
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe determination of 11 sulfonamide antibiotics in water and foods by developing a N-rich magnetic covalent organic framework combined with ultra-high performance liquid chromatography-tandem mass spectrometry†
Ling Ma‡
*ab,
Yue Gu‡ab,
Liqiang Guo*c and
Ke Wang *ab
*ab
aShijiazhuang Center for Disease Control and Prevention, Shijiazhuang 050011, China. E-mail: mamalin001@163.com; wkecdc@163.com
bShijiazhuang Technology Innovation Center for Chemical Poison Detection and Risk Early Warning, Shijiazhuang 050011, China
cWeifang Customs, Weifang 261041, China. E-mail: glq1980@sina.com
First published on 8th July 2024
Abstract
The concentration of antibiotic residues in water and animal-derived foods is low and the matrix is complex, and effective extraction of antibiotic residues in them is a key factor for accurate quantification. It is important to establish a rapid and effective method for the analytical determination of antibiotics in water and foods. In this study, a type of novel magnetic COF (Fe3O4@SiO2@PDE-TAPB-COF) was synthesized and characterized. Moreover, Fe3O4@SiO2@PDE-TAPB-COF combined with ultra-high performance liquid chromatography-tandem mass spectrometry was used to determine the 11 sulfonamide antibiotics (SAs) in water and food. The parameters including pH, adsorption amount, adsorption time, type of elution solvent and elution time were optimized. Under the optimal conditions, the standard curves of 11 SAs showed good linearity (R2 > 0.999) in their respective concentration ranges and had lower detection and quantification limits. The spiked recoveries of the developed MSPE-UPLC-MS/MS method for the 11 SAs in water and foods were 74.3–107.2% and 75.1–102.5%, respectively. And the relative standard deviations (RSDs) were less than 9.56% (n = 7). The results indicated that the method can be used for the determination of SAs in foods and water with low detection limits and high sensitivity.
1. Introduction
In recent years, with the improvement of living standards, people's requirements for food hygiene are becoming higher and higher. Animal food has become the main source of human diet because of its rich protein.1 Therefore, the consumption of veterinary drugs is constantly increasing worldwide. Antibiotics are compounds with antibacterial activity fermented by bacteria and fungi or synthesized by artificial. They are mainly used for disease prevention and treatment and have a great effect in clinical medicine, animal husbandry and aquatic breeding. According to statistics, about 210 million kilograms of antibiotics are used every year in China. Among them, the use of antibiotics in animal husbandry accounted for about 46.1%.2 Antibiotics can be divided into sulfonamides, quinolones, β-lactam, and aminoglycosides based on different structures.3 Among them, sulfonamide antibiotics (SAs) are the longer used class of synthetic antibiotics,4 and their inhibition mechanism is to inhibit bacterial growth by disrupting the metabolism of folic acid in bacteria, which in turn prevents the production of nucleic acid.5 The main effect of SAs is to inhibit the reproduction of bacteria, which is very effective on most Gram-positive bacteria and some Gram-negative bacteria.6,7 SAs are widely used in clinical and livestock breeding fields, but antibiotic contamination is becoming more and more serious due to people's misuse of antibiotics, lack of strict enforcement of rest period regulations and illegal addition of antibiotics to feed. Only a small amount of SAs can be absorbed by humans and animals, and most of them are excreted from the body through feces. If these excreta are fermented and applied to the soil, the residual antibiotics not only affect the microbial species community structure, but also induce microbial resistance and inhibit or enhance the metabolic pathways of microorganisms, etc. When sulfonamide antibiotics enter the soil environment, they will directly affect the longitudinal migration ability of pollutants because of the leaching behavior, which will cause the risk of groundwater pollution.8 Moreover, antibiotics that are not metabolized in animals may also migrate to foodstuffs from these animals, such as meat, eggs, and milk, resulting in the presence of antibiotics residues in these foods.9 Antibiotic residues in foods and water can pose a potential threat to human health. For example, the accumulation of SAs in humans can cause severe allergic reactions, leukocyte generation inhibition, and urinary system damage, etc.10Many countries and regions have established maximum residue limit (MRL) standards for SAs in foods as a way to protect and human health. For example, the United States has established MRL of 100 μg kg−1 for sulfachlorpyridazine and 10–100 μg kg−1 for sulfadimethoxine, and the MRL of SAs in all food-producing species are authorized as 100 μg kg−1 by the European Union.11 The Chinese national food safety standard GB 31650–2019 set the MRL at 100 μg kg−1 for SAs in foods (muscle, liver, milk), but requires that the use of SAs is prohibited during the egg-laying period of animals. Nevertheless, antibiotic residues are not currently included in the environmental quality and pollutant discharge standards, and relevant testing standards have not been established. Given the low limits of SAs in foods and the fact that their contamination of the water environment remains outside the scope of regulation, it is very important to develop the detection methods of SAs residues in water and food samples for food safety monitoring and risk assessment.
