
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and computational studies of new azaheterocyclic coumarin derivatives as anti-Mycobacterium tuberculosis agents targeting enoyl acyl carrier protein reductase (InhA)†
Rasha Z. Batran *a,
Ahmed Sabta,
Jarosław Dziadekb and
Asmaa F. Kassemc
*a,
Ahmed Sabta,
Jarosław Dziadekb and
Asmaa F. Kassemc
aChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo 12622, Egypt. E-mail: rasha_batran@yahoo.com
bLaboratory of Genetics and Physiology of Mycobacterium, Institute of Medical Biology of the Polish Academy of Sciences, Lodz, Poland
cChemistry of Natural and Microbial Products Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo 12622, Egypt
First published on 9th July 2024
Abstract
In this study, we designed and synthesized a series of coumarin derivatives as antitubercular agents targeting the enoyl acyl carrier protein reductase (InhA) enzyme. Among the synthesized compounds, the tetrazole derivative 4c showed the most potent antitubercular effect with a minimum inhibitory concentration value (MIC) of 15 μg mL−1 against Mtb H37Rv and could also inhibit the growth of the mutant strain (ΔkatG). Compound 4c was able to penetrate Mtb-infected human macrophages and suppress the intracellular growth of tubercle bacilli. Moreover, the target derivative 4c showed a potent inhibitory effect against InhA enzyme with an IC50 value of 0.565 μM, which was superior to the reference InhA inhibitor triclosan. Molecular docking of compound 4c within the InhA active site revealed the importance of the 4-phenylcoumarin ring system and tetrazole moiety for activity. Finally, the physicochemical properties and pharmacokinetic parameters of 4c were investigated.
1. Introduction
Tuberculosis (TB), a severe respiratory infectious disease caused by Mycobacterium tuberculosis (Mtb), is considered one of the top ten global causes of mortality and is currently recognized as the world's primary cause of death from a single infectious agent, surpassing HIV/AIDS.1–3 According to the World Health Organization (WHO), it has been estimated that about 10 million new cases of TB were diagnosed in 2021, with approximately 1.5 million deaths.4 The increasing drug resistance of Mycobacterium tuberculosis strains and the limited efficacy of anti-tubercular therapy have contributed to the worsening epidemiological status of TB.5,6 The emerging resistance is also exacerbated by several drawbacks of the traditional anti-TB therapies, such as long duration and use of multiple drugs, e.g. isoniazid (INH), ethambutol, rifampicin, fluoroquinolones and linezolid.7,8 Resistance to rifampicin is due to mutations in the β-subunit of RNA polymerase, whereas resistance to isoniazid primarily arises from mutations in the katG gene9 that encodes the catalase/peroxidase enzyme, which activates the prodrug INH inside the mycobacterial cell and enables it to bind to the active site of enoyl acyl carrier protein reductase (InhA), an essential NADH-dependent enzyme involved in mycolic acid synthesis and plays a crucial role in the survival of mycobacteria.10 Therefore, the inhibition of M. tuberculosis InhA represents a significant approach for the discovery of novel anti-TB drugs that could circumvent the resistance mechanisms exerted by Mtb strains,11–13 e.g. triclosan,14 arylamide,15 diphenyl ether derivatives,16,17 4-hydroxy-2-pyridones18 and pyrrolidine carboxamide analogs.19 The lack of novel remedies coming through the pipeline makes the development of new innovative strategies to cure TB an urgent challenge.Regarding the pharmacophoric features of InhA inhibitors, three InhA key sites were observed to accommodate the inhibitors. Site I includes a tyrosine residue and the ribose group of the NAD factor. Most InhA inhibitors contain an oxygen atom that interacts with the hydroxyl group of the Tyr158 residue and the ribose ring of the NAD cofactor. Site II has a flexible hydrophobic pocket that accommodates long alkyl chains. Extending the inhibitor substrate into this pocket leads to increase in lipophilicity, which provides the benefits of ease in crossing biological membranes and hence enhanced potency. The third pocket, site III, is relatively unexplored, which offers the opportunity for hydrophilic interactions through the phosphate groups of NAD and hydrophobic interactions via Ala198 and Ile202 residues. The undiscovered aspect of the InhA binding pocket offers a considerable chance to modify the physicochemical properties of InhA binding scaffolds.20–22
Coumarin scaffold is a widely recognized naturally derived chemical backbone in the field of medicinal chemistry, exhibiting a diverse array of biological effects, such as anticancer,23,24 anti-inflammatory,25,26 antioxidant,27,28 antimicrobial,29,30 and, particularly, antitubercular, such as InhA inhibitory agents.31–35 In recent years, several coumarin scaffolds have been reported for their potent anti-TB activities (Fig. 1). However, the safety profile of these compounds remains an important issue for the determination of their biological impact; for example, the naturally occurring Calanolide A displayed an anti-TB effect with an MIC value of 3.13 μg mL−1 and relatively low selectivity index (SI = 2.43).34 Similarly, 3-phenyl-4-syrylcoumarin I exhibited potent anti-Mtb effectiveness (MIC = 3.5 μg mL−1) with SI value of 2.85.35
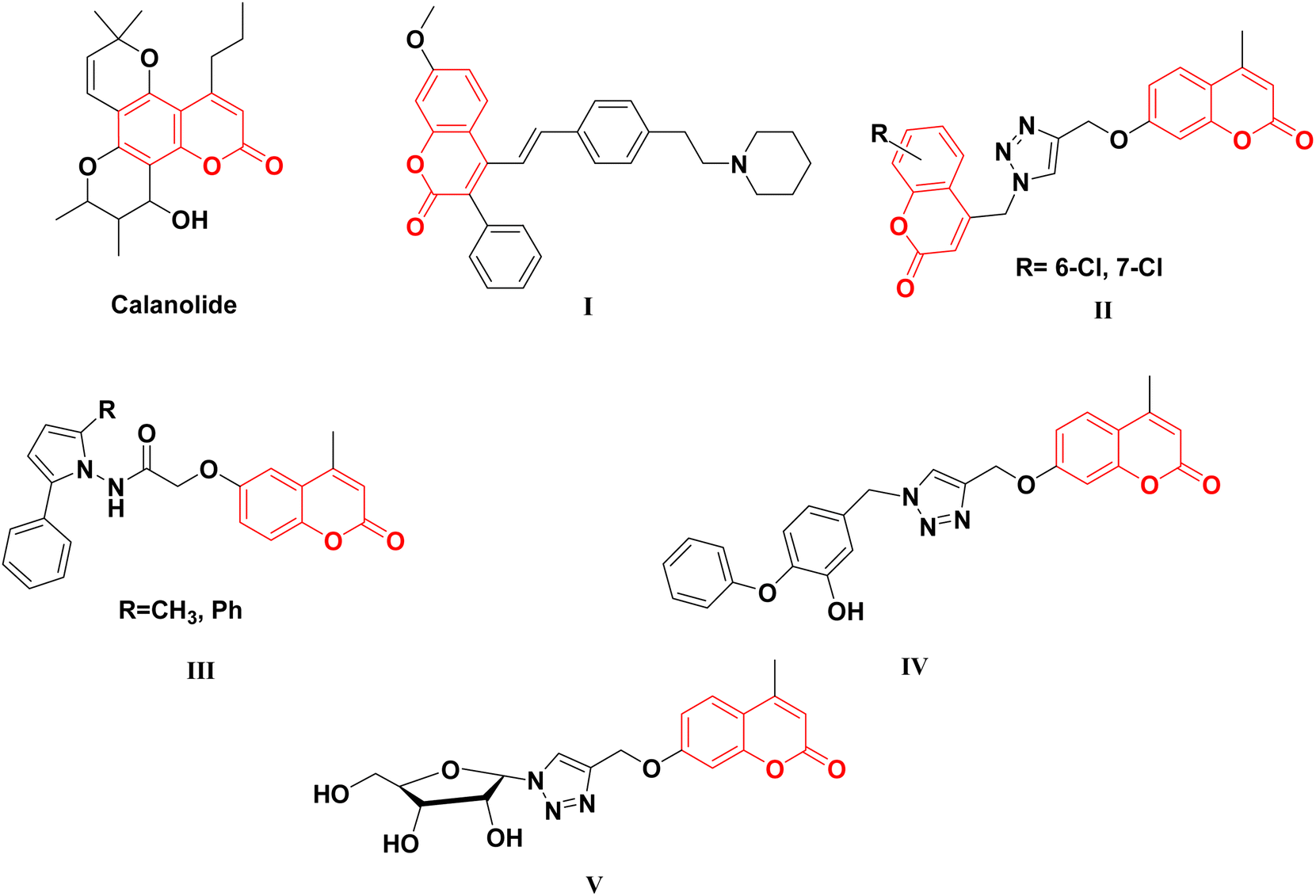 | ||
| Fig. 1 Coumarin-based derivatives as anti-TB agents and InhA inhibitors. | ||
The nature of the coumarin structure facilitates its engagement in various forms of interactions, including hydrogen bonding, hydrophobic interactions, and π–π stacking.36–38 Moreover, the isosteric features of the coumarin scaffold are shared with the bioactive quinoline core, which forms the main skeleton of several FDA-approved antitubercular drugs, such as bedaquiline and fluoroquinolones, making the coumarin core an attractive target for the design and synthesis of anti-TB candidates.
Moreover, nitrogen-containing heterocycles have been recognized as privileged structures owing to their versatile interactions with diverse biological targets.39,40 The five-membered azaheterocyclic molecular scaffolds, such as tetrazoles,41 thiadiazole,42–44 oxadiazole,45 pyrazole41,44,46,47 and pyrrole48 as well as the fused nitrogen-containing heterocycles as indoles,49,50 are encompassed in a vast number of antimycobacterial candidates endowed with InhA inhibitory effects (Fig. 2).
