
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurcumin–polydopamine nanoparticles alleviate ferroptosis by iron chelation and inhibition of oxidative stress damage†
Li Lei ab,
Jiali Yuana,
Qingqing Yangc,
Qiuxia Tua,
Haijun Yud,
Liangzhao Chu*b,
Lei Tang*c and
Chunlin Zhang*a
ab,
Jiali Yuana,
Qingqing Yangc,
Qiuxia Tua,
Haijun Yud,
Liangzhao Chu*b,
Lei Tang*c and
Chunlin Zhang*a
aEngineering Research Center for Molecular Medicine, School of Basic Medical Science, Guizhou Medical University, Guiyang 550025, China
bDepartment of Neurosurgery, The Affiliated Hospital of Guizhou Medical University, Guiyang, Guizhou, China
cGuizhou Provincial Engineering Technology Research Center for Chemical Drug R&D, College of Pharmacy, Guizhou Medical University, Guiyang, 550025, China
dCenter of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
First published on 7th May 2024
Abstract
Ferroptosis, characterized by elevated iron levels and lipid peroxidation (LPO), is a recently identified regulatory mechanism of cell death. Its substantial involvement in ischemic tissue injury, neurodegenerative disorders, and cancer positions ferroptosis inhibition as a promising strategy for managing these diverse diseases. In this study, we introduce curcumin–polydopamine nanoparticles (Cur–PDA NPs) as an innovative ferroptosis inhibitor. Cur–PDA NPs demonstrate remarkable efficacy in chelating both Fe2+ and Fe3+ in vitro along with scavenging free radicals. Cur–PDA NPs were found to efficiently mitigate reactive oxygen species, reduce Fe2+ accumulation, suppress LPO, and rejuvenate mitochondrial function in PC12 cells. Thus, these NPs can act as potent therapeutic agents against ferroptosis, primarily via iron chelation and reduction of oxidative stress.
1. Introduction
Ferroptosis is an iron-dependent type of regulated cell death, distinct from autophagy, necrosis, or apoptosis.1,2 It arises from the excessive accumulation of lethal lipid peroxides driven by iron.3 The Fenton-like reaction, involving iron and lipid peroxides, produces lipid alkoxy and hydroxyl radicals, resulting in elevated levels of reactive oxygen species (ROS).4 This initiates a radical chain reaction, amplifying lipid peroxidation (LPO), targeting biomolecules, and culminating in oxidative stress (OS) and mitochondrial dysfunction.5 Recent comprehensive studies have reported the correlation between ferroptosis and diverse pathological processes, including ischemic tissue injury,6 infectious diseases,7 and neurodegenerative disorders such as Alzheimer's, Huntington's, and Parkinson's diseases, and cancer.8–10 Consequently, pharmacological manipulation of ferroptosis, through induction or inhibition, holds considerable therapeutic promise.11Ferroptosis inhibitors, primarily radical-trapping antioxidants and iron chelators, aim to mitigate OS and safeguard cells.12,13 However, there are significant hurdles in the initial generation of these compounds in clinical advancement due to off-target effects, limited selectivity, and potential toxicity.14 Iron chelators, such as deferoxamine, are hindered by poor absorption and rapid clearance.15–17 Furthermore, certain natural products face challenges of water solubility and chemical stability, impeding their ability to traverse the blood–brain barrier.18,19 To overcome these issues, the distinct properties of various nanomaterials, notably their exceptional biocompatibility and targeting capabilities, have been extensively researched in treating diseases related to ferroptosis inhibition.20,21 Nanoscale structures enhance drug efficacy by increasing drug half-life, improving hydrophobic compound solubility, minimizing potential immunogenicity, and enabling controlled, stimulus-responsive drug release.22 These nanomaterials also precisely control the rate and location of drug release.23,24
Curcumin (Cur), a polyphenolic compound prevalent in turmeric rhizomes, is known for its neuroprotective capabilities, including anti-amyloidogenic effects preventing amyloid protein formation and aggregation, alongside anti-inflammatory, antioxidant, and immunomodulatory properties.25–27 Studies have revealed that the dicarbonyl structure of Cur enables it to bind with Fe2+ and particularly Fe3+, positioning Cur as a potential iron chelator, offering promise in inhibiting ferroptosis.28 The biomedical utility of Cur is constrained by several factors: poor water solubility, a brief half-life, low biological availability, and instability at neutral physiological pH. Therefore, devising techniques to circumvent these limitations represents a critical challenge in contemporary medicine. Polydopamine (PDA) has emerged as a notable solution as it is known for its remarkable biocompatibility, water dispersibility, and stimulus-responsive release properties.29,30 Studies indicate that PDA nanoparticles (NPs), with their reducing functional groups such as catechol and imine, adeptly scavenge various types of ROS.31 Moreover, recent findings have demonstrated the capacity of PDA NPs to chelate and absorb iron ions, contributing to ferroptosis inhibition and mitigating intervertebral disc degeneration.32
In this study, we synthesized Cur–PDA NPs by self-assembling Cur with PDA via encapsulation (Scheme 1a). Cur–PDA NPs successfully resolve the solubility and stability issues inherent to Cur. Using the combined iron chelation and antioxidant properties of Cur and PDA, Cur–PDA NPs efficiently counter OS and impede ferroptosis progression (Scheme 1b). These findings underscore the potential of Cur–PDA NPs in treating ferroptosis-related diseases.
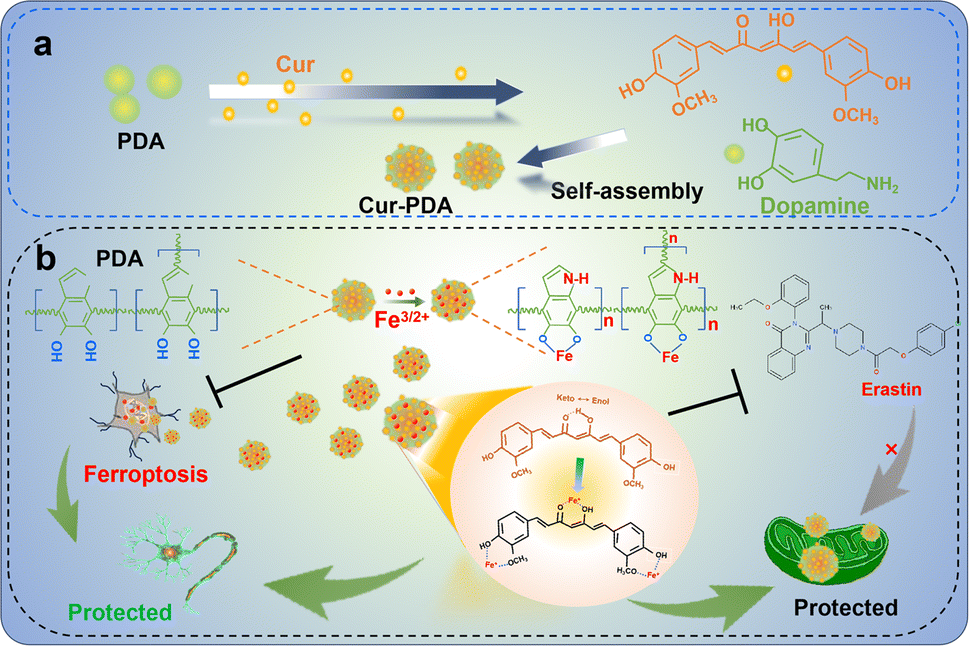 | ||
| Scheme 1 The principle underlying the chelation of iron by curcumin and polydopamine to inhibit oxidative stress, thereby protecting neurons and repairing mitochondrial function. | ||
2. Experimental
2.1 Materials
Curcumin (Cur) and dopamine hydrochloride (DA) all purchased from Aladdin. Maleimide-PEG2k-Fmoc (Fmoc-PEG-MAL) was purchased from Xi'an Ruixi Biological Technology (Xi'an, China). DMEM high glucose medium, trypsin, streptomycin, and glutamine were obtained from Hyclone. Fetal bovine serum (FBS) was supplied by Tianhang Biotech. ABTS reagent 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt was sourced from Shanghai McLean Biochemical Technology Co., Ltd. Propidium iodide/acridine orange staining kit was provided by Biyuntian in China, Shanghai. MTT assay kit, MDA content detection kit, SOD activity assay kit, Prussian blue staining kit, and Fe2+ ion content detection kit were all procured from Solarbio Life Sciences. ROS detection assay kit, BCA protein concentration determination assay kit, and LPO assay kit were acquired from Biyuntian Biotech.2.2 Preparation of Cur–PDA NPs
According to the method described in reference,33 2 mL of NH4OH, 40 mL of ethanol, and 90 mL of deionized water were mixed and added to a round-bottom flask. The mixture was stirred at room temperature for 30 min. Then, 0.5 g of DA was dissolved in 10 mL of deionized water and added to the above mixture (pH = 8.5). Next, 0.098 g of Cur (0.3 mM) and Fmoc-PEG-MAL (20 mg) were dissolved in 2 mL of DMSO and added to the above mixture as well. The reaction was stirred under light-shielded conditions for 24 h. The precipitate was collected by centrifugation at a speed of 4000 rpm, washed with a mixture of DMSO/water (volume ratio: 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and dried for further use. The loading efficiency (LE) and encapsulation efficiency (EE) of Cur was determined using UV-vis spectroscopy according to the following formula: LE (%) = (weight of loaded Cur)/(total weight of Cur–PDA NPs) × 100%. EE (%) = (weight of loaded Cur)/(total dosage weight of Cur) × 100%.
