
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo rhenium compounds with benzimidazole ligands: synthesis and DNA interactions†
Nataliia Shtemenko *abc,
Cristina Galiana-Roselloa,
Ariadna Gil-Martíneza,
Salvador Blascoa,
Jorge Gonzalez-Garcíaa,
Helen Velichkoc,
Oleksandr Holichenkoc,
Olexandr Shtemenkoac and
Enrique García-Españaa
*abc,
Cristina Galiana-Roselloa,
Ariadna Gil-Martíneza,
Salvador Blascoa,
Jorge Gonzalez-Garcíaa,
Helen Velichkoc,
Oleksandr Holichenkoc,
Olexandr Shtemenkoac and
Enrique García-Españaa
aDepartamento de Química Inorgánica, Instituto de Ciencia Molecular (ICMOL), Universitat de València, C/Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: n.shtemenko@i.ua
bOles Honchar National University, Haharina Ave, 72, Dnipro, 49000, Ukraine
cUkrainian State University of Chemical Technology, Haharina Ave, 8, Dnipro, 49005, Ukraine
First published on 20th June 2024
Abstract
Two rhenium compounds: cis-tetrachlorotetrabenzimidazoldirhenium(III) chloride – I and tetrabenzimidazoldioxorhenium(V) – II have been synthesized and characterized. X-ray data are presented for the new complex II. I and II show strong emission that has been used to investigate their interaction with several non-canonical DNA structures. Both compounds have a quenching effect on the fluorescence intensity upon addition of the investigated oligonucleotides; I was more selective for binding G4-than II. Association constant values obtained for I and II with G-quadruplexes reached 106 M−1, which suggests a strong interaction between both complexes and these sequences. FRET-melting assays show that I and II have a rather high level of stabilization of ckit1 and ckit2 quadruplexes. I is toxic against macrophages RAW267.7 only in high concentrations, while complex II shows no toxicity against these cells. I and II accumulate inside cells in different degrees. Molecular dynamic simulation studies have provided insights into the binding modes of II with ckit1 and ckit2 G-quadruplexes. The results obtained show the DNA binding activity of the rhenium complexes and their ability to be players in the anti-cancer fight since they can bind to non-canonical DNA forms in oncogene promoters, accumulate in some cancer cells, and influence the cancer cells microenvironment.
Introduction
The benzimidazole (Benzim) scaffold is an important pharmacophore in anticancer drugs.1–3 Reduced cell viability, migration and invasion, disruption of tubulin polymerization, induction of apoptosis and autophagy, and cell cycle arrest in G1 or G2/M phases, are among the antitumor effects exerted by these pharmacophores. Moreover, it is commonly accepted that metal–organic compounds are often more effective than free organic compounds as antitumor agents.4–6 Accordingly, benzimidazole and its derivatives are promising ligands due to their conjugated π-system, planarity, and capacity to mimic the function of imidazole in proteins and polynucleotides. As a matter of fact, inorganic drug discovery based on ruthenium with benzimidazole ligands has undergone considerable advances over the past two decades, with two representatives of this family currently in active clinical trials.7Several comprehensive reviews highlight the biological effects of different rhenium complexes according to their oxidation states.8,9 The most active rhenium complexes reported to date contain Re(I) carbonyl centers bound to mono-, di-, or tridentate ligands. It is proposed that such compounds induce their cytotoxic effects through covalent binding to DNA bases and/or protein side chains. Also, several rhenium-based compounds have been employed as in vitro and in vivo imaging agents.
It has been proved that several of the rhenium complexes with benzimidazole and nitrogen-containing heterocycles effectively kill cancer cells by triggering necroptosis, a nonapoptotic form of cell death, and induce mitochondrial membrane potential depletion, a possible downstream effect of reactive oxygen species (ROS) production.10–14 One of these complexes was found to be the most potent with IC50 values ranging from 0.1 to 0.4 μM against a panel of cancer cells, values which are 5–70 fold lower than those of cisplatin;15 it accumulates preferentially in mitochondria, depleting mitochondrial membrane potential, and upregulating intracellular ROS, leading finally to necrosis-mediated cancer cell death.
In our previous research, summarized in ref. 15, we provided an overview of the design, and in vitro and in vivo biological activity of the quadruple-bonding cluster dirhenium(III) complexes, preferentially with di and tetra alkyl-carboxylate ligands. Dirhenium(III) clusters had antiradical activity in vitro, anticancer activity in vitro and in vivo, mighty antioxidant, nephro-, hepato-, red blood cells, etc. protective properties and ability to decrease cisplatin-induced toxicity. Consequently, our synthetic activity was aimed at preparing dirhenium(III) complexes with the so-called “biologically active” ligands, such as amino acids, cinnamic, ferulic, and indolyl acetic acids. Thus, we recently synthesized dirhenium(III) cis-dicarboxylates with the α-amino acids residues Asp, Glu, Phe and Tyr and showed their G-quadruplex (G4) stabilization potential, which was much greater for aromatic amino acids.16 We suggest a unique mechanism for molecular DNA recognition for these complexes with the participation of the quadruple bond and aromatic rings of the amino acids. As aromaticity played a positive role in the process of G4 stabilization, it drew attention to investigating the interaction of the benzimidazole dirhenium(III) complexes with G4 quadruplex structures. In this work, we focus our attention on the two representative benzimidazole rhenium compounds, cis-tetrachlorotetrabenzimidazoldirhenium(III) chloride – I and tetrabenzimidazoldioxorhenium(V) – II shown in the Fig. 1.
