
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the active phase of CoO-based thin-film catalysts in order to tune selectivity in CO2 hydrogenation†
Niloofar Mohammadpour *,
Hanna Kierzkowska-Pawlak,
Jacek Balcerzak and
Jacek Tyczkowski
*,
Hanna Kierzkowska-Pawlak,
Jacek Balcerzak and
Jacek Tyczkowski
Department of Molecular Engineering, Faculty of Process and Environmental Engineering, Lodz University of Technology, Wolczanska 213, 93-005 Lodz, Poland. E-mail: niloofar.mohammadpour@dokt.p.lodz.pl
First published on 23rd May 2024
Abstract
In this study, we prepared CoO-based thin films deposited on Kanthal steel wire gauze meshes by plasma-enhanced chemical vapor deposition. X-ray photoelectron spectroscopy (XPS) analysis revealed a structure characterized by a combination of cobalt oxide and metallic cobalt embedded within a carbon matrix. Our primary objective was to gain insights into the roles of Co0 and CoO in CO2 hydrogenation reactions. To achieve this, the performance of the thin-film CoO-based catalyst with an initial atomic ratio of CoO/Co0 at 10.2 was compared with two series of the thin-film catalysts that underwent pre-reduction processes at 350 °C for durations of 30 and 60 minutes, resulting in atomic ratios of CoO/Co0 at 3.1 and 1.1, respectively. Subsequently, catalytic tests were conducted in a continuous flow stirred tank reactor operating at temperatures ranging from 250 °C to 400 °C. Our findings indicate that CoO plays a significant role in activating the CO2 methanation reaction which can be due to the high hydrogen coverage of CoO, while Co0 is the active phase in the reverse water–gas shift reaction. Results highlight the importance of oxidized cobalt for hydrogen adsorption and dissociation in CO2 hydrogenation for CH4 formation.
1 Introduction
Cobalt/cobalt oxide-supported catalysts have been widely applied in CO/CO2 hydrogenation reactions.1,2 The diverse chemical states of cobalt (Co0, Co2+, Co3+) in such catalysts, contribute to distinct behaviour in CO2 hydrogenation reactions,3,4 impacting product distribution.5 However, the exact nature of the active form of cobalt-based catalyst in CO2 hydrogenation remains a subject of ongoing debate among the scientific community.6 Some researchers claim that the metallic form is active phase,7,8 while others argue that the active phase is present in an oxidized state.2,9 These debates can be due to the interplay of factors like particle size, the complex interactions between the active phase and the support material,10,11 and the crystallographic structure of different forms of cobalt.12,13 In addition, different mechanisms can be followed based on the active phase state. Melaet et al.2 synthesized a series of Co catalysts supported on TiO2 and SiO2. Their investigation revealed that cobalt oxide nanoparticles supported on TiO2 exhibited better conversion rates and CH4 selectivity in the CO2 hydrogenation compared to reduced catalysts containing metallic cobalt nanoparticles. Conversely, fully reduced cobalt oxide supported on SiO2 demonstrated increased activity in CO2 hydrogenation and greater selectivity towards CH4. These findings emphasize the significant influence of supports and the chemical state of cobalt in tailoring the distribution of products.Recently, ten Have et al.14 established the hypothesis that cobalt and cobalt oxide can both be active phases by participating in different reaction mechanisms. Based on their findings, metallic cobalt participates in a dissociation pathway via CO formation as an intermediate species. Cobalt oxide, on the other hand, enables an associative route that forms carbonate, formate, and formyl species as intermediates.
The outcomes of some research support the synergetic effect of Co and CoO in the activity of supported cobalt catalysts in CO2 hydrogenation.1,15 Zhao et al.16 prepared self-supported catalysts containing a mixture of Co0 and CoO. These catalysts were subjected to a pre-reduction process at varying temperatures, resulting in different ratios of metal/metal oxide. They found that catalysts with a higher CoO content exhibited superior performance in CO2 hydrogenation due to high CO2 adsorption on CoO sites, which facilitated the formation of formate and carbonate as intermediates for CH4 production. Whereas metallic cobalt played a role in the subsequent hydrogenation step. They concluded that the most effective catalyst for CO2 hydrogenation contains a lot of CoO mixed with Co0. Although in the reported research16 there was no effect of the third solid phase applied as a support, variations in the reduction temperatures can also affect the nanostructure of the active phase, influencing its behaviour during the reaction which was not considered.
In our group, cold plasma-prepared nanocatalysts based on cobalt oxides for CO2 methanation have been developing.9 What makes our research unique is the thin-film form of these catalysts facilitating deposition on various structured supports like fine meshes. This feature offers wide possibilities for practical applications of plasma-prepared catalysts in structured reactors, providing effective heat transfer properties crucial for carrying highly exothermic reactions such as CO2 hydrogenation to methane.9,17 Our previous research demonstrated that the plasma-prepared thin-film CoO-based catalysts are very active in the CO2 methanation reaction.9 Subsequent research aimed to further investigate the structure–activity relationships, revealing that the excellent performance of CoO-based catalysts is attributed to the formation of nanoscale heterojunctions. Here p-type CoO nanocrystallites interact with the n-type carbon matrix, creating spatial charge regions—negative on the CoO and positive on the carbon matrix—which enhance and stabilize catalytic activity towards CH4 production.17
Herein, we have attempted to control the distribution of CH4 and CO products in CO2 hydrogenation by subjecting the CoO-based thin film to the pre-reduction process with different durations, resulting in a change in the oxidation state of cobalt. By controlling the pre-reduction time, we essentially manipulate the relative amounts of CoO and Co0 present on the catalyst surface. In contrast to the most reported studies, we could reasonably assume the absence of interactions between the support and the active phase because the thin film covered uniformly the structured support, preventing its direct contact with the reaction mixture.
