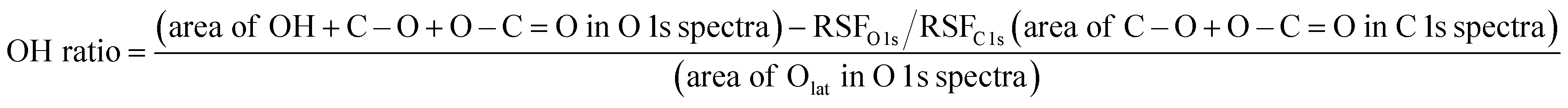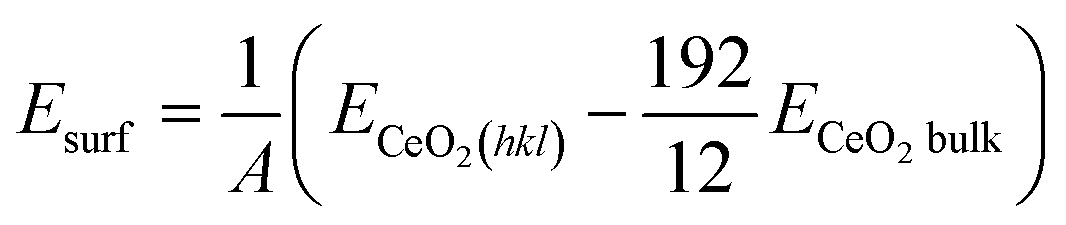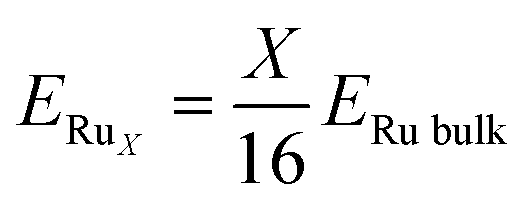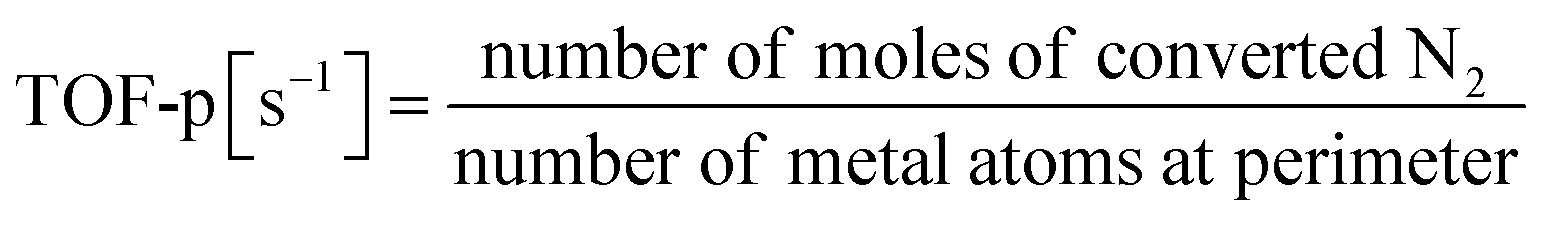DOI:
10.1039/D4RA01457J
(Paper)
RSC Adv., 2024,
14, 9869-9877
Effect of CeO2 support structure on the catalytic performance of ammonia synthesis in an electric field at low temperatures†
Received
25th February 2024
, Accepted 15th March 2024
First published on 25th March 2024
Abstract
Ammonia is an extremely important storage and transport medium for renewable energy, and technology is expected to produce it on demand and onsite using renewable energy. Applying a DC (direct current) to a solid catalyst layer with semiconducting properties makes ammonia synthesis highly efficient, even at low temperatures (approximately 400 K). In this process, oxide supports with semiconducting properties play important roles as metal supports and conduction fields for electrons and protons. The influence of the degree of particle aggregation on the support properties and ammonia synthesis using an electric field was evaluated for CeO2, which is the best material for this purpose because of its semiconducting properties. The results showed that controlling the aggregation structure of the crystalline particles could significantly influence the surface conductivity of protons and electrons; thus, the activity could be largely controlled. The Ru–CeO2 interaction could also be controlled by changing the crystallinity, which suppressed the aggregation of the supported Ru and significantly improved the ammonia synthesis activity using an electric field at low temperatures.
1 Introduction
Recently, hydrogen energy has attracted attention as an alternative energy source to fossil fuels, with the continued research on processes to convert renewable energy into hydrogen energy for storage and transportation.1,2 In particular, ammonia (NH3) is a promising hydrogen carrier because it has a high hydrogen density (17.8 wt%), is carbon-free and is easy to liquefy and handle.3 Because NH3 production using renewable energy must adapt to its temporal and spatial fluctuations, an on-site NH3 production method that can be driven under milder conditions (lower temperature and lower pressure) than that of conventional methods is needed.4 Recently, we have discovered that low-temperature, low-pressure NH3 synthesis can be achieved via a catalytic reaction in an electric field (EF) in which a DC (direct current) is applied to a semiconducting catalyst support.5–8 In this reaction, the applied current induces proton conduction on the surface of the support (surface protonics), which promotes the dissociation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond via proton collisions and intermediate formation, even at low temperatures.5,6 In the NH3 synthesis reaction with an EF via the N2H intermediate, which is formed from the reaction of protons conducted on the support with N2 molecules on the active metal, surface protonics enables a lower reaction temperature. Hydrogen spillover is a similar phenomenon known as proton migration at the catalyst surface, in which protons on the catalyst surface and electrons inside the catalyst are coupled and transferred via a hydrogen concentration gradient on the surface.9–11 In the reaction with an EF, the electrons are forced to move via the applied DC, which can be considered as driving hydrogen spillover.12–15 Thus, the conductivity of protons at the surface of the catalyst support is an important property that affects the reaction with EFs.
N bond via proton collisions and intermediate formation, even at low temperatures.5,6 In the NH3 synthesis reaction with an EF via the N2H intermediate, which is formed from the reaction of protons conducted on the support with N2 molecules on the active metal, surface protonics enables a lower reaction temperature. Hydrogen spillover is a similar phenomenon known as proton migration at the catalyst surface, in which protons on the catalyst surface and electrons inside the catalyst are coupled and transferred via a hydrogen concentration gradient on the surface.9–11 In the reaction with an EF, the electrons are forced to move via the applied DC, which can be considered as driving hydrogen spillover.12–15 Thus, the conductivity of protons at the surface of the catalyst support is an important property that affects the reaction with EFs.
Generally, catalyst supports can regulate the charge of the active metals in the catalytic reaction field and inhibit thermal aggregation owing to the active metal–metal binding.16,17 In the NH3 synthesis reaction, these support properties can be simultaneously expressed with the EF to develop high-performance catalysts. Owing to its oxygen storage, redox, and oxygen ion conductivity, CeO2 is a metal oxide used as a catalyst not only for NH3 synthesis but also for a wide range of reactions, such as automotive exhaust gas purification, water gas shift, steam reforming, and CO oxidation. CeO2 can promote various reactions by changing the molecular adsorption strength and oxygen deficiency concentration by varying the exposure surface.18–30 Although there have been studies on doping heterogeneous cations in CeO2 to change their support properties and improve the performance of NH3 synthesis reactions, no examples of changing the crystalline state of the same oxide to provide new support properties have been reported. In this study, two types of CeO2 (CeO2-p and CeO2-s) with different degrees of crystal aggregation were used. These have almost the same purity but different micro-crystalline structures. These were used to investigate the effect of the support crystal agglomeration on the surface ionic conductivity and ammonia synthesis activity. For the two samples used in this study, CeO2-p had a higher crystal orientation than CeO2-s. The ratio of hydroxyl groups-to-Ce3+ on their surfaces was equal, and the number of spillover carriers was the same. However, AC impedance measurements revealed that CeO2-p had higher proton conductivity than CeO2-s. In addition, CO pulse measurements revealed that CeO2-p had a strong Ru particle–CeO2 interaction, which kept the Ru particles highly dispersed with a longer interface length. Theoretical calculations indicated that this interaction was due to CeO2-p exposing (100) planes with high surface energy. Owing to the concerted effect of high proton conductivity and a longer interface length, CeO2-p was more active than CeO2-s in NH3 synthesis with EFs.