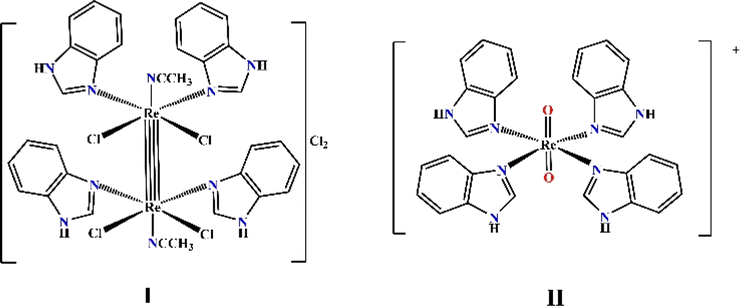 | ||
| Fig. 1 Graphical structure of the Re(III) and Re(V) complexes: [Re2(Benzim)4Cl4(CH3CN)2]Cl2 (I) and [Re(Benzim)4O2]+ (II). | ||
Results
Synthesis and characterization of the complexes
We previously synthesized and fully characterized compound I and its analogue with imidazole ligands.17 The procedure employed for the preparation of both complexes (Fig. 2) is common in the synthesis of the cluster dirhenium(III) compounds and originates from the tetrabutylammonium octachlorodirhenate(III) – (Bu4N)2Re2Cl8.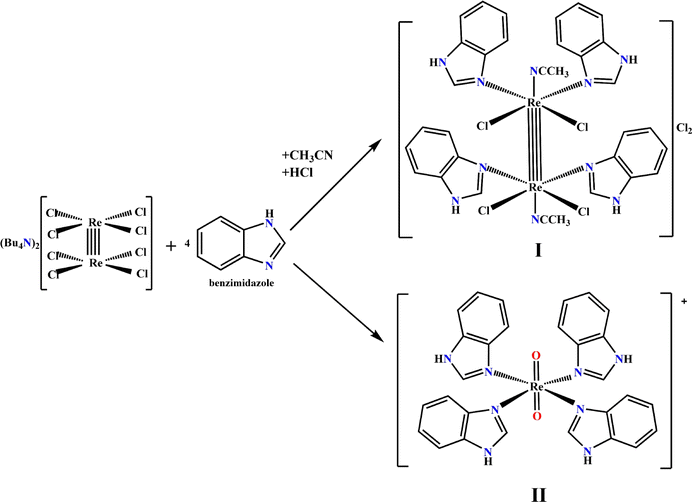 | ||
| Fig. 2 Synthetic route for the preparation of complexes I and II. | ||
The synthesis and characterization data for II are presented in the ESI.† The yield of complex I or II depends on the synthetic procedure employed; I is obtained when HCl is added under an inert atmosphere while II is obtained in the presence of oxygen without adding any acid.
Solid state analysis of II
The crystal structure of II was solved in the P4/mnc space group. The unit cell is formed by four [ReO2(Benzim)4]+ cations and six perrhenate anions. To balance the net charge, each [ReO2(Benzim)4]+ unit must have +1.5 average charge. To explain this situation in a reported similar structure,9,10 it was postulated that there are shared protons between adjacent complex units such that one unit may be formulated as [ReO2(Benzim)4]+ and the other one as [ReO(OH)(Benzim)4]2+. Although in our structure we do not have any direct evidence either of the presence of shared protons or of their location, we have assumed that the protons are in O1 and O2 close to the hydrophilic layer (vide infra), and close to O4 so that they are effectively delocalized between the two positions. Due to the symmetry of the crystal, the hydrogen atoms are located in special positions. The proton close to the O1 (H1A) would be in a special position (0.5, 0.5, 1.0), while the other one (H2A) would be in (0.5, 0.5, 0.78). The distance between these units (in Table 1) can be measured by the distance (O1, O1) [x,y,2 − z] = 2.64(4) Å and (O2, O3) = 2.90(3) Å.| Re1–N1 | 2.111(9) | Re2–N3 | 2.114(10) |
| Re1–O2 | 1.770(17) | Re2–O4 | 1.73(3) |
| Re1–O1 | 1.791(18) | Re2–O3 | 1.745(18) |
| N1–Re1–N1 | 178.2(5) | N3–Re2–N3 | 175.7(5) |
| O2–Re1–N1 | 90.9(2) | O4–Re2–N3 | 87.9(3) |
| O1–Re1–N1 | 89.1(2) | O3–Re2–N3 | 92.1(3) |
The coordination geometry of Re in these units is regular octahedral (see angles in Table 2); the Re–N distances is ca. 2.11 Å and the Re–O distances range from 1.73 to 1.79 Å.
| Composition | C56H49.50N16O18.24Re5 |
|---|---|
| Formula weight/g mol−1 | 2169.41 |
| Size/mm | 0.119 × 0.070 × 0.059 |
| Space group | P4/mnc |
| Unit cell: | |
| • a/Å | 15.974(3) |
| • b/Å | 15.974(3) |
| • c/Å | 27.244(7) |
| • α/degrees | 90 |
| • β/degrees | 90 |
| • γ/degrees | 90 |
| • V/Å3 | 6952(3) |
| Density/g cm−3 | 2.07 |
| Z | 4 |
| μ/mm−1 | 8.75 |
| F000 | 4070 |
| Diffraction limits | 19 ≤ h ≤ −19 |
| 19 ≤ k ≤ −19 | |
| 33 ≤ l ≤ −33 | |
| R(int) | 0.0597 |
| R(sigma) | 0.0176 |
| Reflections | |
| • Collected | 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 620 620 |
| • Unique | 3501 |
| Parameters | 252 |
| Constraints | 0 |
| Restraints | 45 |
| R1 | |
| • Total | 0.0681 |
| • F2 > 2σ(F2) wR2 | 0.0584 |
| • Total | 0.1637 |
| • F2 > 2σ(F2) | 0.1556 |
| Goodness of fit | 1.155 |
The benzimidazole rings adopt a helical conformation with angles of 47.7° between their average planes with respect to the rhenium equatorial plane (measured as O1–Re1–N1–C2 torsion angle).
The complex units are stacked and aligned with the c axis with the benzimidazole moieties equatorially coordinated to the Re atom and the oxygen atoms axially-oriented, giving rise to a layer of cations and anions along that direction (Fig. 3).