2 Experiments
2.1 Catalyst fabrication
The deposition of a CoO-based thin film on the calcined Kanthal steel wire gauze meshes (FeCrAl alloy, TermTech) was carried out in a parallel-plate radio frequency reactor (RF 13.56 MHz) using the plasma-enhanced chemical vapor deposition (PECVD) method, which was described in detail in our previous paper.18 A scheme of the PECVD setup is provided in Fig. S1.† To fabricate the CoO-based thin film, cyclopentadienyl (dicarbonyl) cobalt(I) (CPCo(CO)2, Strem Chemicals) served as the precursor of the deposited material, and argon (99.999%, Linde Gas) was used as the carrier gas.The procedure began with argon etching of supports including four pieces of Kanthal steel meshes, pre-calcined in the air at 900 °C for 48 h to obtain a segregated α-Al2O3,19 with a diameter of 3 cm dedicated for the catalytic test, and similarly thermally pre-treated Kanthal steel plates for XPS (X-ray photoelectron spectroscopy) analysis. Subsequently, the thin film growth occurred over 30 minutes, utilizing a discharge power of 60 W, with argon gas and precursor flow rates set at 0.71 and 0.096 sccm, respectively. The resulting thickness of the as-deposited films was approximately 650–750 nm. The thickness of the films was measured by the interference method, which was explained in detail in the previous paper.20 The deposition procedure was applied on both sides of the meshes and one side of the plates. After synthesizing the catalyst, to have a stable thin film in CO2 methanation, thermal treatment at 400 °C in an argon gas atmosphere for 30 minutes was conducted.17 The samples of thin-film catalysts, which were prepared with this procedure, were named TT-CoO (thermally treated).
2.2 Catalytic test
The catalytic test was conducted in a gradientless continuous flow stirred tank reactor (CSTR) constructed based on the design outlined in reference21 including a heater, a type K thermocouple near the catalyst bed for temperature control, and a stirrer operating at rotational speeds up to 3600 rpm. Schematics of CSTR is shown in Fig. S2.† For each catalytic test, a thin-film catalyst deposited on a piece of mesh with a diameter of 3 cm with a geometric surface area of approximately 10 cm2 was loaded into the CSTR reactor. The reactor underwent helium gas purging (99.999%, Linde Gas) as an initial step. Following the purging process, the catalytic performance of three sets of thin-film catalysts were evaluated.In the first set, the CoO-based catalyst denoted as TT-CoO, was directly tested without any preceding pre-reduction process. A gas mixture containing H2 (99.999%, Linde Gas) and CO2 (99.99%, Linde Gas) in a volumetric 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, with a total flow rate of 50 sccm, was introduced to the reactor. The reactor temperature was gradually increased from an initial setting of 250 °C, with a heating rate of 10 °C min−1. Catalytic performance was assessed over a temperature range of 250–400 °C in increments of 50 °C. At the temperature of 400 °C, by keeping the constant volumetric inlet gas mixture ratio of 4:1, the performance of the catalyst at different inlet gas mixture flow rates of 25, 80, 100, and 125 sccm was evaluated as well. Gas analysis was carried out 15 minutes after reaching each target temperature using gas chromatography (Shimadzu, GCMS-QP2010 SE) equipped with a Carboxen 1010 column and a dielectric-barrier discharge ionization detector (BID). Two analyses were conducted at 20 minute intervals.
:1 ratio, with a total flow rate of 50 sccm, was introduced to the reactor. The reactor temperature was gradually increased from an initial setting of 250 °C, with a heating rate of 10 °C min−1. Catalytic performance was assessed over a temperature range of 250–400 °C in increments of 50 °C. At the temperature of 400 °C, by keeping the constant volumetric inlet gas mixture ratio of 4:1, the performance of the catalyst at different inlet gas mixture flow rates of 25, 80, 100, and 125 sccm was evaluated as well. Gas analysis was carried out 15 minutes after reaching each target temperature using gas chromatography (Shimadzu, GCMS-QP2010 SE) equipped with a Carboxen 1010 column and a dielectric-barrier discharge ionization detector (BID). Two analyses were conducted at 20 minute intervals.
In the second and third sets, two kinds of CoO-based catalysts, namely R-30-TT-CoO and R-60-TT-CoO, derived from the source TT-CoO catalyst, underwent pre-reduction processes at 350 °C in pure hydrogen with a gas flow rate of 100 sccm. The reduction process for R-30-TT-CoO lasted for 30 min, while R-60-TT-CoO underwent a 60 min reduction. Following the reduction process, without allowing the catalyst to be exposed to air, the catalytic tests were conducted using the same procedure as for the TT-CoO catalyst.