At present, the detection methods of SAs mainly include enzyme-linked immunosorbent assays (ELISAs),12 liquid chromatography (LC),13 and liquid chromatography-tandem mass spectrometry (LC-MS/MS).14 Among them, LC-MS/MS is favored by researchers because of its high separation efficiency, short analysis time, high selectivity and low detection limit. Due to the complex matrix composition, the high number of interfering substances, and the low concentration of target compounds in water and food samples, the detection is susceptible to interference from other components, requiring sample pretreatment before determination. The main purpose of sample pretreatment is to separate the target compounds from the matrix, reduce the influence of matrix effects on the results, and increase the concentration of the target compounds in the solution to be measured, thus enhancing the sensitivity of the method, reducing the detection limit, and reducing the damage to the instrument. Compared with pretreatment techniques such as liquid–liquid extraction (LLE),15 solid-phase extraction (SPE),16 and solid-phase microextraction (SPME),17 the magnetic solid-phase extraction (MSPE)18 method, which is simpler and faster to operate, has shown greater promise for application. In the MSPE process, the magnetic adsorbents are dispersed in the solution containing the target analyte by vortexing or shaking. Then the magnetic sorbents with the target analyte adsorbed are separated from the solution by an external magnetic field, and the target analyte is desorbed by a suitable elution solvent to finally achieve the purification and enrichment of the target analyte. The progression of MSPE technology relies on the design and development of solid-phase adsorbent materials. The common solid-phase adsorbent materials currently available are carbon nanotubes (CNTs),19 graphene,20 metal organic framework materials (MOFs),4 covalent organic frameworks (COFs)21 and POP.22,23 COFs has unique crystalline and porous properties, and it has received wide attention from researchers by enabling the precise integration of organic monomers into the crystalline structure through powerful covalent bonds, enabling its high adsorption capacity, large surface area and designability.24 The COFs composed of light elements (S, N, O, C) not only has high crystallinity, high porosity, tunability, but also can show highly stable structure in harsh chemical environments (including boiling water, strong acid and strong alkali, etc.) Because COFs contains the advantages of channel order, tunable structure, easy functionalization, and thermochemical stability, it is widely used in sample separation and enrichment, fluorescence sensing, catalysis, and so on.25,26 Recently, COFs including TPB-DMTP-COFs27 and HP-COF(TpBD)28 have been reported in various extraction methods as the adsorbents. However, its low density also makes it difficult to separate from the matrix, limiting its further application in the field of separation and enrichment. However, the COFs is of light quality, in addition to the time-consuming tedious centrifugal separation, the water dispersion is very poor, greatly reducing the performance of COFs material and limiting its further application in the field of separation and enrichment. The combination of adsorbent and magnetic nanoparticles not only has the inherent advantages of COFs, but also has strong magnetism and recyclability, which solves the difficulty of COFs recovery and becomes an excellent adsorbent in the field of sample pretreatment.29 However, it is still a great challenge with MCOFs as an adsorbent for high-throughout extraction. In the field of antibiotic detection, developing different magnetic COFs materials for high-throughout adsorption is a current research hotspot. Therefore, regardless of the pretreatment technique used to enrich the analyte, the key factor in sample pretreatment is always the adsorbent performance.
SAs contain multiple amine groups and sulfonic acid groups, which can form intermolecular forces with the COF material introduced into the N functional group recognition site. Based on the structure of SAs, a type of magnetic COF (Fe3O4@SiO2@PDE-TAPB-COF) with exposed N-containing groups acting as adsorption sites was designed and synthesized. 1,3,5-Tris(4-aminophenyl)benzene and 2,6-pyridinedicarboxaldehyde were employed to prepare a pyridine-based PDE-TAPB-COF via Schiff base condensation reactions. Fe3O4@SiO2@PDE-TAPB-COF is synthesized with Fe3O4@SiO2 as nucleus and pyridine-based PDE-TAPB-COF as shell. Pyridine-based PDE-TAPB-COF with N-containing heterocyclic ring also can provide specific multiple interaction including hydrophobic, p–π, π–π and hydrogen bonding interactions with SAs. Besides, the pyridine-based material was chosen as the shell due to its high stability, distinguished macrocyclic structure and large p-conjugated system, which can bring in Fe3O4@SiO2@PDE-TAPB-COF with higher porosity, surface area and stability. In addition, pyridine-based COF with N-containing heterocyclic ring also can provide specific multiple interaction including hydrophobic, p–p interaction, electrostatic and hydrogen bonding interactions with SAs. The pyridine structural unit of Fe3O4@SiO2@PDE-TAPB-COF is not only used as an active site but also as a connector for building PDE-TAPB-COF, which effectively avoids the damage of side chains introduced due to functional modification to the pore structure and shielding of active centers.
Fe3O4@SiO2@PDE-TAPB-COF show high adsorption capacity for SAs due to the N-rich pore space. Fe3O4@SiO2@PDE-TAPB-COF combined with UPLC-MS/MS to establish a rapid method for the determination of 11 SAs residues in water and food samples, and apply it to the analytical detection of actual samples.
2. Experimental
2.1 Reagents and materials
All reagents were analytical or superior pure while solvents were of HPLC grade. Sulfamethoxypyridazine, sulfameter, sulfamonomethoxine, sulfachlorpyridazine, sulfadoxine, sulfamethoxazole, sulfisoxazole, sulfabenzamide, sulfadimethoxine, sulfanitran and sulfaquinoxaline were purchased from Dr Ehrenstorfer GmbH. The purity of all the standards was >97.0%. The basic properties of sulfonamide antibiotics are listed in Table S1.† Acetonitrile (Merck) and methanol (Thermo Fisher) are all HPLC grade. Ferric chloride hexahydrate (FeCl3·6H2O), ethylene glycol, sodium acetate trihydrate (CH3COONa·3H2O), ethanol absolute, acetic acid glacial (HAc) and sodium chloride (NaCl) 1,4-dioxane and tetraethoxysilane were analytical grade and purchased from Tianjin Yongda Chemical Reagent Co., Ltd hexanes and formic acid were purchased from Dikma. Polyethylene glycol (PEG-4000) was purchased from Aladdin. 2,6-Pyridinedicarboxaldehyde and 1,3,5-tris(4-aminophenyl)benzene were obtained from Aladdin Biochemicals.Separate standard solutions of 11 SAs were prepared in ACN with a concentration of 1.0 mg mL−1. Then the concentration of each of the above target compounds was diluted 10-fold with ACN. The solvent calibration standard solutions of different concentrations of 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 50 and 100 ng mL−1 were prepared and diluted with compound solvents. Matrix-matched calibration standard solutions were prepared with blank samples containing different analyte contents. All standard solutions were stored at −20 °C.
2.2 Instrumental and analytical conditions
The determination of SAs residues was performed on a UPLC-MS/MS instrument (Exion-TRILPLE QUAD 5500, AB SCIEX, USA), an Agilent ZORBAX XDB-C18 column was used for separation of analytes. The injection volume and flow rate were 3 mL and 0.3 mL min−1, individually. The mobile phase is divided into two components, 0.1% formic acid solution (mobile phase A) and acetonitrile (mobile phase B). A linear-gradient elution program was set as follows: (1) 0–3.5 min, 26% B (v/v); (2) 3.5–3.6 min 26–30% B (v/v); (3) 3.6–5.8 min, 30% B (v/v); (4) 5.8–5.9 min, 30–90% B (v/v); (5) 5.9–7.9 min, 90% B (v/v); (6) 7.9–8.0 min, 90–26% B (v/v) and (7) 8.0–11.0 min, 26% B (v/v). In positive mode, the electrospray ionization (ESI) source was used for ionization analysis and the MRM mode for mass spectrometry. The optimized MRM data acquisitions was listed in Table S2.†A Fourier infrared transform spectra (FT-IR) (Thermo Nicolet Co. Ltd., USA), a D8A X-ray diffractometer (XRD) (Bruker, Germany), Brunauer–Emmett–Teller (BET) (Quantachrome Instruments, USA), scanning electron microscopy (SEM) of an Regulus 8100 instrument (Hitachi, Japan), a SQUID XL-7 vibrating sample magnetometer (Quantum Design, USA), and transmission electron microscope (TEM) of FEI Tecnai G2 F20 (FEI, USA) were used for the characterization of the prepared materials.