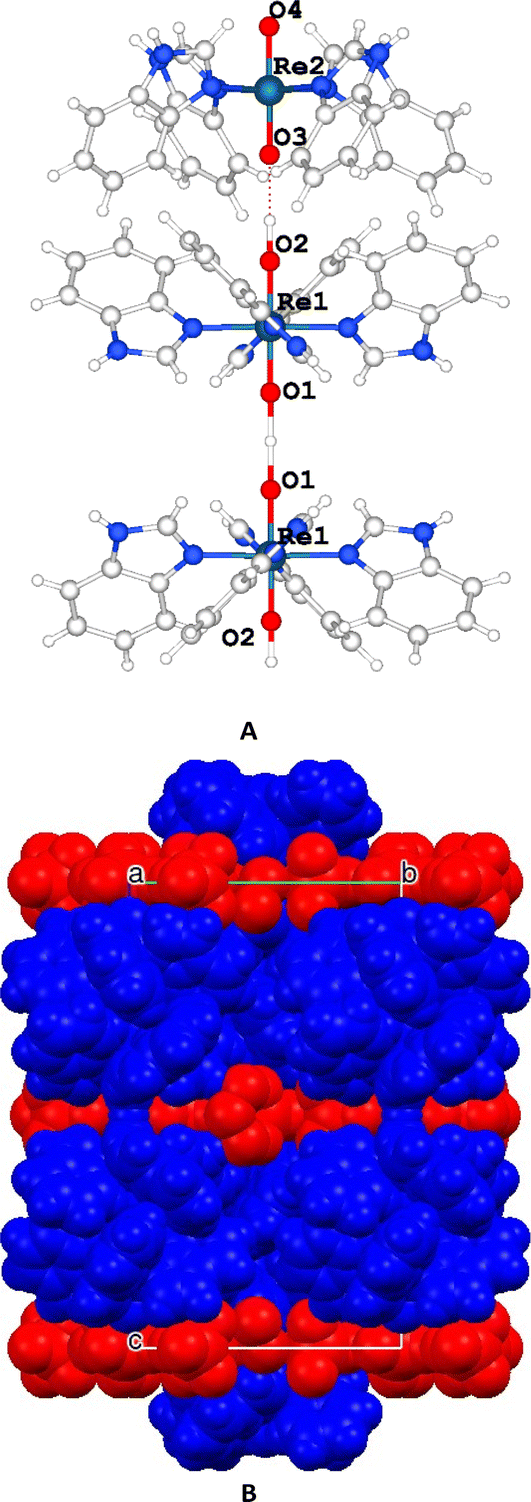 | ||
| Fig. 3 (A) Detail of the complex II. (B) Unit cell packing highlighting the cationic (blue) and the anionic (red) layers. | ||
The [ReO2(Benzim)4]+ units are grouped in pairs, with the hydrophobic part of the benzimidazole moiety facing each other while the hydrophilic part is oriented towards a layer containing perrhenate anions that balances the charge and includes disordered water molecules.
UV-vis and fluorescence properties of the complexes
We then investigated the photophysical properties of both complexes in solution. The intensive absorption maxima for I and II were observed in the range 200–290 nm, which is similar to the benzimidazole spectrum (Fig. 4 and Table 1S in the ESI†).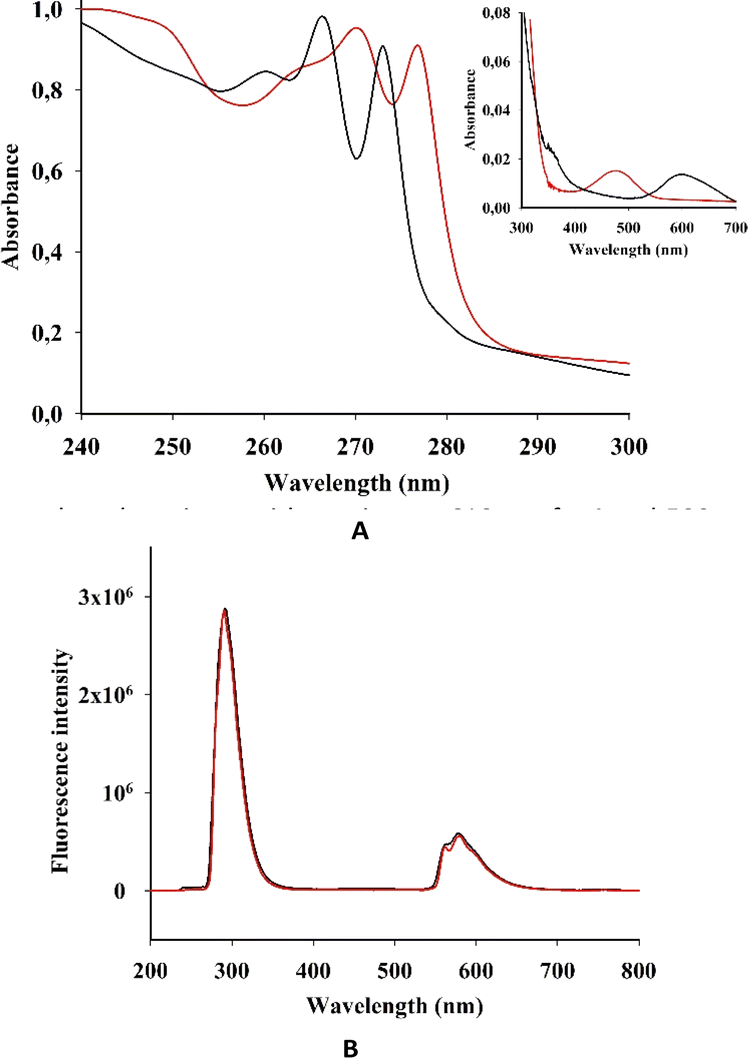 | ||
| Fig. 4 (A) EAS and (B) fluorescent spectra (λexc = 250 nm) of I (black) and II (red) in acetonitrile. (Inset in A) EAS of I and II in the long wave area. | ||
This strong absorption in some transition metal complexes with benzimidazole was mostly attributed to π–π* transitions of the benzimidazole ligands or its derivatives. As we have observed for I and II, metal complexation did not cause a considerable shift in the absorption wavelength of the ligands.18
However, the electronic absorption spectra (EAS) of I and II show a weak peak at 350 nm, which is more pronounced for I. Complexes of benzimidazole with Fe(II), Co(II), Ni(II) and Cu(II) show a rather intense absorption in this area.17 This absorption was ascribed to ligand–metal charge transfers (L → M). Moreover, in the spectra of Mn(II) and Zn(II) complexes, L → M charge transitions were not observed.