:1), and dried for further use. The loading efficiency (LE) and encapsulation efficiency (EE) of Cur was determined using UV-vis spectroscopy according to the following formula: LE (%) = (weight of loaded Cur)/(total weight of Cur–PDA NPs) × 100%. EE (%) = (weight of loaded Cur)/(total dosage weight of Cur) × 100%.
2.3 Characterization of Cur–PDA NPs
We employed transmission electron microscopy (TEM) and scanning electron microscopy (SEM) to identify Cur–PDA NPs particle size and morphology (JEM-2100F, Japan; SU8020, Hitachi, Japan). The hydrodynamic size was quantified utilizing dynamic light scattering (Zeta Plus, Brookhaven, USA). An ultraviolet-visible (UV-vis) near infrared spectrophotometer (UV-3600, Shimadzu, Japan) was used to obtain UV-vis absorption spectra.2.4 Measurement of Cur–PDA NPs ROS scavenging ability
We assessed the antioxidant capacity of Cur–PDA NPs via the 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) assay, following the protocol of the total antioxidant capacity assay kit (Beyotime Technology). Cur–PDA NPs at 25 μg mL−1, 50 μg mL−1, 100 μg mL−1, 200 μg mL−1, and 500 μg mL−1 were introduced into the ABTS solution. After incubation for 10 min at room temperature in the dark, the mixture was passed through a 0.22 μm micro-pore filter. We measured the optical density (OD) at 415 nm utilizing UV-vis spectroscopy (n = 3 per group). Additionally, we determined the ability of Cur–PDA NPs to consume H2O2 using a titanium sulfate colorimetric assay kit (Solarbio Life Sciences). H2O2 (1 mM) was added to 0 μg mL−1, 50 μg mL−1, 100 μg mL−1, and 500 μg mL−1 of Cur–PDA NPs at room temperature for 1 h. Post-incubation, we passed the solution through a 0.22 μm micro-pore membrane. Residual H2O2 was then quantified via measuring the peroxo-titanium complex at OD410 nm (n = 3). Additionally, UV-visible spectroscopy was employed to track the degradation of Cur–PDA NPs in H2O2 solutions of 5 mM and 10 mM. To put it simply, Cur–PDA NPs (100 μg mL−1) were mixed with H2O2 (5 mM and 10 mM). This mixture was stored at room temperature for 24 h in the dark. We subsequently recorded the UV-vis spectra of the solution.2.5 Determination of Cur–PDA NPs iron chelating ability
Iron chelation by Cur–PDA NPs was determined with a kit for iron ion content determination from Solarbio Life Sciences. FeSO4 (100 μM) was added to 25 μg mL−1, 50 μg mL−1, 100 μg mL−1, 200 μg mL−1, and 500 μg mL−1 of Cur–PDA NPs for 1 h at room temperature. Post-incubation, the mixture was filtered via a 0.22 μm micro-pore filter. Fe2+ concentration in the filtrate was then quantified by measuring the OD600 nm of the blue Fe2+-tripyridyltriazine complex (n = 3). The capacity of Cur–PDA NPs to chelate Fe2+ was quantified using the FeSO4 standard curve. Subsequently, to assess the Fe3+ chelation ability, and Fe3+ solution (125 μM) was added to 25 μg mL−1, 50 μg mL−1, 100 μg mL−1, 200 μg mL−1, and 500 μg mL−1 of Cur–PDA NPs for 4 h at room temperature. After incubation, the mixture was filtered via a 0.22 μm micro-pore filter. The filtrate quantity of Fe3+ was determined by measuring the OD520 nm of the 2,2′-bipyridine–Fe3+ complex (n = 3 per group).2.6 Establishment of cell models and exploration of drug action concentrations
The MTT assay was performed to evaluate cell viability. PC12 cells were introduced into a 96-well plate at 1 × 104 cells per well. After 24 h of adhesion, various Cur–PDA NPs concentrations were added for 24 h (cytotoxicity experiments). Following this experiment, cells were further incubated in a high-glucose medium containing MTT (5 mg mL−1) for 4 h. We subsequently removed the supernatant, and 150 μL of DMSO was introduced in each well. The mixture was thoroughly in reacted for 10 min at 180 rpm, 25 °C on a shaker. The OD490 nm was measured with a microplate reader (Infinite M200 Pro multi-mode reader; Tecan Life Sciences, Männedorf, Switzerland). To establish a ferroptosis cell model, PC12 cells were subjected to ferroptosis induction using erastin (5 μM). Following the outcomes of cytotoxicity experiments with Cur–PDA NPs, we selected suitable concentration ranges of Cur–PDA NPs to intervene in this model. This approach was employed to evaluate the Cur–PDA NPs protective effects against this model.2.7 Viability staining for living/dead cells
PC12 (1 × 104 cells) were added in confocal dishes. Post 24 h co-incubation with Cur (20 μg mL−1) and Cur–PDA NPs (40 μg mL−1), the supernatant was replaced with a medium containing calcein/propidum iodide (PI) working buffer, following the kit instructions. The cells were subsequently incubated at 37 °C for 30 min, and then washed with phosphate-buffered saline (PBS). The red/green fluorescence intensity (PI/calcein) indicating live/dead cells in different groups was analyzed using an inverted fluorescence microscope (n = 3). We quantified the integrated optical density (IOD)/4′,6-diamidino-2-phenylindole (DAPI) ratio with Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).2.8 Determination of intracellular glutathione (GSH), malondialdehyde (MDA), and superoxide dismutase (SOD)
