
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoluminescence mechanisms of BF2-formazanate dye sensitizers: a theoretical study†
Parichart Suwannakham ,
Pannipa Panajapo,
Phorntep Promma,
Tunyawat Khrootkaew,
Anyanee Kamkaew and
Kritsana Sagarik*
,
Pannipa Panajapo,
Phorntep Promma,
Tunyawat Khrootkaew,
Anyanee Kamkaew and
Kritsana Sagarik*
School of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima 30000, Thailand. E-mail: kritsana@sut.ac.th; Fax: +66 81 8783994; Tel: +66 81 8783994
First published on 24th June 2024
Abstract
Photodynamic therapy (PDT) is an alternative, minimally invasive treatment for human diseases such as cancer. PDT uses a photosensitizer to transfer photon energy directly to cellular 3O2 to generate 1O2 (Type II), the toxicity of which leads to cancer cell death. In this work, the photoluminescence mechanisms of a BF2-formazanate dye sensitizer (BF2-FORM) and its iodinated derivative (BF2-FORM-D) were studied using complementary theoretical approaches; the photoluminescence pathways in the S1 and T1 states were studied using density functional theory (DFT) and time-dependent (TD)-DFT methods, the kinetic and thermodynamic properties of the pathways using the transition state theory (TST), and the time evolution and dynamics of key processes using non-adiabatic microcanonical molecular dynamics simulations with surface-hopping dynamics (NVE-MDSH). Evaluation of the potential energy surfaces (PESs) in terms of the rotations of the phenyl rings suggested a pathway for the S1 → S0 transition for the perpendicular structure, whereas two pathways were anticipated for the T1 → S0 transition, namely, [T1 → S0]1 occurring immediately after the S1/T1 intersystem crossing (ISC) and [T1 → S0]2 occurring after the S1/T1 ISC and T1 equilibrium structure relaxation, with the T1 → S0 energy gap being comparable to the energy required for 3O2 → 1O2. The PESs also showed that because of the heavy-atom effect, BF2-FORM-D possessed a significantly smaller S1/T1 energy gap than BF2-FORM. The TST results revealed that at room temperature, BF2-FORM-D was thermodynamically more favorable than the parent molecule. Analysis of the NVE-MDSH results suggested that the librational motions of the phenyl rings play an important role in the internal conversion (IC) and ISC, and the S1/T1 ISC and T1 → S0 transitions could be enhanced by varying the irradiation wavelength and controlling the temperature. These findings can be used as guidelines to improve and/or design photosensitizers for PDT.
Introduction
Photosensitization occurs when light at an appropriate wavelength interacts with a photosensitizer, from which the photon energy is transferred onto target molecules.1 Photodynamic therapy (PDT) is an alternative, minimally invasive treatment for various human diseases,2,3 including cancer, rheumatoid arthritis, and psoriasis, that uses only photon energy.1 PDT uses a photosensitizer that releases appropriate energy to generate singlet oxygen (1O2) from triplet oxygen (3O2).4 The cellular oxygen (3O2) absorbs the energy released from the photosensitizer to generate 1O2,5 and the toxicity of 1O2 leads to cell death in cancer, e.g., for 3O2 (3∑g) → 1O2 (1Δg), ΔET1→S1 ∼ 0.97 eV (ref. 5) (∼94 kJ mol−1); in normal cells (healthy tissues), the oxygen levels are in general lower compared to cancer cells, thus resulting in less 1O2 generation and lower cytotoxic effects.6 Effective photosensitizers for therapeutics should have high light absorption coefficients, particularly in the infrared/near-infrared range, to allow for effective tissue penetration. They should exhibit low photobleaching quantum yields, high intersystem crossing (ISC) efficiencies, and low toxicity in the absence of light.7 These include the photodynamic properties, such as appropriate triplet relaxation energy (ΔET1→S0), high phosphorescence quantum yield (ΦT), and long lifetime (τT1) of the triplet state.8The two photochemical mechanisms of PDT involving oxygen molecules are shown in Fig. 1. In Type I, radicals (e.g., OH˙) act as intermediates, transferring electron energy from the photosensitizer to the oxygen derivatives, whereas for Type II, the photosensitizer passes light energy directly to the oxygen molecules.4 In general, phosphorescence is not easily observed because of the interference of fluorescence,8 and triplet excited states are difficult to generate through direct photoexcitation because ISC is symmetrically forbidden. For example, the S1 → T1 transition in chromophores possesses a large singlet–triplet energy gap.9
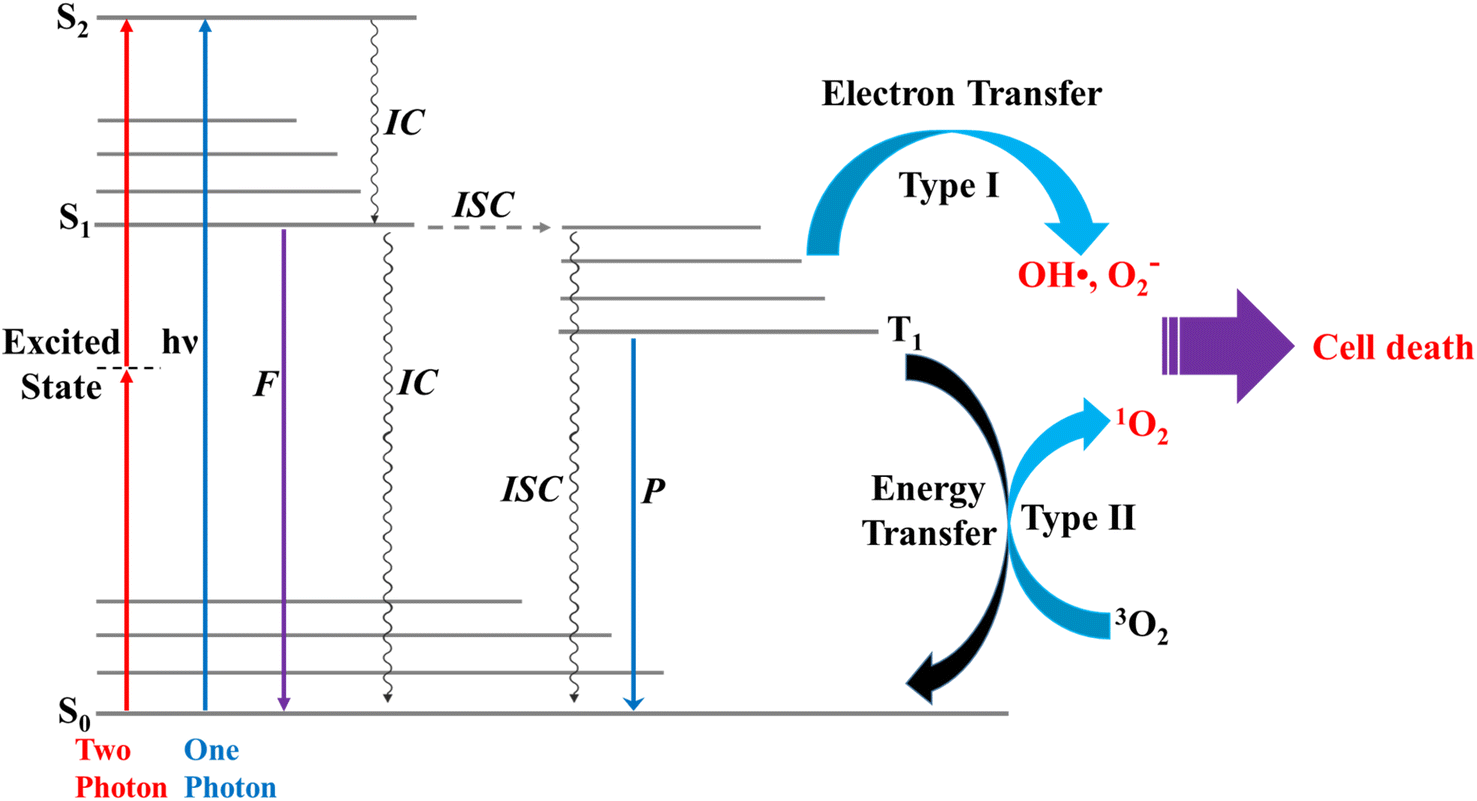 | ||
| Fig. 1 Schematic diagram showing two types of reaction pathways for formation of singlet oxygen from triplet oxygen, 3O2 (3∑g) → 1O2 (1Δg). ISC = intersystem crossing; IC = internal conversion; F = fluorescence; P = phosphorescence. | ||
Theoretical and experimental studies have proposed several strategies to improve the efficiency of ISC and promote the generation of triplet states. These strategies include for example: Heavy atom effect;10 the introduction of heavy atoms like halogen (such as Br and I) into photosensitizers has been shown to increase ISC rates, and the introduction of chalcogen (such as S, Se, or Te) into the photosensitizers could also increase the production of 1O2 upon irradiation due to spin–orbit interactions:11 Molecular design;12 the modification of the structure of photosensitizer molecules can optimize their electronic characteristics to promote ISC and: Sensitizer–sensitizer interactions;13 ISC can also be enhanced through interactions among photosensitizer molecules, whether through complex formation or aggregation.
Boron difluoride (BF2) complexed with π-conjugated organic compounds is widely used as a photosensitizer in PDT, among which boron dipyrromethene (IUPAC = 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, or BODIPY) and its derivatives are the most widely studied.14,15 Such compounds have been reported to possess visible absorption and fluorescence emission between 470 and 550 nm, with a high emission quantum yield (ΦF = 0.60).16 Although BODIPY has a high potential to be a functional component in PDT, attempts have been made to improve ΦT using phenyl and/or heavy-atom substitutions.8
Several experimental methods have been developed to enhance the triplet excitation of photosensitizers by incorporating halogen atoms, such as the iodine (I) or bromine (Br) atoms, into organic aromatic compounds; these heavy atoms could help increase the spin–orbit coupling (SOC) and ISC rate, as well as the 1O2 quantum yield of photosensitizers.17,18 In most cases, the I atom has a higher 1O2 quantum yield (SOQY) than the Br atom.19 The higher ΦT and SOQY for the I atom than the Br atom can be attributed to several factors, such as the higher heavy atom effect, higher radiative decay and lower internal conversion (IC) rates.20–22 These collective factors could lead to an enhancement of ΦT and SOQY in BODIPY-based PDT photosensitizers.