2 Experimental
2.1 Catalyst preparation
All catalysts were supported on two types of CeO2 oxides. CeO2-p, supplied by Mitsui Mining and Smelting Co., Ltd, and CeO2-s, provided by JRC (Japan Reference Catalyst) were used for comparison; both of them have high purity in the CeO2 contents. These two oxides have different migrated crystal structures. Ru particle was loaded onto the two CeO2 particles using an evaporation–drying method with an aqueous solution of Ru(NO3)3, Tanaka Precious Metals. The sample was then stirred 100 rpm for 2 h while deaerating. The sample was then transferred to an evaporating dish, evaporated to dryness on a hot stirrer at about 100 °C and dried in an oven at 120 °C for 24 hours. The resulting powder was treated at 450 °C, 10 °C min−1, 2 h, in H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar = 1:1 (total 100 SCCM). Finally, the catalyst powder was moulded in a pressure moulding machine at 60 kN for 15 min to obtain a particle size of 355–500 μm. As Ru is easily oxidised in air, hydrogen reduction treatment was carried out at 450 °C, 30 min and 10 °C min−1 using hydrogen gas with H2:Ar = 3:1 (Total 240 SCCM) before the activity test.
:Ar = 1:1 (total 100 SCCM). Finally, the catalyst powder was moulded in a pressure moulding machine at 60 kN for 15 min to obtain a particle size of 355–500 μm. As Ru is easily oxidised in air, hydrogen reduction treatment was carried out at 450 °C, 30 min and 10 °C min−1 using hydrogen gas with H2:Ar = 3:1 (Total 240 SCCM) before the activity test.
2.2 Characterisation
The crystal structures of the two CeO2 supports were characterised using powder X-ray diffraction (XRD; Smart Lab III; Rigaku Corp.). Measurements were performed using Cu-Kα radiation at 40 mA and 40 kV. The diffraction peaks in the range 2θ = 25–65° of the sample were measured at a scanning speed of 0.3° min−1. A monochromator was used for the measurements. The average particle size and dispersion of the supported Ru were calculated using CO pulse measurements (BELCAT-II; Microtrac-Bel Corp.). After 50 mg of sample was placed in the sample tube, the reduction was performed under the following conditions: H2:Ar = 3:1 (total 50 mL min−1), 473 or 873 K, and 2 h. The samples were purged with He. The temperature was then lowered to 323 K with He purging (total 50 mL min−1), maintained for 30 min, and stabilised before measurement. The electronic state of Ce on the 3wt%Ru/CeO2-p, and -s, surface and number of surface hydroxyl groups were analysed using X-ray photoelectron spectroscopy (PHI VersaProbe 4; ULVAC-PHI Co.). Charge correction was performed based on Ce 3d at 882.3 eV.31 Before the measurement, a 1 h reduction treatment was conducted in the pre-treatment chamber of the instrument under a flow of N2:H2 = 1:3 (total 100 mL min−1). The samples were transported from the pre-treatment chamber to the measurement chamber without exposure to air. The spectra were measured at ambient temperature and processed to obtain the background spectra using the Proctor–Sherwood–Shirley method.32,33 The surface hydroxyl content was calculated using eqn (1).15| |
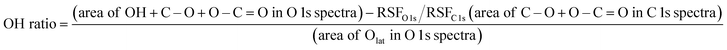 | (1) |
For evaluating surface proton migration ability, the surface ion conductivity was measured using electrochemical impedance spectroscopy (EIS) with a two-electrode, four-wire setup connected via a ZG4 interface to an Alpha-A high-performance frequency analyser (Novocontrol Technologies). Probostat™ (NORECs AS, Norway) sample holder was used for this purpose. All measurements were performed at a flow rate of 40 mL min−1, amplitude of 0.1 V and frequency range of 10 MHz–10 mHz. Before measurements, N2 flowed at 773 K as a pre-treatment to remove the surface-adsorbed species. Pre-treatment was performed until the electrical conductivity attained a steady state. Hydrogen pre-treatment was also performed at 873 K for 12 h at an H2:N2 = 3:1 (total 100 mL min−1). Subsequently, the conductivity was measured at the same gas composition as that in the pre-treatment after a temperature drop to 473 K. The EIS spectra in an H2 atmosphere were measured after the impedance value at a frequency of 1 Hz attained a steady state at the measurement temperature. The discs used for EIS measurements were made of 1.00 g CeO2-p, or -s powder and pressed at 90 kN for 30 min. Following pressing, the discs were pre-sintered at 1173 K and 5 K min−1 for 3 h under an air atmosphere. Subsequently, Pt paste was applied to both sides of the pellets and sintered at 1173 K and 5 K min−1 for 1 h under an air atmosphere. The relative density of the discs obtained was approximately 60% in both cases. These heat treatments for making EIS samples had little effect on their structures of CeO2.
2.3 Theoretical calculations
All ab initio calculations were performed using the Vienna Ab initio Simulation Package (VASP) 6.4.2. The exchange-correlation function was described using the Generalised Gradient Approximation revision Perdew–Burke–Ernzerhof. The electronic states were represented using the projector augmented wave method, and a plane wave basis with a cut-off energy of 400 eV was used for the wave function. The Coulomb interaction of the localised electrons was expressed in Ce 4f with a U parameter of 5 eV. The van der Waals dispersion forces were described using the DFT-D3 of Grimme et al.34–38
All bulk and surface models were structurally optimised under the following conditions: electron occupation was represented using the first-order Methfessel–Paxton method for the Ru bulk and Gaussian method with sigma set to 0.05 for the other models. The k-points were set in reciprocal lattice space, centred at the Γ point every 0.04 Å−1 for the Ru bulk, 5 × 5 × 5 for the CeO2 bulk and 3 × 3 × 1 for all surface models. SCF calculations were performed until the energy difference between successive iterations was <10−8 eV, and structure optimisation was performed until the energy difference between two consecutive structures was <10−5 eV.35–38
For the model construction, first, bulk models of Ru (16 atoms) and CeO2 (12 atoms) were structurally optimised to obtain the lattice constants under these calculation conditions. The structure-optimised CeO2 structure was used to construct a four-layer surface model comprising 192 atoms of CeO2 (111), (110) and (100), introducing a 15 Å vacuum layer in the z-direction. The bottom two layers were fixed and structurally optimised. Cluster(s) of one, four and ten Ru atom(s) were placed on the resulting surface model and fixed to the same atoms for structural optimisation. However, only one Ru atom was considered for CeO2 (110).
All energies were obtained by applying tetrahedral electron occupation with Blöchl's correction to the structure-optimised model for the same k-points as in the structure-optimised SCF calculations, which were performed until the energy difference between successive iterations was <10−6 eV.
The surface (Esurf) and cluster binding (Ebind) energies were calculated as follows:
| |
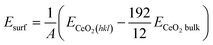 | (2) |
| | |
Ebind = ERuX/CeO2(hkl) − ERuX − ECeO2(hkl)
| (3) |
where
A is the surface index of the CeO
2(
hkl) surface,
ECeO2(hkl) is the energy of the CeO
2(
hkl) surface model,
ECeO2bulk is the energy of the CeO
2 bulk model,
ERuX/CeO2(hkl) is the energy of the CeO
2(
hkl) model carrying the Ru
X atom cluster and
ERuX is the energy of the Ru
X atoms, calculated using the following equation:
| |
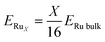 | (4) |
Here,
ERu bulk is the energy of the Ru bulk model and
ERuX is larger than the cluster binding energy because it does not include the surface energy of the Ru cluster itself. Therefore, the positive and negative values of
Ebind did not correspond to the spontaneity of the cluster.
