DOI:
10.1039/D4RA02282C
(Paper)
RSC Adv., 2024,
14, 16649-16660
Investigation of α-Fe2O3 catalyst structure for efficient photocatalytic fenton oxidation removal of antibiotics: preparation, performance, and mechanism†
Received
25th March 2024
, Accepted 13th May 2024
First published on 22nd May 2024
Abstract
Currently, the surface structure modification of photocatalysts is one of the effective means of enhancing their photocatalytic efficiency. Therefore, it is critically important to gain a deeper understanding of how the surface of α-Fe2O3 photocatalysts influences catalytic activity at the nanoscale. In this work, α-Fe2O3 catalysts were prepared using the solvothermal method, and four distinct morphologies were investigated: hexagonal bipyramid (THB), cube (CB), hexagonal plate (HS), and spherical (RC). The results indicate that the hexagonal bipyramid (THB) exhibits the highest degradation activity towards tetracycline (TC), with a reaction rate constant of k = 0.0969 min−1. The apparent reaction rate constants for the cube (CB), hexagonal plate (HS), and spherical (RC) morphologies are 0.0824, 0.0726, and 0.0585 min−1, respectively. In addition, it has been observed that the enhancement of photocatalytic activity is closely related to the increase in surface area, which provides more opportunities for interactions between Fe2+ and holes. The quenching experiments and electron paramagnetic resonance (EPR) results indicate that the ˙O2, ˙OH and h+ contribute mainly to the degradation of TC in the system. This research contributes to a more comprehensive understanding of catalyst surface alterations and their impact on catalytic performance.
1. Introduction
Micropollutants are a growing problem in aquatic environments and pose a major threat to ecosystems.1,2 While conventional water treatment technologies are inefficient and costly, photo-Fenton reaction technology offers a cleaner, more energy-efficient solution that has the potential to increase treatment efficiency and reduce costs.3,4 α-Fe2O3 is a semiconductor catalyst with visible light absorption capabilities, and its catalytic performance is also largely influenced by morphology.5 The material's specific surface area significantly impacts its photocatalytic activity.6 The increase in the surface area of the catalyst enhances contact with degradation substances. This situation can improve material performance by expanding surface area.7
In order to enhance the photocatalytic performance of α-Fe2O3 materials, many attempts have been conducted to improve the material by reducing the size of the catalytic material.8 Many different methods, including hydrolysis method, co-precipitation method, hydrothermal method, solvothermal method, ionic liquid assisted synthesis method, thermal decomposition method and combustion method, have achieved the synthesis of α-Fe2O3 nanocrystals. For instance, Yang et al. used a solvothermal method to systematically size the particles in the range of particle size from ∼170 nm to ∼2 mm by adjusting the concentration of FeCl3 solution, and the synthesized catalysts were subjected to photocatalytic performance and magnetic properties.9,10 Their results showed that these α-Fe2O3 nanoparticles exhibited ferromagnetic or weakly ferromagnetic properties at room temperature. Meanwhile, this catalyst had a good photocatalytic performance for Rhodamine B. In addition, compared to large particle catalysts, the small particle catalyst had a higher photocatalytic degradation efficiency. Xu et al., Zhou et al., and Bharathi reported the effect of morphology on α-Fe2O3.11–13 They prepared nanoparticles with flower, spindle, spherical, octahedral and diamond shapes and assessed their photocatalytic efficiency in degrading rhodamine B and methylene blue. These samples exhibited high specific surface area and porosity demonstrated excellent photocatalytic performance.
Nanostructured iron oxides, including α-Fe2O3, γ-Fe2O3, Fe3O4, α-FeOOH, β-FeOOH, and γ-FeOOH, have extensively been studied as photo-Fenton catalysts.14,15 Hematite (α-Fe2O3) in particular has received extensive study due to its abundance, thermodynamic stability, and environmental friendliness. However, pure α-Fe2O3 has limitations like the short lifespan of photo-induced carrier mobility, limited hole diffusion distance, and decreased efficiency in Fe2+/Fe3+ cycle, which restricts it from being used efficiently in photo-Fenton-like systems. As a result, a challenge remains in finding ways to improve the catalytic activity of α-Fe2O3-based catalysts was evaluated using the photo-Fenton method.
In order to solve the problem, the photocatalytic efficiency of α-Fe2O3 is limited by its very fast photoexcited state life (10–12 s) and short diffusion length (<10 nm).13 In this study, Four unique forms, including hexagonal biconical, cube-shaped, hexagonal plate, nano-spherical, are prepared to further improve the activity of α-Fe2O3 light-Fenton catalyst from morphology regulation (Fig. 1). It is of important theoretical and practical value to construct a new treatment system of micropollutants (MPs) in water source and explore the degradation and removal effect of MPs and its action mechanism. These findings affirm the considerable influence of surface engineering on both the Fenton-like heterogeneous catalysis.efficiency and durability of α-Fe2O3, as explored in this study.11
 |
| | Fig. 1 Schematic diagram depicting the process. | |
2. Materials and methods
2.1 Reagents materials
Sinopharm Chemical Reagent Co., Ltd provided FeCl3·6H2O (ferric chloride hexahydrate) and NaOH (sodium hydroxide) for use 15 tetracycline hydrochloride (TC) was purchased from Beijing Soleibao Technology Co., Ltd. Polyvinylpyrrolidone (PVPK30), potassium ferrocyanide trihydrat (K4[Fe(CN)6]·3H2O), anhydrous ethanol (C2H6O), urea (CO(NH2)2), peroxide hydrogen (H2O2 30%), ferric nitrate nonahydrate, hydrazine hydrate (Fe(NO3)3·9H2O), sodium acetate anhydrous (N2H4·H2O), and isopropanol (C2H3NaO3) purchased from Chengdu Kelong Chemical Co., Ltd. Disodium EDTA was purchased from Guangdong Guanghua Technology Co., Ltd. L-Ascorbic acid, and sodium carboxymethyl cellulose (CMC) were purchased from Shanghai McLean Biochemical Co., Ltd. Sodium azide (NaN3) and tetracycline hydrochloride (TC 96%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. All chemicals and substances. The analytical level that can be obtained from commercial sources is used in the study.