BF2-formazanate (3,3-difluoro-2,4-diphenyl-2,3-dihydro-1,2,4λ4,5,3λ4-tetrazaborinine, BF2-FORM in this work) is a fluorophore with several applications in microscopy. The structure of BF2-FORM, shown in Fig. 2, consists of a BF2 group coupled to a chelating N-donor ligand, forming a stable six-membered heterocyclic ring. BF2-FORM exhibits outstanding photophysical properties such as large molar extinction coefficients and high ΦF, which are generally in the far-red or near-infrared region. Therefore, BF2-FORM has significant potential in cell imaging and PDT applications.23,24
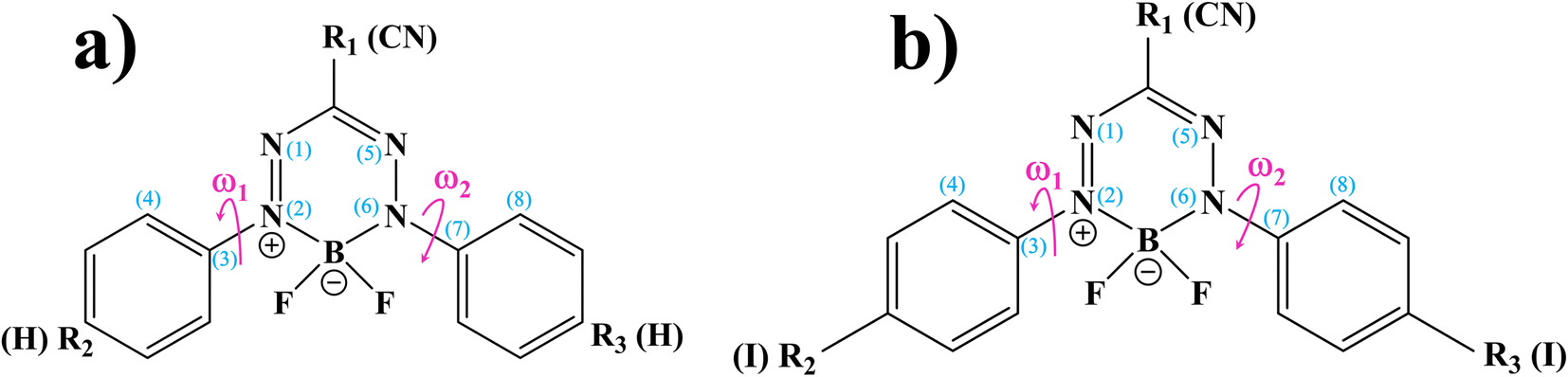 | ||
| Fig. 2 (a) and (b) Structures and numbering systems of BF2-FORM and BF2-FORM-D dye sensitizers, respectively. BF2-FORM = 3,3-difluoro-2,4-diphenyl-2,3-dihydro-1,2,4λ4,5,3λ4-tetrazaborinine; BF2-FORM-D = 3,3-difluoro-2,4-bis(4-iodophenyl)-2,3-dihydro-1,2,4λ4,5,3λ4-tetrazaborinine-6-carbonitrile; ω1 and ω2 = dihedral angles used in the calculations of the potential energy surfaces. | ||
The optical properties of BF2-FORM are strongly affected by the electron-donating substituents at R2 and R3 (Fig. 2), leading to an increase in ΦF and a red-shift in the emission spectra. The strong red-shift could be attributed to a smaller HOMO–LUMO energy gap upon the substitutions.6 For example, in the case of F-BODIPY, the introduction of two ethyl groups at the C2 and C8 positions led to a red-shifted absorption spectra compared to the parent molecule due to a decrease in the HOMO–LUMO energy gap and large charge transfer interaction within the molecule.25 Because electron-withdrawing substituents at R1 could also affect the photophysical properties, the differences in optical properties between Ph-, CN-, and NO2-substituted BF2-FORM are attributed primarily to the electron-withdrawing nature of the substituents (e.g., NO2 > CN ≫ Ph).26
In our previous study,24 the photochemical properties of BF2-FORM-based photosensitizers were experimentally and theoretically studied in the electronic ground (S0), lowest singlet, and triplet excited states (S1 and T1) using density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods with the Becke, 3-Parameter, Lee–Yang–Parr (B3LYP) hybrid functional and 6-311G basis sets. A comparison of the experimental and theoretical results showed that the DFT/B3LYP/6-311G and TD-DFT/B3LYP/6-311G methods could provide insight into the PDT mechanisms and confirmed the effect of heavy-atom substituents (I and Br at R2 and R3) on the ISC rate.
In this study, complementary theoretical approaches were applied to investigate the photoluminescence mechanisms of BF2-FORM to improve its efficiency as a photosensitizer in PDT. Because theoretical and experimental studies24 revealed that substitutions of R2 and R3 at the phenyl rings by I and R1 at the heterocyclic ring by CN can significantly enhance the S1/T1 ISC with high ΦT, both BF2-FORM and its iodinated derivative [3,3-difluoro-2,4-bis(4-iodophenyl)-2,3-dihydro-1,2,4λ4,5,3λ4-tetrazaborinine-6-carbonitrile, BF2-FORM-D in this work] were selected as model molecules.
Theoretical studies focusing on Type II mechanism began with calculations of the equilibrium structures and energetic and spectroscopic properties of BF2-FORM and BF2-FORM-D in the S0, S1, and T1 states. The potential energy surfaces (PESs) for the S1 → S0, S1/T1, and T1 → S0 transitions were computed using the nudged elastic band (NEB) method, from which the kinetics and thermodynamics of the photoluminescence pathways were studied using the transition state theory (TST). Emphasis was placed on the probabilities of IC and ISC, as well as on the effect of the electron-withdrawing substituents on the photophysical properties. Furthermore, to explore the possibility of increasing photoluminescence, non-radiative relaxation, and effect of molecular dynamics on the S1 → S0 transition and S1/T1 ISC were studied using non-adiabatic microcanonical molecular dynamics simulations with surface-hopping dynamics (NVE-MDSH). The theoretical results are discussed in comparison with available theoretical and experimental data.
Computational methods
Quantum chemical calculations
All the DFT and TD-DFT calculations were performed using the TURBOMOLE 7.50 software package.27 For the TD-DFT method, the Tamm–Dancoff approximation (TDA) was applied to avoid singlet instabilities in the lowest singlet and triplet state calculations. The DFT and TD-DFT methods with the B3LYP functionals were chosen based on several benchmarking calculations in photochemical reactions,28,29 including BODIPY-based photosensitizers.9 For example, our benchmarks against the complete active space multiconfigurational second-order perturbation theory (CASPT2) method revealed that for the photodissociation and formation of glycine,28,29 the characteristic structures and energies on the S0 and S1 PESs obtained from the DFT/B3LYP and TD-DFT/B3LYP methods were in good agreement with the CASPT2 results, and DFT/B3LYP/6-311G calculations on the I2-IR783-Mpip photosensitizer6 revealed the equilibrium structures, energetics and absorption spectra comparable with experimental data; the 6-311G basis set was also used successfully in a theoretical study on photoinduced charge separation-charge recombination in BODIPY compounds.30 The performance of the DFT/B3LYP method with various sizes of the basis sets was discussed in detail using boron-doped triazine based covalent organic framework as a model molecule in ref. 31.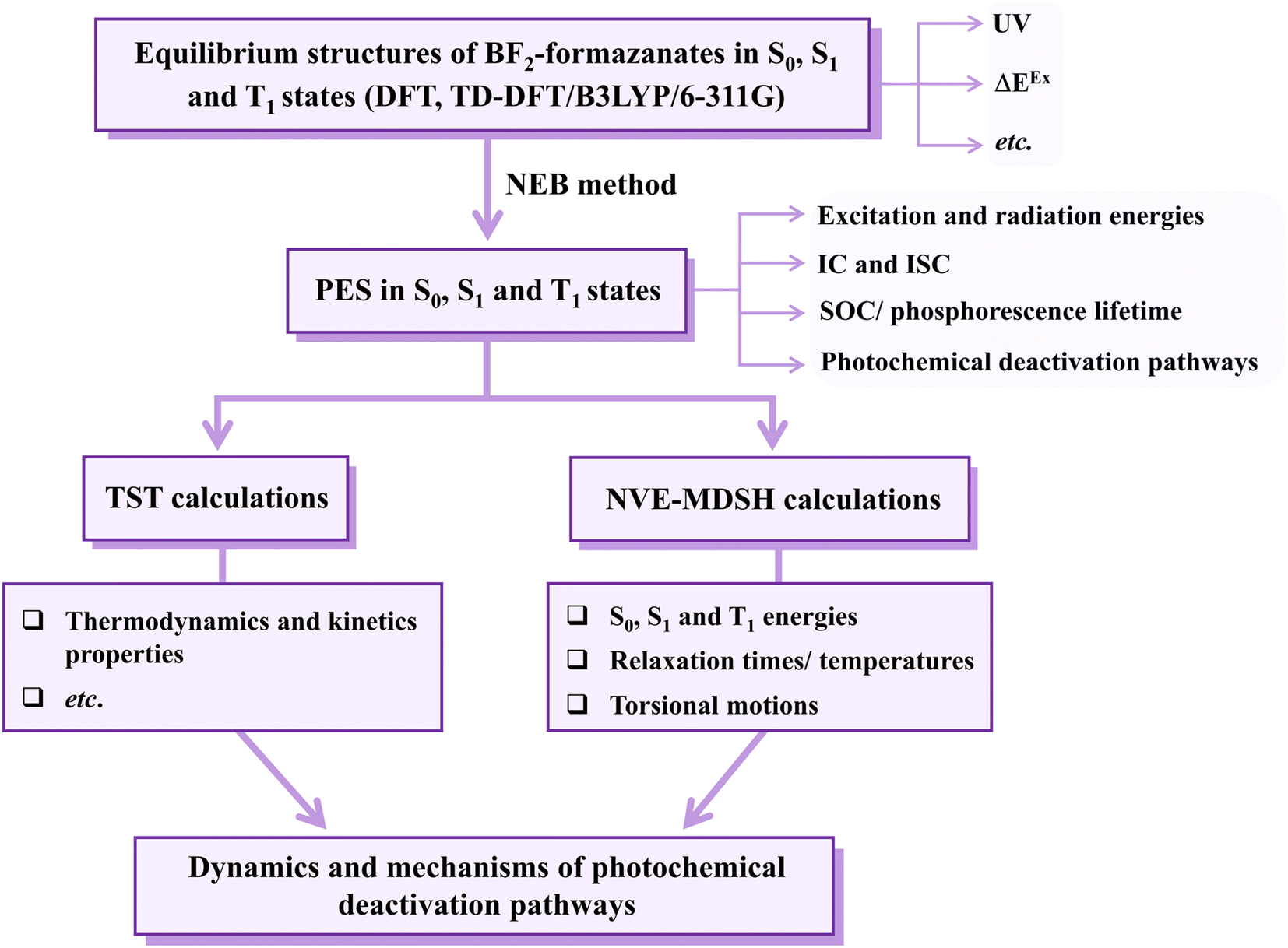 | ||
| Fig. 3 The theoretical strategy and methods used to study photoluminescence mechanisms of the BF2-formazanate dye sensitizers. PES = potential energy surface; ΔEEx = excitation energy; IC and ISC = internal conversion and intersystem crossing; TST = transition state theory; NVE-MDSH = non-adiabatic microcanonical molecular dynamics simulations with surface hopping dynamics. | ||
| (I)* → (II)* → [S1 → S0] | (1) |
| (I)* → (II)* ⇌ (III)‡,§ → S1/T1 ISC → [T1 → S0]1 | (2) |
| (I)* → (II)* ⇌ (III)‡,§ → S1/T1 ISC → (IV) → [T1 → S0]2 | (3) |
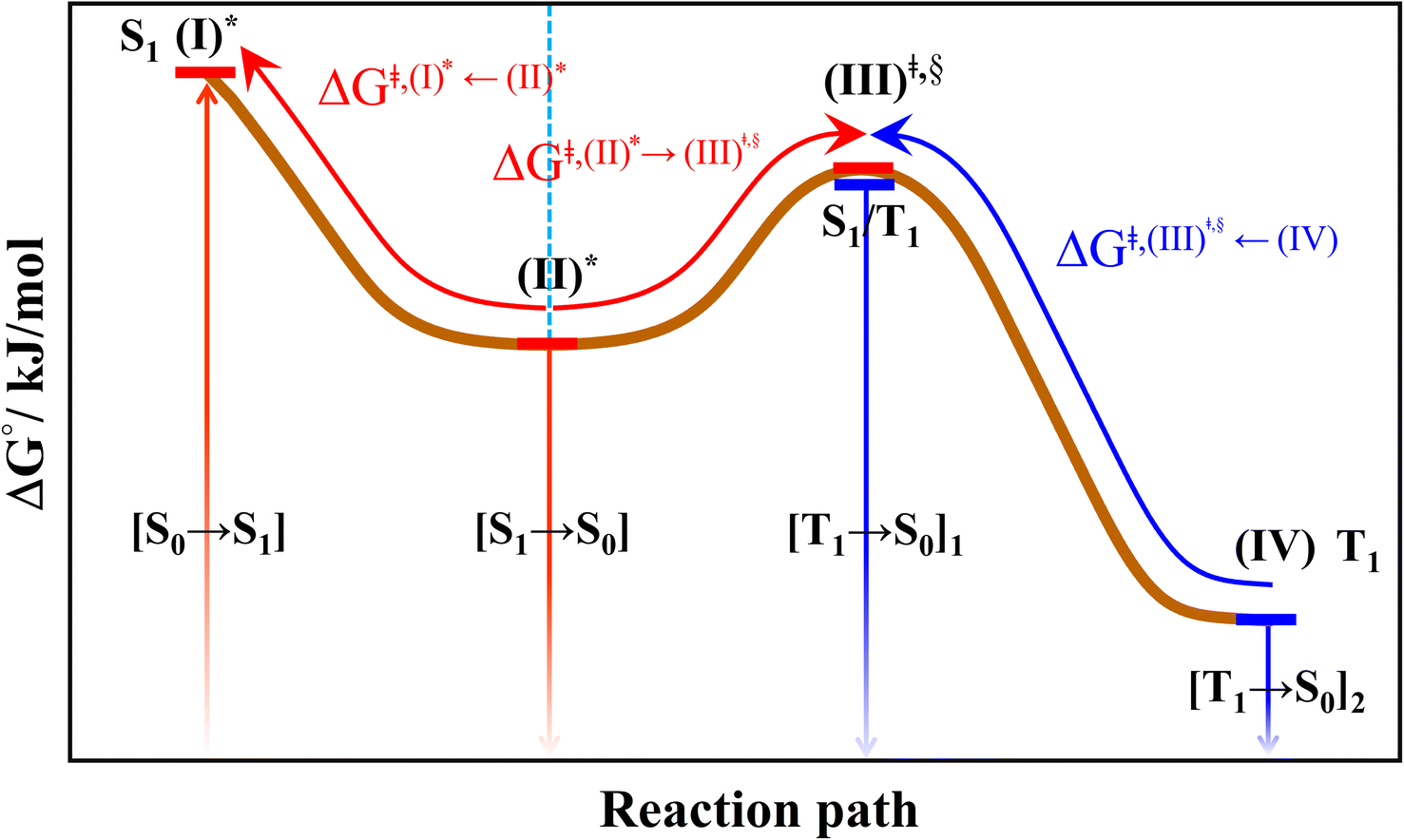 | ||
| Fig. 4 The hypothesized photochemical deactivation pathways ((I)* → (II)* ⇌ (III)‡,§ → (IV)) used in the present study. | ||
The equilibrium structures of BF2-FORM and BF2-FORM-D in the S0, S1, and T1 states obtained from DFT/B3LYP/6-311G and TD-DFT/B3LYP/6-311G geometry optimizations were used in the PES calculations.