2.3 Synthesis of magnetic adsorbents
Highly dispersed spherical magnetic nanoparticles of Fe3O4 were prepared according to the solvothermal method with minor modification.30 Briefly, 60 mL of ethylene glycol and 1.62 g of FeCl3·6H2O were added to a 100 mL conical flask to form a clear yellow solution under vigorous magnetic stirring at room temperature. Then 2.0 g of PEG-4000 and 7.2 g of CH3COONa·3H2O were added to the solution sequentially with stirring for about 30 min until the solution turned brownish-yellow to prevent particle aggregation. Subsequently, the resulting solution was transferred to an autoclave and calcined for 8 hours at 200 °C. Finally, the products were repeatedly washed three times with absolute ethanol and pure water and dried in a vacuum oven for 4 h at 60 °C.The obtained Fe3O4 magnetic nanoparticles were further coated with a SiO2 layer through the sol–gel method with some modifications.31 Briefly, Fe3O4 NPs (100 mg) were added to a mixture of 80 mL of ethanol and 20 mL of ultrapure water and dispersed by sonication. Subsequently, aqueous ammonia solution (1.5 mL, 25 wt%) and TEOS (1.0 mL) were added and stirred at 30 °C for 24 h. The resulting products were washed several times with absolute ethanol and ultrapure water, and vacuum dried under 60 °C for 4 h.
The synthesis method of rich magnetic COFs was according to a published procedure with some changes.26 Fe3O4@SiO2 (150 mg) was added to 1,4-dioxane (30 mL) and ultrasonic dispersed for 5 min. Then, 2,6-pyridine dicarboxaldehyde (101 mg, 0.75 mmol) and 1,3,5-tris(4-aminobenzene)benzene were added to the solvent, stirring for 5 min at room temperature, acetic acid 0.5 mL (0.5 mL, 12 mol L−1) drops to the solution, stirring at room temperature for 2 h after 4.5 mL acetic acid, 70 °C in the reaction for 24 h. The product was washed three times alternately with 1,4-dioxane and ethanol, designated Fe3O4@SiO2@PDE-TAPB-COF.
2.4 Procedure of MSPE
The MSPE program is shown in Fig. 1. 10 mg Fe3O4@SiO2@PDE-TAPB-COF was added into the 10 mL sample solution. Subsequently, the solution was vortexed for 20 min to accelerate the adsorption of the adsorbent to the sulfonamide antibiotics. Afterwards, the sorbents were collected with an external magnet and the supernatant was discarded. The sulfonamide antibiotics were desorbed from the sorbents with 4.0 mL of acetonitrile under vigorous vortex for 5 min, and the eluate was collected by magnetic separation. Twice such replicate elutions were required for desorption of the sulfonamide antibiotics from the sorbents. The collected eluate (8.0 mL acetonitrile) was vortexed and mixed, and then 4.0 mL of the eluate was dried at 40 °C under N2. Finally, under the initial gradient, the residues were redissolved through a 1.0 mL mobile phase and vortexed for 1 min and then filtered through a 0.20 μm membrane. The redissolved sample was injected into the UPLC-MS/MS system for analysis. In addition, Fe3O4@SiO2@PDE-TAPB-COF was recycled by washing two times with 8.0 mL of acetonitrile under vigorously shaking for 5 min.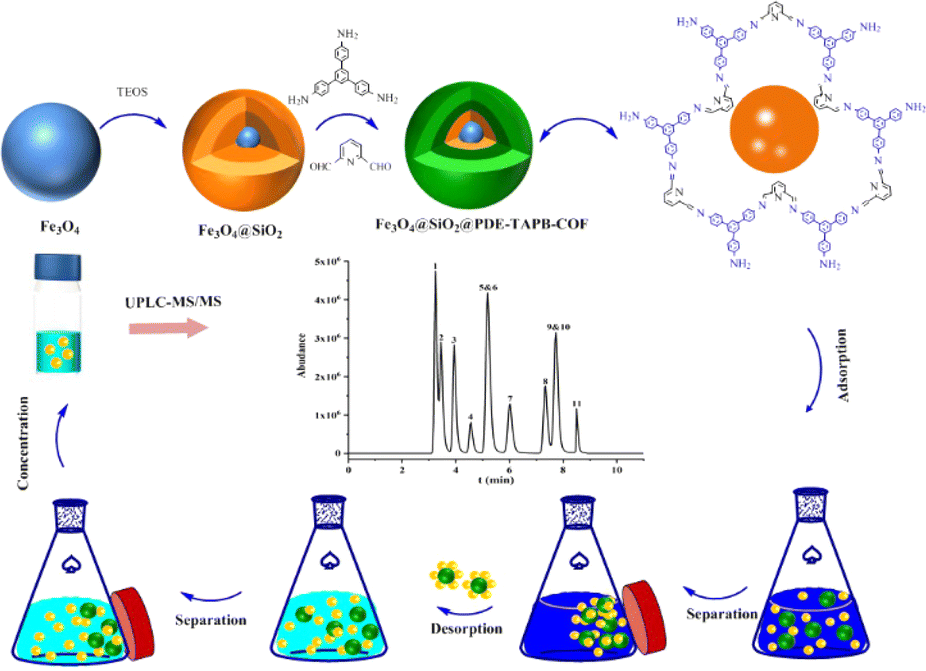 | ||
| Fig. 1 Illustration of the preparation and application of Fe3O4@SiO2@PDE-TAPB-COF. | ||
2.5 Sample preparation
Water samples were collected in the relevant rivers and filtered through 0.45 μm nylon filter membranes. Afterwards, 2 g of NaCl was added to the water sample and was evenly mixed by vortexing and adjusted to pH 4.0 using formic acid, stored in a refrigerator at 4 °C, and then subjected to the MSPE process. Egg samples were prepared by stirring whole eggs (without any added solvent) at 800 rpm for 5 min. The cake samples were crushed and sealed in food bags. For SAs extraction, the water sample (2.00 g) was added to 10 mL of acetonitrile solution, vortexed and sonicated for 10 min. However, for cake samples, 2 mL of water should be added, mixed and vortex, 10 mL of acetonitrile should be added, sonicated for 10 min, and 1.5 g of sodium chloride should be added, vortex and mixed evenly. After centrifugation for 10 min, the supernatant was removed and degraded with 4 mL of hexane three times. Then, the extract (5 mL) was dried under 40 °C of nitrogen blowing. After adding 10 mL ultrapure water, the mixture was vortexed and sonicated. Afterwards, 2 g NaCl was added, vortexed and mixed, and the pH of the sample solution was adjusted to 4.0 using formic acid for MSPE.3. Result and discussion
3.1 Characterization of the magnetic materials
 | ||
| Fig. 2 The SEM of (A) Fe3O4, (B) Fe3O4@SiO2@PDE-TAPB-COF; the TEM of (C) Fe3O4, (D) Fe3O4@SiO2@PDE-TAPB-COF. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration.26 Thus, it is concluded that PDE-TAPB-COF was grafted successfully on the Fe3O4@SiO2 surface.