2.2 Synthesis of α-Fe2O3
The hexagonal biconical α-Fe2O3 (THB) was synthesized via a modified hydrothermal method following a previously reported protocol, mixing K4[Fe(CN)6]·3H2O(0.337 g), polyvinylpyrrolidone (PVP-K30)(0.5 g) and 0.3 ml N2H4 H2O (85%) dissolved in 40 ml of sodium carboxymethyl cellulose (CMC, 300–800 MPas, 1.25 g L−1) solution; the blend was magnetically agitated at ambient temperature for 30 min. Subsequently, the solution got transferred to a 60 ml autoclave made of stainless steel and coated with Teflon, then heated at 160 °C for 6 hours. After reaching room temperature, the products were harvested through centrifugation at 4000 rpm for 10 min and rinsed five times with deionized water and once with ethanol. Finally, a maroon powder of α-Fe2O3 was obtained after drying at 80 °C for 4 hours and further drying in air. The powder was then calcined at 300 °C for 3 hours. These steps were taken to produce high-quality α-Fe2O3 powder, which can be used for various applications in industry and research.
A hexagonal plate-shaped α-Fe2O3 (HS) was synthesized as follows: 1.365 g FeCl3·6H2O and 34 mmol of sodium acetate were dissolved in 35 ml ethanol. After adding 2.5 ml deionized water and magnetic stirring at ambient temperature for 0.5 h, the solution got transferred to a 60 ml stainless steel autoclave lined with Teflon and heated at 180 °C for 12 hours. After cooling, the samples were collected through centrifugation at 4000 revolutions per minute (rpm) for 10 min. Subsequently, they underwent three washes with deionized water and ethanol. Finally, the powdered orange substance α-Fe2O3 was obtained after being dried at 50 to 60 °C. This method of synthesis was reported in a previous study16–18
Nano spherical α-Fe2O3 (RC) was prepared as follows.17 Firstly, 10 mmol of Fe(NO3)3·9H2O and 36 mmol of CO(NH2)2 were dissolved in 60 ml of deionized water and magnetically stirred at ambient temperature for 30 min. The solution was placed into a 60 ml Teflon-coated stainless-steel autoclave and then was heated at 120 °C for 8 hours in an oven. After equilibrating to ambient temperature, the samples were spun at 4000 rpm for 10 min using a centrifuge, Followed by several rinses with deionized water and ethanol. The α-Fe2O3 was finally obtained in the form of dark red powder, which was dried for 3 hours at 50 °C and then further calcined at 500 °C for 3 hours in air.
The synthesis procedure of cube-shaped α-Fe2O3 (CB) was similar to that of hexagonal Biconical α-Fe2O3 (THB), except that the amount of K4[Fe(CN)6]·3H2O and N2H4·H2O (85%) add is changed from 0.337 g to 0.253 g and 0.3 ml to 0.9 ml, white other conditions remain unchanged.
2.3 Characterization methods
XRD analysis and phase analysis were performed using the XRD-6000 X-ray diffractor from Shimzu Co., Ltd. The operating conditions of the instrument are: Cu K (=0.154 nm) is a radiation source, the acceleration voltage is 40 kV, the current is 50 mA, step length is 0.02°, and scanning angle is 10–80°. The catalyst was analyzed using a Zeiss Sigma 300 scanning electron microscope to examine its microstructure. A Fourier Transform Infrared Spectrometer (FT-IR) (Bruker TENSOR27) was used to analyze the groups and bonds contained in the catalyst sample material based on the characteristic vibrational frequencies of the chemical bonds, and thus to characterize the composition and structure of the samples. The catalysts were tested by pressing the catalysts with potassium bromide (KBr). A solid UV-visible absorption spectrometer (UV-27001, SHIMADZU) was used. The solid catalyst is pressed into the sample pool and illuminated on the sample with ultraviolet-visible light. After the light reaches enough energy, the internal energy form of the molecule changes, and the electron energy level transition of the electron in the molecule changes. The FEI Tecnai G2 F20 S-Twin 200 kV transmission electron microscope allows for the observation of the internal structure of the catalyst sample and microscopic particles. A high-resolution transmission electron microscope (HRTEM) and scanning electron microscope can accurately analyze crystal lattice structure and surface parameters, providing a comprehensive study on catalyst morphology and structure. The EPR technique (JESFA300, Japan) detected the ˙OH radicals generated by the α-Fe2O3 catalyst.
This experiment was prepared using Thermo Fisher Scientific K-Alpha X-ray photoelectron spectroscopy elemental composition and oxidation states of the catalyst sample. The test conditions were radiation source: Al target, energy: 1486.8 eV, spot: 30–600 μm, voltage: 15 kV, current: 10 mA, vacuum: 2 × 10−9 mbar; step length: 0.05 eV.
2.4 Evaluation of adsorption properties
The adsorption performance of the α-Fe2O3-x photocatalyst was evaluated by adding equal amounts of α-Fe2O3-x catalyst within equal amounts of TC solution with different concentration gradients.19 Generally, 50 mg α-Fe2O3-x photocatalyst is added to 100 ml aqueous TC solution and placed in a constant temperature (25 °C) shaker to set a frequency oscillation of 180 rpm. Then, a 3 ml sample was taken at 15 min intervals for the first 90 min, and finally the last sample was taken at 120 min. After taking the sample, put the centrifuge tube equipped with the sample symmetrically into the centrifuge, 8000 rpm, and run for 5 min to remove for the test. The filtrate concentration was measured using UV-visible spectrophotometry (UV-27001, Shimadzu) by monitoring the change in the maximum absorption band at 357 nm (TC).