Theoretical studies have shown that ISC in aromatic organic compounds can be mediated by intramolecular motions35 such as molecular rotation or twist;36 thus, reaction pathway optimization began with the S0 → S1 vertically excited precursor (I)*, from which the PESs for the rotations of the torsional angles ω1 and ω2 (Fig. 2) were constructed. Reaction pathway optimization was performed using the NEB method37 with limited-memory Broyden–Fletcher-Goldfarb-Shanno (LBFGS) optimizers included in the ChemShell software package.38 In this work, to search for minimum energy pathways connecting the initial and final structures on the PESs, approximately 10 intermediate images (including the saddle points) along the pathways were optimized. In the NEB calculations,37 the gradients on the reaction pathway were calculated based on the spring forces acting on local tangents between each image and on the true forces acting perpendicular to the local tangents.
Because SOC plays an important role in ISC and phosphorescence, the singlet–triplet energy gaps along the S1 PES were computed using the TD-DFT/B3LYP/6-311G method and the geometries obtained from the NEB calculations. The energy gaps were refined using the RICC2/aug-cc-pVDZ method, from which the phosphorescence lifetimes were computed. SOC was computed using TURBOMOLE 7.50 based on the effective spin-orbital mean field approximation,39 in which the mean field two-electron contribution was computed from the Hartree–Fock density.
Kinetics and thermodynamics of reaction pathways
To study the kinetics and thermodynamics of the proposed photoluminescence pathways, quantized vibrational rate constants (kQ-vib) were calculated over a temperature range of 300–550 K. This range of temperatures includes the standard human body temperature of 310 K. The kQ-vib values were computed using eqn (4),40 in which ΔE‡,ZPC is the zero-point energy-corrected barrier, obtained by including the zero-point correction energy (ΔEZPE) to the energy barriers obtained from the NEB method (ΔE‡).
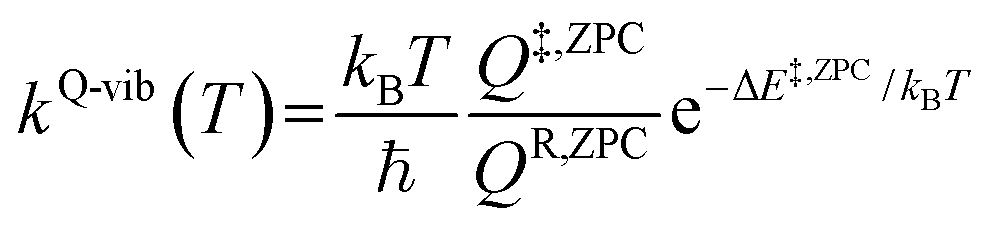 | (4) |
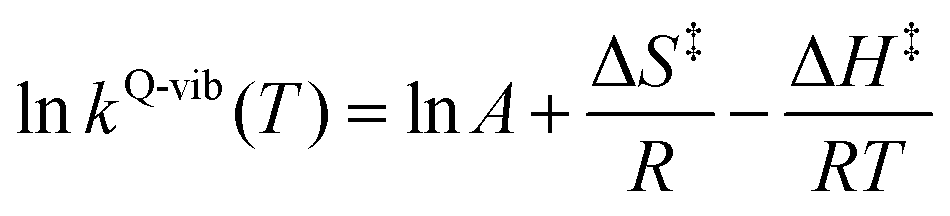 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kQ-vib and 1000/T.
kQ-vib and 1000/T.
Based on the photoluminescence pathways shown in Fig. 4, the thermodynamics of the consecutive reaction pathway (I)* → (II)* ⇌ (III)‡,§ → (IV) were studied, in which (II)* ⇌ (III)‡,§ → (IV) was assumed to be in a quasi-equilibrium. The thermodynamic property of interest was the total Gibbs free energy (ΔG°,tot) of the reactions, computed using ΔG‡ obtained from the TST method. ΔG°,tot was computed by dividing the consecutive reaction pathway into two single steps, namely, (I)* → (II)* and (II)* ⇌ (III)‡,§ → (IV). For (I)* → (II)*, 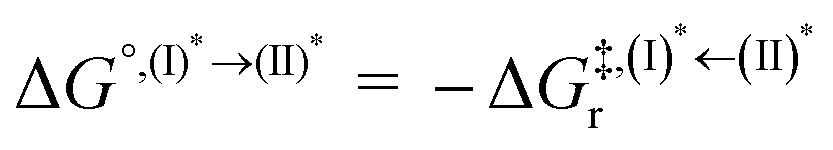 , whereas
, whereas 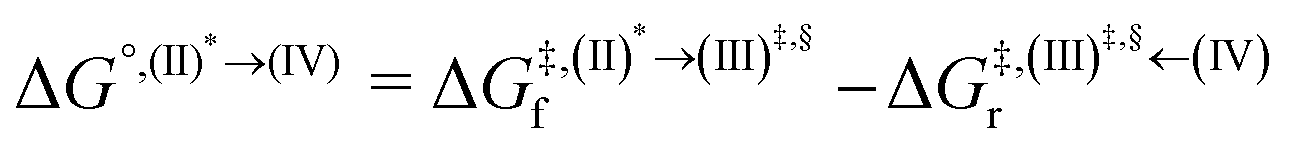 for (II)* ⇌ (III)‡,§ → (IV) and ΔG°,tot = ΔG°,(I)*→(II)* + ΔG°,(II)*→(IV). All the kinetic and thermodynamic properties were computed using the DL-FIND program41 included in the ChemShell software package.38
for (II)* ⇌ (III)‡,§ → (IV) and ΔG°,tot = ΔG°,(I)*→(II)* + ΔG°,(II)*→(IV). All the kinetic and thermodynamic properties were computed using the DL-FIND program41 included in the ChemShell software package.38
Because the (I)* → (II)* relaxation on the S1 PES is exothermic (ΔH° < 0) and thermal energy is required for (II)* ⇌ (III)‡,§ → (IV), the thermodynamic spontaneity could be studied. The spontaneous temperature (Ts), below which the transition structure (III)‡,§ at the S1/T1 intersection is spontaneously formed from (I)*, was obtained from the plot of 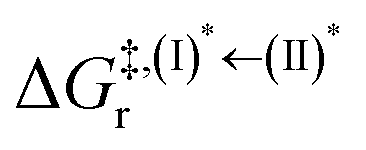 and
and 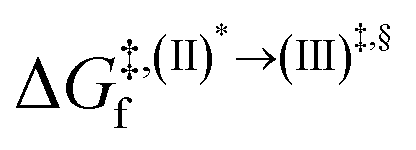 versus temperature; (I)* → (II)* ⇌ (III)‡,§ is spontaneous when
versus temperature; (I)* → (II)* ⇌ (III)‡,§ is spontaneous when 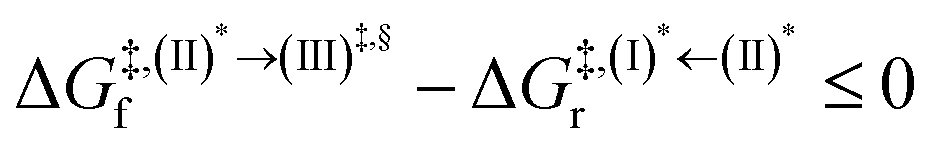 . In other words, the formation of (III)‡,§ is spontaneous when T ≤ Ts.
. In other words, the formation of (III)‡,§ is spontaneous when T ≤ Ts.
Surface-hopping molecular dynamics simulations
Because non-radiative relaxation reduces the emission quantum efficiency, to study the non-radiative S1 → S0 relaxation in BF2-FORM-D, NVE-MDSH simulations were performed. Because the calculations were computationally intensive, NVE-MDSH simulations were conducted using DFT and TD-DFT methods with a smaller (DZP) basis set, except for the iodine atoms, for which the larger TZVPall basis set42 was used to consider the heavy-atom effect. Fifty initial configurations were generated based on the Wigner distribution, from which NVE-MDSH simulations were performed over a time span of ∼4 ps using the TURBOMOLE 7.50 software package.The integration of Newton's equations of motion was conducted using the Verlet algorithm with a timestep of 0.5 fs, which was confirmed in our previous studies to be sufficient to study photochemical processes.28 The characteristic dynamics in the S1 states were categorized, and representative reactions were chosen and investigated in detail. To study the possibility of increasing the S1/T1 ISC, irradiation wavelengths, intramolecular motions, and temperatures, as well as the probabilities for the S1/T1 ISC and T1 → S0 transition, were analyzed in detail for BF2-FORM-D.