2.3 Morphology analysis
The morphology of the fabricated thin films was evaluated using a scanning electron microscope (SEM) – specifically, the Thermo Fisher Scientific Apreo 2 S LoVac. All SEM micrographs were recorded in high vacuum mode using an electron energy of 3.5 keV. The tested samples did not have sputtered coatings.2.4 XPS measurement
To examine the surface composition and elemental chemical state of thin films after pre-reduction without exposure to the external environment, a reduction process of TT-CoO films on Kanthal plates was conducted in a high-temperature gas reaction cell (Cat-Cell, Kratos Analytical Ltd), combined with an XPS spectrometer, to mimic the pre-reduction processes performed in CSTR reactor.The analysis was conducted using the AXIS Ultra spectrometer from Kratos Analytical Ltd, with a monochromatic Al-Kα X-ray source (at 1486.6 eV). During the high-resolution measurements, the anode's power was set at 180 W, and the hemispherical electron energy analyser was operated with a pass energy of 20 eV. A charge neutralizer was employed for all measurements to maintain charge balance. The XPS spectra were calibrated using the carbon C 1s line maximum set at 284.4 eV, for the sp2 carbon assignment. Shirley's function was used to subtract the background from the XPS data and the spectral deconvolution was carried out using the Kratos Vision 2 program (Version 2.2.10, Kratos Analytical Ltd). As mentioned earlier, thin films for Cat-Cell reduction and XPS measurements were deposited onto Kanthal steel plates and were inserted into the plasma reactor alongside the meshes during the deposition process. The XPS analysis of all tested catalysts in the reaction (TT-CoO, R-30-TT-CoO, and R-60-TT-CoO), after the catalytic test in the CO2 hydrogenation reaction were done as well.
3 Result and discussion
3.1 Morphology of thin-film catalysts
Fig. 1 illustrates the morphology of the thin film catalysts. Fig. S3 and S4† show the pattern of Kanthal steel meshes, and the morphology of calcined meshes, respectively. Fig. S4† reveals a characteristic morphology of the calcined Kanthal steel support. The surface is extensively covered by a thin layer of sharp, needle-like features. These features can be identified as alumina (α-Al2O3).9,19 The plasma deposited thin film based on CoO develops on the needle-like structure of the support, forming grains that resemble cauliflower. After subjecting a TT-CoO catalyst to a reduction process, no significant changes in the surface morphology are observed. | ||
| Fig. 1 SEM morphology of TT-CoO, R-30-TT-CoO, and R-60-TT-CoO thin-film catalysts. | ||
It is worth mentioning, that in all catalysts, thin film precisely covers the wire of the fine mesh, exhibiting strong adherence to the structured support, as outlined in patents,22,23 providing evidence of its effectiveness. As we have also shown in previous studies, CoO-based thin films tested in long-term catalytic runs show high stability, which, among other aspects, confirms their strong adhesion to the support.9 Moreover, due to the low thickness of the thin film, the mesh remains unblocked. This ensures unobstructed gas flow through the catalytic system, a key requirement for effective structured catalysts.
3.2 Chemical structure of the surface