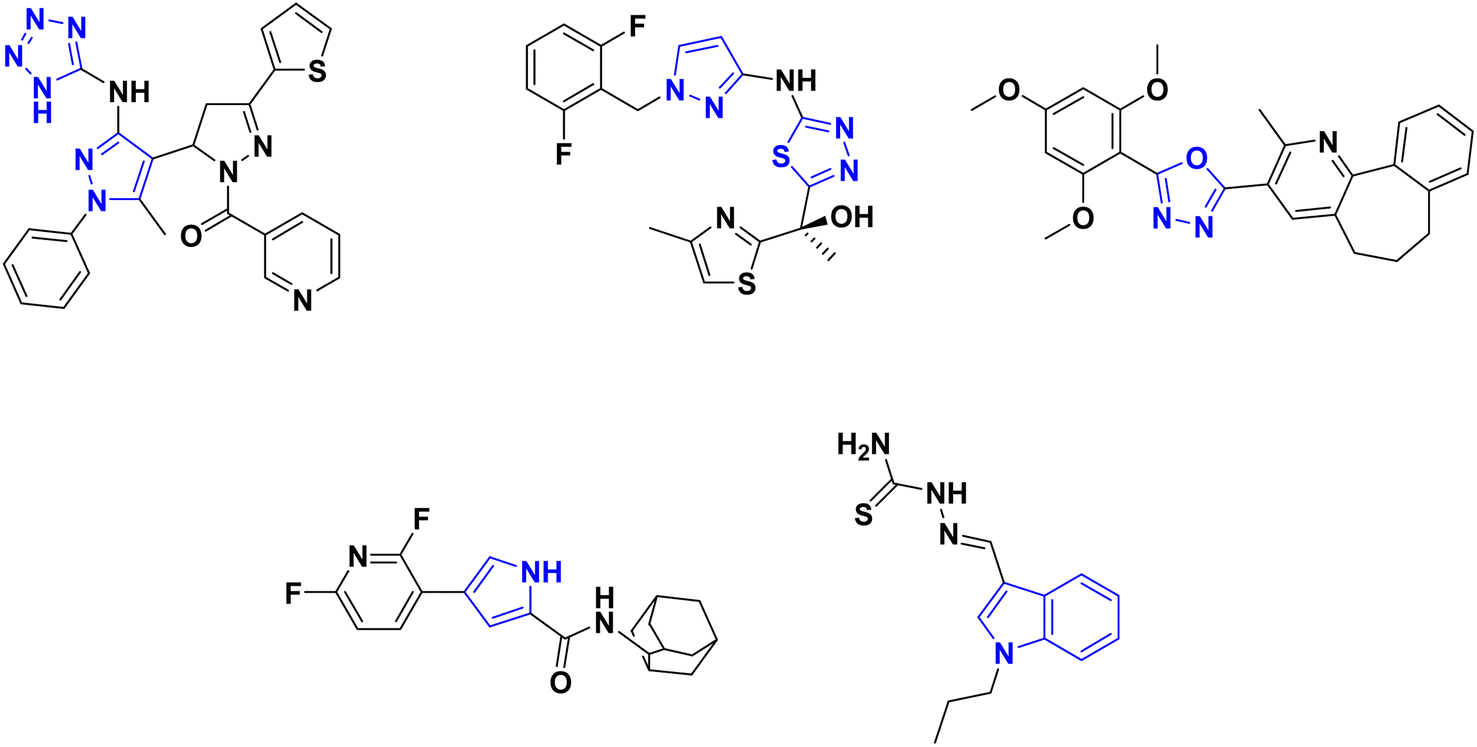 | ||
| Fig. 2 Azaheterocyclic compounds showing antimycobacterial and InhA inhibition effects. | ||
A molecular hybridization strategy was used in drug design and development based on the combination of different pharmacophoric moieties to afford a new hybrid entity with possibly improved affinity and efficacy and reduced undesirable side effects. In this investigation, we aimed to study the potential antitubercular effects of new azaheterocyclic coumarin hybrids against the wild type Mtb H37Rv and the mutant strain (ΔkatG), targeting InhA and taking their safety profile into concern. The design of the target derivatives was rationally established by replacing the quinoline core of the FDA-approved antimycobacterial drugs, e.g. bedaquiline, with the coumarin isostere conserving the common pharmacophoric features of triclosan InhA inhibitor (Fig. 3). Moreover, molecular docking studies were performed to evaluate the modes of interactions between the potent derivatives and the target enzyme. The pharmacokinetic parameters of the most active compound were also studied.
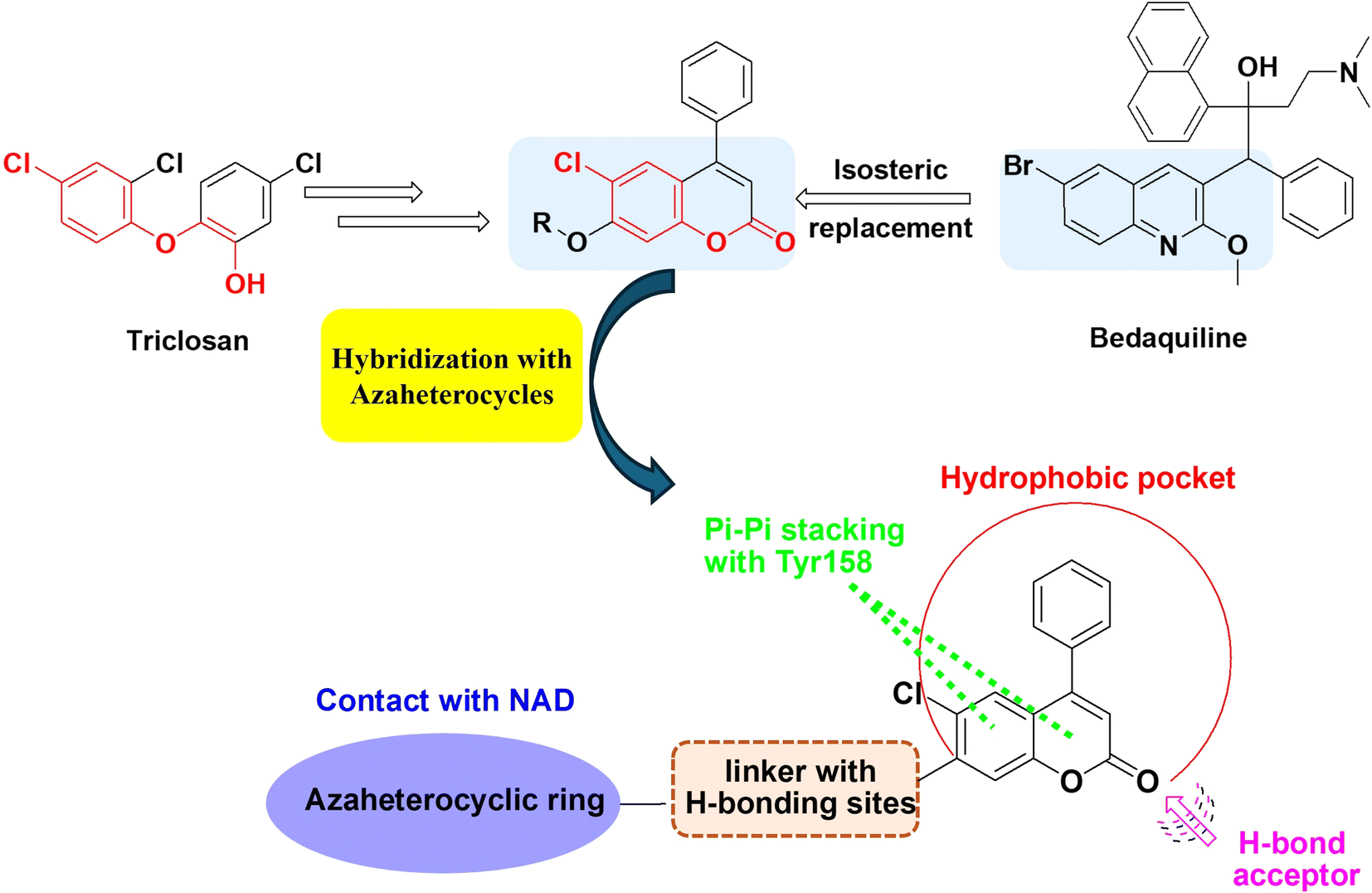 | ||
| Fig. 3 Design strategy for the synthesis of coumarin-azaheterocycle hybrids and possible interaction with InhA active site. | ||
2. Results and discussion
2.1. Chemistry
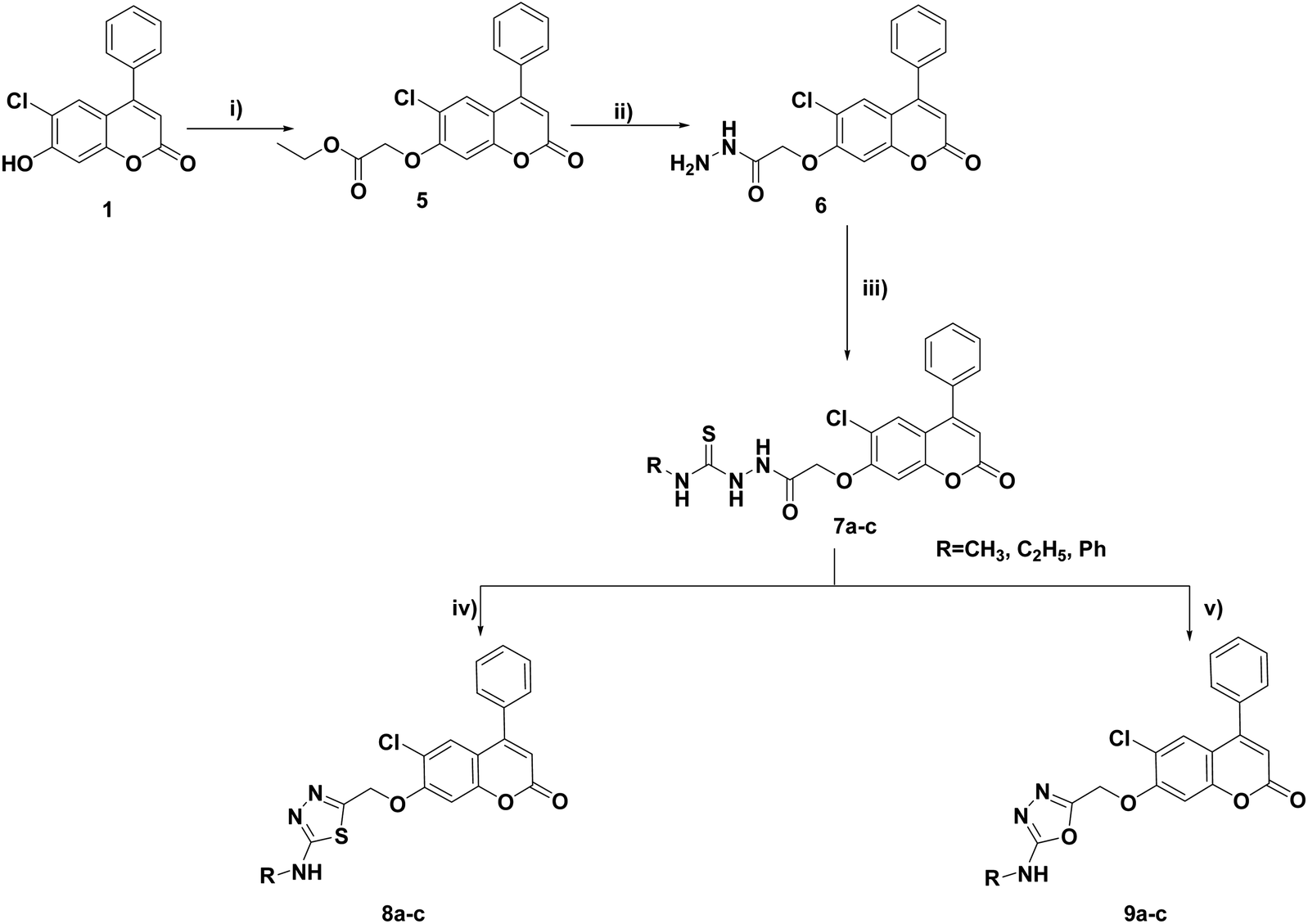
The synthesis of the target compounds 3, 4a–c, 8a–c, 9a–c and 10–17 was outlined in Schemes 1–4. Condensation of the starting compound 6-chloro-7-hydroxy-4-phenylcoumarin with 2-chloroacetonitrile in dry acetonitrile in the presence of anhydrous potassium carbonate afforded the corresponding 7-oxyacetonitrile derivative 2. The heterocyclization