
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCancer cell membrane-coated nanoparticles: a promising anti-tumor bionic platform
Qiuyan Guo a,
Shengmei Wanga,
Rubing Xua,
Yingnan Tang*b and
Xinhua Xia*a
a,
Shengmei Wanga,
Rubing Xua,
Yingnan Tang*b and
Xinhua Xia*a
aSchool of Pharmacy, Hunan University of Chinese Medicine, Changsha, Hunan 410208, China. E-mail: xiaxinhua001@hnucm.edu.cn
bSchool of Pharmacy, Hunan Vocational College of Science and Technology, Changsha, Hunan 410208, China
First published on 2nd April 2024
Abstract
Nanoparticle (NP) drug delivery systems have shown promise in tumor therapy. However, limitations such as susceptibility to immune clearance and poor targeting in a complex intercellular environment still exist. Recently, cancer cell membrane-encapsulated nanoparticles (CCM-NPs) constructed using biomimetic nanotechnology have been developed to overcome these problems. Proteins on the membrane surface of cancer cells can provide a wide range of activities for CCM-NPs, including immune escape and homologous cell recognition properties. Meanwhile, the surface of the cancer cell membrane exhibits obvious antigen enrichment, so that CCM-NPs can transmit tumor-specific antigen, activate a downstream immune response, and produce an effective anti-tumor effect. In this review, we first provided an overview of the functions of cancer cell membranes and summarized the preparation techniques and characterization methods of CCM-NPs. Then, we focused on the application of CCM-NPs in tumor therapy. In addition, we summarized the functional modifications of cancer cell membranes and compiled the patent applications related to CCM-NPs in recent years. Finally, we proposed the future challenges and directions of this technology in order to provide guidance for researchers in this field.
1 Introduction
Cancer is a leading cause of human death worldwide,1 according to the International Agency for Research on Cancer (IARC), there were approximately 19 million new cancer cases and almost 10 million cancer deaths in 2020.2 New cancer cases are expected to increase by 47% in 2040 compared to those in 2020.3 With this growing global burden, cancer prevention has become one of the most significant public health challenges of the 21st century. Current cancer treatments mainly include surgical interventions, radiotherapy, and chemotherapy. However, each therapy has limited efficacy in treating cancer. For instance, surgical resection cannot be applied to all patients, and it often results in incomplete removal of tumor tissue and a high recurrence rate.4,5 Radiotherapy and chemotherapy, while targeting fast-growing cancer cells, may also harm normal tissues.6 Tumor recurrence and metastasis remain challenging, especially for patients diagnosed with advanced disease.7 Therefore, alternative treatment methods are needed. In the last 20 years, the use of nanoparticles (NPs) in nanomedicine has advanced, offering new potential strategies for cancer treatment and diagnosis.8,9 Compared to conventional anti-cancer drugs, nanocarriers have shown better drug solubility, improved drug bioavailability, and a prolonged blood circulation time. First-generation nanomedicines, such as the liposomal formulation of adriamycin (Doxil®/Caelyx®) and NP albumin-bound paclitaxel (Abraxane®), have been clinically used for decades as the first-line treatment for Kaposi's sarcoma and metastatic breast cancer, respectively.10 However, conventional NPs are limited in vivo by various physiological and cellular barriers, such as the systemic circulation, tumor microenvironment (TME), cell membranes, cellular internalization, and intracellular transport,11,12 which they must penetrate to reach the desired tumor sites.13 Statistical analysis has shown that the targeting accumulation efficiency of NPs in solid tumors is only 0.7%.14 Although the targeting inefficiency can be offset by increasing the drug dose, this often causes severe toxicity.To address these issues, researchers introduced polyethylene glycol (PEG) to the nanocarriers to enhance systemic circulation. The principle is that the conjugated PEG chains form a hydrophilic corona on the NP surface, reducing the contact of the NP with plasma proteins as a result of spatial site resistance. This avoids uptake by the reticuloendothelial system (RES) and increases the blood circulation time of the NP.15 To improve tumor targeting, a variety of ligands such as folic acid, peptides or antibodies are further modified on the surface of NPs. Ligand modification offers several benefits in tumor targeting, including improved uptake and internalization of nanocarriers by target cells,16,17 enhanced penetration into tumor tissues,18 and selective modulation of specific receptor-mediated signaling pathways.19,20 While both strategies offer significant advantages in enhancing nanocarrier functionality, other challenges remain. Studies of PEG-associated immune responses have reported that injecting partially PEGylated NPs into mice produces PEG-specific IgM antibodies that bind to subsequently administered PEGylated NPs, leading to enhanced liver uptake and eliminating the expected long circulation time properties.21,22 Therefore, the immune response to PEG strongly limits its use as a PEGylated drug carrier. In addition, when NPs enter the body, blood proteins adsorb onto NP surface, forming a protein barrier that hinders the interaction between the ligand and its target, resulting in lower targeting efficiency in vivo compared to that in vitro.23 Increasing the ligand density generally improves cellular uptake.24 However, this relationship is not linear, and having too many ligands on an NP surface can reduce the targeting and receptor-binding abilities.25,26 Additionally, the high affinity between the ligand and the receptor creates a “binding site barrier” that negatively affects the tissue penetration ability of NPs.27
The ultimate goal of using NPs is the successful delivery of therapeutic agents to the tumor tissue, which requires high tumor targeting and an extended circulation time. In 2011, Zhang et al.28 reported a new drug carrier consisting of biodegradable polymeric NPs wrapped in natural red blood cell (RBC) membranes. Compared to PEGylated NPs, the elimination half-life (t1/2) of RBC membrane encapsulated NPs was prolonged by more than 2-fold. Since then, cell membrane coating NPs has garnered considerable attention.29,30 The cell membrane is the basic component of the cell, multiple cellular functions, such as cell–environment interactions, self-recognition, and signal transduction are regulated by cell membranes.31 By directly transferring the cell membrane onto the NP surface, all biological components are retained on the final wrapped NP, giving the NP specific functions akin to those of the source cell membrane.32 To date, a wide range of cell membranes, including RBC, cancer cell, white blood cell, stem cell, bacterial, and platelet membranes have been employed to modify NPs surfaces.33–35 Compared to other cell types, although cancer cells are notorious, they are easy to culture in vitro and obtain membrane materials. Cancer cell membrane-encapsulated NPs (CCM-NPs) possess multiple biological functions (Fig. 1) and are commonly employed as nanocarriers. Firstly, cancer cell membranes express the Cluster of Differentiation 47 (CD47) protein on their surface, which interacts with a receptor called signal-regulated protein alpha (SIRPα) on macrophages, sending a “don't eat me” signal to the macrophages and thereby protecting themselves from engulfment.36 Therefore, CCM-NPs can achieve immune escape.37,38 In addition, cancer cell membranes are rich in cellular adhesion molecules, such as E-cadherin, N-cadherin, EpCAM, Thomsen–Friedenreich (TF) antigen, galectin-3, which are involved in intercellular interactions, cell adhesion and migration, and homotypic cell recognition.39–42 The first studies in this branch showed that CCM-NPs are taken up by tumor cells 40 and 20 times more efficiently than erythrocyte membrane-coated NPs and naked NPs, respectively.43 Moreover, adding a cancer cell membrane coating to NPs increased their stability and reduced the adsorption of serum proteins onto the NPs surface.44 Interestingly, the cancer cell membrane surface exhibits significant antigen enrichment. Dendritic cells (DCs) specifically uptake tumor antigens, and mature DCs serve as professional antigen-presenting cells (APCs) to initiate different subpopulations of antigen-specific T cells, enabling a comprehensive attack on tumor cells.45 In recent years, CCM-NPs have been widely used in the treatment of many types of tumors.46–49
 | ||
| Fig. 1 The multiple biological functions of cancer cell membrane-coated nanoparticles (CCM-NPs). (A) During systemic delivery, CD47 on membrane surface interacts with signal-regulated protein alpha (SIRPα) receptors on macrophages, sending a “don't eat me” signal to macrophages, which allows them to escape from macrophage phagocytosis. (B) CCM-NPs retain homotypic adhesion molecules, allowing CCM-NPs to target homologous cancer cells. (C) CCM-NPs deliver cancer cell surface antigens to immature dendritic cells (DCs), DCs specifically take up tumor antigens on CCM-NPs, and mature DCs act as specialized antigen-presenting cells (APCs) to T cells, stimulating the activation and proliferation of the latter, which can then initiate tumor cell death by detecting antigens on the tumor. | ||
In this paper, we provide a comprehensive review of recent CCM-NPs research, including the preparation, characterization, and applications of CCM-NPs in tumor therapy. We also summarize the surface modification methods of CCM-NPs, and finally discuss the challenges and prospects of developing this technology.
2 Preparation of CCM-NPs
CCM-NPs synthesis involves three steps: (1) extracting the cancer cell membrane, (2) preparation of the NP core, and (3) fusing the membrane with the NP core (Fig. 2). | ||
| Fig. 2 General preparation method of CCM-NPs. Cancer cell membranes are extracted and then wrapped around a synthetic nanomaterial core by extrusion, sonication, microfluidic electroporation, or flash nanocomplexation techniques. | ||
2.1 Extraction of cancer cell membrane
Membrane extraction involves the removal of intracellular components while preserving all functional membrane surface proteins. To prevent protein denaturation, all cell membrane extractions are conducted in an ice bath. Cell membrane extraction from source cells involves cell separation, lysis, centrifugation, and membrane harvesting. Initially, a sufficient number (200–300 millions) of tumor cells are cultured in vitro, washed with phosphate buffered saline (PBS), and lysed in a hypotonic solution. Divalent ions such as MgCl, KCl are typically added to the hypotonic lysis solutions to preserve membrane stability and minimize functional proteins loss.50,51 Subsequently, the cells in the solution are broken down using homogenization or freeze–thaw methods.52,53 The resulting solution is then differentially centrifuged to eliminate the nucleus and other organelles, leaving behind the cell membrane.54 Finally, the membrane can be microextruded through polycarbonate membranes of varying pore sizes to obtain vesicles of the desired size.55 Typically, these extracted cell membranes are stored at 4 °C or −80 °C, and the lyophilized membrane material is rehydrated in ultrapure water or PBS with a pH of 7.4 before use.53,56,572.2 Preparation of NP cores
NP cores serve as the core components of CCM-NPs, and their intrinsic properties enhance the functionality of CCM-NPs for diagnostic, drug delivery, and therapeutic applications. Ideally, NPs are spherical or non-spherical particle dispersions with a particle size ranging from 10 to 200 nm.58 This ensures that they are small enough to extravasate from tumor vasculature while being large enough to avoid leakage into renal capillaries.59 Therefore, the type, size, and shape of the nano core as well as the choice of surfactant play critical roles in determining the nature of NPs.It has been shown that particles in the range of 50–200 nm in diameter are selectively internalized by cells through clathrin-mediated endocytosis, with enhanced permeability and retention, while avoiding elimination.61 However, this is only an optimal range and different types of NPs or acting cells may produce different therapeutic effects. For example, in the Peretz et al. study, the uptake of 90 nm gold NPs in neck cancer cells (A431) was stronger than that of 5, 30, and 150 nm NPs.62 In another study, spherical mesoporous silica NPs with a diameter of 50 nm showed the highest cellular uptake in HeLa cells.63 The optimal size can therefore be determined depending on the type of NPs or the therapeutic purpose, but some factors are broadly applicable. For example, NPs smaller than 100 nm or even as low as 5 nm can enhance the penetration ability of tumors compared to large-sized particles.64,65 However, the renal filtration barrier as a whole has an effective size cutoff of about 10 nm, and NPs smaller than 10 nm are rapidly cleared by the kidney.66 On the other hand, NPs larger than 200 nm in diameter can activate the body's reticuloendothelial filtration system and be rapidly cleared from the bloodstream before eventually accumulating in the liver and/or spleen.67 Whereas particles larger than about 4 μm may get trapped in the smallest capillaries of the body.68 Therefore, to maximize tumor accumulation, both the ability of NPs to effectively penetrate the tumor tissue and the long blood circulation time are required. This can be achieved by optimizing the preparation parameters in practical experiments.
For instance, NPs with a disc shape are more likely to migrate towards the vessel wall and establish greater interaction with vascular endothelial cells.69 Cylindrical filamentous micelles were effective in evading nonspecific uptake by the RES, allowing for continuous circulation for up to one week after intravenous injection.70 Among NPs larger than 100 nm, rod-shaped particles exhibited the highest uptake against human cervical cancer epithelial (HeLa) cells, followed by spheres, cylinders, and cubes.71 A recent study compared three shapes of gold NPs including nanoshells, nanocages, and nanorods, all approximately 45 nm in size, to evaluate their delivery effect on small interfering RNA (siRNA) in tumor cells. It was observed that all three types of NPs were internalized by the cells, however, nanoshells and nanocages demonstrated more efficient siRNA delivery compared to nanorods. This could be attributed to the fact that it takes longer for cells to envelop rod-shaped metal NPs.72 Nevertheless, considering only the impact of NPs shape on their biological properties may yield contradictory results due to the complexity of their interactions with cells.
A study revealed that when different types of surfactants were examined during the preparation of ZnO NPs, the anionic surfactant sodium dodecyl sulfate (SDS) exhibited better stability due to its higher adsorption level on the surface of ZnO.75 Cationic surfactants are primarily nitrogen-containing organic amine derivatives with hydrophobic alkyl chains and hydrophilic ammonium and halogen ions. Cationic nanoparticles enhance adhesion to negatively charged cell surfaces through electrostatic attraction, resulting in higher cellular uptake compared to anionic and nonionic NPs.76 Although cationic surfactants have a strong electrostatic interaction with cells that promotes the disruption of cell membranes and leads to tumor cell death,77,78 their toxicity remains a major obstacle to their widespread practical application. Nonionic surfactants are widely used in nanomedicine due to their high biocompatibility.79 It was found that NPs encapsulated with poloxamer 184 and 188 avoided phagocytosis by macrophages, resulting in enhanced anticancer activity compared to bare NPs. This was due to an increase in cancer cell accumulation and a decrease in liver accumulation.80 Amphoteric ionic surfactants contain equal positive and negative charges, and they achieve stronger hydration through ionic solvation, which reduces interactions with blood components and prolongs the residence time of NPs in the body.81 In a recent study, it was found that NPs functionalized with amphoteric ionic sulfobetaine silanes not only exhibited good colloidal stability and low toxicity but also demonstrated better uptake in HeLa cells.82
The composition of the NPs core is an important consideration overall when designing CCM-NPs, as it is ultimately the payload that gets delivered to the target tissue. The choice of nano core type, size, and shape plays a crucial role in determining the potential therapeutic effect. Therefore, different designs should be developed on a case-by-case basis for practical applications.
2.3 Fusion of cell membrane vesicles with NP cores
The final step in preparing CCM-NPs is to wrap the cell membranes on the synthesized NPs, typically through physical extrusion, sonication, microfluidic electroporation, or flash nanocomplexation (FNC).In physical extrusion, NP cores and cell membrane vesicles are squeezed together several times through polycarbonate membranes with pore sizes ranging from 400 nm to 100 nm. Subsequently, excessive cell membranes were separated by centrifugation and discarded. The core principle is to disrupt cell membranes by mechanical forces generated during the extrusion process and fuse them with NP cores, thus producing uniformly sized CCM-NPs.28 Typically, after mechanical extrusion, the NPs exhibit a faint gray halo surrounding their outer surface. The thickness of this peripheral ring resembles that of a cell membrane.87 This method is straightforward and allows precise control over the particle size of NPs using a polycarbonate membrane while simultaneously preserving the surface protein activity on the cell membrane. In one application, researchers prepared membrane-coated NPs through co-extrusion. The initial protein concentration of the cell membrane and the concentration of membrane proteins on the nanoparticles were determined using a bicinchoninic acid (BCA) protein kit to achieve a 29% coating efficiency of the cell membrane on the NPs with good reproducibility.42 However, this method can be a tedious process, and the cell membrane may remain on the polycarbonate membrane during self-extrusion, leading to material loss and making it unsuitable for large-scale production.88
In sonication method, cell membrane vesicles are co-incubated with NP cores, and then the mixture is homogenized using ultrasound (US) to generate CCM-NPs.89,90 US energy destroys the cell membrane structure, causing the membrane to reorganize around the NP core.91 The sonication method offers simplified operation, enabling effective fusion of cell membrane with NPs within 1–30 minutes while minimizing loss of membrane protein.92,93 The efficiency of membrane coating may be influenced by the duration of US exposure. Comparative analysis conducted by the researchers revealed that a 2 minute US treatment resulted in a higher coating efficiency of 44.16% compared to durations of 30 seconds (29.04%) and 10 minutes (38.07%).94 However, the obtained NPs may exhibit a heterogeneous size distribution using this method. Furthermore, it necessitates the optimization of the US parameters (such as time, power, and frequency) to achieve efficient nuclear-membrane fusion while minimizing protein denaturation. Simultaneously, the sonication method can effectively disrupt the van der Waals interactions attributed to the carbon nanotube itself, thereby potentially rendering it unsuitable for analogous templates.95
Microfluidic electroporation involves the application of an electric field to break the dielectric layer of the cell membrane, creating transient pores through which NPs can enter. This method has been successfully employed to produce RBC membrane-coated magnetic NPs.96 The cell membrane-coated NPs prepared using this method demonstrated enhanced colloidal stability over a period of 15 days compared to the co-extrusion technique, owing to the utilization of microfluidic electroporation which facilitated a more comprehensive coating of the NPs with cellular membranes. However, the equipment requirements are substantial, and the incomplete coverage of the cell membrane exposes the NP surface to ionic buffers, leading to significant aggregation.
Recently, a new technology called FNC has been developed for the preparation of CCM-NPs.94 In the preparation of CCM-NPs, the solution containing nano cores and cell membrane fragments is introduced into different inlets of a multi-inlet vortex mixer. The kinetic energy generated by the multiple inlet jets at a predetermined flow rate transports the cell membrane fragments and nano cores into small turbulent vortex and shear interlayer regions, leading to improved flow convection and faster cell membrane encapsulation. During this process, dynamic mixing effectively disintegrates the cell membrane into smaller fragments and intricately intertwines the components, resulting in a homogeneous coating with a remarkable efficiency of 59.65% for membrane coating. By using a four-inlet vortex mixer, 120 g of biomimetic nanoproducts can be prepared per day. In addition to facilitating enhanced automation, FNC products demonstrate superior dispersibility and particle colloidal stability, as well as coating efficacy of NPs compared to those prepared using conventional sonication-based methods.
To summarize, each method has its own advantages and disadvantages. In practice, it is necessary to choose the appropriate method based on laboratory conditions, purposes, and the nature of the NP core. Although the yield and production time of the various cell membrane coatings have not been accurately reported, under typical laboratory conditions, the extrusion technique yields approximately 5 mg of coated nanoproducts per batch. The sonication method enables the coverage of up to 50 mg of NPs with the cell membrane coating obtained. Additionally, in the preparation of CCM-NPs, the cell membrane was mixed with NPs at a selected weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:5, which is a commonly reported value.97–99 The weight of the cell membrane was determined by quantifying the weight of membrane proteins using the BCA protein kit. Since the protein-to-lipid ratio is 1:1, it can be estimated that the weight of the cell membrane is twice that of the membrane proteins.100 The particle size of CCM-NPs was found to be 5 to 10 nm larger than that of uncoated NPs, which is consistent with the reported thickness of cell membranes. Furthermore, the encapsulation of the cell membrane results in a decrease in NP adsorption by serum proteins. These CCM-NPs exhibit stability for at least 24 hours in various media such as deionized water, PBS, and fetal bovine serum (FBS). Table 1 presents a summary of different preparation strategies for CCM-NPs for direct comparison.