Results and discussion
To discuss characteristic structures of BF2-FORM and BF2-FORM-D, a three-character code is used, e.g., G-[k]eq, 1E-[k]‡, or 3E-[k]§, where G indicates the structure in the S0 state and 1E and 3E indicate the structures in the S1 and T1 states, respectively. The terms […]eq and […]* denote the equilibrium and vertically excited structures, respectively, whereas […]‡ and […]§ represent the transition structure and structure at the S1/T1 intersection, respectively. Different structures on the same PES are labeled as [k]. For example, 1E-[1]* and 1E-[2]eq represent two structures on the S1 PES. Additional symbols are used to represent the characteristic energies on the PES. In the discussion, for example, ΔES0→S1 and ΔE‡ represent the S0 → S1 vertical excitation energy and energy barrier on PES, respectively, whereas ΔES1/T1 represents the energy gap between the S1 and T1 states at or in the vicinity of the S1/T1 intersection. The terms (…)S0→S1, (…)‡ and (…)S1/T1 are used to represent the corresponding energies in the figures.Static properties
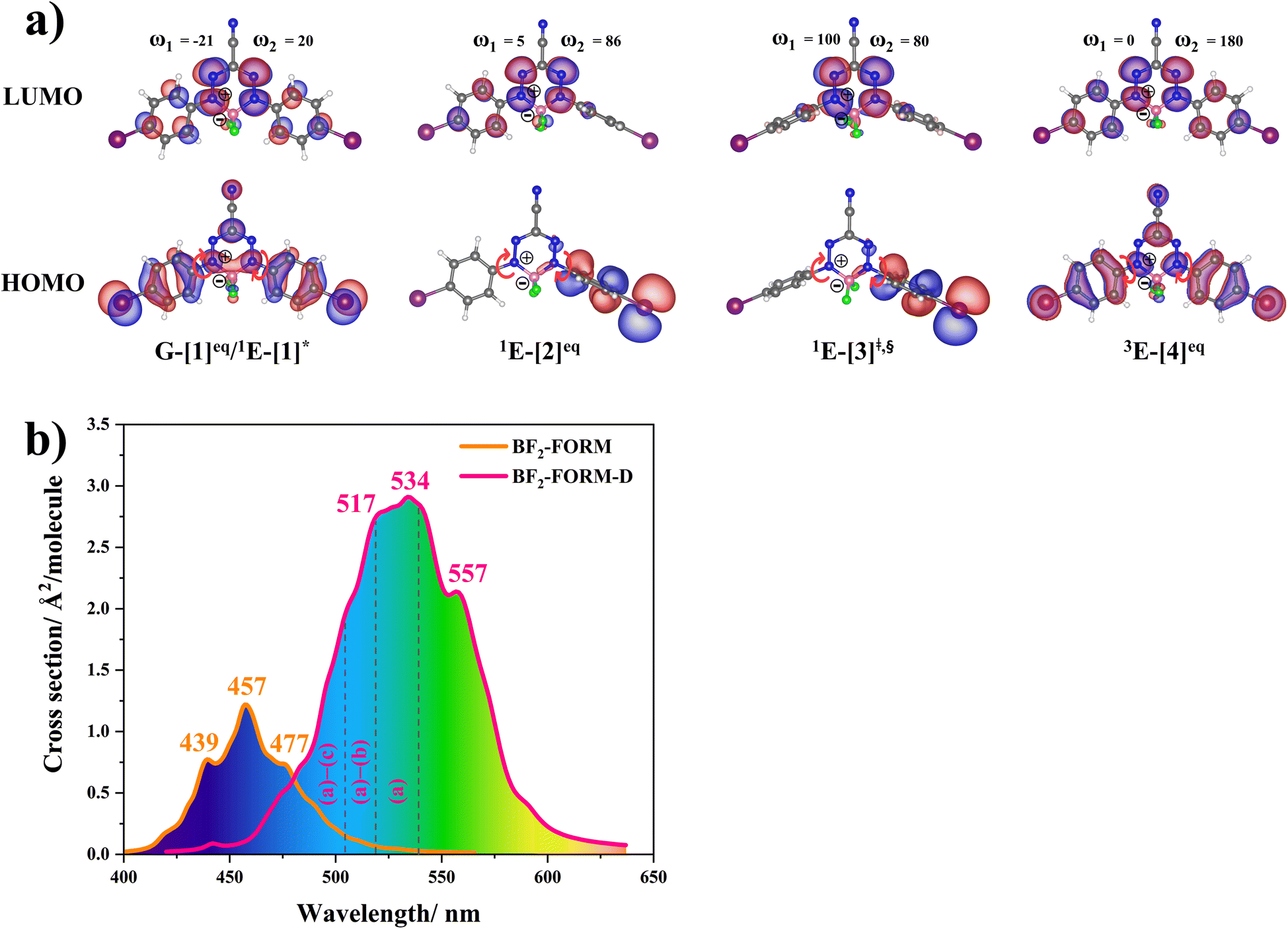 | ||
| Fig. 5 (a) Structures of BF2-FORM-D in the S0, S1 and T1 states, obtained based on DFT/B3LYP/6-311G and TD-DFT/B3LYP/6-311G calculations. The code symbols are explained in the text. (b) Absorption spectra obtained based on 200 Wigner sampled structures. | ||
The equilibrium structures of BF2-FORM and BF2-FORM-D in the S0 and T1 states were virtually identical, as shown by the bent and perfect planar structures G-[1]eq and 3E-[4]eq, respectively, in Fig. 5a. The HOMOs of G-[1]eq and 3E-[4]eq were characterized by a strong π character in the formazanate heterocyclic and phenyl rings, whereas the electron density of the LUMO was highly localized, resulting in a significantly lower degree of conjugation in the S1 and T1 states.
The equilibrium structures of BF2-FORM and BF2-FORM-D in the S1 state were quite different. BF2-FORM was represented by a propeller structure (Fig. S1†) or a twisted bent structure with the same HOMO–LUMO as G-[1]eq, whereas BF2-FORM-D was represented by a perpendicular structure, 1E-[2]eq (Fig. 5a). For 1E-[2]eq, the electron density distributions on the phenyl rings were not symmetrical because of the positive charge on the N(2) atom of the formazanate heterocyclic ring. Comparison of the vertical excitation energies of BF2-FORM and BF2-FORM-D in Tables S1 and S2† shows that the iodine substitutions at the phenyl rings directly affected ΔES0→S1, namely, a strong red-shift is observed for BF2-FORM-D because of a more extensive electron density distribution in HOMO (G-[1]eq) and large charge transfer interaction within the molecule.
For BF2-FORM-D, ΔES0→S1 = 2.42 eV (λS0→S1 = 513 nm) was in good agreement with the absorption spectra obtained based on 200 Wigner-sampled structures, 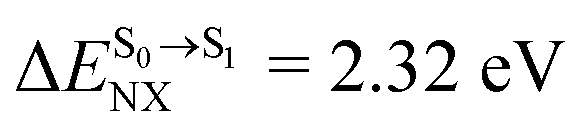 (
(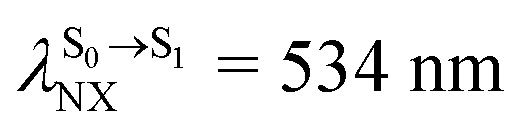 , shown in Fig. 5b) with the fluorescence lifetime,
, shown in Fig. 5b) with the fluorescence lifetime, 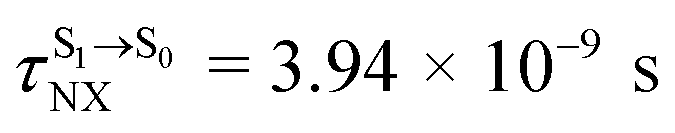 (Table S2†). The RICC2/aug-cc-pVDZ results confirmed the bent structure (G-[1]eq) to possess
(Table S2†). The RICC2/aug-cc-pVDZ results confirmed the bent structure (G-[1]eq) to possess 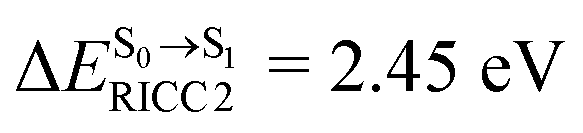
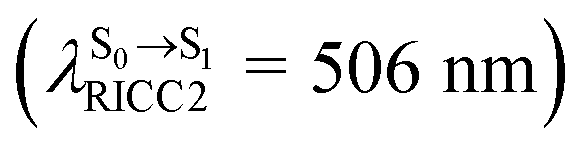 . The calculated excitation energies/wavelengths were in good agreement with the experimental absorption spectra (e.g., in CHCl3,
. The calculated excitation energies/wavelengths were in good agreement with the experimental absorption spectra (e.g., in CHCl3, 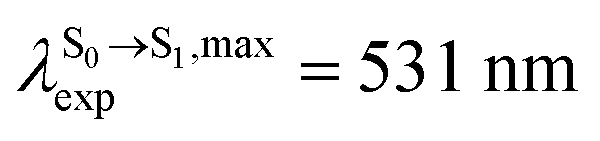 ).24 For BF2-FORM, the vertical excitation energies were higher, where ΔES0→S1 = 2.88 eV (λS0→S1 = 430 nm) and
).24 For BF2-FORM, the vertical excitation energies were higher, where ΔES0→S1 = 2.88 eV (λS0→S1 = 430 nm) and 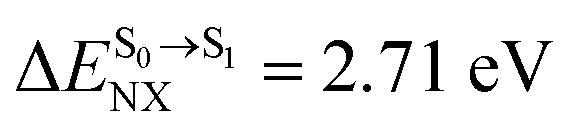 (
(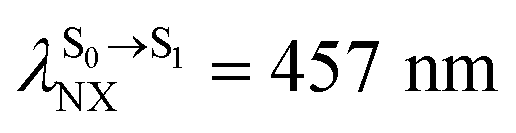 , shown in Fig. 5b) with
, shown in Fig. 5b) with  (Table S1†). The energy values were compatible with the RICC2/aug-cc-pVDZ results,
(Table S1†). The energy values were compatible with the RICC2/aug-cc-pVDZ results, 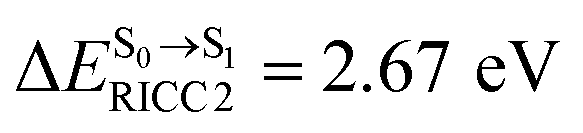 (
(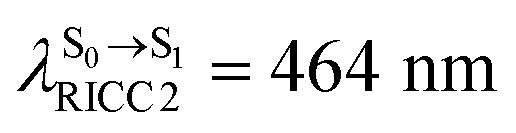 ). The trend of
). The trend of  was in good agreement with the experiment,24 e.g., in toluene,
was in good agreement with the experiment,24 e.g., in toluene, 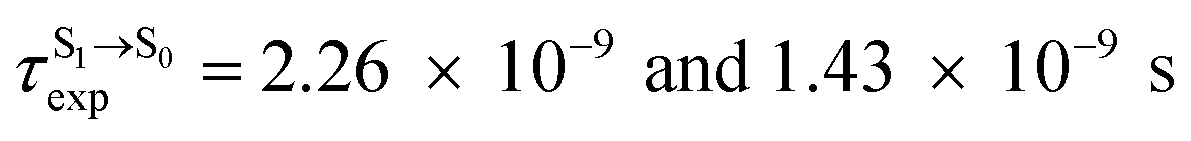 for BF2-FORM-D and BF2-FORM, respectively.
for BF2-FORM-D and BF2-FORM, respectively.