2.4 Activity tests
The activity tests were performed using an atmospheric-pressure fixed-bed flow reactor. A quartz tube (inner diameter: 6 mmϕ, outer diameter: 8 mmϕ) was used for the reaction tube, and a SUS304 rod (diameter: 2 mmϕ) was used for the electrode. The 100 mg catalyst of 3wt%Ru/CeO2-s and 3wt%Ru/CeO2-p was charged in the reactor, and the top and bottom electrodes were inserted to contact the catalyst bed. Moreover, an electric field was applied by passing a DC of 6.0 mA through the catalyst bed with a DC power supply unit. The temperature of the catalyst bed was measured using a thermocouple inserted into the reaction tube, contacting the bottom of the catalyst bed. The response voltage was measured using a digital phosphor oscilloscope (TDS 2001C, Tektronix Inc.). The activity test was conducted under atmospheric pressure, N2:H2 = 1:3 (total 240 mL min−1), thermal catalytic activities were evaluated at 673 K and catalytic activities in the EF were evaluated at 373–473 K. Reduction treatments were performed before each activity test at 723 K for 2 h under the same atmosphere as the activity tests. The ammonia produced was trapped in distilled water, and its concentration was measured using an ion chromatograph (IC-2001; Tosoh Co., Inc.).
3 Results & discussion
3.1 Differences between CeO2-p and CeO2-s crystal structures
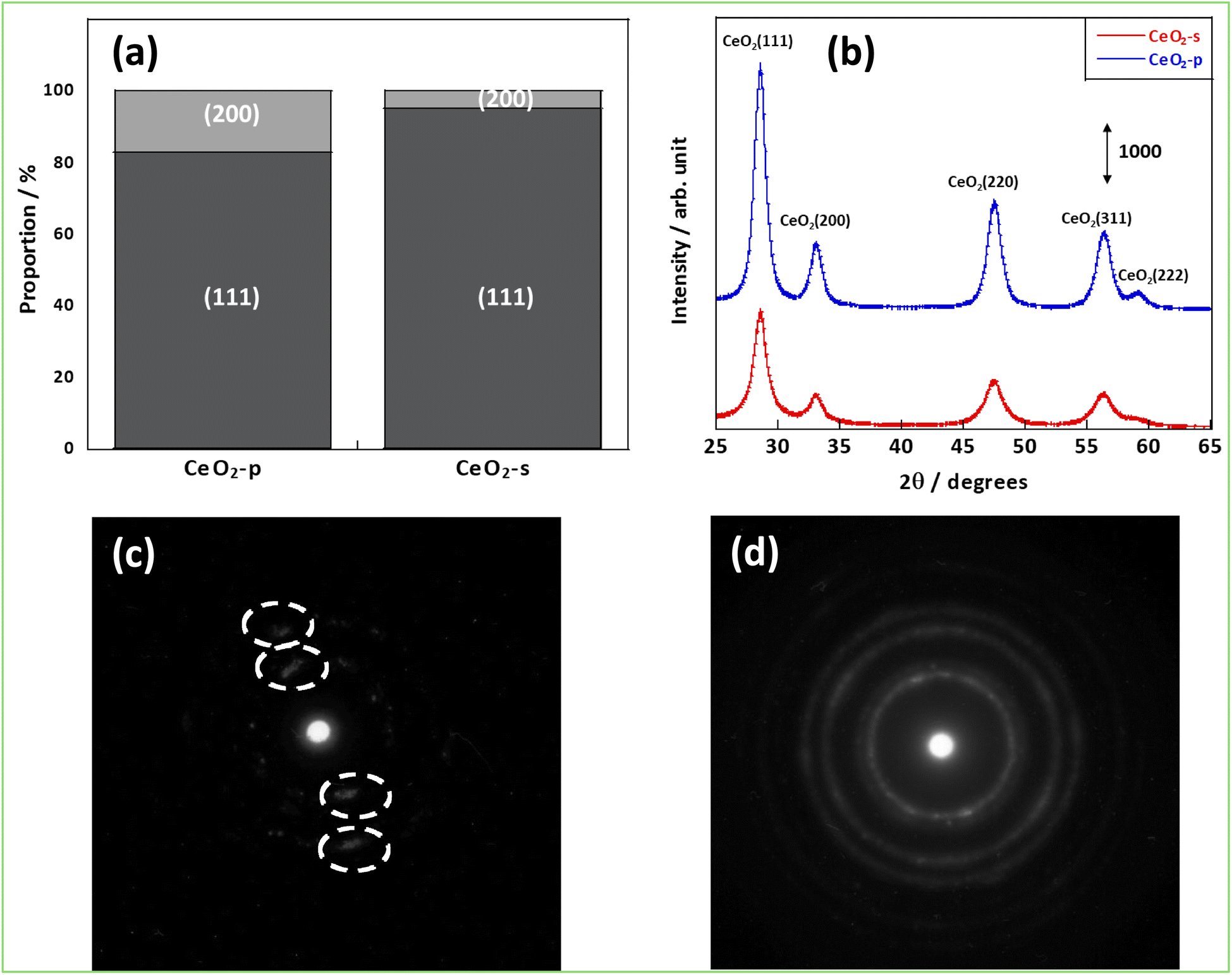
First, the structures of CeO2-p and CeO2-s were evaluated by various methods. Fig. 1(a) shows that the Ce (111) and (200) planes were observed in both samples. Comparing the proportions of these planes between the samples, more Ce (200) planes were exposed on the CeO2-p surface (17.2%) than on the CeO2-s surface. The XRD diffraction spectra were then analysed to confirm each sample's crystallinity and crystal assembly (Fig. 1(b) and Table S1†). No significant peak shifts were observed between the two samples, confirming the absence of crystal stretching caused by the differences in crystallinity. In contrast, focusing on the FWHM, CeO2-p exhibited a higher crystallinity and larger primary crystallite size than CeO2-s. Brunauer–Emmett–Teller surface area measurements demonstrated that CeO2-s had a smaller specific surface area than CeO2-p (Table S2).† Furthermore, the electron diffraction images confirmed that the crystals of CeO2-p were oriented in a specific direction (Fig. 1(c) and (d)). These results suggest that while CeO2-s comprised smaller primary crystallites than CeO2-p, its smaller surface area was due to strong secondary crystallisation.