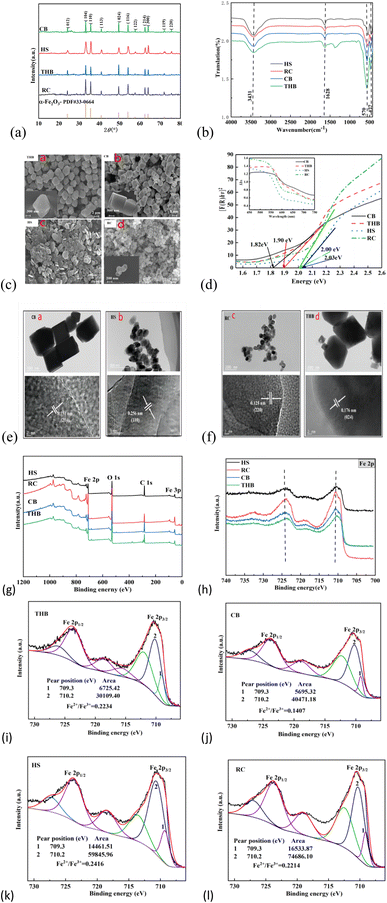
The equilibrium adsorption amount of TC by α-Fe2O3-x catalyst is calculated by the following eqn (1) and (2)7,20
| |
 | (1) |
| |
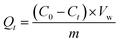 | (2) |
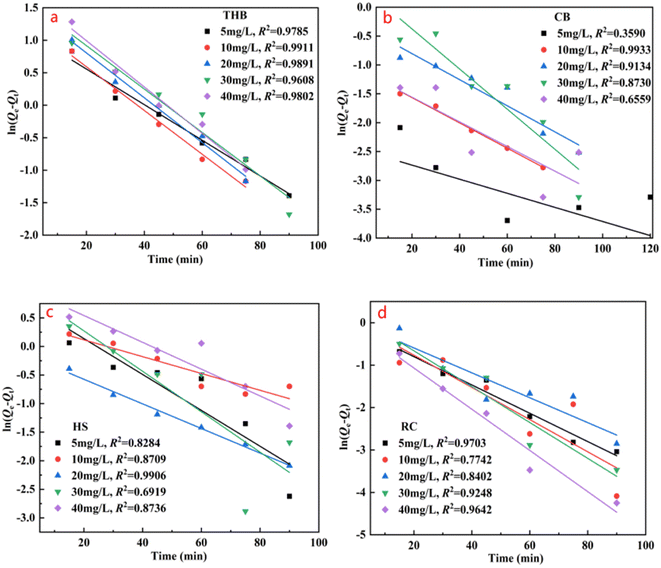
C0, Ce, and Ct are the TC concentrations at the start, equilibrium, and any given time. Vw represents the volume of water in liters, while m (g) denotes the mass of the α-Fe2O3-x catalyst. Fasi's primary and approximate secondary models are frequently utilized in academic literature to depict the link between the contaminant concentration and the duration of contact throughout adsorption. The kinetic fitting model can be expressed in a linear form: (3) and (4)
| |
ln(Qe − Qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − K1t qe − K1t
| (3) |
| |
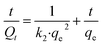 | (4) |
In the equation,
t (min) represents the duration of adsorption, the adsorption capacity of α-Fe
2O
3-
x at the equilibrium time is represented by
qe (mg g
−1), and
k1 and
k2 (min
−1) are the quasi-first order and quasi-second order rate constants, respectively.
21
2.5 Evaluation of photocatalytic performance
The photocatalytic efficiency of α-Fe2O3-x photocatalysts was assessed by degrading TC under 45 W lamp radiation with a 405 nm UV filter in ambient conditions at ambient temperature. The light intensity was stable at 24 μ mol (m−2 s−1), the distance between the illumination source and the reactor was 10 centimeters. A 5.0 mg amount of α-Fe2O3-x photocatalyst was introduced into 100 ml of an aqueous TC solution at a concentration of 20 mg L−1 in one 200 ml Pyrex cylindrical reactor vessel. Afterward, the magnetic suspension was stirred for 0.5 hours in the absence of light to attain equilibrium between adsorption and desorption. The time was then started by adding a certain amount of 30% H2O2 and turning on the UV light source. At set intervals, 3 ml samples were extracted for testing. Since the reaction system was constantly stirred, each sampling was consistent and impartial irrespective of the sampling technique employed. TC concentration in the filtrate was analyzed by tracking shifts in the maximum absorption wavelength (357 nm for TCs) using a Shimadzu UV-Vis spectrometer (UV-27001).
To investigate the impact of light and catalyst and hydrogen peroxide in the reaction, the control experiments without light, catalyst and hydrogen peroxide were set up respectively, and the experimental process was consistent with the above.
In order to obtain a better understanding, a kinetic analysis of tetracycline degradation was performed in the degradation process. According to the literature,5 the degradation of tetracycline basically follows a quasi-primary kinetic model, as depicted in the following eqn (5).
| |
 | (5) |
In the formula,
C0 represents the initial tetracycline concentration,
Ct denotes tetracycline concentration at a given time
t, and
k denotes the reaction rate constant. The concentration ratio at time
t to the initial concentration ratio of
Ct/
C0 was defined as the degradation efficiency. A kinetic fit curve for tetracycline degradation in the corresponding conditions with the reaction time
t as the abscissa and ln(
C0/
Ct) as the ordinate.
2.6 Analytical methods
The concentration of TC was measured using a Shimadzu UV-Vis spectrophotometer (model UV-27001) equipped. The absorption intensity was measured at λ = 357 nm, the peak absorbance wavelength. To investigate ˙OH production by α-Fe2O3-x photocatalysts, we employed DMPO as a spin-trapping agent and TBA as a ˙OH scavenger during the experiment.The detection of ˙OH was done via EPR.