N stretching vibration.26 Thus, it is concluded that PDE-TAPB-COF was grafted successfully on the Fe3O4@SiO2 surface.
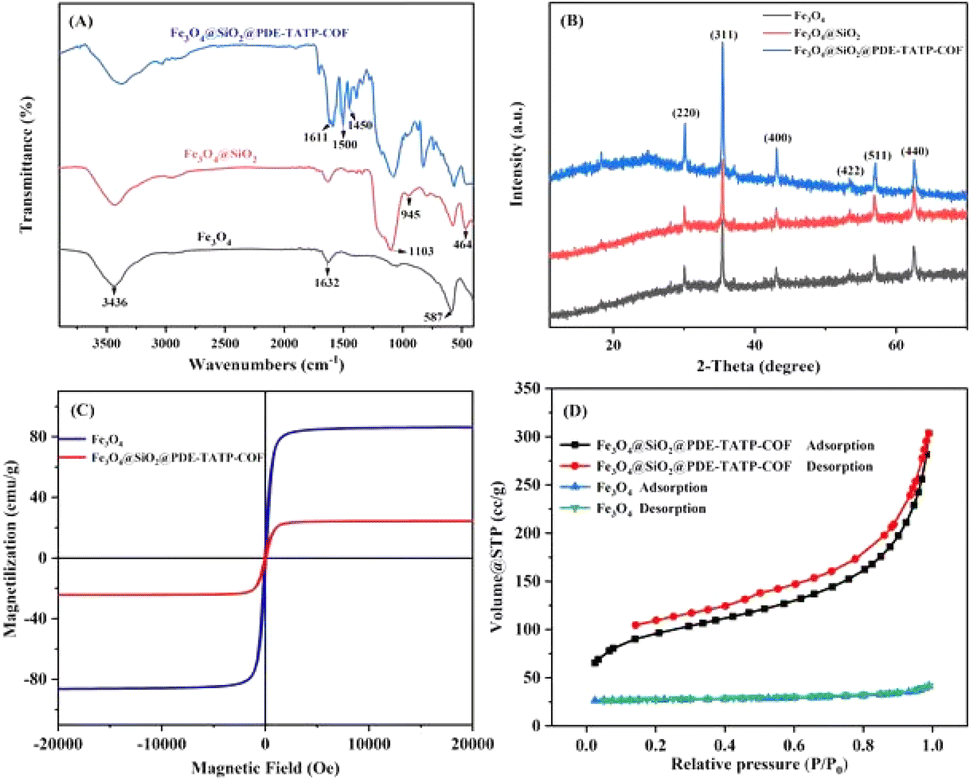 | ||
| Fig. 3 (A) FT-IR spectra of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PDE-TAPB-COF. (B) XRD analysis of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@PDE-TAPB-COF. (C) VSM magnetization curves of Fe3O4 and Fe3O4@SiO2@PDE-TAPB-COF. (D) N2 adsorption–desorption isotherms of Fe3O4, and Fe3O4@SiO2@PDE-TAPB-COF. | ||
3.2 Condition of MSPE
To obtain better extraction efficiency of SAs, the parameters that could affect the MSPE, such as the amount of Fe3O4@SiO2@PDE-TAPB-COF, sample solution pH, ionic concentration, type and dosage of the eluate, extraction time, and desorption time were detailedly optimized. These factors were studied with 10 mL of an aqueous solution containing 20.00 ng mL−1 of SAs.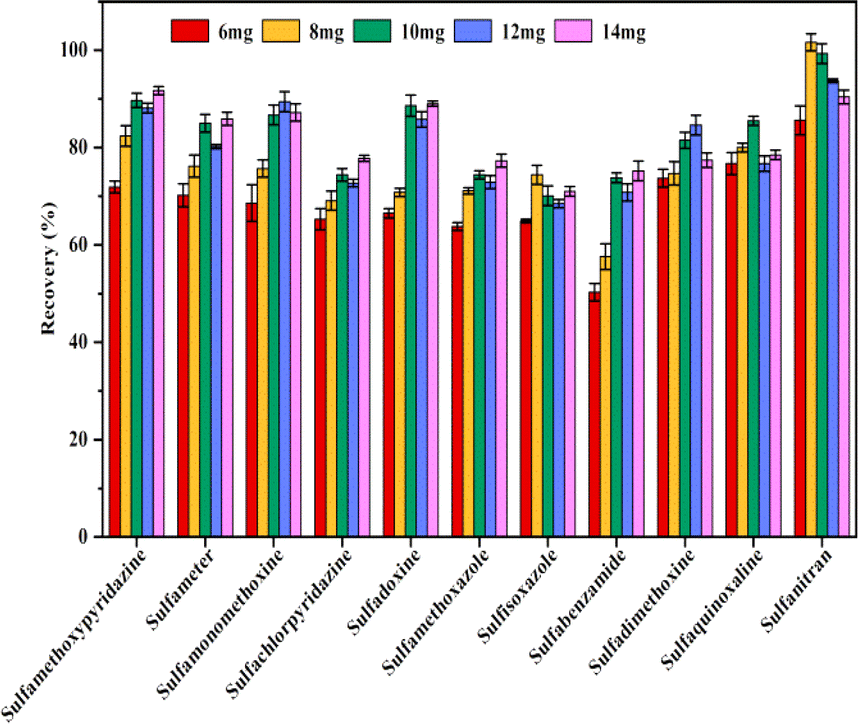 | ||
| Fig. 4 Effect of the amount of adsorbent. | ||
3.3 Adsorption mechanism
The MSPE of SAs on Fe3O4@SiO2 and Fe3O4@SiO2@PDE-TAPB-COF were compared, and evaluated the adsorption mechanisms of Fe3O4@SiO2@PDE-TAPB-COF for SAs (Fig. 5). Fe3O4@SiO2 gave poor extraction for SAs, suggesting that PDE-TAPB-COF played a key role in extraction process. According to the structure of SAs, the target material has a rich π-electron aromatic ring, at least three electron absorbing groups (sulfonic acid group, amino group) and high hydrophobicity. Therefore, there may be various adsorption mechanisms such as intermolecular hydrogen bond (N–S or O–N), π–π filling interaction and hydrophobic interaction in the adsorption relationship between Fe3O4@SiO2@PDE-TAPB-COF and SAs.39–42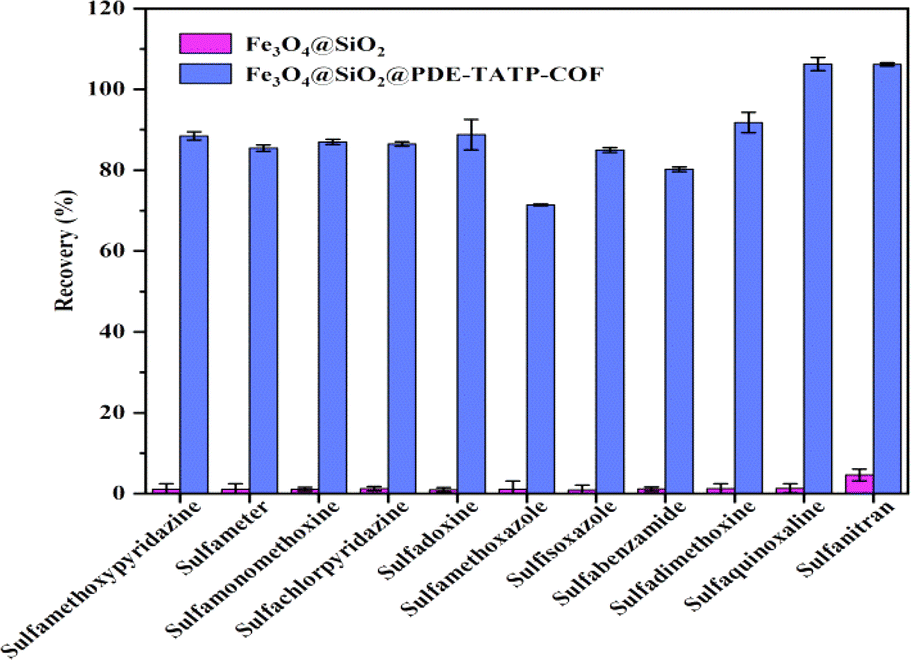 | ||