In our previous study,17 we explored the surface and bulk composition of TT-CoO films and the present paper continues this investigation. The XPS wide spectra of the fresh TT-CoO, R-30-TT-CoO, and R-60-TT-CoO are depicted in Fig. S5.† It should be noted that the thin films produced through the PECVD technique using organometallic precursors include, alongside the metal-based phase, a carbon component commonly referred to as a carbon matrix. Fig. 2 presents the XPS analysis of the Co 2p band, both before (TT-CoO) and after the reduction in the spectrometer's Cat-Cell. The deconvolution of the Co 2p3/2 band reveals two primary asymmetric peaks. The first peak, located in the range of 778.2–778.7 eV, was assigned to Co0.24 The second peak, observed in the range of 780.2–781.0 eV, was attributed to the oxidized state of cobalt (Co2+ in CoO).9,24 Additionally, an intense satellite peak was detected in the range of 785.6–786.2 eV, which serves as a characteristic feature for the recognition and identification of the CoO structure.24,25 As a support of CoO structure presence, in our previous research, XRD analysis of the TT-CoO thin-film catalyst revealed crystallographic planes of CoO (200), CoO (111), CoO (220), CoO (311), and CoO (222), indicative of a cubic structure for CoO with an approximate size of 11 nm. The major planes were CoO (111) and CoO (200).17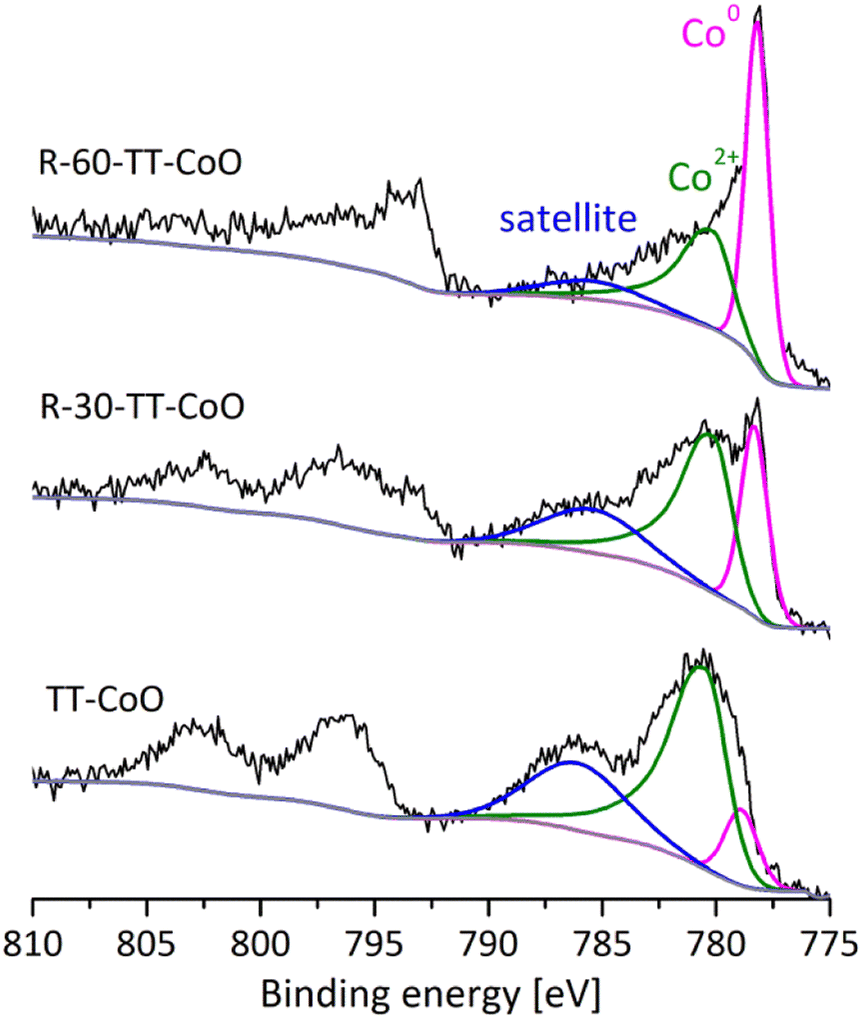 | ||
| Fig. 2 XPS Co 2p spectra of TT-CoO, R-30-TT-CoO, and R-60-CoO thin-film catalysts. | ||
As can be seen in Fig. 2, the pre-reduction process of TT-CoO catalysts affected the chemical state of Co on the surface. For all thin-film catalysts, characteristic features of metallic cobalt (Co0) and cobalt oxide (CoO) are present; however, the reduction process increased the intensity of metallic cobalt peak for R-30-TT-CoO and R-60-TT-CoO compared to TT-CoO. Table 1 shows that for fresh TT-CoO the atomic ratio of CoO/Co0 is 10.2, while after reduction, it decreased to 3.1 and 1.1 for R-30-TT-CoO, and R-60-TT-CoO thin-film catalysts, respectively. Data provided in Table 1 confirmed, that the C/Co atomic ratio decreased slightly after performing the reduction in comparison to the TT-CoO catalyst. On the other hand, the difference in the C/Co ratio between R-30-TT-CoO, and R-60-TT-CoO samples is very close to the experimental uncertainty, while a significant change is observed in the CoO/Co0 ratio. This indicates the impact of pre-reduction duration on the contributions of particular cobalt oxidation states in the three sets of thin-film catalysts, without a simultaneous considerable change in the overall cobalt loading as detailed in Table 1.
| Thin film | H2 reduction condition | Co-loading (%) | C/Co | O/Co | CoO/Co0 |
|---|---|---|---|---|---|
| TT-CoO | — | 13.9 ± 0.1 | 6.4 ± 0.3 | 2.3 ± 0.2 | 10.2 ± 0.9 |
| R-30-TT-CoO | 30 min @ 350 °C | 12.6 ± 0.4 | 5.3 ± 0.3 | 1.6 ± 0.3 | 3.1 ± 0.4 |
| R-60-TT-CoO | 60 min @ 350 °C | 14.1 ± 1.2 | 4.9 ± 0.7 | 0.7 ± 0.1 | 1.1 ± 0.1 |
The XPS spectra of the Co 2p band after applying the catalyst in the CO2 hydrogenation reaction are presented in the ESI (Fig. S6b),† demonstrating that the chemical state of cobalt in the TT-CoO catalyst remains stable after exposure to the reaction gas. The complete stability of the molecular structure as well as nanostructure of TT-CoO films in the CO2 hydrogenation process was confirmed in our resent study.17
However, the chemical state of cobalt in the reduced catalysts (R-30-TT-CoO and R-60-TT-CoO) alters upon exposure to the reaction mixture (H2 + CO2), resulting in partial surface oxidation of cobalt (Table S1†). In both cases, the CoO/Co0 ratio increases in the spent samples compared to the fresh ones.