3. Results and discussion
3.1 Characterization of α-Fe2O3
The XRD analysis identifies the crystal phase and pattern of the sample through X-ray diffraction, as shown in Fig. 2(a). Diffraction peaks at the diffraction angles of 2θ = 24.1°, 33.1°, 35.6°, 40.8°, 49.4°, 54.1°, 57.4°, 62.4°, the peaks at 30.1°, 35.6°, 40.8°, 49.5°, 54.0°, 57.2°, 62.8°, 73.7°, and 76.0° correspond to the characteristic crystal surfaces of α-Fe2O3 at the (012), (104), (110), (113), (024), (116), (122), (214), and (300) planes, respectively.21 The diffraction peaks of all samples align precisely with the characteristic peaks on the standard card, and there are no other impurities, thus proving that the samples of the four shapes tested are pure Fe2O3 crystals of a single phase. Moreover, the catalyst peaks overlap in the XRD spectra, which indicates that the main crystal is Fe2O3.
 |
| | Fig. 2 (a) XRD, (b) FT-IR, (c) SEM, (d) UV-Vis DRS (e) and (f) HRTEM, (g)–(l) XPS. | |
The infrared absorption properties of α-Fe2O3 catalysts with different morphologies were analyzed by FT-IR, and the results are shown in Fig. 2(b).22 From the Fig. 2(b), it can see that all four samples of catalysts have obvious characteristic peaks at 3431 cm−1, 1628 cm−1, 570 cm−1, and 477 cm−1, and the absorption bands of the peaks at 3431 cm−1 may be due to the telescopic vibration of ˙OH, while the absorption peaks at 1628 cm−1 may be due to the bending vibration of water molecules, which may originate from the water in the samples. The absorption peaks at 570 cm−1 and 477 cm−1 may be due to the stretching vibration of the Fe–O bond in α-Fe2O3.10 The results of the FT-IR spectra further proved that the component of α-Fe2O3 is in agreement with the results of the XRD spectra in Fig. 2(a).
The synthesized Fe2O3 catalyst was observed, and the specific results are presented in Fig. 2(c). As evident from the electron scanning microscope (SEM) diagram, the shapes of the different morphology catalysts prepared are hexagonal double cone, cube, hexagonal plate, and spherical, respectively, and their distribution is uniform and the same size.23 The catalyst was consistent with previous studies and was successfully prepared. UV-visible absorption spectroscopy was performed on the four prepared Fe2O3 catalysts with different morphologies, and the results are presented in the Fig. 2(d). According to the results, all Fe2O3 samples have similar absorption edges at 450–600 nm, indicating that the catalyst has light absorption at 450–600 nm. According to the ahν = A(hν − Eg)2n formula, the band gap energy (Eg) for CB, THB, HS, and RC is 1.82, 1.90, 2.00, and 2.03 eV, respectively. The smaller the band gap energy, the more favorable the absorption of visible light. The catalyst band gap energy was narrowed, and the reduction of the band gap could be attributed to the formation of oxygen vacancies in the catalyst, which created a mid-layer band gap in the band gap, the results are in agreement with other reports.24–26 The formation of the mid-layer band gap contributes to the absorption of light and facilitates the separation of photogenerated charge carriers, which is favorable to the photocatalytic activity.
In Fig. 2(e) and (f), the TEM results show that there are obvious lattice stripes on the surface of the catalyst. Through Gatan Digital Micrograph software analysis, the lattice spacing of the four different morphology catalysts is 0.176 nm, 0.151 nm, 0.256 nm, and 0.125 nm, respectively, belonging to the {024} crystal surface of Fe2O3, {214}, {110}, and {220} crystal surfaces, respectively.
The characteristic peaks of Fe 2p, O 1s, C 1s and Fe 3p in the full scan spectrum of the α-Fe2O3 samples, as shown in Fig. 2(g) and (h), and the high-power XPS profile of the Fe 2p of the prepared sample, as shown in Fig. 2(i)–(l). The valence of Fe composition and Fe2+ concentration in mixed-valence α-Fe2O3 samples was further analyzed by high-resolution Fe 2p spectroscopy, with characteristic peaks with binding energies of 710.2 eV and 724.6 eV belonging to Fe3+ and 709.3 eV and 723.2 eV belonging to Fe2+.26 In addition to the major Fe2+ and Fe3+ peaks, higher binding energy Shoulders appear near the 713.4 eV, which may be related to the interaction between Fe2+ and Fe3+. The ratio of Fe2+/Fe3+ in the α-Fe2O3 samples was estimated by the XPS peak area, and the content of Fe2+ was determined qualitatively.
The ASAP2460 physical adsorption analyzer uses the static volume method to measure the specific surface and pore size distribution.27–29 The specific surface was measured by the BET method, in which adsorbed gases and inert carrier gases (N2 and He) were allowed to pass through and saturate the solid adsorbent volume material at the temperature (77.3 K) of a liquid nitrogen bath and then desorbed. The surface area of the material was calculated by measuring the volume of the adsorbed and desorbed gases. The data obtained from the test is shown below Table 1.According to the measured data, the BET surface areas THB, CB, HS, and RC are 19.40 m2 g−1, 13.93 m2 g−1, 5.47 m2 g−1, and 3.23 m2 g−1, respectively. Under the UV radiation, the electrons from the valence band are excited to the conduction band to form electron–hole pairs, and the electrons in the conduction band of α-Fe2O3 are transferred to the catalyst surface to reduce O2 to O2−; on the other hand, the holes in the valence band react with H2O to form hydroxyl radicals. The electrons in the conduction band of α-Fe2O3 are transferred to the surface of the catalyst to reduce O2 to O2−; on the other hand, the holes in the valence band react with H2O to form hydroxyl radicals on the surface of the catalyst. These radicals react with TC to oxidize it, thus degrading the target pollutants. These results indicate that α-Fe2O3-x (THB) synthesized with altered catalyst type has a large specific surface area and abundant mesoporous structure, which provides more active sites and facilitates rapid and effective adsorption/desorption on the surface of catalyst reactants and products. Which further proves that the photocatalytic activity of THB catalysts becomes stronger after changing the catalyst morphology.
Table 1 The particle size distribution results of THB, CB, HS, RC
| Catalyst name |
BET surface area |
BJH desorption cumulative volume of pores |
Adsorption average pore diameter (4V/A by BET) |
Rate of degradation efficiency of TC |
| THB |
19.40 m2 g−1 |
0.15 cm3 g−1 |
30.98 nm |
89.75% |
| CB |
13.93 m2 g−1 |
0.073 cm3 g−1 |
21.31 nm |
87.64% |
| HS |
5.47 m2 g−1 |
0.021 cm3 g−1 |
15.19 nm |
86.05% |
| RC |
3.23 m2 g−1 |
0.0085 cm3 g−1 |
10.46 nm |
80.92% |
3.2 Adsorption properties of α-Fe2O3 catalysts with different morphology
We investigated the adsorption properties and reaction kinetics of the catalyst at different initial TC concentrations (5 mg L−1, 10 mg L−1, 20 mg L−1, 30 mg L−1, and 40 mg L−1) under the condition of adding 0.05 g of catalyst content to 100 ml of solution. The results are presented in Fig. 3. The adsorption equilibrium time varied among the four catalysts, but the amount of adsorption remained constant after 30 minutes. This indicates that all four catalysts reached equilibrium within 30 minutes. Observation reveals that after 30 min of the reaction, the amount of adsorption remained almost unchanged with the prolonged time, indicating that all four catalysts reached the adsorption deadsorption equilibrium almost within 30 min. As depicted in Fig. 3(a), the adsorption capacity of THB reached saturation, increasing gradually with the initial concentration of TC ranging from 5 mg L−1 to 40 mg L−1. The maximum saturated adsorption amount of TC by the THB catalyst was 13.17 mg g−1. As shown in Fig. 3(b), the saturated adsorption capacity of CB increases gradually with the initial TC concentration increasing from 5 mg L−1 to 30 mg L−1. However, when the initial TC concentration further increases from 30 to 40 mg L−1, a plateau is observed. The saturated adsorption amount of CB is unchanged.22 The maximum saturated adsorption amount of TC by the CB catalyst was 7.06 mg g−1. As depicted in Fig. 3(c), the saturation adsorption of HS gradually increases as the initial TC concentration rises from 5 mg L−1 to 30 mg L−1, and then the saturation adsorption of HS gradually decreases as the initial TC concentration rises from 30 mg L−1 to 40 mg L−1. The maximum saturated adsorption amount of TC by the HS catalyst was 6.52 mg g−1. As can be seen in Fig. 3(d), the saturated adsorption amount of RC gradually increases as the initial TC concentration rises from 5 mg L−1 to 20 mg L−1, and then the saturated adsorption amount of RC gradually decreases as the initial concentration of TC increases from 20 mg L−1 to 40 mg L−1. The maximum saturated adsorption amount of TC by the RC catalyst was 3.96 mg g−1.
 |
| | Fig. 3 (a) Hexagonal double cone; (b) cube; (c) hexagonal plate; (d) spherical. | |
3.2.1. Adsorption kinetic studies. For a clearer understanding of the adsorption–desorption process of the catalyst, by mapping the kinetic curve fit to the data in 3.2.1, the quasi-primary kinetic curves and the quasi-secondary kinetic curves are shown in Fig. 4 and 5, respectively. The above data were fitted with eqn (2)–(4) to calculate qe, k1, and k2, the adsorption amount (qe) is 5.51–3.02 mg g−1 (Qe) is 6.47–13.17 mg g−1, the correlation coefficient (R2) is 0.9785–0.9911 in the experiment, and R2 is up to 0.9981–0.9997, the adsorption process is primarily characterized by chemisorption, consistent with its mechanism. Similarly, it is concluded that the TC adsorption of CB, HS, and RC α-Fe2O3 catalysts all conforms to the quasi-secondary dynamics, and the adsorption process is mainly chemical adsorption.
 |
| | Fig. 4 Quasi-primary kinetic curve of adsorption of TC by different topography α-Fe2O3 catalysts. | |
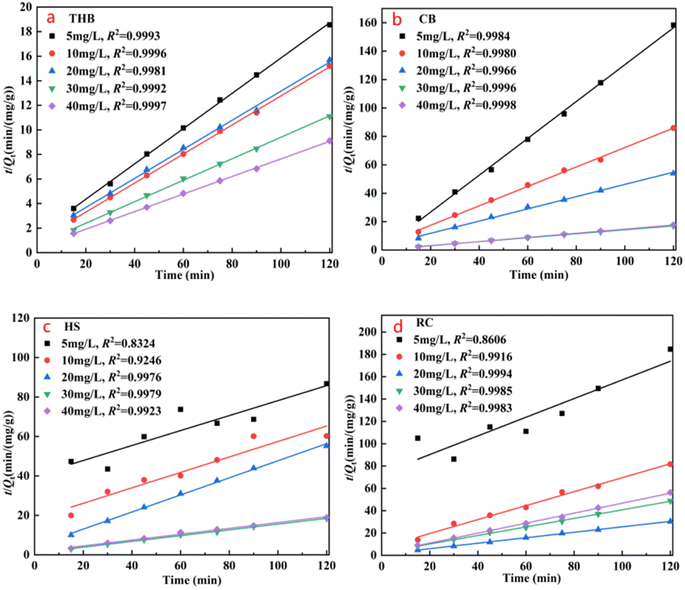 |
| | Fig. 5 Adsorption of TC by nano-α-Fe2O3 catalysts in different morphologies is quasi-secondary kinetic curves. | |
3.3 Catalytic activity of α-Fe2O3
To evaluate the efficiency of the α-Fe2O3 catalyst in photo-Fenton reactions and determine any potential enhancements to its catalytic properties, control experiments without light, catalyst, and hydrogen peroxide were set up, respectively. The α-Fe2O3-x catalyst was added to 0.5 g L−1, tetracycline solution concentration at 20 milligrams per liter, 100 ml, LED UV visible light (wavelength: 405 nm, light intensity: 24 μmol (m−2 s−1)), H2O2 concentration of 48.95 mmol L−1, and the reaction time was 30 min.23 The curves and kinetic changes of the tetracycline degradation effect (Ct/C0) over the course of time are shown in eqn (6). It can be observed from the Fig. 6 that with the increase in reaction time, TC solution will undergo partial degradation without catalyst or H2O2, or light, but the degradation rate is low, and the degradation rate is relatively slow. In the presence of LED visible light irradiation and H2O2, α-Fe2O3/H2O2/LED light degradation TC is significantly improved, TC is rapidly degraded, and the degradation rate is rapidly accelerated. As can be seen in Fig. 6(a), upon addition of catalyst THB, the TC degradation rate and efficiency were 51.75% and 0.0226 min−1, respectively. Without H2O2, in addition, the rate of degradation efficiency of TC was 64.63% and 0.0364 min−1, respectively. When there is no light exposure, the degradation rate and degradation efficiency of TC were 22.53% and 0.0113 min−1, respectively. The rates and degradation rates of TC in THB/H2O2/LED light reactions reached 89.75% and 0.0969 min−1, respectively.
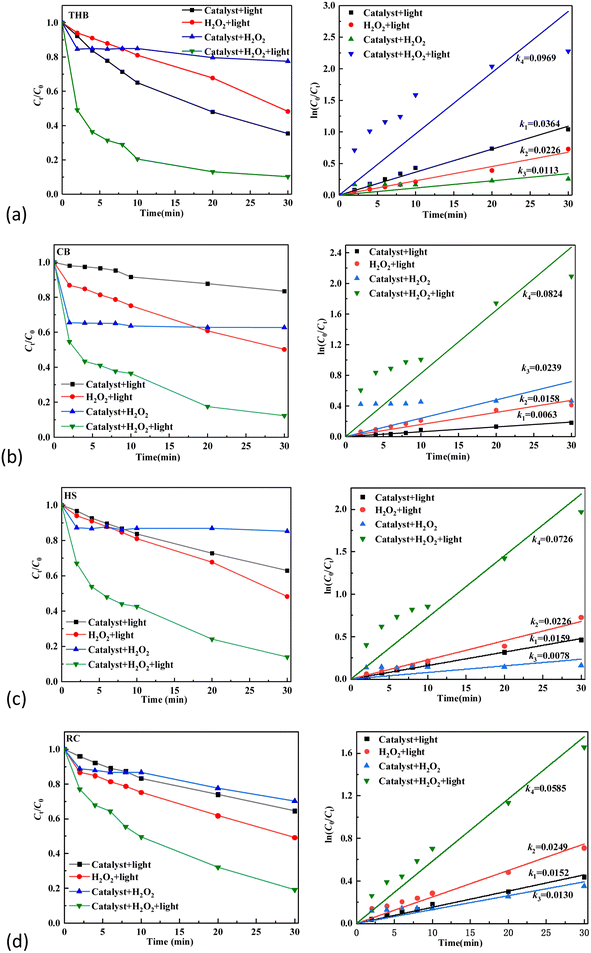 |
| | Fig. 6 Photo-Fenton properties and dynamics of nano-α-Fe2O3 catalysts with different morphology. | |
As illustrated in Fig. 6(b), in the absence of catalyst CB addition, the degradation rate and efficiency of degradation of the TC were 33.80% and 0.0158 min−1, respectively. Without H2O2 in addition, the degradation rate and degradation efficiency of TC were 16.51% and 0.0063 min−1, respectively; when there is no light exposure, the degradation rate and degradation efficiency of TC were 37.29% and 0.0239 min−1, respectively; the TC degradation rate and degradation rate of TC in CB/H2O2/LED light reactions were 87.64% and 0.0824 min−1. As illustrated in Fig. 6(c), the degradation rate of TC is 51.75% and 0.0226 min−1, the degradation rate of TC without H2O2. At 37.03% and 0.0159 min−1, without illumination, the TC degradation rate was 14.79% and 0.0078 min−1, respectively. The TC degradation rate reached 86.05% and 0.0726 min−1 in the HS/H2O2/LED light reaction. As illustrated in Fig. 6(d), upon the addition of catalyst RC, the degradation rate and degradation efficiency of TC were 50.81% and 0.0249 min−1, respectively; without H2O2 in addition, the degradation rate and degradation efficiency of TC were 35.43% and 0.0152 min−1, respectively; when there is no light exposure, the degradation rate and degradation efficiency of TC were 29.70% and 0.0130 min−1, with RC/H2O2/LED light, the TC degradation rate and efficiency reached 80.92% and 0.0585 min−1, respectively.
According to the comparative results in Fig. 7, among the four catalysts of THB, CB, HS, and RC, the THB catalyst had the best effect in the TC degradation, perhaps with more hexagonal double-cone contact surfaces and better catalytic effect. In the α-Fe2O3/H2O2/LED light photo-Fenton system, the UV Fenton reaction improves the conversion rate of Fe2+ and Fe3+, at which point the generation rate of hydroxyl radicals is increased. Meanwhile, H2O2 under UV irradiation generates more hydroxyl radicals. Meanwhile, photogenerated electrons can be directly captured by O2/H2O2, forming ˙O2/˙OH, thus improving the degradation rate of TC. The main reaction equations are as follows (6)–(12):
| | |
α-Fe2O3 + hυ → α-Fe2O3(h+VB + e−CB)
| (6) |
| | |
≡Fe(III) + hυ → ≡Fe(II)
| (7) |
| | |
≡Fe(II) + H2O2 → ≡Fe(III) + OH− + ˙OH
| (8) |
| | |
e− + H2O2 → OH− + ˙OH
| (10) |
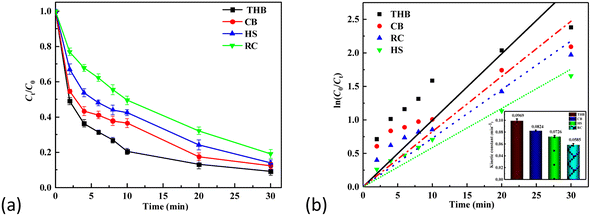 |
| | Fig. 7 (a) and (b), Comparative study of the properties and kinetics of optical Fenton of the four α-Fe2O3 catalysts. | |
3.4 The effect of the pH values on TC degradation
The medium's pH controls the surface electrical properties of the catalyst and the acidity and alkalinity of the reaction medium. To clarify the relationship between pH and its impact on TC degradation, we conducted comparative experimental analyses. As shown in Fig. 8, the pH range reported in our study was 3–11.30,31 It is evident that the degradation of TC in catalyst THB (Fig. 8(a)) significantly with increased alkalinity in the solution. Howere, the degradation rate deteriorated notably beyond pH = 9. In catalyst CB (Fig. 8(b)), the degradation of TC significantly improved with increasing alkalinity in the solution. Howere, the degradation rate deteriorated significantly with increasing pH after pH5 until pH11, where the degradation rates of TC were almost the same between pH5 and pH11. In catalyst HS (Fig. 8(c)), the degradation of TC improved significantly with the increase of alkalinity in the solution, and the best effect was observed at pH11 for 20 min, and pH9 for 30 min. In Catalyst RC (Fig. 8(d)), the degradation of TC significantly improved with the increase of alkalinity in the solution.The most effective results were obtained at pH11. The four catalysts have different factors affecting acidity and alkalinity. It can be assumed that alkaline conditions are more favorable for the hydrolysis of Fe(III), while a low concentration of Fe(III) in solution may hinder the process of the Fenton reaction. Furthermore, hydrogen peroxide is more likely to spontaneously generate hydroxyl radicals under acidic conditions. Both of these factors are more favorable for the degradation of TC, resulting from differences in the main active substances of the four catalysts.
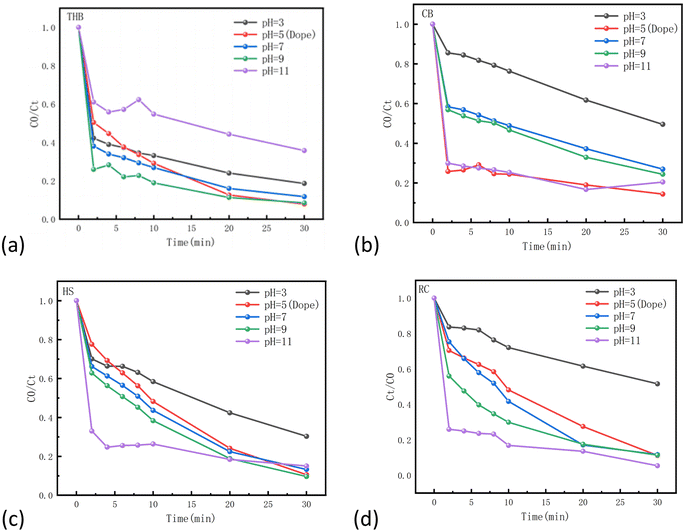 |
| | Fig. 8 Effect of pH on the activity of four different catalysts(THB, CB, HS, RC). | |
3.5 Mechanism of α-Fe2O3/H2O2/visible light system
The experimental steps of radical capture in photocatalytic degradation of pollutants are consistent with photocatalytic degradation. While turning on the lamp source, quickly add 1.36 mol per L IPA (hydroxyl radical trap), 0.05 mol per L ascorbic acid (superoxide radical trap), 0.025 mol per L EDTA-2Na (hole h+ trap) and 2.5 mmol per L sodium azide (single oxygen trap): the trap can quickly react with the reactive radicals produced in the system, forming a competition with the pollutants in the reaction solution, competing for the reactive radicals, thus inhibiting the effective removal of pollutants32–34 (the stability constant of Fe3+ and Fe2+ lgk(steady) are respectively 25.10 and 14.33). Capture experimental phenomena can indirectly prove the active factors in the degradation system of α-Fe2O3/H2O2/visible light system.
As shown in the Fig. 9, when 2.5 mmol per L sodium azide (monotherapy oxygen trapping agent) and 1.36 mol per L isopropanol (hydroxyl radical trapping agent) were added, the inhibitory effect of TC degradation rate was very weak, but 0.025 mol per L EDTA-2Na (hole h + trapping agent) and 0.05 mol per L ascorbic acid (superoxide radical trapping agent) were greatly inhibited, and the removal efficiency of TC was significantly reduced from 89.75% to 24.32% and 9.91% for THB, respectively. Clearing of h+ by the addition of EDTA-2Na significantly reduced the reactivity of THB against TC photodegradation. These results indicate that the predominant active substances in the catalyst HS, THB, RC are OH, h+, and O2−, respectively, while 1O2 makes only a small contribution to the photocatalytic degradation of the TC. Only in the case of the CB catalyst, OH, h+, O2−, and 1O2 all play a role.
 |
| | Fig. 9 TC photocatalytic degradation on the α-Fe2O3-x. In the presence or absence of radical scavengers. | |
The EPR spectra in Fig. 10 showing hydroxyl radicals further confirm that the predominant active species in THB are h+, O2−, OH, and 1O2; in CB are h+, O2−, OH, and 1O2; in RC are h+, O2−, OH, and 1O2; and in HS are h+, O2−, 1O2, and OH. However, it is noteworthy that hydroxyl radicals (OH) do not play a significant role in the photocatalytic degradation of tetracyclines.
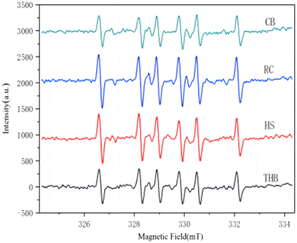 |
| | Fig. 10 EPR plot of hydroxyl radical trapping experiments under light assays. | |
4. Conclusions
In summary, four different morphologies (hexagonal biconical, cubic, hexagonal plate and spherical) were successfully synthesized by varying the reaction conditions of the precursors for the synthesis of α-Fe2O3-x. This combination of morphology engineering and defect modulation of α-Fe2O3-x showed outstanding activity and stability for the degradation of TC in photocatalytic Fenton oxidation systems. Experimental and theoretical results suggest that its unique morphology and mixed valence of Fe2+/Fe3+ are the key factors influencing its excellence. These findings will extend the scope of Fe2o3-based Fenton catalysts through surface engineering and consolidate the fundamental theory of Fenton systems for a wide range of environmental applications.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The study was funded by the Chongqing Science and Technology Innovation and Application Development Special Key Project (CSTB2022TIAD-KPX0128), the the Early Talent Research Program of Chongqing Education Commission, China (CY230920), and Research Program from Chongqing Water Group Co., Ltd (2023Q295, 2022Q247).
References
- J. de Freitas, A. P. G. Ferreira and É. T. G. Cavalheiro, J. Therm. Anal. Calorim., 2023, 1–13 Search PubMed.
- H. Huang, T. Guo, K. Wang, Y. Li and G. Zhang, Sci. Total Environ., 2021, 758, 143957 CrossRef CAS PubMed.
- S. Jia, Y. Wu, H. Zhou, H. Yan, Y. Liao and H. Mao, J. Cleaner Prod., 2023, 426, 139188 CrossRef CAS.
- X. Liu, H. Wang, C. Wang, L. Zhao, H. Pan, Y. Liu, L. Liang, C. Zhao and S. Huang, Bioresour. Technol., 2024, 395, 130357 CrossRef CAS PubMed.
- W. Zhao, C. Liang, B. Wang and S. Xing, ACS Appl. Mater. Interfaces, 2017, 9, 41927–41936 CrossRef CAS PubMed.
- S. A. Bradford, C. Shen, H. Kim, R. J. Letcher, J. Rinklebe, Y. S. Ok and L. Ma, Crit. Rev. Environ. Sci. Technol., 2022, 52, 3753–3762 CrossRef.
- M. Mishra and D.-M. Chun, Appl. Catal., A, 2015, 498, 126–141 CrossRef CAS.
- M. Farahmandjou and F. Soflaee, Phys. Chem. Res., 2015, 3, 191–196 CAS.
- S. Yang, Y. Xu, Y. Sun, G. Zhang and D. Gao, CrystEngComm, 2012, 14, 7915–7921 RSC.
- R. Vinayagam, S. Pai, T. Varadavenkatesan, M. K. Narasimhan, S. Narayanasamy and R. Selvaraj, Surf. Interfaces, 2020, 20, 100618 CrossRef CAS.
- Y. Xu, G. Zhang, G. Du, Y. Sun and D. Gao, Mater. Lett., 2013, 92, 321–324 CrossRef CAS.
- H. Zhou and S. S. Wong, ACS Nano, 2008, 2, 944–958 CrossRef CAS PubMed.
- S. Bharathi, D. Nataraj, K. M. Senthil and Y. Masuda, J. Nanopart. Res., 2013, 15, 1346 CrossRef.
- Y. Zhang, C. Wang, L. Yu, G. Liu, A. Liang, J. Huang, S. Nie, X. Sun, Y. Zhang and B. Shen, Nat. Commun., 2017, 8, 15512 CrossRef CAS PubMed.
- J. He, W. Ma, J. He, J. Zhao and J. C. Yu, Appl. Catal., B, 2002, 39, 119–128 CrossRef.
- L. Gu, Q. Su, W. Jiang, Y. Yao, Y. Pang, W. Ji and C.-T. Au, Catal. Sci. Technol., 2018, 8, 5782–5793 RSC.
- M. Zheng, C. Xing, W. Zhang, Z. Cheng, X. Liu and S. Zhang, Inorg. Chem. Commun., 2020, 119, 108040 CrossRef CAS.
- C. Yin, X. Li, Y. Dai, Z. Chen, D. Yang, R. Liu, W. Zou, C. Tang and L. Dong, Green Chem., 2021, 23, 328–332 RSC.
- J. Zeng, J. Zhong, J. Li and S. Huang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 476–481 CrossRef CAS.
- H. Yin, Y. Zhao, Q. Hua, J. Zhang, Y. Zhang, X. Xu, Y. Long, J. Tang and F. Wang, Front. Chem., 2019, 7, 58 CrossRef CAS PubMed.
- Y. Guo, Q. Liu, X. Tang, H. Liang, G. Li, L. Yang, L. Wang, X. Li and Y. Sun, Chemosphere, 2023, 341, 140114 CrossRef CAS PubMed.
- W. Xu, W. Xue, H. Huang, J. Wang, C. Zhong and D. Mei, Appl. Catal., B, 2021, 291, 120129 CrossRef CAS.
- P. Kumar, N. Thakur, K. Kumar and K. Jeet, Mater. Today: Proc., 2023 DOI:10.1016/j.matpr.2023.01.257.
- Y. Sun, H. Wang, Q. Xing, W. Cui, J. Li, S. Wu and L. Sun, Chin. J. Catal., 2019, 40, 647–655 CrossRef CAS.
- Z. Wang, X. Mao, P. Chen, M. Xiao, S. A. Monny, S. Wang, M. Konarova, A. Du and L. Wang, Angew. Chem., 2019, 131, 1042–1046 CrossRef.
- L. Chen, F. Li, B. Ni, J. Xu, Z. Fu and Y. Lu, RSC Adv., 2012, 26, 10057–10063 RSC.
- Y. Tian and J. Wu, AIChE J., 2018, 64, 286–293 CrossRef CAS.
- N. Davari, M. Farhadian, A. R. S. Nazar and M. Homayoonfal, J. Environ. Chem. Eng., 2017, 5, 5707–5720 CrossRef CAS.
- R. Zouari Ahmed, S. E. Laouini, C. Salmi, A. Bouafia, S. Meneceur, H. A. Mohammed, S. Chihi, F. Alharthi and J. A. A. Abdullah, Biomass Convers. Biorefin., 2023, 1–15 Search PubMed.
- Y. Hu, Y. Li, J. He, T. Liu, K. Zhang, X. Huang, L. Kong and J. Liu, J. Environ. Manage., 2018, 226, 256–263 CrossRef CAS PubMed.
- Y. Chen, L. Liu, L. Zhang, S. Li, X. Zhang, W. Yu, F. Wang, W. Xue, H. Wang and Z. Bian, Appl. Catal., B, 2023, 333, 122775 CrossRef CAS.
- R. W. Field and G. K. Pearce, Adv. Colloid Interface Sci., 2011, 164, 38–44 CrossRef CAS PubMed.
- F. Liu, H. Sun, S. Tao, W. Wei, T. Sun and Peizhe, Chem. Eng. J., 2020, 402, 125477 CrossRef CAS.
- B. Karnik, S. Davies, M. Baumann and S. Masten, Environ. Sci. Technol., 2005, 39, 7656–7661 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Facheng Qiu*a,
Congming Tanga,
Cheng Yang*b,
Zejun Weib and
Anke Dub
*a,
Facheng Qiu*a,
Congming Tanga,
Cheng Yang*b,
Zejun Weib and
Anke Dub













