
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-hydrothermal carbonization of pretreated sludge and polyethylene terephthalate for the preparation of low-nitrogen clean solid fuels†
Ting Yeab,
Le Gouab,
Yue Wangab,
Nan Liuab,
Liyi Dai*ab and
Yuanyuan Wang *ab
*ab
aState Key Laboratory of Petroleum Molecular & Process Engineering, East China Normal University, No. 500 Dongchuan Road, Shanghai 200241, P. R. China. E-mail: ecnu_yywang@163.com; dai_liyi@163.com
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai 200062, P. R. China
First published on 29th May 2024
Abstract
In this work, polyethylene terephthalate (PET) and sewage sludge (SS) were co-hydrothermally carbonized to produce low-nitrogen solid fuels. To minimize the effect of nitrogen, this work introduces a co-hydrothermal carbonization method involving alkali (A), ultrasonic cell disruptor (UCC), and sodium dodecyl sulfate (SDS) for both individual and combined pretreatment of SS and PET. Comparative analysis of the products shows that the combined pretreatment with sodium dodecyl sulfate (SDS) and alkali (A) effectively disrupts the SS cell structure, leading to the loosening of stable extracellular polymeric substances (EPS). This condition is conducive to the release and hydrolysis of proteins during hydrothermal carbonization. Moreover, under conditions where PET serves both as an acid producer and a carbon source, and through parameter optimization at a temperature of 240 °C, reaction time of 2 h, PET addition of 20 wt%, and water addition of 0.6 g cm−3, a high-quality, low-nitrogen clean solid fuel was produced (N: 0.51 wt%, C: 19.10 wt%).
1 Introduction
Since the industrial revolution, the global population has increased, cities have developed, and sewage treatment plants have been constructed, resulting in a rising amount of sewage sludge (SS).1 By 2025, it is estimated that China's annual SS production will exceed 90 million tons (calculated based on 80% moisture content), but approximately 80% of SS remains untreated.2 SS contains numerous components; the primary biochemical components are proteins, carbohydrates, and lipids, among others. It also contains various contaminants, including pathogens, heavy metals, and pharmaceutical residues.3 Hydrothermal carbonization (HTC) technology is an effective method for treating high-moisture organic solid wastes, offering advantages such as water as the reaction medium, no need for drying pre-treatment, and a mild reaction temperature (160 °C to 300 °C).4 The resulting hydrochar material has significant potential as a fuel. HTC disrupts the floc structure of SS under high temperature and pressure, rupturing the cell structure and releasing a large amount of organic matter. These organic materials first undergo dehydration and decarboxylation reactions to produce intermediate products. These intermediates then successively undergo polymerization, condensation, and aromatization reactions, gradually converting into stable carbon structures through subsequent secondary carbonization reactions and integration with solid products.4,5 However, due to the high protein content in SS, the nitrogen—primarily concentrated in proteins—is not readily released during HTC treatment, resulting in a high nitrogen content in the hydrochar.6Current research widely employs catalysts to enhance the denitrogenation efficiency and performance of HTC. Typical catalysts include calcium oxide (CaO)7 and hydrotalcite.8 Furthermore, modifying metal ratios in the catalyst to adjust acidity and basicity sites can effectively reduce nitrogen content. For example, Zhang et al. developed a Ni–Mg–Al layered double oxide catalyst. Acidic sites inhibit the Maillard reaction, whereas basic sites promote the decomposition of extracellular polymeric substances (EPS), yielding a minimum nitrogen content of 1.39 wt%.9 Beyond catalysts, enhancing denitrification efficiency can also be achieved by adding oxidizing agents (fly ash and H2O2)10 and deep eutectic solvents (DESs: ZnCl2 and urea)11 during the HTC process. However, these studies reveal that despite using catalysts, oxidants, or altering solvents in the HTC process, the nitrogen content of hydrochar still exceeds 1 wt%, rendering it too high for fuel applications.6 Moreover, separating the catalyst from the hydrochar poses challenges that may impair combustion performance, and oxidants are rapidly depleted during the HTC process, thereby limiting denitrogenation efficiency.
Co-hydrothermal carbonization (co-HTC) effectively enhances hydrochar fuel performance and mitigates issues associated with single raw materials. Prior studies on co-HTC have primarily concentrated on enhancing the heating value of hydrochar, with processes such as co-HTC of SS with kitchen waste12 and SS with fruit and agricultural waste13 achieving heating values of 22.87 MJ kg−1 and 21.72 MJ kg−1, respectively. However, the nitrogen content in the results from the aforementioned studies generally exceeds 2 wt%. Given the increasing emphasis on environmental concerns, reducing the nitrogen content in hydrochar is essential, alongside improvements to the higher heating value (HHV) using co-HTC technology. For example, Zhang et al. explored the incorporation of acetic acid and ethanol in co-HTC of SS and sawdust to produce clean solid fuels. The findings indicated that acetic acid catalytically deaminates solid nitrogen into liquid form, reducing the nitrogen content to a minimum of 1.35 wt%. Furthermore, ethanol enhances the fixed carbon content and HHV of hydrochar, achieving a maximum HHV of 15.56 MJ kg−1.14 Still, its nitrogen content was also fast approaching 1 wt%, which was not optimal. This is due to the fact that EPS are unique components of SS, in which carbohydrates and proteins are the main components of EPS,15 and the EPS structure of SS is very stable and not easily destroyed in HTC and co-HTC, resulting in the difficulty of releasing nitrogen concentrated on proteins,8 which affects the performance of the finally obtained solid fuels.
Leveraging the ability of acids to enhance denitrogenation efficiency in the HTC process, identifying an additive that acts both as a carbon source and an acid producer in the co-HTC process is a strategic approach to improving the performance of clean solid fuels. It Polyethylene terephthalate (PET) was found to undergo hydrolysis and ester group cleavage under HTC conditions at 240 °C, generating polymer monomers benzene dicarboxylic acid (TPA) and ethylene glycol. This process subsequently created an acidic environment.16 Consequently, co-HTC with SS enhances protein hydrolysis, nitrogen release, and denitrification efficiency in SS. However, protein release is impeded by the stabilized EPS structure of SS. Therefore, in this study, SS underwent pretreatment before co-HTC to destabilize the EPS, facilitating subsequent denitrification during co-HTC. Previous studies have explored co-HTC using pretreated feedstocks. For instance, Xue et al.17 investigated the production of clean solid fuels through co-HTC of straw pretreated with acid and alkali, combined with PVC. Their findings indicated that acid-base pretreatment enhanced co-HTC's synergistic effects and significantly improved the combustion characteristics of the resulting solid products, with the HHV rising from 26.89 MJ kg−1 to 30.83 MJ kg−1. Traditional alkali (A) treatment and ultrasonic cell-crushing (UCC) treatment are more effective in disrupting the EPS of SS.18 The surfactant sodium dodecyl sulfate (SDS), which interacts with cell membrane proteins through hydrophobic interactions to promote the release of proteins and polysaccharides, can also be used for SS pretreatment.19 Additionally, HTC of pretreated SS to produce high-quality clean solid fuels has not yet been reported.
Based on these findings, we investigated the extent of destruction of SS cellular structure by individual and combined pretreatments. We also examined the nitrogen content in the hydrochar of pretreated SS and the forms of nitrogen in the liquid-phase products to hypothesize the impact of SS structure on nitrogen migration during HTC. Subsequently, we produced a high-quality, low-nitrogen solid fuel by co-carbonizing solid waste PET with pretreated SS.
2 Experiment
2.1 Materials
The basic properties of the activated SS (Xi'an Capital Water Co. Ltd in Xi'an, Shanxi Province, China) were as follows: water content of 83.2%, ash content of 48.84%, volatile matter (VM) of 46.59%, fixed carbon (FC) of 4.57%, and stored at 4 °C; PET was purchased from China Resources Chemical Materials Technology Co. Ltd in Changzhou City, Jiangsu Province, China. Sodium hydroxide (NaOH >99%) and sodium dodecyl sulfate (SDS >99%) were purchased from Sinopharm Chemical Reagent Co.2.2 Experimental procedure
NaOH solution (4 mol L−1) adjusted the pH during pretreatment. As using an ultrasonic cell disruptor (S-250D, Branson) can cause an increase in temperature, the pretreatment experiments were not temperature-controlled. To ensure the purity and stability of the samples, after all pretreatments were completed, distilled water was used to wash the samples to remove any reagents added during the reaction. The samples were then centrifuged at 6000 rpm for 10 min using a centrifuge (TG16-WS, Hunan Xiangyi Laboratory Instrument Development Co., Ltd) to obtain wet SS for subsequent HTC treatment.
The co-HTC experiment was conducted by mixing 4 g of feedstock with varying deionized water contents under a high nitrogen concentration, then sealing and heating them individually to the reaction temperature at a stirring speed of 300 rpm. The 4 g represents the total dry basis mass of pretreated SS and PET in varying ratios. Deionized water content was determined by the water amount per unit volume of the reactor, with 30 mL equating to a density of 0.6 g cm−3. The controlled variable method was employed in this experiment to assess the effects of various reaction ratios (PET content: 0, 20, 50, 80, 100 wt%), temperatures (180, 210, 240, 270, 300 °C), durations (0.5, 1, 2, 4 h), and water contents (0.5, 0.6, 0.7, 0.8 g cm−3) on HTC. After the reaction, the reactor was cooled to room temperature using cold water, and the solid and liquid samples were separated by vacuum filtration. Before further product testing, the solid samples were dried in an oven at 105 °C for 18 h, then ground into powder. The liquid samples were stored at 4 °C, requiring thorough shaking and filtration through a 0.45 μm filter membrane at least three times prior to testing. Samples were labeled SP-a-b-c-d to simplify subsequent descriptions, where ‘S’ and ‘P’ represent pretreated SS and PET, respectively. ‘a’ indicates the PET proportion of total mass, ‘b’ the reaction temperature (°C), ‘c’ the reaction time (h), and ‘d’ the water content (g cm−3). SP-0.2-210-1-0.6 denotes a sample with 20 wt% PET, a reaction temperature of 210 °C, a reaction time of 1 h, and a water content of 0.6 g cm−3. All experiments were conducted at least three times to ensure reproducibility and consistency.