| Fig. 5 Comparison of the extraction efficiency among Fe3O4@SiO2, and Fe3O4@SiO2@PDE-TAPB-COF. | ||
Firstly, 2,6-pyridinedicarboxaldehyde and 1,3,5-tris(4-aminophenyl)benzene monomer form a large π–electron system through Schiff base condensation, which forms π–π stacking with SAs. Secondly, the pyridine N-atom in the PDE-TAPB-COF provides a site for binding of SAs, forming intermolecular hydrogen bonding force with SAs. Thirdly, Schiff base formed by condensation of 2,6-pyridinedicarboxaldehyde and 1,3,5-tris(4-aminophenyl)benzene can form p–π conjugate with SAs. Moreover, Fe3O4@SiO2@PDE-TAPB-COF had a high specific surface area and porosity, providing a large number of binding sites, sufficient contact space and transmission channels for the SAs. And compared with other porous adsorbents, Fe3O4@SiO2@PDE-TAPB-COF presented faster adsorption kinetics. To investigate the adsorption selectivity of Fe3O4@SiO2@PDE-TAPB-COF for analytes, it was used for MSPE of three types of analytes including sulfonamides (SAs), nitroimidazoles (NMZs) and quinolones (QNs). As shown in Table S6,† the Fe3O4@SiO2@PDE-TAPB-COF showed the highest extraction recoveries for SAs (78.8–108.7%), while the lowest recoveries for NMZs (0.4–9.5%). By exploring the main adsorption mechanism, it is speculated that π–π conjugation, intermolecular hydrogen bonding and pore size effect are the result of the interaction.