In a 37 °C, 5% CO2 incubator, PC12 cells were pretreated with or without Cur (20 μg mL−1) or Cur–PDA NPs (40 μg mL−1) for 12 h, followed by a 24 h erastin (5 μM) stimulation. Cells from each group were then harvested via centrifugation, and total cellular protein was extracted using cell lysis buffer. GSH, MDA, and SOD levels were quantified using their respective cell count normalization kit (n = 3 for each group).2.9 Detection of intracellular Fe2+, ROS, and lipid peroxides
PC12 cells were seeded at a concentration of 1 × 104 cells in confocal dishes and pre-protected for 12 h using complete medium, with or without Cur (20 μg mL−1) or Cur–PDA NPs (40 μg mL−1). This was followed by a 24 h stimulation with 5 μM erastin in complete medium. The cells were then co-incubated at 37 °C for 30 min with DCFH-DA (10 μM), Liperfluo probes (10 μM), and FerroOrange (1 μM). After triple washing with PBS, intracellular changes in Fe2+ (red fluorescence), ROS (green fluorescence), and lipid peroxides (green fluorescence) were quantified with a fluorescence microscope (Leica DM4000 B; Leica Microsystems).2.10 Iron level detection in C. elegans
Detection of iron levels in C. elegans was conducted using the Iron level detection method. The ability of Cur–PDA NPs to bind with iron was investigated by utilizing the erastin-treated transgenic DA2123 strain nematode model. The nematodes were categorized into four groups: control group, erastin-treated, and groups treated with either free Cur (20 μg mL−1) or Cur–PDA NPs (40 μg mL−1) for a duration of 24 h. Subsequently, all nematodes were exposed to FerroOrange (1 μM) for 1 h and fluorescence microscopy was employed to observe the Fe2+ levels within the nematodes.2.11 Mitochondrial activity evaluation in C. elegans
PD4251 strain nematode model of ferroptosis induced by erastin. Mitochondrial were fluorescently labeled with GFP. The nematodes were categorized into four groups: control, erastin-treated, and groups treated with either free Cur (20 μg mL−1) or Cur–PDA NPs (40 μg mL−1) for 24 h. “The fluorescence microscope was used to assess the mitochondrial levels in the nematodes”.3. Results and discussion
3.1 Characterization of Cur–PDA NPs
Cur–PDA NPs were generated using a previously established method involving the oxidative polymerization of Cur doped with dopamine and maleimide-polyethylene glycol under saturated oxygen conditions (Fig. 1(a)).33 The process took place in a solvent mixture of NH4OH, ethanol, water, and DMSO, with the pH adjusted to 8.5. SEM and TEM demonstrated that Cur–PDA NPs exhibit a uniform spherical morphology with excellent dispersibility (Fig. 1(b) and (c)).34–36 The element distribution in Cur–PDA NPs was analyzed by transmission scanning electron microscope (SEM) images and elemental mapping (Fig. 1(d)). The average hydrodynamic particle size of these NPs is approximately 342 nm, characterized by a narrow size distribution, further affirming the superior water dispersibility of Cur–PDA NPs (Fig. 1(e)). The Fourier-transform infrared spectrum in Fig. 1(f) exhibited a wide absorption attributed to PDA N–H/O–H stretching vibrations (3135–3650 cm−1), with characteristic peaks at 1510 cm−1 and 963 cm−1 corresponding to the –CO and –CH stretching of Cur.37,38 The optical absorption spectrum displayed broadly absorptive properties and a Cur peak at 440 nm (Fig. 1(g)). Measurement of zeta potential indicated that Cur–PDA NPs surface charge was −24.4 ± 2 mV (Fig. S1†). The presented data confirm the successful synthesis of Cur–PDA NPs. We used the calibration curve (Fig. S2†) and evaluated the encapsulation efficiency and loading capacity of Cur within Cur–PDA NPs to be 88.37% ± 0.82% and 58.11% ± 0.65%, respectively.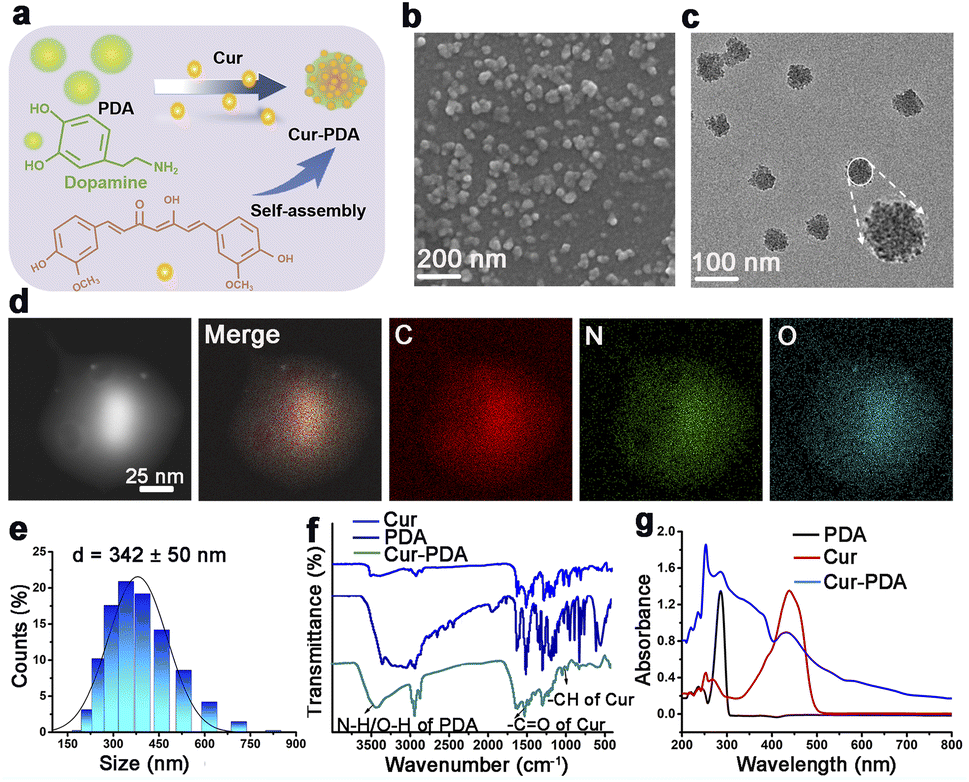 | ||
| Fig. 1 Characterization of Cur–PDA NPs. (a) Schematic of Cur–PDA NPs preparation. (b and c) SEM (b) and TEM (c) images of Cur–PDA NPs. (d) STEM images and elemental mapping of C, N, and O for Cur–PDA NPs at the region. (e) Hydrodynamic particle diameter (average) of Cur–PDA NPs. (f) Fourier-transform infrared spectra of Cur, PDA, and Cur–PDA NPs. (g) Cur–PDA NPs ultraviolet-visible absorption spectrum. | ||
3.2 Iron chelating ability of Cur–PDA NPs