The EAS of I and II have comparatively low absorption in the longer-wavelength regions, with maxima at 610 nm for I and 500 nm for II (Fig. 4).
The absorbances for both I and II are linearly correlated with the concentration, indicating absence of aggregation (see Fig. S1 in the ESI†). In previous spectroscopic studies on transition metal complexes of benzimidazole ligands, the high-energy intense absorption band was assigned to π–π* intraligand transitions, whereas the low-energy absorption band was tentatively assigned to an admixture of metal-to-ligand charge-transfer (MLCT) and ligand-to-ligand charge-transfer (LLCT) transitions.18–20
The absorption in the long-wavelength area for quadruply-bonded dirhenium(III) complexes was described as δ → δ* transitions in the Re–Re centre.14
Both I and II showed a strong emission band at 290 nm when excited at 250 nm (Fig. 4B), which is in accordance with previous research.21
In acetonitrile solutions of I and II at room temperature, the low-energy emission bands were observed from 550 to 635 nm. According to previous investigations on rhenium and dirhenium complexes, the short-lifetime luminescence at higher energy was derived from LLCT while the long-lived phosphorescence at lower energy was from MLCT.22,23
Luminescent properties of I and II require deeper investigations and may be relevant in future applications. For example, the introduction of rhenium complexes and cluster in the structure of metal–organic frameworks (MOFS) leads to effective single-site catalysis for photochemically reducing carbon dioxide to produce a variety of organic compounds.24–27
Evaluation of the G-quadruplex DNA interaction
Initially, we evaluated the interaction of both rhenium complexes I and II with G-quadruplex DNA structures by fluorescence titration experiments in cacodylate buffer (see Fig. 5, S2–S4 in the ESI†).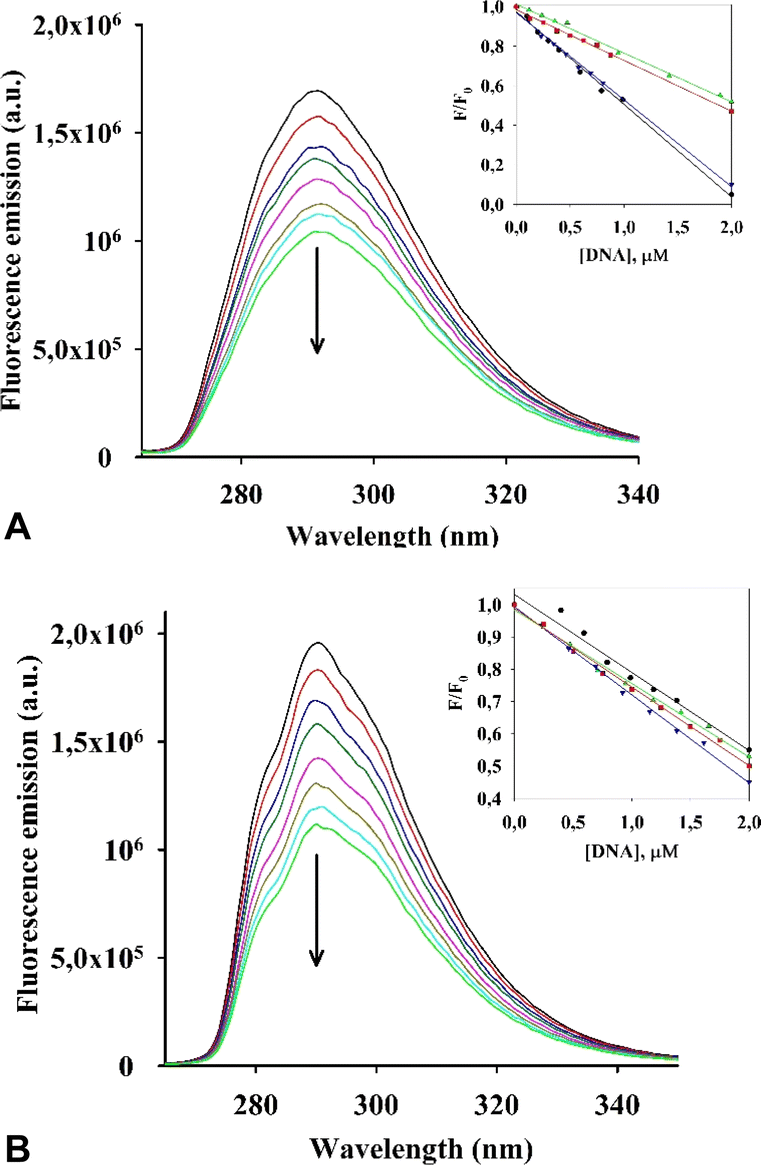 | ||
| Fig. 5 Fluorescent titration spectra of I (A) and II (B) with ckit1 G4 DNA. (Insets) Relative fluorescence intensity (F/F0) of complexes I (A) and II (B) as a function of DNA concentration in the titration experiments. • Bcl; ▼ckit1; ▲ ckit2; ■ cMyc. | ||
Considering our previous research on dirhenium(III) dicarboxylates, which demonstrated a high stabilization effect of ckit1,16 we selected several G4 DNA sequences for our experiment, including those located at the c-Kit promoter region (their sequences are presented in ESI†).
Upon addition of the investigated G4s, both metal complexes had a quenching effect on fluorescence emission. Similar photophysical response was observed for an RNA polymerase I inhibitor when interacting with G4s DNA.28 Results of an analysis of the relative luminescence intensity (F/F0) of complexes I and II as a function of DNA concentration in the titration experiments is shown in Fig. 5A and B. We estimated the values of the binding constants (Kb) from the titration curves, which are shown in Table 3.