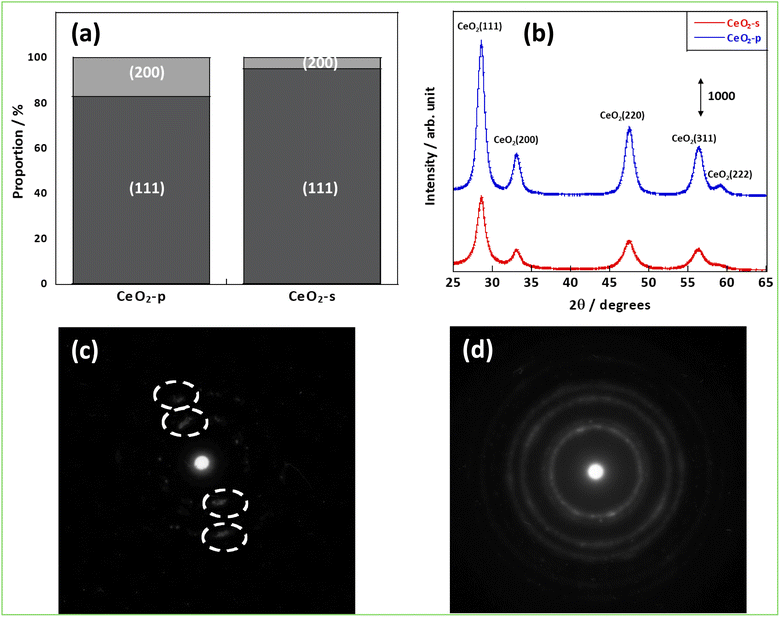 |
| | Fig. 1 Structural analysis results; (a) percentage of surfaces exposed, (b) XRD results, (c) electron diffraction image of CeO2-p, (d) electron diffraction image of CeO2-s. | |
3.2 Evaluation of surface proton conductivity
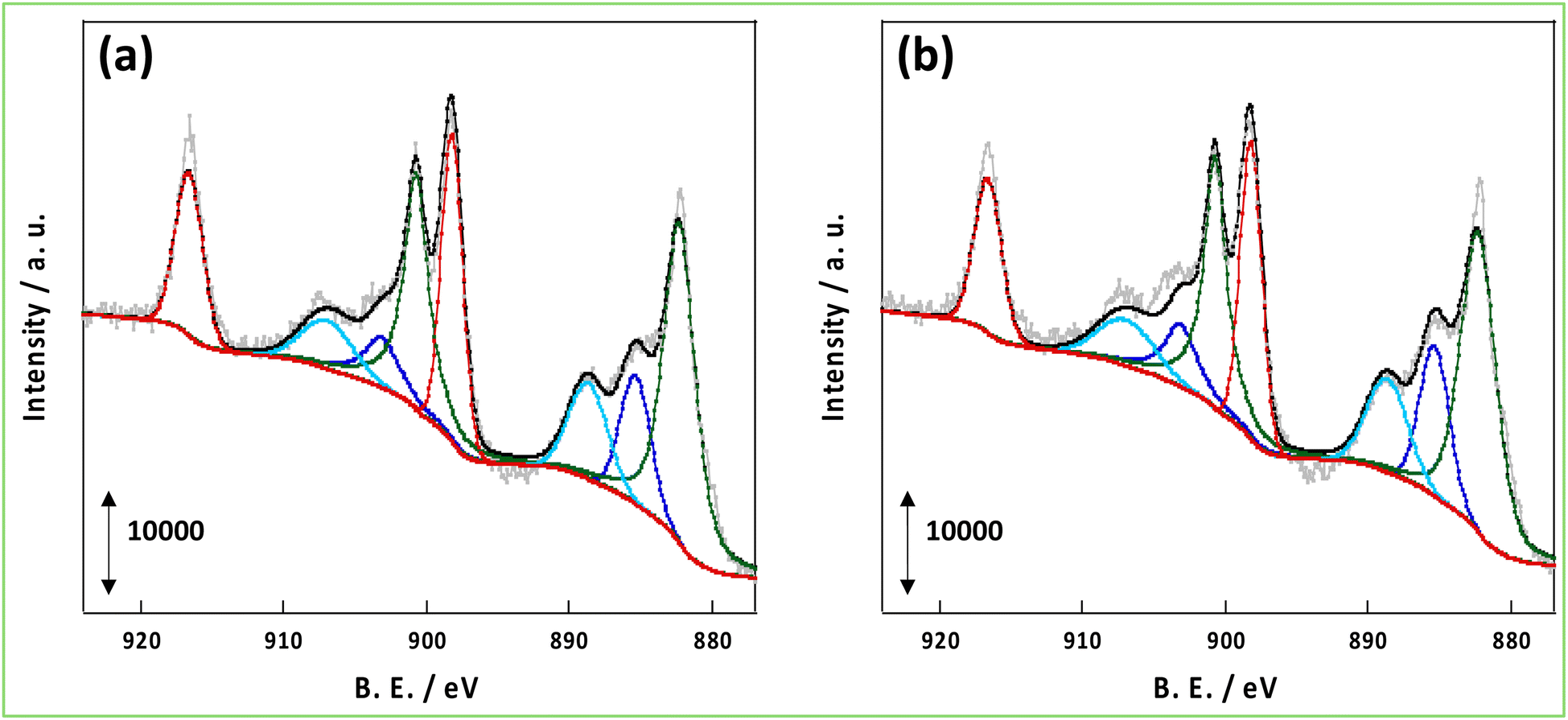
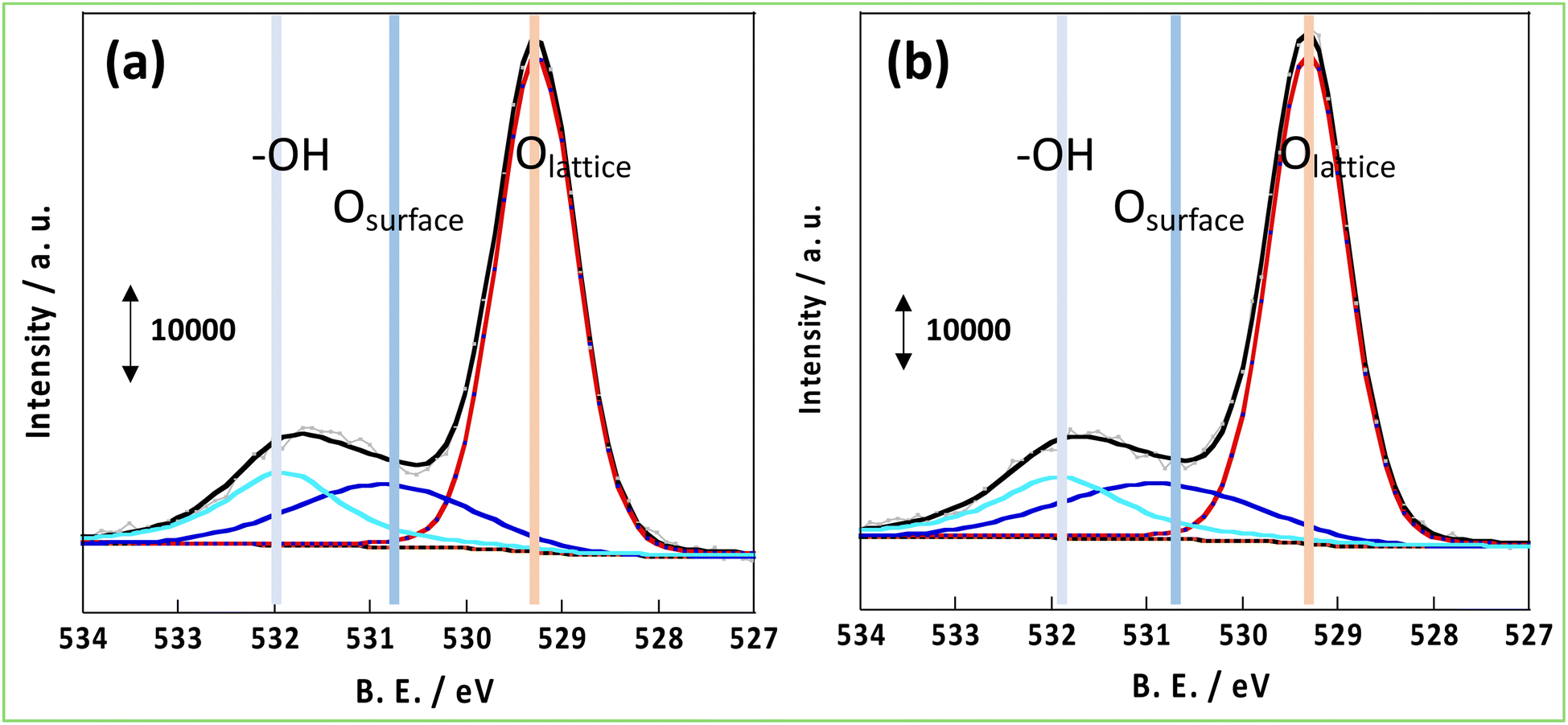
As mentioned in the introduction, hydrogen spillover is thought to be driven by the forced transfer of electrons due to the application of the DC application in the NH3 synthesis reaction with an EF, and the amount and conductivity of protons and electrons are significant. Therefore, these parameters were compared for CeO2-p and CeO2-s. First, to evaluate the number of conduction electrons and H+ carriers on the CeO2-p and CeO2-s surfaces, the XPS spectra of Ce 3d and O 1s were measured following treatment in an NH3 synthetic atmosphere. Moreover, the influence of the degree of crystalline aggregation of the support on the electronic state of CeO2 and the number of surface hydroxyl groups in the reaction atmosphere was investigated. Fig. 2 shows the Ce 3d spectra of each catalyst after the 473 K reduction treatment. The Ce 3d peak shift did not accompany the charge change, and the Ce3+ ratio on the surface remained unchanged, regardless of the degree of crystal assembly (Tables S3 and 4†). The ratio of Ce3+ after the reaction with the EF was also measured, but no change in the electronic state of Ce was observed between the different catalysts (Fig. S3, Tables S5 and 6†). Hydrogen spillover on CeO2 is known to conduct electrons owing to the Ce4+ and Ce3+ redox reactions.39 Therefore, the proportion of Ce3+ corresponded to the carrier concentration of electron conduction at the surface, and this carrier concentration was equal for CeO2-p and CeO2-s. The O 1s spectra showed that the number of surface hydroxyl groups on the two catalysts was similar, so the amount of H+ carriers (i.e. OH groups) did not change with crystallinity (Fig. 3). Therefore, the electrons and protons, which are the species that couple and conduct in the spillover, were aligned in CeO2-p and CeO2-s.
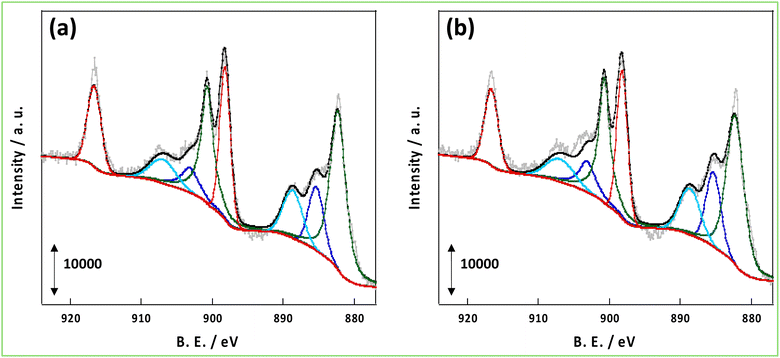 |
| | Fig. 2 XPS spectra of Ce 3d after pre-treatment at 473 K; (a) 3wt%Ru/CeO2-p, (b) 3wt%Ru/CeO2-s. | |
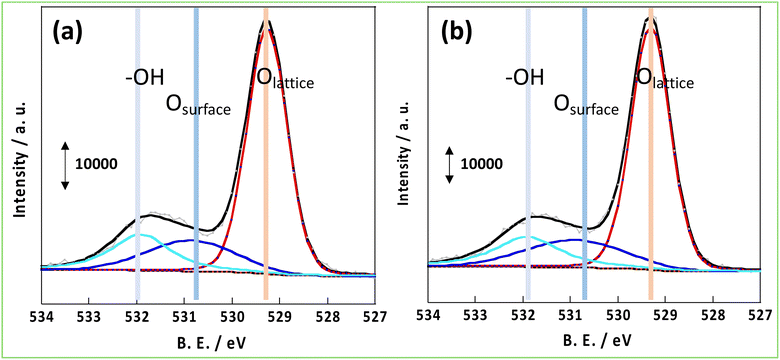 |
| | Fig. 3 XPS spectrum of O 1s after pre-treatment at 473 K; (a) 3wt%Ru/CeO2-p, (b) 3wt%Ru/CeO2-s. | |
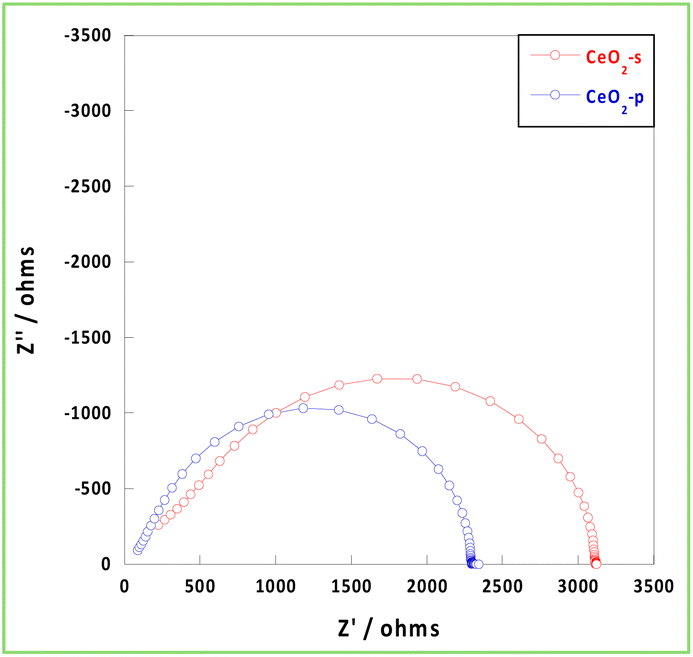
The conductivity of carriers, such as electrons and protons, in the presence of an EF within an NH3 synthetic atmosphere (473 K, H2/N2 = 3/1, total 40 mL min−1) was then investigated using EIS measurements. Generally, hydrogen diffusivity in CeO2 is low, and CeO2 requires considerably high temperatures (>1050 K) to exhibit bulk carrier conduction.40,41 Because the measurement temperature was 473 K, the bulk conduction of hydrogen did not occur, and the carriers were conducted on the surface and grain boundaries via a hopping mechanism. Because the diameter of the circle in the Nyquist plot of the EIS measurement corresponds to the resistivity component, the ionic conductivity of CeO2-p was relatively high at 473 K and under a hydrogen atmosphere (Fig. 4). The resistivity of the grain boundary was higher than that of the bulk material. The inhibition of conduction was more pronounced at low temperatures, as observed in the present study. Space charge layers at the grain boundaries increase the grain boundary resistivity and inhibit ion transport.42,43 The stability of excess charge localisation on the CeO2 surface depended on the face index of the exposed surface.44 Therefore, the more uniform the exposed surfaces and surface orientation of CeO2, the higher the probability of contact between identical surfaces at the grain boundaries. This suppresses the formation of space charge layers and reduces grain boundary resistance. Therefore, CeO2-p exhibited a relatively higher conductivity than CeO2-s because of the alignment of the exposed surfaces and orientations. Thus, the conductivity can be improved by controlling the crystallinity even using the same pure material.
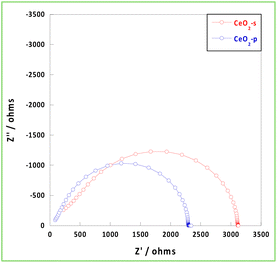 |
| | Fig. 4 Nyquist plot at 473 K under NH3 synthetic atmosphere. | |
3.3 Effect of support structures on CeO2–Ru interaction
The catalyst support binds the impregnated metal as a heterogeneous catalyst, thereby inhibiting the aggregation of active metals during the reaction is crucial for keeping higher dispersion of supported metal. To evaluate the influence of the crystallinity of each support on the support–metal interaction, CO pulse measurements were performed, and the dispersion and average particle size of Ru were observed. Table 1 shows that the Ru particle size in CeO2-s increased nearly five-fold when the pretreatment temperature was increased from 473 to 873 K, whereas in CeO2-p, the increase was approximately two. This indicates that the binding strength of the Ru particles differed between CeO2-s and CeO2-p. The energy of a solid surface is strongly related to particle binding. Therefore, the surface energies and binding energies of the Ru clusters were calculated from first principles for the (111) and (100) surfaces, which were observed in the previous section, and for the (110) surface, which is known as a metastable surface. The results show that the surface energies were of the same order as those previously reported: (111) < (110) < (100). Furthermore, the binding energies of the clusters of one Ru atom on these surfaces were in the order (100) < (111) < (110). Thus, the (100) surface was more Ru-bound than the (111) surface, and CeO2-s was less likely to aggregate Ru particles than CeO2-p because of the higher proportion of (100) surfaces. Furthermore, the (110) surface energy, which was lower than that of the (111) surface, exhibited weaker binding than the (111) surface, indicating that CeO2-s with a (100) surface, instead of a (110) surface, was important for suppressing aggregation. The binding energies were also determined for Ru 4- and 10-atom clusters to investigate the tendency of multiple Ru and O atoms on the surface to bind. The results showed that the trend towards the stronger binding of Ru clusters on the (100) surface than on the (111) surface was maintained, even when the number of binding sites increased (Table 2).