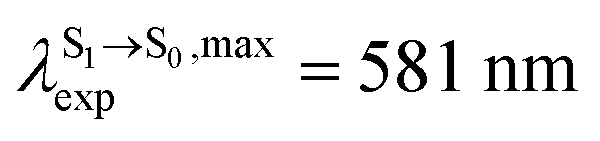 in CHCl3, whereas the latter is within the range observed experimentally,
in CHCl3, whereas the latter is within the range observed experimentally, 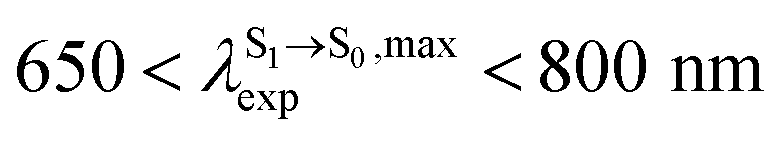 for BF2-FORM-D.24
for BF2-FORM-D.24
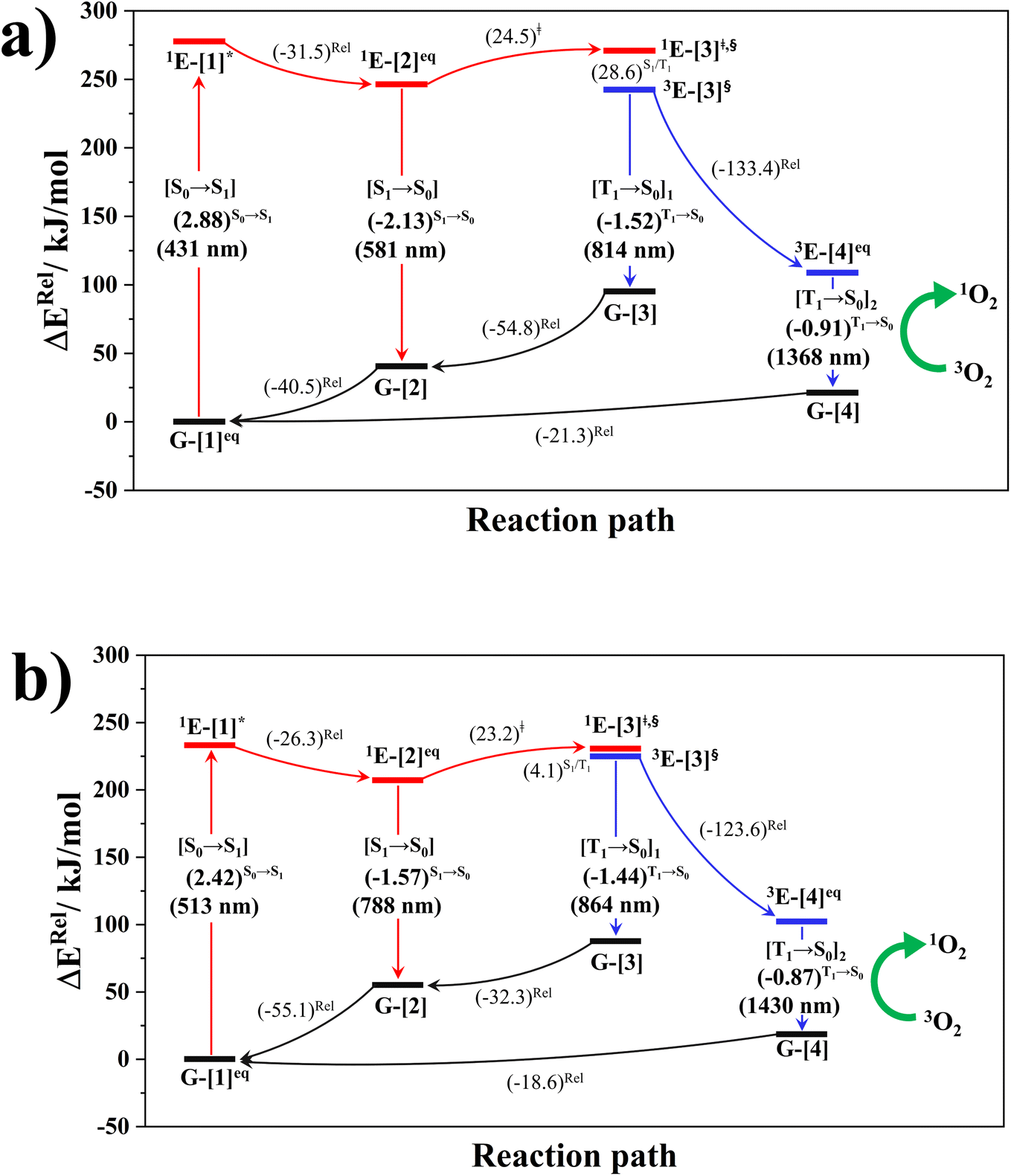 | ||
| Fig. 6 (a) and (b) Proposed photoluminescence pathways for BF2-FORM and BF2-FORM-D dye sensitizers obtained from the DFT/B3LYP/6-311G, TD-DFT/B3LYP/6-311G and NEB methods. Energies are in kJ mol−1 unless specified otherwise. (…)S0→S1 = vertical excitation energy in eV; ΔERel = relative energy with respect to the total energy in the S0 state; (…)Rel = relative energy with respect to the transition structure; (…)‡ = energy barrier; […]* = S0 → S1 vertically excited structure; […]§ = structure at the S1/T1 intersection. | ||
The photoluminescence pathways for BF2-FORM and BF2-FORM-D shown in Fig. 6 further suggest a possibility of the S1/T1 ISC at 1E-[3]‡,§, with the energy barriers for 1E-[2]eq → 1E-[3]‡,§, ΔE‡ = 24.5 and 23.2 kJ mol−1, and S1/T1 energy gaps, ΔES1/T1 = 0.30 and 0.04 eV, respectively. Because ΔES0→S1 of BF2-FORM-D is closer to the center of the visible light spectrum (550 nm) with a smaller ΔES1/T1, BF2-FORM-D is anticipated to be a more effective luminophore, mainly because of the heavy-atom effect. Therefore, subsequent discussions focus on BF2-FORM-D, with the results for BF2-FORM included in parentheses.
For BF2-FORM-D, two deactivation pathways for T1 → S0 transition, [T1 → S0]1 and [T1 → S0]2 in eqn (2) and (3), were observed after the S1/T1 ISC (Fig. 6). [T1 → S0]1 occurred immediately after the S1/T1 ISC, 3E-[3]§ → G-[3] with ΔET1→S0 = −1.44 (−1.52) eV (λT1→S0 = 864 (814) nm), whereas [T1 → S0]2 occurred after the 3E-[3]§ → 3E-[4]eq structural relaxation in the T1 state, 3E-[4]eq → G-[4] with ΔET1→S0 = −0.87 (−0.91) eV (λT1→S0 = 1430 (1368) nm). To confirm ΔET1→S0 obtained from the TD-DFT/B3LYP/6-311G method and to study the phosphorescence lifetimes, RICC2/aug-cc-pVDZ calculations based on the spin–orbit coupling with perturbation theory (SOC-PT-CC2) were made on 3E-[3]§ for [T1 → S0]1 and on 3E-[4]eq for [T1 → S0]2. The values obtained for [T1 → S0]1 are 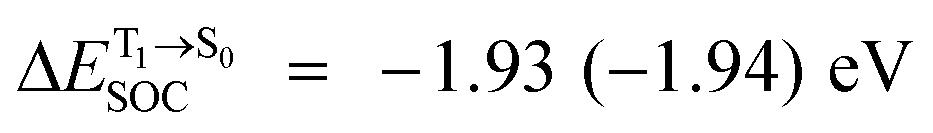 [λ = 642 (639) nm] and
[λ = 642 (639) nm] and  , whereas
, whereas 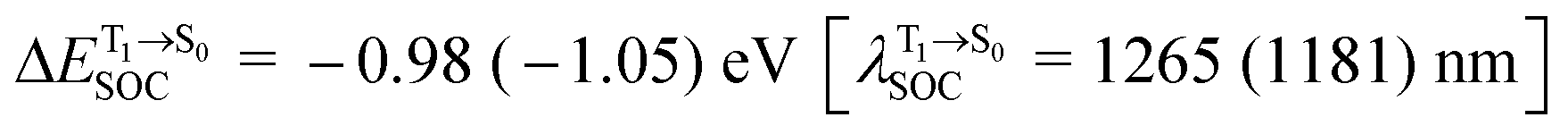 and
and 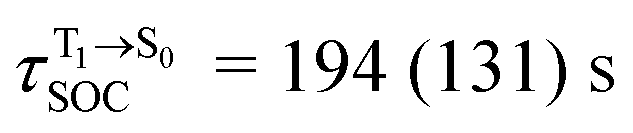 for [T1 → S0]2.
for [T1 → S0]2.
Because ΔET1→S0 and 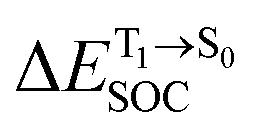 for [T1 → S0]2 are close to the absorption energy for 3O2 (3∑g) → 1O2 (1Δg), ΔET1→S1 ≈ 0.97 eV,5 and
for [T1 → S0]2 are close to the absorption energy for 3O2 (3∑g) → 1O2 (1Δg), ΔET1→S1 ≈ 0.97 eV,5 and 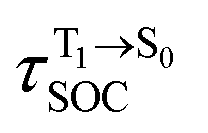 is considerably longer than [T1 → S0]1, [T1 → S0]2 is confirmed to be a key process to drive 3O2 (3∑g) → 1O2 (1Δg), and BF2-FORM-D is thus a better photosensitizer in PDT; experiments have shown that long-lived triplet excited state could promote the formation of 1O2 and increases the efficiency of the PDT.11 Therefore, only the kinetics and thermodynamics of [T1 → S0]2 are discussed further.
is considerably longer than [T1 → S0]1, [T1 → S0]2 is confirmed to be a key process to drive 3O2 (3∑g) → 1O2 (1Δg), and BF2-FORM-D is thus a better photosensitizer in PDT; experiments have shown that long-lived triplet excited state could promote the formation of 1O2 and increases the efficiency of the PDT.11 Therefore, only the kinetics and thermodynamics of [T1 → S0]2 are discussed further.
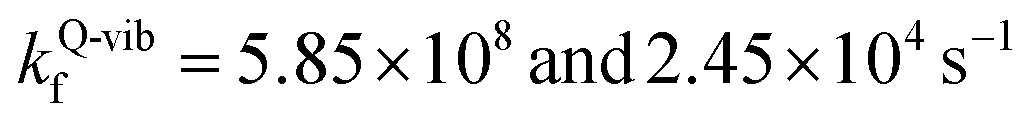 , respectively.
, respectively.Analysis of ΔG°,tot in Fig. 7c shows that although [T1 → S0]2 was thermodynamically favorable for both BF2-FORM-D and BF2-FORM, e.g., at 300 K, ΔG°,tot = −119.5 and −122.5 kJ mol−1, respectively, the rate-determining process (II)* ⇌ (III)‡,§ (1E-[2]eq → 1E-[3]‡,§) was spontaneous at room temperature only for BF2-FORM-D; for BF2-FORM-D, the plot of 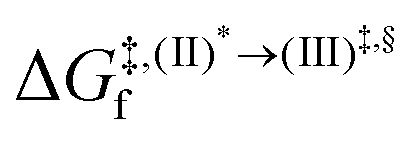 and
and 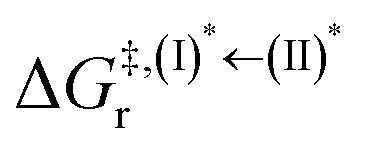 as a function of T (Fig. 7b) showed that (I)* → (II)* ⇌ (III)‡,§ could be spontaneous at T ≤ Ts = 320 K, whereas the same plot did not show Ts for BF2-FORM (Fig. 7a). These results indicate that for [T1 → S0]2, although BF2-FORM was kinetically more favorable, BF2-FORM-D was thermodynamically more favorable because the 1E-[2]eq → 1E-[3]‡,§ could be spontaneous below T = 320 K.
as a function of T (Fig. 7b) showed that (I)* → (II)* ⇌ (III)‡,§ could be spontaneous at T ≤ Ts = 320 K, whereas the same plot did not show Ts for BF2-FORM (Fig. 7a). These results indicate that for [T1 → S0]2, although BF2-FORM was kinetically more favorable, BF2-FORM-D was thermodynamically more favorable because the 1E-[2]eq → 1E-[3]‡,§ could be spontaneous below T = 320 K.