K) of interaction of I and II with G4 DNA obtained by fluorescence titrations
| G4 DNA | logK (I) |
logK (II) |
|---|---|---|
| cMyc | 5.65 ± 0.01 | 6.01 ± 0.01 |
| ckit1 | 6.20 ± 0.01 | 6.13 ± 0.01 |
| ckit2 | 5.81 ± 0.01 | 5.88 ± 0.01 |
| Bcl2 | 6.38 ± 0.01 | 6.20 ± 0.01 |
The values (Kb) obtained for I and II with the G4s cKit1 and Bcl2 are in the order of 106 M−1, which suggests a strong interaction between each of the complexes and these G4s located in the promoter regions of the corresponding oncogenes.
Then, the G-quadruplex (ckit1 and ckit2) stabilization effects of I and II were evaluated by fluorescence resonance energy transfer (FRET) melting assay, which measures the shift in melting temperature (ΔTm) of the folded G-quadruplex structures as a function of ligand concentration. Both I and II showed a rather high level of stabilization of both ckit1 and ckit2 (Fig. 6 and S5†).
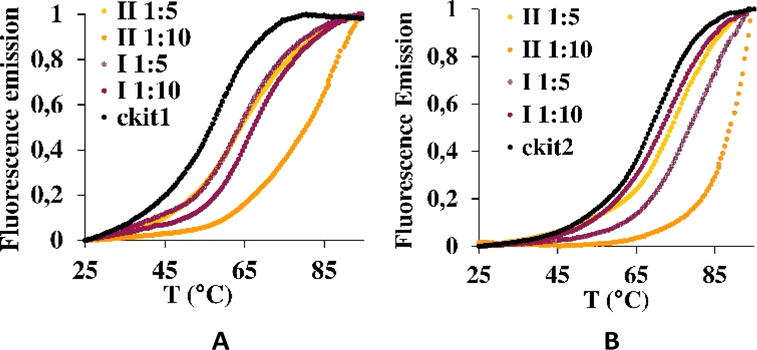 | ||
| Fig. 6 Stabilization curves of oligonucleotides ckit1 (A) and ckit2 (B) in the presence of I and II at the DNA: I and DNA: II 1:5 and 1:10. | ||
The largest stabilization effect was obtained for the interaction of ckit-1 and II. Both compounds did not stabilize ds26 even under 1:20 DNA:complex molar ratio (see Fig. S6†).
It is well-known that the bis-benzimidazole molecule Hoechst is used as for fluorescence DNA staining because of its sequence-specific DNA binding ability. However, the selective recognition of some new benzimidazole systems towards G-quadruplex DNA, their selective cytotoxicity towards some cancer cells and potent telomerase inhibition ability were demonstrated.29 In these works, the authors showed that in contrast to the standard groove binder Hoechst, bivalent metal ions prompted a “switch off” emission effect upon duplex DNA binding. The bivalent metal ions coordinate the nitrogen atoms of the imidazole moieties, forcing the imidazole N–H to stay away from the surface of the minor groove pocket of the duplex DNA, making the ligand ineffective in binding duplex DNA. However, some of these ligands were active in stabilizing G4 DNA with a 100-fold preference for G4 over duplex DNA. We observed similar behavior for compounds I and II because they did not stabilize duplex DNA. Attachment of the tertiary nitrogen atoms of benzimidazole to the rhenium atoms prevented the possibility of interacting with duplex DNA but retained their ability to interact with other non-canonical structures, such as G-quadruplexes.
Metal complexes have demonstrated stronger selectivity for G4 structures than duplex DNA owing to proper coordination features, positive charge, and electron density promoted by the organic ligands.30,31 The named reviews showed that the stabilization of G-quadruplexes can be achieved through extended square aromatic surfaces, increased hydrophobicity, different auxiliary ligands, axially coordinated ligands, and the nature of the metal center. We suppose that the nature of the quadruply-bonded dirhenium core and the aromaticity of the four benzimidazole ligands play a positive role in G4-interactions and selectivity of I and II.
Molecular dynamic simulation performed according to ref. 32 and 33 (see the details in the ESI† Fig. S7 and S8) on the interaction of I with ckit1 shows that cytosine C11 points outwards the G4 structure and bends, accommodating the complex into the groove where it gives a conformation that remains stable in the simulation (Fig. 7).