3.4 Reusability of the adsorbent
Evaluation of the reusability is significant when evaluating the extraction performance of a MSPE adsorbent. From the perspective of application, the cycle stability, regeneration ability and versatility of materials should be considered. The ability of the material to show a high level of reusability in multiple cycles without loss of initial performance is a necessary indicator to measure its potential.39 The reusability of the adsorbent which directly affected the experimental cost was evaluated by carrying out several successive adsorption–desorption tests. In order to eliminate the SAs residue from the previous cycles, Fe3O4@SiO2@PDE-TAPB-COF was washed twice with 4 mL of acetonitrile and then vacuum dried at 60 °C for 4 h before the next use. After multiple cycles, the extraction effect comparison data of Fe3O4@SiO2@PDE-TAPB-COF was shown in Fig. 6. It can be seen that at the spiked concentration of 4 μg L−1, the extraction efficiency of magnetic COFs for 11 SAs decreased after five repetitions, but still reached over 70%. The results demonstrated that Fe3O4@SiO2@PDE-TAPB-COF could be reused at least five times without the significant loss of extraction ability for SAs.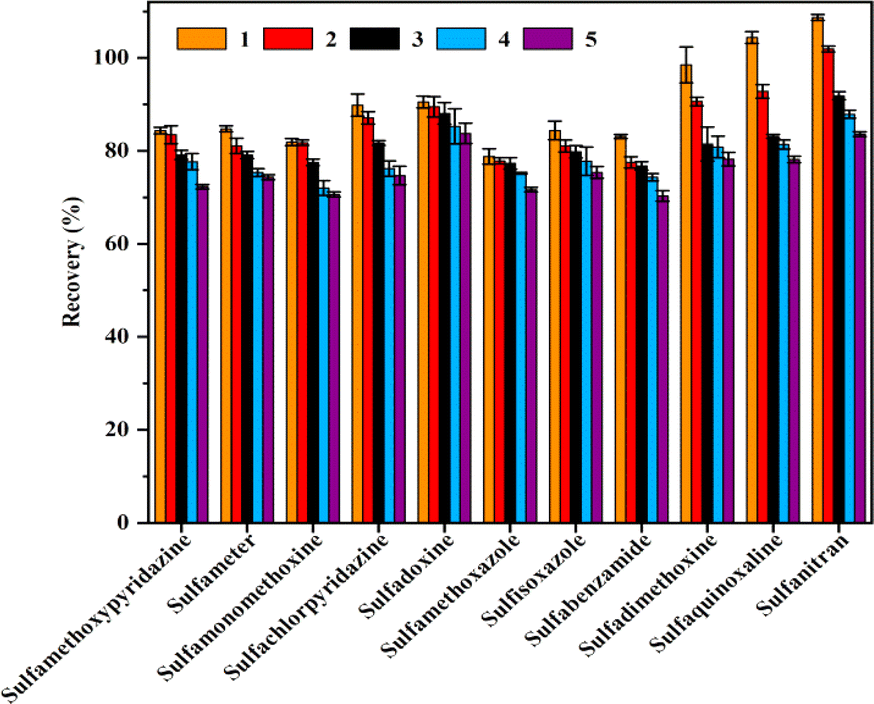 | ||
| Fig. 6 Reusability of Fe3O4@SiO2@PDE-TAPB-COF on the extraction of SAs. | ||
3.5 Matrix effect
Matrix effect (ME) refers to the effect of a component of the sample other than the target on the measured value of the analyte to be measured. Due to the different effects of the matrix on the detection signal response value, the matrix effect may have a matrix enhancement or inhibitory effect on the sample detection signal. The ME can be calculated using the following formula: ME (%) = AX/AS × 100% (AX is the slope of the matrix-matched standard curve; AS is the slope of the solvent standard curve).8 If the ME value was close to 80–120%, the matrix effect can be ignored. The experimental ME results of the 11 SAs in the water and egg samples are shown in Table S3.† The results showed that the ME values of 11 SAs in water and egg samples were 91.36–112.35% and 81.18–115.57%, respectively, which made ignorable matrix effect. Therefore, we chose an external standard method using solvent standard solution for quantitative analysis in this study.3.6 Method validation
To evaluate the performance of the proposed Fe3O4@SiO2@PDE-TAPB-COF-based MSPE, the methodological investigation was performed under the above optimized conditions in combination with UPLC-MS/MS. Standard curves were generated by plotting the areas under the curves of the SAs against their concentrations (0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 50, and 100 ng mL−1). The correlation coefficients (R2) were calculated by linear regression on the standard curves. The limit of detection (LOD) can be calculated by 3 times the standard deviation of the lowest concentration level response/slope of the calibration curve. The limit of quantitation (LOD) can be calculated by 10 times the standard deviation of the lowest concentration level response/slope of the calibration curve.8 As shown in Table 1, all the 11 SAs have a good linear relationship, and all R2 is higher than 0.999. The LODs and LOQs in waters were in the ranges of 0.001–0.078 μg L−1 and 0.003–0.261 μg L−1, while the LODs and LOQs in eggs were 0.01–0.73 μg kg−1 and 0.02–2.44 μg kg−1, respectively.| Compounds | Linearity equation | Linear range (ng mL−1) | R2 | Surface waters | Eggs | ||||
|---|---|---|---|---|---|---|---|---|---|
| LOD (μg L−1) | LOQ (μg L−1) | LOD (μg kg−1) | LOQ (μg kg−1) | ||||||
| Sulfamethoxypyridazine | Y = 299![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 070.0X + 11815.5 070.0X + 11815.5 |
0.1–100 | 0.9999 | 0.002 | 0.007 | 0.01 | 0.04 | ||
| Sulfameter | Y = 317717.0X + 12664.9 |
0.1–100 | 0.9999 | 0.001 | 0.003 | 0.01 | 0.03 | ||
| Sulfamonomethoxine | Y = 283116.0X + 1788.7 |
0.2–100 | 0.9997 | 0.008 | 0.028 | 0.01 | 0.03 | ||
| Sulfachlorpyridazine | Y = 58399.3X + 16467.8 |
0.5–100 | 0.9999 | 0.024 | 0.080 | 0.09 | 0.30 | ||
| Sulfadoxine | Y = 288612.0X + 2709.1 |
0.2–100 | 0.9998 | 0.007 | 0.022 | 0.01 | 0.02 | ||
| Sulfamethoxazole | Y = 92436.4X + 2605.3 |
0.5–100 | 0.9999 | 0.014 | 0.047 | 0.02 | 0.07 | ||
| Sulfisoxazole | Y = 115973.0X + 33.3 |
0.5–100 | 0.9995 | 0.013 | 0.044 | 0.02 | 0.06 | ||
| Sulfabenzamide | Y = 122279.0X + 1214.8 |
0.2–100 | 0.9998 | 0.011 | 0.036 | 0.02 | 0.05 | ||
| Sulfadimethoxine | Y = 222159.0X + 41245.7 |
0.2–100 | 0.9999 | 0.009 | 0.031 | 0.01 | 0.04 | ||
| Sulfaquinoxaline | Y = 76067.6X + 8276.9 |
0.5–100 | 0.9999 | 0.015 | 0.049 | 0.05 | 0.17 | ||
| Sulfanitran | Y = 5893.0X + 2232.8 | 2.0–100 | 0.9998 | 0.078 | 0.261 | 0.73 | 2.44 | ||
To assess the accuracy of the recoveries, 11 SAs at three different concentrations were added separately to blank water and egg samples, and seven parallel samples were run at each concentration. The recoveries of the 11 SAs in water and egg samples are shown in Tables S4 and S5.† The average recovery of 11 SAs was 74.3% to 107.2% in waters at 2.0, 4.0, and 20.0 μg L−1, respectively; the relative standard deviation (RSDs) was low, 1.55% to 8.17%. And the average recovery of 11 SAs was 75.1% to 102.5% in eggs at 10.0, 20.0 and 100.0 μg L−1, respectively; the relative standard deviation (RSDs) was low, 2.18% to 9.56% which all less than 10%. The method had good precision and accuracy and was suitable for the detection of residues of 11 SAs in waters and foods.