Ferroptosis, characterized by iron-dependent LPO, is influenced by various cellular metabolic pathways encompassing redox homeostasis, iron metabolism, mitochondrial function, and the metabolism of amino acids, lipids, and carbohydrates.39 Given the pivotal role of iron in ferroptosis, we studied the iron chelation capabilities of Cur–PDA NPs, particularly toward Fe2+ and Fe3+. Fe3+ forms a pink complex with 2,2′-bipyridine, known as the 2,2′-bipyridine-Fe3+ complex. To determine the chelation ability of Fe3+, we measured the levels of this complex after the addition of Cur–PDA NPs. Following Cur–PDA NPs addition, the OD520 nm of the 2,2′-bipyridine-Fe3+ complex decreased in a concentration-dependent manner, confirming the ability of Cur–PDA NPs to chelate Fe3+ (Fig. 2(a) and (b)). Similarly, Fe2+ forms a blue terpyridyltriazine-Fe2+ complex. The capability of Cur–PDA NPs to chelate Fe2+ ions was assessed by evaluating the OD value of this complex. The graph clearly reveals a concentration-dependent reduction in the characteristic peak of the terpyridyltriazine-Fe2+ complex at 600 nm following Cur–PDA NPs addition (Fig. 2(c) and S3†). This trend effectively confirms the capacity of Cur–PDA NPs to chelate Fe2+.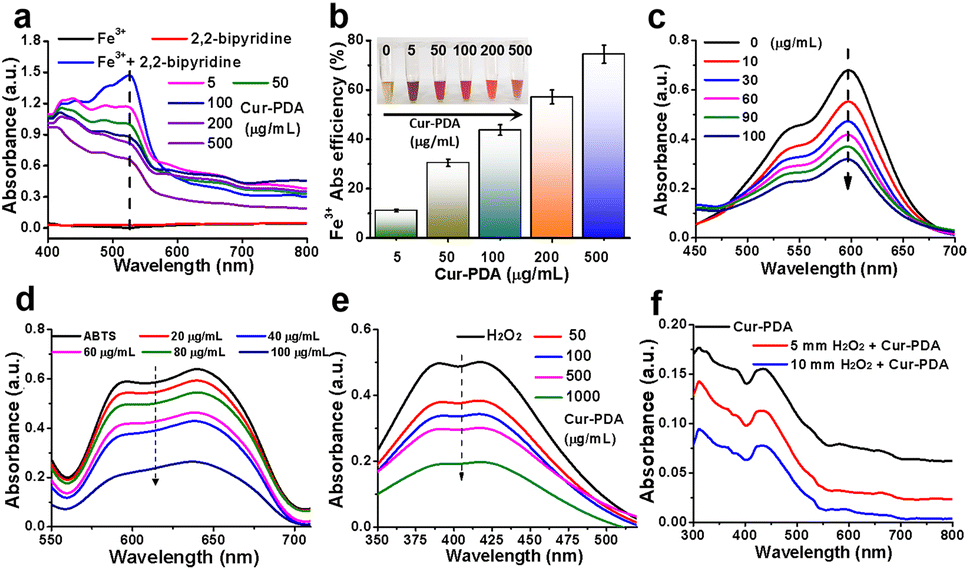 | ||
| Fig. 2 The principle underlying iron chelation by Cur–PDA NPs. (a) UV-vis absorption spectra of the pink 2,2′-bipyridine-Fe3+ complex following Cur–PDA introduction, suggesting that Cur–PDA can chelate Fe3+. (b) Chelation efficiency of Cur–PDA for Fe3+. (c) UV-vis absorption spectra of the blue terpyridyltriazine-Fe2+ complex following the introduction of Cur–PDA NPs, suggesting that Cur–PDA NPs can chelate Fe2+. (d) Antioxidant activity of Cur–PDA NPs for non-enzymatic ABTS method. (e) UV-vis absorption spectra to the titanium sulfate method to assess the H2O2 scavenging capacity of Cur–PDA NPs. (f) Consumption of H2O2 when incubated with Cur–PDA NPs. The data are provided as mean values with corresponding s.d. (n = 3). | ||
3.3 Antioxidant activity of Cur–PDA NPs
Iron-induced OS is closely related to ferroptosis.40 An imbalance between oxidation and antioxidative defenses in the body leads to OS.41 Homeostasis by cellular antioxidants is maintained by regulating oxidation-related non-enzymatic substances and enzymes.42 Non-enzymatic and enzymatic methods can be used to measure the antioxidant activity of antioxidants.43 We employed the widely recognized non-enzymatic ABTS method to preliminarily evaluate the antioxidant capacity of Cur–PDA NPs. When ABTS reacts with potassium persulfate, it is oxidized into the monocationic free radical ABTS˙+, which is positively charged with a maximum absorption at 634 nm, displaying a blue/green color. It can receive an electron from antioxidants to form a stable neutral molecule, leading to a shift in color to a lighter shade.44 The ability of an antioxidant to scavenge ABTS˙+ is indicated using inhibition percentage (%). Fig. 2(d) and S4,† reveals that ABTS˙+ exhibited a concentration-dependent decrease following exposure to varying Cur–PDA NPs concentrations (20–100 μg mL−1). Following this observation, we used the titanium sulfate method to assess the H2O2 scavenging capacity of Cur–PDA NPs. H2O2 reacts with titanium sulfate to form a peroxo-titanium complex that can be inhibited using an antioxidant, as detected by spectrophotometry. With the addition of Cur–PDA NPs at 410 nm, there is a dose-dependent reduction in the characteristic peak of the peroxo-titanium complex (Fig. 2(e)). This trend indicates that the ability of Cur–PDA NPs to deplete H2O2 is concentration-dependent. Moreover, Cur–PDA NPs correspondingly decreased after 24 h of co-incubation with H2O2 (Fig. 2(f)). This suggests that Cur–PDA NPs undergoes biodegradation in the presence of ROS.3.4 Cell viability analysis and cytotoxicity assay
MTT and cytotoxicity assays revealed that Cur maintains a safe concentration range of 0–20 μg mL−1 over 24 h, with an IC50 of 60 μg mL−1 (Fig. S5(a) and (b)†). At a concentration of 100 μg mL−1, Cur–PDA NPs demonstrated minimal toxicity to cells within the same timeframe (Fig. 3(a)). Furthermore, Fig. S5(a),† featuring acridine orange/propidium iodide staining, suggests that Cur–PDA NPs exhibits a lesser impact on cell viability than Cur. All these results indicate that Cur–PDA has good biocompatibility.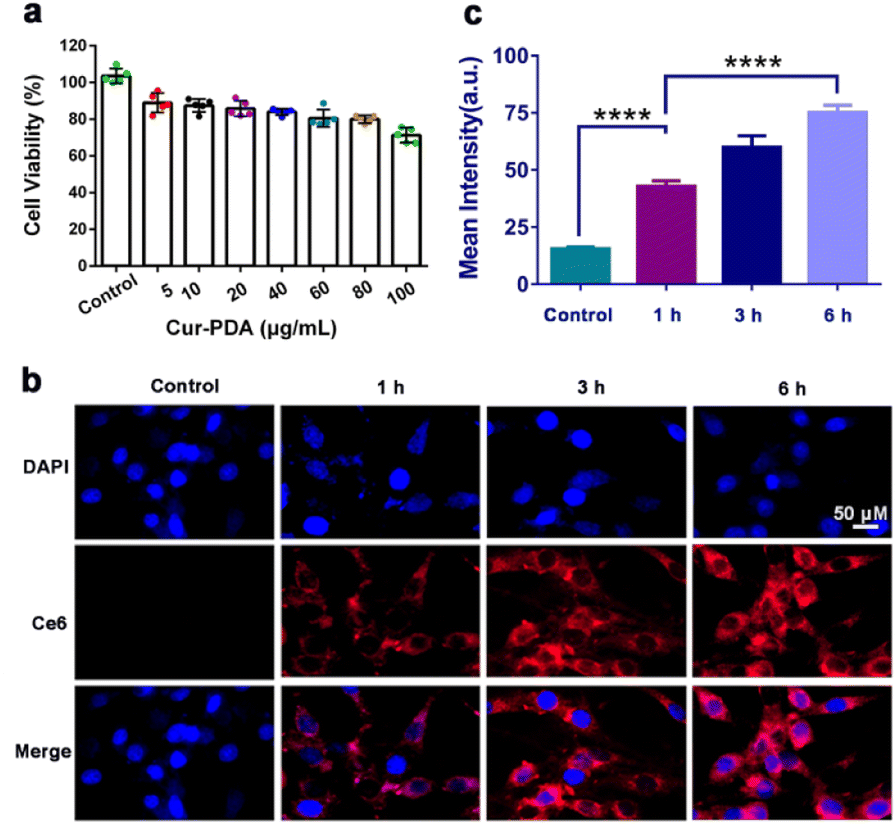 | ||
| Fig. 3 (a) Cell survival rate of PC12 cells following 24 h of treatment with varying Cur–PDA NPs concentrations. (b) Cellular uptake of Cur–PDA@Ce6 NPs, indicating changes in fluorescence intensity of Cur–PDA NPs (10 μg mL−1) in PC12 cells at varying time intervals. (c) Quantitative fluorescence graph of b; n = 3; *p < 0.05; **p < 0.01; ***p < 0.001. | ||
The synthesis of Cur–PDA@Ce6 NPs, through modification with dihydrochlorin (Ce6), enabled subsequent co-incubation with PC12 cells over various time periods. Laser confocal microscopy allowed us to track Ce6 distribution within the cells. We found that the fluorescence intensity of Cur–PDA@Ce6 NPs inside the cells progressively increased (Fig. 3(b) and (c)). Importantly, starting from 6 h post-incubation, Cur–PDA@Ce6 NPs was found to accumulate not only in the cytoplasm but also within the cell nucleus (Fig. 3(c)). Thus, Cur–PDA NPs exhibits superior cell compatibility than Cur and possesses beneficial characteristics in terms of size and cell permeability, laying a robust foundation for its potential in intracellular antioxidant activity and ferroptosis inhibition for therapeutic applications.