Table 1 Ru particle size of each catalyst
| Sample |
Pretreatment temperature/K |
Metal dispersion/% |
Ru particle size/nm |
| 3wt%Ru/CeO2-s |
473 |
52.9 |
2.54 |
| 873 |
11.4 |
11.8 |
| 3wt%Ru/CeO2-p |
473 |
88.1 |
1.52 |
| 873 |
43.9 |
3.05 |
Table 2 Binding energies of Ru atoms and clusters on CeO2 surfaces
| Miller index |
Surface energies/eV Å−2 |
EbindRu1/eV |
EbindRu4/eV |
EbindRu10/eV |
| (111) |
0.12 |
1.69 |
9.15 |
19.6 |
| (110) |
0.18 |
2.01 |
— |
— |
| (100) |
0.25 |
0.8 |
5.01 |
16.4 |
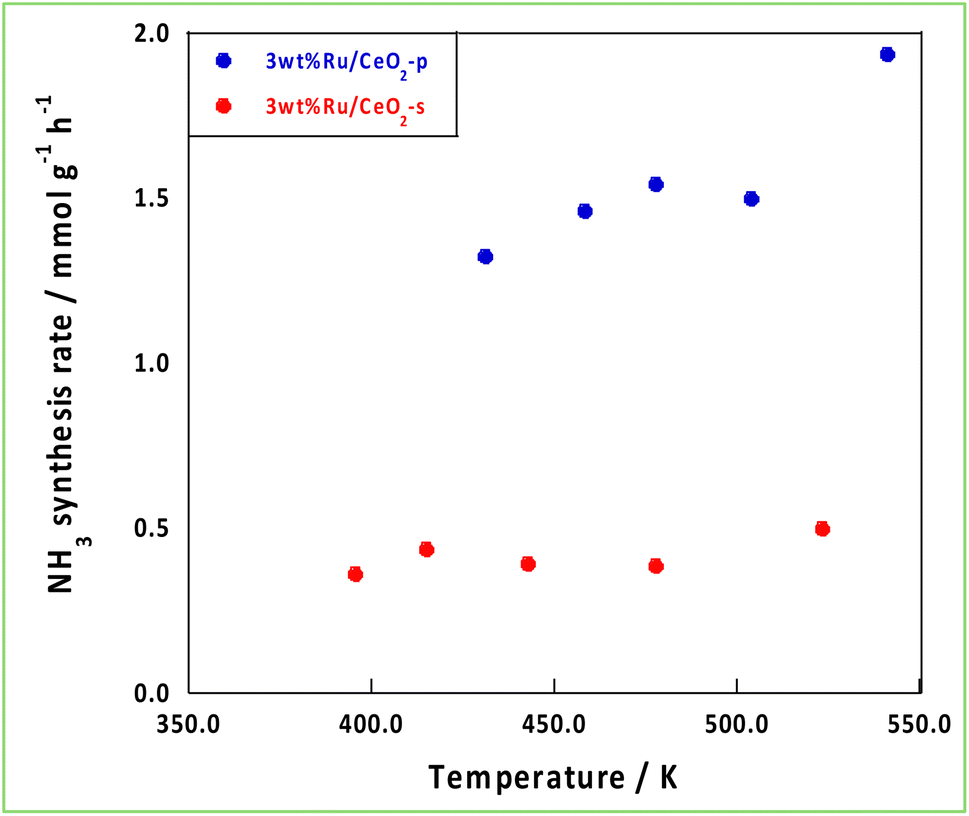
3.4 Impact of support effects on NH3 synthesis with an EF
Based on the characterisation and theoretical calculations described above, improved ionic conductivity and strong CeO2–Ru interactions were achieved by controlling the crystalline assembly of the supports. The catalytic performances of these supports were evaluated by performing NH3 synthesis using an DC-EF. Fig. 5 shows the NH3 synthesis activity in the temperature-change tests in an electric field. Based on these results, NH3 synthesis activity with an EF was enhanced when CeO2-p was used as the catalyst support. The turnover frequency normalised to the metal perimeter (TOF-p), which indicates the catalytic performance at the active sites situated at the metal–support interface, was calculated (eqn (5)). CeO2-p was superior to CeO2-s in NH3 synthesis with an EF because of its high ionic conductivity, even when the effect of Ru was excluded (Table 3).| |
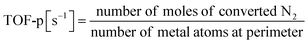 | (5) |
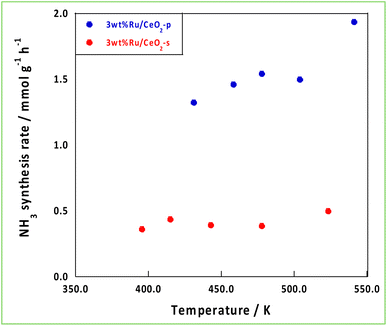 |
| | Fig. 5 Temperature variation test in the EF. | |
Table 3 Comparison of NH3 synthesis activity in the EF and TOF-p
| Sample |
Temperature/K |
Response voltage/kV |
NH3 synthesis rate/mmol g−1 h−1 |
NH3 synthesis rate/mmol g−1 h−1 W−1 |
TOF-p/s−1 |
| 3wt%Ru/CeO2-s |
470 |
0.168 |
0.390 |
0.387 |
0.00832 |
| 3wt%Ru/CeO2-p |
478 |
0.389 |
1.54 |
0.660 |
0.0118 |
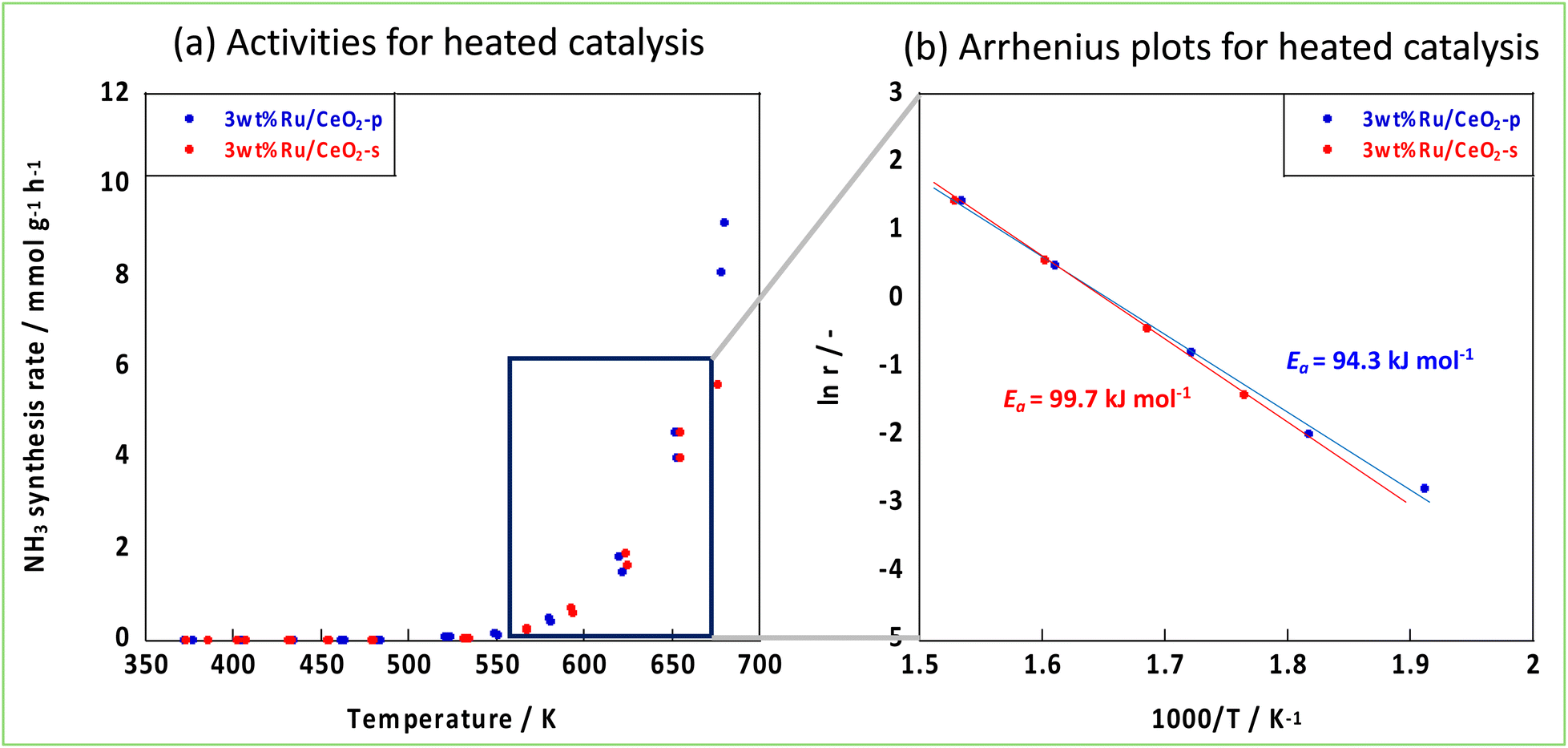
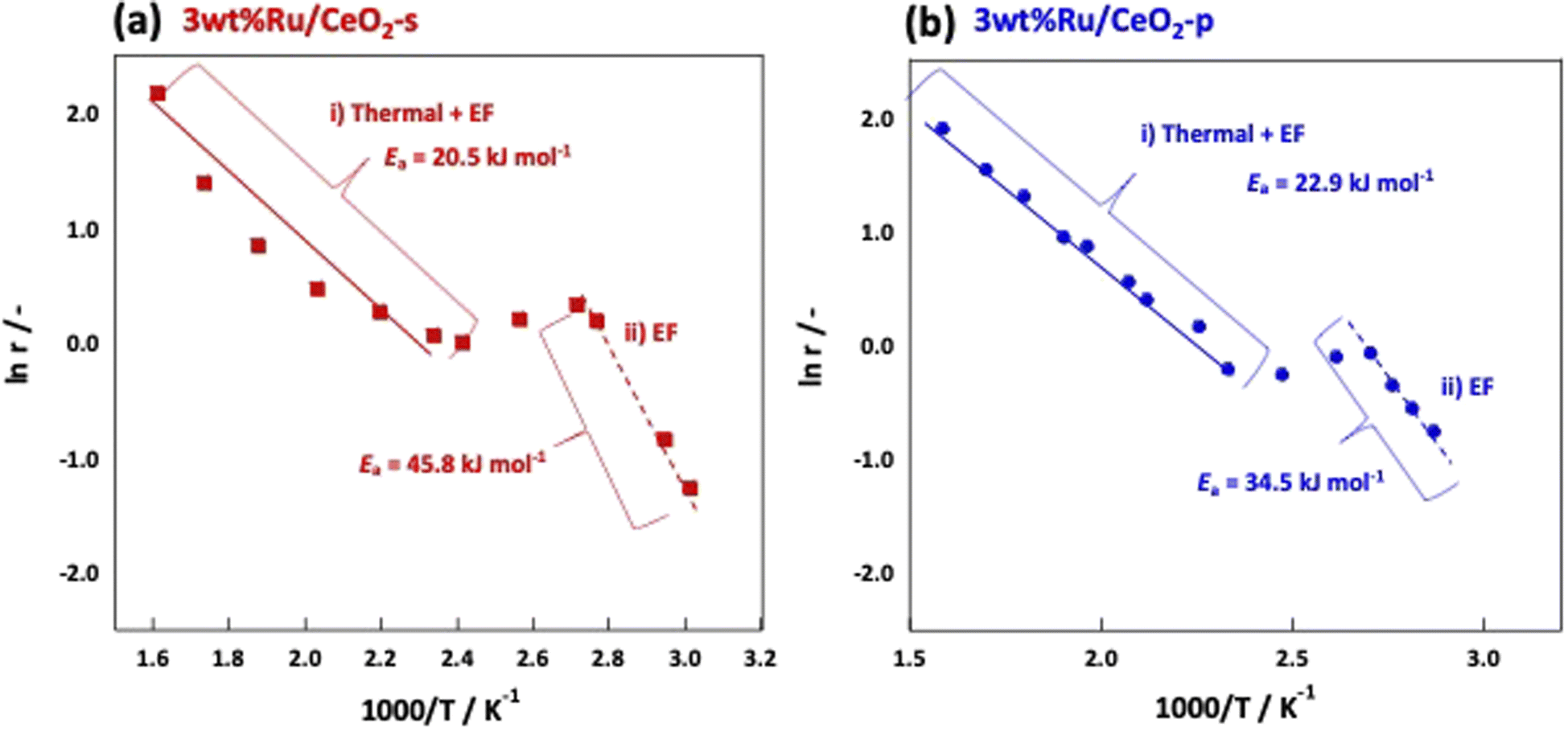
The NH3 reaction with an EF proceeded via a mechanism different from that of the conventional thermal reaction, and the reaction proceeded in parallel with the thermal reaction when an EF was applied. Therefore, to visualise the effect of an EF, we considered both the EF and thermal reactions. Arrhenius plots were made based on the catalytic performances obtained from the catalytic reactions in an electric field and thermal reactions in the temperature range where the thermal reaction was not dominant. Subsequently, the activation energies were compared. Fig. 6 shows the Arrhenius plots generated by the temperature change during the tests. These results demonstrate that the apparent activation energies of the two catalyses were comparable when the effect of the EF was eliminated. However, in the presence of an EF, the Arrhenius plots revealed that CeO2-p proceeded with a lower apparent activation energy than did CeO2-s in the specified temperature range (Fig. 7). This result clearly indicates that the ionic conductivity obtained by controlling the crystal assembly lowered the activation barrier for NH3 synthesis with the EF. Fig. 7 also suggests that the temperature range at which switching from the EF reaction to the thermal reaction is similar for the two catalyses, and the surface hydroxyl groups are uniform during the reaction. As observed in the activity and Arrhenius plots, controlling the crystal assembly improved the surface ionic conductivity and enhanced the formation of N2H intermediates, thus increasing the activity per active site of the reaction and lowering the apparent activation energy.
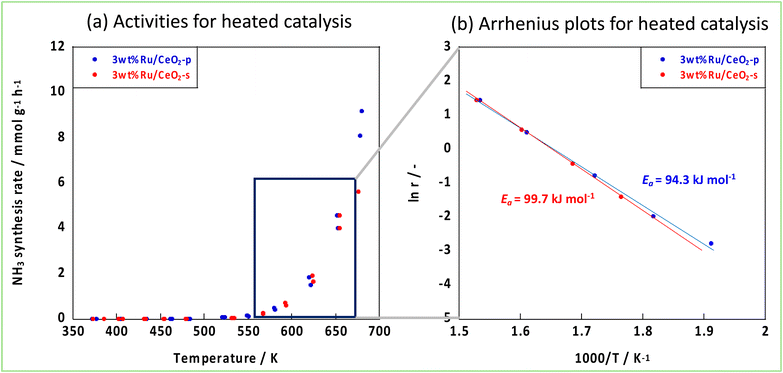 |
| | Fig. 6 Activities of two Ru-catalysts supported on different CeO2 supports, (a) activities for heated catalysis, (b) Arrhenius plots. | |
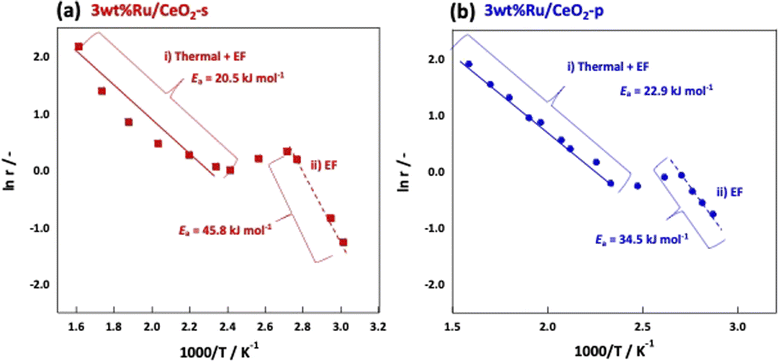 |
| | Fig. 7 Arrhenius plots when an electric field is applied to (a) 3wt%Ru/CeO2-s, (b) 3wt%Ru/CeO2-p. | |
4 Conclusion
Herein, the influences of the degree of crystal aggregation of CeO2 on the support properties and NH3 synthesis activity of an EF were investigated. CeO2-p had a high crystal orientation, and numerous CeO2 (100) planes were exposed. CeO2-p had a high ion conductivity, and it may be attributed to the reduced grain boundary resistance owing to the highly crystalline orientation. This result indicates that the trade-off between ion-conducting carrier quantity and mobility in surface proton conduction under an EF, which can be regarded as current-induced hydrogen spillover, can be promoted by controlling the crystal assembly. Furthermore, Ru aggregation was controlled by suppressing the secondary crystallisation of CeO2. This phenomenon is attributed to the strong binding of Ru to the CeO2 (100) surface, which was exposed in large numbers to the CeO2 crystalline surface. The high proton conductivity and strong Ru–CeO2 interactions obtained by suppressing crystal aggregation resulted in an increase in the activity of NH3 synthesis in an EF at low temperatures.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
All the catalysts used in this study were prepared and supplied by Mitsui Mining and Smelting Co., Ltd. Electron diffraction observations were conducted using equipment (JEM-2100F: Material Characterization Central Laboratory in Waseda University) shared with the MEXT Project for Promoting Public Utilization of Advanced Research Infrastructure (Program for supporting the construction of core facilities) under grant numbers JPMXS0440500022 and JPMXS0440500023. This study was partly conducted using a supercomputer system at the Information Initiative Centre, Hokkaido University, Sapporo, Japan.