Fig. 3 displays C 1s spectra for TT-CoO, R-30-TT-CoO, and R-60-TT-CoO. C 1s feature can be resolved into several peaks at binding energies of 284.6, 285.2–285.4, 286.3–287.2, 287.9–288.5, and 288.6 eV as discussed in detail in the previous paper.17 Peaks at a binding energy of 284.6 and in the range of 285.2–285.4 are assigned to carbon atoms in sp2 and sp3 hybridization, respectively.26,27 Peaks in the range of 286.3–287.2 and 287.9–288.5 eV and 288.6 are attributed to C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and COO–R respectively.17,26,27 The main carbon structure in the studied films is in the form of C (sp2), which can suggest a graphite-like structure of carbon. However, XRD analysis did not reveal the presence of graphite, which indicates the amorphous nature of the carbon matrix.17
O, and COO–R respectively.17,26,27 The main carbon structure in the studied films is in the form of C (sp2), which can suggest a graphite-like structure of carbon. However, XRD analysis did not reveal the presence of graphite, which indicates the amorphous nature of the carbon matrix.17
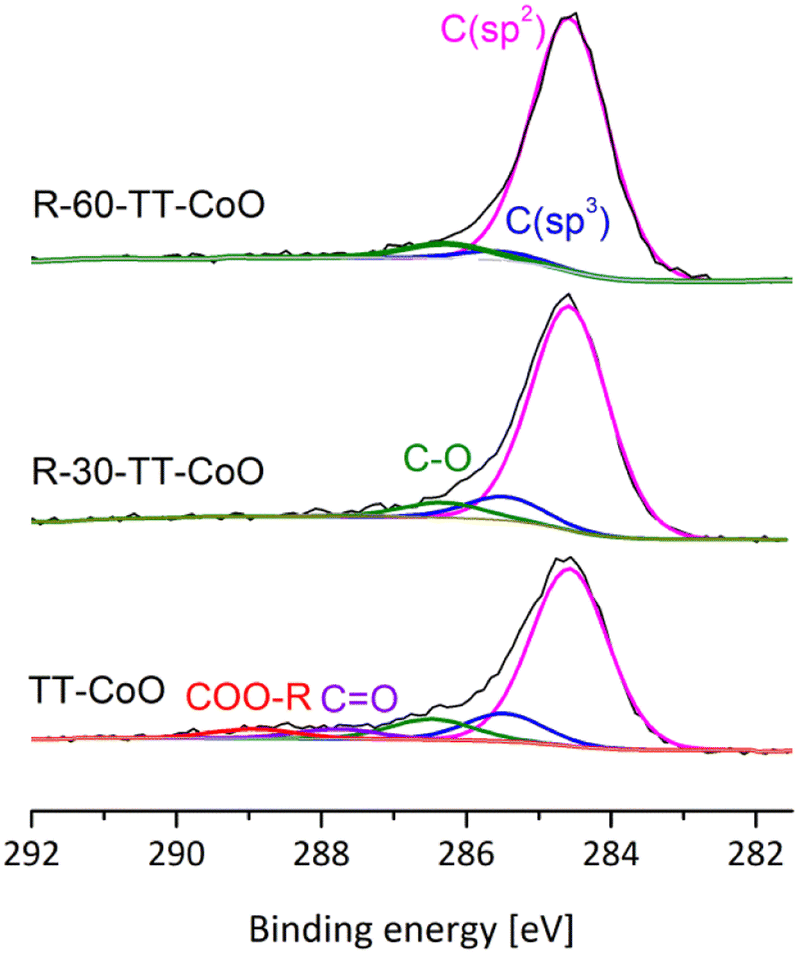 | ||
| Fig. 3 XPS C 1s spectra of TT-CoO, R-30-TT-CoO, and R-60-TT-CoO thin-film catalysts. | ||
The conversion of C (sp3) into C (sp2) on the surface after reduction at 350 °C can be seen. This conversion involves the rearrangement of carbon atoms and the transition from an amorphous sp3-hybridized carbon structure to the sp2-hybridized structure.28 It was shown in our previous publication, that the n-type carbon matrix, containing mostly C (sp2) form, has a significant effect on the stability of the CoO-based catalyst in CO2 hydrogenation.17 No other changes were observed in the carbon structure of the catalysts after the reduction in H2 gas in the XPS spectrometer's Cat-Cell.
The XPS spectra of the C 1s bond of spent catalysts are provided in the ESI (Fig. S6a).† Similar to the Co 2p spectra, the C 1s spectra of the TT-CoO catalyst demonstrate stability. However, for the reduced catalysts, the C 1s spectra reveal the presence of Co–C bonds (cobalt carbide) in the spent catalysts, which are located at lower binding energies of 282.2 eV compared to CC and C–C bonds.29 Therefore, the pre-reduction of CoO-based catalysts and subsequent subjecting them to the reaction mixture, affect the structure of carbon in the carbon matrix.
3.3 Selectivity of CO2 hydrogenation
Fig. 4 shows the comparative performance of TT-CoO, R-30-TT-CoO, and R-60-TT-CoO in CO2 hydrogenation. The catalytic tests were conducted at a constant flow rate of a reaction mixture of 50 sccm using a single piece of catalyst: a round mesh with a geometric surface area of 10 cm2. The results expressed in terms of CO2 conversion (XCO2) and selectivity to methane (SCH4) demonstrate how the pre-reduction process influenced the catalyst's performance compared to a non-reduced TT-CoO. According to Fig. 4a, the pre-reduction process had minimal effect on the CO2 conversion, and by increasing the temperature from 250 to 400 °C, CO2 conversion increased from 3 to approximately 45%. Fig. 4b illustrates that the selectivity to CH4 is strongly affected by the pre-reduction process and at all reaction temperatures, pre-reduced catalysts and a non-reduced TT-CoO catalyst follow different trends. While TT-CoO shows an increase in CH4 selectivity, rising from 65% to 75% as the temperature goes up from 250 °C to 400 °C, R-30-TT-CoO and R-60-TT-CoO catalysts exhibit an initial rise in CH4 selectivity from 60% to 65% and from 50% to 55%, respectively, only in the temperature range of 250–300 °C. However, this is followed by a decline, dropping to 57% and 50%, respectively, as the temperature rises from 300 °C to 400 °C. Therefore, unlike CO2 conversion, CH4 selectivity is affected by the pre-reduction process which results in different contributions of Co0 and CoO on the surface. Fig. 4c confirmed that yields of CH4 at all temperatures follow the order TT-CoO > R-30-TT-CoO > R-60-TT-CoO. In Fig. 4d, the direct effect of the CoO/Co0 ratio on CH4 and CO yield is depicted, showing a decrease in YCH4 from 33% to 22% (an increase in YCO) as the ratio decreases from 10.2 to 1.1. It is apparent that increasing the content of reduced cobalt (Co0) on the surface of the catalyst leads to a decrease in CH4 production. To explain the observed result, examining the intermediate reaction products can be valuable. Recently, it was claimed that CoO more likely favours the formate route (associative mechanism of CO2 methanation), and therefore, the production of CH4 goes through intermediates of carbonate, formate, and formyl species.14 According to this mechanism, CO is not an intermediate that is further hydrogenated towards CH4. However, our research, as depicted in Fig. 5, reveals a contrary observation. When we increased the total flow rate of the reaction mixture in the catalytic test from 25 to 125 sccm at 400 °C, the TT-CoO catalyst with the higher amount of CoO sites exhibited not only decreased XCO2 but also decreased CH4 production, indicating increased selectivity for CO. The observed result supports that CO is an intermediate in the CO2 and H2 reaction and at lower gas flow rates, leading to higher contact times, we observed higher CH4 formation and smaller CO production. Consequently, it is not plausible to claim the CoO-based thin-film catalyst follows an associative pathway in which CO is not an intermediate in CO2 hydrogenation. Therefore, it is more reasonable to explain that the higher CH4 production in catalysts with higher CoO sites is attributed to the activation of H2 on the CoO sites. Consequently, it can be concluded that CoO plays an important role in H2 adsorption and dissociation, thereby promoting CO2 methanation. It is noteworthy that, Li et al.30 by calculating the energy profile of H2 dissociation on different planes of CoO and Co0 showed that H2 dissociation on CoO (111) sites is comparable to Co0 (111), indicating easy dissociation on CoO (111) similar to Co0, both characterized by low barriers. Considering the fact that, as mentioned earlier, the major crystallographic site of cubic CoO in the CoO-based thin-film catalyst is CoO (111), the higher CH4 production on catalysts with higher CoO sites can be attributed to the feasible activation of H2 on these CoO sites. This result stands in contrast to most research that revealed metallic cobalt as an active phase in the hydrogenation step.3,16,31 Instead, our results suggest that CoO (rather than Co0) embedded in a carbon matrix is the active phase for the promotion of the hydrogenation step and CH4 production. The other reason behind the high production of CH4 on CoO sites is related to the enhanced basic nature of CoO sites embedded in a carbon matrix as demonstrated in our previous research.17 According to Gao et al.,32 proper basicity of metal oxides can favour the CO2 hydrogenation to methane. Our findings are also in line with the study by Parastaev et al.,10 who compared Co-supported ceria–zirconia catalysts' behaviour in CO2 