2.3 Analytical methods
Total Organic Carbon (TOC) and Total Carbon (TC) in the liquid products were determined using a TOC analyzer (TOC-L CPN, Shimadzu/Suzhou). hydrochar’s C, H, O, N, and S contents were determined using an elemental analyzer (Vario Micro cube CHNS/UNICUBE). Fourier transform infrared spectroscopy (Nicolet NEXUS 670 Thermo Fisher Scientific) tested functional groups of hydrochar, and the samples were dried entirely before testing. A biomicroscope (FPMRC-Panthera Mc Auldie Industrial Group Ltd) was used to observe the changes before and after the pretreatment of the wet SS structure. The NH4+-N and NO3−-N in the samples were determined using the San plus continuous flow analyzer from Skalar, the Netherlands, in which the Cd–Cu reduction method was used for NO3−-N and the indophenol blue method was used for NH4+-N. Quality control was implemented during the experiments, and the analytical error was less than 5%. Total nitrogen (TN) was analyzed by alkaline potassium persulfate digestion-ultraviolet spectrophotometry (HJ636-2012) with the instrument model TU-1810 PC, and dissolved organic nitrogen (DON) = TN − NH4+ −NO3− in the liquid product, calculated according to Xu et al.20 Shen et al.21 The thermal properties of the samples were analyzed using a thermogravimetric analyzer (TGA/SDTA851e, Mettler Toledo Instruments). The samples were heated up to 800 °C at room temperature (25 °C) with a heating rate of 20 °C min−1 and a flow rate of 50 mL min−1.2.4 Calculation methods
The rules for calculating mass yield (Yield), calorific value (HHV), energy recovery (ER), and denitrification efficiency (ηN) for hydrochar are referenced from Xie et al.22 as follows:
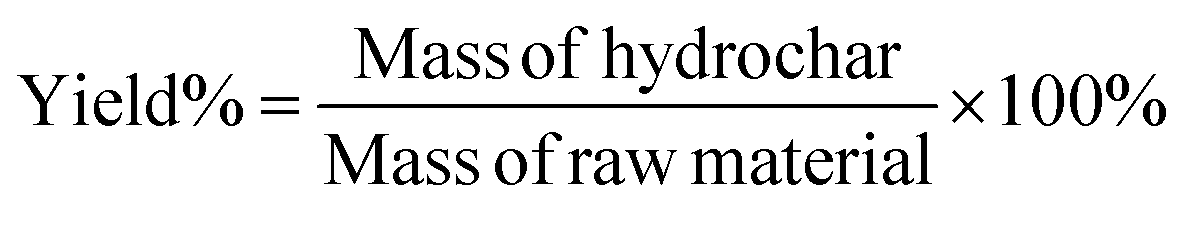 | (1) |
Mass of hydrochar and mass of raw material were both calculated on a dry mass basis. In the HTC experiments, mass of raw materials represented the dry mass of SS, while in the co-HTC experiments, Mass of raw materials comprised the total dry mass of SS and PET, totaling 4 g HHV(c) and HHV(r) represent the calorific value of hydrochar and raw material, respectively.
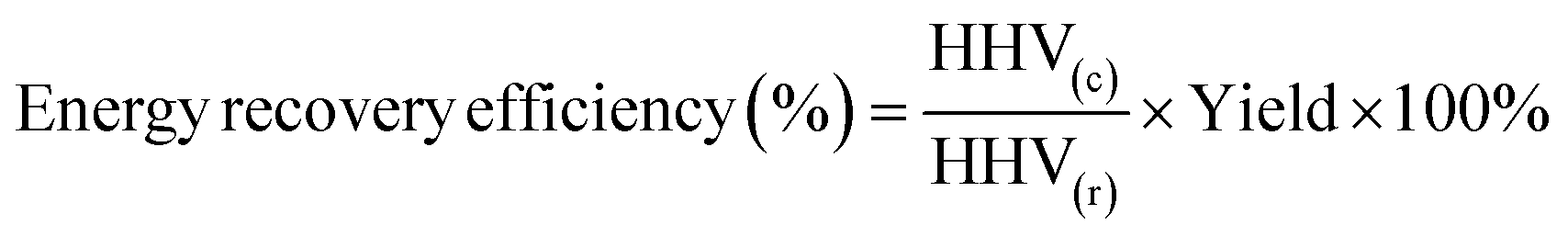 | (2) |
| HHV (MJ kg−1) = 0.335[C] + 1.423[H] − 0.145[N] − 0.154[O] | (3) |
[C], [H], [N], and [O] represent the amount of carbon, hydrogen, nitrogen, and oxygen in the sample (wt%), respectively.
The equation for ηN involved in the HTC experiment is as follows:
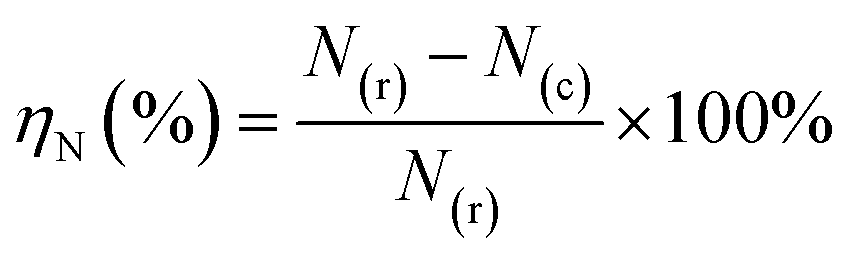 | (4) |
N(r) and N(c) represent the nitrogen content (wt%) in raw material and hydrochar, respectively.
The equation for ηN involved in the co-HTC experiment is as follows:
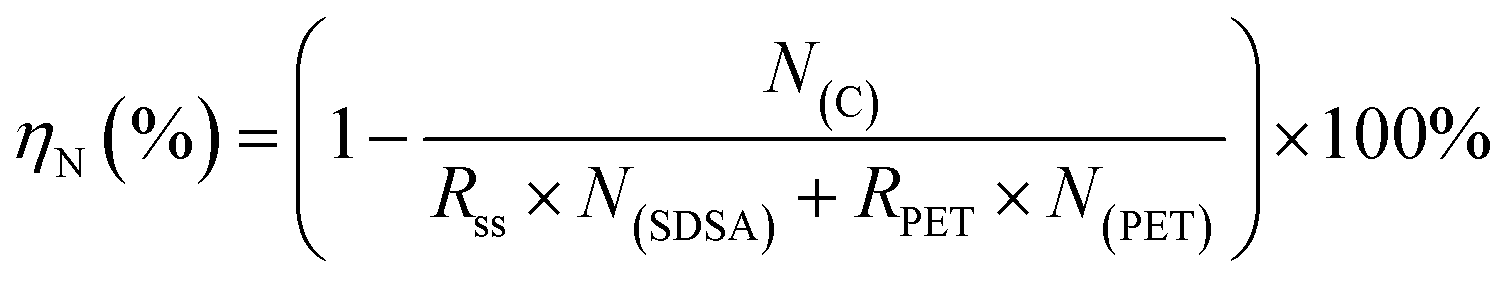 | (5) |
RSDSA and RPET are the percentage of SDSA and PET in the feedstock, respectively; N(SDSA) and N(PET) are the nitrogen content (wt%) in SDSA and PET, respectively.
The TG-DTG curve and the comprehensive combustion index calculation method are based on GB/T 33304-2016, the index (S) is used to evaluate the combustion performance of solid fuels, and the calculation formula is as follows:
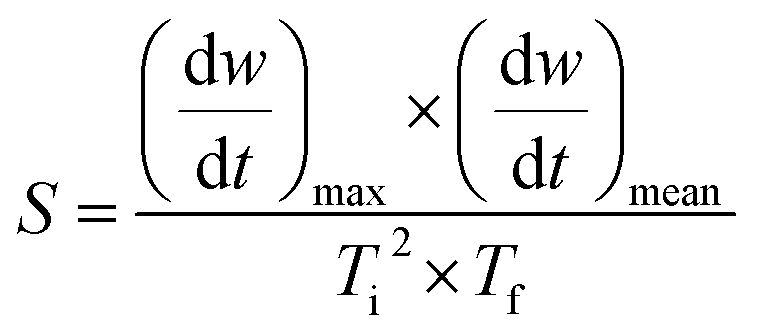 | (6) |
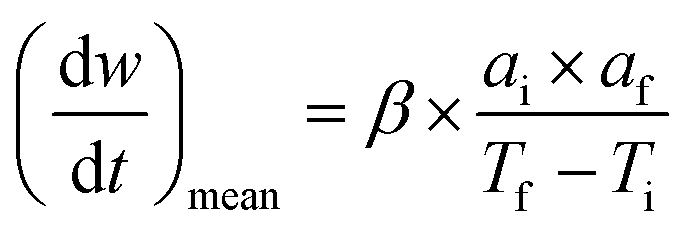 | (7) |
Ti and Tf are the ignition and burnout temperatures, respectively; af and ai are the hydrochar residuals corresponding to Tf and Ti, respectively, (dw/dt)max and (dw/dt)mean are the maximum and average combustion rates, respectively; β is the rate of warming (20 °C min−1)
| Tmass residue = Mmass residue SDSA × Rss + Mmass residue PET × RPET | (8) |
Tmass residue is the theoretical value of combustion residue. Mmass residue SDSA and Mmass residue PET are the combustion residues of SDSA and PET hydrochar obtained by thermogravimetric testing.