References
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- R. H. Lin, Y. Y. Zhao and B. D. Wu, Int. J. Hydrogen Energy, 2020, 45, 20164–20175 CrossRef CAS.
- Y. Sekine and T. Higo, Top. Catal., 2021, 64, 470–480 CrossRef CAS.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- K. Murakami, R. Manabe, H. Nakatsubo, T. Yabe, S. Ogo and Y. Sekine, Catal. Today, 2018, 303, 271–275 CrossRef CAS.
- K. Murakami, Y. Tanaka, S. Hayashi, R. Sakai, Y. Hisai, Y. Mizutani, A. Ishikawa, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki, H. Nakai and Y. Sekine, J. Chem. Phys., 2019, 151, 064708 CrossRef.
- K. Murakami, Y. Tanaka, R. Sakai, Y. Hisai, S. Hayashi, Y. Mizutani, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki and Y. Sekine, Chem. Commun., 2020, 56, 3365–3368 RSC.
- K. Murakami and Y. Sekine, Phys. Chem. Chem. Phys., 2020, 22, 22852 RSC.
- K. Shun, K. Mori, S. Masuda, N. Hashimoto, Y. Hinuma, H. Kobayashi and H. Yamashita, Chem. Sci., 2022, 13, 8137–8147 RSC.
- W. Karim, A. Kleibert, U. Hartfelder, A. Balan, J. Gobrecht, J. A. van Bokhoven and Y. Ekinci, Sci. Rep., 2016, 6, 18818 CrossRef CAS PubMed.
- R. Sakai, K. Murakami, Y. Mizutani, Y. Tanaka, S. Hayashi, A. Ishikawa, T. Higo, S. Ogo, H. Tsuneki, H. Nakai and Y. Sekine, ACS Omega, 2020, 5(12), 6846–6851 CrossRef CAS PubMed.
- Y. Tanaka, K. Murakami, S. Doi, K. Ito, K. Saegusa, Y. Mizutani, S. Hayashi, T. Higo, H. Tsuneki, H. Nakai and Y. Sekine, RSC Adv., 2021, 11, 7621–7626 RSC.
- K. Murakami, Y. Mizutani, H. Sampei, A. Ishikawa and Y. Sekine, J. Chem. Phys., 2021, 154, 164705 CrossRef CAS PubMed.
- K. Murakami, Y. Mizutani, H. Sampei, A. Ishikawa, Y. Tanaka, S. Hayashi, S. Doi, T. Higo, H. Tsuneki, H. Nakai and Y. Sekine, Phys. Chem. Chem. Phys., 2021, 23, 4509–4516 RSC.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- J. G. Nunan, H. J. Robota, M. J. Cohn and S. A. Bradley, J. Catal., 1992, 133, 309–324 CrossRef CAS.
- D. Andreeva, V. Idakiev, T. Tabakova, L. Ilieva, P. Falaras, A. Bourlinos and A. Travlos, Catal. Today, 2002, 72, 51–57 CrossRef CAS.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108, 20240–20246 CrossRef CAS.
- I. I. Soykal, H. Sohn and U. S. Ozkan, ACS Catal., 2012, 2, 2335–2348 CrossRef CAS.
- I. I. Soykal, B. Bayrama, H. Sohna, P. Gawadea, J. T. Miller and U. S. Ozkan, Appl. Catal., A, 2012, 449, 47–58 CrossRef CAS.
- S. Carrettin, P. Concepcion, A. Corma, J. M. L. Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- Q. Li, Y. Zhang, G. Chen, J. Fan, H. Lan and Y. Yang, J. Catal., 2010, 273, 167–176 CrossRef CAS.
- C. Li, Y. Shi, Z. Zhang, J. Ni, X. Wang, J. Lin, B. Lin and L. Jiang, J. Energy Chem., 2021, 60, 403–409 CrossRef CAS.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- M. Nolan and G. W. Watson, J. Phys. Chem. B, 2006, 110, 16600–16606 CrossRef CAS PubMed.
- B. Lin, Y. Liu, L. Heng, X. Wang, J. Ni, J. Lin and L. Jiang, Ind. Eng. Chem. Res., 2018, 57, 9127–9135 CrossRef CAS.
- Z. Ma, S. Zhao, X. Pei, X. Xionga and B. Hu, Catal. Sci. Technol., 2017, 7, 191 RSC.
- N. Nakano, M. Torimoto, H. Sampei, R. Yamashita, R. Yamano, K. Saegusa, A. Motomura, K. Nagakawa, H. Tsuneki, S. Ogo and Y. Sekine, RSC Adv., 2022, 12, 9036–9043 RSC.
- A. Proctor and P. M. A. Sherwood, Anal. Chem., 1982, 54(1), 13–19 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B, 1972, 5(12), 4709 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. D. Krcha, A. D. Mayernick and M. J. Janik, J. Catal., 2012, 293, 103–115 CrossRef CAS.
- H. T. Chen and J. G. Chang, J. Chem. Phys., 2010, 132, 214702 CrossRef PubMed.
- A. D. Mayernick and M. J. Janik, J. Phys. Chem. C, 2008, 112(38), 14955–14964 CrossRef CAS.
- M. B. Watkins, A. S. Foster and A. L. Shluger, J. Phys. Chem. C, 2007, 111(42), 15337–15341 CrossRef CAS.
- A. Beck, D. Kazazis, Y. Ekinci, X. Li, E. A. M. Gubler, A. Kleibert, M. G. Willinger, L. Artiglia and J. A. van Bokhoven, ACS Nano, 2023, 17, 1091–1099 CrossRef CAS PubMed.
- Y. Nigari, K. Kawamura, T. Kawada, J. Mizusaki and M. Ishigame, J. Electrochem. Soc., 1999, 146, 2948–2953 CrossRef.
- T. Matsuda, R. Ishibashi, Y. Koshizuka, H. Tsuneki and Y. Sekine, Chem. Commun., 2022, 58, 10789–10792 RSC.
- X. Guo and J. Maier, J. Electrochem. Soc., 2001, 148, E121 CrossRef CAS.
- X. Guo, W. Sigle and J. Maier, J. Am. Ceram. Soc., 2003, 86, 77–87 CrossRef CAS.
- P. P. Bailac, P. G Lustemberg and M. V. G. Pirovano, J. Phys.: Condens. Matter, 2021, 33, 504003 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Reika Nakayamaa,
Takuma Higo
a,
Reika Nakayamaa,
Takuma Higo