of compound 2 to the corresponding tetrazole derivative 3 proceeded via a reaction with sodium azide and ammonium chloride in DMF. The reaction of tetrazole derivative 3 with 2-(dimethylamino)ethyl chloride hydrochloride, 2-(diethylamino)ethyl chloride hydrochloride, and/or 4-(2-chloroethyl) morpholine hydrochloride in dry acetonitrile in the presence of anhydrous potassium carbonate yielded the corresponding dimethylaminoethyl, diethylaminoethyl and morpholinoethyl tetrazole derivatives 4a–c, respectively (Scheme 1).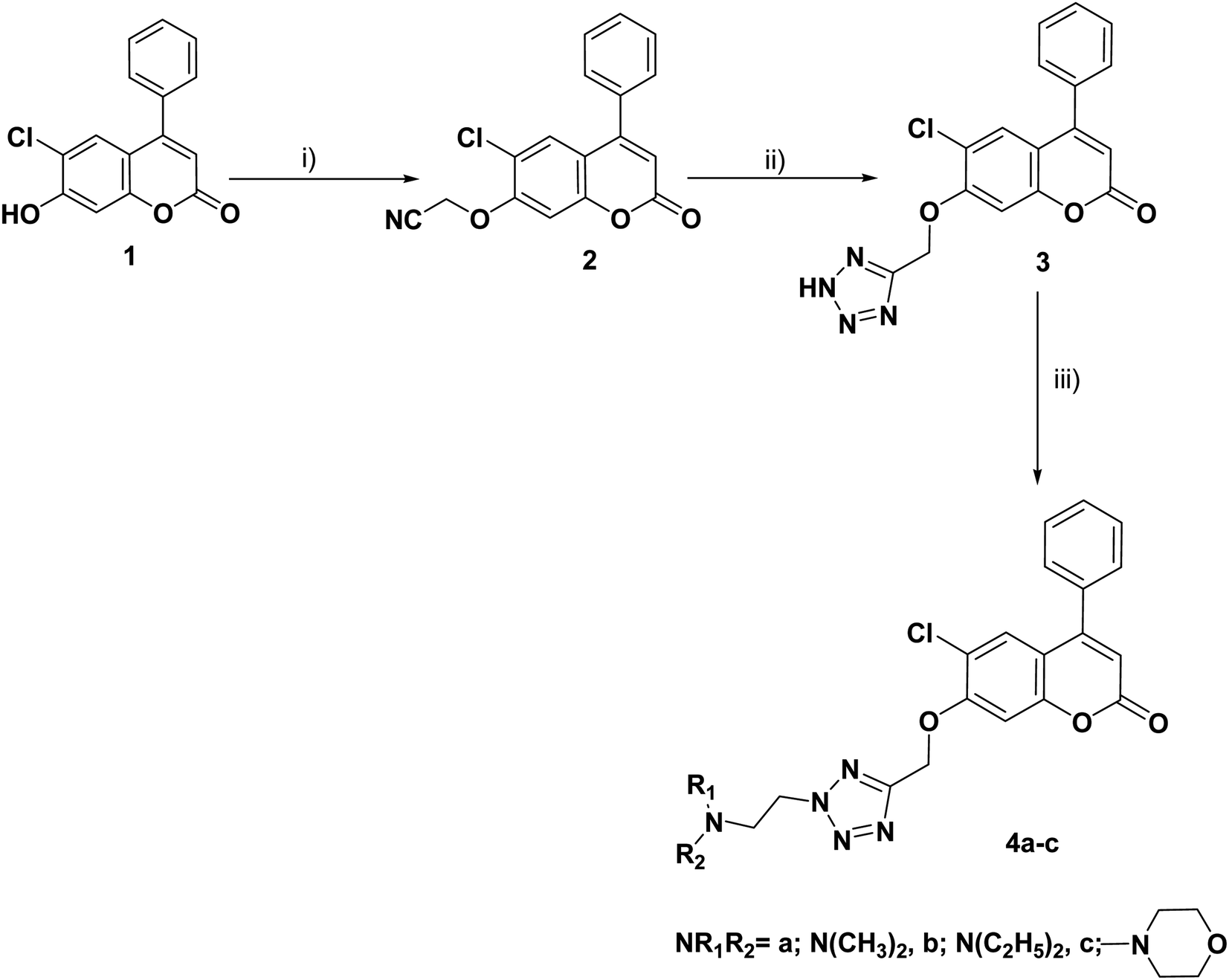 | ||
| Scheme 1 Reagents and conditions: (i) ClCH2CN, K2CO3, CH3CN, reflux, 8 h; (ii) NH4Cl, NaN3, DMF, 120 °C; 7 h (iii) ClCH2CH2NR1R2·HCl, K2CO3, CH3CN, reflux, 8–10 h. | ||
 | ||
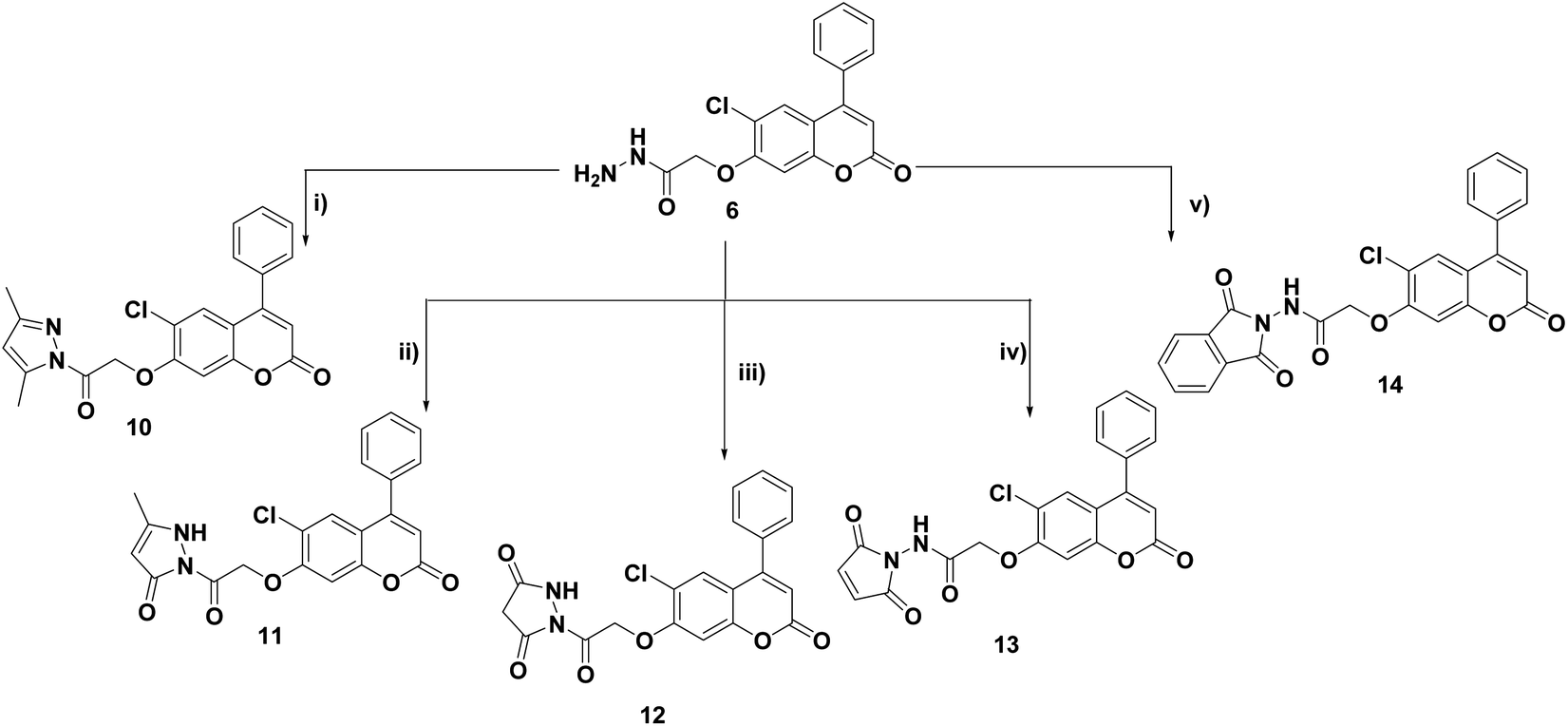
| Scheme 2 Reagents and conditions: (i) C2H5OCOCH2Br, anhydrous K2CO3, acetone, reflux, 8–10 h (ii) NH2NH2·H2O, EtOH, RT, 24 h; (iii) RNCS, EtOH, reflux, 5–7 h; (iv) H2SO4, 0 °C, 30 min; (v) pyridine, reflux, 10–14 h. | ||
 | ||
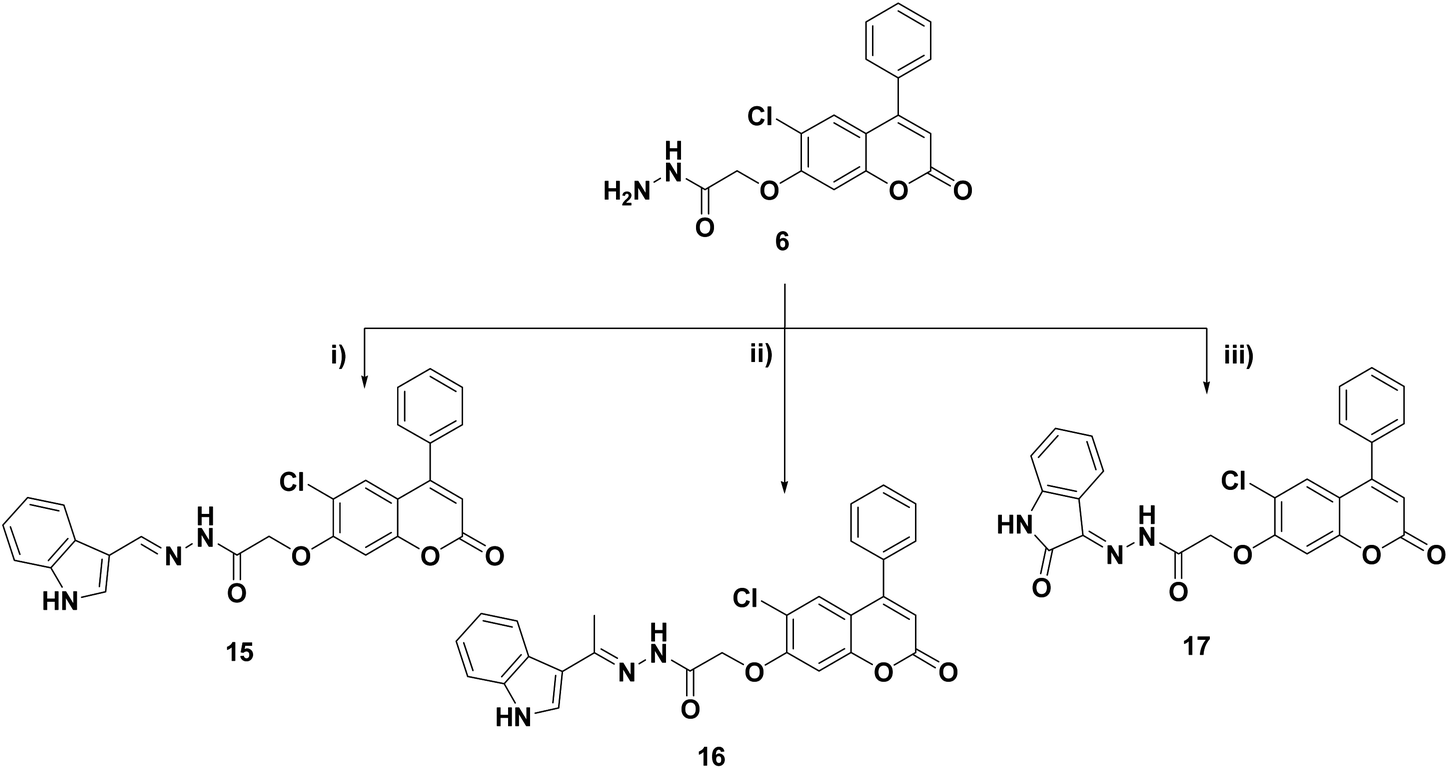
| Scheme 3 Reagents and conditions: (i) CH3COCH2COCH3, EtOH, reflux, 8 h; (ii) CH3COCH2COOC2H5, EtOH, reflux, 10 h; (iii) CH2(COOC2H5)2, AcOH, reflux, 12 h; (iv) maleic anhydride, AcOH, reflux, 6 h; (v) phthalic anhydride, AcOH, reflux, 8 h. | ||
 | ||
| Scheme 4 Reagents and conditions: (i) 3-indole carbaldehyde, EtOH, AcOH, 6 h, reflux; (ii) 3-acetylindole, EtOH, AcOH, reflux, 6 h; (iii) isatin, EtOH, AcOH, reflux, 8 h. | ||
Cyclization of the key thiosemicarbazides 7a–c in conc. sulfuric acid at 0 °C and refluxing pyridine afforded the corresponding alkyl or phenylamino-1,3,4-thiadiazoles 8a–c and 1,3,4-oxadiazole derivatives 9a–c, respectively (Scheme 2).