3.5 Clearance of intracellular iron deposition and ferroptosis inhibition
The two hallmark features of ferroptosis are iron ion accumulation and LPO.45 This form of cell death is initiated by an imbalance between the generation and elimination of lipid ROS in cells.46 This imbalance results in a depletion of cellular antioxidant capacity and a subsequent increase in lipid ROS, which stimulates iron ion accumulation. Thus, agents that either directly inhibit LPO or reduce cellular iron levels can mitigate ferroptosis.47 The impact of Cur–PDA NPs on the intracellular Fe2+ levels in erastin-treated cells was observed by using FerroOrange. As shown in Fig. 4(a) and (b), the content of Fe2+ increased in the erastin-treated groups. However, and the level of Fe2+ decreased after Cur treatment, but Cur–PDA NPs diminished the intracellular Fe2+ content to normal levels. In addition, Perls'solution and eosin staining shows that fewer blue areas in the Cur–PDA NPs-treated cells compared with erastin treatment (Fig. 4(c)). The quantitative assessment of the blue (positively stained) pixels presented in Fig. S6,† unveiled that the Cur–PDA NPs exhibited an iron elimination efficiency that surpassed free Cur by 4.5 times.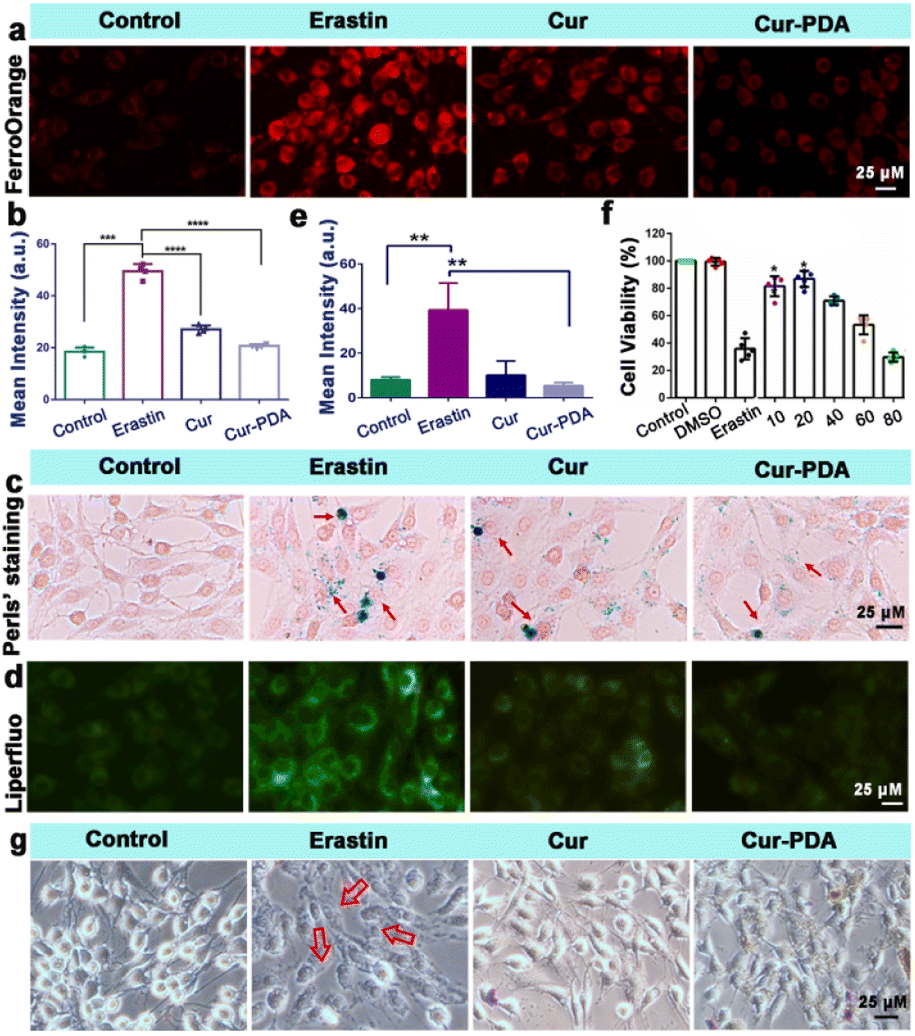 | ||
| Fig. 4 (a) FerroOrange staining of PC12 cells treated with or without erastin, Cur (5 μg mL−1) and Cur–PDA NPs (10 μg mL−1). (b) Quantitative fluorescence graph of (a). Cell survival rate of PC12 cells following 24 h of treatment with varying Cur–PDA NPs concentrations. (c and d) Perls' (c) and Liperfluo (d) staining PC12 cells treated with or without erastin, Cur (5 μg mL−1) and Cur–PDA NPs (10 μg mL−1). (e) Fluorescence intensity of Liperfluo in each group. (f) The cytotoxicity of 5 μM erastin to cells with or without different concentrations of Cur–PDA NPs. (g) An inverted microscope of morphological alterations in the cells incubated with DMEM, erastin, Cur, Cur–PDA NPs for 24 h; n = 3; *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Next, Liperfluo probe indicates erastin treatment increases intracellular LPO; however, Cur–PDA NPs reduces the production of lipid peroxides (Fig. 4(d) and (e)), exhibiting significantly enhanced efficacy compared to Cur alone. Consistent with earlier observations, the ferroptosis induction in PC12 cells using erastin allowed us to verify, via the MTT assay, that Cur–PDA NPs (20 μg mL−1) effectively and significantly mitigates cell death (Fig. 4(f)). Following erastin treatment, we observed notable morphological alterations in the cells using an inverted microscope, including diminished protrusions, shortened synapses, and a transition to a more rounded cell shape (Fig. 4(g)).48 Subsequent treatments with Cur and Cur–PDA NPs (20 μg mL−1 each) resulted in varying degrees of reversal of these abnormal morphological changes.
3.6 Clearance of ROS for mitochondrial function protection
Mitochondria play a central role in ferroptosis, supplying specific lipid precursors through fatty acid metabolism and glutaminolysis.49–51 Mitochondria-induced lipid peroxides can instigate mitochondrial LPO and damage ex vivo, fueled by OS.52 This process interrupts the regulation of mitochondrial iron homeostasis, culminating in ferroptosis.53 Mitigation of ferroptosis also results in a corresponding reduction in mitochondrial damage. In our experiments with erastin-induced ferroptosis in PC12 cells, Cur–PDA NPs (10 μg mL−1) effectively inhibited cell death. To elucidate the mechanism underlying the inhibition of ferroptosis by Cur–PDA NPs, we assessed changes in intracellular ROS levels and mitochondrial activity in the erastin-induced ferroptosis model following Cur–PDA NPs intervention. Our findings indicated a substantial clearance of intracellular ROS, with a rate of 93% ± 2.6% (Fig. 5(a) and S7(a)†). Concurrently, mitochondrial activity exhibited signs of recovery, a trend more pronounced than that observed with Cur alone. Moreover, levels of GSH—a critical thiol-containing molecule in mitochondria vital for antioxidant defense and iron balance—also increased (Fig. 5(c)).54 This increase in GSH levels indicated a positive correlation with the restoration of mitochondrial activity.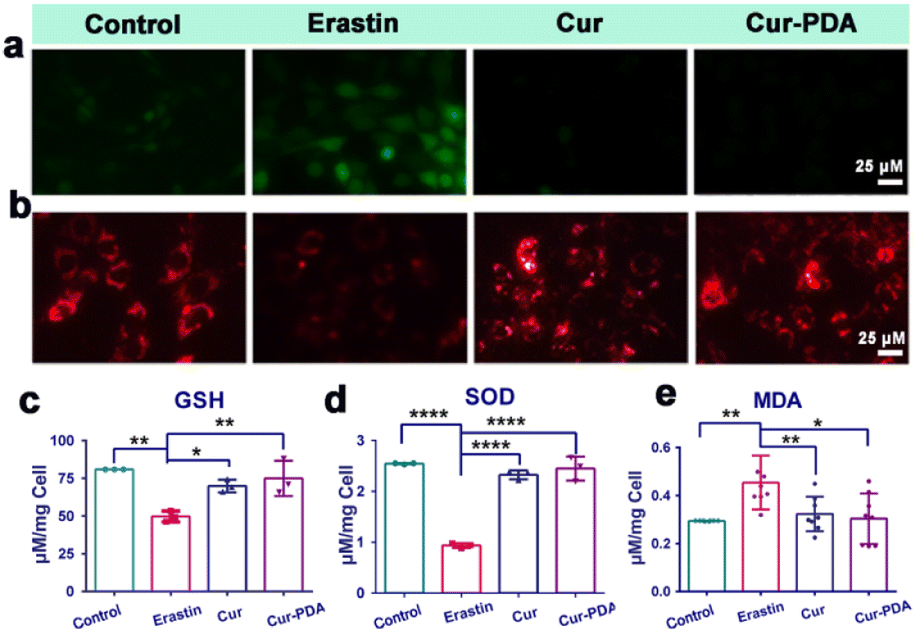 | ||
| Fig. 5 (a) The effects of Cur (5 μg mL−1) and Cur–PDA NPs (10 μg mL−1) on ROS scavenging. (b) The effects of Cur (5 μg mL−1) and Cur–PDA NPs (10 μg mL−1) on the recovery of mitochondrial activity induced by erastin. (c) Quantitatively analyzed intracellular levels of GSH. (d) Quantitatively analyzed levels of intracellular SOD. (e) Quantitatively analyzed levels of intracellular MDA. Data are represented as mean ± SD; n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
To further evaluate the antioxidant potential of Cur–PDA NPs, we monitored alterations in SOD and MDA levels. SOD contributes crucially to the antioxidant defense system in the body, scavenging free radicals to mitigate OS.55 Its activity serves as an indirect marker of OS damage in cells. Our experiments revealed a significant decrease in SOD activity in erastin-treated cells compared to the normal control group (Fig. 5(d)). However, following treatment with Cur–PDA NPs, SOD activity was found to increase, indicating the capacity of Cur–PDA NPs to rejuvenate antioxidant enzyme activity and, consequently, reduce OS levels. Intracellular MDA, an LPO byproduct, serves as an indirect indicator of OS severity in cells. We observed a significant increase in MDA levels in the erastin-induced ferroptosis group compared to the control group, indicating enhanced LPO (Fig. 5(e)). However, treatment with Cur–PDA NPs resulted in reduced MDA levels, implying its effectiveness in decreasing cellular LPO and thereby reducing OS. Altogether, these findings highlight the antioxidative activity of Cur–PDA NPs, which is facilitated by scavenging intracellular ROS and modulating biomolecules such as MDA and SOD. This activity effectively mitigates OS damage and promotes mitochondrial functional recovery, thereby attenuating ferroptosis progression.
3.7 Inhibition of ferroptosis in an erastin-induced nematode model
Mitigating OS is imperative; however, this alone does not directly prevent ferroptosis triggered by excessive iron accumulation.56 Consequently, our investigation extended to assessing iron content in Caenorhabditis elegans. Cur–PDA NPs was found to effectively facilitate the clearance of Fe2+ in these models (Fig. 6(a)). Quantitative analysis using FerroOrange fluorescence revealed a substantial clearance rate of Fe2+, reaching as high as 65.5% (Fig. 6(b)). In normal PD4251 strain nematodes, mitochondria are evenly distributed and abundant.57 Conversely, erastin treatment weakens mitochondrial fluorescence significantly, and their count notably decreases, indicating severe mitochondrial damage caused by erastin-induced ferroptosis (Fig. 6(c)). Although Cur treatment leads to a marginal improvement in mitochondrial numbers, the effect is limited. It is noteworthy that mitochondrial fluorescence intensifies in Cur–PDA NPs-treated nematodes, and the count along the body wall is restored to levels comparable to the normal group (Fig. 6(c) and (d)). We speculate that PDA encapsulation enhances the absorption of Cur in the nematodes, and the inherent antioxidative properties and iron chelation potential of PDA enhance the therapeutic effects of Cur–PDA NPs. This segment of the study reveals that Cur–PDA NPs can chelate iron, clear iron deposits in both cells and nematodes, resist ferroptosis, and restore mitochondrial function.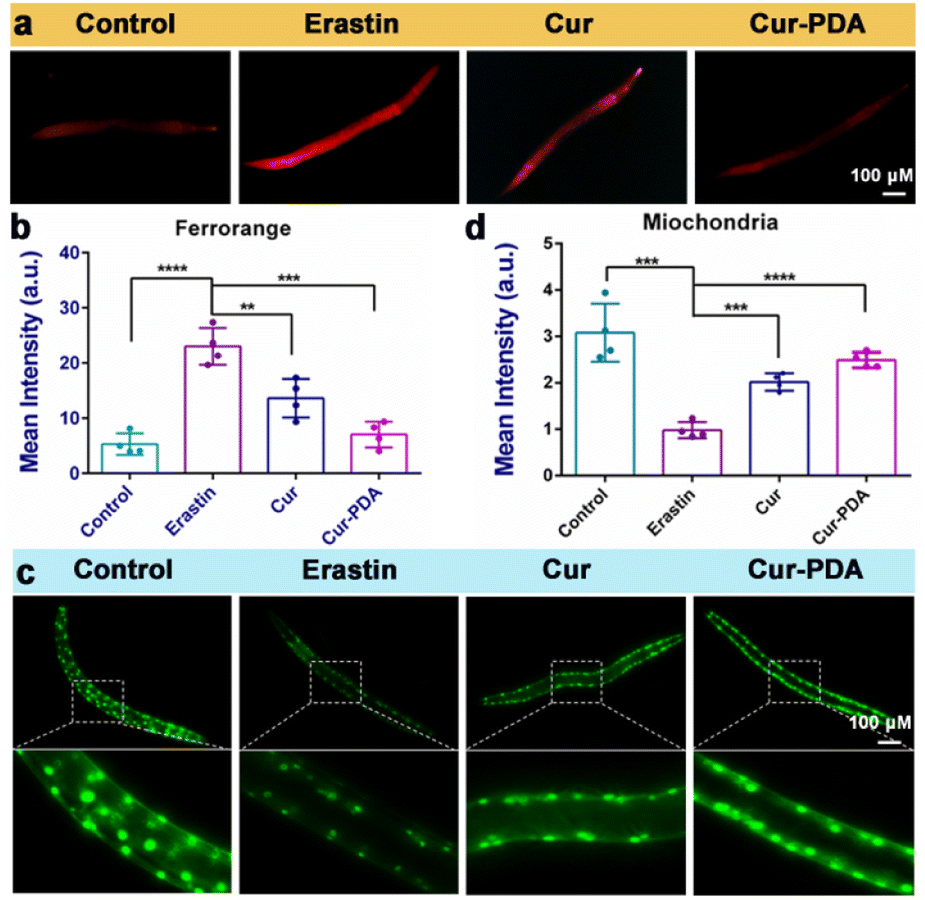 | ||
| Fig. 6 (a and b) FerroOrange staining (a) and quantitative analysis of FerroOrange fluorescence (b) erastin-induced clearance of Fe2+ in nematodes with the treatment of Cur (5 μg mL−1) and Cur–PDA NPs (10 μg mL−1). (c) Changes in the number and fluorescence intensity of mitochondria in nematodes following erastin-induced (5 μM) damage. (d) Quantitative mitochondrial fluorescence analysis from (c). Data are represented as mean ± SD; n = 3, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
4. Conclusions
Ferroptosis is a form of cell death indicated by iron-dependent LPO and governed by a myriad of cellular metabolic pathways, including redox homeostasis, iron metabolism, mitochondrial activity, and the metabolism of amino acids, lipids, and carbohydrates. Moreover, ferroptosis is influenced by various disease-related signaling pathways. In addressing the challenges associated with Cur, such as poor water solubility, instability, and rapid metabolism, we have developed Cur–PDA NPs as an innovative inhibitor of ferroptosis. Our in vitro studies crucially reveal that Cur–PDA NPs serves as a potent therapeutic agent against erastin-induced ferroptosis. It achieves this through antioxidation and iron chelation, thereby preserving cellular homeostasis and mitigating OS-induced ferroptosis in PC12 cells. In particular, Cur–PDA NPs reduces Fe2+ accumulation, offering a novel therapeutic strategy for diseases associated with ferroptosis and paving the way for future clinical applications of Cur.Author contributions
LL: performed the methodology, and investigation, JY: performed the methodology, and investigation, QT: contributed validation of work, QY: contributed validation of work, HY: contributed to, conceptualization, and supervision, LT: contributed validation of work, LC: contributed to, conceptualization, and supervision, CZ: contributed to, conceptualization, and, supervision. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Lei, J. Yuan, and Q. Yang contributed equally to this study. Financial support from the National Natural Science Foundation of China (82160234 and 32360241), Guizhou Provincial Science and Technology Projects (ZK[2023]293), the Elexible Talents Program of Guizhou Medical University (RN21045), Science and technology research project of traditional Chinese medicine and ethnic medicine of Guizhou Province Administration of Traditional Chinese Medicine (QZYY-2024-010), Cultivation program of Guizhou Medical University (21NSFCP24), and the National College Students Innovation and Entrepreneurship Training Program (202210660044, S202310660032, S202310660039) is acknowledged. We thank the Molecular Medicine Engineering Research Center of Guizhou Medical University, the Laboratory Animal Engineering Technology Center of Guizhou Province, and the Key Laboratory of Pathogenesis and Drug Research of Common Chronic Diseases in Guizhou Province for their support.Notes and references
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison 3rd and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- W. S. Yang, R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell, Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- J. Y. Cao and S. J. Dixon, Cell. Mol. Life Sci., 2016, 73, 2195–2209 CrossRef CAS PubMed.
- X. Wu, Y. Li, S. Zhang and X. Zhou, Theranostics, 2021, 11, 3052–3059 CrossRef CAS PubMed.
- X. Teng, X. Xiong, X. Sha, Y. Lei, Y. Diao, J. Liu, Y. Tian, L. Liu and J. Zhong, Front. Cell. Infect. Microbiol., 2023, 13, 1103471 CrossRef CAS PubMed.
- Y. Wang, M. N. Lv and W. J. Zhao, Ageing Res. Rev., 2023, 91, 102035 CrossRef CAS PubMed.
- Z. L. Wang, L. Yuan, W. Li and J. Y. Li, Trends Mol. Med., 2022, 28, 258–269 CrossRef CAS PubMed.
- B. Hassannia, P. Vandenabeele and T. Vanden Berghe, Cancer Cell, 2019, 35, 830–849 CrossRef CAS PubMed.
- Y. Gu, Y. Li, J. Wang, L. Zhang, J. Zhang and Y. Wang, Eur. J. Med. Chem., 2023, 247, 115015 CrossRef CAS PubMed.
- L. Wang, R. Xu, C. Huang, G. Yi, Z. Li, H. Zhang, R. Ye, S. Qi, G. Huang and S. Qu, Gen. Psychiatry, 2023, 36, e101072 CrossRef CAS PubMed.
- Y. Fan, Y. Zhang, K. Shi, S. Cheng, D. Pei and X. Shu, Cell Death Dis., 2022, 13, 1000 CrossRef CAS PubMed.
- M. Guo, T. Peng, C. Wu, X. Pan and Z. Huang, Bioengineering, 2023, 10, 727 CrossRef CAS PubMed.
- G. Jones, S. K. Goswami, H. Kang, H. S. Choi and J. Kim, Nanomedicine, 2020, 15, 1341 CrossRef CAS PubMed.
- H. Kang, M. Han, J. Xue, Y. Baek, J. Chang, S. Hu, H. Nam, M. J. Jo, G. El Fakhri, M. P. Hutchens, H. S. Choi and J. Kim, Nat. Commun., 2019, 10, 5134 CrossRef PubMed.
- S. H. Park, R. S. Kim, W. R. Stiles, M. Jo, L. Zeng, S. Rho, Y. Baek, J. Kim, M. S. Kim, H. Kang and H. S. Choi, Adv. Sci., 2022, 9, 15 Search PubMed.
- H. Yang, F. Zeng, Y. Luo, C. Zheng, C. Ran and J. Yang, Molecules, 2022, 27, 3879 CrossRef CAS PubMed.
- L. Luo, H. Wang, W. Tian, X. Li, Z. Zhu, R. Huang and H. Luo, Theranostics, 2021, 11, 9937–9952 CrossRef CAS PubMed.
- Q. Jiang, K. Wang, X. Zhang, B. Ouyang, H. Liu, Z. Pang and W. Yang, Small, 2020, 16, e2001704 CrossRef PubMed.
- L. Luo, H. Wang, W. Tian, X. Li, Z. Zhu, R. Huang and H. Luo, Theranostics, 2021, 11, 9937–9952 CrossRef CAS PubMed.
- P. Makvandi, M. Chen, R. Sartorius, A. Zarrabi, M. Ashrafizadeh, F. Dabbagh Moghaddam, J. Ma, V. Mattoli and F. R. Tay, Nano Today, 2021, 40, 101279 CrossRef CAS PubMed.
- M. A. Luzuriaga, A. Shahrivarkevishahi, F. C. Herbert, Y. H. Wijesundara and J. J. Gassensmith, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1735 CAS.
- C. Cortez-Jugo, E. Czuba-Wojnilowicz, A. Tan and F. Caruso, Adv. Healthc. Mater., 2021, 10, e2100574 CrossRef PubMed.
- C. Park, J. Lei and J. O. Kim, Chemosphere, 2023, 310, 136910 CrossRef CAS PubMed.
- H. Yan, P. Li, X. Jiang, X. Wang, Y. Hu, Y. Zhang, R. Su and W. Su, Biomater. Adv., 2022, 139, 213040 CrossRef PubMed.
- H. Chen, Z. Li, J. Xu, N. Zhang, J. Chen, G. Wang and Y. Zhao, Oxid. Med. Cell. Longev., 2023, 2023, 6896790 Search PubMed.
- M. L. Alfieri, T. Weil, D. Y. W. Ng and V. Ball, Adv. Colloid Interface Sci., 2022, 305, 102689 CrossRef CAS PubMed.
- L. Wang, Z. Wang, Y. Pan, S. Chen, X. Fan, X. Li, G. Chen, Y. Ma, Y. Cai, J. Zhang, H. Yang, W. Xiao and M. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 19975–19987 CrossRef CAS PubMed.
- Y. Zhang, X. Ren, Y. Wang, D. Chen, L. Jiang, X. Li, T. Li, M. Huo and Q. Li, ACS Appl. Mater. Interfaces, 2021, 13, 53671–53682 CrossRef CAS PubMed.
- X. Bao, J. Zhao, J. Sun, M. Hu and X. Yang, ACS Nano, 2018, 12, 8882–8892 CrossRef CAS PubMed.
- X. Yang, Y. Chen, J. Guo, J. Li, P. Zhang, H. Yang, K. Rong, T. Zhou, J. Fu and J. Zhao, Adv. Sci., 2023, 10, e2207216 CrossRef PubMed.
- L. Lei, Q. Tu, X. Zhang, S. Xiang, B. Xiao, S. Zhai, H. Yu, L. Tang, B. Guo, X. Chen and C. Zhang, Chem. Eng. J., 2023, 461, 141606 CrossRef CAS.
- X. Yang, L. Ai, J. Yu, G. I. N. Waterhouse, L. Sui, J. Ding, B. Zhang, X. Yong and S. Lu, Sci. Bull., 2022, 67, 1450–1457 CrossRef CAS PubMed.
- R. Fu, H. Song, X. Liu, Y. Zhang, G. Xiao, B. Zou, G. I. N. Waterhouse and S. Lu, Chin. J. Chem., 2023, 41, 1007–1014 CrossRef CAS.
- B. Wang, Z. Wei, L. Sui, J. Yu, B. Zhang, X. Wang, S. Feng, H. Song, X. Yong, Y. Tian, B. Yang and S. Y. Lu, Light Sci. Appl., 2022, 11, 172 CrossRef CAS PubMed.
- Y. Liu, S. Lu, L. L. Wu, L. Yang, L. Yang and J. Wang, Cell Death Dis., 2023, 14, 519 CrossRef CAS PubMed.
- Z. Song, W. Yuan, R. Zhu, S. Wang, C. Zhang and B. Yang, Int. J. Biol. Macromol., 2017, 96, 192–199 CrossRef CAS PubMed.
- J. Li, S. Zheng, Y. Fan and K. Tan, Cell Death Dis., 2023, 14, 628 CrossRef PubMed.
- S. Yao, M. Pang, Y. Wang, X. Wang, Y. Lin, Y. Lv, Z. Xie, J. Hou, C. Du, Y. Qiu, Y. Guan, B. Liu, J. Wang, A. P. Xiang and L. Rong, Redox Biol., 2023, 67, 102871 CrossRef CAS PubMed.
- W. Zhang, H. Yu, Q. Lin, X. Liu, Y. Cheng and B. Deng, Aging, 2021, 13, 10659–10671 CrossRef CAS PubMed.
- X. Yu, Q. Su, T. Shen, Q. Chen, Y. Wang and W. Jia, Mar. Drugs, 2020, 18, 490 CrossRef CAS PubMed.
- L. Goodla, M. Manubolu, K. Pathakoti, T. Jayakumar, J. R. Sheu, M. Fraker, P. B. Tchounwou and P. R. Poondamalli, Int. J. Environ. Res. Public Health, 2019, 16, 1440 CrossRef CAS PubMed.
- B. Enaru, G. Dreţcanu, T. D. Pop, A. Stănilă and Z. Diaconeasa, Antioxidants, 2021, 10, 1967 CrossRef CAS PubMed.
- Y. Yu, Y. Yan, F. Niu, Y. Wang, X. Chen, G. Su, Y. Liu, X. Zhao, L. Qian, P. Liu and Y. Xiong, Cell Death Discov., 2021, 7, 193 CrossRef CAS PubMed.
- Y. Xie, W. Hou, X. Song, Y. Yu, J. Huang, X. Sun, R. Kang and D. Tang, Cell Death Differ., 2016, 23, 369–379 CrossRef CAS PubMed.
- Y. Wang, S. Wu, Q. Li, H. Sun and H. Wang, Adv. Sci., 2023, 10, e2300325 CrossRef PubMed.
- Y. Liu, L. He, B. Liu, Y. Ying, J. Xu, M. Yu, J. Dang and K. Liu, iScience, 2022, 25, 104533 CrossRef CAS PubMed.
- J. J. Jiang, G. F. Zhang, J. Y. Zheng, J. H. Sun and S. B. Ding, Front. Pharmacol, 2022, 13, 876550 CrossRef CAS PubMed.
- H. Wang, C. Liu, Y. Zhao and G. Gao, Eur. J. Cell Biol., 2020, 99, 151058 CrossRef CAS PubMed.
- M. Gao, J. Yi, J. Zhu, A. M. Minikes, P. Monian, C. B. Thompson and X. Jiang, Mol. Cell, 2019, 73, 354–363 CrossRef CAS PubMed.
- M. Martín-Fernández, V. Arroyo, C. Carnicero, R. Sigüenza, R. Busta, N. Mora, B. Antolín, E. Tamayo, P. Aspichueta, I. Carnicero-Frutos, H. Gonzalo-Benito and R. Aller, Antioxidants, 2022, 11, 2217 CrossRef PubMed.
- T. Issitt, E. Bosseboeuf, N. De Winter, N. Dufton, G. Gestri, V. Senatore, A. Chikh, A. M. Randi and C. Raimondi, iScience, 2019, 11, 205–223 CrossRef CAS PubMed.
- X. Shi, B. Reinstadler, H. Shah, T. L. To, K. Byrne, L. Summer, S. E. Calvo, O. Goldberger, J. G. Doench, V. K. Mootha and H. Shen, Nat. Commun., 2022, 13, 2483 CrossRef CAS PubMed.
- Y. Zhang, X. Li and X. Li, J. Inflamm. Res., 2021, 14, 5551–5566 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Trends Cell Biol., 2016, 26, 165–176 CrossRef CAS PubMed.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02336f |
| This journal is © The Royal Society of Chemistry 2024 |