:1–1:5, which is a commonly reported value.97–99 The weight of the cell membrane was determined by quantifying the weight of membrane proteins using the BCA protein kit. Since the protein-to-lipid ratio is 1:1, it can be estimated that the weight of the cell membrane is twice that of the membrane proteins.100 The particle size of CCM-NPs was found to be 5 to 10 nm larger than that of uncoated NPs, which is consistent with the reported thickness of cell membranes. Furthermore, the encapsulation of the cell membrane results in a decrease in NP adsorption by serum proteins. These CCM-NPs exhibit stability for at least 24 hours in various media such as deionized water, PBS, and fetal bovine serum (FBS). Table 1 presents a summary of different preparation strategies for CCM-NPs for direct comparison.
| Method | Principle | Yield (%) | Production condition | Ref. |
|---|---|---|---|---|
| Physical extrusion | Mechanical forces promote the fusion of the cell membrane with the NP cores | 29 | Extruded 10–20 times through a polycarbonate membrane | 101 and 102 |
| Sonication | Ultrasound energy promotes the reorganization of the cell membrane around the NP cores | 29.04–44.16 | Ultrasound 30 s to 30 min | 90, 91 and 94 |
| Microfluidic electroporation | The electric field creates transient pores in the cell membrane to promote the entry of NP cores | — | Pulse voltage, duration and flow rate were 50 V, 200 μs and 20 μL min−1 | 96 |
| FNC | Kinetic energy promotes the encapsulation of NP cores by the cell membrane | 59.65 | Flow rate of 90–150 mL min−1 | 94 |
3 Validation of CCM-NPs
It is necessary to verify that cancer cell membranes are successfully coated with NPs, including physical and biological characteristics, such as the size of the nanoparticles after coating, zeta potential and appearance structure, and surface proteins on the membrane (Fig. 3).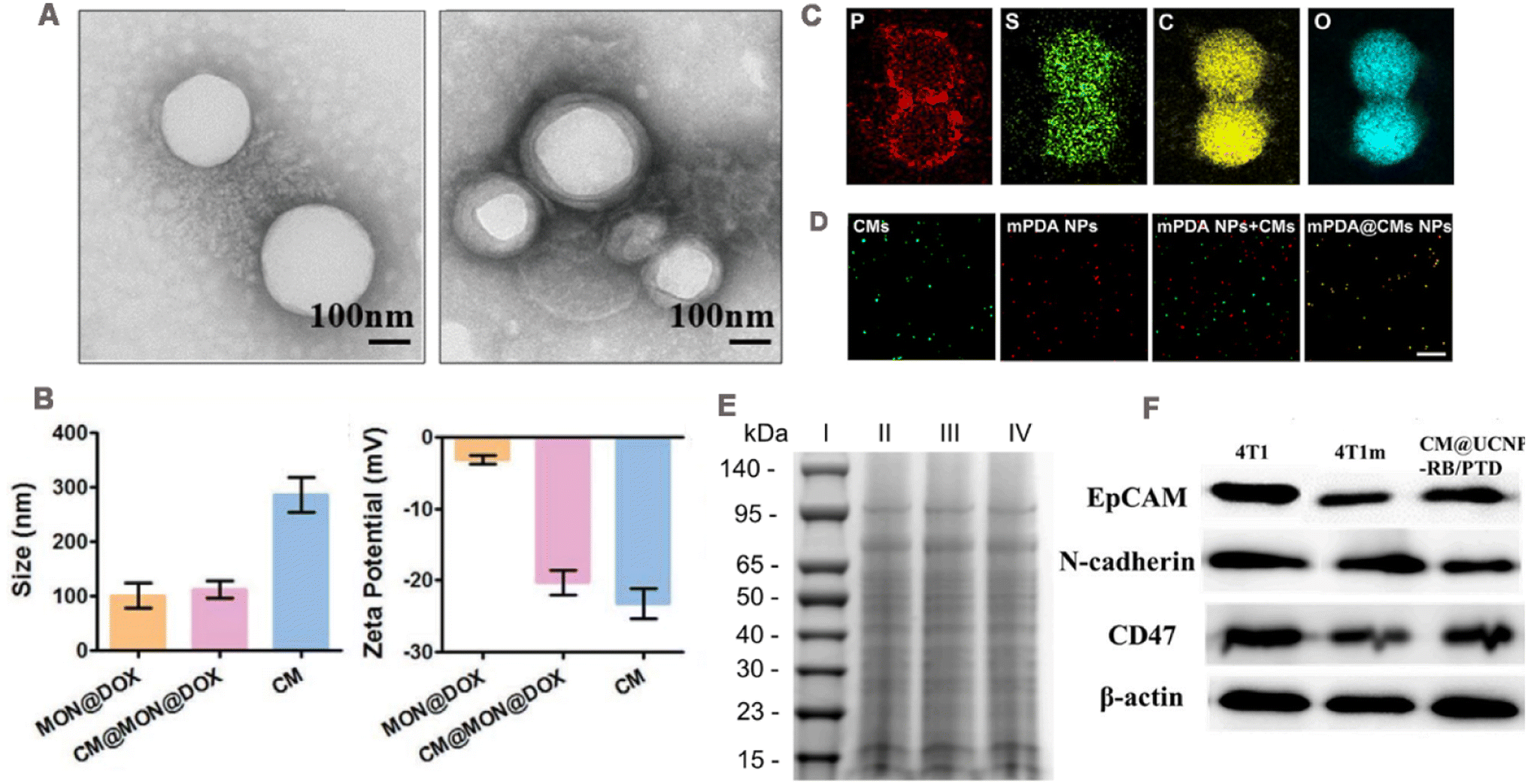 | ||
| Fig. 3 (A) TEM images of NP cores before and after cell membrane encapsulation. Adapted with permission from ref. 103. Copyright © 2022 The Author(s). (B) Size and zeta potential of CM@MON@DOX, MON@DOX, and cancer cell membrane (CM), respectively. Adapted with permission from ref. 104. Copyright © 2020 Wiley-VCH GmbH. (C) TEM elemental mapping images of mPDA@CMs NPs. (D) Fluorescence images of mPDA NPs, cancer cell membrane, a mixture of mPDA NPs and cancer cell membrane, and the fused mPDA@CMs NPs determined by confocal microscopy; green fluorescence is from DiO and red fluorescence is from ICG. Adapted with permission from ref. 105. Copyright © 2021. Published by Elsevier Ltd. (E) Representative SDS-PAGE results, (I) Marker, (II) CT26 cell lysates, (III) CCM vehicles, and (IV) H@PLA@CCM. Adapted with permission from ref. 106. Copyright © 2023 Elsevier Inc. All rights reserved. (F) Western blotting analysis showing membrane-specific protein markers with epitope-modulating properties. Adapted with permission from ref. 37. Copyright © 2021 Elsevier B.V. All rights reserved. | ||
3.1 Physical characteristics
Transmission electron microscopy (TEM) is usually used to observe the morphology of CCM-NPs. Compared with the NPs that are not cell membrane-encapsulated, CCM-NPs exhibit a core–shell structure (Fig. 3A),103 and the thickness of the shell is consistent with that of the cell membrane. The hydrodynamic diameter and zeta potential of CCM-NPs are commonly measured using dynamic light scattering (DLS). Shao et al.104 reported that the hydrodynamic diameter of CCM-NPs was slightly larger than that of uncoated NPs, and the zeta potential of CCM-NPs was equivalent to that of the cell membrane (Fig. 3B). To verify the successful coating of the cell membrane on the nanoparticle surface, Huang et al.105 used element mapping analysis and fluorescence co-localization. Since the cell membrane is mainly composed of phospholipid bilayer, the element mapping image (Fig. 3C) shows the presence of P, S, C and O element on the NPs coated by the cell membrane, proving that the membrane was successfully modified on the NPs. In fluorescence co-localization, the cancer cell membrane was labeled with a lipophilic fluorescent dye 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and then coated on the NPs loaded with the fluorescence dye indocyanine green (ICG), and the co-localization of DiO and ICG could be seen (Fig. 3D). Furthermore, successful cell membrane encapsulation could be confirmed through Fourier transform infrared spectroscopy. The characteristic absorption patterns of the amide bond, phosphate, and carbohydrate regions of the cancer cell membrane in the nanostructure indicate successful attachment of the membrane.913.2 Protein characteristics
The retention of cell membrane surface proteins determines whether the wrapped NPs can perform their specific functions. Therefore, membrane protein characterization is typically required, including the determination of protein profiles and specific protein concentrations. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is commonly employed to analyze protein profiles on the surfaces of CCM-NPs. The protein distributions observed in the CCM-NPs and cancer cell membrane profiles closely resemble those of the source cancer cell, indicating that membrane proteins are mostly well retained during the cell membrane extraction and coating process (Fig. 3E).106 Moreover, the presence of functional membrane proteins can be verified through western blotting. As shown in Fig. 3F, homologous targeting-related and immune escape-related proteins were detected on both CCM-NPs and extracted membranes, and showed a similar extent to the source cancer cell membranes.37 These results indicate that the membrane protein components of cancer cell membrane are successfully retained during the preparation of CCM-NPs.4 CCM-NPs in anti-tumor therapies
CCM-NPs have promising applications due to their biocompatibility, extended cycle life, and isotype-targeting ability. In recent years, CCM-NPs have been successfully employed in various anti-tumor therapies, encompassing drug delivery, photothermal therapy, photodynamic therapy, sonodynamic therapy, chemodynamic therapy, tumor imaging and immunotherapy (Table 2).| Disease | NP Core | Application | Cargo | t1/2/t′1/2 (h) | Cancer cells | Ref. |
|---|---|---|---|---|---|---|
| a t1/2: circulation half-life of the NPs core, t′1/2: circulation half-life of CCM-NPs. | ||||||
| Liver cancer | PLGA | Drug delivery | DOX | — | HepG2 | 107 |
| Liver cancer | MOF | Drug delivery, starvation therapy | GOx, AQ4N | — | HepG2 | 108 |
| Liver cancer | Liposomes | PTT, drug delivery | DOX, ICG | — | HepG2 | 98 |
| Breast cancer | Prussian blue | PTT, drug delivery | Lonidamine, DL-menthol | — | 4T1 | 125 |
| Breast cancer | Human serum albumin | PDT, drug delivery | PFTBA, ICG | — | 4T1 | 127 |
| Breast cancer | Zr-MOF | PDT, drug delivery | Apatinib, MnO2 | 3.5/4 | 4T1 | 99 |
| Breast cancer | HMTNPs | SDT, drug delivery | HCQ | 8.7/12.3 | MCF-7 | 141 |
| Breast cancer | Cu–Zn protoporphyrin IX nanoscale coordination polymers | CDT, drug delivery | Cu2+, ZnPPIX | — | MDA-MB-231 | 151 |
| Breast cancer | PLGA | Tumor imaging | Ag2Te quantum dots | 1.6/7.4 | 4T1 | 42 |
| Breast cancer | UCNPs | Tumor imaging | — | — | MDA-MB-231 | 172 |
| Breast cancer | PLGA | Immunotherapy, PTT | Prussian blue NPs, DTX, imiquimod | — | 4T1 | 178 |
| Cervical cancer | PLGA | Drug delivery | PTX; siRNA | — | HeLa | 55 |
| Colon cancer | Metallic bismuth | PTT | — | 4/11.5 | CT26 | 97 |
| Colon cancer | C-doped TiO2 | SDT, drug delivery | Tirapazamine | — | CT26 | 142 |
| Colon cancer | PLA | SDT | Hemoglobin | 0.75/3.23 | CT26 | 106 |
| Colorectal cancer | Fe3O4 | Tumor imaging, chemotherapy | Lycorine hydrochloride | — | HT29 | 171 |
| Melanoma | Hollow mesoporous silica | PDT, drug delivery | Ce6, GOx, CPPO, PFC | — | B16–F10 | 128 |
| Melanoma | PLGA | Immunotherapy | — | — | B16–F10 | 43 |
| Melanoma | Aluminum phosphate | Immunotherapy | CpG | — | B16–F10 | 175 |
| Melanoma | Hollow copper sulfide | Tumor imaging, PTT | DOX, ICG | — | B16–F10 | 168 |
| Osteosarcoma | Mesoporous Fe3O4 | CDT, starvation therapy, PTT | PFP, GOx | — | K7M2 osteosarcoma | 38 |
| Lung cancer | PLGA | Tumor imaging, PTT | PFCE, ICG | —/9.8 | A549 | 87 |
4.1 Drug delivery
Systemic administration of therapeutic drugs is the most common method in cancer treatment, but most of the drugs have problems such as poor targeting and low bioavailability. The drug was encapsulated in the NP core, which was then coated by the cancer cell membrane. CCM-NPs significantly improve the bioavailability of drugs and achieve precise targeting by exploiting the characteristics of homologous targeting and phagocytic escape of cancer cell membrane.In one study, HepG2 cell membranes were coated onto the surface of PLGA NPs (Fig. 4A).107 This HepM-PLGA NPs had good immunocompatibility. The internalization of HepM-PLGA NPs in RAW264.7 was reduced by about 75% compared with uncoated PLGA NPs. The homotypic binding ability of nanoparticles was then assessed by CLSM and flow cytometry. When HepM-PLGA NPs, human normal liver cells (L02 cells) membrane-encapsulated NPs (L02M-PLGA) and naked PLGA NPs were co-incubated with HepG2 and L02 cells, respectively, the fluorescence intensity of HepM-PLGA NPs in HepG2 cells was 4–5-fold higher than that higher than that of the other two groups. However, no significant NP fluorescence was observed in L02 cells. HepG2 and L02 cells were mixed and co-cultured with HepM-PLGA NPs, which selectively targeted HepG2 cells but not L02 cells. Furthermore, when incubated with different cell lines, the uptake of HepM-PLGA NPs in HepG2 cells was significantly better than that in other cell lines. These results revealed that cancer cell membrane coating endowed NPs with self-recognition ability. Using doxorubicin (DOX) as the model drug, and the loading content was determined to be 38.88 μg mg−1. The toxicity of DOX-HepM-PLGA NPs to HepG2 cells was stronger than that of uncoated nanoparticles and free drugs due to the affinity of cell membrane coating to source cells. In the nude mouse hepatocellular carcinoma solid tumor model, the fluorescence of tumor region in the DOX-HepM-PLGA NPs treated was stronger than that in DOX-PLGA NPs (Fig. 4B). After 11 days of treatment, the formulation showed excellent tumor growth control (Fig. 4C–E). During treatment, the weight of nude mice did not change significantly compared with the control group, indicating the safety of the platform (Fig. 4F).
 | ||
| Fig. 4 (A) Design strategy of cancer cell membrane biomimetic nanoparticles HepM-PLGA. (B) Fluorescence image of HepG2 tumor-bearing nude mice 11 days after the intravenous injection of Dox-HepM-PLGA and its counterparts. (C) Photos of the tumors extracted from the nude mice bearing the HepG2 tumor 11 days after the intravenous injection of DOX-HepM-PLGA and its counterparts. (D) Weights of the tumors extracted from the nude mice in (C). (E) Quantitative results of the HepG2 tumor relative volumes during chemotherapy. (F) Body weights of the nude mice during chemotherapy. Adapted with permission from ref. 107. Copyright © The author(s). | ||
In addition to delivering single drugs, CCM-NPs are also used for the co-delivery of multiple drugs to achieve synergistic therapy. For example, Xu et al.55 encapsulated paclitaxel (PTX) and siRNA in PLGA and coated Heal cell membrane to obtain a bionic nanosystem with dual drug loading (Si/PNPs@HeLa). The resulting NPs had drug loading of 2.3% and 58.8 (μg/10 mg) for PTX and siRNA respectively. SDS-PAGE and western blotting analysis showed that membrane markers were better retained on Si/PNPs@HeLa. In vitro, HeLa cell membrane-encapsulated NPs were internalized by HeLa cells more efficiently than bare NPs and had little binding capacity to other types of tumor cells. In addition, owing to the high expression of CD47 in the membrane, Si/PNPs@HeLa uptake in RAW264.7 cells was reduced 3-fold. Similarly, in HeLa tumor-bearing mouse models, the accumulation of HeLa cell membrane-coated NPs within tumors was 3-fold higher than that of bare NPs. Meanwhile, the t1/2 of the HeLa cell membrane-coated NPs was 2.2 times longer than that of the bare NPs. Compared with other groups, Si/PNPs@HeLa group achieved a tumor volume inhibition rate of 83.6% and effective co-delivery of siRNA and PTX without side effects in major organs.
To maximize the delivery of highly active therapeutic agents to tumor tissues, CCM-NPs with cascade responsiveness have also been designed. In a study, metal–organic framework ZIF-8 nanocarriers loaded with glucose oxidase (GOx) and banoxantrone (AQ4N) were encapsulated using HepG2 cell membrane to create biomimetic nanoreactors (AQ4N/GOx@ZIF-8@CM).108 GOx is a naturally occurring protein oxidoreductase enzyme that converts intra-tumor glucose and oxygen into gluconic acid and H2O2, thus disrupting the supply of glucose and oxygen within the tumor for starvation therapy.109 AQ4N is a prodrug that is activated to cytotoxic AQ4N under hypoxic conditions.110 The loadings of GOx and AQ4N in AQ4N/GOx@ZIF-8@CM were approximately 123 μg mg−1 and 36 μg mg−1, respectively. For in vitro anticancer evaluation, HepG2 cells were treated with different groups. The AQ4N/GOx@ZIF-8@CM treatment group had the lowest cell viability and the highest apoptotic rate due to the homologous recognition of the biomimetic nanoreactor and the cascade between GOx and AQ4N. Subsequently, they were evaluated using a tumor-bearing mouse model and fluorescence imaging revealed significant aggregation of AQ4N/GOx@ZIF-8@CM in the tumor tissue with sustained fluorescence intensity for over 48 hours. The inhibition of tumor growth was approximately 80% over a 21 day period, whereas single prodrug treatment or starvation exhibited only a moderate effect on inhibiting tumor growth. These findings suggest that the employed cascade significantly enhanced the tumor response.
4.2 Phototherapy
Noninvasive and selective therapy has always been a research hotspot in the field of anti-tumor. Phototherapy has been widely explored in the treatment of tumors because of its unique advantages of simplicity and high efficacy. According to the therapeutic mechanism, phototherapy can be divided into photothermal therapy (PTT) and photodynamic therapy (PDT). PTT utilizes a photothermal agent to generate vibrational heat under near-infrared (NIR) laser irradiation, which raises the temperature at the tumor site and induces tumor cell death.11 Unlike PTT, which “burns” cancer cells by photothermal heating, PDT is the induction of photosensitizers under light irradiation to produce cytotoxic reactive oxygen species (ROS), leading to cell death and tissue destruction.111Currently, highly effective phototherapeutic agents based on conventional PTT and PDT have been synthesized and studied. For instance, anthocyanin dyes such as ICG,112 IR780,113 and IR820114 are extensively employed as photothermal agents and photosensitizers due to their capacity for generating 1O2 upon near-infrared excitation. Among them, ICG is a blood volume determination dye approved by the FDA. A range of photosensitizers, such as hematoporphyrin, 5-aminolevulinic acid, verteporfin, and phthalocyanine, have been employed in clinical practice for PDT.115 However, the inherent phototoxicity and limited selectivity of conventional phototherapy drugs continue to hinder their clinical application. The integration of nanomaterials with photothermal agents or photosensitizers shows promise in enhancing the efficacy of phototherapy while mitigating its adverse effects. Moreover, certain NPs that exhibit robust NIR absorption, such as gold NPs,116 carbon-based nanomaterials,117 silicon NPs,118 and transition metal oxides119 have emerged as promising photothermal agents for phototherapy. This section provides a comprehensive overview of the utilization of CCM-NPs in phototherapy wherein the integration of cancer cell membranes with these photothermally responsive NPs enables precise and targeted treatment.
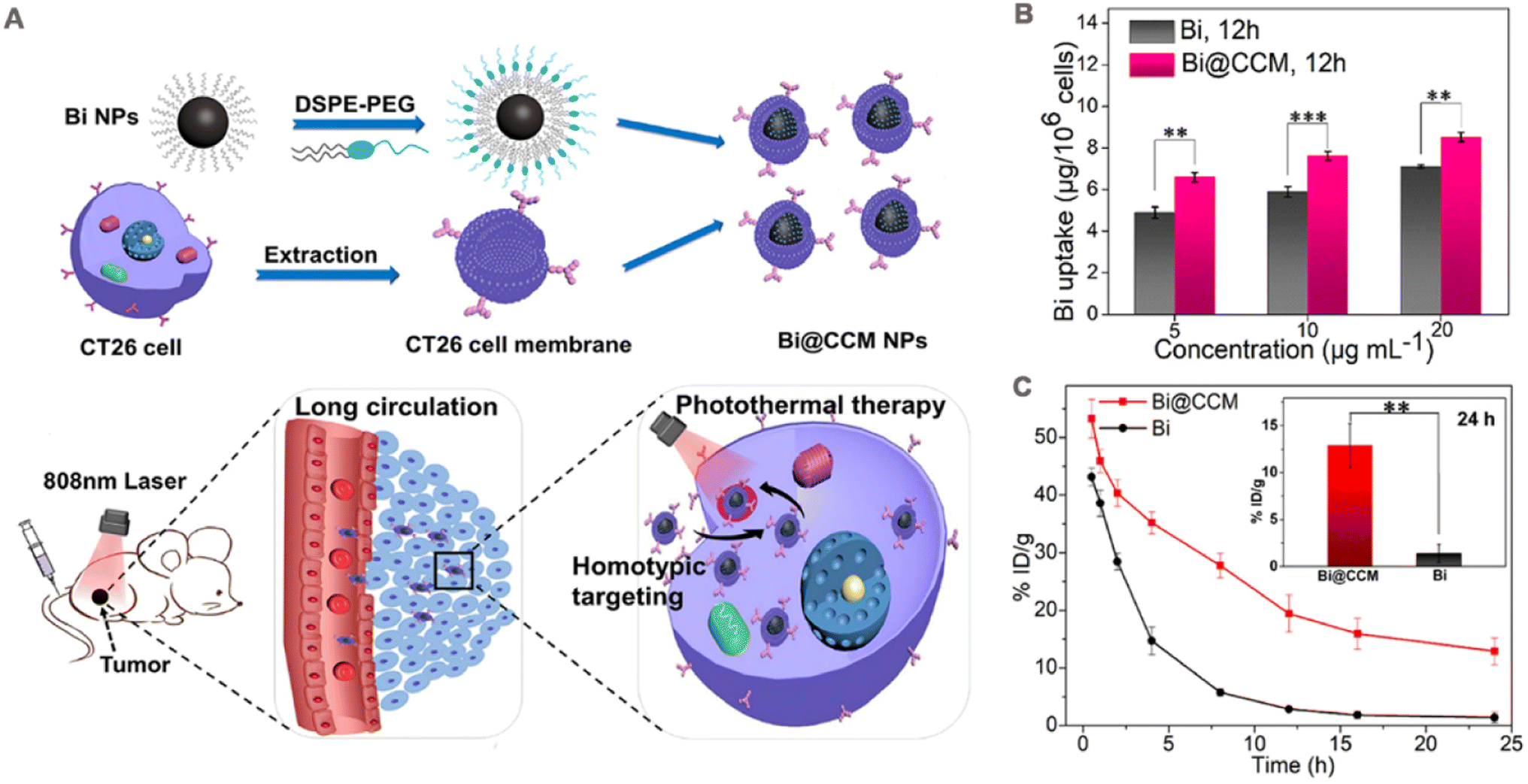 | ||
| Fig. 5 (A) Schematic diagram of cell membrane-coated bismuth metal nanoparticles (Bi@CCM NPs) for enhanced photothermal therapy. (B) Different concentrations of Bi and Bi@CCM with CT26 cancer cell uptake analysis after 12 h of incubation. (C) In vivo blood retention at 24 h after intravenous injection of Bi and Bi@CCM. Adapted with permission from ref. 97. Copyright © 2021 Elsevier Inc. | ||
PTT alone cannot kill cancer cells outside the irradiated area, and residual cancer cells carry the risk of recurring or causing metastasis. Therefore, PTT is usually used in combination with other therapies, such as chemotherapy, PDT, and immunotherapy to achieve a long-lasting anti-tumor effect. Photodynamic therapy and immunotherapy are introduced in subsequent section. This section mainly describes the treatment modality of PTT combined with chemotherapy. In one study. Sun et al.98 prepared thermosensitive liposomes coated with DOX and ICG loaded HepG2 cells (ICG-DOX-HepM-TSL) for the treatment of recurrent tumors. The photothermal conversion efficiency of the liposomes remained unaffected by cell membrane encapsulation, as their temperature increased to approximately 60 °C under 808 nm laser irradiation at a power density of 1.41 W cm−2. Meanwhile, the HepG2 cell membrane coating significantly augmented the in vitro interaction between ICG-DOX-HepM-TSL and HepG2 cells. The loading content of DOX and ICG in ICG-DOX-HEPM-TSL was 41.32 μg mg−1 and 34.83 μg mg−1, respectively. Upon laser irradiation, ICG effectively converted the incident light into thermal energy, leading to the disruption of liposomal shell integrity, thereby enhancing the release rate of DOX and eliciting potent cytotoxicity. Subcutaneous injection of HepG2 cells into nude mice was performed to establish a solid tumor of hepatocellular carcinoma, enabling the evaluation of its anti-tumor efficacy. Upon irradiation, the administration of ICG-DOX-HepM-TSL resulted in a remarkable 70% reduction in the volume of recurrent tumors observed in nude mice over a period of 13 days, whereas both the PBS group and nude NPs group exhibited significant increases in tumor volume.
PTT induces tumor cell death while potentially causing indirect damage to normal tissues at high temperatures,120 so mild temperature (≤45 °C) PTTs have been developed.121,122 However, heat shock proteins (HSPs) overexpressed by tumor cells induce heat resistance to PTT under mild hyperthermia conditions.123,124 To solve the problem of heat resistance of PTT, Shu et al.125 loaded hollow mesoporous Prussian blue nanoparticles with lonidamine (which can inhibit the expression of HSPs) and DL-menthol (which acts as a plugging agent and controls the release of lonidamine) and encapsulated them with 4T1 cancer cell membrane to obtain biomimetic nano platform (PBLM@CCM NPs). The system was exposed to a 793 nm laser for 5 minutes, resulting in a significant temperature increase of approximately 20 °C, whereas the PBS group exhibited only a modest increase of 3.6 °C. The drug loading efficiency of lonidamine in PBLM@CCM NPs was about 11.3%, and the release of lonidamine was temperature dependent. Due to the introduction of cell membrane, PBLM@CCM NPs showed significant binding to 4T1 cells. After laser irradiation (793 nm, 0.8 W cm−2, 300 s), the average temperature of the tumor treated with PBLM@CCM increased rapidly from 33.4 °C to 43.8 °C, indicating the anti-tumor effect of PTT. At 21 days of treatment, the tumor weight of the PBLM@CCM NPs + laser group was about 0.17 g, and the inhibition rate was 77.9%, which was nearly 2.5 times higher than that of the unloaded lonidamine group.
To achieve oxygen delivery and effective PDT, human serum albumin (HSA) was used as a carrier loaded with ICG and perfluorotributylamine (PFTBA) and subsequently coated with 4T1 cell membrane to obtain the nanoprobe (CCm-HSA-ICG-PFTB) (Fig. 6A).127 In this study, bare HSA-ICG-PFTBA released 70% ICG in serum at 12 h after dialysis, which was 3.5-fold higher than CCm-HSA-ICG-PFTBA (20% release), indicating that cell membrane coating was able to enhance the stability of nanoprobes. PFTBA has a large O2 retention capacity can provide oxygen for PDT treatment, which is further enhanced by the targeting property of the membrane coating that allows more NPs to enter the cells. In vitro cytotoxicity assay showed that CCm-HSA-ICG-PFTBA exhibited the strongest cytotoxicity under NIR irradiation (Fig. 6B). In vivo, CCm-HSA-ICG-PFTBA effectively localized to the tumor site and persisted for 48 h (Fig. 6C). Oxygen concentration was measured in isolated tumor sections, and immunofluorescence staining of the hypoxia probe showed that the hypoxic area of the tumor shrank 10-fold 24 h after the injection of CCm-HSA-ICG-PFTBA. Finally, in a mouse 4T1 tumor model, the CCm-HSA-ICG-PFTBA combined with NIR irradiation showed slow tumor growth (Fig. 6D) and lowest tumor weight by 14 days post treatment (Fig. 6E), which prevented tumor progression better than uncoated cell membrane NPs.
 | ||
| Fig. 6 (A) Illustration of the biomimetic oxygen-delivery nanoprobe. It was cancer cell membrane-coated indocyanine green-doped perfluorocarbon (CCm–HSA–ICG–PFTBA) for homologous targeting and improving oxygen concentration at tumor sites. 18F-FMISO PET/CT imaging was performed to measure the hypoxia in vivo. CCm-HSA-ICG-PFTBA was injected into 4T1 xenografts and then photodynamic therapy was performed. Tumor volume was measured to evaluate the therapeutic efficacy enhancement. (B) Cell viability after treatment with CCM-HSA-ICG-PFTBA, HSA-ICG-PFTBA, and HSA-ICG with or without near-infrared (NIR) laser irradiation (n = 5). (C) In vivo fluorescence images of 4T1 xenografts after injection of CCM-HSA-ICG-PFTBA, HSA-ICG-PFTBA, HSA-ICG, and saline at different time points. Red circles indicate tumor sites. (D) Relative tumor volumes of mice after irradiation with CCM-HSA-ICG-PFTBA, HSA-ICG-PFTBA, HSA-ICG, saline, and NIR laser. (E) Tumor weight after 14 days of treatment. Adapted with permission from ref. 127. Copyright © The Author(s), 2021. | ||
A bionanoreactor (bio-NRs) based on chemiluminescent resonance energy transfer (CRET) has been developed for the combination treatment of PDT and starvation therapy.128 The photosensitizers chlorine e6 (Ce6) and GOx were modified on the surface of hollow mesoporous silica NPs (HMSNs). Then, bis[2,4,5-trichloro-6-(pentyloxycarbonyl)phenyl] oxalate (CPPO) and perfluorohexane (PFC) were co-loaded into the cavities of the HMSNs, which were then coated with B16–F10 cell membrane to obtain bio-NRs. Owing to the homologous adhesion and immune escape properties of tumor cell membranes, bio-NRs are able to target tumors and gradually accumulate at the tumor site. These are then used for synergistic anti-tumor therapy with PDT and starvation therapy through the following three modes: (1) Ce6 is excited by the energy of the reaction between CPPO and H2O2 in the cell, and CRET generates ROS for PDT. (2) GOx catalyzes the conversion of glucose to H2O2, which puts the cell in a starved state and in turn provides H2O2 to enhance ROS production. (3) PFC has a large O2 retention capacity, which enables the NPs to carry oxygen, and the O2 released after entering the cells improves the hypoxic state of the tumor and accelerates glucose oxidation to enhance ROS generation. In the lung metastasis mouse model, the lung metastases in the oxygen-carrying bio-NRs group completely disappeared. In addition, bio-NRs treated mice had 100% survival within 30 days, while all other groups showed obvious tumor metastasis and different degrees of weight loss.
PDT shuts down the vascular system during treatment, however tumor cells lacking blood supply activate the expression of vascular endothelial growth factor leading to tumor angiogenesis and causing tumor recurrence or metastasis.129,130 In addition, the high concentration of glutathione (GSH) in tumors has a powerful scavenging effect on ROS generated during PTD, thus affecting PDT efficacy. Recently, a biomimetic metal–organic framework (MOF) nanoplatform (aMMTm) has been developed to enhance PDT therapy.99 In this biomimetic nanosystem, photosensitive porphyrin-type Zr-MOF was used as a carrier loaded with the anti-angiogenesis inhibitor apatinib, then wrapped with a layer of MnO2 as a shell, and finally coated with 4T1 cell membranes on the surface of MnO2-coated nanoparticles (aMM). In this case, MnO2 can act as a GSH scavenger and reduce the removal of ROS. To test the hypothesis that MnO2 depletes GSH in tumors, the nanoparticles were added to 4T1 cells, and the MnO2-coated nanoparticles decreased GSH levels in 4T1 cells by more than 50% compared with drug-loaded nanoparticles (aM) without MnO2 coating. In addition, the release of apatinib in aMMTm was gradually elevated with the addition of GSH. These results suggest that the MnO2 shell effectively depleted GSH in tumor cells and effectively triggered GSH-dependent drug release. The homo-binding ability of cell membrane and PDT effect combined with accelerated drug release properties make aMMTm the highest cytotoxic to 4T1 cells under light irradiation. In vivo, the bare MOF NPs had a t1/2 of only 0.4 h and were quickly cleared from the blood, while aMMTm had a prolonged t1/2 of 3.5 h and enhanced tumor accumulation. Finally, in 4T1-bearing tumor models, the aMMTm + light group effectively inhibited tumor growth, whereas both the NPs unloaded with apatinib and aMMTm without light groups demonstrated ineffective anti-tumor effects.
4.3 Sonodynamic therapy
Sonodynamic therapy (SDT) is a novel cancer treatment strategy that uses low-intensity US. US as an external trigger source to excite the acoustic sensitizer to produce ROS to kill the tumor. US is a mechanical wave with high tissue penetration to reach deep into the tumor.131 Therefore, SDT has better potential for deep tumor treatment than PDT. Sonosensitizers play a pivotal role in the therapeutic efficacy of SDT. Currently, the reported repertoire of sonosensitizers predominantly includes both organic and inorganic variants. Organic acoustic sensitizers, such as hemoporfin,132 hemoglobin molecules,133 and erythrosine B,134 have demonstrated their efficacy in SDT against tumors. Additionally, the small molecule dyes ICG and IR780 have shown a strong response to ultrasound and are considered promising candidates for acoustic sensitization.135,136 Certain inorganic nanomaterials, including TiO2 NPs137 and carbon nanomaterials,138 demonstrate significant in vitro cancer cytotoxicity when exposed to ultrasound irradiation. As a result, they have been used as inorganic acoustic sensitizers or carriers for ultrasound sensitizers with promising applications in SDT.Wen et al.106 obtained H@PLA@CCM by encapsulating cell membranes on poly(lactic acid) (PLA) polymer NPs loaded with the acoustic sensitizer hemoglobin (Fig. 7A). The fluorescence intensity of the monooxygen fluorescence probe SOSG confirmed that H@PLA@CCM has a highly efficient 1O2 generation ability under US irradiation, which increased with the irradiation time. Owing to its isoform-binding property, the platform preferentially entered homologous CT26 cells, while uptake was not obvious in 4T1 cells. When H@PLA@CCM was incubated with CT26 cells stained with the fluorescent probe, a significant enhancement of intracellular green fluorescence was observed under US irradiation, confirming the presence of a large amount of intracellular ROS. By contrast, cells treated with US irradiation or H@PLA@CCM alone showed almost no fluorescence (Fig. 7B). Owing to efficient cellular uptake and ROS generation capacity, H@PLA@CCM significantly induced apoptosis in CT26 cells under US irradiation. In vivo, H@PLA@CCM had a significantly prolonged t1/2 than uncoated H@PLA (3.23 h vs. 0.75 h). CT26 tumor-bearing mice were injected with different groups of drugs separately. At the end of treatment on day 16, the tumor inhibition rate of 83.9% was achieved in the H@PLA@CCM + US group compared with those in the other control groups (Fig. 7C).
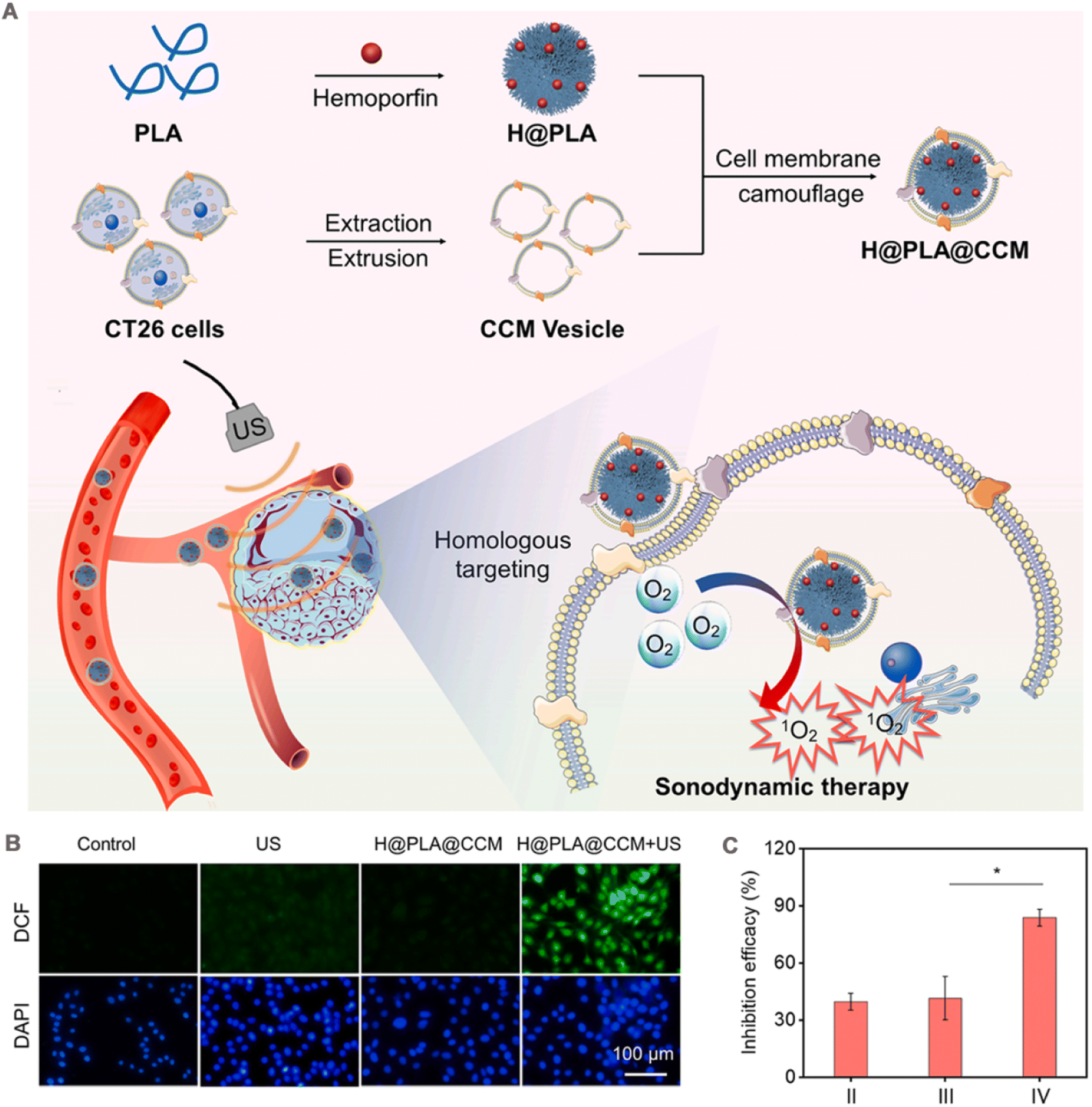 | ||
| Fig. 7 (A) A scheme of preparation processes of H@PLA@CCM and the illustration of homologous tumor-targeted SDT driven by H@PLA@CCM and US irradiation. (B) Intracellular ROS level induced by the H@PLA@CCM and/or US. (C) The inhibition efficacy of (II) H@PLA + US, (III) H@PLA@4T1CCM + US, (IV) H@PLA@CT26CCM + US. Adapted with permission from ref. 106. Copyright © 2023 Elsevier Inc. | ||
Mitochondria are often considered a major target of SDT, and SDT-induced oxidative stress in injured tumor cells tends to activate the mitochondrial autophagy process, which then protects tumor cells from oxidative stress by eliminating damaged mitochondria and attenuating apoptotic cell death, thereby reducing the efficacy of SDT.139,140 In a study, Feng et al.141 loaded the autophagy inhibitor hydroxychloroquine sulfate (HCQ) into hollow mesoporous titanium dioxide NPs (HMTNPs), which were subsequently encapsulated by cancer cell membrane to obtain a bionic nanoplatform (CCM-HMTNPs/HCQ). Owing to the retention of membrane surface antigens on the surface of cancer cell membranes, CCM-HMTNPs/HCQ showed enhanced immune escape and homologous tumor accumulation and bypassed heterologous tumors. The hollow mesoporous titanium dioxide NP core endowed CCM-HMTNPs/HCQ with good SDT efficacy and induced apoptosis in tumor cells. In this system, the loading efficiency of HCQ was calculated as 46.4%. US irradiation disrupted the cell membrane coating and triggered the release of HCQ, which further inhibited SDT-induced protective autophagy in cancer cells, thereby weakening the resistance of cancer cells to SDT. In addition, HCQ improved vascular function and alleviated tumor hypoxia, further enhancing the SDT effect. This combined strategy of SDT killing and autophagy inhibition induced significant ROS generation, autophagic vesicle accumulation, and apoptosis. In MCF-7 tumor-bearing nude mice, the final volume/initial volume ratio (v/v0) of CCM-HMTNPs/HCQ + US group was 1.71 ± 0.11, which was significantly smaller than that of CCM-HMTNPs + US group (3.68 ± 0.14) and HCQ group (4.87 ± 0.22).
Similar to PDT, the therapeutic efficacy of SDT can be limited by the hypoxic microenvironment of the tumor. In contrast to the common approach of increasing the oxygen content in the tumor, Ning et al.142 exploited the hypoxia in the tumor by wrapping CT26 cell membranes around C–TiO2 hollow nanoshells (HNSs) containing tirapazamine (TPZ) to obtain a bionic drug delivery system (C-TiO2/TPZ@CM). In the treatment process, C–TiO2/TPZ@CM perfectly utilized the cell membrane coating to achieve efficient homologous tumor cell targeting. At the same time, SDT induced anoxic microenvironment, and TPZ was activated in anoxic environment to produce high cytotoxic free radicals, which synergically enhanced the killing effect on tumors.
4.4 Chemodynamic therapy
Chemodynamic therapy (CDT) based on Fenton/Fenton-like response has emerged as a novel and minimally invasive approach for cancer treatment, initially proposed by Bu et al. in 2016.143 The discovery of the Fenton reaction came from the British scientist Henry J. Fenton, and the main reaction process is: The chain reaction between Fe2+ and H2O2 catalyzes the formation of the highly harmful ˙OH under acidic conditions.144 ˙OH is the most potent oxidizing reactive oxygen species, capable of inducing apoptosis in tumor cells through DNA damage and protein inactivation.145 In recent years other metal ion (e.g., Cu2+ and Mn2+) mediated Fenton-like reactions have also been developed for CTD enhancement.146,147 In general, the accumulation of H2O2 and the acidic pH in the TME confer high selectivity to CTD for cancer therapy. However, despite the higher concentration of H2O2 (100 μM to 1 mM) in the TME compared to normal tissues, it remains insufficient for sustained ˙OH production.148 Hence, improving the therapeutic efficacy of CDT, by increasing the level of intra-tumor H2O2, and improving the hypoxic environment are essential.Wang et al.38 constructed an adaptive nanoplatform (M-mFeP@O2-G) for synergistic enhancement of CDT by encapsulating cancer cell membrane on mesoporous Fe3O4 nanoparticles loaded with perfluoropentane (PFP) and GOx. The assembly process of the bionic system is demonstrated in Fig. 8A. In this system, the camouflage of cancer cell membranes allowed the nanoparticles to precisely target to the tumor site and enhanced the immune escape ability of the nanoparticles. After reaching the tumor site, the M-mFeP@O2-G nanoparticles released the iron ions to generate ˙OH via the Fenton reaction. Under 808 nm laser irradiation, the photothermal conversion efficiency of M-mFeP@O2-G reached 36.83%, and more Fe ions were released, accelerating the Fenton reaction. GOx consumes the glucose in the tumor cells and kills the tumor cells through starvation therapy. It also produces a large amount of H2O2, which further enhances the Fenton reaction. In addition, PFP carries O2, and under laser irradiation M-mFeP@O2-G releases O2 as the temperature increases, relieving tumor cell hypoxia and providing O2 for the cascade reaction, thus enhancing CDT. The preparation process and in vivo process of M-mFeP@O2-G is shown in Fig. 8B. K7M2 osteosarcoma model was established by subcutaneous injection of K7M2 cells into the right/lower limb of BALB/c mice. On the 22nd day, the relative mean tumor volume growth was slower in the M-mFeP@O2-G group + light-induced group than in the other groups (Fig. 8C). The tumor inhibition rate was 90.50%, whereas the tumor growth inhibition rates for the non-laser group M-mFeP@O2-G and the non-coated group mFeP@O2-G were 68.68% and 51.72%, respectively (Fig. 8D). Furthermore, there was no significant change in the body weight of mice during the experiment (Fig. 8E), indicating that the MmFeP@O2-G nanoparticles exhibited favorable in vivo safety.
 | ||
| Fig. 8 (A) Schematic illustration of the preparation of M-mFeP@O2-G nanoparticles. (B) Schematic diagram of the mechanisms of M-mFeP@O2-G nanoparticles for tumor-specific cascade reactions via enhanced CDT after intravenous injection. (C) The average relative tumor volume vs. time curve after administration of the different NPs to mice. (D) The tumor growth inhibition (TGI) rates on day 22. (E) Mouse body weight changes after different treatments. Adapted with permission from ref. 38. Copyright © 2022 The Authors. | ||
The highly toxic ˙OH produced by ROS-based CDT during treatment is also cleared by GSH. Therefore, the efficiency of CDT can also be improved if GSH is consumed. However, the GSH elimination method can only partially inhibit the antioxidant capacity of cancer cells. When cancer cells are stimulated by excessive ROS, they overexpress heme oxygenase-1 (HO-1), and HO-1 metabolites can clear ROS to form a system with high antioxidant capacity, thereby reducing efficacy.149,150 Based on this, biomimetic nanoscale coordination polymer NPs (CCPPM) that simultaneously deplete GSH and inhibit HO-1 activity were prepared.151 During their preparation, Cu2+ and HO-1 competitive inhibitor Zn protoporphyrin IX (ZnPPIX) coordinated to form a coordination polymer (CCP). To increase its solubility, CCP was modified with polyvinylpyrrolidone (PVP) to obtain PVP-modified CCP (CCPP), which was then coated with MDA-MB-231 cell membrane. CCPPM was taken up by MDA-MB-231 via endocytosis, intracellular GSH reacted with CCPPM via redox reactions to generate Cu+, and the generated Cu+ converted endogenous H2O2 into cytotoxic ˙OH. Meanwhile, GSH induced the catabolism of CCPPM, and the catabolized Cu2+ generated Cu+ with GSH, which enhanced the generation of –OH. In addition, ZnPPIX released from CCPPM inhibited HO-1 activity and reduced the tolerance of cancer cells to oxidative stress. In vitro, CCPPM incubated with MDA-MB-231 cells for 12 h significantly increased intracellular ROS levels, decreased GSH/GSSG ratio, and inhibited HO-1 activity by 80%. When injected intravenously into MDA-MB-231 hormonal mice, CCPPM showed enhanced accumulation at tumor sites and anti-tumor effects compared to PBS and bare CCPP, and minimal tumor volume was observed after 17 days of treatment. Tumors were collected at the end of treatment, and malondialdehyde, a product of lipid peroxidation, was higher in tumor cells of the CCPPM-treated group than in other control groups, with the lowest HO-1 activity and GSH/GSSG ratio. In addition, significant side effects were observed during the treatment period.
4.5 Tumor imaging
Tumor imaging plays an important role in the early detection and staging of tumors for diagnosis.152,153 This advancement significantly enhances disease management through timely identification, treatment, and prevention. Common biological imaging techniques encompass NIR fluorescence imaging, photoacoustic (PA) imaging, and magnetic resonance imaging (MRI).In one study, in order to achieve real-time tumor monitoring and accurate tumor surgical guidance, Ag2Te quantum dots were self-assembled with PLGA NPs and then wrapped with 4T1 cell membrane to obtain membrane camouflage nanoparticles (CPQDs) (Fig. 9A).42 The platform demonstrates exceptional fluorescence brightness and remarkable stability within the NIR II window. The fluorescence intensity of the colloidal quantum dots (CPQDs) remains at 97% even under continuous exposure to an 808 nm laser, while the ICG only maintains a fluorescence intensity of 24% (Fig. 9B). Furthermore, the penetration depth of CPQDs (≈7 mm) was more than twice that of ICG (3 mm) under the same conditions (Fig. 9C). The pharmacokinetic study was performed in ICR mice, which were intravenously injected with PBS solution containing unmodified PQDs, PEG-coated control (PPQDs) and CPQDs. According to the quantitative analysis of Ag+, the t1/2 of CPQDs was 7.4 h, while those of PQDs and PPQDs were 1.6 h and 6 h, respectively (Fig. 9D). PQDs, PPQDs and CPQDs were intravenously injected into nude mice subcutaneously transplanted with 4T1 tumor for in vivo fluorescence imaging. The whole-body NIR II fluorescence images were collected at 48 h. CPQDs showed higher fluorescence intensity in the tumor (Fig. 9E). This study, confirms that fluorescence imaging mediated by membrane-encapsulated nanoparticles holds great promise as a cancer treatment strategy.
 | ||
| Fig. 9 (A) Schematic illustration of the nanobioprobe preparation and application. QDs: Ag2Te quantum dots; PLGA: poly(lactic-co-glycolic acid); PQDs: NIR II fluorescent assembly of Ag2Te QDs and PLGA; CVs: cell membrane-derived vesicles; CPQDs: CVs-camouflaged PQDs; FL: fluorescence. (B) The photostability of the PQDs, PPQDs, CPQDs, and ICG in terms of FL intensities under continuous 808 nm laser irradiation. (C) NIR II images of the CPQDs and ICG for different tissue thicknesses. (D) Concentration–time profiles of the PQDs, PPQDs, and CPQDs in the blood as measured by the Ag+ content. (E) In vivo NIR II FL imaging of 4T1 tumor-bearing mice injected with PQDs, PPQDs, and CPQDs over the course of 48 p.i. Adapted with permission from ref. 42. Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In one study, the melanoma B16 F10 cell membranes were used to camouflage hollow copper sulfide NPs loaded with DOX and ICG (ID-HCuSNP@B16 F10) by Wu et al..168 In the ID-HCuSNP@B16 F10 system, CuS NPs can serve as both a photothermal agent and a contrast agent for PA imaging. With the inclusion of DOX and ICG, chemotherapy, PTT, and PA imaging can be simultaneously performed. The loading efficiency of ID-HCuSNP@B16 F10 for ICG and DOX was 98% and 85%, respectively. After laser irradiation, the release of both drugs increased. Compared with HCuSNPs, ID-HCuSNP@B16F10 NPs exhibited a higher temperature at the same irradiation time, indicating that the loading of ICG produced an additional photothermal effect. This was further evaluated in the B16F10 tumor model. Mice were given intravenous injections of IDHCuSNP@B16F10, ID-HCuSNPs. Accumulation of ID-HCuSNPs@B16F10 in tumors was much higher than that in the IDHCuSNP group, and a strong local PA signal could be observed in the tumor area 4 h after injection. After 14 days of treatment, the tumor volume of the ID-HCuSNP@B16F10 + laser treatment group was significantly reduced.
Li et al.171 prepared magnetic iron oxide nanoparticles (LH-Fe3O4@M) coated with colorectal cancer cell membrane and loaded with lycorine hydrochloride for homologous targeting, MRI and chemotherapy. The T1 and T2 relaxation values measured by the MRI scanner demonstrated that LH-Fe3O4@M exhibited an relaxation rate (r2/r1) > 10, indicating a pronounced T2 effect when used as a contrast agent. HT29 tumor-bearing mouse tumor model was established for in vivo T2-weighted imaging. Cross-sectional images of the tumor showed a distinct MR signal within the tumor at LH-Fe3O4@M 20 h after injection. Moreover, the degree of contrast enhancement of negative signal in the tumor area was greater than that in the PBS group and PEG modified Fe3O4 (Fe3O4@PEG), indicating that the cell membrane coating guided LH-Fe3O4@M to further accumulate at the tumor site. The chemotherapeutic drug, lycorine hydrochloride, exhibited a loading efficiency of 32.68% in LH-Fe3O4@M. Subsequently, this system was employed in nude mice with HT29 tumors for a duration of 20 days, wherein LH-Fe3O4@M demonstrated remarkable efficacy in chemotherapy and achieved a substantial tumor ablation rate.
In a recent study, triple-negative breast cancer MDA-MB-231 cell membrane was used to camouflage Gd3+-doped UCNPs.172 This CCm231-UCNPs were designed to enable tumor visualization by combining upconversion luminescence (UCL), MRI, and positron emission tomography (PET), and further distinguish different subtypes of breast cancer. In this multimodal imaging, UCL can reach deep into the tissue by using NIR laser as an excitation source, avoiding autofluorescence of biological tissues. MRI provides high spatial resolution. PEF has high sensitivity and unlimited detection depth. In vivo, NPs were injected into MDA-MB-231 tumor-bearing nude mice. UCL imaging, MRI, and PET imaging showed that, compared with the erythrocyte membrane-coated UCNPs (RBCm-UCNPs) and UCNPs groups, the CCm231-UCNPs group exhibited high uptake by the tumor and low uptake by the liver. Subsequently, CCm231-UCNPs were injected into MDA-MB-231 and MCF-7 tumor-bearing mice, the accumulation of CCm231-UCNPs in MDA-MB-231 tumors was higher than that in MCF-7 tumors through three imaging modalities, effectively differentiating different subtypes of breast cancer. In another complex system, A549 cancer cell membrane camouflaged nanoprobes (AM-PP@ICGNPs) containing perfluoro-15-crown-5-ether (PFCE) and ICG demonstrated accurate tumor diagnosis and PTT effects.87 In this system, PFCE is an excellent 19F MRI reagent, ICG is used for for NIR fluorescence and PA imaging. Thus, the probe can be imaged in three modes. The ICG content in PP@ICGNPs was about 1.2%, and the temperature of the solution containing AM-PP@ICGNPs rose to 56.5 °C under continuous 765 nm NIR laser irradiation, demonstrating the photothermal effect of AM-PP@ICGNPs. Lung cancer A549 cell membrane coating significantly promoted PP@ICGNPs tumor targeting and retention. The location and distribution of AM-PP@ICGNPs within the tumor were comprehensively observed by three-mode imaging. Furthermore, tumor volume was reduced by 86% in the AM-PP@ICGNPs group in response to ICG-induced PTT.
4.6 Immunotherapy
Tumor immunotherapy has become an important option for tumor treatment because it stimulates the immune system to kill tumor.173,174 CCM-NPs serve as drug carriers for tumor immunotherapy, deliver tumor-specific antigens, and activate downstream immune responses for anti-tumor effects. Therefore, modulating of tumoral immunity via CCM-NPs is an effective tumor treatment method.In 2014, Fang et al.43 wrapped mouse melanoma B16–F10 cell membrane around PLGA NPs by physical extrusion (Fig. 10A) and demonstrated that the tumor-associated antigen glycoprotein 100 of melanoma was present on the CCNPs. When bound to the immune adjuvant monophosphoryl lipid A (MPLA), CCNPs induced the maturation of DCs, with significant upregulation of the maturation markers CD40, CD80, and CD86 (Fig. 10B). When CCNPs with MPLA were added to DCs and co-cultured with splenocytes from transgenic pmel-1 mice, the splenocytes clustered significantly around the DCs (Fig. 10C), and produced significantly higher levels of the cytokine interferon-gamma (IFNγ) (Fig. 10D). This strategy has also been applied in tumor vaccines. Gan et al.175 prepared CpG loaded and B16–F10 cancer cell membrane-encapsulated aluminum phosphate NPs (APMC), and demonstrated their effectiveness as a vaccine. Due to the coating of cancer cell membrane, APMC carry a comprehensive tumor antigen, showing specific anti-tumor immune function. Different formulations to bone marrow-derived DCs (BMDCs) were added in vitro, APMC and free cell membrane + CpG significantly promoted the maturation of BMDCs, which possibly due to the co-binding of the cancer cell membrane and immune adjuvant to promote the immune response. In vivo, APMC was efficiently delivered to mouse lymph nodes and significantly increased co-uptake of tumor antigen and CpG by lymph node resident APCs. The immune response to NPs was tested by measuring the cytokines secreted by T cells and lymph node cells. The results showed that APMC promoted higher levels of cytokine secretion than the other NPs. In addition, the tumor prevention and treatment effects of the vaccine were tested in B16–F10 tumor-bearing mice. The results of the prophylactic mouse model showed that the mean tumor volume at day 20 in the APMC treatment group was only about 200 mm3, compared with more than 2000 mm3 in the PBS group. Similar results were obtained in the antitumor model, and the APMC group had the highest median survival time of 30 days. Finally, safety evaluation revealed no organ toxicity or inflammatory reactions caused by APMC.
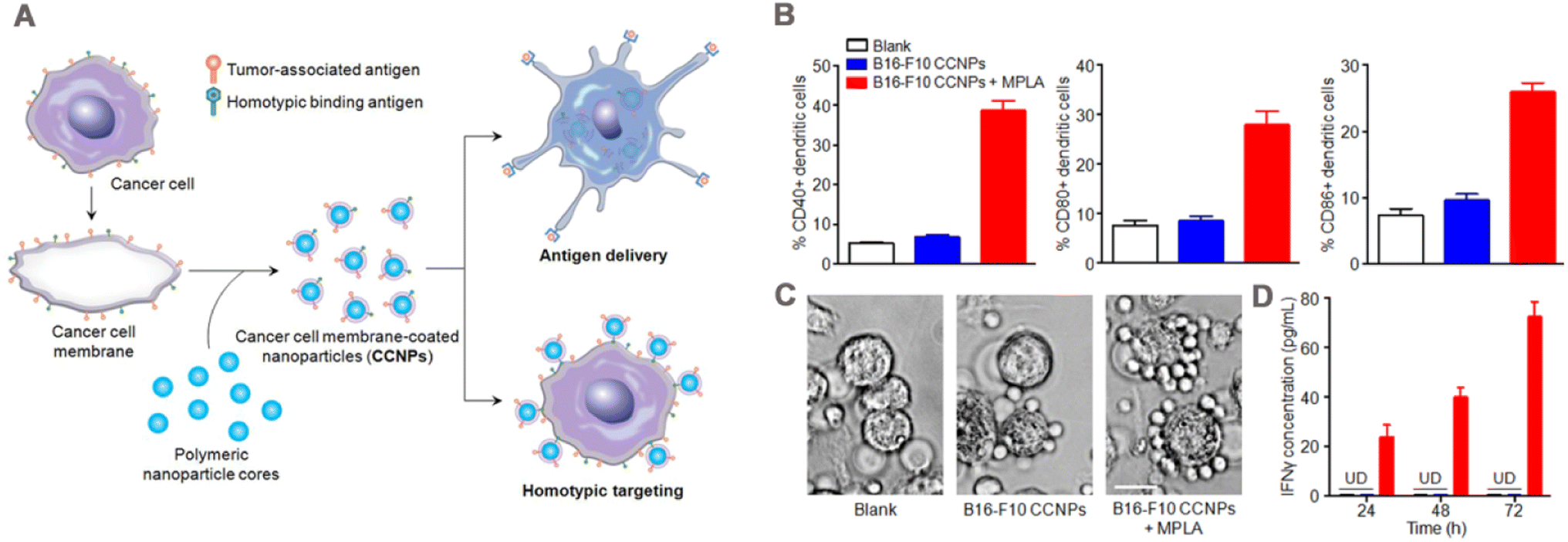 | ||
| Fig. 10 (A) Preparation of cancer cell membrane-coated NPs and mechanism of their application in immunization. Surface of cancer cell membrane is rich in antigen and isotype adhesion molecules, which are retained on NPs after coating to deliver antigens to immune cells and target homologous tumor cells. (B) Dendritic cells were incubated with blank solution, B16–F10 cells coated NPs (B16–F10 CCNPs), or B16–F10 CCNPs with MPLA as adjuvant for 48 h. Cells were then immunostained with CD11c antibody as a DC marker and CD40, CD80, or CD86 antibody as a maturation marker, and the maturation of DCs was analyzed by flow cytometry. (C) Phase-contrast microscopy images of DCs treated with blank solution, CCNPs, or MPLA for 24 h and splenocytes from pmel-1 transgenic mice after co-culture for 72 h. T lymphocytes were clustered around DCs. (D) Specific response of IFNγ to melanoma-associated gp100 antigen was detected by ELISA at 24, 48 and 72 h after co-culture. Adapted with permission from ref. 43, copyright© 2014 American Chemical Society, unless otherwise noted. | ||
Despite the initial success of emerging immunotherapies, solid tumors are often immunosuppressive, leading to inefficient and resistant anti-tumor immune responses. A rational combination of multiple therapeutic modalities may become an effective strategy in the fight against cancer. Currently immunotherapy is often used in combination with chemotherapy, PTT or PDT for the treatment of tumors. Certain chemotherapy agents such as docetaxel (DTX) can promote the polarization of tumor-growing M2-like tumor-associated macrophages into tumor-inhibiting M1-like macrophages, effectively reverse the immunosuppressive TME.176 PTT locally kills tumors and induces dead cells to release tumor-associated antigens thereby activating the immune response.177 Chen et al.178 used cancer cell membrane-encapsulated PLGA nanospheres (M@P-PDR) with a core loaded with Prussian blue NPs, and a shell encapsulated with DTX and the immune adjuvant imiquimod (R837). Cancer cell membrane encapsulation enhanced tumor targeting and accumulation of nanospheres, and the accumulation at the tumor site was 2.49 times higher in the M@P-PDR group than in the unencapsulated group (P-PDR) after 8 h of injection in vivo. The Prussian blue NPs acted as photothermal conductive agents, and under laser irradiation, the PTT effect was triggered, which in combination with DTX induces tumor eradication in situ. The results of ex vivo and in vivo immunoreactivity showed that the DC maturation level was significantly higher in the M@P-PDR + laser group compared with that in the membrane-encapsulated NPs + laser group without R837 and that in the M@P-PDR group, suggesting that the integration of PTT with R837 has a stronger ability to promote DC maturation. Further, the addition of DTX promoted the repolarization of M2-like macrophages to M1 mode. This bionic nanoplatform combined with chemotherapy, PTT, and immune adjuvant effectively inhibited the growth and metastasis of the primary tumor, and at the end of treatment, there was no significant tumor recurrence. In addition, by monitoring the survival rate of mice in each group, the mice in the M@P-PDR + laser group all survived without significant tumor recurrence within 57 days.
5 Surface modification of CCM-NPs
CCM-NPs effectively deliver drugs and attack targets on different types of tumor. However, single-cell types have inadequate molecular functionality of the membrane, reducing the drug delivery efficiency to the tumor sites. To produce multifunctional CCM-NPs, additional functions are usually required. Current strategies for functionalizing CCM-NPs include physical, chemical, and genetic modification and membrane fusion (Fig. 11, Table 3).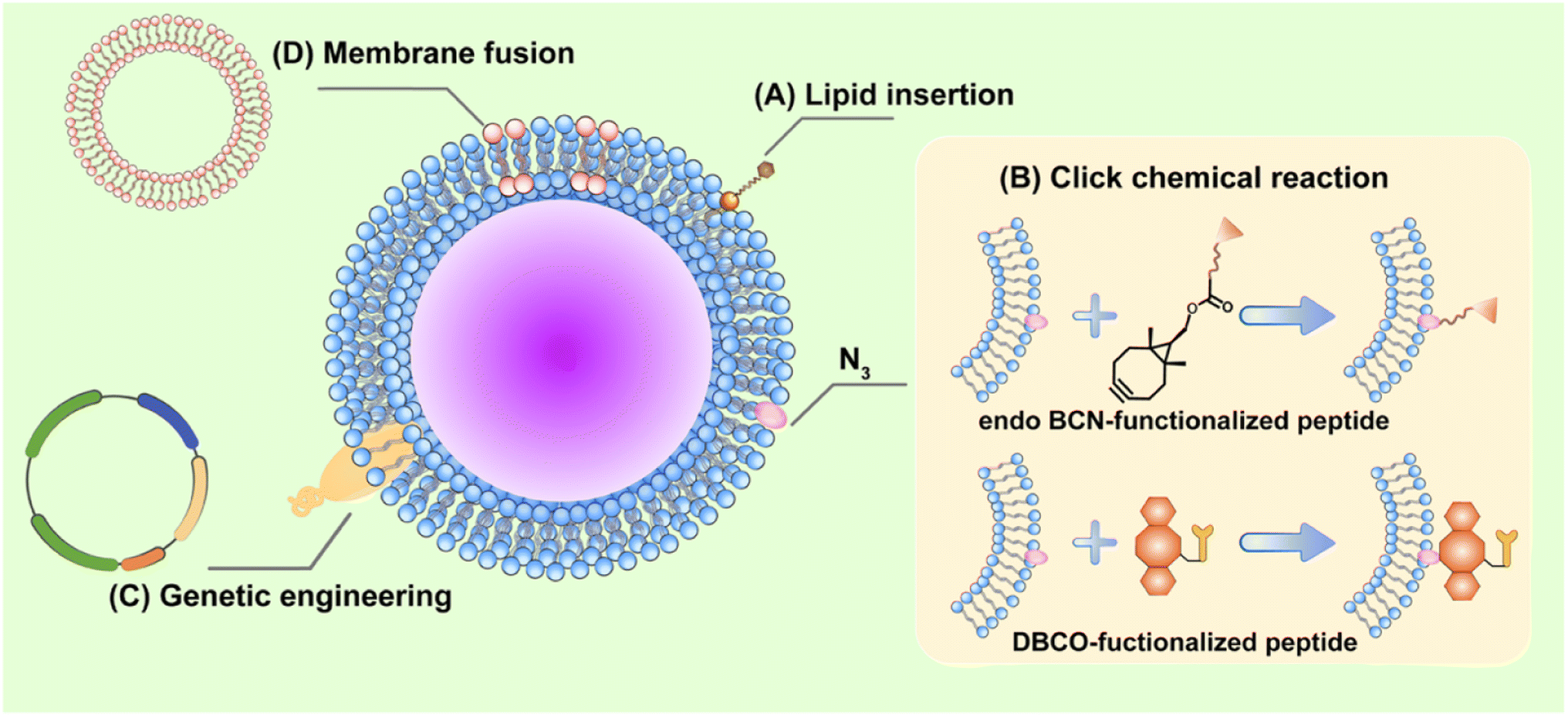 | ||
| Fig. 11 Strategies for functionalization of CCM-NPs. (A) Physical engineering: functional lipid molecules anchored to cell membrane through their affinity for cell membrane. (B) Chemical engineering: N3 modified on the membrane binds to endo BCN or DBCO modified compound via click chemical reaction. (C) Genetic engineering: required products are expressed on cell membrane surface through gene transcription and translation. (D) Cell membrane fusion: two types of cell membranes are fused, allowing NPs to co-express surface features of two different cell types. | ||
| Modified molecule | Modification strategies | Membrane source | NP core | Outcomes | Ref. |
|---|---|---|---|---|---|
| Mannose | Lipid insertion | B16-OVA | PLGA | Increasing the uptake of BMDCs and triggering DC maturation more effectively | 179 |
| DSPE-PEG | Lipid insertion | MCF-7 | PLGA | Reducing nonspecific binding between NPs and serum proteins | 39 |
| cRGD | Click chemistry reaction | GL261 | CaCO3 | Binding to αvβ3 integrin and promoting blood–brain barrier penetration | 180 |
| Anti-CD205 | Click chemistry reaction | 4T1 | Fe3O4 | Targeting CD8+ dc promotes an effective immune response | 181 |
| M2pep peptide | Genetic engineering | KPC | PLGA | Targeting tumor-associated macrophages significantly reduced the percentage of M2-like macrophages | 182 |
| Ovalbumin and CD80 | Genetic engineering | Wild-type B16–F10 | PLGA | Enhancing the activation of antigen-specific T cells and triggering immune responses | 183 |
| — | Cell membrane fusion | RBCs, B16–F10 | Hollow copper sulfide | Enhancing circulation in vivo and inhibiting tumor growth | 185 |
| — | Cell membrane fusion | Bacteria, B16–F10 | Hollow polydopamine | Ability to stimulate the maturation of dendritic cells and enhance anti-tumor | 100 |
5.1 Physical modification
Physical modifications are relatively mild and mainly based on the lipid structure and fluidity of the cell membrane. Lipid insertion is a common and simple modification method, in which the functional part linked to the lipid can be spontaneously integrated into the phospholipid bilayer via hydrophobic interactions (Fig. 11A).In one study, Yang et al.179 loaded R837 (an anti-toll-like receptor 7 agonist) into PLGA NPs and then coated the NPs with B16-OVA cancer cell membrane to obtain NP-R@M, which was subsequently modified with mannose on the cell membrane by lipid-anchoring method. In this NP-R@M–M system, mannose was believed to specifically target APCs. The cancer cell membranes provide tumor-specific antigens, and R837 is an immune adjuvant to enhance tumor immunotherapy. Owing to the additional PEG chains anchored on the NP surface, the DLS analysis showed that the particle size of NP-R@M increased after mannose modification. In vitro studies have shown significantly enhanced uptake of NP-R@M–M by BMDCs and more efficient triggering of DC maturation. After injection into the left foot of mice, NP-R@M–M had a much higher retention rate in the lymph nodes than NP-R@M. Finally, in mice immunized three times at 1 week intervals with different formulations and then attacked with B16-OVA melanoma cells on day 7 after the last injection, NP-R@M–M showed the strongest anti-tumor efficacy and triggered the upregulation of IFN-γ production, demonstrating powerful tumor prevention. In addition, the lipid tails in polyethylene glycol lipid derivatives (e.g., DSPE-PEG2000) can easily be inserted into the vesicle's membrane layer. PEG-modified cell membranes can be obtained by physically extruding cell membranes with DSPE-PEG2000 through a 220 nm polycarbonate membrane.39 This modification has been shown to reduce non-specific binding between NPs and serum proteins and protect them from phagocytosis, thereby prolonging their t1/2 in vivo.
5.2 Chemical modification
A common in chemical engineering strategies is the modification of cell membranes with azide (N3) using intrinsic biosynthesis, which can be conjugated with functional compounds via clicking chemical reactions (Fig. 11B).Zhao et al.180 designed CaCO3 NPs with positive targeting effects that could penetrate the blood–brain barrier. First, tumor cells were pretreated with N-azi-doacetylmannosamine-tetraacylated to enable N3 to attach to the cell surface. The membrane was then co-extruded with CaCO3 NPs loaded with mRNA (mRNA@CaCO3 NPs) to form the membrane-coated CaCO3 NPs (mRNA@CMCaCO3 NPs). Subsequently, the arg-gy-asp (cRGD) is produced between N3 on the surface of the cell membrane and the alkynyl group of the pre-synthesized endo-bicyclo[6.1.0]nonyne (BCN)-cRGD (endo-BCN-cRGD) and attached to the mRNA@CM-CaCO3 NPs surface, resulting in mRNA@cRGD-CM-CaCO3 NPs. In this system, cRGD bound to the integrin receptor αvβ3, which is overexpressed in tumor neovasculogenesis, promoting the blood–brain barrier penetration of NPs. The membrane coatings may further improve the targeting ability of NPs after crossing the blood–brain barrier. In vitro and in vivo targeting assays confirmed that compared to mRNA@CM-CaCO3 NPs, mRNA@cRGD-CM-CaCO3 NPs were heavily internalized by glioma cells, showing a good anti-tumor effect. Similarly, Li et al.181 designed cancer cell membrane containing N3 to coat the surface of Fe3O4 magnetic nanoclusters (MNCs). Then dibenzocyclooctyne (DBCO)-modified anti-CD205 was spliced with N3 on the cell membrane by a click reaction to form anti-CD205-modified cancer cell membrane-encapsulated CpG oligodeoxynucleotide (CpG-ODN)-loaded MNC (A/M/C-MNC). Anti-CD205 modification directs more MNCs to CD8+ DCs, promoting an effective immune response, and the camouflaged cancer cell membrane acts as an antigen reservoir, further promoting the effective presentation of related antigens. Five tumor models were established, and A/M/C-MNC showed preventive and therapeutic effects.
Chemical modifications can display new functional groups on cell membranes that can confer a wider range of functions to NPs. However, chemical modification involves many chemical reactions, and the activity of over-modified membrane proteins may be disrupted. Therefore, the reaction conditions should be controlled during the preparation process.
5.3 Genetic modification
Gene modification involves expressing a desired protein or peptide on the cell membrane surface via viral or non-viral transduction to achieve a targeted or therapeutic function (Fig. 11C). Compared to natural cells, genetically modified cells have good reproducibility and modifiable functions and are commonly used in tumor vaccines and immunotherapy.Wang et al.182 transfected pancreatic cancer KPC cells with lentivirus encoding M2pep, a peptide targeting M2 macrophages, and confirmed the presence of M2pep on KPC cell membranes by CLSM visualization. Then, M2pep-expressing KPC cell membranes (KMCM) were encapsulated on the surface of gemcitabine-loaded PLGA NPs, and biomimetic NPs (PG@KMCM) were synthesized for co-targeting of macrophages and tumors. Co-incubation with macrophages in vitro resulted in rapid internalization of PG@KMCM, which significantly induced M2 macrophage death and reduced the macrophage M2/M1 population ratio compared to NPs (PG@KCM) not encapsulated with M2pep-modified cell membranes, suggesting that the modification of M2pep enhanced nanomedicine delivery in macrophages. In the KPC tumor mouse model, owing to the homotypic binding of cancer cell membrane coating and macrophage targeting by M2pep, PG@KMCM showed enhanced tumor accumulation and prolonged retention time compared to PG@KCM and non-membrane-coated NPs, which effectively augmented the efficacy of gemcitabine and led to a dramatic reduction in tumor size. Subsequently, the percentage of macrophages in the tumor was detected, and flow cytometry results showed that PG@KMCM significantly reduced the percentage of M2-like macrophages, confirming the effectiveness of the PG@KMCM nanosystems as a dual tumor cell and macrophage targeting therapy for tumor treatment. In addition, PG@KMCM combined with PD-L1 immune checkpoint inhibitor treatment was able to effectively reprogram the TME and kill cancer cells, thereby increasing the overall therapeutic potential.
In a recent study, Jiang et al.183 used viral transfection to overexpress two different proteins in the cell membrane of the wild-type B16-F10 (B16-WT) murine melanoma cell line. The first is the cytoplasmic form of ovalbumin (OVA), which provides a wide range of immune tools for antigen modeling. The other is the co-stimulatory marker CD80, which binds to the CD28 receptor on T cells, thereby promoting the activation of homologous T cells. Cell line B16-CD80/OVA was obtained, and then the cell membrane of B16-CD80/OVA was coated on the nanoparticles to prepare [CD80/OVA] NPs. Western blotting analysis and flow cytometry confirmed the expression of the OVA protein and the co-stimulatory marker CD80 in B16-CD80/OVA cells. Incubated in vitro with OT-I splenocytes, [CD80/OVA] NPs showed enhanced antigen-specific T cell activation. To verify the distribution of the CD80/OVA NPs in vivo, an OT-I mouse model was established, and after subcutaneous injection, immunofluorescence images showed a large amount of CD80/OVA NPs fluorescence near the CD8+ T cells. Then, in a C57BL/6 mouse model of adoptive OT-I spleen cells, the CD80/OVA NPs significantly upregulated the CD69 activation marker of adoptively transferred CD8+ T cells and the level of IFNγ secreted by the lymph node cells. Finally, an immunoactive tumor model was developed. Compared with the other groups, the CD80/OVA NPs demonstrated better preventive and anti-tumor effects, with the slowest tumor growth and longest survival in mice.
Genetic engineering typically involves integrating exogenous genetic material into the genome of the target cell, followed by its combination with nanomaterials, which exhibit a high degree of stability. However, this process is relatively complex.
5.4 Cell membrane fusion
In addition to directly modifying the membrane surface or CCM-NPs, preparing hybrid cell membranes by fusing two different types of cell membranes is a common means of expanding the functions of cell membranes (Fig. 11D). In 2017, Zhang et al.184 prepared novel hybrid membrane-coated NPs by mixing RBC membrane with platelet membrane. The hybrid membrane could exhibit the properties of both types of cells, opening the door for producing biomimetic NPs with different hybrid functions. Recently, cancer cell membranes have been hybridized with several types of cell membranes for use in tumor therapy.In one study, Wang et al.185 fused membrane materials from RBCs and B16–F10 cells, coated on DOX-loaded hollow copper sulfide nanoparticles (DCuS@[RBC–B16] NPs). To prepare the mixed membrane, the B16–F10 cell membrane solution was mixed with the erythrocyte membrane solution at a mass ratio of 1:1. And, the [RBC–B16] membrane was then obtained by sonication for 10 minutes at 37 °C. To validate the fusion, two different dyes were added to B16–F10 cell membrane to form a Förster resonance energy transfer (FRET) pair, and the fluorescence gradually decreased at 670 nm as the number of erythrocytes increased, suggesting that the intercalation of the two membrane materials reduced the FRET interactions on the original B16–F10 cell membrane. At the protein level, the characteristic proteins on both RBC membrane and B16 cell membrane were retained in the hybridized membrane. The researchers also labeled the B16–F10 cell membrane with DiO, labeled the red cell membrane with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), and then prepared a hybrid membrane coated on the nanoparticles. CLSM images showed overlapping signals of DiO and DiI, all of which demonstrated the successful fusion of the two cell membranes. DCuS@[RBC–B16] NPs exhibited self-recognition in vivo, with higher accumulation at the tumor site compared to the other groups. Under NIR irradiation, the tumor inhibition rate of DCuS@[RBC–B16] NPs reached almost 100%. In addition to mammalian cell membranes, the bacterial outer membrane vesicles (OMV), a natural vesicles secreted by Gram-negative bacteria that contains many pathogen-associated molecular patterns, has been used as a drug carrier, vaccine delivery agent, and cancer immunotherapeutic agent.186 Wang et al.100 used a similar method to mix OMVs derived from Escherichia coli DH5α with B16–F10 cell membranes at a weight ratio of 1:1, resulting in the formation of hybrid membranes (OMV-CC), which were then coated on hollow polydopamine (HPDA) NPs to obtain HPD@[OMV-CC] NPs. In this system, OMV acts as an immunotherapeutic agent. In the in vivo distribution, HPDA@[OMV-CC] NPs accumulated abundantly on tumor tissues and lymph nodes. The tumor inhibition rate of the HPDA@[OMV-CC] NPs + NIR irradiation-treated group was about 99.9%.