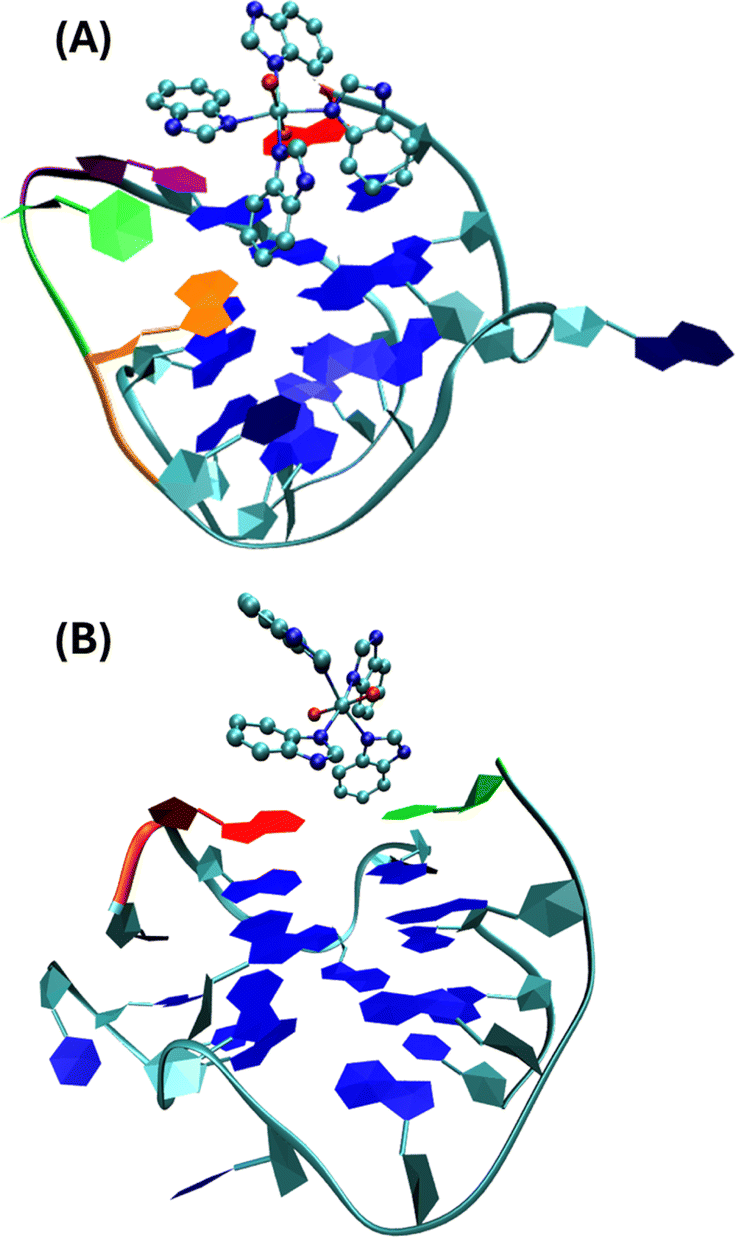 | ||
| Fig. 7 Stable conformations of ckit1 (A) and ckit2 (B) with II. Color code: adenine-1 (red), guanine-10 (orange), cytosine-11 (green), thymine-12 (purple). | ||
This final conformation is stabilized by face-to-face π-stacking between one of the benzimidazole rings and thymine 12, corroborating past results obtained for benzimidazole-carbazole ligands and this G4.34 We also found the complex to be very close and in persistent proximity to the aromatic moieties of guanine-10 and adenine-1. The simulation studies with ckit2 show a similar interaction with complex II. II interacts with adenine-13 via face-to-face π-stacking as well as with cytosine-1. The complex is formed at 2.2 ns and stays stable until the end of the simulation. These in silico studies provided insight into the mode of binding of II to G-quadruplexes formed in the oncogene promoters. The c-Kit proto-oncogene encodes a transmembrane tyrosine kinase receptor, which participates, once activated by endogenous ligands, in a broad range of physiological processes, including cell proliferation, migration, maturation and survival.35 Overexpression and/or mutations of c-Kit gene have been implicated in a number of human cancers, including gastrointestinal stromal tumors (GIST), pancreatic cancer, leukemia and melanoma. In the proximal promoter of c-Kit, three guanine (G)-rich regions able to fold into G4s structures have been identified. The putative G4-forming sequences within the c-Kit promoter named ckit1, ckit* and ckit2, are closely clustered and separated from each other by only a few nucleotides.28,36,37 ckit1 G4 contains unique structural features; an isolated guanine that is involved in the formation of the G-quartet core and a stem-loop comprising five nucleotides. The stabilization of G4s in appropriate sites of its promoter region via the use of c-Kit -selective small-molecule ligands provides a good strategy to suppress c-Kit activity.32 Such ligands have been identified. Members of the isoalloxazine family of ligands demonstrated a binding preference for c-Kit over telomeric G4s and inhibited c-Kit oncogene expression; (i) a naphthalene diimide derivative showed stabilization of c-Kit G4s accompanied by reduction of encoded protein levels, leading to an arrest of cell growth in patient-derived gastrointestinal tumor cells; (ii) benzo[a]phenoxazines and quinazolines. Interestingly, some quinazoline derivatives bind and stabilize c-Kit G4s with high selectivity, inhibit the transcription and expression of c-Kit, and exhibit significant cytotoxicity on the GIST cell line HGC-27. They showed ΔT1/2 = 11.3 °C and 13.4 °C for ckit1, and 13.6 °C and 14.3 °C for ckit2 in the FRET experiments (DNA:ligand = 1:5).29 These values agree with our results for II and ckit1: ΔT1/2 = 13.8 °C under molar ratio DNA:ligand 1:5.
Assessment of the cell activity of the rhenium complexes
Tumor-associated macrophages (TAM) contribute to the tumor progression at different levels, including promoting genetic instability, nurturing cancer stem cells, paving the way to metastasis, and taming protective adaptive immunity.38–40 TAM are major players in the connection between inflammation and cancer; thus, TAM-centered therapeutic strategies have the potential to complement and synergize together with chemotherapy and immunotherapy.In our experiments, we used Raw 264.7, which is a transformed murine monocyte macrophage cell line. In our experiments the cells of macrophages had an unregulated growth that reached 126% in comparison to the control (Fig. 8 and Table S2†).