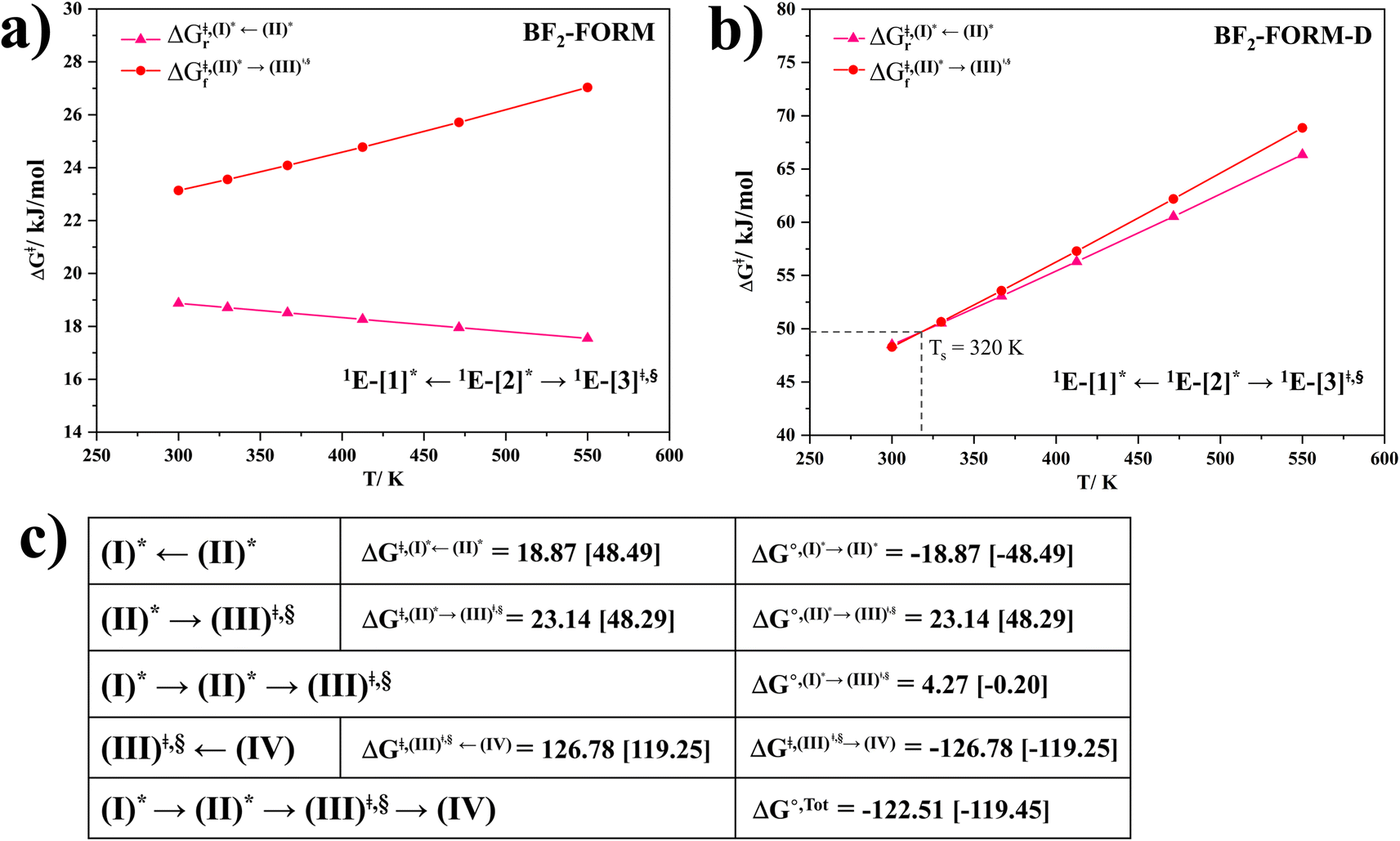 | ||
Fig. 7 (a) and (b) Plots of 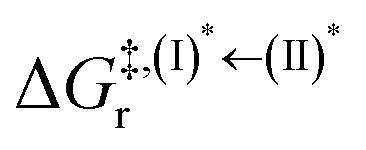 and and 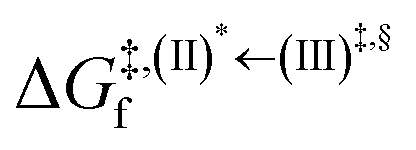 as a function of T for BF2-FORM and BF2-FORM-D, respectively. Ts = spontaneous temperature below which the reaction is spontaneous. (c) Gibbs free energies for the photochemical deactivation processes (Fig. 4) for BF2-FORM at 300 K. […] = values for BF2-FORM-D. as a function of T for BF2-FORM and BF2-FORM-D, respectively. Ts = spontaneous temperature below which the reaction is spontaneous. (c) Gibbs free energies for the photochemical deactivation processes (Fig. 4) for BF2-FORM at 300 K. […] = values for BF2-FORM-D. | ||
Surface-hopping dynamics
In order to enhance the photoluminescence quantum efficiency, it is important to study the non-radiative S1 → S0 relaxation process in BF2-FORM-D and the possibility of increasing the S1/T1 ISC. Because the NVE-MDSH simulations applied in this work considered only the non-radiative S1 → S0 relaxation ([S1 → S0] in Fig. 6), to explore the possibility of the S1/T1 ISC (1E-[3]‡,§ → 3E-[3]§ in Fig. 6), the time evolutions of the total energies in the S0, S1, and T1 states, temperatures, and dihedral angles ω1 and ω2 were extracted from the NVE-MDSH simulations and considered in the dynamic analysis. Characteristic results were selected as examples and are shown in Fig. 8.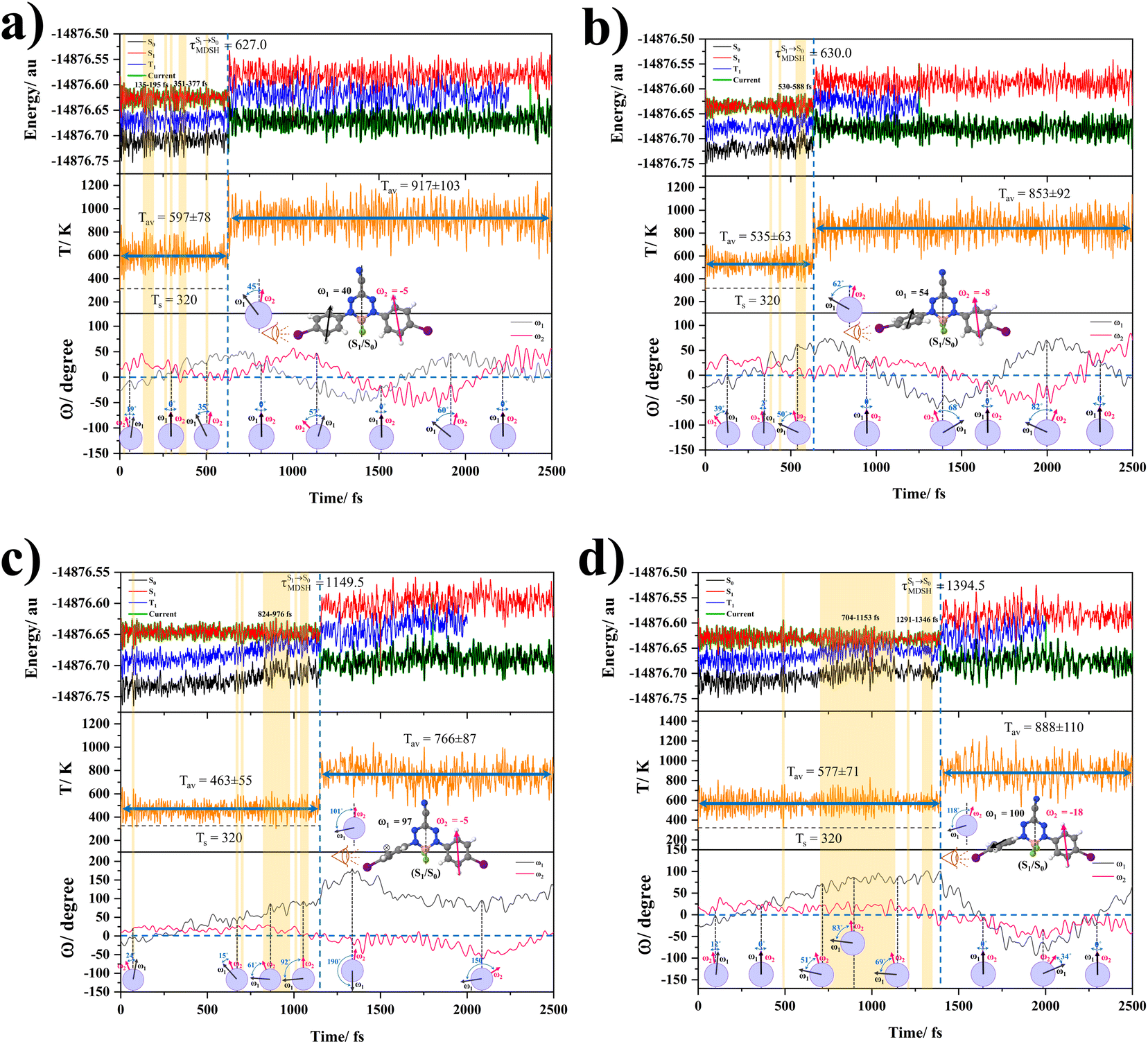 | ||
Fig. 8 Examples of time evolutions of energies in the S0, S1 and T1 states, temperatures, and dihedral angles ω1 and ω2, obtained from NVE-MDSH simulations on BF2-FORM-D.  The Newman projections at the bottom of the figures are used to differentiate the anti- and non-synchronous large-amplitude librational motions, anti-L-ALM and non-L-ALM, respectively. (a) and (b) anti-L-ALM. (c) and (d) non-L-ALM. The Newman projections at the bottom of the figures are used to differentiate the anti- and non-synchronous large-amplitude librational motions, anti-L-ALM and non-L-ALM, respectively. (a) and (b) anti-L-ALM. (c) and (d) non-L-ALM. 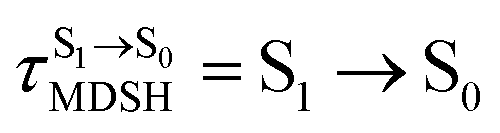 surface hopping time; Tav = the average temperature; ω1 and ω2 = dihedral angles; Ts = spontaneous temperature; (S1/S0) = structure at surface hopping time; Tav = the average temperature; ω1 and ω2 = dihedral angles; Ts = spontaneous temperature; (S1/S0) = structure at  . . | ||
The time evolutions of ω1 and ω2 suggest two characteristic motions of the phenyl rings; large- (L-ALM) and small-amplitude librational motions (S-ALM). L-ALM is characterized by ω1 and ω2 varying over a wide range, whereas S-ALM can be considered fine structures of L-ALM. To study the effect of L-ALM on the non-radiative S1 → S0 relaxation and on the S1/T1 ISC, two vectors were defined on the phenyl rings. Newman projections of these two vectors and ΔωMDSH = |ω1,MDSH − ω2,MDSH| acquired from the NVE-MDSH simulations are shown in Fig. 8.