Cyclocondensation of the hydrazide intermediate 6 with different β-dicarbonyl compounds, namely, acetylacetone, ethylacetoacetate and diethylmalonate, yielded the corresponding 3,5-dimethylpyrazole 10, 3-methylpyrazol-5-one 11 and pyrazolidine-3,5-dione 12, respectively. The target imides, 2,5-dioxo-2H-pyrrole 13 and 1,3-dioxoisoindoline 14, were obtained by the heterocyclization of hydrazide 6 with different acid anhydrides as maleic and/or phthalic anhydride in acetic acid, respectively (Scheme 3).
Refluxing the intermediate hydrazide 6 with different indole compounds, namely, 3-indole carbaldehyde, 3-acetyl indole and/or isatin, in ethyl alcohol using a catalytic amount of glacial acetic acid afforded the corresponding Schiff's bases 15–17 (Scheme 4). The structures of the newly synthesized target compounds were confirmed by applying analytical and spectral methods, such as IR, MS, 1H NMR and 13C NMR.
2.2. Biological activity
2.2.1.1 Antitubercular effects and cytotoxicity of the synthesized target compounds. All the target azaheterocyclic coumarin hybrids were assessed for their activity towards the tubercle bacilli, M. tuberculosis and nontuberculous mycobacteria M. abscessus. Primary screening showed that most of the tested compounds suppressed the growth of M. tuberculosis but not M. abscessus at 125 μg mL−1. The minimal inhibitory concentration (MIC) was investigated for the active compounds against M. tuberculosis using MABA (microplate alamar blue assay) (Tables 1 and S1†). The tetrazole derivatives 4b and 4c showed the lowest MIC value of 15 μg mL−1. The methylamino-1,3,4-oxadiazol compound 9a inhibited the growth of Mtb H37Rv, displaying an MIC value of 31.25 μg mL−1. The tetrazoles 3 and 4a, thiadiazoles 8b,c and their oxadiazole congeners 9b,c in addition to the pyrazoles 11 and 12 and imide compounds 13 and 14 exhibited MIC values ranging from 62.5 to 125 μg mL−1. Although the relationship between the structure of the target compounds and their anti-Mtb activity is not very clear, some points could still be concluded. The elongation of the dialkylamino side chain of the tetrazole derivatives could enhance the antitubercular effects; for example, diethylamino and morpholino derivatives 4b and 4c demonstrated more potent inhibition effects than the dimethylamino derivative 4a. Similarly, ethylamino and phenylamino-thiadiazole compounds 8b and 8c showed moderate potency when compared to the methylamino derivative 8a, which was devoid of any antitubercular effect. On the contrary, the presence of long alkylamino chains or bulky groups on the oxadiazole derivatives showed reduced anti-TB effects; for example, the ethylamino-oxadiazole 9b and phenylamino-oxadiazole 9c derivatives were less potent than the methylamino-oxadiazole derivative 9a. For the pyrazole-linked coumarin derivatives, methylpyrazol-5-one compound 11 was the most active, while the activity was dramatically reduced for 3,5-dioxopyrazole compound 12 and completely disappeared for 3,5-dimethylpyrazole derivative 10. Moreover, compounds with acetohydrazide linker 15–17 lost any activity towards Mtb.
| Compounds | Mtb − MIC [μg mL−1] | IC50-L929 [μg mL−1] | IC50/MIC − Mtb |
|---|---|---|---|
| 3 | 62.5 | ND | ND |
| 4a | 62.5 | ND | ND |
| 4b | 15 | 31.25 | 2 |
| 4c | 15 | 125 | 8 |
| 8b | 62.5 | ND | ND |
| 8c | 62.5 | ND | ND |
| 9a | 31.25 | 62.5 | 2 |
| 9b | 62.5 | ND | ND |
| 9c | 62.5 | ND | ND |
| 11 | 62.5 | ND | ND |
| 12 | 125 | ND | ND |
| 13 | 62.5 | ND | ND |
| 14 | 62.5 | ND | ND |
| INH | 0.05 | — | — |
| ETH | 0.25–16 (ref. 52) | — | — |
Compounds showing inhibition potential at concentrations ≤31.25 μg mL−1 were assessed for their cytotoxicity against mouse fibroblast cell line L929 using the MTT assay according to international standards (ISO 10993-5:2009(E)).51 The determined IC50 value was about 8-fold higher for compound 4c and 2 times higher for compounds 4b and 9a (Table 2). Considering the high selectivity index (SI) ratio of 4c compared to derivatives 4b and 9a as well as to previously reported coumarin compounds,34,35 compound 4c was chosen for further biological assays.
| Compounds | ΔkatG MIC [μg mL−1] |
|---|---|
| 4c | 15 |
| INH | 200 |
2.2.1.2 Antimycobacterial activity of 4c against ΔkatG mutant. Given that InhA enzyme is the target for the reference INH and the compounds tested in this study, the efficacy of the promising compound 4c was assessed against ΔkatG mutant strain lacking the functional KatG enzyme, which shows resistance to INH. The promising anti-mycobacterial activity observed against the ΔkatG strain suggests that 4c could be a potential candidate for future tuberculosis treatments (Table 2).
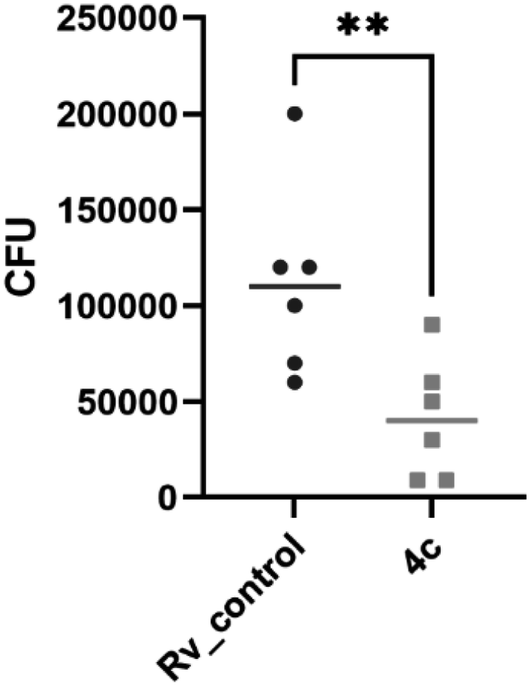
2.2.1.3 Evaluation of the bactericidal activity of the target compound 4c. The bactericidal effect was evaluated for the promising compound 4c by assessing the viability of bacteria exposed to the tested compound and by determining the number of colony-forming units (CFU). Compound 4c displayed about 60% decrease in the viability of tubercle bacilli after 7 and 14 days of incubation at 25 μg mL−1 concentration and about 90% decrease at 75 μg mL−1 (Fig. 4).
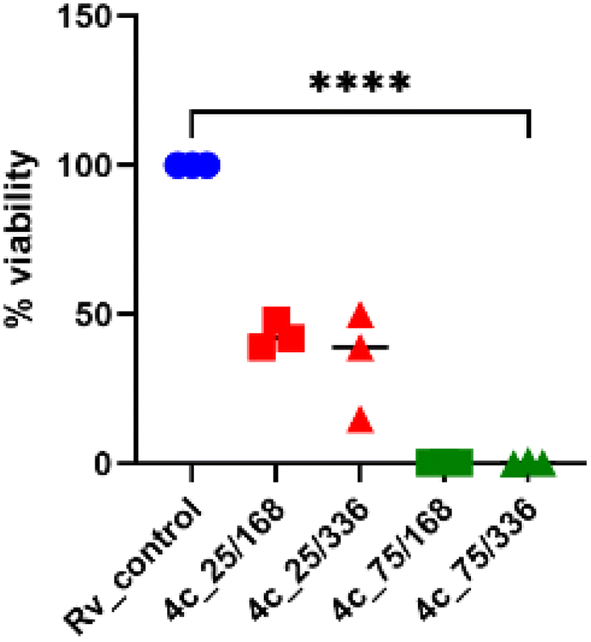 | ||
| Fig. 4 Bactericidal effect of 4-phenylcoumarin 4c. The tested compound was used at concentrations of 25 and 75 μg mL−1 for 7 (168 h) and 14 days (336 h), respectively. The number of viable bacteria was assessed through CFU analysis. Ordinary one-way ANOVA test was applied to compare control untreated M. tuberculosis (Rv_control) to bacilli treated with the tested compound at indicated concentrations and time points. The adjusted p value was <0.0001 for each pair comparison. The statistical analysis and graph were prepared using GraphPad Prism 9 version 9.3.1. | ||
2.2.1.4 Assessment of the antimycobacterial activity of 4c inside human macrophages. Potential antimycobacterial drugs should be able to penetrate human macrophages to suppress the multiplication of M. tuberculosis that is located inside these phagocytic cells without causing lysis. Thus, the promising compound 4c was evaluated for its activity towards tubercle bacilli inside human macrophages and cytotoxicity to human monocyte-derived macrophages (MDMs) in concentrations 2× and 4× MIC (Table S1†). Compound 4c showed a significant decrease in the number of viable bacilli (p = 0.0041) compared to the untreated control (Fig. 5 and Table S2†), which proved its capability to cross the cell membrane.
 | ||
| Fig. 5 Human monocyte-derived macrophages (MDMs) infected with M. tuberculosis and treated with compound 4c in a 2× MIC concentration. Unpaired t test was applied to compare a number of intracellular M. tuberculosis (control) to bacilli treated with compound 4c. The adjusted p value was determined as p = 0.0041. The statistical analysis was performed and the graph was prepared using GraphPad Prism 9 version 9.3.1. | ||
2.2.1.5 Assessment of the effectiveness of 4c against mycobacterial biofilms. M. tuberculosis can adhere to surfaces and develop biofilms that are important factors for antimicrobial resistance.53 The potential new anti-tuberculosis agents should exhibit bactericidal effects against intra- or extracellular planktonic bacilli and against biofilm-forming bacteria. The bactericidal effect of compound 4c on the Mtb biofilm formation was identified using a resazurin-based assay to determine the viability of the tubercle bacilli in tested and control untreated Mtb biofilm cultures. The presence of compound 4c in the 2× MIC concentration decreased the biofilm viability by approximately 10% (p < 0.0001) (Fig. 6). Therefore, it seems that the tested compound 4c has limited efficacy against biofilms formed by Mtb, which may result from its greater bacteriostatic (MIC value) than bactericidal activity.
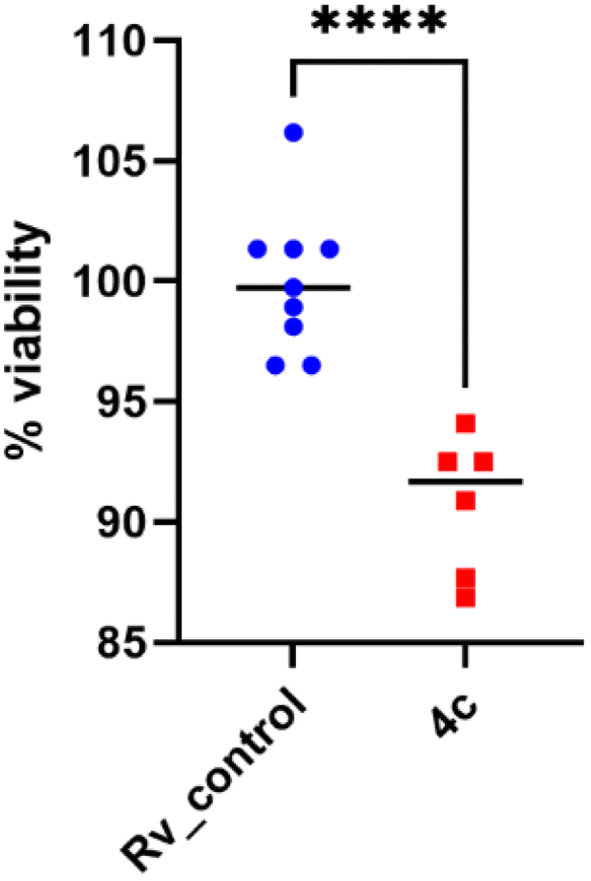 | ||
| Fig. 6 Effect of compound 4c on the mature biofilm composed of M. tuberculosis. Data were compared using unpaired t test. **** depicts the values with significant differences at p < 0.0001. The statistical analysis was performed and the graph was prepared using GraphPad Prism 9 version 9.3.1. | ||
| Compound | IC50 (μM) |
|---|---|
| 4c | 0.565 ± 0.02 |
| Triclosan | 0.731 ± 0.03 |
2.3. Molecular docking
To predict the binding mode of the most active compound 4c within the specified InhA active site, a molecular docking study was performed in which the co-crystallized ligand (PDB ID: 4TZK)19 was used as a reference to compare the docking results of the examined compound 4c. Docking was carried out in the presence of NAD cofactor, which participated in the recognition and binding of the co-crystalized ligand. The docking process was validated by redocking the co-crystallized ligand. The RMSD value of the redocked ligand was 0.5699 Å, which is a precise prediction of the favorable position. The co-crystallized ligand was able to create two hydrogen bonds with Tyr158 and Met161, as well as several hydrophobic interactions, and displayed a docking score of −9.5 kcal mol−1 (Fig. 7).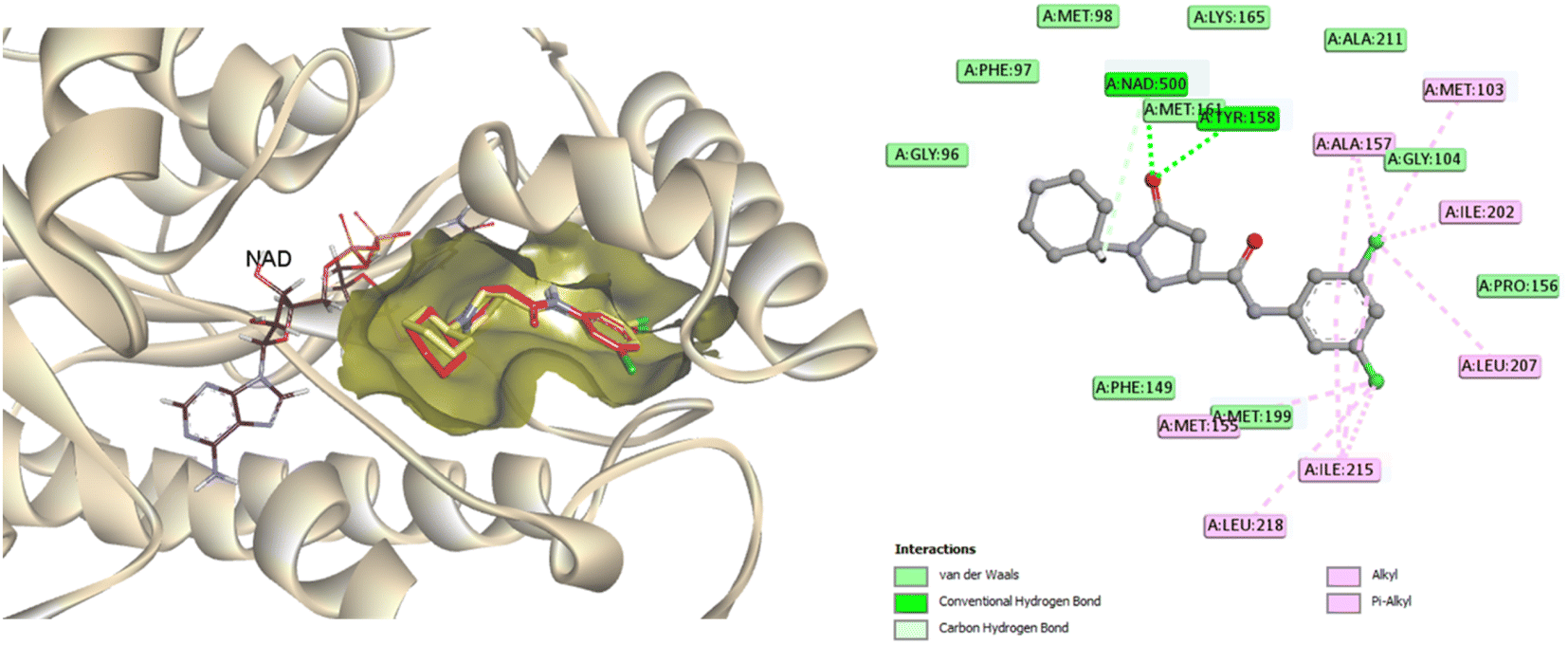 | ||
| Fig. 7 2D interaction map and 3D overlay plot in the InhA protein active site (PDB ID: 4TZK) between co-crystallized (red) and redocked (yellow) ligands showing an RMSD of 0.5699 Å. | ||
The most active compound, 4c, occupied the active binding site in contact with the NAD, as demonstrated in Fig. 8, and displayed a strong binding affinity of −9.2 kcal mol−1, which is about 0.3 kcal mol−1 less than that of the co-crystallized ligand (−9.5 kcal mol−1). The coumarin core was observed to form π–π stacking interaction with Tyr158. The 4-phenylcoumarin system was found to interact hydrophobically with the non-polar pocket of the InhA active site, which includes Met103, Phe149, Gly104, Pro156, Met155, Tyr158, Ala157, Ile215, Ile202, Met199 and Leu218. Furthermore, the coumarin carbonyl oxygen atom was able to form an H-bonding interaction with the Gly104 residue. Compound 4c and Tyr158 established one more H-bond via the tetrazole ring's nitrogen. As shown in Fig. 8, the tetrazole ring also established a π–sulfur interaction with Met161. Furthermore, 4c maintained hydrophobic contact with NAD through its 2-morpholinoethyl tetrazole tail. In conclusion, 4c displayed a strong binding affinity for the active site of InhA, matching the co-crystallized ligand that could clarify the modes of interaction of the recently synthesized candidates and contribute to the creation of new anti-tuberculosis agents.
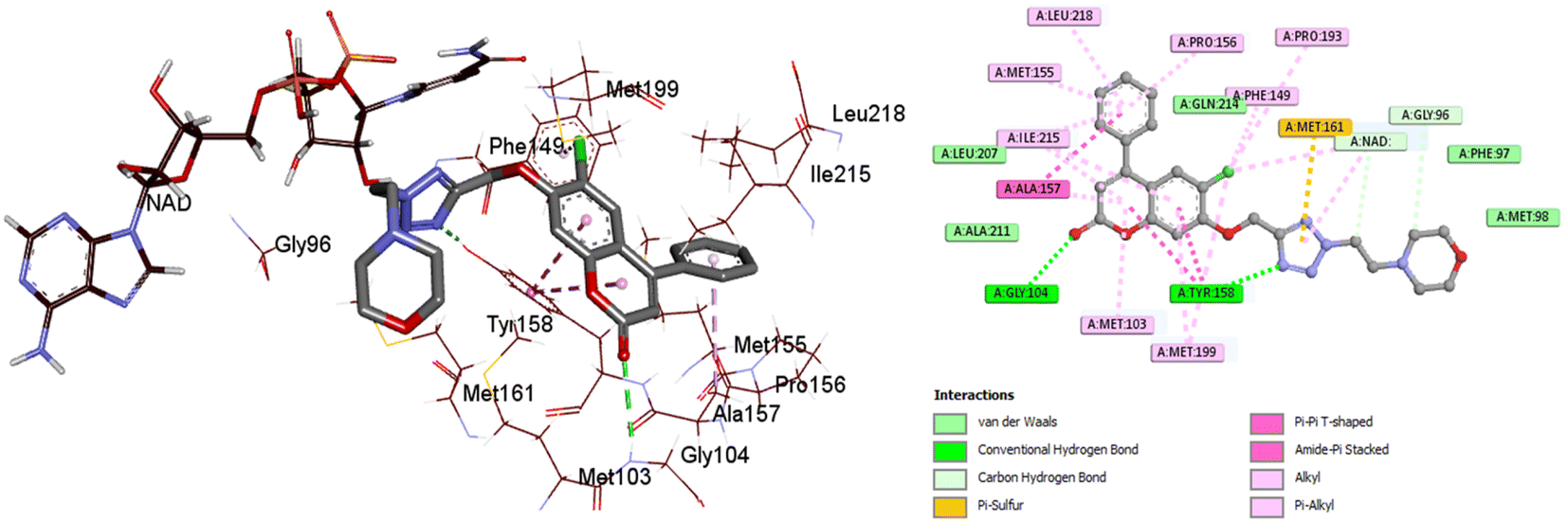 | ||
| Fig. 8 2D and 3D interaction maps between InhA residues and ligand 4c. | ||
2.4. Pharmacokinetics and physicochemical properties of compound 4c
The drug-likeness properties and parameters of the promising compound 4c were predicted using the free web tool SwissADME, and some in silico descriptors and parameters were also calculated. The target compound 4c displayed high gastrointestinal absorption with a moderate solubility profile and 0.55 bioavailability value, and it also showed no PAINS alerts. Furthermore, the investigated derivative was identified as a non-carcinogenic non-biodegradable compound (Table 4).| Compd | #Heavy atoms | #Rotatable bonds | #H-bond acceptors | #H-bond donors | MR | TPSA | WLOGP |
|---|---|---|---|---|---|---|---|
| 4c | 33 | 7 | 8 | 0 | 126.36 | 95.51 | 2.48 |
| Compd | SOL class | GI absorption | Carcinogenicity | Lipinski #violations | Bioavailability score | PAINS #alerts | Biodegradation |
|---|---|---|---|---|---|---|---|
| 4c | Moderately soluble | High | No | 0 | 0.55 | 0 | No |
3. Conclusion
In this investigation, we designed and synthesized a series of coumarin derivatives bearing different azaheterocyclic rings to assess their effects against TB. The coumarin-tetrazole hybrid compound 4c showed the highest potency against Mtb H37Rv, with an MIC value of 15 μg mL−1 and a favorable safety profile toward L929. It also inhibited the growth of the mutant strain (ΔkatG) that is resistant to the reference drug INH. Moreover, compound 4c was able to suppress the multiplication of M. tuberculosis inside human professional phagocytes and displayed low cytotoxicity against host cells. Compound 4c was further evaluated for its inhibitory activity against InhA and exhibited significant activity at low micromolar concentration (IC50 = 0.565 μM). The binding mode of 4c within InhA binding pocket was also investigated via molecular docking, which revealed that 4c exhibited a strong binding affinity comparable to the reference compound (ΔG = −9.2 kcal mol−1), forming potential interactions with essential amino acids within the enzyme active site. Additionally, compound 4c revealed favorable pharmacokinetic properties and obeyed Lipinski's rule. These findings confirm that compound 4c could be considered a promising antitubercular candidate endowed with characteristic inhibitory properties against the MTB InhA enzyme.4. Materials and methods
4.1. Chemistry
All melting points were uncorrected and measured using the electrothermal IA 9000 apparatus. Infrared spectra (IR) were measured using a JASCO FT/IR-4100 spectrometer with KBr discs. The nuclear magnetic resonance spectra 1H NMR (400 MHz) and 13C NMR spectra (100 MHz) were recorded using a Bruker spectrometer with TMS as the internal standard. The mass spectrum was carried out on the Direct Inlet part of the mass analyzer in the Thermo Scientific GCMS model ISQ. The reactions were followed by TLC (silica gel, aluminum sheets 60 F254, Merck) using chloroform–methanol (9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 v/v) as an eluent and sprayed with iodine-potassium iodide reagent. The purity of the newly synthesized compounds was assessed by TLC and elemental analysis and was found to be higher than 95%. The key intermediates 5, 6 and 7a–c were previously synthesized.54
:0.5 v/v) as an eluent and sprayed with iodine-potassium iodide reagent. The purity of the newly synthesized compounds was assessed by TLC and elemental analysis and was found to be higher than 95%. The key intermediates 5, 6 and 7a–c were previously synthesized.54
Yield 89%, mp 170–1 °C. Anal. calcd for C17H10ClNO3 (311.72): C, 65.50; H, 3.23; N, 4.49. Found: C, 65.59; H, 3.31; N, 4.59. IR (cm−1, KBr): 3053 (CH aromatic stretching), 2917 (CH aliphatic stretching), 2362 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1720 (C
N), 1720 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching). 1H NMR (DMSO-d6, δ, ppm): 5.47 (2H, s, OCH2), 6.43 (1H, s, H-3 coumarin), 7.42–7.61 (7H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 55.36, 103.42, 114.03, 114.43, 116.21, 118.22, 127.75, 128.91, 129.53, 130.45, 134.37, 154.12, 154.28, 154.60, 159.78. MS m/z (R.A. %): 311, 313 (M+, M++2) (41.05, 12.61%), 115 (100.00%).
O stretching). 1H NMR (DMSO-d6, δ, ppm): 5.47 (2H, s, OCH2), 6.43 (1H, s, H-3 coumarin), 7.42–7.61 (7H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 55.36, 103.42, 114.03, 114.43, 116.21, 118.22, 127.75, 128.91, 129.53, 130.45, 134.37, 154.12, 154.28, 154.60, 159.78. MS m/z (R.A. %): 311, 313 (M+, M++2) (41.05, 12.61%), 115 (100.00%).
:1) to give compound 3.Yield 85%, mp 259–60 °C. Anal. calcd for C17H11ClN4O3 (354.75): C, 57.56; H, 3.13; N, 15.79. Found: C, 57.66; H, 3.19; N, 15.86. IR (cm−1, KBr): 3424 (NH stretching), 3105 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1679 (CO stretching). 1H NMR (DMSO-d6, δ, ppm): 5.66 (2H, s, OCH2), 6.25 (1H, s, H-3 coumarin), 7.01–7.58 (7H, m, Ar–H), 11.50 (1H, brs, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 56.50, 104.36, 112.02, 117.47, 127.42, 128.80, 129.44, 130.27, 135.08, 154.19, 154.80, 157.09, 160.13. MS m/z (R.A. %): 354, 356 (M+, M++2) (2.83, 0.90%), 128 (100.00%).
4.1.3.1 7-((2-(2-(Dimethylamino)ethyl)-2H-tetrazol-5-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 4a. Yield 76%; mp 87–8 °C. Anal. calcd for C21H20ClN5O3 (425.87): C, 59.23; H, 4.73; N, 16.44. Found: C, 59.31; H, 4.82; N, 16.52. IR (cm−1, KBr): 3067 (CH aromatic stretching), 2919, 2851 (CH aliphatic stretching), 1725 (C
O stretching). 1H NMR (CDCl3, δ, ppm): 2.38 (6H, s, 2CH3N), 2.83–2.86 (2H, t, CH2N), 4.19–4.22 (2H, t, CH2S), 5.48 (2H, s, OCH2), 6.25 (1H, s, H-3 coumarin), 6.93–7.54 (7H, m, Ar–H). 13C NMR (CDCl3, δ, ppm): 31.93, 47.82, 57.51, 68.11, 101.44, 112.75, 117.63, 127.50, 128.27, 129.01, 129.82, 134.73, 154.27, 154.65, 155.07, 155.82, 160.86. MS m/z (R.A. %): 425, 427 (M+, M++2) (27.7%, 9.1%), 167 (100.00%).
4.1.3.2 7-((2-(2-(Diethylamino)ethyl)-2H-tetrazol-5-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 4b. Yield 75%, mp 91–2 °C. Anal. calcd for C23H24ClN5O3 (453.92): C, 60.86; H, 5.33; N, 15.43. Found: C, 60.94; H, 5.39; N, 15.52. IR (cm−1, KBr): 3061 (CH aromatic stretching), 2921, 2852 (CH aliphatic stretching), 1717 (C
O stretching). 1H NMR (CDCl3, δ, ppm): 1.28 (6H, m, 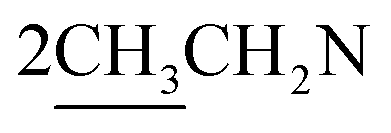 ), 2.78 (4H, m,
), 2.78 (4H, m, 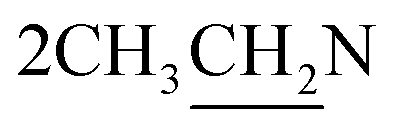 ), 3.31 (2H, t, CH2N), 4.54 (2H, t, CH2S), 5.51 (2H, s, OCH2), 6.30 (1H, s, H-3 coumarin), 6.99–7.57 (7H, m, Ar–H). 13C NMR (CDCl3, δ, ppm): 14.12, 45.51, 46.03, 57.51, 68.11, 101.44, 112.86, 119.34, 127.50, 128.28, 129.06, 129.90, 134.95, 154.27, 154.94, 156.98, 160.62. MS m/z (R.A. %): 453, 455 (M+, M++2) (66.69%, 26.03%), 209 (100.00%).
), 3.31 (2H, t, CH2N), 4.54 (2H, t, CH2S), 5.51 (2H, s, OCH2), 6.30 (1H, s, H-3 coumarin), 6.99–7.57 (7H, m, Ar–H). 13C NMR (CDCl3, δ, ppm): 14.12, 45.51, 46.03, 57.51, 68.11, 101.44, 112.86, 119.34, 127.50, 128.28, 129.06, 129.90, 134.95, 154.27, 154.94, 156.98, 160.62. MS m/z (R.A. %): 453, 455 (M+, M++2) (66.69%, 26.03%), 209 (100.00%).
4.1.3.3 7-((2-(2-Morpholinoethyl)-2H-tetrazol-5-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 4c. Yield 74%, mp 137–8 °C. Anal. calcd for C23H22ClN5O4 (467.9): C, 59.04; H, 4.74; N, 14.97. Found: C, 59.11; H, 4.83; N, 15.07. IR (cm−1, KBr): 3057 (CH aromatic stretching), 2920, 2851 (CH aliphatic stretching), 1723 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 2.57 (4H, m, 2 CH2N morpholine), 2.82 (2H, t, CH2N), 4.33–4.36 (4H, t, 2 CH2O morpholine), 3.58–3.60 (2H, t, CH2S), 5.51 (2H, s, OCH2), 6.34 (1H, s, H-3 coumarin), 7.35–7.60 (7H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 54.00, 56.83, 60.06, 66.53, 68.31, 102.83, 112.75, 112.90, 118.28, 128.84, 129.50, 130.37, 134.89, 154.48, 154.58, 157.00, 160.06. MS m/z (R.A. %): 467, 469 (M+, M++2) (2.93%, 1.03%), 59 (100.00%).
4.1.4.1 7-((5-(Methylamino)-1,3,4-thiadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 8a. Yield 79%, mp 177–8 °C. Anal. calcd for C19H14ClN3O3S (399.85): C, 57.07; H, 3.53; N, 10.51; S, 8.02. Found: C, 57.16; H, 3.61; N, 10.59; S, 8.12. IR (cm−1, KBr): 3407 (NH stretching), 3107 (CH aromatic stretching), 2918, 2853 (CH aliphatic stretching), 1720 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 2.89 (3H, s, CH3), 5.58 (2H, s, OCH2), 6.36 (1H, s, H-3 coumarin), 7.36–7.84 (8H, m, Ar–H and NH). 13C NMR (DMSO-d6, δ, ppm): 31.78, 66.20, 103.55, 113.34, 113.41, 118.39, 127.24, 128.83, 129.46, 130.36, 134.78, 152.35, 154.16, 154.39, 155.85, 159.89, 171.44. MS m/z (R.A. %): 399, 401 (M+, M++2) (100.00%, 29.86%).
4.1.4.2 7-((5-(Ethylamino)-1,3,4-thiadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 8b. Yield 86%, mp 142–3 °C. Anal. calcd for C20H16ClN3O3S (413.88): C, 58.04; H, 3.90; N, 10.15; S, 7.75. Found: C, 58.12; H, 3.99; N, 10.22; S, 7.85. IR (cm−1, KBr): 3442 (NH stretching), 3107 (CH aromatic stretching), 2918, 2850 (CH aliphatic stretching), 1722 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 1.16–1.19 (3H, t, CH3), 3.29–3.31 (2H, m, CH2), 5.58 (2H, s, OCH2), 6.36 (1H, s, H-3 coumarin), 7.36–7.95 (8H, m, Ar–H and NH). 13C NMR (DMSO-d6, δ, ppm): 14.62, 39.45, 66.16, 103.59, 113.38, 113.47, 118.39, 127.27, 128.85, 129.49, 130.38, 134.80, 152.24, 154.17, 154.43, 155.86, 159.91, 170.42. MS m/z (R.A. %): 413, 415 (M+, M++2) (6.58%, 2.16%), 114 (100.00%).
4.1.4.3 7-((5-(Phenylamino)-1,3,4-thiadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 8c. Yield 92%, mp 64–5 °C. Anal. calcd for C24H16ClN3O3S (461.92): C, 62.40; H, 3.49; N, 9.10; S, 6.94. Found: C, 62.48; H, 3.57; N, 9.19; S, 7.02. IR (cm−1, KBr): 3447 (NH stretching), 3056 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1716 (C
O stretching). 1HNMR (DMSO-d6, δ, ppm): 5.69 (2H, s, OCH2), 6.37 (1H, s, H-3 coumarin), 6.99–7.63 (12H, m, Ar–H), 10.49 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 66.06, 103.65, 113.42, 113.66, 116.96, 118.03, 118.40, 122.62, 127.10, 127.30, 128.84, 129.48, 129.56, 130.38, 134.78, 140.80, 141.00, 142.07, 154.18, 154.41, 154.55, 155.80, 159.91, 166.28. MS m/z (R.A. %): 461, 463 (M+, M++2) (19.86%, 7.23%), 57 (100.00%).
4.1.5.1 7-((5-(Methylamino)-1,3,4-oxadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 9a. Yield 80%, mp 259–60 °C. Anal. calcd for C19H14ClN3O4 (383.79): C, 59.46; H, 3.68; N, 10.95. Found: C, 59.55; H, 3.73; N, 11.05. IR (cm−1, KBr): 3445 (NH stretching), 3133 (CH aromatic stretching), 2917, 2850 (CH aliphatic stretching), 1677 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 2.86 (3H, s, CH3), 5.58 (2H, s, OCH2), 6.26 (1H, s, H-3 coumarin), 7.01–7.59 (7H, m, Ar–H), 11.55 (1H, s, NH, D2O exchangeable). 13CNMR (DMSO-d6, δ, ppm): 31.78, 66.24, 103.55, 113.36, 113.41, 118.39, 127.20, 128.85, 129.46, 130.34, 134.78, 152.35, 154.16, 154.38, 155.83, 159.89, 171.46. MS m/z (R.A. %): 383, 385 (M+, M++2) (53.32%, 16.68%), 306 (100.00%).
4.1.5.2 7-((5-(Ethylamino)-1,3,4-oxadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 9b. Yield 80%, mp 222–3 °C. Anal. calcd for C20H16ClN3O4 (397.81): C, 60.38; H, 4.05; N, 10.56. Found: C, 60.44; H, 4.11; N, 10.64. IR (cm−1, KBr): 3442 (NH stretching), 3107 (CH aromatic stretching), 2918, 2850 (CH aliphatic stretching), 1679 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 1.26–1.29 (3H, t, CH3), 3.29–3.31 (2H, m, CH2), 5.54 (2H, s, OCH2), 6.26 (1H, s, H-3 coumarin), 7.29–7.59 (7H, m, Ar–H), 11.55 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 13.78, 37.84, 61.69, 103.59, 112.01, 113.51, 118.26, 127.45, 128.88, 129.46, 130.29, 134.80, 147.32, 154.20, 154.42, 155.52, 157.03, 159.90, 160.12. MS m/z (R.A. %): 397, 399 (M+, M++2) (39.59%, 13.15%), 145 (100.00%).
4.1.5.3 7-((5-(Phenylamino)-1,3,4-oxadiazol-2-yl)methoxy)-6-chloro-4-phenyl-2H-chromen-2-one 9c. Yield 82%, mp 225–6 °C. Anal. calcd for C24H16ClN3O4 (445.85): C, 64.65; H, 3.62; N, 9.42. Found: C, 64.75; H, 3.70; N, 9.51. IR (cm−1, KBr): 3424 (NH stretching), 3063 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1716 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 5.56 (2H, s, OCH2), 6.25 (1H, s, H-3 coumarin), 7.01–7.81 (12H, m, Ar–H), 11.55 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 66.07, 103.65, 113.40, 113.56, 116.96, 118.03, 118.41, 122.62, 127.10, 127.28, 128.84, 129.49, 129.55, 130.38, 134.78, 140.80, 141.00, 142.06, 154.18, 154.42, 154.55, 155.80, 159.90, 166.30. MS m/z (R.A. %): 445, 447 (M+, M++2) (12.45%, 3.99%), 167 (100.00%).
Yield 90%, mp 205–6 °C. Anal. calcd for C22H17ClN2O4 (408.83): C, 64.63; H, 4.19; N, 6.85. Found: C, 64.69; H, 4.27; N, 6.94. IR (cm−1, KBr): 3118 (CH aromatic stretching), 2917, 2850 (CH aliphatic stretching), 1728, 1609 (CO stretching). 1HNMR (DMSO-d6, δ, ppm): 2.25 (3H, s, CH3-pyrazole), 2.48 (3H, s, CH3-pyrazole), 5.81 (2H, s, OCH2), 6.28 (1H, s, CH-pyrazole), 6.34 (1H, s, H-3 coumarin), 7.38–7.61 (7H, m, Ar–H). 13C NMR (DMSO-d6, δ, ppm): 14.01, 67.79, 103.26, 111.80, 113.21, 113.33, 118.24, 127.22, 128.87, 129.50, 130.37, 134.91, 144.25, 153.06, 154.23, 154.47, 156.53, 159.98, 167.35. MS m/z (R.A. %): 408, 410 (M+, M++2) (100.00%, 35.40%).
Yield 86%, mp 160–161 °C. Anal. calcd for C21H15ClN2O5 (410.81): C, 61.40; H, 3.68; N, 6.82. Found: C, 61.48; H, 3.77; N, 6.90. IR (cm−1, KBr): 3434 (NH stretching), 3107 (CH aromatic stretching), 2918, 2851 (CH aliphatic stretching), 1731, 1695, 1610 (CO stretching). 1H NMR (DMSO-d6, δ, ppm): 3.37 (3H, s, CH3-pyrazole), 5.28 (2H, s, OCH2), 6.34 (1H, s, CH-pyrazole), 6.36 (1H, s, H-3 coumarin), 7.15–7.61 (7H, m, Ar–H), 10.78 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 16.79, 66.74, 102.83, 112.98, 113.20, 118.24, 127.26, 128.86, 129.51, 130.38, 134.93, 148.76, 154.15, 154.55, 156.46, 159.99, 168.61, 169.92. MS m/z (R.A. %): 410, 412 (M+, M++2) (40.19%, 12.89%), 139 (100.00%).
Yield 82%, mp > 300 °C. Anal. calcd for C20H13ClN2O6 (412.78): C, 58.19; H, 3.17; N, 6.79. Found: C, 58.27; H, 3.26; N, 6.87. IR (cm−1, KBr): 3250 (NH stretching), 3063 (CH aromatic stretching), 2919, 2851 (CH aliphatic stretching), 1714, 1638, 1603 (CO stretching). 1H NMR (DMSO-d6, δ, ppm): 3.71 (2H, s, CH2 pyrazole), 4.34 (2H, s, OCH2), 6.62 (1H, s, H-3 coumarin), 6.63–7.53 (7H, m, Ar–H), 12.00 (1H, s, NH pyrazole, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 55.71, 61.61, 103.98, 107.34, 115.37, 116.59, 118.78, 127.39, 135.81, 136.15, 141.88, 142.87, 145.70, 147.76, 149.81, 158.80, 160.99, 161.13, 161.95. MS m/z (R.A. %): 412, 414 (M+, M++2) (13.56%, 4.18%), 345 (100.00%).
4.1.9.1 2-(6-Chloro-2-oxo-4-phenyl-2H-chromen-7-yloxy)-N-(2,5-dioxo-2H-pyrrol-1(5H)-yl) acetamide 13. Yield 82%, mp 221–2 °C. Anal. calcd for C21H13ClN2O6 (424.79): C, 59.38; H, 3.08; N, 6.59. Found: C, 59.46; H, 3.15; N, 6.68. IR (cm−1, KBr): 3423 (NH stretching), 3061 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1714, 1605 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 4.84 (2H, s, OCH2), 6.21 (1H, s, H-3 coumarin), 7.00–7.59 (10H, m, Ar–H, CH pyrrole and NH). 13C NMR (DMSO-d6, δ, ppm): 67.29, 111.59, 112.73, 117.74, 118.27, 127.27, 128.77, 129.43, 130.24, 134.90, 135.14, 154.29, 154.88, 156.94, 157.80, 160.02, 160.23. MS m/z (R.A. %): 424, 426 (M+, M++2) (15.19%, 5.55%), 127 (100.00%).
4.1.9.2 2-(6-Chloro-2-oxo-4-phenyl-2H-chromen-7-yloxy)-N-(1,3-dioxoisoindolin-2-yl) acetamide 14. Yield 88%, mp 277–8 °C. Anal. calcd for C25H15ClN2O6 (474.85): C, 63.23; H, 3.18; N, 5.90. Found: C, 63.29; H, 3.26; N, 5.96. IR (cm−1, KBr): 3426 (NH stretching), 3063 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1736, 1671, 1604 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 5.14 (2H, s, OCH2), 6.39 (1H, s, H-3 coumarin), 7.26–8.01 (11H, m, Ar–H), 11.09 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 66.98, 103.23, 113.41, 113.58, 118.43, 124.33, 127.32, 128.85, 129.52, 129.85, 130.41, 134.80, 135.86, 154.12, 154.49, 156.20, 159.98, 165.33, 166.94. MS m/z (R.A. %): 474, 476 (M+, M++2) (7.10%, 2.56%), 209 (100.00%).
4.1.10.1 N′-((1H-Indol-3-yl)methylene)-2-(6-chloro-2-oxo-4-phenyl-2H-chromen-7-yloxy) acetohydrazide 15. Yield 84%, mp > 300 °C. Anal. calcd for C26H18ClN3O4 (471.89): C, 66.18; H, 3.84; N, 8.90. Found: C, 66.26; H, 3.93; N, 8.99. IR (cm−1, KBr): 3428, 3250 (NH stretching), 3061 (CH aromatic stretching), 2918 (CH aliphatic stretching), 2851 (CH = azomethine stretching), 1717, 1677 (C
O stretching), 1609 (CN azomethine stretching). 1H NMR (DMSO-d6, δ, ppm): 5.53 (2H, s, OCH2), 6.34 (1H, s, H-3 coumarin), 7.12–8.17 (12H, m, Ar–H), 8.23 (1H, s, CHN), 11.43 (1H, s, NH, D2O exchangeable), 11.58 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 66.86, 103.07, 111.75, 112.30, 112.95, 118.27, 121.04, 122.37, 123.10, 124.55, 127.12, 128.86, 129.51, 130.36, 131.12, 134.96, 137.55, 142.12, 145.54, 154.18, 154.58, 157.17, 160.02, 162.49, 167.39. MS m/z (R.A. %): 471, 473 (M+, M++2) (51.35%, 16.83%), 295 (100.00%).
4.1.10.2 N′-(1-(1H-Indol-3-yl)ethylidene)-2-(6-chloro-2-oxo-4-phenyl-2H-chromen-7-yloxy) acetohydrazide 16. Yield 89%, mp 268–9 °C. Anal. calcd for C27H20ClN3O4 (485.92): C, 66.74; H, 4.15; N, 8.65. Found: C, 66.84; H, 4.22; N, 8.74. IR (cm−1, KBr): 3372, 3114 (NH stretching), 3059 (CH aromatic stretching), 2917, 2850 (CH aliphatic stretching), 1715, 1680 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 2.33 (3H, s, CH3), 5.55 (2H, s, OCH2), 6.36 (1H, s, H-3 coumarin), 7.13–8.28 (12H, m, Ar–H), 10.71 (1H, s, NH, D2O exchangeable), 11.49 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 15.05, 67.41, 103.01, 112.11, 112.95, 115.16, 118.24, 120.85, 122.65, 123.74, 124.78, 127.15, 128.86, 129.06, 129.52, 130.38, 134.94, 137.63, 149.03, 154.17, 154.59, 157.10, 160.03, 168.15. MS m/z (R.A. %): 485, 487 (M+, M++2) (20.31%, 6.30%), 102 (100.00%).