methanation. They performed a reduction in hydrogen gas at different temperatures, and they observed less hydrogen adsorbed on the surface with fewer CoO sites, resulting in lower CH4 production. Additionally, metallic cobalt is likely an active phase for CO production through the reverse-water–gas-shift (RWGS) reaction. As recently shown,33 CO is an intermediate that has been detected on the metallic cobalt site of a zirconium oxide-based cobalt catalyst. The present results shown in Fig. 5 further confirm CO as an intermediate.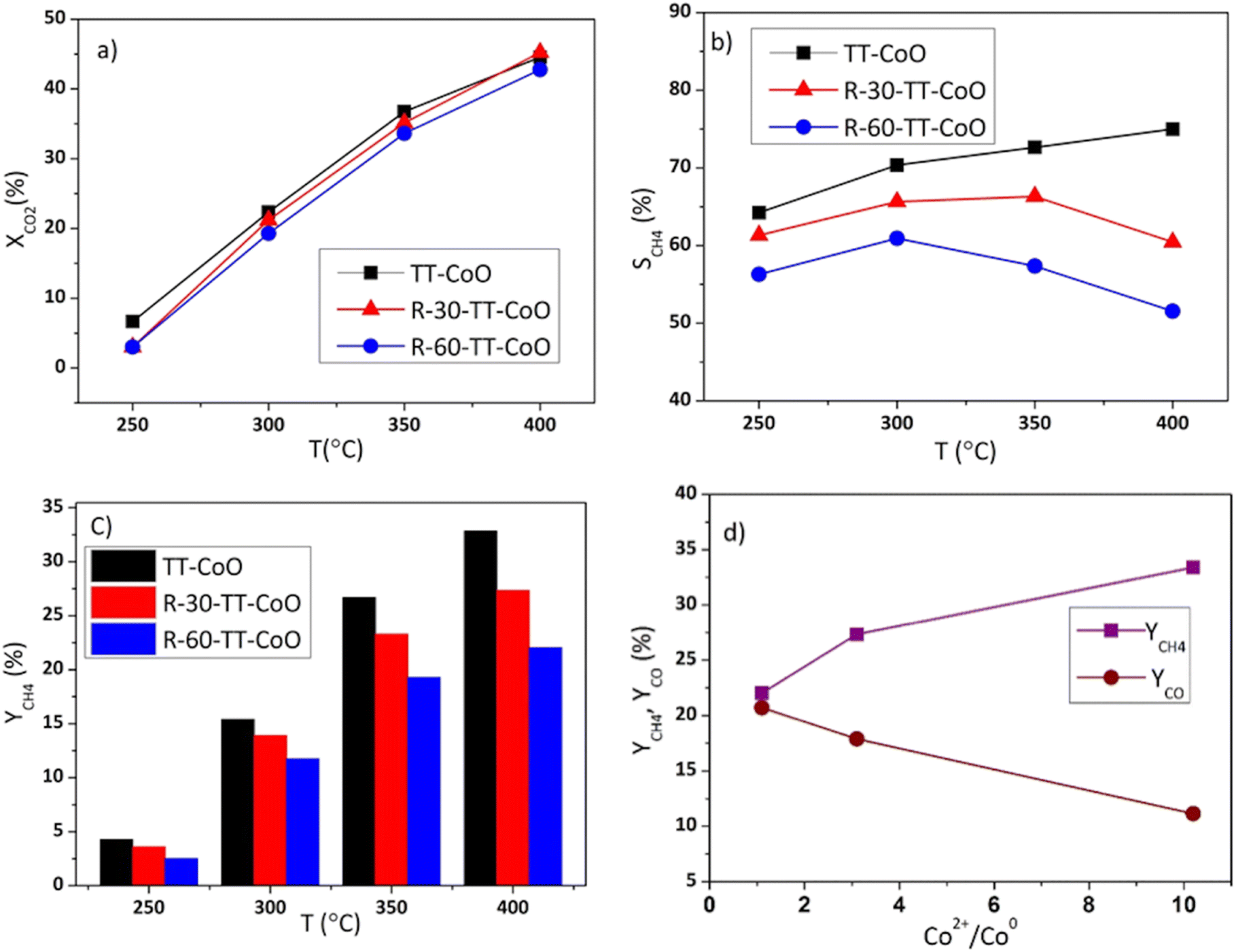 | ||
| Fig. 4 CO2 hydrogenation performance of TT-CoO, R-30-CoO, and R-60-TT-CoO (a) CO2 conversion (XCO2) as a function of temperature (b) CH4 selectivity (SCH4) as a function of temperature (CO selectivity is SCO = 1 − SCH4) (c) CH4 yield (YCH4) at different temperatures (d) CH4 and CO yields (YCH4, YCO) as a function of the chemical state of cobalt at 400 °C. Reaction conditions: P = 0.1 MPa, total gas flow rate of 50 sccm, H2/CO2 = 4. | ||
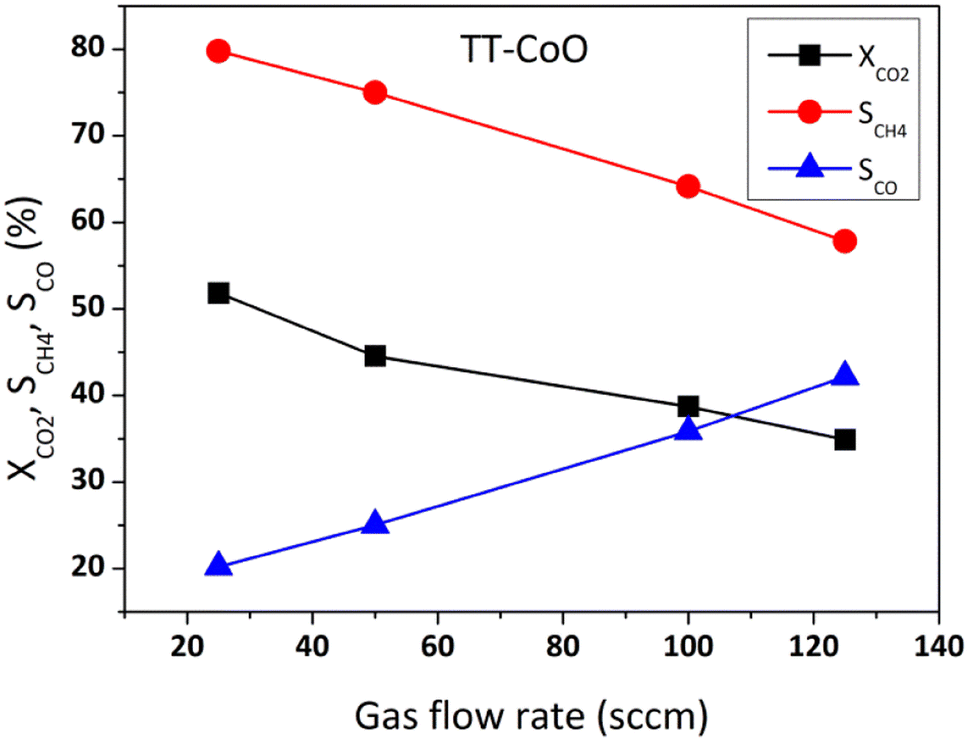 | ||
| Fig. 5 CO2 hydrogenation performance of TT-CoO catalyst at different gas flow rates at 400 °C. Reaction conditions: P = 0.1 MPa, H2/CO2 = 4. | ||
These are the first conclusions of the study however, to establish a more conclusive explanation of the role of metallic and oxidized cobalt in the active phases and their participation in the CO2 hydrogenation reaction, additional experiments on H2 adsorption and dissociation to gain deeper insight into the catalyst's behaviour are needed.
4 Conclusions
CoO-based thin-film catalysts at different ratios of CoO/Co0 were fabricated on the structured support and applied in CO2 hydrogenation. Our results indicate that the catalyst with more CoO sites is more active toward CH4, whereas the catalysts with Co0 sites promote more RWGS reaction. The observed performance of the catalysts can directly be related to the chemical state of cobalt because there is no significant change in the chemical state of carbon in the carbon matrix after the reduction process. Furthermore, because of the thin-film form of the catalysts, the effect of support is eliminated. The results highlight the feasibility of controlling the chemical state of cobalt-based catalysts toward desired products and the importance of the active phase for H2 activation in the CO2 hydrogenation reaction for the production of CH4. In addition, the approach of using plasma-prepared thin films on structured reactors offers a powerful strategy for catalyst development, enabling precise control over the active phase while leveraging the advantages of structured supports, such as improved mass and heat transfer in practical applications.Author contributions
Niloofar Mohammadpour: writing – original draft, conceptualization, formal analysis, investigation. Hanna Kierzkowska-Pawlak: conceptualization, review & editing, resource methodology, supervision, project administration. Jacek Balcerzak: review & editing, formal analysis, investigation. Jacek Tyczkowski: resource methodology, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank PhD candidate Bartosz Panek and Dr Aleksandra Kędzierska-Sar for performing SEM measurements.References
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J. Kim, X. Meng and F. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- G. Melaet, W. T. Ralston, C.-S. Li, S. Alayoglu, K. An, N. Musselwhite, B. Kalkan and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 2260–2263 CrossRef CAS PubMed.