In summary, several types of CCM-NPs have been developed and demonstrated excellent anti-tumor effects. The functionality of CCM-NPs is mainly achieved by membrane coatings, in addition to other functions provided by the NP core, such as drug loading, photothermal, photodynamic, chemodynamic, sonodynamic, or imaging functions. To achieve the best anti-cancer effect, combinations of various approaches are often used. For example, drug delivery can be coupled with PTT, SDT to achieve on-demand drug release. In immunotherapy, only relying on antigens on the surface of tumor cells may not be sufficient to induce an anti-tumor adaptive immune response, so it needs to be combined with immune adjuvants. In short, these CCM-NPs have excelled in drug delivery, noninvasive treatments, tumor imaging, and have also brought great success to immunotherapy.
6 Patents of CCM coating technology
With the development and application of CCM-NPs, an increasing number of related patent applications have been filed in the past decade. Table 4 lists the patent applications for CCM-NPs in recent years. These applications highlight the gradual popularization of bionic drug designs.| Patent number | Patent title | Applicants | Filling year |
|---|---|---|---|
| CN110559448 | Targeted siRNA delivery bionic nanoparticle as well as preparation method and application thereof | Guangzhou Medical University | 2019 |
| US20210338583 | Cell membrane lipid-extracted nanoparticles for selective targeting image analysis and cancer therapy | Robert B. Campbell | 2019 |
| CN111821283 | Zinc glutamate-coated Prussian blue nanoparticles with triphenylphosphine-clonidamine coated with cancer cell membrane and preparation method thereof | Huaqiao University | 2020 |
| CN112603999 | Tumor microenvironment response type nanoparticle based on biomimetic engineering as well as preparation method and application thereof | Chongqing Medical University | 2020 |
| CN112494495 | Preparation method of cancer cell membrane chimeric liposome nano drug delivery system | Tianjin Medical University | 2020 |
| CN113368079 | Cancer cell membrane-coated drug-loaded lignin nanoparticles as well as preparation method and application thereof | Jinan University | 2021 |
| CN112791062 | Targeting nanomaterial with cell membrane coated with Au–Fe3O4 as well as preparation method and application of targeting nanomaterial | Qilu University of Technology | 2021 |
| CN113876691 | Hydrogel encapsulating target lncRNA pvt1 nanoparticles and preparation method and application thereof | Union Hospital, Tongji Medical College, Huazhong University of Science and Technology | 2021 |
| CN113648289 | Arginine deiminase lipid nanoparticles wrapped by lung cancer cell membrane and preparation method of arginine deiminase lipid nanoparticles | Chongqing Medical University | 2021 |
| CN113694216 | Nano diagnosis and treatment agent as well as preparation method and application thereof | Shenzhen University | 2021 |
| CN114146064 | Genetically engineered cell membrane bionic nano-microsphere with pancreatic cancer microenvironment targeting and method thereof | Zhejiang University | 2021 |
| CN114569578 | Preparation and application of bionic nanoparticles based on double-drug co-assembly and having phototherapy and chemotherapy functions | Fuzhou University | 2022 |
| CN114949213 | Collagenase functionalized bionic drug-loaded gold nanocage as well as preparation method and application thereof | Zhejiang University | 2022 |
7 Conclusion and perspectives
Bionanotechnology based on camouflaged natural cell membranes may overcome the poor stability, short elimination times, and systemic side effects of unmodified drugs. Due to the specific surface proteins, cancer cell membrane-encapsulation technology achieves safe and efficient therapeutic effects by fully replicating these surface protein structures from cells to NPs, conferring significant immunocompatibility. CCM-NPs can specifically target homogeneous tumor sites. In addition, because cancer cells have an unlimited ability to multiply and do not require complex culture conditions, it is easy to obtain large amounts of cell membranes. CCM-NPs have attracted extensive attention in drug delivery, immunotherapy, vaccine application, and tumor imaging research in recent years, achieving good experimental results. This paper reviews the preparation, characterization, and application of CCM-NPs, the successful development of CCM-NPs demonstrates that cancer cell membranes are high-quality donors and that CCM-NPs are a promising platform for nanomedicine.CCM-NPs have unique biological and interfacial properties, which also pose some challenges to its clinical translation. First, CCM-NPs are usually administered intravenously for anti-tumor therapy. However, nuclear and genetic material in cancer cells may pose a carcinogenic risk if not completely removed, thus inevitably raising concerns among patients about whether the use of CCM-NPs will introduce tumors into the body. In addition, tumor cell lysates are composed of complex components, many of which are endogenous non-tumor associated antigenic substances. On the contrary, cancer cell membranes have a higher proportion of tumor antigens and can more effectively act as a variety of tumor antigens to mimic the cancer-specific immune response.187 Therefore, in the process of preparing CCM-NPs, it is necessary to obtain a high purity of cancer cell membrane. Secondly, various strategies, such as co-extrusion and ultrasound, have been developed to fuse cancer cell membrane with NPs. However, different experimental responses have slightly different parameters, such as the number of extrusions, ultrasound time, and ultrasound frequency. Moreover, the relevant studies are laboratory-based, so the wrapping efficiency at a large production scale cannot be guaranteed. Therefore, there is a need to optimize the preparation process and standards of unified CCM-NPs for large-scale production. Third, the models utilized in most experiments involving CCM-NPs are based on two-dimensional (2D) systems with immortalized cancer cell lines, which hinders the reconstruction of the unique physicochemical properties of the human TME.188 Although in vivo animal models offer a better option for overcoming the limitations of 2D models, they are costly and time-consuming. Additionally, immunosuppressed mice derived from humanized xenografts lack the tumor immune microenvironment and do not fully represent cancer patients.189 Therefore, using advanced research models such as the three-dimensional (3D) organ-on-chip platform to simulate the key structural and functional characteristics of the tumor microenvironment in vivo would be a good choice. These organ-on-a-chip models integrate 3D cell culture, tissue engineering, and microfluidic technologies to replicate the dynamic and pathophysiological response processes of TME in real-time monitoring mode.190,191 This model has been applied to various tumors, enabling the study of tumor development in a more closely aligned TME and providing a more realistic reflection of the dynamic changes of drugs in vivo and their effects on organs.192–195 Furthermore, membrane modification is an effective strategy to develop a multifunctional delivery platform for CCM-NPs. At present, the main strategies for membrane modification include physical, chemical and genetic modification, or fusion with other types of cell membranes. Although significant results have been achieved in related studies, the appropriate reaction conditions should be controlled during cellular functionalization, especially chemical reactions, to prohibit using reagents that impair cellular activity and ensure that the membrane protein activity is not disturbed. Finally, ensuring the CCM-NPs stability is key in clinical conversion. During prolonged storage, the cell membrane lipid components may be exposed to oxidation or CCM-NPs contamination by pyrogens and viruses. Therefore, developing quality standards for CCM-NPs stability or freezing agents that do not destroy the cell membrane components may be helpful.
In conclusion, despite ongoing challenges in achieving clinical translation, CCM-NPs possess undeniable natural advantages and great potential in anti-tumor applications. With continuous research, CCM-NPs will become a promising nano-delivery platform and play an important role in biomedical fields.
Abbreviation
| IARC | International Agency for Research on Cancer |
| NPs | Nanoparticles |
| TME | Tumor microenvironment |
| PEG | Polyethylene glycol |
| RES | Reticuloendothelial system |
| CCM-NPs | Cancer cell membrane-encapsulated NPs |
| CD47 | Cluster of Differentiation 47 |
| SIRPα | Signal-regulated protein alpha |
| TF | Thomsen–Friedenreich |
| DCs | Dendritic cells |
| APCs | Antigen-presenting cells |
| PBS | Phosphate buffered saline |
| FBS | Fetal bovine serum |
| siRNA | Small interfering RNA |
| PLGA | Poly(lactic-co-glycolic acid) |
| MOFs | Metal–organic frameworks |
| UCNPs | Upconversion NPs |
| US | Ultrasound |
| RBC | Red blood cell |
| FNC | Flash nanocomplexation |
| BCA | Bicinchoninic acid |
| TEM | Transmission electron microscopy |
| DLS | Dynamic light scattering |
| DiO | 3,3′-Dioctadecyloxacarbocyanine perchlorate |
| ICG | Indocyanine green |
| SDS-PAGE | Sulfate-polyacrylamide gel electrophoresis |
| DOX | Doxorubicin |
| GOx | Glucose oxidase |
| PTT | Photothermal therapy |
| NIR | Near-infrared |
| ROS | Reactive oxygen species |
| PDT | Photodynamic therapy |
| HSA | Human serum albumin |
| PFTBA | Perfluorotributylamine |
| Ce6 | Chlorine e6 |
| CRET | Chemiluminescence resonance energy transfer |
| CPPO | Bis[2,4,5-trichloro-6-(pentyloxycarbonyl)phenyl] oxalate |
| PFC | Perfluorohexane |
| GSH | Glutathione |
| SDT | Sonodynamic therapy |
| PLA | Poly(lactic acid) |
| HNSs | Hollow nanoshells |
| HMTNPs | Hollow mesoporous titanium dioxide nanoparticles |
| HCQ | Hydroxychloroquine sulfate |
| TPZ | Tirapazamine |
| CDT | Chemodynamic therapy |
| PFP | Perfluoropentane |
| HO-1 | Heme oxygenase-1 |
| ZnPPIX | Zn protoporphyrin IX |
| PA | Photoacoustic |
| MRI | Magnetic resonance imaging |
| SBR | Signal background ratio |
| UCL | Upconversion luminescence |
| PET | Positron emission tomography |
| PFCE | Perfluoro-15-crown-5-ether |
| gp100 | Glycoprotein 100 |
| IFNγ | Interferon-gamma |
| DTX | Docetaxel |
| MNCs | Magnetic nanoclusters |
| OVA | Ovalbumin |
| DiI | 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate |
| FRET | Förster resonance energy transfer |
| OMV | Bacterial outer membrane vesicles |
| HPDA | Hollow polydopamine |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
Author contributions
The manuscript was written through the contributions of all authors. Guo Qiuyan conceived and wrote the review, the production and modification of related figures; Tang Yingnan completed the collation of relevant literature; Wang Shengmei and Xu Rubing made comments and suggestions on the original manuscript; Xia Xinhua revised the original manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the [Natural Science Foundation of China #1] under Grant [number 81573621]; [Key Discipline Project on Chinese Pharmacology of Hunan University of Chinese Medicine #2] under Grant [number 202302]; and [Science and Technology Innovation Program of Hunan Province #3] under Grant [number 2021RC4064].References
- F. Bray, M. Laversanne and E. Weiderpass, et al., The ever-increasing importance of cancer as a leading cause of premature death worldwide, Cancer, 2021, 127(16), 3029–3030 CrossRef PubMed.
- GLOBOCAN, http://gco.iarc.fr/today/home, accessed 17 Mar 2023 Search PubMed.
- R. L. Siegel, K. D. Miller and H. E. Fuchs, et al., Cancer Statistics, Ca-Cancer J. Clin., 2021, 71(1), 7–33 CrossRef PubMed.
- P. McGale, C. Taylor and C. Correa, et al., Effect of radiotherapy after mastectomy and axillary surgery on 10-year recurrence and 20-year breast cancer mortality: meta-analysis of individual patient data for 8135 women in 22 randomised trials, Lancet, 2014, 383(9935), 2127–2135 CrossRef CAS PubMed.
- R. Vinay and V. KusumDevi, Potential of targeted drug delivery system for the treatment of bone metastasis, Drug Delivery, 2016, 23(1), 21–29 CrossRef CAS PubMed.
- Q. Gao, G. Zhou and S. J. Lin, et al., How chemotherapy and radiotherapy damage the tissue: Comparative biology lessons from feather and hair models, Exp. Dermatol., 2019, 28(4), 413–418 CrossRef PubMed.
- X. Zheng, Y. Zhao and Y. Jia, et al., Biomimetic co-assembled nanodrug of doxorubicin and berberine suppresses chemotherapy-exacerbated breast cancer metastasis, Biomaterials, 2021, 271, 120716 CrossRef CAS PubMed.
- N. Amreddy, A. Babu and R. Muralidharan, et al., Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery, Adv. Cancer Res., 2018, 137, 115–170 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai and Z. Dai, et al., Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer, Chem. Soc. Rev., 2019, 48(7), 2053–2108 RSC.
- N. Bertrand, J. Wu and X. Xu, et al., Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- Q. Jiang, Y. Liu and R. Guo, et al., Erythrocyte-cancer hybrid membrane-camouflaged melanin nanoparticles for enhancing photothermal therapy efficacy in tumors, Biomaterials, 2019, 192, 292–308 CrossRef CAS PubMed.
- A. Ahmed, S. Sarwar and Y. Hu, et al., Surface-modified polymeric nanoparticles for drug delivery to cancer cells, Expert Opin. Drug Delivery, 2021, 18(1), 1–24 CrossRef CAS PubMed.
- Y. H. Cheng, C. He and J. E. Riviere, et al., Meta-Analysis of Nanoparticle Delivery to Tumors Using a Physiologically Based Pharmacokinetic Modeling and Simulation Approach, ACS Nano, 2020, 14(3), 3075–3095 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares and Q. Dai, et al., Analysis of nanoparticle delivery to tumours, Nat. Rev. Mater., 2016, 1(5), 1–12 Search PubMed.
- W. Xue, Y. Liu and N. Zhang, et al., Effects of core size and PEG coating layer of iron oxide nanoparticles on the distribution and metabolism in mice, Int. J. Nanomed., 2018, 13, 5719–5731 CrossRef CAS PubMed.
- R. Xu, K. Zhang and J. Liang, et al., Hyaluronic acid/polyethyleneimine nanoparticles loaded with copper ion and disulfiram for esophageal cancer, Carbohydr. Polym., 2021, 261, 117846 CrossRef CAS PubMed.
- J. Wang, L. Zhang and H. Xin, et al., Mitochondria-targeting folic acid-modified nanoplatform based on mesoporous carbon and a bioactive peptide for improved colorectal cancer treatment, Acta Biomater., 2022, 152, 453–472 CrossRef CAS PubMed.
- Y. Wang, Y. Xie and J. Li, et al., Tumor-Penetrating Nanoparticles for Enhanced Anticancer Activity of Combined Photodynamic and Hypoxia-Activated Therapy, ACS Nano, 2017, 11(2), 2227–2238 CrossRef CAS PubMed.
- H. Wang, F. Zhang and H. Wen, et al., Tumor- and mitochondria-targeted nanoparticles eradicate drug resistant lung cancer through mitochondrial pathway of apoptosis, J. Nanobiotechnol., 2020, 18(1), 8 CrossRef CAS PubMed.
- X. Meng, Y. Zhao and B. Han, et al., Dual functionalized brain-targeting nanoinhibitors restrain temozolomide-resistant glioma via attenuating EGFR and MET signaling pathways, Nat. Commun., 2020, 11(1), 594 CrossRef CAS PubMed.
- T. Ishida, X. Wang and T. Shimizu, et al., PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner, J. Controlled Release, 2007, 122(3), 349–355 CrossRef CAS PubMed.
- K. Shiraishi, M. Hamano and H. Ma, et al., Hydrophobic blocks of PEG-conjugates play a significant role in the accelerated blood clearance (ABC) phenomenon, J. Controlled Release, 2013, 165(3), 183–190 CrossRef CAS PubMed.
- V. Mirshafiee, M. Mahmoudi and K. Lou, et al., Protein corona significantly reduces active targeting yield, Chem. Commun., 2013, 49(25), 2557–2559 RSC.
- F. Gu, L. Zhang and B. A. Teply, et al., Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(7), 2586–2591 CrossRef CAS PubMed.
- J. F. Stefanick, J. D. Ashley and T. Kiziltepe, et al., A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes, ACS Nano, 2013, 7(4), 2935–2947 CrossRef CAS PubMed.
- Z. Poon, S. Chen and A. C. Engler, et al., Ligand-clustered “patchy” nanoparticles for modulated cellular uptake and in vivo tumor targeting, Angew. Chem., Int. Ed. Engl., 2010, 49(40), 7266–7270 CrossRef CAS PubMed.
- G. P. Adams, R. Schier and A. M. McCall, et al., High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules, Cancer Res., 2001, 61(12), 4750–4755 CAS.
- C. M. Hu, L. Zhang and S. Aryal, et al., Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(27), 10980–10985 CrossRef CAS PubMed.
- Z. He, Y. Zhang and N. Feng, Cell membrane-coated nanosized active targeted drug delivery systems homing to tumor cells: A review, Mater. Sci. Eng., C, 2020, 106, 110298 CrossRef CAS PubMed.
- M. Zhang, S. Cheng and Y. Jin, et al., Membrane engineering of cell membrane biomimetic nanoparticles for nanoscale therapeutics, Clin. Transl. Med., 2021, 11(2), e292 CrossRef PubMed.
- S. Zalba and T. L. Ten Hagen, Cell membrane modulation as adjuvant in cancer therapy, Cancer Treat. Rev., 2017, 52, 48–57 CrossRef CAS PubMed.
- Z. Chen, P. Zhao and Z. Luo, et al., Cancer Cell Membrane-Biomimetic Nanoparticles for Homologous-Targeting Dual-Modal Imaging and Photothermal Therapy, ACS Nano, 2016, 10(11), 10049–10057 CrossRef CAS PubMed.
- L. Zhu, Y. Zhong and S. Wu, et al., Cell membrane camouflaged biomimetic nanoparticles: Focusing on tumor theranostics, Mater. Today Bio, 2022, 14, 100228 CrossRef CAS PubMed.
- H. Gong, Q. Zhang and A. Komarla, et al., Nanomaterial Biointerfacing via Mitochondrial Membrane Coating for Targeted Detoxification and Molecular Detection, Nano Lett., 2021, 21(6), 2603–2609 CrossRef CAS PubMed.
- X. Z. Liu, Z. J. Wen and Y. M. Li, et al., Bioengineered Bacterial Membrane Vesicles with Multifunctional Nanoparticles as a Versatile Platform for Cancer Immunotherapy, ACS Appl. Mater. Interfaces, 2023, 15(3), 3744–3759 CrossRef CAS PubMed.
- M. H. Kershaw and M. J. Smyth, Immunology. Making macrophages eat cancer, Science, 2013, 341(6141), 41–42 CrossRef CAS PubMed.
- F. Jin, J. Qi and D. Liu, et al., Cancer-cell-biomimetic Upconversion nanoparticles combining chemo-photodynamic therapy and CD73 blockade for metastatic triple-negative breast cancer, J. Controlled Release, 2021, 337, 90–104 CrossRef CAS PubMed.
- Y. Wang, D. Wang and Y. Zhang, et al., Tumor Microenvironment-Adaptive Nanoplatform Synergistically Enhances Cascaded Chemodynamic Therapy, Bioact. Mater., 2023, 22, 239–253 CAS.
- H. Tian, Z. Luo and L. Liu, et al., Cancer cell membrane-biomimetic oxygen nanocarrier for breaking hypoxia-induced chemoresistance, Adv. Funct. Mater., 2017, 27(38), 1703197 CrossRef.
- T. Y. Na, L. Schecterson and A. M. Mendonsa, et al., The functional activity of E-cadherin controls tumor cell metastasis at multiple steps, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(11), 5931–5937 CrossRef CAS PubMed.
- S. K. Khaldoyanidi, V. V. Glinsky and L. Sikora, et al., MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions, J. Biol. Chem., 2003, 278(6), 4127–4134 CrossRef CAS PubMed.
- J. J. Zhang, Y. Lin and H. Zhou, et al., Cell Membrane-Camouflaged NIR II Fluorescent Ag(2) Te Quantum Dots-Based Nanobioprobes for Enhanced In Vivo Homotypic Tumor Imaging, Adv. Healthcare Mater., 2019, 8(14), e1900341 CrossRef PubMed.
- R. H. Fang, C. M. Hu and B. T. Luk, et al., Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery, Nano Lett., 2014, 14(4), 2181–2188 CrossRef CAS PubMed.
- Z. Liu, F. Wang and X. Liu, et al., Cell membrane-camouflaged liposomes for tumor cell-selective glycans engineering and imaging in vivo, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(30), e2022769118 CrossRef CAS PubMed.
- W. L. Liu, M. Z. Zou and T. Liu, et al., Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells, Nat. Commun., 2019, 10(1), 3199 CrossRef PubMed.
- D. Nie, Z. Dai and J. Li, et al., Cancer-Cell-Membrane-Coated Nanoparticles with a Yolk-Shell Structure Augment Cancer Chemotherapy, Nano Lett., 2020, 20(2), 936–946 CrossRef CAS PubMed.
- Y. Wu, R. Zhu and M. Zhou, et al., Homologous cancer cell membrane-camouflaged nanoparticles target drug delivery and enhance the chemotherapy efficacy of hepatocellular carcinoma, Cancer Lett., 2023, 558, 216106 CrossRef CAS PubMed.
- W. Zhang, M. Yu and Z. Xi, et al., Cancer Cell Membrane-Camouflaged Nanorods with Endoplasmic Reticulum Targeting for Improved Antitumor Therapy, ACS Appl. Mater. Interfaces, 2019, 11(50), 46614–46625 CrossRef CAS PubMed.
- Q. Zhao, X. Sun and B. Wu, et al., Construction of homologous cancer cell membrane camouflage in a nano-drug delivery system for the treatment of lymphoma, J. Nanobiotechnol., 2021, 19(1), 8 CrossRef CAS PubMed.
- Y. Qu, B. Chu and X. Wei, et al., Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Multiple Myeloma Based on Bone Marrow Homing, Adv. Mater., 2022, 34(46), e2107883 CrossRef PubMed.
- M. Chen, M. Chen and J. He, Cancer cell membrane cloaking nanoparticles for targeted co-delivery of doxorubicin and PD-L1 siRNA, Artif. Cells, Nanomed., Biotechnol., 2019, 47(1), 1635–1641 CrossRef CAS PubMed.
- J. Gao, F. Wang and S. Wang, et al., Hyperthermia-Triggered On-Demand Biomimetic Nanocarriers for Synergetic Photothermal and Chemotherapy, Adv. Sci., 2020, 7(11), 1903642 CrossRef CAS PubMed.
- Y. Wu, X. Chang and G. Yang, et al., A Physiologically Responsive Nanocomposite Hydrogel for Treatment of Head and Neck Squamous Cell Carcinoma via Proteolysis-Targeting Chimeras Enhanced Immunotherapy, Adv. Mater., 2023, e2210787 CrossRef PubMed.
- B. Qiao, Y. Luo and H. B. Cheng, et al., Artificial Nanotargeted Cells with Stable Photothermal Performance for Multimodal Imaging-Guided Tumor-Specific Therapy, ACS Nano, 2020, 14(10), 12652–12667 CrossRef CAS PubMed.
- C. Xu, W. Liu and Y. Hu, et al., Bioinspired tumor-homing nanoplatform for co-delivery of paclitaxel and siRNA-E7 to HPV-related cervical malignancies for synergistic therapy, Theranostics, 2020, 10(7), 3325–3339 CrossRef CAS PubMed.
- W. Zhang, C. Liu and Z. Liu, et al., A Cell Selective Fluoride-Activated MOF Biomimetic Platform for Prodrug Synthesis and Enhanced Synergistic Cancer Therapy, ACS Nano, 2022, 16(12), 20975–20984 CrossRef CAS PubMed.
- J. Li, D. Huang and R. Cheng, et al., Multifunctional Biomimetic Nanovaccines Based on Photothermal and Weak-Immunostimulatory Nanoparticulate Cores for the Immunotherapy of Solid Tumors, Adv. Mater., 2022, 34(9), e2108012 CrossRef PubMed.
- F. Alexis, E. Pridgen and L. K. Molnar, et al., Factors affecting the clearance and biodistribution of polymeric nanoparticles, Mol. Pharm., 2008, 5(4), 505–515 CrossRef CAS PubMed.
- H. Yang and H. A. Clark, Size-tunable DNA-based micelles for deep tumor penetration, Chem, 2019, 5(7), 1687–1689 CAS.
- N. Hao, L. Li and Q. Zhang, et al., The shape effect of PEGylated mesoporous silica nanoparticles on cellular uptake pathway in Hela cells, Microporous Mesoporous Mater., 2012, 162, 14–23 CrossRef CAS.
- F. Zhao, Y. Zhao and Y. Liu, et al., Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials, Small, 2011, 7(10), 1322–1337 CrossRef CAS PubMed.
- V. Peretz, M. Motiei and C. N. Sukenik, et al., The effect of nanoparticle size on cellular binding probability, J. At. Mol. Phys., 2012, 2012, 404536 Search PubMed.
- F. Lu, S.-H. Wu and Y. Hung, et al., Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles, Small, 2009, 5(12), 1408–1413 CrossRef CAS PubMed.
- Y. L. Su, T. W. Yu and W. H. Chiang, et al., Hierarchically targeted and penetrated delivery of drugs to tumors by size-changeable graphene quantum dot nanoaircrafts for photolytic therapy, Adv. Funct. Mater., 2017, 27(23), 1700056 CrossRef.
- A. Kumar, S. Huo and X. Zhang, et al., Neuropilin-1-targeted gold nanoparticles enhance therapeutic efficacy of platinum (IV) drug for prostate cancer treatment, ACS Nano, 2014, 8(5), 4205–4220 CrossRef CAS PubMed.
- J. E. Zuckerman, C. H. J. Choi and H. Han, et al., Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(8), 3137–3142 CrossRef CAS PubMed.
- S. A. Kulkarni and S.-S. Feng, Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery, Pharm. Res., 2013, 30, 2512–2522 CrossRef CAS PubMed.
- K. Müller, D. A. Fedosov and G. Gompper, Margination of micro-and nano-particles in blood flow and its effect on drug delivery, Sci. Rep., 2014, 4(1), 4871 CrossRef PubMed.
- S. Guo, C. Xu and H. Yin, et al., Tuning the Size, Shape, and Structure of RNA Nanoparticles for Favorable Cancer Targeting and Immunostimulation 1, RNA Nanotechnol. Ther., 2022, 543–562 Search PubMed.
- J. H. Park, G. von Maltzahn and L. Zhang, et al., Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting, Small, 2009, 5(6), 694–700 CrossRef CAS PubMed.
- S. E. Gratton, P. A. Ropp and P. D. Pohlhaus, et al., The effect of particle design on cellular internalization pathways, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(33), 11613–11618 CrossRef CAS PubMed.
- E. Morgan, D. Wupperfeld and D. Morales, et al., Shape matters: gold nanoparticle shape impacts the biological activity of siRNA delivery, Bioconjugate Chem., 2019, 30(3), 853–860 CrossRef CAS PubMed.
- J. K. Ferri and K. J. Stebe, Which surfactants reduce surface tension faster? A scaling argument for diffusion-controlled adsorption, Adv. Colloid Interface Sci., 2000, 85(1), 61–97 CrossRef CAS PubMed.
- L. L. Schramm, E. N. Stasiuk and D. G. Marangoni, 2 Surfactants and their applications, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2003, 99, 3–48 RSC.
- K. Anand, S. Varghese and A. Krishnamoorthy, Role of surfactants on the stability of nano-zinc oxide dispersions, Part. Sci. Technol., 2017, 35, 67–70 CrossRef CAS.
- L. W. C. Ho, Y. Liu and R. Han, et al., Nano-cell interactions of non-cationic bionanomaterials, Acc. Chem. Res., 2019, 52(6), 1519–1530 CrossRef CAS PubMed.
- E. Fröhlich, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int. J. Nanomed., 2012, 5577–5591 CrossRef PubMed.
- A. M. Silva, C. Martins-Gomes and T. E. Coutinho, et al., Soft cationic nanoparticles for drug delivery: Production and cytotoxicity of solid lipid nanoparticles (SLNs), Appl. Sci., 2019, 9(20), 4438 CrossRef CAS.
- H. Cortés, H. Hernández-Parra and S. A. Bernal-Chávez, et al., Non-ionic surfactants for stabilization of polymeric nanoparticles for biomedical uses, Materials, 2021, 14(12), 3197 CrossRef PubMed.
- H. Liao, S. Zhao and H. Wang, et al., Self-assembly of retinoid nanoparticles for melanoma therapy, Int. J. Nanomed., 2019, 7963–7973 CrossRef CAS PubMed.
- Q. Jin, Y. Chen and Y. Wang, et al., Zwitterionic drug nanocarriers: A biomimetic strategy for drug delivery, Colloids Surf., B, 2014, 124, 80–86 CrossRef CAS PubMed.
- E. Drijvers, J. Liu and A. Harizaj, et al., Efficient endocytosis of inorganic nanoparticles with zwitterionic surface functionalization, ACS Appl. Mater. Interfaces, 2019, 11(42), 38475–38482 CrossRef CAS PubMed.
- M. Pinto, V. Silva and S. Barreiro, et al., Brain drug delivery and neurodegenerative diseases: Polymeric PLGA-based nanoparticles as a forefront platform, Ageing Res. Rev., 2022, 79, 101658 CrossRef CAS PubMed.
- W. J. Goh, S. Zou and C. K. Lee, et al., EXOPLEXs: Chimeric Drug Delivery Platform from the Fusion of Cell-Derived Nanovesicles and Liposomes, Biomacromolecules, 2018, 19(1), 22–30 CrossRef CAS PubMed.
- X. Zhen, P. Cheng and K. Pu, Recent Advances in Cell Membrane-Camouflaged Nanoparticles for Cancer Phototherapy, Small, 2019, 15(1), e1804105 CrossRef PubMed.
- J. Estelrich, M. J. Sánchez-Martín and M. A. Busquets, Nanoparticles in magnetic resonance imaging: from simple to dual contrast agents, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- S. Li, W. Jiang and Y. Yuan, et al., Delicately Designed Cancer Cell Membrane-Camouflaged Nanoparticles for Targeted (19)F MR/PA/FL Imaging-Guided Photothermal Therapy, ACS Appl. Mater. Interfaces, 2020, 12(51), 57290–57301 CrossRef CAS PubMed.
- J. X. Cai, J. H. Liu and J. Y. Wu, et al., Hybrid Cell Membrane-Functionalized Biomimetic Nanoparticles for Targeted Therapy of Osteosarcoma, Int. J. Nanomed., 2022, 17, 837–854 CrossRef CAS PubMed.
- Z. Li, H. Cai and Z. Li, et al., A tumor cell membrane-coated self-amplified nanosystem as a nanovaccine to boost the therapeutic effect of anti-PD-L1 antibody, Bioact. Mater., 2023, 21, 299–312 CAS.
- F. Li, T. Chen and F. Wang, et al., Enhanced Cancer Starvation Therapy Enabled by an Autophagy Inhibitors-Encapsulated Biomimetic ZIF-8 Nanodrug: Disrupting and Harnessing Dual Pro-Survival Autophagic Responses, ACS Appl. Mater. Interfaces, 2022, 14(19), 21860–21871 CrossRef CAS PubMed.
- J. Wang, J. Sun and W. Hu, et al., A Porous Au@Rh Bimetallic Core-Shell Nanostructure as an H(2) O(2) -Driven Oxygenerator to Alleviate Tumor Hypoxia for Simultaneous Bimodal Imaging and Enhanced Photodynamic Therapy, Adv. Mater., 2020, 32(22), e2001862 CrossRef PubMed.
- D. Zheng, J. Zhou and L. Qian, et al., Biomimetic nanoparticles drive the mechanism understanding of shear-wave elasticity stiffness in triple negative breast cancers to predict clinical treatment, Bioact. Mater., 2023, 22, 567–587 CAS.
- Y. Wu, X. Chang and G. Yang, et al., A Physiologically Responsive Nanocomposite Hydrogel for Treatment of Head and Neck Squamous Cell Carcinoma via Proteolysis-Targeting Chimeras Enhanced Immunotherapy, Adv. Mater., 2023, 35(12), e2210787 CrossRef PubMed.
- H. Hu, C. Yang and F. Zhang, et al., A Versatile and Robust Platform for the Scalable Manufacture of Biomimetic Nanovaccines, Adv. Sci., 2021, 8(15), 2002020 CrossRef CAS PubMed.
- R. Arrigo, R. Teresi and C. Gambarotti, et al., Sonication-Induced Modification of Carbon Nanotubes: Effect on the Rheological and Thermo-Oxidative Behaviour of Polymer-Based Nanocomposites, Materials, 2018, 11(3), 383 CrossRef PubMed.
- L. Rao, B. Cai and L. L. Bu, et al., Microfluidic Electroporation-Facilitated Synthesis of Erythrocyte Membrane-Coated Magnetic Nanoparticles for Enhanced Imaging-Guided Cancer Therapy, ACS Nano, 2017, 11(4), 3496–3505 CrossRef CAS PubMed.
- X. Ren, S. Yang and N. Yu, et al., Cell membrane camouflaged bismuth nanoparticles for targeted photothermal therapy of homotypic tumors, J. Colloid Interface Sci., 2021, 591, 229–238 CrossRef CAS PubMed.
- Y. Sun, W. Zhai and X. Liu, et al., Homotypic cell membrane-cloaked biomimetic nanocarrier for the accurate photothermal-chemotherapy treatment of recurrent hepatocellular carcinoma, J. Nanobiotechnol., 2020, 18(1), 60 CrossRef CAS PubMed.
- H. Min, J. Wang and Y. Qi, et al., Biomimetic Metal-Organic Framework Nanoparticles for Cooperative Combination of Antiangiogenesis and Photodynamic Therapy for Enhanced Efficacy, Adv. Mater., 2019, 31(15), e1808200 CrossRef PubMed.
- D. Wang, C. Liu and S. You, et al., Bacterial Vesicle-Cancer Cell Hybrid Membrane-Coated Nanoparticles for Tumor Specific Immune Activation and Photothermal Therapy, ACS Appl. Mater. Interfaces, 2020, 12(37), 41138–41147 CrossRef CAS PubMed.
- J. J. Zhang, Y. Lin and H. Zhou, et al., Cell membrane-camouflaged NIR II fluorescent Ag2Te quantum dots-based nanobioprobes for enhanced in vivo homotypic tumor imaging, Adv. Healthcare Mater., 2019, 8(14), 1900341 CrossRef PubMed.
- M. Wu, T. Mei and C. Lin, et al., Melanoma cell membrane biomimetic versatile CuS nanoprobes for homologous targeting photoacoustic imaging and photothermal chemotherapy, ACS Appl. Mater. Interfaces, 2020, 12(14), 16031–16039 CrossRef CAS PubMed.
- Y. Wang, L. Zhang and G. Zhao, et al., Homologous targeting nanoparticles for enhanced PDT against osteosarcoma HOS cells and the related molecular mechanisms, J. Nanobiotechnol., 2022, 20(1), 83 CrossRef CAS PubMed.
- D. Shao, F. Zhang and F. Chen, et al., Biomimetic Diselenide-Bridged Mesoporous Organosilica Nanoparticles as an X-ray-Responsive Biodegradable Carrier for Chemo-Immunotherapy, Adv. Mater., 2020, 32(50), e2004385 CrossRef PubMed.
- X. Huang, L. Chen and Y. Lin, et al., Tumor targeting and penetrating biomimetic mesoporous polydopamine nanoparticles facilitate photothermal killing and autophagy blocking for synergistic tumor ablation, Acta Biomater., 2021, 136, 456–472 CrossRef CAS PubMed.
- M. Wen, Y. Zhao and P. Qiu, et al., Efficient sonodynamic ablation of deep-seated tumors via cancer-cell-membrane camouflaged biocompatible nanosonosensitizers, J. Colloid Interface Sci., 2023, 644, 388–396 CrossRef CAS PubMed.
- X. Liu, Y. Sun and S. Xu, et al., Homotypic Cell Membrane-Cloaked Biomimetic Nanocarrier for the Targeted Chemotherapy of Hepatocellular Carcinoma, Theranostics, 2019, 9(20), 5828–5838 CrossRef CAS PubMed.
- F. Shao, Y. Wu and Z. Tian, et al., Biomimetic nanoreactor for targeted cancer starvation therapy and cascade amplificated chemotherapy, Biomaterials, 2021, 274, 120869 CrossRef CAS PubMed.
- X. Wan, L. Song and W. Pan, et al., Tumor-Targeted Cascade Nanoreactor Based on Metal-Organic Frameworks for Synergistic Ferroptosis-Starvation Anticancer Therapy, ACS Nano, 2020, 14(9), 11017–11028 CrossRef CAS PubMed.
- W. Zhang, C. Zhang and C. Yang, et al., Photochemically-driven highly efficient intracellular delivery and light/hypoxia programmable triggered cancer photo-chemotherapy, J. Nanobiotechnol., 2023, 21(1), 11 CrossRef CAS PubMed.
- Z. Guo, Y. Liu and X. Cheng, et al., Versatile biomimetic cantharidin-tellurium nanoparticles enhance photothermal therapy by inhibiting the heat shock response for combined tumor therapy, Acta Biomater., 2020, 110, 208–220 CrossRef CAS PubMed.
- W. Li, J. Yang and L. Luo, et al., Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death, Nat. Commun., 2019, 10(1), 3349 CrossRef PubMed.
- C. Jiang, H. Cheng and A. Yuan, et al., Hydrophobic IR780 encapsulated in biodegradable human serum albumin nanoparticles for photothermal and photodynamic therapy, Acta Biomater., 2015, 14, 61–69 CrossRef CAS PubMed.
- T. Jin, D. Cheng and G. Jiang, et al., Engineering naphthalimide-cyanine integrated near-infrared dye into ROS-responsive nanohybrids for tumor PDT/PTT/chemotherapy, Bioact. Mater., 2022, 14, 42–51 CAS.
- J. Liu, J. Shi and W. Nie, et al., Recent Progress in the Development of Multifunctional Nanoplatform for Precise Tumor Phototherapy, Adv. Healthcare Mater., 2021, 10(1), e2001207 CrossRef PubMed.
- L. Arellano-Galindo, E. Villar-Alvarez and A. Varela, et al., Hybrid Gold Nanorod-Based Nanoplatform with Chemo and Photothermal Activities for Bimodal Cancer Therapy, Int. J. Mol. Sci., 2022, 23(21), 13109 CrossRef CAS PubMed.
- W. Miao, G. Shim and S. Lee, et al., Structure-dependent photothermal anticancer effects of carbon-based photoresponsive nanomaterials, Biomaterials, 2014, 35(13), 4058–4065 CrossRef CAS PubMed.
- W. Xu, C. Pang and C. Song, et al., Black porous silicon as a photothermal agent and immunoadjuvant for efficient antitumor immunotherapy, Acta Biomater., 2022, 152, 473–483 CrossRef CAS PubMed.
- T. Xu, X. Zhu and L. Yang, et al., Defective transition metal hydroxide-based nanoagents with hypoxia relief for photothermal-enhanced photodynamic therapy, J. Mater. Chem. B, 2021, 9(4), 1018–1029 RSC.
- S. Sharma, N. Shrivastava and F. Rossi, et al., Nanoparticles-based magnetic and photo induced hyperthermia for cancer treatment, Nano Today, 2019, 29, 100795 CrossRef CAS.
- Z. Zhou, N. Jiang and J. Chen, et al., Selectively down-regulated PD-L1 by albumin-phenformin nanoparticles mediated mitochondrial dysfunction to stimulate tumor-specific immunological response for enhanced mild-temperature photothermal efficacy, J. Nanobiotechnol., 2021, 19(1), 375 CrossRef CAS PubMed.
- Z. Jiang, T. Li and H. Cheng, et al., Nanomedicine potentiates mild photothermal therapy for tumor ablation, Asian J. Pharm. Sci., 2021, 16(6), 738–761 CrossRef PubMed.
- G. Zhang, W. Cheng and L. Du, et al., Synergy of hypoxia relief and heat shock protein inhibition for phototherapy enhancement, J. Nanobiotechnol., 2021, 19(1), 9 CrossRef CAS PubMed.
- Y. Zhao, N. Liu and P. Liu, et al., Robust boron nanoplatform provokes potent tumoricidal activities via inhibiting heat shock protein, Asian J. Pharm. Sci., 2022, 17(5), 728–740 CrossRef PubMed.
- X. Shu, Y. Chen and P. Yan, et al., Biomimetic nanoparticles for effective mild temperature photothermal therapy and multimodal imaging, J. Controlled Release, 2022, 347, 270–281 CrossRef CAS PubMed.
- H. Wang, Y. Chao and J. Liu, et al., Photosensitizer-crosslinked in situ polymerization on catalase for tumor hypoxia modulation & enhanced photodynamic therapy, Biomaterials, 2018, 181, 310–317 CrossRef CAS PubMed.
- H. Fang, Y. Gai and S. Wang, et al., Biomimetic oxygen delivery nanoparticles for enhancing photodynamic therapy in triple-negative breast cancer, J. Nanobiotechnol., 2021, 19(1), 81 CrossRef CAS PubMed.
- Z. Yu, P. Zhou and W. Pan, et al., A biomimetic nanoreactor for synergistic chemiexcited photodynamic therapy and starvation therapy against tumor metastasis, Nat. Commun., 2018, 9(1), 5044 CrossRef PubMed.
- D. Yue, X. Cai and M. Fan, et al., An Alternating Irradiation Strategy-Driven Combination Therapy of PDT and RNAi for Highly Efficient Inhibition of Tumor Growth and Metastasis, Adv. Healthcare Mater., 2021, 10(8), e2001850 CrossRef PubMed.
- A. Sharma, J. F. Arambula and S. Koo, et al., Hypoxia-targeted drug delivery, Chem. Soc. Rev., 2019, 48(3), 771–813 RSC.
- A. Ma, H. Chen and Y. Cui, et al., Metalloporphyrin Complex-Based Nanosonosensitizers for Deep-Tissue Tumor Theranostics by Noninvasive Sonodynamic Therapy, Small, 2019, 15(5), e1804028 CrossRef PubMed.
- Y. Zhao, M. Wen and N. Yu, et al., Design and synthesis of cancer-cell-membrane-camouflaged hemoporfin-Cu(9)S(8) nanoagents for homotypic tumor-targeted photothermal-sonodynamic therapy, J. Colloid Interface Sci., 2023, 637, 225–236 CrossRef CAS PubMed.
- M. Yuan, S. Liang and Y. Zhou, et al., A Robust Oxygen-Carrying Hemoglobin-Based Natural Sonosensitizer for Sonodynamic Cancer Therapy, Nano Lett., 2021, 21(14), 6042–6050 CrossRef CAS PubMed.
- N. Yumita, K. Kawabata and K. Sasaki, et al., Sonodynamic effect of erythrosin B on sarcoma 180 cells in vitro, Ultrason. Sonochem., 2002, 9(5), 259–265 CrossRef CAS PubMed.
- T. G. Nguyen Cao, J. H. Kang and J. Y. You, et al., Safe and Targeted Sonodynamic Cancer Therapy Using Biocompatible Exosome-Based Nanosonosensitizers, ACS Appl. Mater. Interfaces, 2021, 13(22), 25575–25588 CrossRef CAS PubMed.
- L. Zhang, H. Yi and J. Song, et al., Mitochondria-Targeted and Ultrasound-Activated Nanodroplets for Enhanced Deep-Penetration Sonodynamic Cancer Therapy, ACS Appl. Mater. Interfaces, 2019, 11(9), 9355–9366 CrossRef CAS PubMed.
- D. G. You, V. G. Deepagan and W. Um, et al., ROS-generating TiO2 nanoparticles for non-invasive sonodynamic therapy of cancer, Sci. Rep., 2016, 6, 23200 CrossRef CAS PubMed.
- B. Geng, J. Hu and Y. Li, et al., Near-infrared phosphorescent carbon dots for sonodynamic precision tumor therapy, Nat. Commun., 2022, 13(1), 5735 CrossRef CAS PubMed.
- F. Qu, P. Wang and K. Zhang, et al., Manipulation of Mitophagy by “All-in-One” nanosensitizer augments sonodynamic glioma therapy, Autophagy, 2020, 16(8), 1413–1435 CrossRef CAS PubMed.
- C. Gao, C. H. T. Kwong and Q. Wang, et al., Conjugation of Macrophage-Mimetic Microalgae and Liposome for Antitumor Sonodynamic Immunotherapy via Hypoxia Alleviation and Autophagy Inhibition, ACS Nano, 2023, 17(4), 4034–4049 CrossRef CAS PubMed.
- Q. Feng, X. Yang and Y. Hao, et al., Cancer Cell Membrane-Biomimetic Nanoplatform for Enhanced Sonodynamic Therapy on Breast Cancer via Autophagy Regulation Strategy, ACS Appl. Mater. Interfaces, 2019, 11(36), 32729–32738 CrossRef CAS PubMed.
- S. Ning, X. Dai and W. Tang, et al., Cancer cell membrane-coated C-TiO(2) hollow nanoshells for combined sonodynamic and hypoxia-activated chemotherapy, Acta Biomater., 2022, 152, 562–574 CrossRef CAS PubMed.
- C. Zhang, W. Bu and D. Ni, et al., Synthesis of Iron Nanometallic Glasses and Their Application in Cancer Therapy by a Localized Fenton Reaction, Angew. Chem., Int. Ed. Engl., 2016, 55(6), 2101–2106 CrossRef CAS PubMed.
- Z. Tang, P. Zhao and H. Wang, et al., Biomedicine Meets Fenton Chemistry, Chem. Rev., 2021, 121(4), 1981–2019 CrossRef CAS PubMed.
- U. S. Srinivas, B. W. Q. Tan and B. A. Vellayappan, et al., ROS and the DNA damage response in cancer, Redox Biol., 2019, 25, 101084 CrossRef CAS PubMed.
- L. H. Fu, Y. Wan and C. Qi, et al., Nanocatalytic Theranostics with Glutathione Depletion and Enhanced Reactive Oxygen Species Generation for Efficient Cancer Therapy, Adv. Mater., 2021, 33(7), e2006892 CrossRef PubMed.
- F. Gao, M. Sun and J. Zhang, et al., Fenton-like reaction and glutathione depletion by chiral manganese dioxide nanoparticles for enhanced chemodynamic therapy and chemotherapy, J. Colloid Interface Sci., 2022, 616, 369–378 CrossRef CAS PubMed.
- H. Gao, Z. Cao and H. Liu, et al., Multifunctional nanomedicines-enabled chemodynamic-synergized multimodal tumor therapy via Fenton and Fenton-like reactions, Theranostics, 2023, 13(6), 1974–2014 CrossRef CAS PubMed.
- P. S. Yang, Y. C. Hsu and J. J. Lee, et al., Heme Oxygenase-1 Inhibitors Induce Cell Cycle Arrest and Suppress Tumor Growth in Thyroid Cancer Cells, Int. J. Mol. Sci., 2018, 19(9), 2502 CrossRef PubMed.
- A. Loboda, A. Jozkowicz and J. Dulak, HO-1/CO system in tumor growth, angiogenesis and metabolism - Targeting HO-1 as an anti-tumor therapy, Vasc. Pharmacol., 2015, 74, 11–22 CrossRef CAS PubMed.
- Y. Zhang, F. Wang and L. Shi, et al., Nanoscale coordination polymers enabling antioxidants inhibition for enhanced chemodynamic therapy, J. Controlled Release, 2023, 354, 196–206 CrossRef CAS PubMed.
- R. Weissleder and M. J. Pittet, Imaging in the era of molecular oncology, Nature, 2008, 452(7187), 580–589 CrossRef CAS PubMed.
- R. Weissleder, Molecular imaging in cancer, Science, 2006, 312(5777), 1168–1171 CrossRef CAS PubMed.
- Z. Wang, X. Wang and J. B. Wan, et al., Optical imaging in the second near infrared window for vascular bioimaging, Small, 2021, 17(43), 2103780 CrossRef CAS PubMed.
- F. Ding, J. Feng and X. Zhang, et al., Responsive optical probes for deep-tissue imaging: Photoacoustics and second near-infrared fluorescence, Adv. Drug Delivery Rev., 2021, 173, 141–163 CrossRef CAS PubMed.
- S. Qu, Q. Jia and Z. Li, et al., Chiral NIR-II fluorescent Ag(2)S quantum dots with stereospecific biological interactions and tumor accumulation behaviors, Sci. Bull., 2022, 67(12), 1274–1283 CrossRef CAS PubMed.
- Y. Chen, W. Meng and M. Chen, et al., Biotin-decorated hollow gold nanoshells for dual-modal imaging-guided NIR-II photothermal and radiosensitizing therapy toward breast cancer, J. Mater. Chem. B, 2023, 11(41), 10003–10018 RSC.
- Q. Ding, J. Zhao and H. Zhang, et al., Enantiomeric NIR-II Emitting Rare-Earth-Doped Ag(2) Se Nanoparticles with Differentiated In Vivo Imaging Efficiencies, Angew. Chem., Int. Ed. Engl., 2022, 61(45), e202210370 CrossRef CAS PubMed.
- Y. Ding, B. Park and J. Ye, et al., Surfactant-Stripped Semiconducting Polymer Micelles for Tumor Theranostics and Deep Tissue Imaging in the NIR-II Window, Small, 2022, 18(6), e2104132 CrossRef PubMed.
- B. D. Zheng, J. Ye and Y. Y. Huang, et al., Phthalocyanine-based photoacoustic contrast agents for imaging and theranostics, Biomater. Sci., 2021, 9(23), 7811–7825 RSC.
- Q. Fu, R. Zhu and J. Song, et al., Photoacoustic Imaging: Contrast Agents and Their Biomedical Applications, Adv. Mater., 2019, 31(6), e1805875 CrossRef PubMed.
- J. Geng, W. Li and L. P. Smaga, et al., Damage-responsive microcapsules for amplified photoacoustic detection of microcracks in polymers, Chem. Mater., 2018, 30(7), 2198–2202 CrossRef CAS.
- S. Li, K. H. Lui and T. H. Tsoi, et al., pH-responsive targeted gold nanoparticles for in vivo photoacoustic imaging of tumor microenvironments, Nanoscale Adv., 2019, 1(2), 554–564 RSC.
- S. Shi, H. Li and X. Zheng, et al., Visualization system based on hierarchical targeting for diagnosis and treatment of hepatocellular carcinoma, Mater. Today Bio, 2022, 16, 100398 CrossRef CAS PubMed.
- A. De la Zerda, C. Zavaleta and S. Keren, et al., Carbon nanotubes as photoacoustic molecular imaging agents in living mice, Nat. Nanotechnol., 2008, 3(9), 557–562 CrossRef CAS PubMed.
- C. Guo, J. Sun and J. Dong, et al., A natural anthocyanin-based multifunctional theranostic agent for dual-modal imaging and photothermal anti-tumor therapy, J. Mater. Chem. B, 2021, 9(36), 7447–7460 RSC.
- C. A. Wood, S. Han and C. S. Kim, et al., Clinically translatable quantitative molecular photoacoustic imaging with liposome-encapsulated ICG J-aggregates, Nat. Commun., 2021, 12(1), 5410 CrossRef CAS PubMed.
- M. Wu, T. Mei and C. Lin, et al., Melanoma Cell Membrane Biomimetic Versatile CuS Nanoprobes for Homologous Targeting Photoacoustic Imaging and Photothermal Chemotherapy, ACS Appl. Mater. Interfaces, 2020, 12(14), 16031–16039 CrossRef CAS PubMed.
- A. Farzin, S. A. Etesami and J. Quint, et al., Magnetic Nanoparticles in Cancer Therapy and Diagnosis, Adv. Healthcare Mater., 2020, 9(9), e1901058 CrossRef PubMed.
- Z. Wu, L. Dai and K. Tang, et al., Advances in magnetic resonance imaging contrast agents for glioblastoma-targeting theranostics, Regener. Biomater., 2021, 8(6), rbab062 CrossRef PubMed.
- J. Li, C. Lin and Y. Zhu, et al., Colorectal cancer cell membrane biomimetic ferroferric oxide nanomaterials for homologous bio-imaging and chemotherapy application, Med. Oncol., 2023, 40(11), 322 CrossRef CAS PubMed.
- H. Fang, M. Li and Q. Liu, et al., Ultra-sensitive Nanoprobe Modified with Tumor Cell Membrane for UCL/MRI/PET Multimodality Precise Imaging of Triple-Negative Breast Cancer, Nano-Micro Lett., 2020, 12(1), 62 CrossRef CAS PubMed.
- L. B. Kennedy and A. K. S. Salama, A review of cancer immunotherapy toxicity, Ca-Cancer J. Clin., 2020, 70(2), 86–104 CrossRef PubMed.
- Y. Yang, Cancer immunotherapy: harnessing the immune system to battle cancer, J. Clin. Invest., 2015, 125(9), 3335–3337 CrossRef PubMed.
- J. Gan, G. Du and C. He, et al., Tumor cell membrane enveloped aluminum phosphate nanoparticles for enhanced cancer vaccination, J. Controlled Release, 2020, 326, 297–309 CrossRef CAS PubMed.
- L. Chen, L. Zhou and C. Wang, et al., Tumor-Targeted Drug and CpG Delivery System for Phototherapy and Docetaxel-Enhanced Immunotherapy with Polarization toward M1-Type Macrophages on Triple Negative Breast Cancers, Adv. Mater., 2019, 31(52), e1904997 CrossRef PubMed.
- L. Rong, Y. Zhang and W. S. Li, et al., Iron chelated melanin-like nanoparticles for tumor-associated macrophage repolarization and cancer therapy, Biomaterials, 2019, 225, 119515 CrossRef CAS PubMed.
- Q. Chen, L. Zhang and L. Li, et al., Cancer cell membrane-coated nanoparticles for bimodal imaging-guided photothermal therapy and docetaxel-enhanced immunotherapy against cancer, J. Nanobiotechnol., 2021, 19(1), 449 CrossRef CAS PubMed.
- R. Yang, J. Xu and L. Xu, et al., Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination, ACS Nano, 2018, 12(6), 5121–5129 CrossRef CAS PubMed.
- P. Zhao, Y. Tian and Y. Lu, et al., Biomimetic calcium carbonate nanoparticles delivered IL-12 mRNA for targeted glioblastoma sono-immunotherapy by ultrasound-induced necroptosis, J. Nanobiotechnol., 2022, 20(1), 525 CrossRef CAS PubMed.
- F. Li, W. Nie and F. Zhang, et al., Engineering Magnetosomes for High-Performance Cancer Vaccination, ACS Cent. Sci., 2019, 5(5), 796–807 CrossRef CAS PubMed.
- M. Wang, Q. Hu and J. Huang, et al., Engineered a dual-targeting biomimetic nanomedicine for pancreatic cancer chemoimmunotherapy, J. Nanobiotechnol., 2022, 20(1), 85 CrossRef CAS PubMed.
- Y. Jiang, N. Krishnan and J. Zhou, et al., Engineered Cell-Membrane-Coated Nanoparticles Directly Present Tumor Antigens to Promote Anticancer Immunity, Adv. Mater., 2020, 32(30), e2001808 CrossRef PubMed.
- D. Dehaini, X. Wei and R. H. Fang, et al., Erythrocyte-Platelet Hybrid Membrane Coating for Enhanced Nanoparticle Functionalization, Adv. Mater., 2017, 29(16), 201606209 CrossRef PubMed.
- D. Wang, H. Dong and M. Li, et al., Erythrocyte-Cancer Hybrid Membrane Camouflaged Hollow Copper Sulfide Nanoparticles for Prolonged Circulation Life and Homotypic-Targeting Photothermal/Chemotherapy of Melanoma, ACS Nano, 2018, 12(6), 5241–5252 CrossRef CAS PubMed.
- L. P. Jahromi and G. Fuhrmann, Bacterial extracellular vesicles: Understanding biology promotes applications as nanopharmaceuticals, Adv. Drug Delivery Rev., 2021, 173, 125–140 CrossRef CAS PubMed.
- S. Li, X. Feng and J. Wang, et al., Multiantigenic nanoformulations activate anticancer immunity depending on size, Adv. Funct. Mater., 2019, 29(49), 1903391 CrossRef CAS.
- M. Kapałczyńska, T. Kolenda and W. Przybyła, et al., 2D and 3D cell cultures–a comparison of different types of cancer cell cultures, Arch. Med. Sci., 2018, 14(4), 910–919 Search PubMed.
- S. Green, M. S. Dam and M. N. Svendsen, Mouse avatars of human cancers: the temporality of translation in precision oncology, Hist. Philos. Life Sci., 2021, 43(1), 27 CrossRef PubMed.
- B. Zhang, A. Korolj and B. F. L. Lai, et al., Advances in organ-on-a-chip engineering, Nat. Rev. Mater., 2018, 3(8), 257–278 CrossRef.
- X. Liu, Q. Su and X. Zhang, et al., Recent advances of organ-on-a-chip in cancer modeling research, Biosensors, 2022, 12(11), 1045 CrossRef CAS PubMed.
- H. L. Lanz, A. Saleh and B. Kramer, et al., Therapy response testing of breast cancer in a 3D high-throughput perfused microfluidic platform, BMC Cancer, 2017, 17, 1–11 CrossRef PubMed.
- S. J. Kerns, C. Belgur and D. Petropolis, et al., Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies, eLife, 2021, 10, e67106 CrossRef CAS PubMed.
- E. Kromidas, A. Geier and A. Weghofer, et al., Immunocompetent PDMS-Free Organ-on-Chip Model of Cervical Cancer Integrating Patient-Specific Cervical Fibroblasts and Neutrophils, Adv. Healthcare Mater., 2023, e2302714 Search PubMed.
- L. Jiang, H. Khawaja and S. Tahsin, et al., Microfluidic-based human prostate-cancer-on-chip, Front. Bioeng. Biotechnol., 2024, 12, 1302223 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |