∼3 kcal mol−1). On the contrary, the torsional energy barrier to reach the point of conical intersection in the S1 state for the three analogues are significantly less. Indeed, the energy for the two conformations at β = 0° and β = 90° are nearly degenerate in the excited state, as shown in Fig. 6. This indicates that in the excited state, both conformations are inter-convertible to each other through a small energy barrier at room temperature. However, for 3c, the barrier is slightly higher (see Fig. S17, ESI†) compared to that in 3a and 3b, probably due to the presence of extended conjugation in the alkyl chain of 3c. The S1 potential energy curve of all three analogues has a torsional energy barrier that is dependent on the solvent polarity. The torsional energy barriers of the S0 and S1 potential energy curves of 3a, 3b and 3c calculated in four solvents are listed in Table 4. It is seen that the torsional energy barrier in the S1 potential energy curve increases as the solvent polarity decreases as acetonitrile > methanol > ethyl acetate > cyclohexane. Methanol, although less polar (ε = 32.7) than acetonitrile (ε = 35.688), the torsional energy barrier is less in methanol than in acetonitrile due to the effect of hydrogen bonding formed by methanol with the chromophores. Thus, the trend of the torsional energy barrier seen here accords with the trend of decrease in fluorescence intensity in the four solvents. In a more polar solvent, the Frank–Condon (FC) state quickly reaches the point of conical intersection through a low torsional energy barrier while in a more polar solvent, movement of the FC state is sluggish due to a higher torsional energy barrier. Furthermore, along the dihedral angle β (N3–C4–C7–C8), the energy barrier between the S0 and the S1 state at β = 90° is significantly less (Table 4) indicating the strong coupling between both states. Hence, a conical intersection is observed at that point. Therefore, coupling between the S0 and S1 states at β = 90° leads to the internal conversion (IC) and this excited state phenomenon may be suggested to be the reason for isomerization, which has been experimentally observed for molecule 3a (Fig. S7–S12†). In addition to the exocyclic CC bond, the three chromophores have another CC bond (dihedral angle γ) about which isomerization is also possible. Therefore, in order to understand the possibility of internal conversion by isomerization about the dihedral angle γ (C2–C20–C21–C22), ground and excited state potential energy curves were calculated for 3a (Fig. 6). As seen in Fig. 6, torsional motion about the dihedral angle γ (C2–C20–C21–C22) and the conical intersection are observed. However, as the S1 state of 3a decays through the conical intersection to the ground state, it decays to a higher energy ground state compared to that in the initial ground state with γ = 180°. Thus the Z,Z-isomer obtained by torsional motion along the dihedral angle γ with β,γ = 0° becomes unstable compared to the most stable Z,E-isomer with β = 0° and γ = 180°.
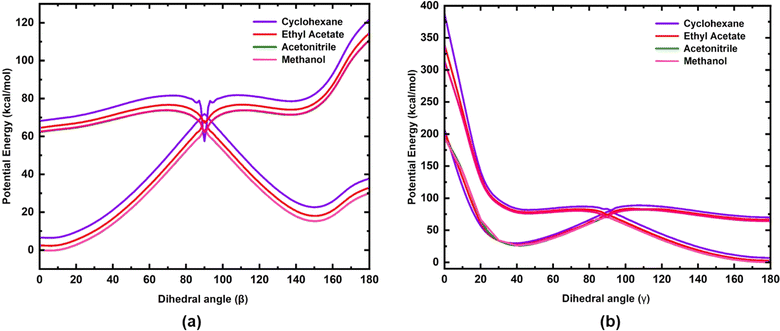 |
| | Fig. 6 Potential energy curves of the S0 and S1 electronic states of 3a for rotation along (a) the dihedral angle β (N3–C4–C7–C8) and (b) dihedral angle γ (C2–C20–C21–C22). | |
Table 4 The energy barrier (in kcal mol−1) for the RFP analogues 3a, 3b and 3c for rotation along the dihedral angle β (N3–C4–C7–C8) in four solvents in both the ground and excited states
| |
Analogues |
Cyclohexane |
Ethyl acetate |
Acetonitrile |
Methanol |
| Ground state |
3a |
65.20 |
65.76 |
63.44 |
63.51 |
| 3b |
63.19 |
62.69 |
59.93 |
59.93 |
| 3c |
63.94 |
63.69 |
62.63 |
62.69 |
| Excited state |
3a |
10.67 |
2.82 |
0.50 |
0.44 |
| 3b |
4.14 |
3.14 |
0.31 |
0.25 |
| 3c |
6.84 |
5.96 |
4.71 |
4.64 |
4.5 Conformational stability of the products of photoisomerization reaction
There are two pathways for photoisomerization-mediated internal conversion of the three RFP chromophore analogues. One is through photoisomerization of the Z,E-isomer to the E,E-isomer by torsional motion along the C(4)C(7) bond having Z-stereochemistry (torsional motion along β). The other pathway is photoisomerization of the Z,E-isomer to the Z,Z-isomer by torsional motion along the C(20)C(21) bond having E-stereochemistry (torsional motion along γ). The schematic representation of the photoisomerization reaction is shown in Scheme 3 and the energies of the ground state and excited state in four different solvents are tabulated in Table 5.
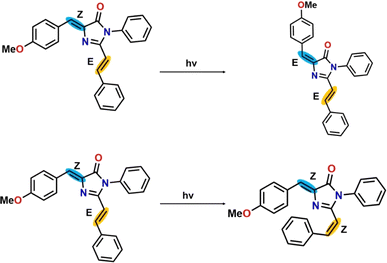 |
| | Scheme 3 Representative photoisomerization reaction for RFP chromophore analogue 3a. | |
Table 5 Conformation energies (in kcal mol−1) for the photoisomerization reaction for the conversion of the Z,E-isomer to the Z,Z-isomer and the Z,E-isomer to the E,E-isomer
| |
Cyclohexane |
Ethyl acetate |
Acetonitrile |
Methanol |
| Z,E to Z,Z-isomer |
| Ground state |
199.93 |
199.43 |
192.90 |
192.96 |
| Excited state |
319.16 |
273.85 |
250.76 |
250.63 |
|
| Z,E to E,E-isomer |
| Ground state |
30.50 |
31.12 |
30.06 |
30.12 |
| Excited state |
53.65 |
50.14 |
48.38 |
48.38 |
In the case of photoisomerization, the change of energy in the ground state for the conversion of the Z,E-isomer to the Z,Z-isomer is ∼200 kcal mol−1, corresponding to a photon of wavelength ∼ 143 nm, which falls in the far-UV region. The Z,Z-isomer formed after irradiation will not be stable due to the steric hindrance exerted by the two bulky phenyl groups. While, in the excited state, the change of energy for photoconversion of the Z,E-isomer to the Z,Z-isomer is even more than 200 kcal mol−1 compared to its ground state. Hence, photoisomerization of the Z,E-isomer to the Z,Z-isomer is energetically highly unfavourable. On the other hand, changes in the energy for photoisomerization of the Z,E-isomer to the E,E-isomer in the ground state and excited state are ∼30 kcal mol−1 and ∼50 kcal mol−1, respectively, which are much lower than those for the photoisomerization of the Z,E-isomer to Z,Z-isomer. Thus, compared to photoisomerization of the Z,E-isomer to Z,Z-isomer, isomerization to the E,E-isomer is energetically highly favorable. Thus, the photoisomerization of the Z,E-isomer is regioselective for energetic reasons, and destabilization of the Z,Z-isomer by van der Waals repulsion between the methoxy-substituted benzene ring and the benzene ring present in the extended conjugated part. This observation clearly accords with the photoisomerization of the exocyclic CC bond, computationally observed by Olsen and Smith.13 Furthermore, the change of energy involved in photoisomerization is dependent on solvent polarity. For example, in acetonitrile and cyclohexane, the change of energy involved in the excited state as the Z,E-isomer isomerizes to the E,E-isomer is 48.38 kcal mol−1 and 53.65 kcal mol−1, respectively. Therefore, photoisomerization is more favourable in more polar solvents than in less polar solvents.
5. Conclusion
The nature of the electronic transitions, which occur in the RFP chromophore analogues is π → π*. The dominant electronic transitions are HOMO → LUMO and HOMO → LUMO + 1. While the orbital contribution of the HOMO → LUMO transitions is very similar in all three chromophores, the orbital contribution of the HOMO → LUMO + 1 transition increases from 3a to 3c through 3b. Analysis of the frontier molecular orbitals suggests intramolecular charge transfer from the methoxy-substituted benzylidene moiety to the extended conjugation part through the imidazolinone ring. Calculation of the potential energy curves shows the decay of the S1 state through a conical intersection. Decay of the FC state through internal conversion by conical intersection is solvent's polarity dependent. The decrease of the torsional energy barrier in the S1 state with increasing solvent polarity accords with the observed decrease of fluorescence intensity with increasing solvent polarity. Photoisomerization of the Z,E-isomer occurs preferentially through the exocyclic CC bond (Z-stereochemistry) to form the E,E-isomer because of the involvement of a small change of energy. The computational data strongly support the experimentally observed regioselective photoisomerization through the exocyclic CC bond (Z-stereochemistry), which triggers internal conversion. Furthermore, the computational data also support the experimentally observed decrease of fluorescence quantum of the RFP chromophore analogues with increasing solvent polarity. In the native red fluorescent protein, the torsional motion along the exocyclic CC bond is prevented by the network of hydrogen bonds present in the protein barrel structure thus ensuring loss of the excited state energy primarily through the radiative pathway.
Conflicts of interest
There are no conflicts of interest among the authors.
Acknowledgements
The authors acknowledge the Supercomputing facility ‘PARAM-Ishan’ and National Supercomputing Mission (NSM) for providing computing resources of ‘PARAM Kamrupa’ at IIT Guwahati, which is implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India. HPB also thanks DST for the INSPIRE Fellowship (No. DST/INSPIRE Fellowship/2017/IF170899). The three RFP chromophore analogues were synthesized in the laboratory of Prof. Gurunath Ramanathan, Department of Chemistry, IIT Kanpur. BKR acknowledges the fruitful discussion and help received from Prof. Gurunath Ramanathan for this research.
References
- K. A. Lukyanov, Fluorescent proteins for a brighter science, Biochem. Biophys. Res. Commun., 2022, 633, 29–32 CrossRef CAS PubMed.
- M. A. Ehrmann, C. H. Scheyhing and R. F. Vogel, In vitro stability and expression of green fluorescent protein under high pressure conditions, Lett. Appl. Microbiol., 2001, 32, 230–234 CrossRef CAS PubMed.
- R. Tsien, The green fluorescent protein, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- R. N. Day and M. W. Davidson, The fluorescent protein palette: tools for cellular imaging, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- D. M. Shcherbakova, O. M. Subach and V. V. Verkhusha, Red Fluorescent Proteins: Advanced Imaging Applications and Future Design, Angew. Chem., Int. Ed., 2012, 51, 10724–10738 CrossRef CAS PubMed.
- R. E. Campbell, O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias and R. Y. Tsien, A monomeric red fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7877–7882 CrossRef CAS PubMed.
- R. Ando, H. Hama, M. Yamamoto-Hino, H. Mizuno and A. Miyawaki, An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12651–12656 CrossRef CAS PubMed.
- A. F. Fradkov, Y. Chen, L. Ding, E. V. Barsova, M. V. Matz and S. A. Lukyanov, Novel fluorescent protein from Discosoma coral and its mutants possesses a unique far-red fluorescence, FEBS Lett., 2000, 479, 127–130 CrossRef CAS PubMed.
- R. Heim, D. C. Prasher and R. Y. Tsien, Wavelength mutations and posttranslational autoxidation of green fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12501–12504 CrossRef CAS PubMed.
- G. S. Baird, D. A. Zacharias and R. Y. Tsien, Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11984–11989 CrossRef CAS PubMed.
- D. Yarbrough, R. M. Wachter, K. Kallio, M. V. Matz and S. J. Remington, Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0 Å resolution, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 462–467 CrossRef CAS PubMed.
- X. He, A. F. Bell and P. J. Tonge, Synthesis and spectroscopic studies of model red fluorescent protein chromophores, Org. Lett., 2002, 4, 1523–1526 CrossRef CAS PubMed.
- S. Olsen and S. C. Smith, Radiationless decay of red fluorescent protein chromophore models via twisted intramolecular charge-transfer states, J. Am. Chem. Soc., 2007, 129, 2054–2065 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, Conformationally locked chromophores as models of excited-state proton transfer in fluorescent proteins, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- B. Lama and M. Sarma, Unraveling the mechanistic pathway for the dual fluorescence in green fluorescent protein (GFP) chromophore analogue: a detailed theoretical investigation, J. Phys. Chem. B, 2022, 126, 9930–9944 CrossRef CAS PubMed.
- S. Rafiq, B. K. Rajbongshi, N. N. Nair, P. Sen and G. Ramanathan, Excited state relaxation dynamics of model green fluorescent protein chromophore analogs: Evidence for cis–trans isomerism, J. Phys. Chem. A, 2011, 115, 13733–13742 CrossRef PubMed.
- A. Follenius-Wund, M. Bourotte, M. Schmitt, F. Iyice, H. Lami, J.-J. Bourguignon, J. Haiech and C. Pigault, Fluorescent Derivatives of the GFP Chromophore Give a New Insight into the GFP Fluorescence Process, Biophys. J., 2003, 85, 1839–1850 CrossRef CAS PubMed.
- R. Gepshtein, D. Huppert and N. Agmon, Deactivation Mechanism of the Green Fluorescent Chromophore, J. Phys. Chem. B, 2006, 110, 4434–4442 CrossRef CAS PubMed.
- Q. Zhang, X. Chen, G. Cui, W.-H. Fang and W. Thiel, Concerted asynchronous hula-twist photoisomerization in the S65T/H148D mutant of green fluorescent protein, Angew. Chem., Int. Ed., 2014, 53, 8649–8653 CrossRef CAS PubMed.
- S. Kojima, H. Ohkawa, T. Hirano, S. Maki, H. Niwa, M. Ohashi, S. Inouye and F. I. Tsuji, Fluorescent properties of model chromophores of tyrosine-66 substituted mutants of Aequorea green fluorescent protein (GEP), Tetrahedron Lett., 1998, 39, 5239–5242 CrossRef CAS.
- M. Z. Badr, H. A. H. El-Sherief and M. E. Tadros, Studies on 2-Methyl- and 2-Phenyl-4-arylmethylene-2-imidazolin-5-ones and Related Compounds, Bull. Chem. Soc. Jpn., 1982, 55, 2267–2270 CrossRef CAS.
- B. K. Rajbongshi and G. Ramanathan, Dominant π⋯π interaction in the self assemblies of 4-benzylidene imidazolin-5-one analogues, J. Chem. Sci., 2009, 121, 973–982 CrossRef CAS.
- G. Bhattacharjya, S. S. Agasti and G. Ramanathan, Solvent free Lewis acid catalyzed vinylogous condensation, ARKIVOC, 2006, 2006, 152–161 Search PubMed.
- B. K. Rajbongshi, N. N. Nair, M. Nethaji and G. Ramanathan, Segregation into chiral enantiomeric conformations of an achiral molecule by concomitant polymorphism, Cryst. Growth Des., 2012, 12, 1823–1829 CrossRef CAS.
- K. A. K. Chidvilas, P. Mani, S. S. K. Lyer, B. K. Rajbongshi, and R. Gurunath, in 2009 34th IEEE Photovoltaic Specialists Conference (PVSC), 2009, pp. 001069–001072 Search PubMed.
- W. H. Melhuish, Quantum efficiencies of fluorescence of organic substances: effect of solvent an concentration of the fluorescent solute, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- R. Bauernschmitt and R. Ahlrichs, Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: characterization and correction of the time-dependent local density approximation ionization threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertu
![[s with combining tilde]](https://www.rsc.org/images/entities/char_0073_0303.gif) and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes, Chem. Phys., 1982, 65, 239–245 CrossRef.
and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes, Chem. Phys., 1982, 65, 239–245 CrossRef. - M. Cossi, V. Barone, R. Cammi and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Ab initio study of ionic solutions by a polarizable continuum dielectric model, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical, New York,
United States, 5th edn, 1989 Search PubMed.
- B. Mennucci and J. Tomasi, Continuum solvation models: a new approach to the problem of solute's charge distribution and cavity boundaries, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- J. Tomasi, B. Mennucci and E. Cancès, The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level, J. Mol. Struct., 1999, 464, 211–226 CrossRef CAS.
- E. Cancès, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci, E. Cancès and J. Tomasi, Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, An sp-hybridized all-carboatomic ring, cyclo[18]carbon: electronic structure, electronic spectrum, and optical nonlinearity, Carbon, 2020, 165, 461–467 CrossRef CAS.
- B. G. Levine and T. J. Martínez, Isomerization Through Conical Intersections, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Nemukhin, Potential Energy Landscape of the Electronic States of the GFP Chromophore in Different Protonation Forms: Electronic Transition Energies and Conical Intersections, J. Chem. Theory Comput., 2010, 6, 2377–2387 CrossRef CAS PubMed.
- N. Minezawa and M. S. Gordon, Optimizing conical intersections of solvated molecules: the combined spin-flip density functional theory/effective fragment potential method, J. Chem. Phys., 2012, 137, 034116 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc., 2016 Search PubMed.
- B. K. Rajbongshi, P. Sen and G. Ramanathan, Twisted intramolecular charge transfer in a model green fluorescent protein luminophore analog, Chem. Phys. Lett., 2010, 494, 295–300 CrossRef CAS.
- V. Voliani, R. Bizzarri, R. Nifosì, S. Abbruzzetti, E. Grandi, C. Viappiani and F. Beltram, Cis−Trans photoisomerization of fluorescent-protein chromophores, J. Phys. Chem. B, 2008, 112, 10714–10722 CrossRef CAS PubMed.
- Y. Yamaguchi, Y. Matsubara, T. Ochi, T. Wakamiya and Z. Yoshida, How the π conjugation length affects the fluorescence emission efficiency, J. Am. Chem. Soc., 2008, 130, 16442 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Sheikh Abdullah
*a,
Sheikh Abdullah