- A. Efremova, T. Rajkumar, Á. Szamosvölgyi, A. Sápi, K. Baán, I. Szenti, J. Gómez-Pérez, G. Varga, J. Kiss, G. Halasi, Á. Kukovecz and Z. Kónya, J. Phys. Chem. C, 2021, 125, 7130–7141 CrossRef CAS.
- S. Liu, Y. He, W. Fu, J. Chen, J. Ren, L. Liao, R. Sun, Z. Tang, C. Mebrahtu and F. Zeng, J. CO2 Util., 2023, 67, 102322 CrossRef CAS.
- T. Fan, H. Liu, S. Shao, Y. Gong, G. Li and Z. Tang, J. Phys. Chem. Lett., 2021, 12, 10486–10496 CrossRef CAS PubMed.
- A. V. Puga, Catal. Sci. Technol., 2018, 8, 5681–5707 RSC.
- G. Zhou, T. Wu, H. Xie and X. Zheng, Int. J. Hydrogen Energy, 2013, 38, 10012–10018 CrossRef CAS.
- Z. Liu, X. Gao, B. Liu, W. Song, Q. Ma, T. Zhao, X. Wang, J. W. Bae, X. Zhang and J. Zhang, Appl. Catal., B, 2022, 310, 121303 CrossRef CAS.
- H. Kierzkowska-Pawlak, J. Tyczkowski, J. Balcerzak and P. Tracz, Catal. Today, 2019, 337, 162–170 CrossRef CAS.
- A. Parastaev, V. Muravev, E. H. Osta, T. F. Kimpel, J. F. M. Simons, A. J. F. van Hoof, E. Uslamin, L. Zhang, J. J. C. Struijs, D. B. Burueva, E. V. Pokochueva, K. V. Kovtunov, I. V. Koptyug, I. J. Villar-Garcia, C. Escudero, T. Altantzis, P. Liu, A. Béché, S. Bals, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2022, 5, 1051–1060 CrossRef CAS.
- J. Díez-Ramírez, P. Sánchez, V. Kyriakou, S. Zafeiratos, G. Marnellos, M. Konsolakis and F. Dorado, J. CO2 Util., 2017, 21, 562–571 CrossRef.
- W. Li, X. Nie, H. Yang, X. Wang, F. Polo-Garzon, Z. Wu, J. Zhu, J. Wang, Y. Liu, C. Shi, C. Song and X. Guo, Appl. Catal., B, 2022, 315, 121529 CrossRef CAS.
- C. Yang, S. Liu, Y. Wang, J. Song, G. Wang, S. Wang, Z. Zhao, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 11242–11247 CrossRef CAS PubMed.
- I. C. ten Have, J. J. G. Kromwijk, M. Monai, D. Ferri, E. B. Sterk, F. Meirer and B. M. Weckhuysen, Nat. Commun., 2022, 13, 324 CrossRef PubMed.
- K. An, S. Zhang, J. Wang, Q. Liu, Z. Zhang and Y. Liu, J. Energy Chem., 2021, 56, 486–495 CrossRef CAS.
- K. Zhao, M. Calizzi, E. Moioli, M. Li, A. Borsay, L. Lombardo, R. Mutschler, W. Luo and A. Züttel, J. Energy Chem., 2021, 53, 241–250 CrossRef CAS.
- N. Mohammadpour, H. Kierzkowska-Pawlak, J. Balcerzak, P. Uznański and J. Tyczkowski, Catalysts, 2024, 14, 38 CrossRef CAS.
- J. Tyczkowski, H. Kierzkowska-Pawlak, R. Kapica, J. Balcerzak and J. Sielski, Catal. Today, 2019, 337, 44–54 CrossRef CAS.
- J. Łojewska, A. Kołodziej, T. Łojewski, R. Kapica and J. Tyczkowski, Appl. Catal., A, 2009, 366, 206–211 CrossRef.
- J. Tyczkowski, R. Kapica, M. Kozanecki, H. Kierzkowska-Pawlak, J. Sielski, T. Aoki and H. Mimura, Mater. Des., 2022, 222, 111095 CrossRef CAS.
- D. Nazimek, M. Kusmierz and P. Kirszensztejn, Pol. J. Appl. Chem., 2006, 50, 41–53 CAS.
- J. Tyczkowski, R. Kapica, J. Łojewska and A. Kołodziej, Poland Pat., PL217586(B1), 2014.
- J. Tyczkowski, H. Kierzkowska-Pawlak and R. Kapica, Poland Pat., PL429641(A1), 2022 Search PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- G. Speranza and N. Laidani, Diamond Relat. Mater., 2004, 13, 451–458 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- Z. Sun, X. Shi, X. Wang and Y. Sun, Diamond Relat. Mater., 1999, 8, 1107–1113 CrossRef CAS.
- Q. Fan, Z. Guo, Z. Li, Z. Wang, L. Yang, Q. Chen, Z. Liu and X. Wang, ACS Appl. Electron. Mater., 2019, 1, 444–453 CrossRef CAS.
- K. Li, X. Li, L. Li, X. Chang, S. Wu, C. Yang, X. Song, Z.-J. Zhao and J. Gong, JACS Au, 2023, 3, 508–515 CrossRef CAS PubMed.
- Y. Lian, T. Fang, Y. Zhang and B. Liu, J. Catal., 2019, 379, 46–51 CrossRef CAS.
- X. Gao, P. Cai, Z. Wang, X. Lv and S. Kawi, Top. Catal., 2023, 66, 299–325 CrossRef CAS.
- T. H. Nguyen, H. B. Kim and E. D. Park, Catalysts, 2022, 12, 212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02355b |
| This journal is © The Royal Society of Chemistry 2024 |