For BF2-FORM-D, the Newman projections and ΔωMDSH reveal two types of L-ALM, namely, anti- and non-synchronous L-ALM, abbreviated anti-L-ALM and non-L-ALM, respectively. Because the librational motions of ω1 and ω2 are coupled in the S1 state, anti-L-ALM is characterized by 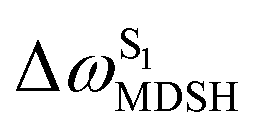 varying uniformly across a narrow range, e.g.,
varying uniformly across a narrow range, e.g., 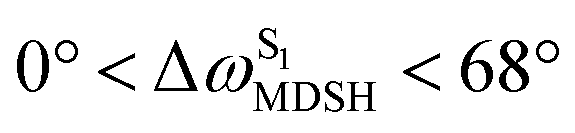 , shown in Fig. 8a and b, whereas
, shown in Fig. 8a and b, whereas 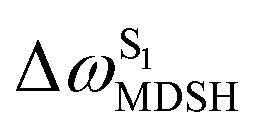 for non-L-ALM varies across a wider range, e.g.,
for non-L-ALM varies across a wider range, e.g., 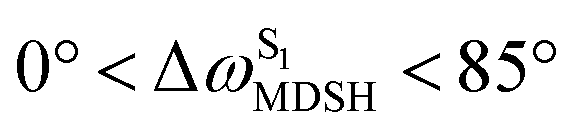 , shown in Fig. 8c and d. Analysis of the NVE-MDSH results shows that short S1 → S0 surface-hopping times
, shown in Fig. 8c and d. Analysis of the NVE-MDSH results shows that short S1 → S0 surface-hopping times 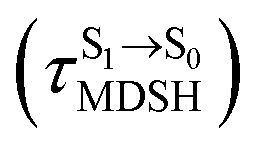 are associated with anti-L-ALM (e.g.,
are associated with anti-L-ALM (e.g., 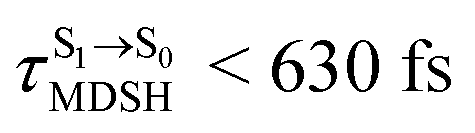 , shown in Fig. 8a and b), whereas non-L-ALM dominates for long
, shown in Fig. 8a and b), whereas non-L-ALM dominates for long 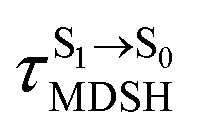 (e.g.,
(e.g., 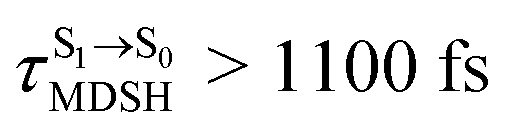 , shown in Fig. 8c and d).
, shown in Fig. 8c and d).
Because the NVE-MDSH simulations do not directly account for S1 → S0 fluorescence and T1 → S0 phosphorescence, further structural, energetic, and dynamic analyses must be performed for BF2-FORM-D. Based on the hypothesis that the non-radiative S1 → S0 relaxation occurs in an ultrashort 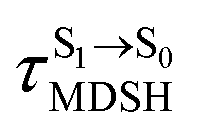 , the probability of the S1 → S0 fluorescence
, the probability of the S1 → S0 fluorescence 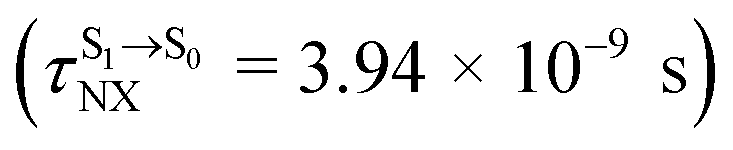 , S1/T1 ISC and T1 → S0 phosphorescence
, S1/T1 ISC and T1 → S0 phosphorescence  could increase when
could increase when 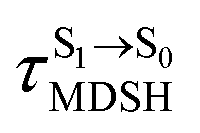 is sufficiently long. Therefore, the factors that could affect the length of
is sufficiently long. Therefore, the factors that could affect the length of 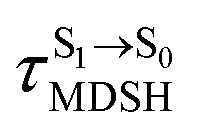 were studied during NVE-MDSH simulations. Likewise, because the S1/T1 energy degeneration is one of the preconditions for the S1/T1 ISC and T1 → S0 phosphorescence, the factors affecting the duration of the S1/T1 energy degeneration were monitored during NVE-MDSH simulations.
were studied during NVE-MDSH simulations. Likewise, because the S1/T1 energy degeneration is one of the preconditions for the S1/T1 ISC and T1 → S0 phosphorescence, the factors affecting the duration of the S1/T1 energy degeneration were monitored during NVE-MDSH simulations.
The S1/T1 energy degenerations are clearly seen in Fig. 8c and d (yellow stripes), for which long S1/T1 degeneration times 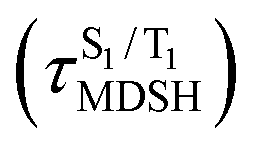 are associated with non-L-ALM. The time evolutions of the S1 and T1 total energies also suggest that for non-L-ALM, each
are associated with non-L-ALM. The time evolutions of the S1 and T1 total energies also suggest that for non-L-ALM, each 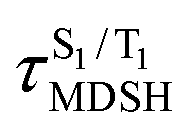 could span between
could span between 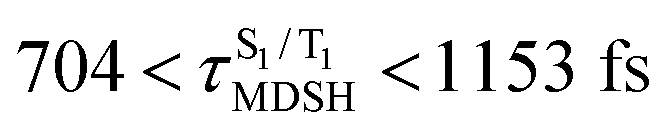 , compared with
, compared with 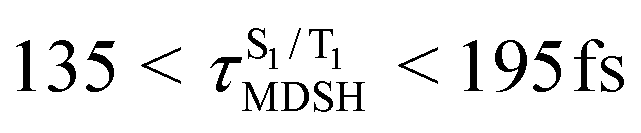 for anti-L-ALM. In other words, the higher the probability of the non-L-ALM mode, the higher probability of the S1/T1 ISC and T1 → S0 phosphorescence.
for anti-L-ALM. In other words, the higher the probability of the non-L-ALM mode, the higher probability of the S1/T1 ISC and T1 → S0 phosphorescence.
To study the effect of the absorbed radiation energy (the S0 → S1 excitation energy) on the non- and anti-L-ALM modes, the correlations among ΔES0→S1 of the Wigner-sampled structures, average temperatures in the S1 state 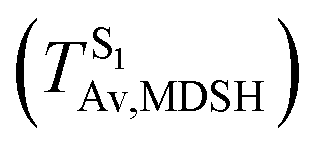 , S1 → S0 surface-hopping times
, S1 → S0 surface-hopping times 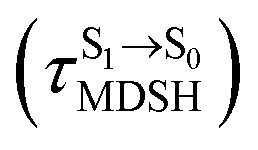 , and ΔωMDSH of the structures at
, and ΔωMDSH of the structures at 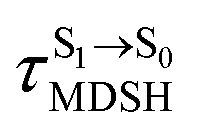
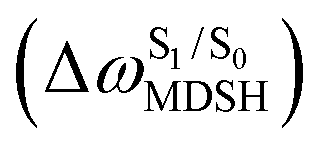 obtained from all NVE-MDSH simulations are plotted in Fig. 9. The results show that for anti-L-ALM,
obtained from all NVE-MDSH simulations are plotted in Fig. 9. The results show that for anti-L-ALM, 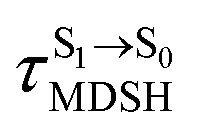 can be categorized into two groups, namely,
can be categorized into two groups, namely, 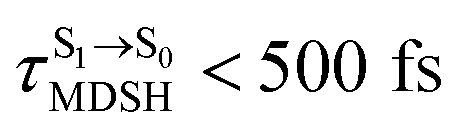 and
and 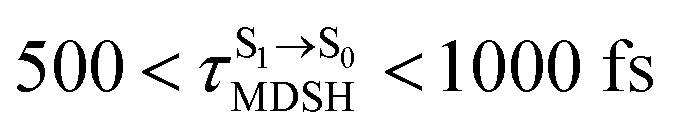 , whereas
, whereas 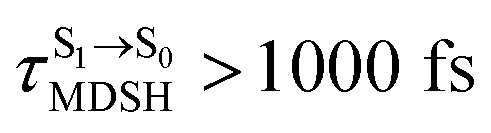 is for non-L-ALM; these are indicated as (a), (b), and (c) in Fig. 9a, respectively.
is for non-L-ALM; these are indicated as (a), (b), and (c) in Fig. 9a, respectively.
 | ||
Fig. 9 (a) Correlation between 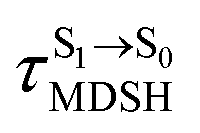 and ΔES0→S1. (b) Correlation between and ΔES0→S1. (b) Correlation between 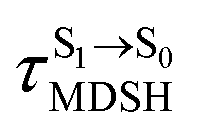 and and 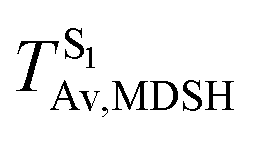 . (c) Correlation between . (c) Correlation between 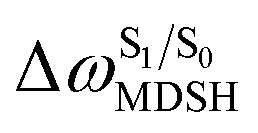 and ΔES0→S1. and ΔES0→S1. 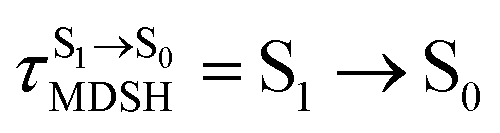 surface hopping time; ΔES0→S1 = S0 → S1 excitation energy; surface hopping time; ΔES0→S1 = S0 → S1 excitation energy; 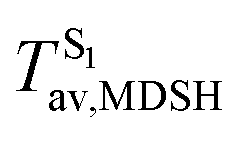 = average temperature in the S1 state; = average temperature in the S1 state; 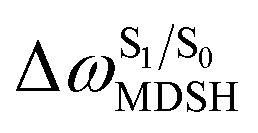 = different between ω1 and ω2 at = different between ω1 and ω2 at 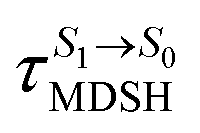 . . | ||
It appears in Fig. 9a that anti-L-ALM (a) occurs exclusively in the absorbed radiation energy range of 2.30 < ΔES0→S1 < 2.40 eV, whereas anti-L-ALM (a) and (b), and non-L-ALM (c) could be concurrently found when ΔES0→S1 > 2.40 eV. The asymptotic behaviors of ΔES0→S1, 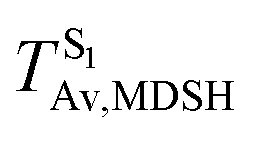 , and
, and 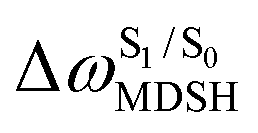 in Fig. 9 suggest that to increase the probability of long S1 → S0 surface-hopping time, the absorbed radiation energy should be ΔES0→S1 ≈ 2.67 eV or λabs ≈ 464 nm, corresponding to blue light source (Fig. 5b) with
in Fig. 9 suggest that to increase the probability of long S1 → S0 surface-hopping time, the absorbed radiation energy should be ΔES0→S1 ≈ 2.67 eV or λabs ≈ 464 nm, corresponding to blue light source (Fig. 5b) with 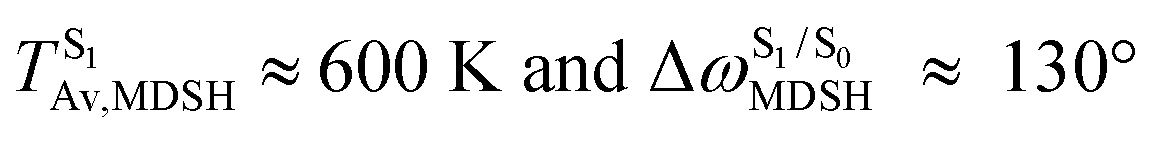 .
.