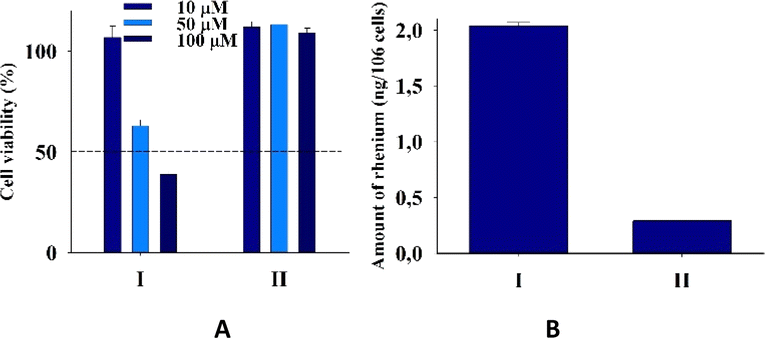 | ||
| Fig. 8 (A) Cell viability of cells Raw 264.7 upon addition of I and II. (B) Amount (ng) of rhenium in cells Raw 264.7 upon 24 h of incubation with 20 μM I and II for two independent experiments. | ||
II did not practically affect the proliferative activity of the macrophages in contrast to I, which decreased the cell viability at 50 and 100 μM concentrations to 65 and 39%, respectively. The accumulation of rhenium in the cells that were treated with I was very significant and seven times larger than that in cells treated with II. The ability of the rhenium compound I to accumulate in macrophages has an essential biological significance described here for the first time. It is well-known that cellular uptake plays an important role in the bioactivity of drugs, and the internalization and accumulation of metal-based drugs into cancer cells is crucial for their therapeutic effect. So, such an essential uptake of I in TAM is a very important property of the dirhenium compound, which is the basis of the future investigation of their anti-cancer properties. Also, our understanding of the significance of the “metallic” crosstalk between macrophages and cancer cells is still primitive.41
Furthermore, Raw 264.7 cells changed their morphology under the influence of both rhenium compounds, with stronger effects associated with the influence of I (Fig. S9†). The size of round cells under influence of I increased and the shape changed to more elongated forms. The macrophage changes in cell shape are associated with their activation.42
For example, changing morphology of Raw 264.7 was connected with production of cysteine proteases, responsible for cancer growth and neovascularization.43 It is possible to suppose that under the influence of the rhenium compounds, some metabolic changes in the cells of macrophages took place with the following activation, which also has biological significance. In our previous works,15 we showed that some dirhenium(III) carboxylates led to decrease in the growth of tumors in the experiments in vivo, and its joint administration with cisplatin stopped tumor growth. This synergistic effect of the combinational treatment was explained by antioxidant properties of the quadruply bonded dirhenium compounds, which led to decreasing ROS production caused by the cisplatin-triggered malignation process. The results of this work with macrophages where I was accumulated in the cells following change of their morphology may be another explanation of the synergism of dirhenium(III) complexes with cisplatin, i.e. by influence of I on the immune system of xenografts. The obtained results may present a useful strategy of complementing antiproliferative activity of some agents with the secreted molecules of activated macrophage cells as inhibitors of metastasis to obtain some additive effects that may be a new contribution to the combinational therapy of cancer.
We see perspectives of our future research in synthetic activity, aimed to involve new ligands to the dirhenium(III) fragment; investigation of photophysical properties of the dirhenium(III) compounds; further development in the area of DNA and RNA recognition with photoactivated molecules; interaction of the dirhenium(III) clusters with cancer cells with deepening into biochemical characteristics of the cancer cells and their environment; and in preclinical experiments to apply the elaborated combinational therapeutic procedures.
Conclusions
To our knowledge, the G4-quadruplex binders on the base of the rhenium(III) clusters, were first described in our work.16 Here we have demonstrated that introduction of benzimidazoles (a route to increasing aromaticity) to the rhenium core (dirhenium(III) or dioxo rhenium(V)) has been a good strategy for producing effective and selective DNA binders. The strong binding activity of I and II towards G4 quadruplexes showed their significance for DNA recognition in medicinal chemistry. Our primary finding in cell studies revealed a unique uptake of dirhenium(III) cluster in tumor associated macrophages that may offer insights into new approaches for combinational therapy and all together provide evidence of the importance of investigations in this direction.Author contributions
Conceived and designed the experiments: EG-E, JG-G, NS, OS, SB; performed the experiments: NS, OS, OH, HV, CGR, AG-M, SB; analyzed the data: NS, OS, JG-G, CGR, AG-M; contributed reagents\materials\tools: JG-G, SB, OH, HV, CGR, AG-M, SB; wrote the paper: NS, OS, JG-G, SB, EG-E.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Spanish Ministry for Science and Innovation, The National Research Agency and FEDER funds from the EU (grants PID2019-110751RB-I00, PID2019-108643GA-I00, and CEX2019-000919) and the Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital of the Generalitat Valenciana for project PROMETEO (Grant CIPROM/2021/030 and CIDEGENT/2018/015). This contribution is also based upon work from COST Action CA18202, NECTAR – Network for Equilibria and Chemical Thermodynamics Advanced Research, supported by COST (European Cooperation in Science and Technology). NS and OS express great thanks to Prof. Enrique García-España and to authorities of the University of Valencia for their acceptance of NS and OS as coworkers of UV during war in Ukraine.References
- S. Yadav, B. Narasimhan and H. Kaur, Anticancer Agents Med. Chem., 2016, 16, 1403–1425 CrossRef CAS PubMed.
- N. Shrivastava, M. J. Naim, M. J. Alam, F. Nawaz, S. Ahmed and O. Alam, Arch. Pharm. Chem. Life Sci., 2017, 350, e1700040 Search PubMed.
- D.-S. Son, E.-S. Lee and S. E. Adunyah, Immune Netw., 2020, 20, e29–e49 CrossRef PubMed.
- A. Kanwal, F. A. Saddique, S. Aslam, M. Ahmad, A. F. Zahoor and N.-A. Mohsin, Pharm. Chem. J., 2018, 51, 1068–1077 CrossRef CAS.
- D. Hernández-Romero, S. Rosete-Luna, A. López-Monteon, A. Chávez-Piña, N. Pérez-Hernández, J. Marroquín-Flores, A. Cruz-Navarro, G. Pesado-Gómez, D. Morales-Morales and R. Colorado-Peralta, Coord. Chem. Rev., 2021, 439, 213930–213981 CrossRef.
- V. T. Nguyen, T. K. C. Huynh, G. T. T. Ho, T. H. A. Nguye, A. N. T. Le, D. Q. Dao, T. V. T. Mai, L. K. Huynh and T. K. D. Hoang, R. Soc. Open Sci., 2022, 9, 220659–220676 CrossRef CAS PubMed.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and K. P. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- E. Palma, J. F. Santos, C. Fernandes and A. Paulo, Chem.–Eur. J., 2024, 30, e202303591 CrossRef CAS PubMed.
- S. Belanger, S. Fortin and A. L. Beauchamp, Inorg. Chem., 1996, 35, 7836–7844 CrossRef CAS.