3.7 Method applications in real samples
This study was applied to the detection of 11 sulfonamide antibiotic residues in 15 water collected from relevant rivers in Shijiazhuang and 15 eggs and cakes purchased in local markets and supermarkets. In two waters and one cake samples, sulfamonomethoxine was detected at 1.34 μg L−1, 1.37 μg L−1 and 2.40 μg kg−1, respectively. And sulfachlorpyridazine was observed in two water at 27.39 μg L−1 and 2.25 μg L−1. Moreover, SAs in other real samples were all lower than the LOQs.3.8 Comparison with other methods
According to the reported results, different sample pretreatment techniques combined with chromatograph-MS detection samples were analyzed and compared, and the results are shown in Table 2 compared with PLE and DLLME, this method consumed less organic solvent and had lower detection limits. In addition, this method had shorter extraction time compared with HFRLM, SPE and SPME, and the sorbents could be directly separated from the sample solution under the action of an applied magnetic field, which made the operation process simpler and did not require other equipments. Moreover, the method can detect 11 sulfonamide antibiotics simultaneously in water and food samples, while the maximum number of SAs detected by other methods was 10 and the sample type was relatively homogeneous. Therefore, an effective method for measuring SAs in water and food samples based on the combination of Fe3O4@SiO2@PDE-TAPB-COF with UPLC-MS/MS.| Method | Materials | Extraction time (min) | Matrix (numbers of SAs) | Linearity | Recovery (%) | LOD | References |
|---|---|---|---|---|---|---|---|
| a HFRLM: hollow fiber renewal liquid membrane.b PLE: pressurized liquid extraction.c SPME: solid-phase microextraction.d DLLME: dispersive liquid–liquid microextraction. | |||||||
| HFRLM-LC-MS/MSa | — | 40 | Honey samples (5) | 16–500 μg kg−1 | 80.9–103.1 | 5.1–27.4 μg kg−1 | 40 |
| PLE-SPE-LC-MS/MSb | — | 16 | Biosolids (4) | 31.6–500 μg L−1 | 90.4–99.9 | 0.6–4.2 ng g−1 | 41 |
| SPE-HPLC-MS | HP-TpBD | 60 | Meat (5) | 0.5–200 μg kg−1 | 85.0–119.9 | 0.10–0.23 μg kg−1 | 28 |
| SPME-UPLC-MS/MSc | PBAPM | 200 | Tap, lake and river water (10) | 0.05–200 μg L−1 | 82.0–105.4 | 0.54–4.5 ng L−1 | 17 |
| DLLME-UPLC-MS/MSd | — | 1 | Powdered milk-based infant formulas (7) | 5–500 μg kg−1 | 82.6–100.1 | 0.5–1.5 μg kg−1 | 42 |
| MSPE-UPLC-MS/MS | Fe3O4@SiO2@PDE-TAPB-COF | 20 | Surface water and egg samples (11) | 0.1–100 ng mL−1 | 74.3–107.2 | 0.003–0.261 μg L−1 | This work |
| 0.01–0.73 μg kg−1 | |||||||
4. Conclusion
Sulfonamide antibiotics are often added to animal feeds, and therefore SAs may be present in animal-derived foods and environmental media as residues, albeit at low levels. In this study, a novel adsorbent Fe3O4@SiO2@PDE-TAPB-COF was prepared, which had remarkable features such as large specific surface area and strong magnetic properties, and had excellent extraction effect on sulfonamide antibiotics. Then, an MSPE-UPLC-MS/MS method was developed for the simultaneous determination of 11 SAs in water and food samples with simple operation, low detection limit and high accuracy. The method provides a basis for the detection of sulfonamide antibiotics in other similar or even simpler samples and further improves the monitoring system for sulfonamide antibiotics.Data availability
All relevant data are within the paper. All data supporting the findings of this study are available within the paper and its ESI.†Author contributions
Ling Ma: conceptualization, formal analysis, methodology, writing – original draft, funding acquisition. Yue Gu: data curation, formal analysis, methodology. Liqiang Guo: methodology, supervision, conceptualization. Ke Wang: conceptualization, funding acquisition, project administration, resources, writing-review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81903322) and S&T Program of Hebei (223777116D).References
- W. Duan, H. Cui, X. Jia and X. Huang, Sci. Total Environ., 2022, 820, 153178 CrossRef CAS PubMed.
- W. Cheng, J. Li, Y. Wu, L. Xu, C. Su, Y. Qian, Y.-G. Zhu and H. Chen, J. Hazard. Mater., 2016, 304, 18–25 CrossRef CAS PubMed.
- S. Quaik, A. Embrandiri, B. Ravindran, K. Hossain, N. A. Al-Dhabi, M. V. Arasu, S. Ignacimuthu and N. Ismail, J. King Saud Univ., Sci., 2020, 32, 1300–1305 CrossRef.
- L. Han, P. Qin, M. Li, D. Li, M. Mu, Y. Gao, S. Zhu, M. Lu and Z. Cai, Chem. Eng. J., 2023, 456, 140969 CrossRef CAS.
- G. Feng, W. Zou and Y. Zhong, J. Hazard. Mater. Adv., 2022, 5, 100045 CrossRef CAS.
- M. Zhu, X. Pang, J. Wan, X. Xu, X. Wei, R. Hua, X. Zhang, Y. Wang and X. Yang, Ecotoxicol. Environ. Saf., 2022, 243, 113979 CrossRef CAS PubMed.
- Y. Wang, W.-B. Jiao, J.-T. Wang, G.-F. Liu, H.-L. Cao and J. Lü, Bioresour. Technol., 2019, 277, 128–135 CrossRef CAS PubMed.
- S. Li, C. Zhang, H.-X. Tang, Y. Gu, A.-J. Guo, K. Wang and K.-Q. Lian, J. Food Drug Anal., 2023, 31, 73–84 CrossRef PubMed.
- L. M. Chiesa, L. DeCastelli, M. Nobile, F. Martucci, G. Mosconi, M. Fontana, M. Castrica, F. Arioli and S. Panseri, Lwt, 2020, 131 Search PubMed.
- Y. Dai, N. Wu, L.-e. Liu, F. Yu, Y. Wu and N. Jian, Food Chem., 2023, 404 CAS.