3 Results and discussion
3.1 Effect of different pretreatment on sludge properties
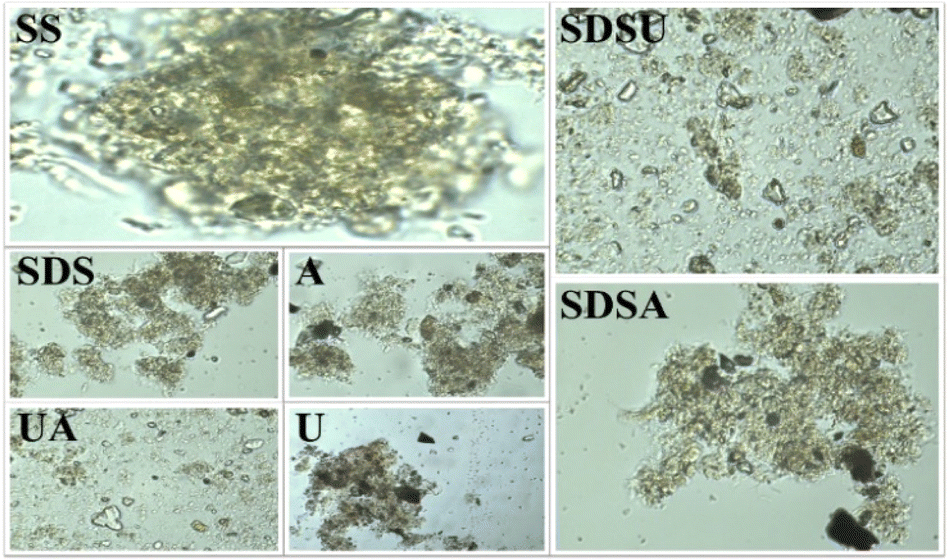 | ||
| Fig. 1 Microscopic picture of SS before and after pretreatment. | ||
| Sample name | Ultimate analysis results (wt%) | H/C | N/C | O/C | HHV (MJ kg−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| C | H | N | S | O | |||||
| SS | 23.28 ± 0.24 | 4.09 ± 0.07 | 3.76 ± 0.06 | 0.90 ± 0.02 | 23.99 ± 0.26 | 2.11 | 0.14 | 0.77 | 9.37 |
| SDS | 17.53 ± 0.16 | 3.02 ± 0.09 | 2.75 ± 0.05 | 0.33 ± 0.01 | 22.76 ± 0.24 | 2.07 | 0.13 | 0.97 | 6.27 |
| A | 19.65 ± 0.16 | 3.22 ± 0.07 | 2.85 ± 0.06 | 0.36 ± 0.01 | 22.01 ± 0.22 | 1.97 | 0.12 | 0.84 | 7.37 |
| U | 22.45 ± 0.24 | 3.42 ± 0.04 | 3.01 ± 0.05 | 0.44 ± 0.02 | 24.30 ± 0.24 | 1.83 | 0.11 | 0.81 | 8.21 |
| SDSA | 22.13 ± 0.22 | 3.91 ± 0.08 | 2.51 ± 0.06 | 0.78 ± 0.02 | 22.22 ± 0.18 | 2.12 | 0.10 | 0.75 | 9.19 |
| UA | 10.52 ± 0.11 | 1.97 ± 0.02 | 1.15 ± 0.03 | 0.42 ± 0.01 | 15.04 ± 0.26 | 2.25 | 0.09 | 1.07 | 3.85 |
| SDSU | 13.80 ± 0.10 | 2.34 ± 0.06 | 1.46 ± 0.02 | 0.42 ± 0.01 | 15.20 ± 0.17 | 2.03 | 0.09 | 0.83 | 5.40 |
| PET | 61.10 ± 0.37 | 3.78 ± 0.07 | 0 | <d. l. | 33.49 ± 0.34 | 0.74 | 0 | 0.41 | 20.69 |
To enhance the effectiveness, we combined the three individual methods in two to pretreat the SS. Observations from Fig. 1 show that after SDSA pretreatment, the SS morphology alters, with flocculent material beginning to disaggregate and detach, thus loosening the structure. Following SDSU and UA pretreatments, the morphology of the SS is completely transformed, breaking down into numerous small fragments without any flocculent material, indicating almost complete disintegration of the EPS and cell walls. The extent of destruction escalates sequentially. Despite SDSA and UA sharing alkaline conditions, the cell structure is more profoundly disrupted in UA, presenting lower carbon and nitrogen contents and N/C ratio. This is attributed to the shear forces generated by ultrasound in UCC, which can decompose the EPS in SS, releasing proteins, carbohydrates, and cells into the alkaline environment and thus accelerating the hydrolysis rate. This leads to a significant dissolution of organic matter in the liquid phase.24 Comparing the values before and after the different pretreatments in Table 1, it can be seen that both N content and N/C decreased, but the effects were different. For example, the release of N content in SS by individual pretreatment is not as good as that by combined pretreatment methods. The O and N content of SDSU and UA in the combined pretreatment methods decreased the most, which is consistent with the change in SS cell structure in Fig. 1.
Observations from Table 1 reveal that SDS, A, U, and SDSA, with undamaged cell walls, exhibited minimal changes in O content, while UA and SDSU, characterized by severe cell wall disruption and EPS structural breakdown, showed reduced O content. This suggests that intact cell walls inhibit the deep dehydration of SS. However, the O/C values for UA and SDSU increased, likely due to a more pronounced loss of proteins and lipids, which have higher O/C values, compared to polysaccharides. The S content in SS primarily originates from proteins,8 which substantiates that various pretreatments predominantly degrade the EPS and cell wall structures of SS, resulting in the release of proteins, thus facilitating hydrolysis and reducing N and S levels. In summary, combined pretreatments are more effective than individual ones in terms of structural destruction and the release of N content. Consequently, we proceeded with HTC of SS following combined pretreatments to identify the optimal method for producing hydrochar with reduced N content.
Observing Table S1,† at the same temperature, the pretreated SS liquid phase exhibited the highest TC values with SDSA-210 at 9.97 g L−1. This effect is attributed to the fact that during the pre-treatment stage, SDSA induces a transition of the EPS in the SS from a stable to an unstable state, with a moderate degree of destruction, thereby preventing significant loss of organic matter. Consequently, in subsequent HTC processes, further destruction of the EPS occurs, facilitating the release and hydrolysis of recalcitrant proteins, which results in increased TC and reduced N content. The relatively low TC values of UA-210 (7.79 g L−1) and SDSU-210 (7.91 g L−1) were due to UA and SDSU inflicting severe damage to the SS's EPS, including cell wall rupture, leading to early loss of internal organic matter and precluding further losses in the liquid phase during HTC.
Table S1† displays the denitrification efficiency (ηN) of both the original and pre-treated SS during HTC across varying reaction temperatures. As the reaction temperature increased, the ηN correspondingly increased. The ηN of AU and SDSU was lower than that of the original SS due to the severe damage these treatments inflicted on the SS structure, resulting in significant nitrogen loss during the pre-treatment stage. Conversely, SDSA, which only moderately disrupted the SS structure, had the highest ηN (SDSA-270: 65.74%). This indicates that ηN during HTC is closely associated with the degree of SS damage incurred during the pre-treatment stage; moderate damage can enhance ηN in subsequent HTC processes, whereas severe damage may lead to excessive nitrogen loss, thereby reducing ηN. Fig. 2 displays the overall ηN for the combined pre-treatment and HTC process. Overall, the ηN achieved by the combined pre-treatment and HTC process was significantly higher than that observed with the HTC process alone.