- S. Belanger, S. Fortin and A. L. Beauchamp, Can. J. Chem., 1997, 75, 37–45 CrossRef CAS.
- A. F. Cotton, E. V. Dikarev and M. A. Petrukhina, Inorg. Chim. Acta, 2002, 334, 67–70 CrossRef.
- K. Suntharalingam, S. Awuah, P. M. Bruno, T. C. Johnstone, F. Wang, W. Lin, Y.-R. Zheng, J. E. Page, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 2967–2974 CrossRef CAS PubMed.
- S. Das, P. Joshi and M. Patra, Inorg. Chem., 2023, 62, 19720–19733 CrossRef CAS PubMed.
- N. I. Shtemenko and A. V. Shtemenko, Ukr. Biochem. J., 2017, 89, 5–30 CrossRef.
- N. Shtemenko, I. Pont, Yu. Husak, A. Golichenko, S. Blasco, A. Shtemenko and E. García-España, J. Inorg. Biochem., 2021, 225, 111605–111621 CrossRef CAS PubMed.
- O. V. Velichko, O. A. Golichenko and O. V. Shtemenko, Odessa National University Herald: Chemistry, 2019, 24, 26–38 Search PubMed.
- H. Kırpık and M. Kose, J. Mol. Struct., 2020, 1200, 1270–1273 CrossRef.
- A. Y.-Y. Tam, W. H. Lam, Dr K. M. Wong, N. Zhu, W.-W. Vivian and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4562–4576 CrossRef CAS PubMed.
- M. Gillard, H. Bonnet, R. Larita, H. Yacoub, J. Dejeu, E. Defrancq and B. Elias, Bioconjugate Chem., 2023, 34, 414–421 CrossRef CAS PubMed.
- J. P. L. Roque, M. T. S. Rosado, R. Fausto and I. Reva, J. Org. Chem., 2023, 88, 2884–2897 CrossRef CAS PubMed.
- B. G. Alberding, M. H. Chisholm, J. C. Gallucci, T. L. Gustafson, C. R. Reed and C. Turro, Dalton Trans., 2010, 39, 11587–11593 RSC.
- L. D. Ramos, R. N. Sampaio, F. F. de Assis, K. T. de Oliveira, P. Homem-de-Mello, A. O. T. Patrocinio and K. P. M. Frin, Dalton Trans., 2016, 45, 11688–11698 RSC.
- R. Huang, Y. Peng, C. Wang, Z. Shi and W. Lin, Eur. J. Inorg. Chem., 2016, 27, 4358–4362 CrossRef.
- Z. Su, B. yu, J. Feng, M. Zhong, X. Li and J. Shi, Catalysts, 2023, 13, 1510 CrossRef CAS.
- Y. M. Litvinova, Y. M. Gaylufin, K. A. Kovalenko, D. G. Samsonenko, J. van Leusen, I. V. Korolkov, V. P. Fedin and Y. V. Mironov, Inorg. Chem., 2018, 57, 2072–2084 CrossRef CAS PubMed.
- W. N. Yang, B. Ruan, L.-Ch. Tsai, N. Ma, D. Shi, T. Jiang and F.-C. Tsai, ECS J. Solid State Sci. Technol., 2021, 10, 056003–056015 CrossRef.
- S. Mazzini, R. Gargallo, L. Musso, F. De Santis, A. Aviñó, L. Scaglioni, R. Eritja, M. Di Nicola, F. Zunino, A. Amatulli and S. Dallavalle, Int. J. Mol. Sci., 2019, 20, 4927–4944 CrossRef CAS PubMed.
- B. Maji and S. Bhattacharya, Chimia, 2013, 67, 39–43 CrossRef CAS PubMed.
- T. Kench and R. Vilar, Annu. Rep. Med. Chem., 2020, 54, 485–515 CAS.
- P. M. Toro, M. Saldias and G. Valenzuela-Barra, Curr. Med. Chem., 2023, 30, 573–600 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. H. Kaulage, B. Maji, S. Pasadi, A. Ali, S. Bhattacharya and K. Muniyappa, Eur. J. Med. Chem., 2018, 148, 178–194 CrossRef CAS PubMed.
- A. Kotar, R. Rigo, C. Sissi and J. Plavec, Nucleic Acids Res., 2019, 47, 2641–2653 CrossRef CAS PubMed.
- L. Savva and S. N. Georgiades, Molecules, 2021, 26, 841–868 CrossRef CAS PubMed.
- X. Wang, C. X. Zhou, J. W. Yan, J. Q. Hou, S. B. Chen, T. M. Ou, L. Q. Gu and Z. S. Huang, ACS Med. Chem. Lett., 2013, 4, 909–914 CrossRef CAS PubMed.
- G. Solinas, G. Germano, A. Mantovani and P. Allavena, J. Leukocyte Biol., 2009, 5, 1065–1073 CrossRef PubMed.
- A. Mantovani, F. Marchesi, A. Malesci, L. Laghi and P. Allavena, Nat. Rev. Clin. Oncol., 2017, 14, 399–416 CrossRef CAS PubMed.
- Q. Qi, Q. Wang, Y. Li, D. Z. Silva, M. E. L. Ruiz, R. Ouyang, B. Liu and Y. Miao, Molecules, 2023, 28, 2733–2756 CrossRef CAS PubMed.
- M. Serra, A. Columbano, U. Ammarah, M. Mazzone and A. Menga, Front. Oncol., 2020, 10, 646 CrossRef PubMed.
- H.-W. Nam, J. Bae, Y.-W. Kim, H.-H. An, S.-H. Kim, K.-Y. Kim, S.-N. Yu, B.-B. Park, S.-Y. Lee and S.-C. Ahn, Ann. Clin. Lab. Sci., 2020, 50, 739–746 CAS.
- P. Collery, D. Desmaële, A. Harikrishnan and V. Veena, Curr. Pharm. Des., 2023, 29, 2396–2407 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2310987. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02669a |
| This journal is © The Royal Society of Chemistry 2024 |