- J. Ma, S. Fan, L. Sun, L. He, Y. Zhang and Q. Li, Food Sci. Hum. Wellness, 2020, 9, 363–369 CrossRef.
- W. Jiang, Z. Wang, R. C. Beier, H. Jiang, Y. Wu and J. Shen, Anal. Chem., 2013, 85, 1995–1999 CrossRef CAS.
- T. Yao and K. Du, Food Chem., 2020, 331 Search PubMed.
- C. Zhang, S. Li, J. Wu, T. Ping, L. Ma, K. Wang and K. Lian, Food Chem., 2023, 403, 134442 CrossRef CAS.
- K. Fikarová, B. Horstkotte, D. Machián, H. Sklenářová and P. Solich, Talanta, 2021, 221, 121427 CrossRef PubMed.
- Y. Ning, Y. Ye, W. Liao, Y. Xu, W. Wang and A.-j. Wang, Food Chem., 2022, 397, 133831 CrossRef CAS PubMed.
- W. Ma, H. Chen, L. Fan, Y. Bai, H. Hou and Q. Hu, Microchem. J., 2024, 198, 110164 CrossRef CAS.
- I. S. Ibarra, J. M. Miranda, J. A. Rodriguez, C. Nebot and A. Cepeda, Food Chem., 2014, 157, 511–517 CrossRef CAS PubMed.
- A. N. M. Nasir, N. Yahaya, N. N. M. Zain, V. Lim, S. Kamaruzaman, B. Saad, N. Nishiyama, N. Yoshida and Y. Hirota, Food Chem., 2019, 276, 458–466 CrossRef CAS PubMed.
- M. Shirani, E. Parandi, H. R. Nodeh, B. Akbari-adergani and F. Shahdadi, Food Chem., 2022, 373 Search PubMed.
- Y.-F. Ma, F. Yuan, X.-H. Zhang, Y.-L. Zhou and X.-X. Zhang, Analyst, 2017, 142, 3212–3218 RSC.
- S. Dutta, W. Mandal, A. V. Desai, S. Fajal, G. K. Dam, S. Mukherjee and S. K. Ghosh, Mol. Syst. Des. Eng., 2023, 8, 1483 RSC.
- W. Mandal, S. Fajal, P. Samanta, S. Dutta, M. M. Shirolkar, Y. D. More and S. K. Ghosh, ACS Appl. Polym. Mater., 2022, 4, 8633 CrossRef CAS.
- T. Wang, H. Xie, Y. Cao, Q. Xu and N. Gan, J. Chromatogr. A, 2022, 1685, 463614 CrossRef CAS PubMed.
- Z. Liu, J. Wang, T. Duan, Y. Guo, W. Liu, X. Yang, Q. Wu and Z. Wang, J. Chromatogr. A, 2022, 1679, 463387 CrossRef CAS PubMed.
- G. Das, T. Prakasam, M. A. Addicoat, S. K. Sharma, F. Ravaux, R. Mathew, M. Baias, R. Jagannathan, M. A. Olson and A. Trabolsi, J. Am. Chem. Soc., 2019, 141, 19078–19087 CrossRef CAS PubMed.
- L. Wen, L. Liu, X. Wang, M.-L. Wang, J.-M. Lin and R.-S. Zhao, J. Chromatogr. A, 2020, 1625, 461275 CrossRef CAS PubMed.
- R. Shen, L. Huang, R. Liu and Q. Shuai, J. Chromatogr. A, 2021, 1655, 462518 CrossRef CAS PubMed.
- J. Zhang, Z. Chen, S. Tang, X. Luo, J. Xi, Z. He, J. Yu and F. Wu, Anal. Chim. Acta, 2019, 1089, 66–77 CrossRef CAS.
- Y. Zhao, R. Wu, H. Yu, J. Li, L. Liu, S. Wang, X. Chen and T. W. D. Chan, J. Chromatogr. A, 2020, 1610, 460543 CrossRef CAS.
- C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H.-J. Gao, Nanoscale, 2011, 3, 701–705 RSC.
- Q.-B. Fu, H.-L. Jiang, L.-Q. Qiao, X. Sun, M.-L. Wang and R.-S. Zhao, J. Chromatogr. A, 2020, 1630, 461534 CrossRef CAS PubMed.
- Y. Yang, J. Miao, Z. Yin, W. Hao, H. Shi, L. Ma, T. Shi and C. Pettinari, Bioinorg. Chem. Appl., 2022, 2022, 1–15 CrossRef PubMed.
- M. Payam, H. Kargar and M. Fallah-Mehrjardi, Inorg. Chem. Commun., 2022, 145, 109951 CrossRef CAS.
- L. Chen, M. Zhang, F. Fu, J. Li and Z. Lin, J. Chromatogr. A, 2018, 1567, 136–146 CrossRef CAS PubMed.
- M. Zhang, J. Li, C. Zhang, Z. Wu, Y. Yang, J. Li, F. Fu and Z. Lin, J. Chromatogr. A, 2020, 1615, 460773 CrossRef CAS.
- L. Qiao, C. Yu, R. Sun, Y. Tao, Y. Li and Y. Yan, J. Chromatogr. A, 2021, 1652, 462372 CrossRef CAS.
- M. Saad Aldoori, M. Merdivan and A. Altınışık Tağaç, Microchem. J., 2023, 184, 108165 CrossRef CAS.
- Y. Gao, C. Zhao, Q. Tan, M. Gao, G. Chen, R. Zhai, X. Huang, X. Xu, G. Liu, J. Wang, Y. Zhang and D. Xu, Microchem. J., 2022, 174, 107103 CrossRef CAS.
- G. C. Bedendo, I. C. S. F. Jardim and E. Carasek, J. Chromatogr. A, 2010, 1217, 6449–6454 CrossRef CAS PubMed.
- A. Pamreddy, M. Hidalgo, J. Havel and V. Salvadó, J. Chromatogr. A, 2013, 1298, 68–75 CrossRef CAS PubMed.
- H. Shaaban, A. Mostafa, A. M. Alqarni, R. Alsultan, Z. A. shehab, Z. Aljarrash, W. Al-Zawad, S. Al-Kahlah and M. Amir, J. Food Compos. Anal., 2023, 117, 105137 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02530j |
| ‡ Equal first author: Ling Ma and Yue Gu contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |