
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis and evaluation of novel ALK inhibitors containing the sulfoxide structure†
Han Yao,
Yuanyuan Ren,
Feng Wu,
Jiadai Liu,
Longcai Cao,
Ming Yan * and
Xingshu Li*
* and
Xingshu Li*
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: lixsh@mail.sysu.edu.cn
First published on 31st May 2024
Abstract
With ceritinib as the lead, a series of novel compounds containing the sulfoxide structure were synthesized and evaluated as anaplastic lymphoma kinase inhibitors. Among them, compounds 18a–d exhibited excellent anti-proliferation activities on H2228 EML4-ALK cancer cell lines with 14–28 nM of the IC50 values. In xenograft mouse models, 18a–d inhibited tumor growth with an excellent inhibitory rate of 75.0% to 86.0% at the dosage of 20 mg kg−1 as compared to 72.0% of the reference ceritinib. Using 18d as a representative, which exhibited the best in vivo results, we carried out mechanistic studies such as anti-colony formation, induced tumor cell apoptosis, ALK kinase protein phosphorylation in H2228 tumor cells, and molecular docking. All these results indicate that compound 18d is a good anti-tumor lead compound and worthy of further study.
Introduction
Anaplastic lymphoma kinase (ALK) is a highly conserved tyrosine receptor kinase (TRK).1 Research shows that the abnormal expression of ALK, such as gene mutation, rearrangement, and amplification, is closely related to a variety of human malignant tumors (such as neuroblastoma and non-small cell lung cancer).2,3 In patients with non-small cell lung cancer (NSCLC), about 5% are accompanied by ALK and EML4 (echinoderm microtubule-associated protein-like 4) gene fusion mutation, which activates ALK and multiple downstream signal pathways, resulting in rapid cell proliferation and differentiation, finally leading to the occurrence of tumors.4,5 Thus, ALK is one of the hot targets in anti-tumor drug development. In 2011, the first drug for NSCLC treatment with ALK-positive crizotinib was approved by FDA.6 Subsequently, second-generation ALK inhibitors with stronger specificity and affinity for ALK kinase, such as ceritinib7 and brigatinib,8 were also launched successively. Although they are indispensable in saving the lives of cancer patients, drug resistance inevitably occurs after long-term use due to various reasons.9,10 Therefore, it is of great significance to develop new drug molecules with good anti-tumor activity and minimal side effects.Sulfoxide compounds also exhibit chirality when their sulfur atoms are linked with two different groups.11 Although most chiral research focuses on chiral carbon compounds, chiral sulfoxides have also received extensive attention in the development of some important drugs.12,13 For example, omeprazole, a drug for treating stomach diseases, has a sulfoxide structure and was marketed in the form of a racemate in the early stages for clinical use. The (−)-isomer of omeprazole is mainly metabolized by CYP3A4, whereas the (+)-isomer is mainly metabolized by CYP2C19. Considering the obvious expression difference of the CYP2C19 metabolic enzyme in the population, esomeprazole, the (−)-isomer of omeprazole, was launched in 2001 as a sole component14 to ensure the safety of individual drug use and is still among the top 200 globally sold drugs. In addition, potassium ion channel inhibitor aprikalim,15 antiplatelet aggregation molecule OPC-29030,16,17 glutathione synthesis inhibitor L-butyronine sulfoximine,18 and anticancer drug AZD6738 (ref. 19) are all chiral sulfoxide derivatives (Fig. 1).
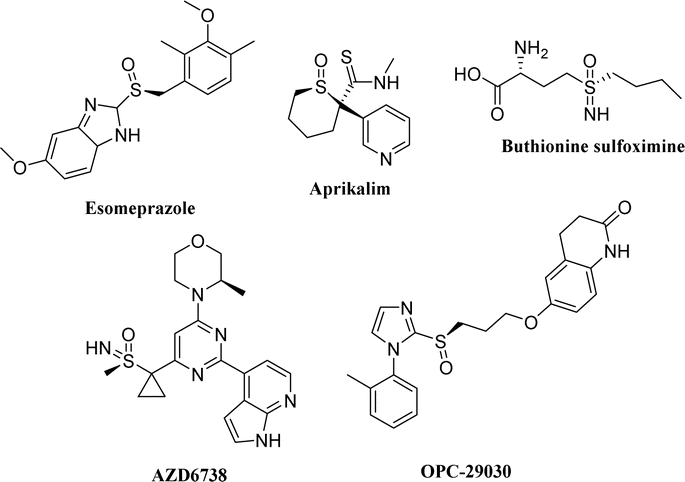 | ||
| Fig. 1 Drugs containing sulfoxide. | ||
From the structures of second-generation ALK inhibitors such as ceritinib, brigatinib, TAE684,20 and ASP-3026,21 we found that they share similar skeletal structures, such as pyrimidine ring cores and aniline moiety linked with cyclic aliphatic amino groups (Fig. 2). Investigations indicated that ceritinib has higher metabolic stability and fewer toxic side effects as compared to molecules such as ASP-3026 with amino groups at the 1- and 4-positions of the phenyl ring.12 Considering drug design and structure–activity studies, small changes in molecular structure often lead to huge differences in their activities, therefore, we chose ceritinib for further structural modification, hoping to get a new lead compound with better activity and fewer side effects.
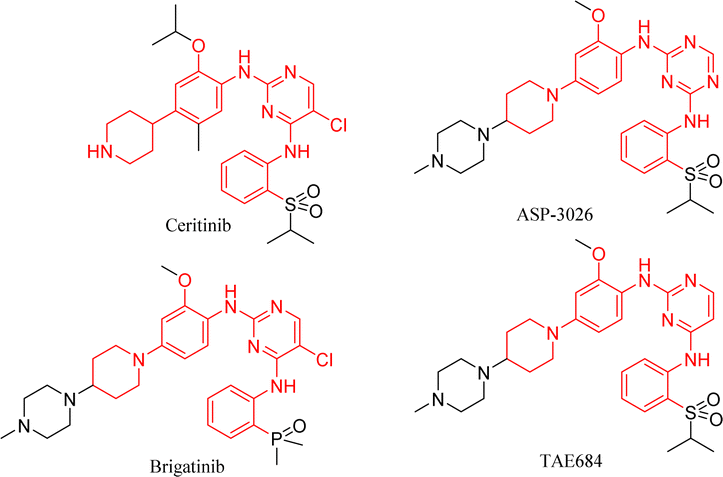 | ||
| Fig. 2 Representative ALK inhibitors. | ||
Our modification strategy for new ALK inhibitors is shown in Fig. 3. Firstly, considering the characteristics and potential chirality of sulfoxide groups, we replaced the sulfone structure in ceritinib with sulfoxide and simultaneously screened the effects of different alkyl groups R3 on activity. Halogen atoms, such as fluorine, have strong electronegativity, small atomic radius, and good lipophilicity;22 therefore, introducing fluorine atoms into drug molecules can adjust the lipophilicity of molecules, further affecting the affinity, pharmacokinetics, and bioavailability of drug molecules with targets.23,24 Therefore, we substituted fluorine and chlorine in the m-position of the sulfoxide group, expecting candidate compounds with superior activity. We also explored the effects of other structural sites, such as different alkoxy groups (R1), on the benzene ring, different groups on the nitrogen atom of piperidine (R2), and alkyl substitution on the benzene ring (R4), on anti-tumor activity.
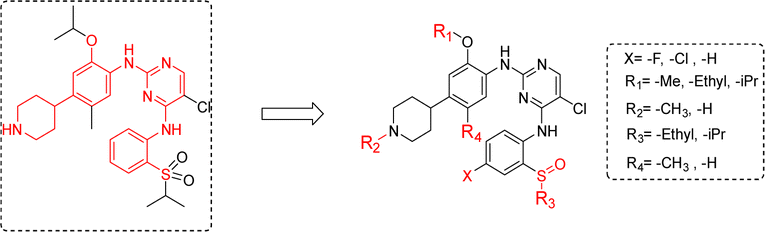 | ||
| Fig. 3 The modification strategy of the new ALK inhibitors. | ||
Experimental
Chemistry
The general method for the synthesis of intermediates 2a–b. Starting material 1 (10 mmol) was dissolved in 20 mL of sodium hydroxide aqueous solution (5 mol L−1), heated to reflux, and monitored with TLC. After the reaction was completed, hydrochloric acid (1 mol L−1) was added to adjust the pH to 6–7. The precipitate was filtered and dried to obtain the product.
2-Amino-5-fluorobenzenethiol (2a). Yellow-green solid. 50.3% yield. 1H NMR (400 MHz, chloroform-d) δ 7.01–6.82 (m, 2H), 6.67 (dd, J = 8.5, 4.7 Hz, 1H), 4.19 (s, 2H).
2-Amino-5-chlorobenzenethiol (2b). Light-green solid. 58.7% yield. 1H NMR (400 MHz, chloroform-d) δ 7.23–6.95 (m, 2H), 6.65 (d, J = 8.6 Hz, 1H), 4.32 (s, 2H).
The general method for the synthesis of intermediates 3a–d. The intermediate 2a–c (10 mmol) was dissolved in methanol (20 mL), and then potassium hydroxide solid (20 mmol, 5.6 g), and bromoethane or 2-bromopropane (12 mmol) were added in turn. The reaction mixture was stirred overnight at room temperature. After TLC confirmed that the reaction was complete, the solvent was removed under a vacuum, and 15 mL of water was added, followed by extraction with dichloromethane (30 mL × 2). The combined organic phase was washed with brine, dried with anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain the crude product, which was purified by silica gel column chromatography (petroleum ether as eluent
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 30–20:1) to obtain the intermediates 3a–d.
:ethyl acetate = 30–20:1) to obtain the intermediates 3a–d.
2-(Ethylthio)aniline (3a). Light yellow oil. 95.5% yield. 1H NMR (400 MHz, chloroform-d) δ 7.41 (dd, J = 7.6, 1.3 Hz, 1H), 7.14 (td, J = 8.0, 1.5 Hz, 1H), 6.80–6.62 (m, 2H), 4.37 (s, 2H), 2.79 (q, J = 7.3 Hz, 2H), 1.26 (t, J = 7.3 Hz, 3H).
2-(Isopropylthio)aniline (3b). Light yellow oil. 91.2% yield. 1H NMR (400 MHz, chloroform-d) δ 7.41 (dd, J = 7.6, 1.3 Hz, 1H), 7.14 (td, J = 8.0, 1.5 Hz, 1H), 6.80–6.62 (m, 2H), 4.33 (s, 2H), 3.06–3.09 (m, 1H), 1.29–1.26 (d, J = 7.3 Hz, 6H).
2-(Ethylthio)-4-fluoroaniline (3c). Light yellow oil. 87.9% yield. 1H NMR (400 MHz, CDCl3) δ 7.05–6.89 (m, 2H), 6.72–6.53 (m, 1H), 4.74 (s, 2H), 3.40–2.94 (m, 2H), 1.22 (t, J = 7.5 Hz, 3H).
4-Chloro-2-(ethylthio)aniline (3d). Light yellow oil. 85.3% yield. 1H NMR (400 MHz, chloroform-d) δ 7.00–6.59 (m, 2H), 6.42–6.23 (m, 1H), 4.53 (s, 2H), 3.05–2.77 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H).
The general method for the synthesis of intermediates 4a–d. With the synthesis of intermediate 4a as an example, 3a (10 mmol, 1.53 g) was dissolved in 5 mL of acetic acid, and then 30% hydrogen peroxide solution (1.25 g, 11 mmol) was slowly added at 0 °C. The mixture was stirred at the same temperature for 30 minutes, and then at room temperature for 4–5 hours. Sodium hydroxide (3.5 g) in crushed ice was added to the reaction system and stirred for 5 minutes. The mixture was extracted twice with 30 mL of dichloromethane. The organic phase was combined, washed with brine, and dried with anhydrous sodium sulfate. The solvent was removed under a vacuum and purified by silica gel column chromatography (petroleum ether
:ethyl acetate = 8:1 to 2:1) to afford 1.56 g of yellowish oil product. Yield: 92%.1H NMR (400 MHz, DMSO-d6) δ 7.27 (d, J = 7.9 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 6.71 (dd, J = 18.1, 8.1 Hz, 2H), 5.63 (s, 2H), 2.94 (tq, J = 13.2, 6.6 Hz, 2H), 1.06 (t, J = 7.6 Hz, 3H).
The general method for the synthesis of intermediates 5a–d. With the synthesis of intermediate 5a as an example, to the solution of intermediate 4a (10 mmol, 1.7 g) in dry DMF (30 mL), sodium hydride (60 wt%, 25 mmol, 1 g, 2.5 eq.) was added at 0 °C. The mixture was stirred for 15 minutes, and 2,4,5-trichloropyrimidine (20 mmol, 3.62 g) in dry DMF (10 mL) was added dropwise within 1 hour. The mixture was stirred at room temperature overnight. Water 30 mL was added, and the mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic phase was washed with water and brine, dried over anhydrous sodium sulfate, concentrated to obtain the crude product, which was purified in silica gel column chromatography (petroleum ether
:ethyl acetate = 10:1 to 4:1) to obtain 1.92 g of light-yellow solid product. Yield: 61%. 1H NMR (400 MHz, chloroform-d) δ 11.03 (s, 1H), 8.62 (d, J = 8.5 Hz, 1H), 8.25 (d, J = 1.3 Hz, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.31 (dd, J = 7.7, 1.7 Hz, 1H), 7.19 (t, J = 7.5 Hz, 1H), 3.29–2.81 (m, 2H), 1.27 (td, J = 7.6, 1.2 Hz, 3H).
The general method for the synthesis of intermediates 7a–d. With the synthesis of intermediate 7b as an example, caesium carbonate (30 mmol, 9.77 g) was added to a solution (25 mL) of starting material 6 (20 mmol, 3.78 g) in isopropanol. The mixture was refluxed for 10 hours. After the reaction was completed, the solvent was removed under a vacuum. Water (20 mL) was added to the residue and then extracted with dichloromethane (30 mL × 2). The combined organic phase was washed with brine and dried with anhydrous sodium sulfate. The solvent was removed to obtain 3.8 g of the orange solid product. Yield: 83%. 1H NMR (400 MHz, chloroform-d) δ 7.72 (d, J = 7.5 Hz, 1H), 7.09 (d, J = 7.9 Hz, 1H), 4.63 (dt, J = 13.8, 6.1 Hz, 1H), 2.36 (d, J = 7.9 Hz, 3H), 1.41 (t, J = 7.0 Hz, 6H).
The general method for the synthesis of intermediates 8a–d. With the synthesis of intermediate 8b as an example, to the solution of intermediate 7b (10 mmol, 2.29 g) in 1,4-dioxane (30 mL), potassium carbonate solid (25 mmol, 3.46 g), bis triphenylphosphine palladium(II) chloride (210.6 mg, 0.3 mmol), 4-pyridine boric acid (12 mmol, 1.48 g), and 10 mL of water were added in turn. The reaction was refluxed under nitrogen protection for 8 hours. After the reaction finished, the solvent was removed under a vacuum. Water was added to the residue and the mixture was extracted with dichloromethane (40 mL × 3). The combined organic phase was washed with brine and dried with anhydrous sodium sulfate. The solvent was removed under a vacuum to obtain the crude product, which was purified by silica gel column chromatography (petroleum ether
:ethyl acetate:dichloromethane/8:2:1) to obtain 2.55 g of a yellow solid. Yield: 93%. 1H NMR (400 MHz, chloroform-d) δ 8.61 (d, J = 6.1 Hz, 2H), 7.27 (d, J = 6.1 Hz, 2H), 6.68 (d, J = 18.5 Hz, 2H), 4.53 (p, J = 6.0 Hz, 1H), 2.21 (s, 3H), 1.38 (d, J = 6.1 Hz, 6H).
The general method for the synthesis of intermediates 9a–d and 10a–d. The syntheses of intermediates 9b and 10b are used as examples. To a solution of intermediate 8b (10 mmol, 2.72 g) in 15 mL acetonitrile, iodomethane (15 mmol, 2.15 g) was added. After stirring at 50 °C for 3 hours, the solvent was removed under a vacuum. The crude product 9b was used directly for the next step.
To a solution of crude product 9b in 20 mL of methanol, sodium borohydride (40 mmol, 1.51 g) was added in batches in an ice bath. The reaction was stirred for half an hour, and then at room temperature for 2 hours. A saturated ammonium chloride solution was added to the reaction system to quench the reaction, the solvent was removed under reduced pressure. Water (30 mL) was added to the residue and then extracted with dichloromethane (30 mL × 3). The combined organic phase was washed with brine and dried with anhydrous sodium sulfate. The solvent was removed under a vacuum and the crude product was purified by column chromatography (dichloromethane:methanol = 50:1 to 20:1) to obtain 2.1 g of yellowish oily product. Yield: 72%. 1H NMR (400 MHz, chloroform-d) δ 7.61 (s, 1H), 6.81 (s, 1H), 5.60 (s, 1H), 4.59 (dq, J = 12.1, 6.2 Hz, 1H), 3.10 (q, J = 2.8 Hz, 2H), 2.66 (t, J = 5.6 Hz, 2H), 2.41 (d, J = 16.5 Hz, 4H), 2.24 (s, 3H), 1.64 (s, 1H), 1.36 (d, J = 6.0 Hz, 6H).
The general method for the synthesis of intermediates 11a–d. With the synthesis of intermediate 11b as an example, tin(IV) chloride dihydrate (50 mmol, 10.11 g) was added to the solution of intermediate 10b (10 mmol, 2.6 g) in dichloromethane/methanol (40 mL, v/v, 1
:1). The mixture was stirred, and hydrochloric acid (6.7 mL) was added in drops. After the reaction was stirred and refluxed at 50 °C for 5 hours, ammonia water was used to adjust the pH to 5, and then to 9 with solid sodium carbonate. This was followed by filtration with diatomite and washing the filter cake with a mixture of dichloromethane:methanol = 1:1. The solvent was removed under a vacuum to afford the crude product, which was purified by column chromatography (dichloromethane:methanol = 50:1 to 20:1) to obtain a bright yellow solid product. 1H NMR (400 MHz, chloroform-d) δ 7.61 (s, 1H), 6.81 (s, 1H), 5.60 (s, 1H), 4.59 (m, J = 12.1, 6.2 Hz, 1H), 3.10 (q, J = 2.8 Hz, 2H), 2.66 (t, J = 5.6 Hz, 2H), 2.41 (d, J = 16.5 Hz, 4H), 2.24 (s, 3H), 1.64 (s, 1H), 1.36 (d, J = 6.0 Hz, 6H).
The general method for the synthesis of intermediates 12a–d. With the synthesis of intermediate 12b as an example, to a solution of intermediate 11b (5 mmol, 1.45 g) in methanol (5 mL), 10% palladium carbon (150 mg) was added. The reaction was carried out at 50 °C in a hydrogen atmosphere at a pressure of 40 bar. After 24 h, the temperature was decreased to room temperature. The mixture was filtered with diatomaceous earth and the cake was washed twice with anhydrous ethanol (10 mL × 2). The solvent was removed, and the crude product was purified with silica gel column chromatography to obtain a light-yellow oil-free substance.
1H NMR (400 MHz, chloroform-d) δ 6.73 (s, 1H), 6.53 (s, 1H), 4.44 (p, J = 6.1 Hz, 1H), 3.62 (s, 2H), 2.98 (d, J = 11.5 Hz, 2H), 2.60 (p, J = 8.1, 7.6 Hz, 1H), 2.34 (s, 3H), 2.21 (s, 3H), 2.07 (dd, J = 15.0, 11.2 Hz, 2H), 1.83–1.64 (m, 4H), 1.33 (d, J = 6.1 Hz, 6H).
The general procedure for the synthesis of target compounds 18a–l. With the synthesis of intermediate 18b as an example, to a solution of intermediate 12b (1 mmol, 262 mg, 1 eq.) in 10 mL of isopropanol, intermediate 5b (2 mmol, 630 mg, 2 eq.) and trifluoroacetic acid (2 mmol, 2 eq.) were added. The reaction was heated and refluxed for 8 hours, and isopropanol was removed under a vacuum. Then, a 10% sodium hydroxide aqueous solution was added to adjust the pH to 10, and dichloromethane (30 mL × 3) was used for extraction. The combined organic phase was washed with brine, dried with anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (ethyl acetate
:petroleum ether = 1:2–3:1) to obtain a yellowish solid.
4-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N5-(2-(isopropylsulfinyl)phenyl)pyrimidine-2,5-diamine (18a). Pale yellow solid, yield: 39.8%, HPLC (peak area normalization method) purity 97.08%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.481 min. 1H NMR (400 MHz, chloroform-d) δ 10.25 (s, 1H), 8.59 (d, J = 8.3 Hz, 1H), 8.10 (d, J = 28.3 Hz, 2H), 7.52 (s, 2H), 7.38 (d, J = 7.7 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 6.82 (s, 1H), 4.65–4.44 (m, 1H), 3.39 (dq, J = 13.7, 6.7 Hz, 1H), 3.21 (d, J = 5.5 Hz, 2H), 2.89–2.72 (m, 3H), 2.20 (s, 3H), 1.77–1.56 (m, 5H), 1.42 (d, J = 6.9 Hz, 3H), 1.38 (d, J = 6.0 Hz, 6H), 1.15 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.54, 155.61, 155.03, 144.69, 140.89, 138.05, 132.03, 128.30, 127.56, 126.76, 126.29, 123.86, 122.45, 120.72, 111.00, 105.77, 71.44, 52.63, 47.40, 38.50, 33.85, 22.28, 18.92, 16.50, 16.11. HR-ESI-MS for C28H36ClN5O2S ([M + Na]+) calcd: 564.2170; Found: 564.2161. FT-IR (KBr) v(–N–H) 3413.05 cm−1; v(–C–H): 2972.59 cm−1, 2928.43 cm−1; v(–S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O): 1111.09 cm−1; v(–C–Cl) 734.06 cm−1. Elemental analysis: analysis calculated for C28H36ClN5O2S, C, 62.03; H, 6.69; Cl, 6.54; N, 12.92; O, 5.90; S, 5.91; observed, C, 61.86, H, 6.75, N, 13.06, S, 5.98.
O): 1111.09 cm−1; v(–C–Cl) 734.06 cm−1. Elemental analysis: analysis calculated for C28H36ClN5O2S, C, 62.03; H, 6.69; Cl, 6.54; N, 12.92; O, 5.90; S, 5.91; observed, C, 61.86, H, 6.75, N, 13.06, S, 5.98.
4-Chloro-N2-(2-isopropoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)-N5-(2-(isopropylsulfinyl)phenyl)pyrimidine-2,5-diamine (18b). Pale yellow solid, yield: 48.1%, HPLC (peak area normalization method) purity 98.00%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.497 min. 1H NMR (400 MHz, chloroform-d) δ 10.25 (s, 1H), 8.59 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 28.4 Hz, 2H), 7.51 (s, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 6.82 (s, 1H), 4.53 (p, J = 6.1 Hz, 1H), 3.38 (p, J = 6.8 Hz, 1H), 3.00 (d, J = 8.5 Hz, 2H), 2.66 (q, J = 9.3, 8.1 Hz, 1H), 2.36 (s, 3H), 2.19 (s, 3H), 2.15–2.01 (m, 2H), 1.83–1.71 (m, 4H), 1.42 (d, J = 6.9 Hz, 3H), 1.37 (d, J = 6.0 Hz, 6H), 1.15 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.53, 155.59, 155.02, 144.66, 140.90, 137.57, 132.02, 128.28, 127.50, 126.83, 126.27, 123.84, 122.42, 120.67, 110.73, 105.75, 71.29, 56.59, 52.63, 46.54, 37.38, 33.00, 22.27, 18.92, 16.49, 16.10. HR-ESI-MS for C29H38ClN5O2S ([M + H]+) calcd: 556.2508; found: 556.2579. FT-IR (KBr) v(–N–H) 3416.35 cm−1; v(–C–H): 2972.95 cm−1, 2933.48 cm−1; v(–SO): 1112.45 cm−1 v(–C–Cl) 760.35 cm−1. Elemental analysis: analysis calculated for C29H38ClN5O2S, C, 62.63; H, 6.89; Cl, 6.37; N, 12.59; O, 5.75; S, 5.76; observed, C, 62.56, H, 7.01, N, 13.46, S, 5.98.
4-Chloro-N5-(2-(ethylsulfinyl)phenyl)-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18c). Pale yellow solid, yield: 40.2%, HPLC (peak area normalization method) purity 97.26%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.046 min. 1H NMR (400 MHz, chloroform-d) δ 10.06 (s, 1H), 8.54 (d, J = 8.3 Hz, 1H), 8.08 (d, J = 36.3 Hz, 2H), 7.57–7.33 (m, 3H), 7.16 (t, J = 7.5 Hz, 1H), 6.80 (s, 1H), 4.54 (p, J = 6.0 Hz, 1H), 3.13 (ddd, J = 43.9, 12.9, 7.4 Hz, 4H), 2.77 (ddt, J = 11.2, 6.0, 2.9 Hz, 3H), 2.18 (s, 3H), 1.75 (d, J = 9.4 Hz, 5H), 1.36 (d, J = 6.1 Hz, 6H), 1.26 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.55, 155.70, 155.09, 144.68, 140.48, 138.00, 132.14, 127.81, 127.56, 127.27, 126.78, 123.97, 122.89, 120.67, 110.99, 105.75, 71.46, 47.36, 47.29, 38.46, 33.77, 22.28, 18.91, 7.58. HR-ESI-MS for C27H34ClN5O2S ([M–H]−) calcd: 526.2049; found: 526.2109. FT-IR (KBr) v(–N–H) 3414.36 cm−1; v(–C–H): 2974.32 cm−1, 2931.76 cm−1; v(–SO): 1111.54 cm−1 v(–C–Cl) 753.04 cm−1. Elemental analysis: analysis calculated for C27H34ClN5O2S, C, 61.41; H, 6.49; Cl, 6.71; N, 13.26; O, 6.06; S, 6.07; observed, C, 60.86, H, 6.44, N, 13.36, S, 6.28.
4-Chloro-N5-(2-(ethylsulfinyl)phenyl)-N2-(2-isopropoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18d). Pale yellow solid, yield: 44.3%, HPLC (peak area normalization method) purity 97.00%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 0.936 min. 1H NMR (400 MHz, chloroform-d) δ 9.98 (s, 1H), 8.53–8.35 (m, 1H), 8.00 (d, J = 37.2 Hz, 2H), 7.57–7.36 (m, 2H), 7.32 (d, J = 7.6 Hz, 1H), 7.09 (t, J = 7.5 Hz, 1H), 6.72 (s, 1H), 4.50 (dq, J = 12.1, 6.0 Hz, 1H), 3.26 (d, J = 9.9 Hz, 2H), 3.17–2.93 (m, 2H), 2.74–2.58 (m, 1H), 2.45 (s, 3H), 2.34 (t, J = 12.2 Hz, 2H), 2.08 (s, 3H), 1.91 (d, J = 12.0 Hz, 2H), 1.73 (t, J = 12.9 Hz, 2H), 1.29 (d, J = 6.0 Hz, 6H), 1.17 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.51, 155.55, 155.30, 144.77, 138.80, 137.86, 131.92, 129.84, 128.33, 127.30, 126.80, 126.64, 125.51, 120.68, 110.74, 105.64, 71.30, 56.58, 47.52, 46.54, 37.38, 33.01, 32.97, 22.26, 18.92, 7.42. and HR-ESI-MS for C28H36ClN5O2S ([M + H]+) calcd: 514.0815; found: 514.0994. FT-IR (KBr) v(–N–H) 3415.67 cm−1; v(–C–H): 2973.92 cm−1, 2934.78 cm−1; v(–SO): 1111.85 cm−1 v(–C–Cl) 775.55 cm−1. Elemental analysis: analysis calculated for C28H36ClN5O2S, C, 62.03; H, 6.69; Cl, 6.54; N, 12.92; O, 5.90; S, 5.91; observed, C, 61.86, H, 6.84, N, 12.76, S, 6.28.
4-Chloro-N5-(2-(ethylsulfinyl)phenyl)-N2-(2-methoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18e). Pale yellow solid, yield: 50.1%, HPLC (peak area normalization method) purity 98.34%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.253 min. 1H NMR (400 MHz, chloroform-d) δ 10.10 (s, 1H), 8.55 (d, J = 8.3 Hz, 1H), 8.14 (s, 1H), 8.04 (s, 1H), 7.51 (dd, J = 16.4, 9.2 Hz, 2H), 7.45–7.35 (m, 1H), 7.17 (t, J = 7.9 Hz, 1H), 6.81 (s, 1H), 3.86 (s, 3H), 3.11–3.02 (m, 2H), 2.49 (s, 3H), 2.29 (t, J = 12.0 Hz, 3H), 2.21 (s, 3H), 2.03–1.88 (m, 3H), 1.82 (t, J = 11.6 Hz, 3H), 1.27 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.58, 155.69, 155.04, 146.75, 140.53, 136.83, 132.14, 127.65, 127.28, 126.70, 123.84, 122.85, 120.74, 117.17, 108.20, 107.68, 106.03, 56.26, 55.76, 47.30, 45.70, 36.89, 32.11, 18.90, 8.97, 7.59. HR-ESI-MS for C26H32ClN5O2S ([M + H]+) calcd: 532.0753; found: 532.0714.
4-Chloro-N5-(2-(ethylsulfinyl)-4-fluorophenyl)-N2-(2-methoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18f). Pale yellow solid, yield: 47.7%, HPLC (peak area normalization method) purity 98.26%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 0.811 min. 1H NMR (400 MHz, chloroform-d) δ 9.41 (s, 1H), 8.45–8.34 (m, 1H), 8.13 (s, 1H), 7.92 (s, 1H), 7.46 (s, 1H), 7.23 (d, J = 7.4 Hz, 2H), 6.81 (s, 1H), 3.86 (s, 3H), 3.11 (td, J = 13.5, 5.5 Hz, 4H), 2.94 (q, J = 7.4 Hz, 1H), 2.42 (s, 3H), 2.19 (s, 3H), 1.95–1.73 (m, 6H), 1.28 (d, J = 7.5 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.56, 157.56 (d, J1C–F = 251.49 Hz), 157.08, 157.08, 155.83, 146.76, 136.89, 135.56 (d, J4C–F = 2.9 Hz), 131.58 (d, J3C–F = 5.6 Hz), 126.73 (t, J3C–F = 10.9), 120.69, 118.81 (d, J2C–F = 22.1 Hz), 113.73 (d, J2C–F = 24.6 Hz), 107.69, 105.58, 55.83, 47.47, 46.08, 37.52, 31.80, 18.90, 7.17. HR-ESI-MS for C26H31ClFN5O2S ([M + Na]+) calcd: 564.2170; found: 564.2194.
4-Chloro-N2-(2-ethoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)-N5-(2-(ethylsulfinyl)-4-fluorophenyl)pyrimidine-2,5-diamine (18g). Pale yellow solid, yield: 49.3%, HPLC (peak area normalization method) purity 99.00%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 0.814 min. 1H NMR (400 MHz, chloroform-d) δ 9.39 (s, 1H), 8.38 (dd, J = 8.8, 4.6 Hz, 1H), 8.13 (s, 1H), 7.91 (s, 1H), 7.50 (s, 1H), 7.22 (dd, J = 10.5, 7.9 Hz, 2H), 6.80 (s, 1H), 4.08 (q, J = 6.9 Hz, 2H), 3.10 (ddt, J = 21.2, 12.7, 7.3 Hz, 4H), 2.69 (td, J = 11.3, 6.7 Hz, 1H), 2.39 (s, 3H), 2.17 (s, 3H), 1.94–1.66 (m, 6H), 1.44 (t, J = 6.9 Hz, 3H), 1.27 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.58, 157.58 (d, J1C–F = 249.47), 157.11, 155.84, 145.88, 137.58, 135.50, 131.66, 126.75 (d, J3C–F = 7.1 Hz), 126.52 (d, J3C–F = 9.4 Hz), 120.55, 118.81 (d, J2C–F = 22.2 Hz), 113.70 (d, J2C–F = 24.5 Hz), 108.82, 105.32, 64.27, 56.49, 47.46, 46.35, 37.31, 32.72, 18.89, 14.99, 7.15. HR-ESI-MS for C27H33ClN5O2S ([M + H]+) calcd: 546.2100; found: 546.2191.
4-Chloro-N5-(2-(ethylsulfinyl)-4-fluorophenyl)-N2-(2-methoxy-5-methyl-4-(piperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18h). Pale yellow solid, yield: 38.2%, HPLC (peak area normalization method) purity 95.07%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.040 min. 1H NMR (400 MHz, chloroform-d) δ 9.44 (s, 1H), 8.49–8.31 (m, 1H), 8.14 (s, 1H), 7.95 (s, 1H), 7.47 (s, 1H), 7.24 (d, J = 7.3 Hz, 2H), 6.80 (s, 1H), 3.89 (s, 3H), 3.37 (d, J = 13.5 Hz, 3H), 3.18–3.02 (m, 2H), 2.95–2.76 (m, 3H), 2.20 (s, 3H), 1.91–1.73 (m, 4H), 1.28 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.52, 157.61 (d, J1C–F = 254.5), 157.05, 155.80, 146.72, 137.52, 135.59, 126.66 (t, J3C–F = 9.1), 126.38, 120.70, 118.83 (d, J2C–F = 22.3 Hz), 113.72 (d, J2C–F = 24.5 Hz), 108.30, 107.74, 105.49, 56.42, 55.75, 47.46, 46.21, 37.22, 32.57, 18.89, 9.55, 7.19. HR-ESI-MS for C25H29ClFN5O2S ([M + H]+) calcd: 518.1787; found: 518.1794.
4-Chloro-N5-(4-chloro-2-(ethylsulfinyl)phenyl)-N2-(2-isopropoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18i). Pale yellow solid, yield: 45.4%, HPLC (peak area normalization method) purity 96.62%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.472 min. 1H NMR (400 MHz, chloroform-d) δ 9.85 (s, 1H), 8.49 (d, J = 8.8 Hz, 1H), 8.14 (s, 1H), 7.96 (s, 1H), 7.64–7.35 (m, 3H), 6.83 (s, 1H), 4.65–4.43 (m, 1H), 3.29–3.07 (m, 2H), 3.07–2.96 (m, 2H), 2.68 (p, J = 8.6, 8.1 Hz, 1H), 2.36 (s, 3H), 2.20 (s, 3H), 2.11 (dd, J = 13.6, 5.0 Hz, 2H), 1.79 (qd, J = 7.8, 5.2, 4.8 Hz, 4H), 1.37 (d, J = 6.1 Hz, 6H), 1.30 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.51, 155.55, 155.30, 144.77, 138.80, 137.86, 131.92, 129.84, 128.33, 127.30, 126.80, 126.64, 125.51, 120.68, 110.74, 105.64, 71.30, 56.58, 47.52, 46.54, 37.38, 33.01, 22.26, 18.92, 7.42. HR-ESI-MS for C28H35Cl2N5O2S ([M + H]+) calcd: 576.2144; found: 576.2033.
4-Chloro-N5-(2-(ethylsulfinyl)-4-fluorophenyl)-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18j). Pale yellow solid, yield: 33.7%, HPLC (peak area normalization method) purity 97.28%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.135 min. 1H NMR (400 MHz, chloroform-d) δ 9.39 (s, 1H), 8.38 (dd, J = 8.8, 4.8 Hz, 1H), 8.13 (s, 1H), 7.93 (s, 1H), 7.59 (s, 1H), 7.27–7.16 (m, 2H), 6.81 (s, 1H), 4.56 (dt, J = 12.0, 6.0 Hz, 1H), 3.29 (d, J = 9.5 Hz, 2H), 3.11 (dd, J = 14.4, 6.9 Hz, 3H), 2.83 (t, J = 12.1 Hz, 3H), 2.17 (s, 3H), 1.86–1.61 (m, 4H), 1.38 (d, J = 6.0 Hz, 6H), 1.33–1.29 (m, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.38, 157.61 (d, J1C–F = 258.56), 156.91, 155.72, 148.24, 140.30, 135.85, 130.78 (d, J3C–F = 5.2 Hz), 126.98, 126.68 (d, J3C–F = 7.0 Hz), 119.22, 118.59 (t, J2C–F = 22.2 Hz), 113.72 (d, J2C–F = 24.5 Hz), 108.67, 105.66, 56.22, 55.63, 47.48, 46.19, 41.63, 33.45, 33.35, 7.30. HR-ESI-MS for C27H33ClFN5O2S ([M + H]+) calcd: 546.2100; found: 546.2157.
4-Chloro-N5-(2-(ethylsulfinyl)phenyl)-N2-(2-methoxy-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18k). Pale yellow solid, yield: 50.3%, HPLC (peak area normalization method) purity 95.53%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 0.937 min. 1H NMR (400 MHz, chloroform-d) δ 10.15 (s, 1H), 8.51 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 8.7 Hz, 1H), 8.11 (s, 1H), 7.54–7.45 (m, 2H), 7.41–7.32 (m, 1H), 7.16 (t, J = 7.9 Hz, 1H), 6.79–6.71 (m, 2H), 3.88 (s, 3H), 3.26 (d, J = 11.4 Hz, 2H), 3.11 (ddd, J = 45.1, 13.0, 7.5 Hz, 2H), 2.63–2.51 (m, 1H), 2.49 (s, 3H), 2.34 (t, J = 12.9 Hz, 2H), 1.93 (dd, J = 29.6, 11.9 Hz, 4H), 1.24 (d, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 157.58, 155.69, 155.04, 146.75, 140.53, 136.83, 132.14, 127.65, 127.28, 126.70, 123.84, 122.85, 120.74, 117.17, 107.68, 106.03, 56.26, 55.76, 47.30, 45.70, 36.89, 32.11, 18.90, 8.97, 7.59. HR-ESI-MS for C25H30ClN5O2S ([M + H]+) calcd: 500.1882; found: 500.1942.
4-Chloro-N5-(2-(ethylsulfinyl)-4-fluorophenyl)-N2-(2-isopropoxy-5-methyl-4-(1-methylpiperidin-4-yl)phenyl)pyrimidine-2,5-diamine (18l). Pale yellow solid, yield: 33.2%. HPLC (peak area normalization method) purity 99.46%, HPLC conditions: 1 mL min−1 gradient elution MeCN
:H2O = 90:10 → 10:90 (0.1% Et3N in each component), tR = 1.446 min. 1H NMR (400 MHz, chloroform-d) δ 9.38 (s, 1H), 8.47–8.34 (m, 1H), 8.13 (s, 1H), 7.92 (s, 1H), 7.51 (s, 1H), 7.26–7.17 (m, 2H), 6.82 (s, 1H), 4.53 (dt, J = 12.1, 5.9 Hz, 1H), 3.19–2.95 (m, 4H), 2.74–2.59 (m, 1H), 2.36 (s, 3H), 2.16 (s, 3H), 2.14–2.04 (m, 2H), 1.83–1.70 (m, 4H), 1.37 (d, J = 6.0 Hz, 6H), 1.27 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, chloroform-d) δ 159.56, 157.55 (d, J1C–F = 243.4), 157.09, 155.82, 144.66, 137.71, 135.58 (d, J4C–F = 2.9 Hz), 131.64 (d, J3C–F = 6.1 Hz), 127.35, 126.73 (t, J3C–F = 7.07 Hz), 120.52, 118.82 (d, J2C–F = 22.1 Hz), 113.72 (d, J2C–F = 24.5 Hz), 110.69, 105.27, 71.29, 56.56, 47.46, 46.51, 37.36, 32.97, 22.25, 18.92, 7.18. HR-ESI-MS for C28H35ClFN5O2S ([M + H]+) calcd: 560.2257; found: 560.2352.
Biology
Cell origin. The H2228 cell line, a human non-small cell lung cancer cell line, was bought from Shanghai Cell Bank, Chinese Academy of Sciences. The BaF3/ALK cell line was from Kyinno Biotechnology Co. LTD (Beijing, China). For human normal cell lines, EA.hy926, HK-2, and L02 were also obtained from Shanghai Cell Bank, Chinese Academy of Sciences.
Cell culture. For cell culture, H2228, HK-2, L02, and BaF3/ALK cell lines were maintained in RPIM 1640 medium supplemented with 10% FBS (fetal bovine serum). For the H2228 cell culture, we added 1% sodium pyruvate (100 mM, Gibco) and GlutaMAX (1X, Gibco) as a nutritional supplement. EA.hy926 cells were cultivated in DMEM medium, with 10% FBS added. All the above cell lines were in an atmosphere of 37 °C, 5% CO2, and saturated humidity.
Other biology experiments such as cytotoxicity assays, kinase inhibition assays, cell colony formation, etc., are displayed in the ESI† section.
Results and discussion
Chemistry
The synthesis of target compounds 18a–l and the corresponding intermediates is shown in Schemes 1–4. Starting from benzo[d]thiazol-2-amine derivatives with chlorine or fluorine atoms substituted on the benzene ring, hydrolysis was carried out in the presence of NaOH at reflux temperatures to yield 2-amino-5-fluorobenzenethiol (2a) or 2-amino-5-chlorobenzenethiol (2b), which reacted with brominated alkanes to give thioether intermediates 3a–d. Oxidation of 3a–d with hydrogen peroxide yielded the sulfoxide intermediates 4a–d. The reaction of 4a–d with 2,4,5-trichloropyrimidine in the presence of sodium hydride in N,N-dimethylformamide (DMF) yielded the key intermediates 5a–d with pyrimidine structures (Scheme 1).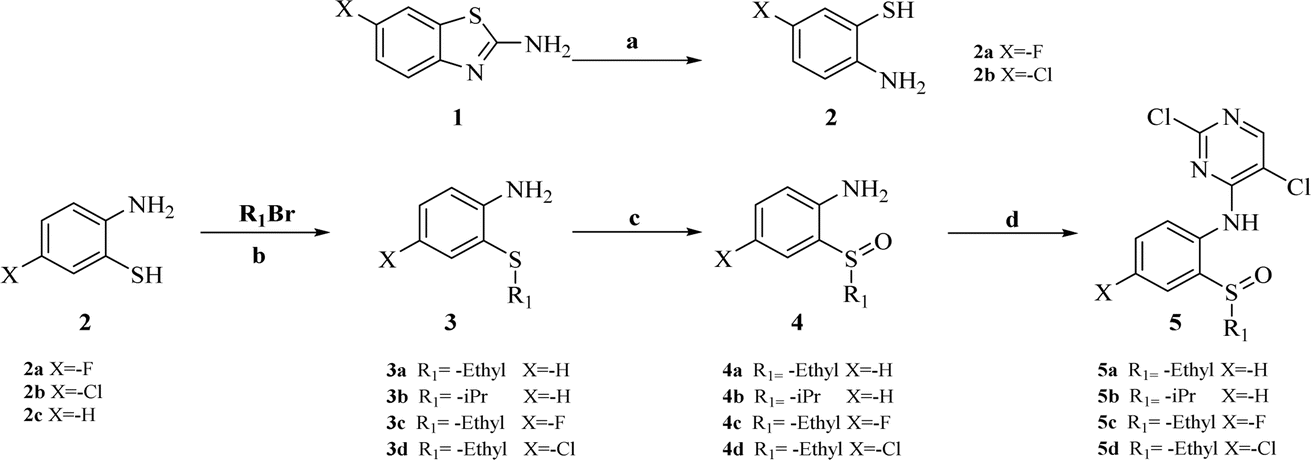 | ||
| Scheme 1 Synthesis of intermediates 2a, 2b, and 5a–d. Reagents and conditions: (a) NaOH (aq., 2 mol L−1), reflux; (b) NaOH, MeOH, rt. (c) AcOH, H2O2, 0 °C. (d) NaH, 2,4,5-trichloropyrimidine, DMF, 0 °C. | ||
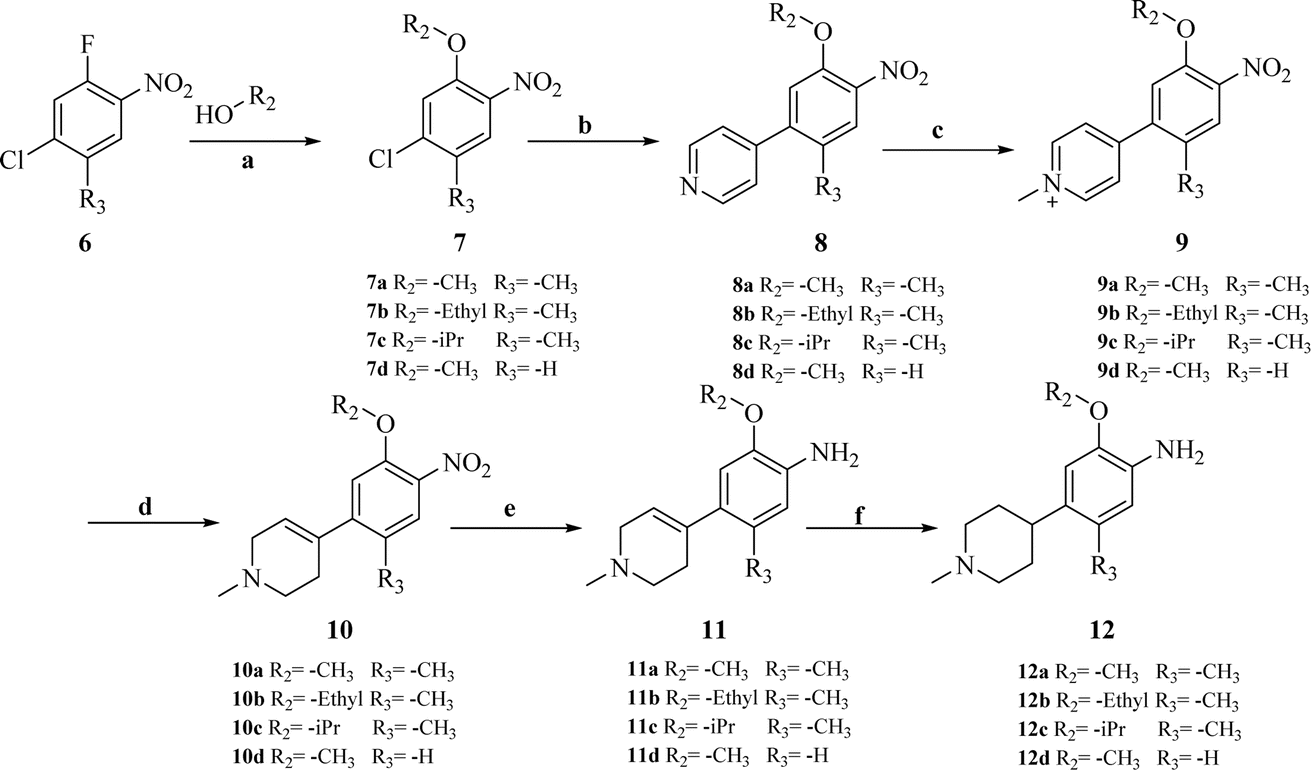 | ||
| Scheme 2 Synthesis of intermediates 12a–d. Reagents and conditions: (a) K2CO3, reflux. (b) K2CO3, pyridin-4-ylboronic acid, Pd(PPh3)Cl2, dioxane, H2O. (c) MeCN, MeI. (d) MeOH, NaBH4, 0 °C. (e) SnCl2, HCl, MeOH/DCM = 1:1, reflux. (f) 10% Pd/C, H2, 40 bar, 50 °C. | ||
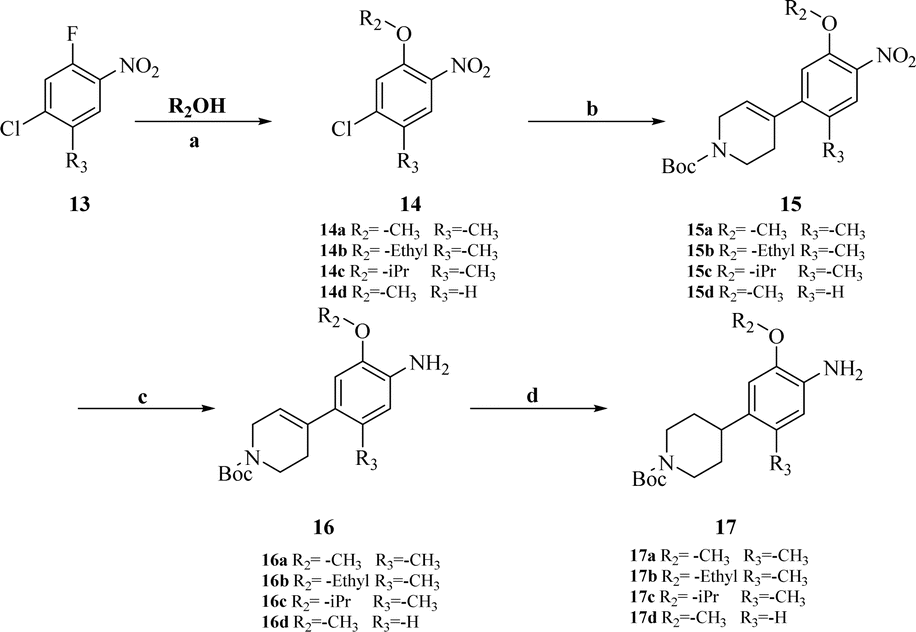 | ||
| Scheme 3 Synthesis of intermediates 17a–c. Reagents and conditions: (a) K2CO3, reflux. (b) K2CO3, (1-(tert-butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid Pd(PPh3)Cl2, dioxane, H2O. (c) SnCl2, HCl, MeOH/DCM = 1:1, reflux. (d) 10% Pd/C, H2, 40 bar, 50 °C. | ||
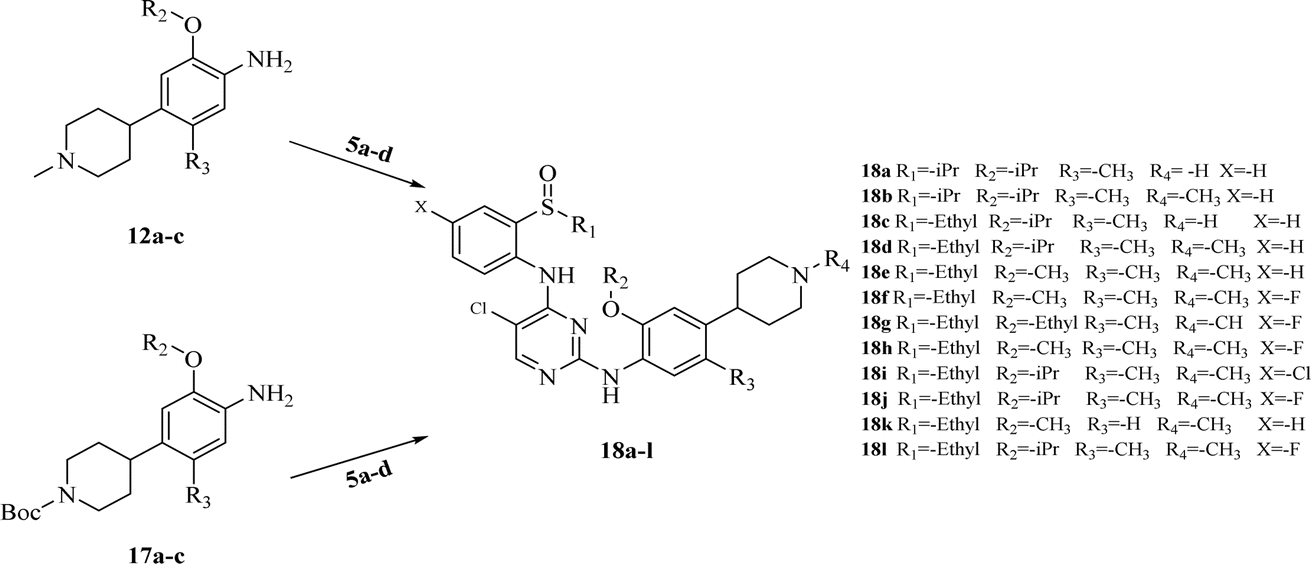 | ||
| Scheme 4 Synthesis of target compounds 18a–l. Reagents and conditions: TFA, isopropyl alcohol, reflux. | ||
Other key intermediates, 4-(1-methylpiperidin-4-yl)aniline derivatives, were prepared according to the route shown in Scheme 2. Compound 2-chloro-4-fluoro-5-nitrobenzene or 1-chloro-5-fluoro-2-methyl-4-nitrobenzene (6) was refluxed in different alcohols in the presence of potassium carbonate to obtain intermediates 7a–d. Suzuki coupling of 7a–d with pyridin-4-ylboronic acid was catalyzed by Pd(PPh3)Cl2, providing intermediates 8a–d, which reacted with iodomethane to afford intermediates 9a–d.
After reduction by sodium borohydride, 9a–d were converted into 1-methyl-4-(4-nitrophenyl)-1,2,3,6-tetrahydropyridine derivatives (10a–d), and then, reduced by tin(II) chloride under acidic conditions to obtain aniline derivatives 11a–d. Hydrogenation of 11a–d under palladium carbon ultimately yielded the key intermediates 12a–d.
Intermediates 17a–d were used to synthesize compounds containing the piperidine moiety, such as compounds 18b and 18c. Except for some differences in starting materials, the synthesis of 17a–d was similar to that of 12a–d.
After obtaining the above key intermediates, the target products 18a–l can be obtained by the reaction of intermediates 5a–d with intermediates 12a–d or 17a–d catalyzed by trifluoroacetic acid in isopropanol at reflux temperature.
Biology
|
|||||||
|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | R4 | X | H2228 IC50 EML4-ALK(nM) | BaF3/NMP-ALK IC50 (nM) |
| a Experiments repeated independently at least three times. | |||||||
| 18a | iPr | iPr | Me | H | H | 28 ± 8 | 19 ± 9 |
| 18b | iPr | iPr | Me | Me | H | 21 ± 6 | 15 ± 3 |
| 18c | Et | iPr | Me | H | H | 19 ± 7 | 11 ± 3 |
| 18d | Et | iPr | Me | Me | H | 14 ± 6 | 9 ± 4 |
| 18e | Et | Me | Me | Me | H | 44 ± 12 | 33 ± 10 |
| 18f | Et | Me | Me | Me | F | 37 ± 11 | 31 ± 13 |
| 18g | Et | Et | Me | Me | F | 33 ± 7 | 29 ± 12 |
| 18h | Et | Me | Me | H | F | 58 ± 16 | 47 ± 15 |
| 18i | Et | iPr | Me | Me | Cl | 81 ± 28 | 77 ± 15 |
| 18j | Et | iPr | Me | H | F | 63 ± 8 | 56 ± 4 |
| 18k | Et | Me | H | Me | H | 57 ± 6 | 24 ± 5 |
| 18L | Et | iPr | Me | Me | F | 31 ± 10 | 28 ± 8 |
| Ceritinib | — | — | — | — | — | 25 ± 010 | 18 ± 1 |
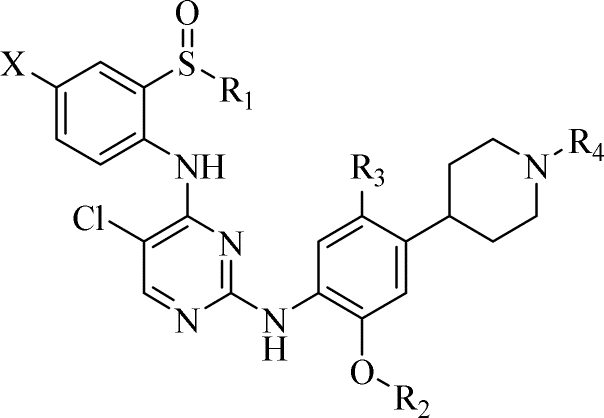
The structure–activity relationship (SAR) analysis of the target compound indicated the following: (1) when R1 is ethyl instead of isopropyl, the anti-proliferation effect shows little increase; (2) keeping other structures unchanged, when R2 is methyl, ethyl, and isopropyl, the efficacy increases sequentially, which may disclose that a group occupying a certain space is preferable here; (3) when R3 is methyl, its activity is superior to that of hydrogen-substituted compounds on the cell level; (4) the structural difference between compound 18b and ceritinib is only that the former includes a sulfoxide group, while the latter contains a sulfone structure. However, compound 18d has better antiproliferative activity against both ALK-overexpressing cells as compared to the reference, indicating that the sulfoxide group is more conducive to improving anti-tumor cell proliferation activity; (5) compounds with fluorine and chlorine atom substituents on the benzene ring exhibit a significant decrease in activity; (6) compounds with R4 as a -methyl substituent display better inhibition activity than that with hydrogen substitution.
Due to the superior anti-proliferative activity of compounds 18a–d, we conducted an ALK kinase inhibition activity assay, also with ceritinib as the reference. The results in Table 2 show that 18b–d exhibited better activity than ceritinib, which is consistent with the results of anti-proliferation; a slight difference lies with the R4 group. It seems that 1-methylpiperidine is better at the kinase level. Based on the above anti-tumor cell proliferation activity and kinase inhibition activity tests, we selected 18d as the representative for further mechanism research.
| Compound | IC50 of ALK kinase |
|---|---|
| a Experiments repeated independently at least three times. | |
| 18a | 3 ± 0.7 |
| 18b | 1.4 ± 0.2 |
| 18c | 0.4 ± 0.03 |
| 18d | 1.2 ± 0.4 |
| Ceritinib | 2.4 ± 0.8 |
000 cells for each well to form spheroids. 18d presented an inhibitory effect on H2228 spheroids with an IC50 of 44.7 nM, which is more than 2 times that of ceritinib.Clone formation reflects the in vitro proliferation ability of tumor cells and their dependence on a colony, which can serve as a supplement to an in vitro anti-tumor assay.25 H2228 cells and BaF3/ALK cells are adherent and suspended cells, respectively. Based on the cell growth characteristics, we used plate cloning and soft agar cloning methods, respectively, for these two types of tumor cells.26 By processing the colony formation results using corresponding data analysis methods, it was found that 18d can effectively inhibit the colony formation of two types of tumor cells and present a dose-dependent effect (Fig. 4). By the action of compound 18d at the concentration of 15 nM, the clone inhibition rates of H2228 and BaF3/ALK cells reached over 70% and over 90%, respectively, and the inhibition of BaF3/ALK cells was more effective, which corresponds with the results in Table 1.
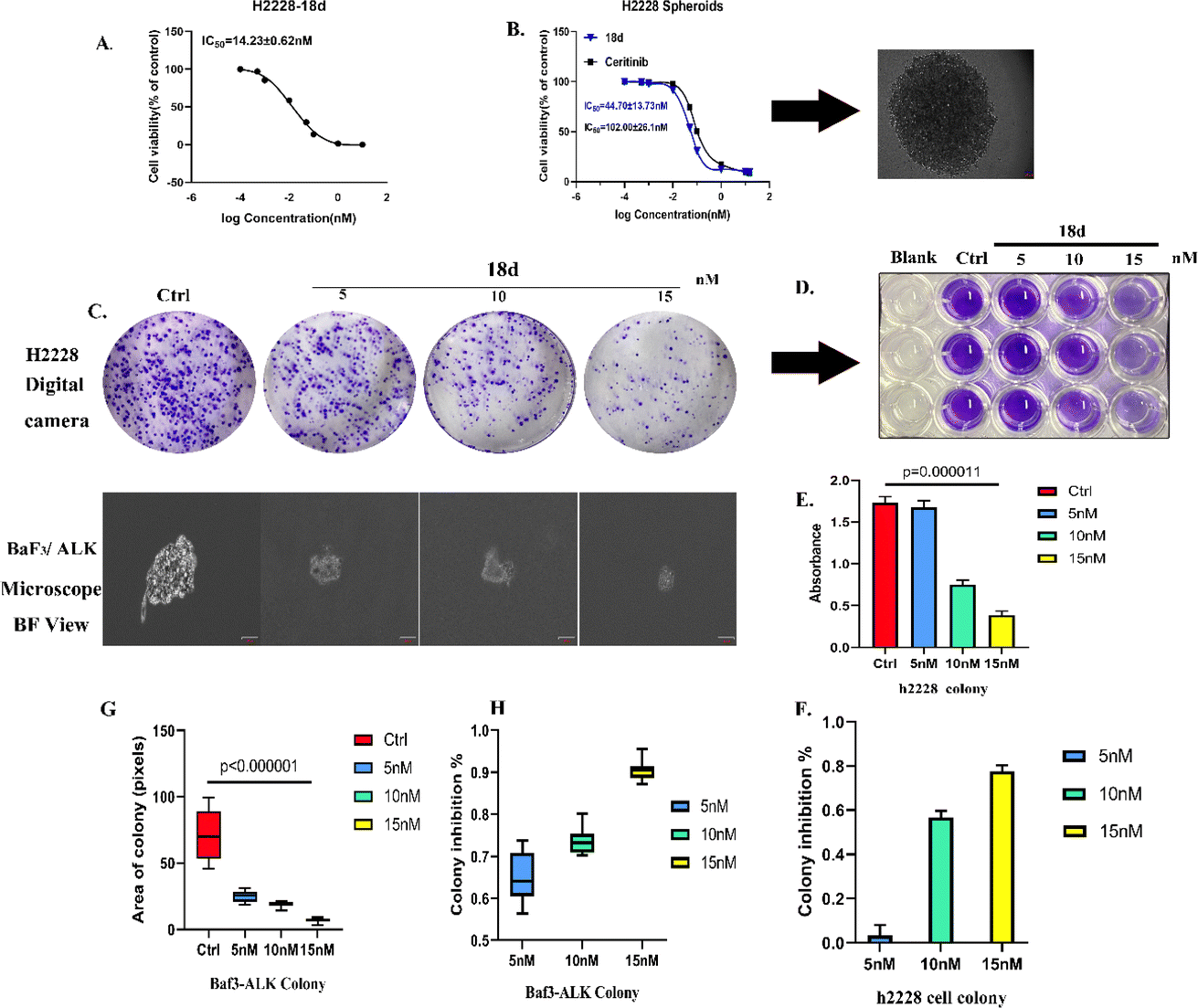 | ||
| Fig. 4 (A) The concentration of 18d and the H2228 monolayer model viability curve. (B) The concentration of 18d or ceritinib and the H2228 spheroid model viability curve; the photograph shows the spheroids under a 4× objective. (C) Representative images of H2228 plate colony formation and BaF3/ALK soft agar colony formation results; the scale bar represents 50 μm and the colony formation images of Baf3 cells were obtained using a 20× objective microscope. (D) 33% acetic acid eluted the color of H2228 cells stained with crystal violet. (E) The absorbance values of each group of H2228 after elution using a microplate reader. (F) The inhibitory effects of compound 18d at different concentrations calculated using absorbance values of the H2228 cell colony. (G) Ten randomly selected fields of view under a 20× objective; photographs of the formed colony cell clusters were taken and image J was used to calculate the area of the colony sphere. A plot of the size of the colony clusters formed by Baf3-ALK cells under different concentrations of 18d intervention. (H) The colony inhibition rates of 18d at various concentrations based on the colony area. The data was shown as mean ± SD and the unpaired two-way t-test (assuming all data with the same scatter) was used to determine the levels of significance between comparable samples. p > 0.05 was not significant (ns). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
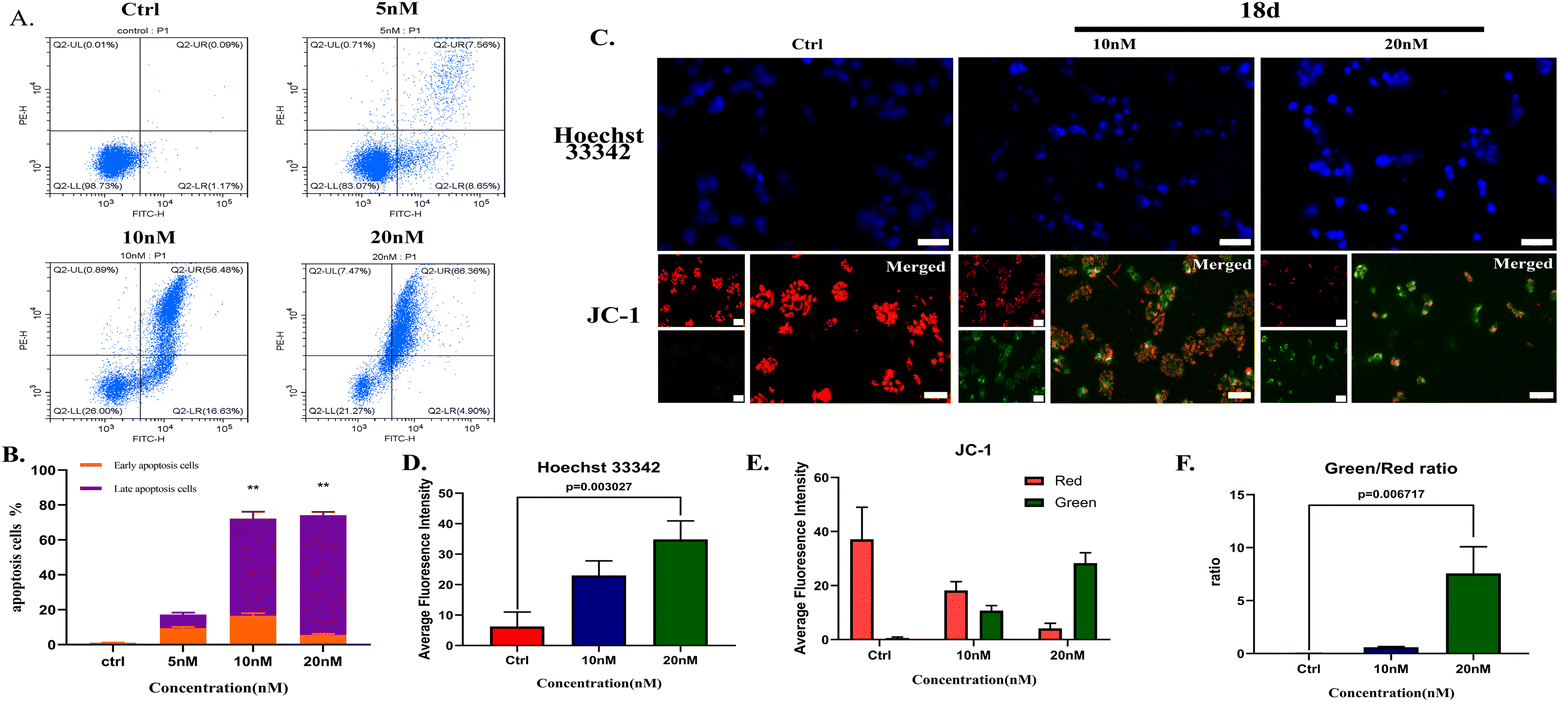 | ||
| Fig. 5 (A & B) Flow cytometry results and statistics of PI/FITC double staining after treatment with different concentrations of 18d. (C) H2228 cells cultured with or without 18d for 48 h, then stained with Hoechst 33342 and photographed by fluorescence microscopy; the scale bar is 50 μm. (D) After JC-1 staining, H2228 cells were observed and photographed using the 10× objective of the fluorescence microscope under the U, B, and G channels; the scale bar is 100 μm. (E) The average fluorescence intensity of green and red channels of each group. (F) The green/red ratio of each group. The data are shown as the mean ± SD, and the unpaired two-way t-test (assuming all data with the same scatter) was used to determine the levels of significance between comparison samples. p > 0.05 was not significant (ns). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
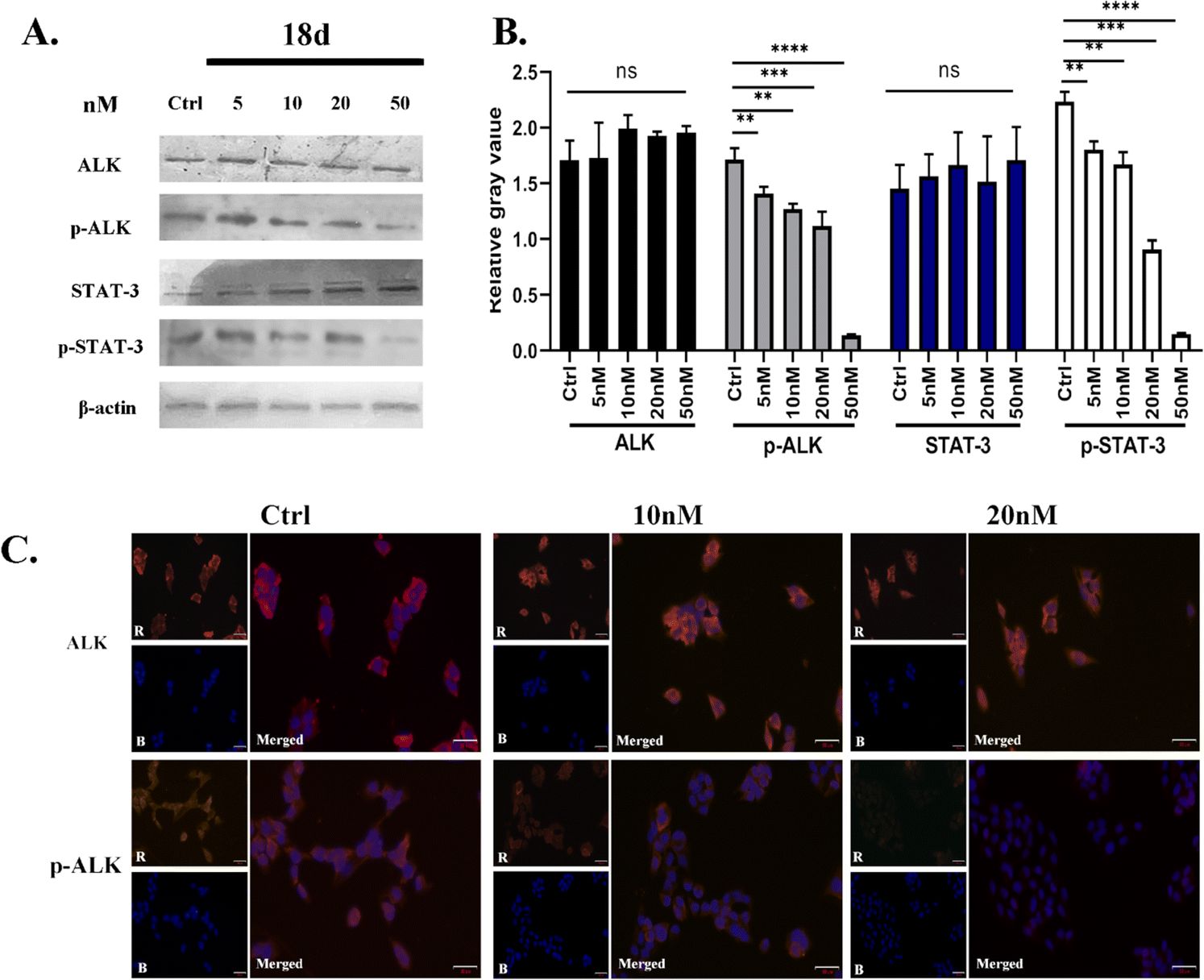 | ||
| Fig. 6 (A & B) Changes in ALK, p-ALK, STAT-3, and p-STAT-3 protein bands in H2228 cells. (C) The changes in ALK and p-ALK in H2228 cells in the presence of 18d. The scale bar is 50 μm. The data are shown as the mean ± SD, and the unpaired two-way t-test (assuming all data with the same scatter) was used to determine the levels of significance between comparison samples. p > 0.05 was not significant (ns). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
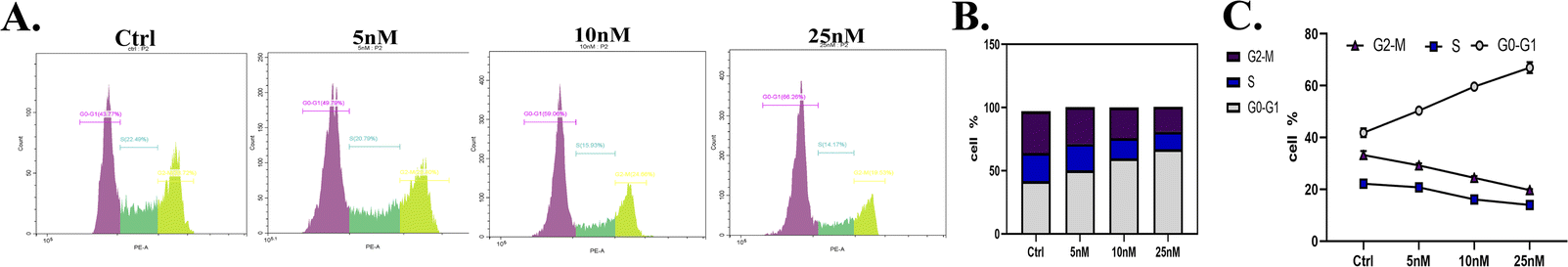 | ||
| Fig. 7 (A) The cell cycle of H2228 cells after treatment with different doses of 18d. (B) The ratio of each cell stage in the whole cell cycle. (C) The ratio changes at each stage after the intervention with different concentrations of 18d. | ||
 | ||
| Fig. 8 (A) Experimental procedures and time points. (B) Images of tumors in each group. The scale bar is 1 cm. (C) The changes in the tumor volumes of nude mice in each group. The data were analyzed by the unpaired t-test (GraphPad Prism 8), and there were significant differences between the control group and 18a–d, P < 0.0001. (D) The weights of tumors in each group. (E) Growth inhibition rate (TGI) of tumors in each group. (F) The body weight of each group after treatment. (G) Control and 18d H&E staining results for the kidney, liver, and lung. The scale bar is 50 μm. The data are shown as the mean ± SD, and the unpaired two-way t-test (assuming all data with the same scatter) was used to determine the levels of significance between comparison samples. p > 0.05 was not significant (ns). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
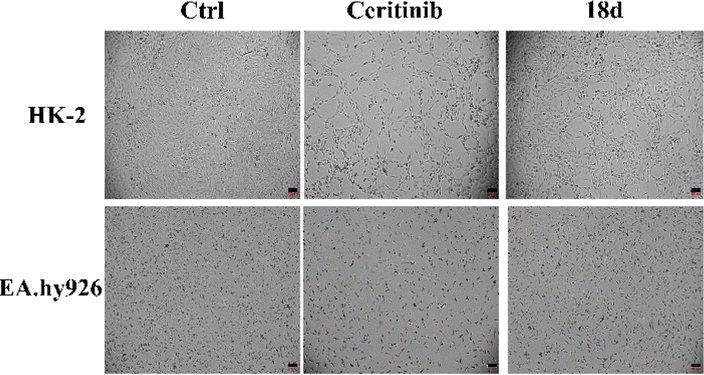 | ||
| Fig. 9 HK-2 and EA.hy926 cell morphology when cultured with ceritinib or 18d (both 1 μM), the scale bar is 100 μm. | ||
| Compound 18a IC50 μM | Ceritinib IC50 μM | ||
|---|---|---|---|
| a The test results were obtained from at least three independent experiments.b EA.hy926 are activated by 10 mg mL−1 VEGF-containing medium. | |||
| EA.hy926 (quiescent) | 6.47 ± 0.95 | EA.hy926 (quiescent) | 2.44 ± 0.34 |
| EA.hy926b (active) | 8.92 ± 1.89 | EA.hy926 (active) | 3.42 ± 0.56 |
| HK-2 | 5.79 ± 0.72 | HK-2 | 1.44 ± 0.25 |
| L02 | >10 | L02 | 3.42 ± 0.56 |
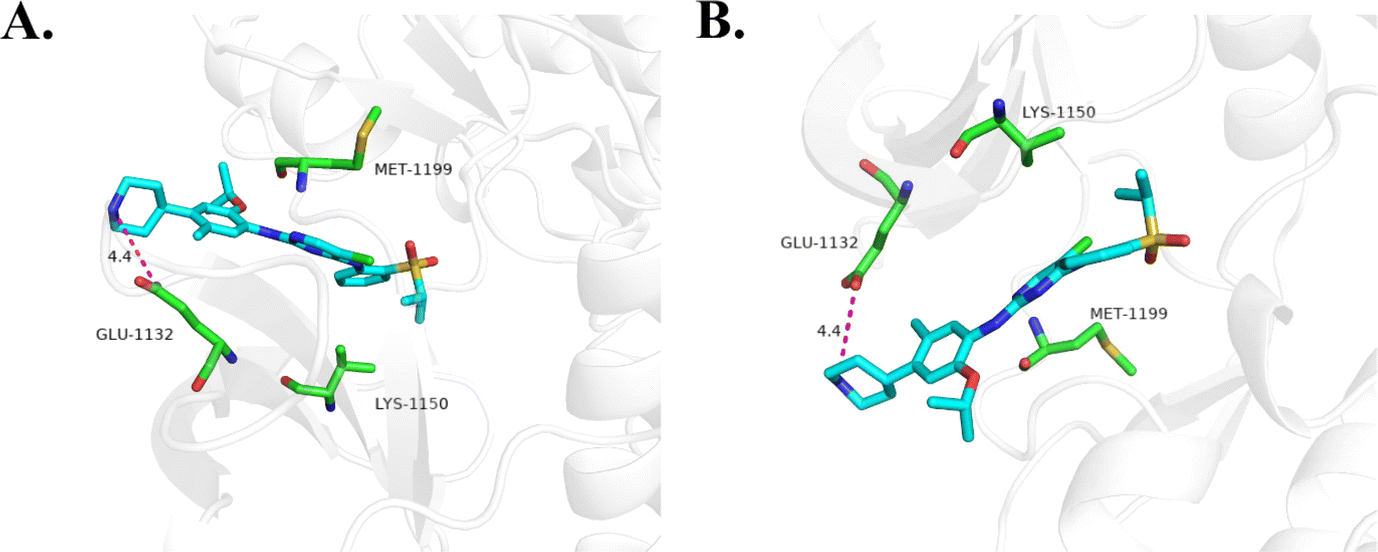 | ||
| Fig. 10 (A) Molecular docking of 18d with ALK kinase. (B) Molecular docking of ceritinib with ALK kinase. | ||
Conclusion
To develop new anti-cancer compounds with better activity and fewer side effects, we took the second-generation ALK inhibitor ceritinib as the lead and adopted strategies such as structural transformation and configuration control to synthesize a series of compounds with the pyrimidine core and sulfoxide group. Using CCK8 and MTT methods, Ba/F3-ALK, and the human non-small cell lung cancer cell line NCI-H2228, the anti-proliferative activity of the synthesized compounds was assayed with ceritinib as the reference. The results showed that most compounds exhibited good anti-tumor activity, and among them, 18a–d showed slightly better activity than the reference. We conducted anti-tumor experiments on compounds 18a–d in H2228 cell-modeled nude mice, and the results showed that the in vivo activities of all four compounds were superior to those of positive drugs. We also conducted the assay of the optimal compounds on human normal cell activity, and the results showed that the optimal compounds had lower IC50 values of anti-proliferative activity compared to the reference. The mechanism study showed that 18d could result in cell apoptosis through the mitochondrial pathway. Moreover, 18d also arrested the cancer cell cycle in the G0-G1 phase. In the cell signal pathway, 18d could reduce the content of activated p-ALK, and p-STAT-3, without affecting ALK and STAT-3. In addition, preliminary studies on the toxicity of compound 18d revealed that its toxicity toward several normal human cells was lower than that of the positive drug ceritinib. All these results indicate that compounds 18a–d are good anti-tumor lead compounds and are worthy of further study.Statistical analysis
All images were quantified by image J and the data analysis was conducted using GraphPad Prism 8. All experiments were repeated three times independently unless specifically indicated. The data are shown as the mean ± SD, and the unpaired two-way t-test (assuming all data with the same scatter) was used to determine the levels of significance between comparison samples. p > 0.05 was not significant (ns). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.Animal ethics
All experiments were carried out in accordance with the ethical requirements of laboratory animals. Laboratory animal use license number: SYXK (Guangdong) 2020-0102. Animal test certificate number: 00279323. After the end of the experiment, the animal carcasses were treated uniformly and harmlessly.Author contributions
Han Yao is responsible for biology evaluation and animal experiment. Yuanyuan Ren is responsible for compound synthesis. Feng Wu carrys on structural characterization and elucidation of compounds. Jiadai Liu undertakes the docking part. Longcai Cao undertakes statistical analysis. Ming Yan provides financial support and overall planning. Xingshu Li propses the design and idea of the whole study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by National Natural Science Foundation of China, grant number 82061128001.References
- J. J. Lin, G. J. Riely and A. T. Shaw, Targeting ALK: Precision Medicine Takes on Drug Resistance, Cancer Discovery, 2017, 7(2), 137–155 CrossRef CAS PubMed.
- T. Li, S. E. Stayrook and Y. Tsutsui, et al., Structural basis for ligand reception by anaplastic lymphoma kinase, Nature, 2021, 600(7887), 148–152 CrossRef CAS PubMed.
- A. V. Reshetnyak, P. Rossi and A. G. Myasnikov, et al., Mechanism for the activation of the anaplastic lymphoma kinase receptor, Nature, 2021, 600(7887), 153–157 CrossRef CAS PubMed.
- N. Haratake, G. Toyokawa and T. Seto, et al., The mechanisms of resistance to second- and third-generation ALK inhibitors and strategies to overcome such resistance, Expert Rev. Anticancer Ther., 2021, 21(9), 975–988 CrossRef CAS PubMed.
- S. M. Barrows, K. Wright and C. Copley-Merriman, et al., Systematic review of sequencing of ALK inhibitors in ALK-positive non-small-cell lung cancer, Lung Cancer, 2019, 10, 11–20 CAS.
- X. Du, Y. Shao and H. F. Qin, et al., ALK-rearrangement in non-small-cell lung cancer (NSCLC), Thorac. Cancer, 2018, 9(4), 423–430 CrossRef CAS PubMed.
- R. Chiarle, C. Martinengo and C. Mastini, et al., The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination, Nat. Med., 2008, 14(6), 676–680 CrossRef CAS PubMed.
- T. H. Marsilje, W. Pei and B. Chen, et al., Synthesis, structure–activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials, J. Med. Chem., 2013, 56(14), 5675–5690 CrossRef CAS PubMed.
- S. Peters, D. R. Camidge and A. T. Shaw, et al., Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer, N. Engl. J. Med., 2017, 377(9), 829–838 CrossRef CAS PubMed.
- D. R. Camidge, H. R. Kim and M. J. Ahn, et al., Brigatinib versus Crizotinib in Advanced ALK Inhibitor-Naive ALK-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial, J. Clin. Oncol., 2020, 38(31), 3592–3603 CrossRef CAS PubMed.
- R. Bentley, Role of sulfur chirality in the chemical processes of biology, Chem. Soc. Rev., 2005, 34(7), 609–624 RSC.
- W. R. F. Goundry, B. Adams and H. Benson, et al., Development and Scale-up of a Biocatalytic Process To Form a Chiral Sulfoxide, Org. Process Res. Dev., 2017, 21(1), 107–113 CrossRef CAS.
- T. Matsui, Y. Dekishima and M. Ueda, Biotechnological production of chiral organic sulfoxides: current state and perspectives, Appl. Microbiol. Biotechnol., 2014, 98(18), 7699–7706 CrossRef CAS PubMed.
- H. J. Federsel, Facing chirality in the 21st century: approaching the challenges in the pharmaceutical industry, Chirality, 2003, 15, S128–S142 CrossRef CAS PubMed.
- K. Okamoto and T. Nishino, Mechanism of inhibition of xanthine oxidase with a new tight binding inhibitor, J. Biol. Chem., 1995, 270(14), 7816–7821 CrossRef CAS PubMed.
- M. Matsugi, R. Shimada and S. Ohata, et al., The improved synthesis of OPC-29030, a platelet adhesion inhibitor via diastereoselective oxidation of chiral non-racemic sulfide, Chem. Pharm. Bull., 2002, 50(11), 1511–1513 CrossRef CAS PubMed.
- Y. Ozeki, Y. Nagamura and H. Ito, et al., An anti-platelet agent, OPC-29030, inhibits translocation of 12-lipoxygenase and 12-hydroxyeicosatetraenoic acid production in human platelets, Br. J. Pharmacol., 1999, 128(8), 1699–1704 CrossRef CAS PubMed.
- C. L. Defty and J. R. Marsden, Melphalan in regional chemotherapy for locally recurrent metastatic melanoma, Curr. Top. Med. Chem., 2012, 12(1), 53–60 CrossRef CAS PubMed.
- AstraZeneca UK Limited and AstraZeneca AB. Swed, Morpholinio Pyrimidines and Their Use in Therapy, WO2011154737A1, 2011 Search PubMed.
- A. T. Shaw, T. M. Bauer and F. De Marinis, et al., First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer, N. Engl. J. Med., 2020, 383(21), 2018–2029 CrossRef CAS PubMed.
- K. Iikubo, Y. Kondoh and I. Shimada, et al., Discovery of N-{2-Methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}-N′-[2-(propane-2-sulfonyl)phenyl]-1,3,5-triazine-2,4-diamine (ASP3026), a Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor, Chem. Pharm. Bull., 2018, 66(3), 251–262 CrossRef PubMed.
- J. Wang, M. Sánchez-Roselló and J. L. Aceña, et al., Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011), Chem. Rev., 2014, 114(4), 2432–2506 CrossRef CAS PubMed.
- S. Purser, P. R. Moore and S. Swallow, et al., Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37(2), 320–330 RSC.
- R. Peters, Carbon-fluorine compounds: chemistry, biochemistry and biological activities, Ciba Found. Symp., 1971, 2, 1–7 CAS.
- X. Zhang, T. Yang, X. Jin, K. Lin, X. Dai, T. Gao, G. Huang, M. Fan, L. Ma, Z. Liu and J. Cao, Synthesis and biological evaluation of cytotoxic activity of novel podophyllotoxin derivatives incorporating piperazinyl-cinnamic amide moieties, Bioorg. Chem., 2022, 123, 105761, DOI:10.1016/j.bioorg.2022.105761.
- Q. Dou, H. N. Chen, K. Wang, K. Yuan, Y. Lei, K. Li, J. Lan, Y. Chen, Z. Huang, N. Xie, L. Zhang, R. Xiang, E. C. Nice, Y. Wei and C. Huang, Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer, Cancer Res., 2016, 76(15), 4457–4469, DOI:10.1158/0008-5472.CAN-15-2887.
- X. Wang, X. Lu and R. Zhu, et al., Betulinic Acid Induces Apoptosis in Differentiated PC12 Cells via ROS-Mediated Mitochondrial Pathway, Neurochem Res, 2017, 42, 1130–1140, DOI:10.1007/s11064-016-2147; J. Lopez and S. W. Tait, Mitochondrial apoptosis: killing cancer using the enemy within, Br. J. Cancer, 2015, 112(6), 957–962, DOI:10.1038/bjc.2015.85.
- Y. Lu, Z. Fan and S. J. Zhu, et al., A new ALK inhibitor overcomes resistance to first- and second-generation inhibitors in NSCLC, EMBO Mol. Med., 2022, 14(1), e14296, DOI:10.15252/emmm.202114296.
- T. Pan, Y. Dan and D. Guo, et al., Discovery of 2,4-pyrimidinediamine derivatives as potent dual inhibitors of ALK and HDAC, Eur. J. Med. Chem., 2021, 224, 113672, DOI:10.1016/j.ejmech.2021.113672.
- E. F. Mesaros, T. V. Thieu and G. J. Wells, et al., Strategies to mitigate the bioactivation of 2-anilino-7-aryl-pyrrolo[2,1-f][1,2,4]triazines: identification of orally bioavailable, efficacious ALK inhibitors, J. Med. Chem., 2012, 55(1), 115–125, DOI:10.1021/jm2010767.
- We encourage the citation of primary research over review articles, where appropriate, in order to give credit to those who first reported a finding. Find out more about our commitments to the principles of San Francisco Declaration on Research Assessment (DORA).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01556h |
| This journal is © The Royal Society of Chemistry 2024 |