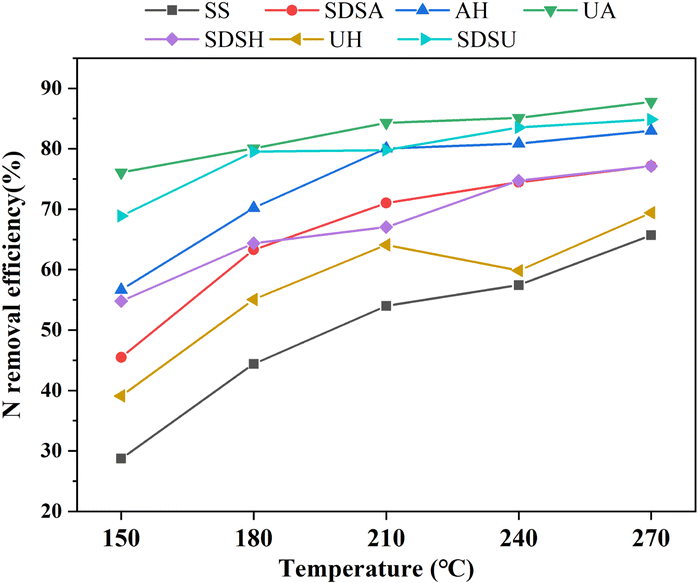 | ||
| Fig. 2 Nitrogen removal efficiency of sludge treated by combined pretreatment and hydrothermal carbonization. | ||
Combined with Table S1,† the N and S contents of pretreated SS hydrochar are lower than that of the original SS hydrochar, so the combustion of hydrochar with low N and S contents can vastly reduce the pollution of the environment. Among them, UA-270 has the lowest N content (0.46 wt%) and the highest ηN (87.8%), and the efficiency is improved by 22.1% compared with ηN (65.7%) of SS-270 at the same temperature. The effects of SDSU-270 (N: 0.57 wt%, ηN: 84.8%) and SDSA-270 (N: 0.86 wt%, ηN: 77.1%) were also good. However, observing Table S1,† it is found that the C content of the hydrochar from UA and SDSU is much lower than that of the hydrochar from the original SS, even to a single digit, while the C content of the hydrochar from SDSA decreased very little. Therefore, if one wants to obtain low-nitrogen and high-carbon hydrochar for use as fuel, one should not only pursue low nitrogen but also moderately damage the SS cell structure to prevent excessive loss of carbon content. Therefore, considering all factors, SDSA is the preferred pre-treatment method before subsequent co-HTC.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –CN– functional groups,20,29 respectively, and the intensity of the peaks of all hydrochars diminishes with increasing temperature. It suggests that elevated temperature promotes dehydration and decarboxylation, which enhances the dehydration reaction between hydroxyl-containing sugars and carboxyl-containing proteins or organic polymers, such as organic acids, in the SS; the carbonyl group is converted to CO2 for release into the gas phase.30 As a result, the content of oxygen-containing functional groups such as hydroxyl group, carboxyl group and carbonyl group in the hydrochar decreases, the H and O content in the hydrochar does not decrease, and the values of H/C and O/C decrease, which contributes to the gradual enhancement of the aromaticity of the hydrochar. The decrease of –CN– functional groups indicates that proteins and amino acids are further decomposed at high temperatures, nitrogen-containing substances are transferred to the liquid phase, and the N content of hydrochar decreases.31 All these changes are consistent with the trend of changes in Table S1.† However, comparing SS, the intensity of the peaks where UA and SDSU are located in –OH becomes weaker, which is consistent with the previous elemental analysis, suggesting that the cell wall prevents SS from deep dehydration.
O and –CN– functional groups,20,29 respectively, and the intensity of the peaks of all hydrochars diminishes with increasing temperature. It suggests that elevated temperature promotes dehydration and decarboxylation, which enhances the dehydration reaction between hydroxyl-containing sugars and carboxyl-containing proteins or organic polymers, such as organic acids, in the SS; the carbonyl group is converted to CO2 for release into the gas phase.30 As a result, the content of oxygen-containing functional groups such as hydroxyl group, carboxyl group and carbonyl group in the hydrochar decreases, the H and O content in the hydrochar does not decrease, and the values of H/C and O/C decrease, which contributes to the gradual enhancement of the aromaticity of the hydrochar. The decrease of –CN– functional groups indicates that proteins and amino acids are further decomposed at high temperatures, nitrogen-containing substances are transferred to the liquid phase, and the N content of hydrochar decreases.31 All these changes are consistent with the trend of changes in Table S1.† However, comparing SS, the intensity of the peaks where UA and SDSU are located in –OH becomes weaker, which is consistent with the previous elemental analysis, suggesting that the cell wall prevents SS from deep dehydration.
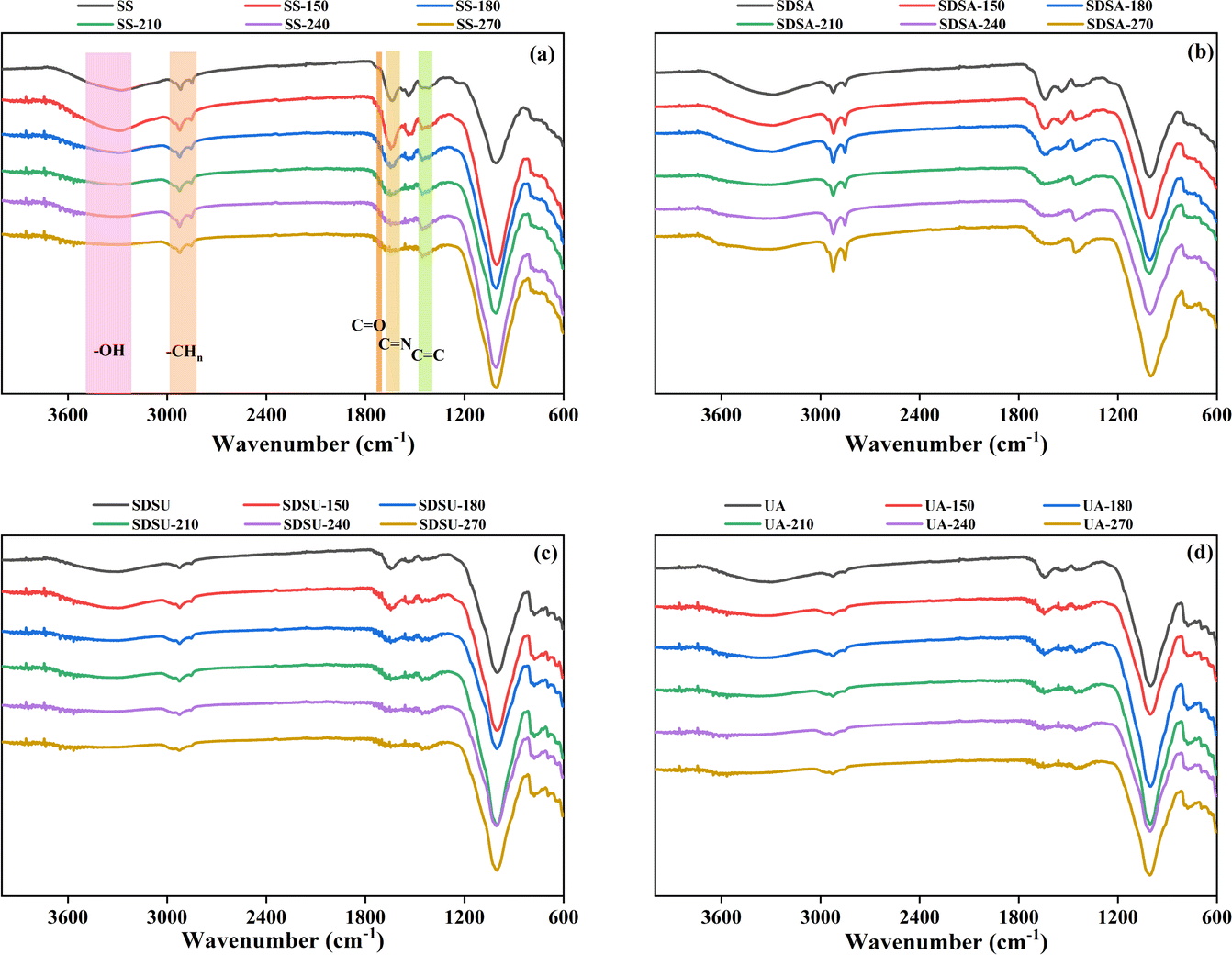 | ||
| Fig. 3 FT-IR spectra of hydrochars: (a) SS; (b) SDSA; (c) SDSU; (d) UA. | ||
The CC functional group at 1460 cm−1 attributed to the aromatic moiety, where only the original SS and SDSA showed a slight enhancement in intensity with increasing temperature, suggesting that the intermediates degraded at high temperatures further generate aromatic compounds through the aromatization reaction to enhance the carbonation.32 And compared with SS, the peak intensity of SDSA at 1460 cm−1 is deeper, while the peaks of UA and SDSU are weaker, which suggests that proper disruption of the EPS structure of SS promotes the arylation reaction of the HTC process. The absorption peaks of aliphatic compounds were located at 2920 cm−1 and 2850 cm−1,33 and compared with the original SS, the intensity of the peaks of SDSA deepened here, while the intensity of the peaks of UA and SDSU, which had been severely damaged by the EPS structure, decreased, which indicated that the appropriate destruction of the EPS structure of the SS was favorable for the generation of aliphatic compounds. It was also observed that the peaks of SDSA were enhanced with increasing temperature, and the peaks of UA and SDSU were weakened with increasing temperature, and it was hypothesized that the EPS structure was responsible for the generation of aliphatic compounds in hydrochar.
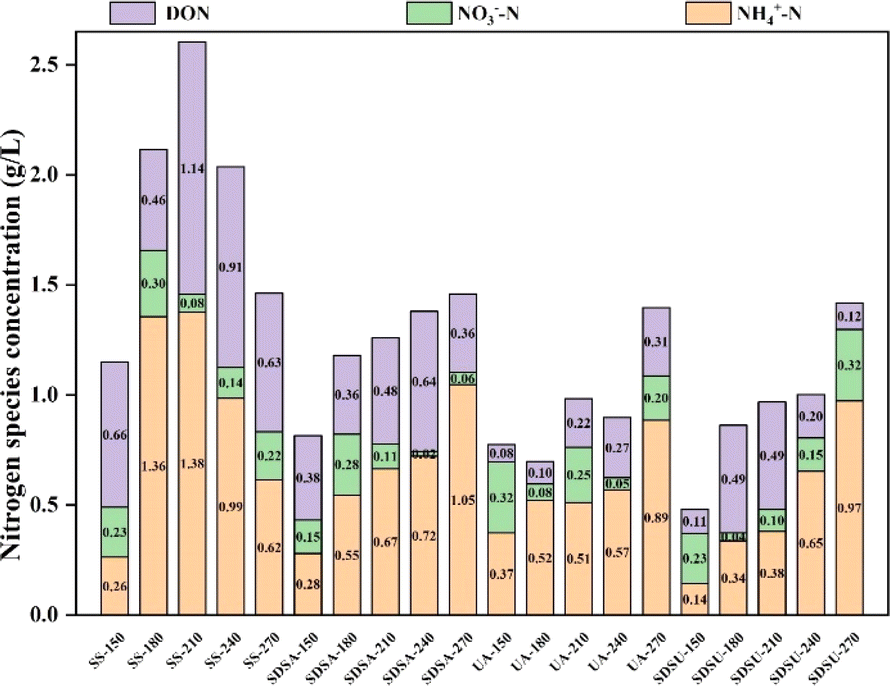 | ||
| Fig. 4 Morphological distribution of nitrogen in liquid products. | ||
Compared to the original SS, the liquid-phase DON and TN contents in the pretreated SS are significantly lower, particularly in UA and SDSU samples, which exhibit the most extensive cell structure damage and the lowest TN content. As indicated in Table 1 and Fig. 1, SDSA released fewer nitrogen-containing substances during pretreatment, which involved moderate structural damage. The ηN during the HTC process remained comparable to that of the original SS, yet the TN released in the liquid phase was lower. Consequently, it is hypothesized that SDSA pretreatment results in a greater conversion of nitrogen-containing substances to the gas phase during the HTC process, thereby increasing the proportion of nitrogen in the gas phase.
3.2 Yield and denitrogenation efficiency of SDSA co-hydrothermal carbonization with PET
Table 2 demonstrates the results of elemental analysis and yield after co-HTC of different ratios of PET with SDSA, and the rest of the results of the effect of temperature, reaction time, and water content on hydrochar are shown in Table S3 (In the ESI).† Carbon content increased, and nitrogen content decreased after co-HTC compared to SDSA alone. As the PET ratio increased, the nitrogen content decreased more significantly, and the denitrification efficiency increased, which is due to the hydrolysis reaction of PET. Kang et al.38 demonstrated that PET undergoes major hydrolysis during HTC, generating benzene dicarboxylic acid (TPA) and ethylene glycol. To verify this conclusion, pH values of the liquid phase products were tested, as presented in Table S4 (In the ESI).† In alignment with this conclusion, the pH value decreased progressively with the addition of PET, indicative of an escalating acid production from PET. TPA creates an acidic environment that favors protein hydrolysis and contributes to enhanced deamination, which reduces nitrogen content in hydrochar. Combining Tables 1 and 2, it is observed that the O content is elevated after HTC of PET alone because the solid product of PET hydrolysis is a mixture of PET residues and TPA,39 thus also leading to a higher O content of co-HTC compared to SDSA, and the calorific value content did not increase significantly, only by 1.84 MJ kg−1. It was observed that the yield from water-thermal carbonization of PET alone was significantly higher than that from SDSA.| Sample name | EA results of hydrochars (wt%) | Yield (wt%) | HHV (MJ kg−1) | ηN (%) | ||||
|---|---|---|---|---|---|---|---|---|
| C | H | N | S | O | ||||
| a PET loading proportion (wt%) (240 °C, 2 h, water loading amount = 0.6 g cm−3). | ||||||||
| SS-240-2-0.6 | 16.74 ± 0.22 | 2.16 ± 0.06 | 1.48 ± 0.03 | 0.41 ± 0.01 | 14.36 ± 0.16 | 70.24 ± 0.35 | 6.26 | 60.64 |
| SP-0-240-2-0.6 | 12.23 ± 0.32 | 1.59 ± 0.02 | 0.78 ± 0.02 | 0.24 ± 0.01 | 10.95 ± 0.16 | 74.68 ± 0.24 | 4.57 | 68.92 |
| SP-0.2-240-2-0.6 | 19.10 ± 0.37 | 1.72 ± 0.03 | 0.51 ± 0.01 | 0.15 ± 0.01 | 15.31 ± 0.36 | 72.38 ± 0.34 | 6.41 | 74.60 |
| SP-0.5-240-2-0.6 | 37.38 ± 0.48 | 2.80 ± 0.06 | 0.31 ± 0.01 | 0.09 ± 0.01 | 21.22 ± 0.22 | 69.46 ± 0.38 | 13.19 | 75.30 |
| SP-0.8-240-2-0.6 | 51.53 ± 0.82 | 3.41 ± 0.02 | 0.10 ± 0.01 | 0.03 ± 0.01 | 28.62 ± 0.37 | 69.32 ± 0.36 | 17.70 | 80.08 |
| SP-1-240-2-0.6 | 59.44 ± 0.62 | 3.74 ± 0.06 | 0 | 0 | 35.05 ± 0.42 | 80.47 ± 0.21 | 19.84 | |
However, in the co-HTC process, contrary to expectations, the yield decreased as the PET ratio increased. This reduction in yield is attributed to the increased acidity in the environment at 240 °C, as the PET ratio rises, leading to excessive decomposition of organic matter in SDSA. Furthermore, TPA, produced by PET through self-catalysis, acts as an acidic catalyst for its own hydrolysis. However, while the TPA produced is insufficient to catalyze the hydrolysis of PET itself, the hydrolysis of organic matter in SS results in the production of additional organic acids (the pH of the SS liquid phase in Table S4† is less than 7).40 These acids serve as acidic catalysts, further promoting the excessive decomposition of organic matter and reducing the yield in co-HTC.
Observe the TOC, TC, and TIC values of the liquid phase in Table S4.† The TC values produced by SDSA and water-thermal carbonization of PET alone are 8.79 g L−1 and 23.12 g L−1, respectively. According to the proportion, the theoretical TC values produced by SP-0.2-240-2-0.6 and SP-0.5-240-2-0.6 are 11.66 g L−1 and 15.96 g L−1, respectively, but the actual values increased by 3.72 g L−1 and 6.07 g L−1, respectively. This is also due to the stacking effect of SDSA and PET acid production, which causes a large amount of organic matter to dissolve in the liquid phase, increasing the amount of TC in the liquid phase. Therefore, in order not to allow more carbon content to be lost and to consider the conditions of optimal yield, we subsequently chose to study the data with a PET addition of 20 wt% and a hydrochar yield of 72.38 wt%.
In the study of reaction temperature, reaction time, and water content, before 240 °C, the decomposition rate of PET increased dramatically. The TPA produced acted as an acidic catalyst, accelerating both the hydrolysis of nitrogenous organic matter in SDSA and its own degradation. This not only reduced the nitrogen content in the hydrochar but also led to a decrease in yield. However, between 240–270 °C, the nitrogen content did not decrease continuously. This was attributed to the SS-rich carbohydrates producing large amounts of reducing sugars under high-temperature conditions. These sugars could undergo a Maillard reaction with protein-N, forming nitrogen-containing heterocyclic compounds like pyridines, pyrroles, and amines, which were transferred to the solid phase, thereby increasing its nitrogen content. At 300 °C, both the nitrogen and carbon contents decreased once more. This decrease was due to the decomposition of proteins stabilized at this temperature, which allowed the nitrogen concentrated in proteins to be transferred to the liquid phase.41
The influence of reaction time on the nitrogen content of hydrochar was minimal between 0.5 h and 2 h; however, extending the reaction time beyond 2 h led to an increase in nitrogen content. This increase may be attributed to a prolonged reaction time intensifying the Maillard reaction, which results in the formation of stable nitrogen-containing organic compounds in the solid phase.42 However, at a reaction time of 4 h, the prolonged high temperatures lead to the degradation of some stable nitrogen-containing organic compounds, resulting in a subsequent decrease in nitrogen content. This degradation partially accounts for the observed reduction in yield at extended reaction times.
It was observed that TC increased with higher reaction temperatures and longer reaction times, primarily due to the hydrolysis of more organic matter under these conditions, which enhanced the TC content in the liquid phase. However, at a temperature of 300 °C and a reaction time of 4 h, although the C content in the hydrochar was relatively low, the TC content in the liquid phase did not follow the expected trend. This discrepancy is speculated to result from the conversion of some carbohydrates in the water phase to the gas phase at high temperatures.43 Therefore, to optimize for low nitrogen and high carbon content, the conditions of 240 °C and 2 h were identified as the most suitable. In the study on water loading, neither the C content in hydrochar nor the TC content in the liquid phase showed significant changes. However, the lowest nitrogen content in hydrochar was achieved at a water loading of 0.6 g cm−3.
Subsequent to a series of experiments, it was determined that at a temperature of 240 °C, a reaction time of 2 h, PET addition of 20 wt%, and a water loading of 0.6 g cm−3, both carbon content (19.10 wt%) and ER efficiency were enhanced, yielding high-quality, low-nitrogen solid fuel (0.51 wt%). Although under these conditions, the calorific value of the resulting hydrochar (6.41 wt%) showed a marginal increase compared to that of the original SS hydrochar (Table 2: 6.26 wt%), the improvements in carbon and nitrogen content were significant.
3.3 Analysis of hydrochar coalification degree
The Van Krevelen plot depicted in Fig. 5 illustrates the degree of coalification of the hydrochar. With an increase in PET content from 0 to 50 wt%, there is a noticeable decrease in H/C and O/C value, indicative of enhanced dehydration and decarboxylation. This suggests a higher degree of coalification and an increase in the energy content of the hydrocha.44 The observed changes are due, firstly, to the addition of PET, which increases the C content in hydrochar more significantly than the O content. Secondly, the increased production of TPA by PET accelerates the hydrolysis of the protein fraction in SDSA, promoting dehydration and decarboxylation. Among the effects of reaction temperature, time, and water content, time has minimal impact on H/C value but significantly reduces O/C value. This reduction in O/C value is attributed to longer durations intensifying the Maillard reaction, where increasing amounts of reducing sugars and peptides or amino acids form heterocyclic macromolecules, thereby raising the C content in the hydrochar. Elemental analysis corroborates a trend of increasing C content over time, whereas the O content remains relatively unchanged, thus leading to a decrease in the overall O/C ratio. With increasing reaction temperatures, both H/C and O/C value in hydrochar decrease due to enhanced dehydration and decarboxylation, thereby improving the fuel performance of the hydrochar. An increase in temperature accelerates the hydrolysis of PET ester groups, releasing more TPA and facilitating the release and hydrolysis of proteins and stubborn macromolecules in SDSA. These components decompose more readily at higher temperatures, thereby enhancing decarboxylation and dehydration. As the water content increases, the H/C and O/C value of hydrochar typically rise; however, an inverse relationship is observed, where the lowest H/C and O/C value occur at a water content of 0.6 g cm−3.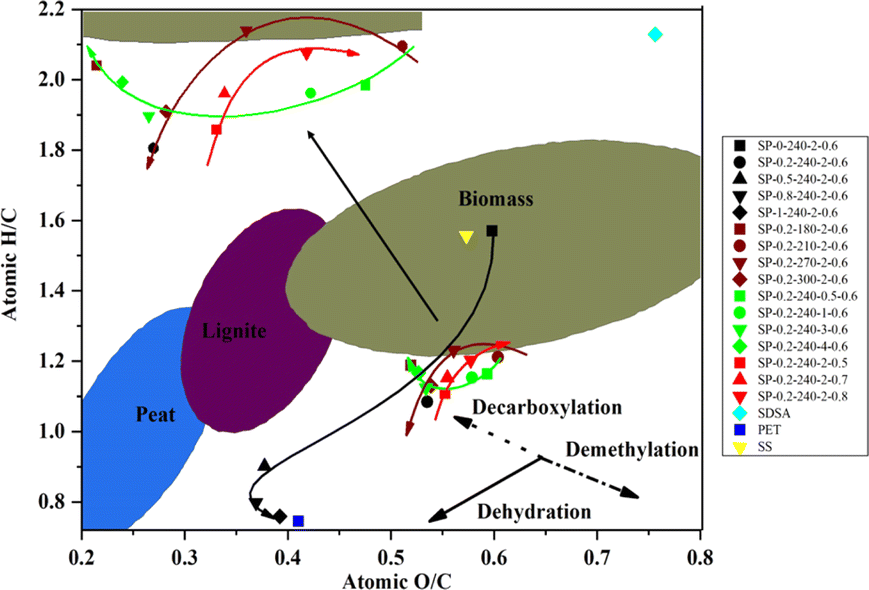 | ||
| Fig. 5 Van Krevelen diagram for hydrochar. | ||
3.4 FT-IR analysis of hydrochar
Fig. 6 displays the FT-IR spectra of hydrochar. In the FT-IR spectrum of hydrochar from PET alone, shown in Fig. 6a, the vibrational peaks at 1570 cm−1 and 1510 cm−1 correspond to the characteristic skeletal absorption peaks of benzene rings, while the peak near 3100 cm−1 is indicative of unsaturated hydrocarbons. These features suggest the presence of an aromatic structure in the product. The absorption peaks at 3063 cm−1 (C–H), 1675 cm−1 (CO), and 1280 cm−1 (C–O) are characteristic spectral bands of the aromatic dicarboxylic acid in TPA, as well as the unique vition of 1,4-disubstituted benzene ring at 1137 cm−1 and 1018 cm−1.29 These peaks all indicate that the solid product of PET hydrolysis is a mixture of PET residues and TPA.39 As the proportion of PET increases, the peaks in the hydrochar FT-IR spectra deepen, attributed to the increased content of the solid product post-PET hydrolysis. This product shows a rising number of functional groups, including –CO– and unsaturated hydrocarbons, which correspond to increases in the O and C content, consistent with elemental analyses. However, at a PET addition of 20 wt%, the spectral bands typical of the aromatic dicarboxylic acids associated with TPA were absent, indicating that the TPA produced by PET hydrolysis did not transfer to the solid phase at this ratio.
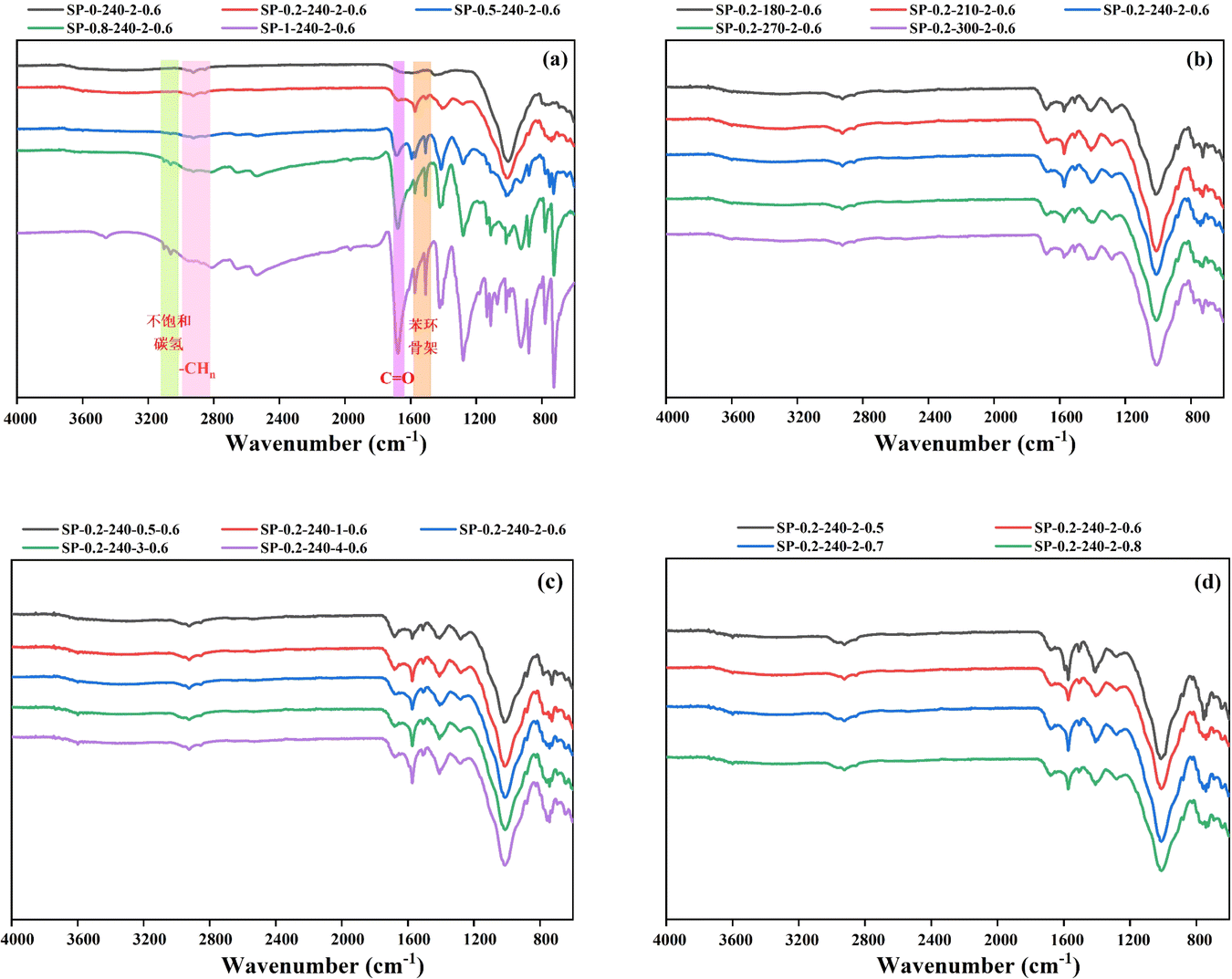 | ||
| Fig. 6 FT-IR spectra of hydrochar: (a) different PET additions; (b) different reaction temperatures; (c) different reaction times; (d) different water additions. | ||
The peaks located at 2920 cm−1 and 2850 cm−1 are symmetric and asymmetric methylene-aliphatic absorption peaks,33 which show signs of gradual disappearance when the PET addition is greater than 80 wt%. This stems from the PET hydrochar product's almost no aliphatic structure and more functional groups reflecting unsaturated hydrocarbons.45 When the PET addition was 20 wt%, there was hardly any change in the aliphatic structure content. The peak located at 1000 cm−1 may be due to a large amount of ash leading to the creation of Si–O stretching. The reason is that the intensity of the peak located at 1000 cm−1 is gradually weakened as the proportion of PET increases, so it may be due to a large amount of ash leading to the creation of Si–O stretching, which suggests that the ash content will be retained in the SS after the HTC, and a portion of the ash content will be SiO2.46 The peak located at 1645 cm−1 was attributed to the stretching vibration of the amide group –CN–.20 The peaks located at 1645 cm−1 were all significantly weakened along with the increase of PVC addition. Even the absorption peaks finally almost disappeared when the PET addition was 80 wt%, indicating that the nitrogen removal efficiency increased with the rise of PET addition.
Fig. 6b shows that the peaks of the characteristic skeleton of the benzene ring at 1570 cm−1 and 1510 cm−1 were found to deepen gradually as the reaction temperature was increased to 240 °C. When the temperature continues to increase, there is a tendency to weaken instead. It indicates that the generation of aromatic compounds is more favorable at moderate temperatures. From Fig. 6c, it can be observed that the peaks at the characteristic skeleton of the benzene ring also gradually deepen with the extension of the reaction time, indicating that the increase of the reaction time is also favorable for the generation of aromatic compounds.32 The intensity of the peaks at 1700 cm−1 (CO) tends to diminish with the extension of the reaction time, which indicates that the extension of the time promotes decarboxylation reaction, and that the intensity of the peaks at 3450 cm−1 (–OH) diminishes within a short period, which is not significant for this effect. The intensity of the peak at 3450 cm−1 (–OH) was weakened in a short period of time, and the effect of time extension was not obvious. The intensity of the peak at 3450 cm−1 (–OH) decreased in a short period of time, and the effect of time extension was not obvious. From Fig. 6c, it is evident that the peaks corresponding to the benzene ring skeleton deepen as the reaction time extends, indicating that prolonged reaction time favors the formation of aromatic compounds. The intensity of the peaks at 1700 cm−1 (CO) diminishes as the reaction time increases, suggesting that extended reaction time promotes decarboxylation. Similarly, the intensity of the peaks at 3450 cm−1 (–OH) decreases initially, but this reduction is not significant over time. Analysis of spectrograms across various reaction times indicates that the series of reactions affecting hydrochar functional groups can occur rapidly. Fig. 6d illustrates the impact of water content on functional groups, showing that increased water content has minimal effect on dehydration and decarboxylation reactions, as reflected by stable H/C and O/C value, consistent with Table S3.† However, the figure also reveals that a moderate water content enhances the characteristic skeletal peaks of the benzene ring at 1570 cm−1 and 1510 cm−1.
3.5 Analysis of combustion properties of hydrochar
Fig. 7 shows the TG and DTG curves of the hydrochar, and Table S5 (In the ESI)† organizes the combustion properties of the hydrochar. Fig. 7 can be divided into three stages, with temperatures less than 200 °C for the dehydration stage, 200–400 °C for the de-volatilization stage, and 400–620 °C for the stationary carbon combustion stage,47 and temperatures higher than 600 °C being the combustion phase, which is associated with volatile inorganic decomposition. In the second stage, mainly a large number of volatiles are burned, as shown in Fig. 7a; the HTC of PET alone has more volatiles, the volatile fraction weight loss rate is higher, and co-HTC with SDSA increases the volatiles. Therefore, from Fig. 7a, it is observed that the volatile fraction weight loss rate increases sequentially with increasing proportion of PET. At this stage, lower Tm generally means a faster decomposition reaction and higher reactivity.48 Table S5† shows that the Tm temperature range of the hydrochar is roughly 345–370 °C, which is lower than that of the hydrochar of SDSA alone (Tm = 402.48 °C). This observation suggests that the co-HTC of SDSA and PET are more likely to produce hydrochar with a lower maximum burning rate temperature. In the third stage, the main combustion is of fixed carbon, and the weight loss rate increases with the increase of PET proportion, indicating an increase in fixed carbon content and further carbonization. Moreover, with the rise in proportion, the peak of volatile matter gradually shifts to the fixed carbon combustion stage. Higher fixed carbon and lower volatiles in hydrochar mean better combustion performance because of less intense flames and more stable flames.49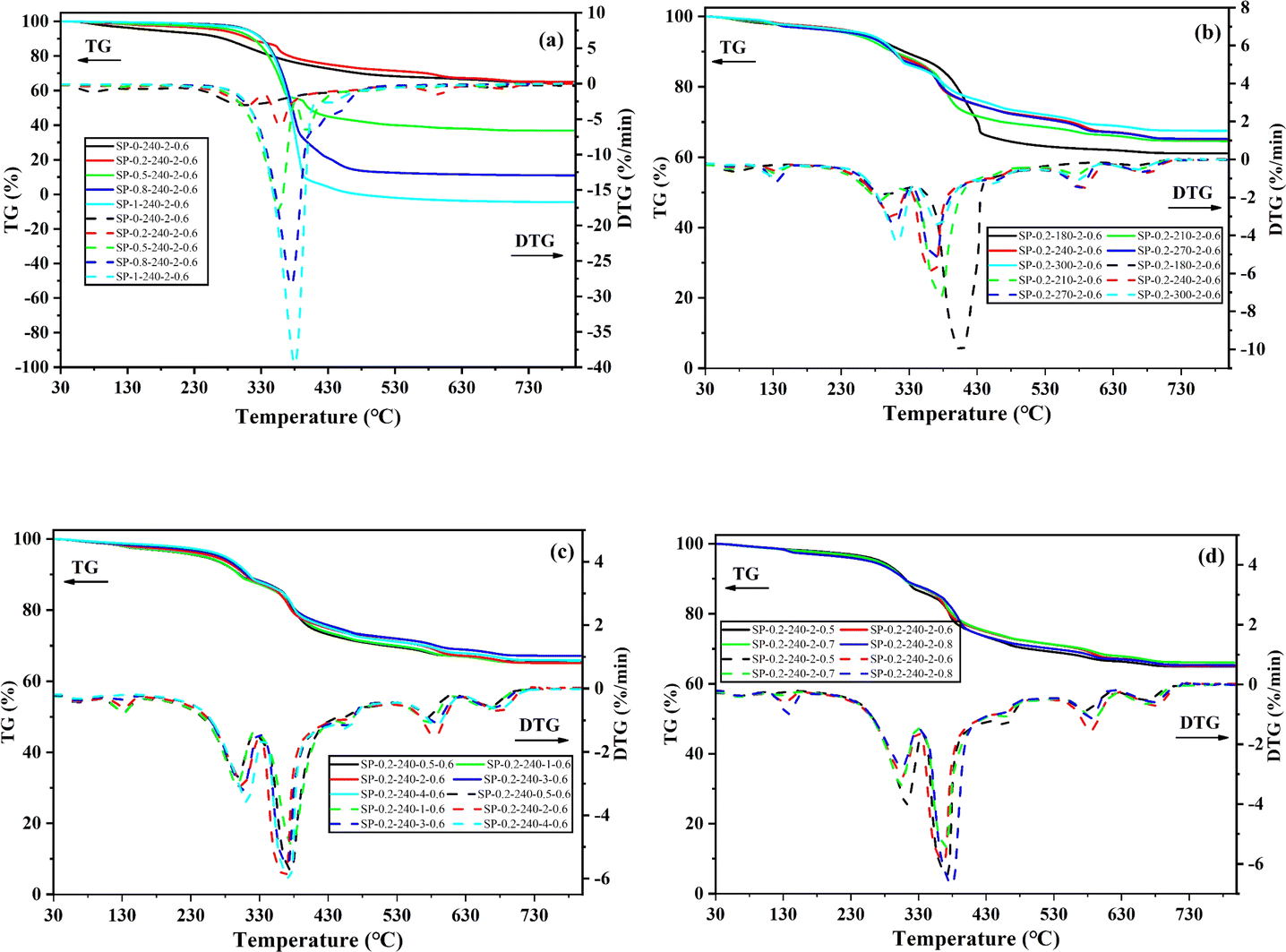 | ||
| Fig. 7 TG-DTG curves of hydrochar. (a) Different PET additions; (b) different reaction temperatures; (c) different reaction times; (d) different water additions. | ||
In addition, when using hydrochar as a solid fuel, the ignition temperature (Ti) and the burnout temperature (Tf) are important parameters as combustion properties, and the Ti ignition temperature determines the ease of ignition of the fuel. Observing Table S5,† the Ti of hydrochar increases significantly with increasing PET content, which suggests that the potential fire and explosion risk of hydrochar is reduced by co-HTC treatment,50 allowing safer handling, storage, and transportation.51 The Tf value reflects the fuel content and combustion performance. Higher Tf values indicate more unburned fuel and less efficient combustion. When the Tf value is low, it indicates that the fuel is more likely to burn out at lower temperatures and combustion is more efficient.52 Combined with the integrated combustibility index (S), the higher S value with the increase of PET ratio indicates better combustion performance. Then, a series of results suggest that the hydrochar of co-HTC exhibits higher Ti, lower Tm, and Tf, along with a high fixed carbon content and a high S value. Therefore, the hydrochar obtained from co-HTC is expected to be used as a high-performance fuel. However, its volatile content increases with increasing ratio, and high volatiles can lead to flame instability, which can increase heat loss during combustion.53 Therefore, it can be seen from Fig. 7a that the volatile weight loss rate is lower, and the combustion rate of fixed carbon is higher when the PET addition is 20 wt%. In conclusion, co-HTC with low PET addition (20 wt%) can convert SS into solid fuel with enhanced combustion performance. In conclusion, the low addition (20 wt%) PET co-HTC with SS is more favorable to enhance the solid fuel combustion performance.
Observe Fig. 7b; with the increase of temperature, the rate of weight loss of volatile fraction is gradually decreasing; this is because organic matter, with the temperature rise, a large number of hydrolysis, volatile fraction of the material to reduce. With the increase in temperature, the stationary carbon combustion stage as a whole shifted to the right, tending to the high-temperature stage, indicating that it is favorable to the formation of heavy fixed carbon. Fig. 7c and d display the TG-DTG data corresponding to varying reaction times and water loadings, respectively. As reaction time or water content increases, the rates of volatiles and solids initially show little change. However, excessively long reaction times or high water contents lead to an increased rate of volatiles and a decreased rate of fixed carbon. At a reaction time of 2 h and a water content of 0.6 g cm−3, the combustion stage for fixed carbon exhibits the highest rate with fewer volatiles, enhancing the fuel properties.
4 Conclusion
Pretreatment altered the physicochemical properties of SS, resulting in the production of high-quality clean solid fuel (N: 0.51 wt%, C: 19.10 wt%) through co-HTC of SDSA-pretreated SS with PET. Compared to the co-HTC process using original SS, the ηN of the pretreated SS increased by 14%, reaching 74.6%. The yield, carbon content, and calorific value of hydrochar increased. The enhancement of ηN was primarily attributed to two factors: firstly, the loosening of the EPS structure in pretreated SS, which facilitated the release and hydrolysis of protein-N during co-HTC, and secondly, the degradation of PET under co-HTC conditions that produced TPA, enhancing the protein hydrolysis process. Together, these factors led to enhanced protein hydrolysis and deamination reactions, thereby transferring more nitrogen from the solid to the liquid phase. Therefore, this study demonstrates that SDSA pretreatment can significantly enhance the combustion performance of co-HTC solid products, offering an effective processing route for the efficient recycling of SS and PET, and the future production of high-quality clean solid fuels.Author contributions
Ting Ye: conceptualization, validation, methodology, investigation, writing – original draft, writing – review & editing. Le Gou: validation, methodology, investigation. Yue Wang: validation, methodology, investigation. Nan Liu: validation, methodology, investigation. Liyi Dai: supervision, funding acquisition, writing – review & editing. Yuanyuan Wang: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the National Key R&D Program of China (2021YFE0104900), the National Natural Science Foundation of China (22078103, 22378133).Notes and references
- B. A. Mohamed and L. Y. Li, Environ. Chem. Lett., 2022, 21, 153–182 CrossRef.
- L. Feng, J. Luo and Y. Chen, Environ. Sci. Technol., 2015, 49, 4781–4782 CrossRef CAS PubMed.
- W. Ma, G. Du, J. Li, Y. Fang, L. a. Hou, G. Chen and D. Ma, Waste Manage., 2017, 59, 371–378 CrossRef CAS PubMed.
- S. Yu, J. He, Z. Zhang, Z. Sun, M. Xie, Y. Xu, X. Bie, Q. Li, Y. Zhang, M. Sevilla, M.-M. Titirici and H. Zhou, Adv. Mater., 2024, 36, 2307412 CrossRef CAS.
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 RSC.
- L. Wang, Y. Chang and A. Li, Renewable Sustainable Energy Rev., 2019, 108, 423–440 CrossRef CAS.
- C. He, K. Wang, Y. Yang, P. N. Amaniampong and J.-Y. Wang, Environ. Sci. Technol., 2015, 49, 6872–6880 CrossRef CAS PubMed.
- Z.-X. Xu, H. Song, P.-J. Li, X. Zhu, S. Zhang, Q. Wang, P.-G. Duan and X. Hu, J. Hazard. Mater., 2020, 398, 122833 CrossRef CAS.
- B. Zhang, J. Wang, Z. Xu, S. Wu, R. Luque and H. Zhang, J. Cleaner Prod., 2023, 386, 135880 CrossRef CAS.
- X. Wang, Y. Shen, X. Liu, T. Ma, J. Wu and G. Qi, Chemosphere, 2022, 290, 133209 CrossRef CAS PubMed.
- Z. Xu, X. Ma, J. Liao, S. M. Osman, S. Wu and R. Luque, ACS Sustain. Chem. Eng., 2022, 10, 4258–4268 CrossRef CAS.
- C. Zheng, X. Ma, Z. Yao and X. Chen, Bioresour. Technol., 2019, 285, 121347 CrossRef CAS PubMed.
- C. He, Z. Zhang, C. Ge, W. Liu, Y. Tang, X. Zhuang and R. Qiu, Waste Manage., 2019, 100, 171–181 CrossRef CAS PubMed.
- Q. Zhang, K. Mu, J. Han, L. Qin, B. Zhao and L. Yi, Energy, 2023, 278, 128012 CrossRef CAS.
- C. Feng, T. Lotti, R. Canziani, Y. Lin, C. Tagliabue and F. Malpei, Sci. Total Environ., 2021, 753, 142051 CrossRef CAS.
- D. Carta, G. Cao and C. D'Angeli, Environ. Sci. Pollut. Res., 2003, 10, 390–394 CrossRef CAS PubMed.
- Y. Xue, L. Bai, M. Chi, X. Xu, Z. Chen, K. Yu and Z. Liu, J. Environ. Chem. Eng., 2022, 10, 106975 CrossRef CAS.
- J. Gao, Y. Wang, Y. Yan and Z. Li, J. Cleaner Prod., 2021, 295, 126288 CrossRef CAS.
- L.-F. Wang, B.-C. Huang, L.-L. Wang, Y. Min and H.-Q. Yu, Sci. Total Environ., 2018, 633, 198–205 CrossRef CAS.
- Z.-X. Xu, H. Song, S. Zhang, S.-Q. Tong, Z.-X. He, Q. Wang, B. Li and X. Hu, Energy, 2019, 187, 115972 CrossRef CAS.
- Y. Shen, S. Yu, S. Ge, X. Chen, X. Ge and M. Chen, Energy, 2017, 118, 312–323 CrossRef CAS.
- L. Xie, L. Gou, Y. Wang and L. Dai, Sci. Total Environ., 2022, 804, 150094 CrossRef CAS PubMed.
- L. Xie, L. Gou, D. Xu, K. Kapusta, L. Dai and Y. Wang, Sci. Total Environ., 2023, 866, 161354 CrossRef CAS PubMed.
- X. Tian, A. P. Trzcinski, L. L. Lin and W. J. Ng, J. Environ. Chem. Eng., 2016, 4, 4801–4807 CrossRef CAS.
- L. Leng, L. Yang, S. Leng, W. Zhang, Y. Zhou, H. Peng, H. Li, Y. Hu, S. Jiang and H. Li, Sci. Total Environ., 2021, 756, 143679 CrossRef CAS PubMed.
- Z. Wang, Y. Zhai, T. Wang, C. Peng, S. Li, B. Wang, X. Liu and C. Li, Environ. Pollut., 2020, 260, 114067 CrossRef CAS PubMed.
- M. Hejna, K. Świechowski and A. Białowiec, Materials, 2023, 16, 6903 CrossRef CAS PubMed.
- J. Yang, Q. He and L. Yang, Appl. Energy, 2019, 250, 926–945 CrossRef CAS.
- J. V. Valh, B. Vončina, A. Lobnik, L. F. Zemljič, L. Škodič and S. Vajnhandl, Text. Res. J., 2019, 90, 1446–1461 CrossRef.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281 RSC.
- S. Yu, X. Dong, P. Zhao, Z. Luo, Z. Sun, X. Yang, Q. Li, L. Wang, Y. Zhang and H. Zhou, Nat. Commun., 2022, 13, 3616 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- C. Zhang, X. Ma, T. Huang, Y. Zhou and Y. Tian, Bioresour. Technol., 2020, 314, 123676 CrossRef CAS PubMed.
- H. Liu, G.-Q. Luo, H.-Y. Hu, Q. Zhang, J.-K. Yang and H. Yao, J. Hazard. Mater., 2012, 235–236, 298–306 CrossRef CAS PubMed.
- V. Koskue, S. Freguia, P. Ledezma and M. Kokko, J. Environ. Chem. Eng., 2021, 9, 106286 CrossRef CAS.
- S. M. Changi, J. L. Faeth, N. Mo and P. E. Savage, Ind. Eng. Chem. Res., 2015, 54, 11733–11758 CrossRef CAS.
- A. A. Peterson, R. P. Lachance and J. W. Tester, Ind. Eng. Chem. Res., 2010, 49, 2107–2117 CrossRef CAS.
- M. J. Kang, H. J. Yu, J. Jegal, H. S. Kim and H. G. Cha, Chem. Eng. J., 2020, 398, 125655 CrossRef CAS.
- Z. Xu and X. Bai, Chemosphere, 2022, 297, 134203 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem.–A Euro. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- Y. Chen, L. Tian, T. Liu, Z. Liu, Z. Huang, H. Yang, L. Tian, Q. Huang, W. Li, Y. Gao and Z. Zhang, Waste Manage., 2023, 162, 8–17 CrossRef CAS PubMed.
- T. Wang, Y. Zhai, Y. Zhu, C. Li and G. Zeng, Renewable Sustainable Energy Rev., 2018, 90, 223–247 CrossRef CAS.
- P. J. Valdez and P. E. Savage, Algal Res., 2013, 2, 416–425 CrossRef.
- P. McKendry, Bioresour. Technol., 2002, 83, 47–54 CrossRef CAS PubMed.
- C. Peng, Y. Zhai, A. Hornung, C. Li, G. Zeng and Y. Zhu, ACS Sustain. Chem. Eng., 2018, 6, 9461–9469 CrossRef CAS.
- C. He, A. Giannis and J.-Y. Wang, Appl. Energy, 2013, 111, 257–266 CrossRef CAS.
- S. Cheng, Y. Qiao, J. Huang, W. Wang, Z. Wang, Y. Yu and M. Xu, Proc. Combust. Inst., 2019, 37, 2715–2722 CrossRef CAS.
- H. Ahn, D. Kim and Y. Lee, Renewable Energy, 2020, 147, 957–968 CrossRef CAS.
- X. Chen, X. Ma, X. Peng, Y. Lin and Z. Yao, Bioresour. Technol., 2018, 249, 900–907 CrossRef CAS PubMed.
- M. Xu and C. Sheng, Energy Fuels, 2011, 26, 209–218 CrossRef.
- X. Zhuang, H. Zhan, Y. Huang, Y. Song, X. Yin and C. Wu, Bioresour. Technol., 2018, 267, 17–29 CrossRef CAS PubMed.
- X. Liu, Y. Zhai, S. Li, B. Wang, T. Wang, Y. Liu, Z. Qiu and C. Li, J. Hazard. Mater., 2020, 388, 122084 CrossRef CAS PubMed.
- Z. Liu, A. Quek, S. Kent Hoekman and R. Balasubramanian, Fuel, 2013, 103, 943–949 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02165g |
| This journal is © The Royal Society of Chemistry 2024 |