These results suggest that the absorbed radiation energy and intramolecular librational motions govern the non-radiative S1 → S0 relaxation process and time, and non-L-ALM could increase the probability of fluorescence, S1/T1 ISC and phosphorescence. Because the TST results suggest that the S1/T1 ISC is thermodynamically favorable at Ts < 320 K and because the asymptotic average temperature for long S1 → S0 surface-hopping time is 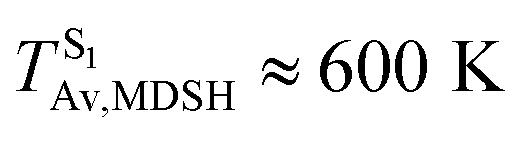 , to thermodynamically enhance the probability of photoluminescence, a temperature control mechanism is required.
, to thermodynamically enhance the probability of photoluminescence, a temperature control mechanism is required.
Conclusion
Photodynamic therapy (PDT) is a promising medical treatment for a range of human diseases, in which one of the key factors in its effectiveness is how well the photosensitizer can transfer photon energy to the target molecules. In this study, to improve the efficiency of photosensitizers for PDT, theoretical methods were used to study the photoluminescence mechanisms of BF2-formazanate dye (BF2-FORM) and its iodinated derivative (BF2-FORM-D) to investigate the heavy-atom effect. To complete this mechanistic study, complementary theoretical approaches were applied to investigate three important issues; (1) luminescence pathways in the S1 and T1 states, studied using DFT and TD-DFT methods, (2) kinetic and thermodynamic properties of the proposed pathways, studied using the TST method, and (3) time evolution and dynamics of the key processes, studied using NVE-MDSH simulations.The DFT/B3LYP/6-311G and TD-DFT/B3LYP/6-311G results showed that in the S0 and T1 states, the equilibrium structures of BF2-FORM and BF2-FORM-D were similar, represented by bent and perfect planar structures, respectively, whereas in the S1 state, the equilibrium structure of BF2-FORM took a propeller structure and that of BF2-FORM-D a perpendicular structure. The HOMOs of the equilibrium structures in the S0 state were characterized by strong π character at the formazanate heterocyclic and phenyl rings, whereas electron density distribution in LUMOs was localized, and iodine substitutions at the phenyl rings led to a strong red-shift of ΔES0→S1 close to the center of the visible light spectrum (green light with maximum intensity at 520 < λS0→S1 < 532 nm).
The PESs for rotations of the dihedral angles ω1 and ω2 suggested two mechanisms for T1 → S0 transition: [T1 → S0]1 occurring immediately after S1/T1 ISC and [T1 → S0]2 occurring after S1/T1 ISC and T1 equilibrium structure relaxation. [T1 → S0]2 transition (ΔET1→S0) is in the near IR range and close to the absorption energy for 3O2 (3∑g) → 1O2 (1Δg). Because ΔES0→S1 of BF2-FORM-D is closer to the center of the visible light spectrum and ΔES1→T1 is significantly smaller than BF2-FORM, the iodinated derivative is anticipated to be a more effective luminophore, mainly owing to the heavy-atom effect. The RICC2/aug-cc-pVDZ (SOC-PT-CC2) calculations confirmed these findings and further suggested that the phosphorescence lifetime of [T1 → S0]2 of BF2-FORM-D is significantly longer than that of BF2-FORM.
Based on the PESs obtained using the DFT/B3LYP/6-311G, TD-DFT/B3LYP/6-311G, and NEB methods, TST calculations confirmed that [T1 → S0]2 of BF2-FORM-D is thermodynamically favorable below the spontaneous temperature (Ts = 320 K). The time evolutions of the dihedral angles ω1 and ω2 obtained from the analysis of NVE-MDSH simulations suggested that anti-L-ALM underlies ultrafast non-radiative S1 → S0 relaxation, whereas non-L-ALM could enhance the probability of S1/T1 ISC and photoluminescence. Therefore, to delay non-radiative S1 → S0 relaxation and increase the probability of photoluminescence, anti-L-ALM of the phenyl rings should be promoted. Analysis of the NVE-MDSH results suggested that the photoluminescence quantum yield could also be enhanced by varying the irradiation wavelength, for example, by using a blue light source to promote phosphorescence. Because efficient delivery of the photosensitizer to target cells is also a critical factor in PDT, our upcoming theoretical study will examine the photodynamic properties of BF2-FORM-D when incorporated into a specific nanostructure. These results will serve as a foundation for future theoretical and experimental research, to optimize and/or design effective PDT photosensitizers.
Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the high-performance computer facilities provided by the National e-Science project of the National Electronics and Computer Technology Centre (NECTEC), and the National Science and Technology Development Agency (NSTDA). This work was supported by (I) Suranaree University of Technology (SUT), (II) Thailand Science Research and Innovation (TSRI), and (III) National Science, Research, and Innovation Fund (NSRF). This research has received funding support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research, and Innovation (PMU-B) [grant number B13F660060] and Thailand Toray Science Foundations (TTSF for Prof. Dr Kritsana Sagarik).References
- B. P. Chan, Tissue Eng., Part B, 2010, 16, 509–522 CrossRef CAS PubMed.
- J. Piskorz, W. Porolnik, M. Kucinska, J. Dlugaszewska, M. Murias and J. Mielcarek, ChemMedChem, 2021, 16, 399–411 CrossRef CAS PubMed.
- Y. Zhang, Z. Yang, X. Zheng, L. Yang, N. Song, L. Zhang, L. Chen and Z. Xie, Dyes Pigm., 2020, 178, 108348 CrossRef CAS.
- A. Hak, V. R. Shinde and A. K. Rengan, Photodiagn. Photodyn. Ther., 2021, 33, 102205 CrossRef CAS PubMed.
- M. J. Davies, Biochem. Biophys. Res. Commun., 2003, 305, 761–770 CrossRef CAS PubMed.
- S. Siriwibool, N. Kaekratoke, K. Chansaenpak, K. Siwawannapong, P. Panajapo, K. Sagarik, P. Noisa, R. Y. Lai and A. Kamkaew, Sci. Rep., 2020, 10, 1283 CrossRef CAS PubMed.
- A. Escudero, C. Carrillo-Carrión, M. C. Castillejos, E. Romero-Ben, C. Rosales-Barrios and N. Khiar, Mater. Chem. Front., 2021, 5, 3788–3812 RSC.
- X. F. Zhang, X. Yang, K. Niu and H. Geng, J. Photochem. Photobiol., A, 2014, 285, 16–20 CrossRef CAS.
- M. W. Baig, M. Pederzoli, M. Kývala, L. Cwiklik and J. Pittner, J. Phys. Chem. B, 2021, 125, 11617–11627 CrossRef PubMed.
- H. S. Kim, J. Y. Lee, S. Shin, W. Jeong, S. H. Lee, S. Kim, J. Lee, M. C. Suh and S. Yoo, Adv. Funct. Mater., 2021, 31, 2104646 CrossRef CAS.
- L. Xu, K. Zhou, H. Ma, A. Lv, D. Pei, G. Li, Y. Zhang, Z. An, A. Li and G. He, ACS Appl. Mater. Interfaces, 2020, 12, 18385–18394 CrossRef CAS PubMed.
- B. Hao, J. Wang, C. Wang, K. Xue, M. Xiao, S. Lv and C. Zhu, Chem. Sci., 2022, 13, 4139–4149 RSC.
- Z. Meng, H. Xue, T. Wang, B. Chen, X. Dong, L. Yang, J. Dai, X. Lou and F. Xia, J. Nanobiotechnol., 2022, 20, 344 CrossRef CAS PubMed.
- H. G. Knaus, T. Moshammer, H. C. Kang, R. P. Haugland and H. Glossmann, J. Biol. Chem., 1992, 267, 2179–2189 CrossRef CAS PubMed.
- A. Treibs and F. H. Kreuzer, Adv. Cycloaddit., 1968, 718, 208–223 CAS.
- S. Zhu, N. Dorh, J. Zhang, G. Vegesna, H. Li, F. T. Luo, A. Tiwari and H. Liu, J. Mater. Chem., 2012, 22, 2781–2790 RSC.
- S. Gan, S. Hu, X. L. Li, J. Zeng, D. Zhang, T. Huang, W. Luo, Z. Zhao, L. Duan, S. J. Su and B. Z. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 17327–17334 CrossRef CAS PubMed.
- Y. Xiang, Y. Zhao, N. Xu, S. Gong, F. Ni, K. Wu, J. Luo, G. Xie, Z. H. Lu and C. Yang, J. Mater. Chem. C, 2017, 5, 12204–12210 RSC.
- J. Zou, P. Wang, Y. Wang, G. Liu, Y. Zhang, Q. Zhang, J. Shao, W. Si, W. Huang and X. Dong, Chem. Sci., 2019, 10, 268–276 RSC.
- E. Y. Gül, E. A. Karataş, H. A. Doğan, Ö. F. Karataş, B. Çoşut and E. T. Eçik, ChemMedChem, 2023, 18, e202200439 CrossRef PubMed.
- E. Y. Gül, M. Erdem, H. H. Kazan and E. T. Eçik, New J. Chem., 2023, 47, 17469–17480 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- N. Sharma, S. M. Barbon, T. Lalonde, R. R. Maar, M. Milne, J. B. Gilroy and L. G. Luyt, RSC Adv., 2020, 10, 18970–18977 RSC.
- T. Khrootkaew, S. Wangngae, K. Chansaenpak, K. Rueantong, W. Wattanathana, P. Pinyou, P. Panajapo, V. Promarak, K. Sagarik and A. Kamkaew, Chem.–Asian J., 2023, e202300808 Search PubMed.
- A. R. Chaudhry, S. Muhammad, B. U. Haq, A. Laref, A. Shaari and M. A. Gilani, Chem. Phys., 2019, 527, 110488 CrossRef.
- G. N. Lipunova, T. G. Fedorchenko and O. N. Chupakhin, Russ. J. Gen. Chem., 2019, 89, 1225–1245 CrossRef CAS.
- TURBOMOLE V 7.5 2020, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, BIOVIA.TURBOMOLE@3ds.com, V7.5 edn., 2019.
- J. Nirasok, P. Panajapo, P. Promma, P. Suwannakham and K. Sagarik, J. Photochem. Photobiol., A, 2023, 436, 114354 CrossRef CAS.
- P. Panajapo, P. Suwannakham, P. Promma and K. Sagarik, R. Soc. Open Sci., 2024, 11, 231957 CrossRef.
- W. Hu, M. Liu, X. F. Zhang, Y. Wang, Y. Wang, H. Lan and H. Zhao, J. Phys. Chem. C, 2019, 123, 15944–15955 CrossRef CAS.
- A. Allangawi, H. Sajid, K. Ayub, M. A. Gilani, M. S. Akhter and T. Mahmood, Comput. Theor. Chem., 2023, 1120, 113990 CrossRef.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro Jr, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, J. Chem. Theory Comput., 2022, 18, 6851 CrossRef CAS PubMed.
- M. Barbatti, G. Granucci, M. Ruckenbauer, F. Plasser, R. C. Otero, J. Pittner and H. Lischka, NEWTON-X: A Package for Newtonian Dynamics Close to the Crossing Seam. Version 2, 2016, http://www.newtonx.org.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- D. R. Sanjuán, A. F. Monerris, I. F. Galván, P. Farahani, R. Lindh and Y. J. Liu, SPR-Photochemistry, 2017, 44, 16–60 Search PubMed.
- R. Ahmed and A. K. Manna, J. Phys. Chem. A, 2022, 126, 6594–6603 CrossRef CAS PubMed.
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc., New Jersey, 2009 Search PubMed.
- ChemShell, a Computational Chemistry Shell, http://www.chemshell.org.
- B. H. Paris, C. Hättig and C. V. Wüllen, J. Chem. Theory Comput., 2016, 12, 1892–1904 CrossRef PubMed.
- J. E. House, Principles of Chemical Kinetics, Kindle Edition, Academic Press, USA, 2nd edn, 2007 Search PubMed.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02240h |
| This journal is © The Royal Society of Chemistry 2024 |