4.1.10.3 2-(6-Chloro-2-oxo-4-phenyl-2H-chromen-7-yloxy)-N′-(3-oxoindolin-2-ylidene) acetohydrazide 17. Yield 86%, mp > 300 °C. Anal. calcd for C25H16ClN3O5 (473.86): C, 63.37; H, 3.40; N, 8.87. Found: C, 63.44; H, 3.49; N, 8.96. IR (cm−1, KBr): 3434, 3250 (NH stretching), 3061 (CH aromatic stretching), 2918 (CH aliphatic stretching), 1726, 1698, 1606 (C
O stretching). 1H NMR (DMSO-d6, δ, ppm): 5.16, 5.62 (2H, 2s, OCH2), 6.36 (1H, s, H-3 coumarin), 6.94–7.61 (11H, m, Ar–H), 11.27 (1H, s, NH, D2O exchangeable), 13.53 (1H, s, NH, D2O exchangeable). 13C NMR (DMSO-d6, δ, ppm): 67.41, 103.27, 111.66, 113.05, 118.33, 120.12, 121.53, 123.12, 127.25, 128.87, 129.52, 130.40, 132.56, 134.84, 142.12, 154.19, 154.45, 159.96, 162.85, 167.39. MS m/z (R.A. %): 473, 475 (M+, M++2) (23.72%, 8.97%), 209 (100.00%).
4.2. Biology
4.2.1.1 MIC determination. The minimal inhibitory concentration (MIC) for M. tuberculosis H37Rv and M. abscessus was determined in liquid 7H9/OADC (Middlebrook, Difco, Baltimore, MD, United States) media supplemented with various concentrations of the tested chemical agents. The compounds were dissolved in dimethyl sulfoxide (DMSO) and added directly to the growth medium. The final concentration of DMSO in the medium never exceeded 0.1% (vol/vol), and DMSO did not affect the growth of the bacilli. To define the MIC value, the Microplate Alamar Blue Assay (MABA test) was applied, as described by Franzblau et al.55 The susceptibility of the tested strains was assessed based on the change in color from blue to pink based on visual inspection. Wells containing only bacteria, medium, or compound were used as controls in this experiment, and the MABA test was repeated independently three times.
4.2.1.2 In vitro cytotoxicity assay. Cytotoxicity assays were performed precisely according to international standards (ISO 10993-5:2009(E)) using L929 cells and the MTT assay. Additionally, for the selected compounds, cytotoxicity was determined against human monocyte-derived macrophages, but in this case, the cells were treated with the tested compounds for 48 h.
4.2.1.3 Bactericidal effect. The bacterial activity of the tested compounds was verified by measuring the optical density (OD600) of M. tuberculosis H37Rv cultures exposed to the selected compounds and by determining the number of colony-forming units (CFU). To obtain OD600 = 0.1, the M. tuberculosis H37Rv culture was diluted by adding liquid 7H9/OADC (Middlebrook, Difco, Baltimore, MD, United States) medium supplemented with 0.05% Tween 80. The appropriate amounts of compounds were added at the following concentrations: 25 μg mL−1 and 75 μg mL−1 for 4c (2 repeats for each concentration). A bottle containing the culture without any compound represented the control for this experiment. The culture bottles were incubated at 37 °C. The optical density was measured after 7 and 14 days. The number of colony-forming units was determined by utilizing a solid 7H10/OADC (Middlebrook, Difco, Baltimore, MD, United States) medium with 0.5% glycerol. Mycobacteria from the bottles were diluted in liquid 7H9/OADC medium with 0.05% Tween 80 and cultured using prepared plates on day 1 and subsequently on days 7 and 14 of the experiment. After three to five weeks of incubation at 37 °C, the number of colonies (CFU) was counted.
4.2.1.4 Biofilm formation. The biofilm of M. tuberculosis was developed as described previously with minor modifications.56 M. tuberculosis was cultured to an OD600 of 1 in 7H9/OADC supplemented with 0.05% tyloxapol. Next, the inoculum was added at a ratio of 1
:100 v/v to Sauton's medium and dispensed into each well of a 24-well plate (2.5 mL per well), covered with a lid, protected with parafilm, and incubated in humidity at 37 °C for 5 weeks. Once the biofilm developed, the medium was replaced with fresh one supplemented with 0.1% casitone and the compounds in various concentrations, and it was further incubated at 37 °C for 48 h. The viability of the bacilli was determined by fluorescence measurements (excitation: 550 nm, emission: 590 nm) in the presence of resazurin (375 μl of 0.02% resazurin per well) after 90 min of incubation using the multimode microplate reader SpectraMax® i3 (Syngen). The results were expressed as the percent viability compared to the untreated M. tuberculosis biofilm.
4.2.1.5 Preparation of human MDMs, and evaluation of the bactericidal effect of the compounds on intracellularly growing tubercle bacilli. Human monocytes were isolated from commercially available (Regional Blood Donation Station, Lodz, Poland) and freshly prepared buffy coats from healthy human blood donors.57,58 After washing the cultures of differentiated human monocyte-derived macrophages (MDMs) extensively to remove any nonadherent cells, they were rested overnight and incubated with a culture medium supplemented with various concentrations of the tested compounds. The viability of the macrophages was assessed after 48 h of incubation using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO, United States). Additionally, the MDMs were infected with tubercle bacilli at an MOI of 1
:10, as described by Korycka-Machala et al.59 The bacteria outside the cells were removed by washing with a culture medium two hours after infection. Then, the samples were incubated for 1 h in a medium with 1 g per L gentamicin (Sigma, St. Louis, MO, United States) and rinsed three times with Iscove's medium with 2% human AB serum (Sigma, St. Louis, MO, United States). Next, a culture medium with or without (control) the test compounds at a concentration of 2× MIC was added to independent cultures of the infected macrophages, followed by incubation at 37 °C for 48 h under a humidified atmosphere of 10% CO2-90% air. Finally, the macrophages were lysed with 0.1% SDS, and the number of CFUs (colony forming units) was determined as previously described.60
4.3. Molecular modeling
From the RCSB PDB, the structural coordinates of the M. tuberculosis InhA protein were obtained in pdb format.19 Except for NAD, non-protein moieties, including water, were removed, and hydrogen atoms were added using AutoDockTools.61 The 2D and 3D geometric structures of 4c were built using a Marvin Sketch. In the current docking study, AutoDock Vina was chosen to predict protein–ligand interactions and binding affinities.62 The grid box size was 22.5 Å × 17.0 Å × 16.7 Å, with a position determined by the co-crystallized ligand's center (x, y, x; 9.6, 32.2, 60.7). Discovery Studio Visualizer was utilized for the analysis and visualization of the docking results.634.4. Pharmacokinetics and physicochemical properties
The free SwissADME web tool available from the Swiss Institute of Bioinformatics (SIB) and admetSAR 2.0 internet server were used to investigate some properties of the newly synthesized promising derivative 4c. The structure of the compound was converted to a SMILES notation and then submitted to the online server for calculation.Ethical approval
The study was conducted according to the appropriate ethical standards as required by guidance from The Medical Research Ethics Committee (MREC), Egypt, ethical clearance certificate no. 7445062022.Data availability
The data supporting this article are included in ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
JD was supported by the Ministry of Science and Higher Education, POL-OPENSCREEN, DIR/WK/2018/06 and National Science Centre, Poland UMO-2023/49/B/NZ7/01421.References
- J. Chakaya, M. Khan, F. Ntoumi, E. Aklillu, R. Fatima, P. Mwaba, N. Kapata, S. Mfinanga, S. E. Hasnain, P. D. M. C. Katoto, A. N. H. Bulabula, N. A. Sam-Agudu, J. B. Nachega, S. Tiberi, T. D. McHugh, I. Abubakar and A. Zumla, Global Tuberculosis Report 2020 - Reflections on the Global TB burden, treatment and prevention efforts, Int. J. Infect. Dis., 2021, 113, S7–S12 CrossRef CAS PubMed.
- J. Furin, H. Cox and M. Pai, Tuberculosis, Lancet, 2019, 393, 1642–1656 CrossRef PubMed.
- M. Pai, M. A. Behr, D. Dowdy, K. Dheda, M. Divangahi, C. C. Boehme, A. Ginsberg, S. Swaminathan, M. Spigelman, H. Getahun, D. Menzies and M. Raviglione, Tuberculosis, Nat. Rev. Dis. Prim., 2016, 2, 16076 CrossRef PubMed.
- WHO, Global tuberculosis report, 2022, https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 Search PubMed.
- J. B. Lynch, Multidrug-resistant Tuberculosis, Med. Clin. North Am., 2013, 97, 553–579 CrossRef PubMed.
- C. Lange, K. Dheda, D. Chesov, A. M. Mandalakas, Z. Udwadia and C. R. Horsburgh Jr, Management of drug-resistant tuberculosis, Lancet, 2019, 394, 953–966 CrossRef CAS PubMed.
- C. A. Peloquin and G. R. Davies, The Treatment of Tuberculosis, Clin. Pharmacol. Ther., 2021, 110, 1455–1466 CrossRef PubMed.
- Y. He, A. Fan, M. Han, Y. Zhang, Y. Tong, G. Zheng and S. Zhu, New perspectives on the treatment of mycobacterial infections using antibiotics, Appl. Microbiol. Biotechnol., 2020, 104, 4197–4209 CrossRef CAS PubMed.
- T. M. Walker, P. Miotto, C. U. Köser, P. W. Fowler, J. Knaggs, Z. Iqbal, M. Hunt, L. Chindelevitch, M. Farhat, D. M. Cirillo, I. Comas, J. Posey, S. V. Omar, T. E. Peto, A. Suresh, S. Uplekar, S. Laurent, R. E. Colman, C. M. Nathanson, M. Zignol, A. S. Walker, CRyPTIC Consortium, Seq&Treat Consortium, D. W. Crook, N. Ismail and T. C. Rodwell, The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: A genotypic analysis, Lancet Microbe, 2022, 3, e265–e273 CrossRef CAS PubMed.
- M. S. Prasad, R. P. Bhole, P. B. Khedekar and R. V. Chikhale, Mycobacterium enoyl acyl carrier protein reductase (InhA): A key target for antitubercular drug discovery, Bioorg. Chem., 2021, 115, 105242 CrossRef CAS PubMed.
- R. J. Kinsella, D. A. Fitzpatrick, C. J. Creevey and J. O. McInerney, Fatty acid biosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptive evolution, and gene duplication, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10320–10325 CrossRef CAS PubMed.
- W. Zhai, F. Wu, Y. Zhang, Y. Fu and Z. Liu, The Immune Escape Mechanisms of Mycobacterium Tuberculosis, Int. J. Mol. Sci., 2019, 20, 340 CrossRef PubMed.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, The challenge of new drug discovery for tuberculosis, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- S. L. Parikh, G. Xiao and P. J. Tonge, Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid, Biochemistry, 2000, 39, 7645–7650 CrossRef CAS PubMed.
- X. He, A. Alian and P. R. Ortiz de Montellano, Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides, Bioorg. Med. Chem., 2007, 15, 6649–6658 CrossRef CAS PubMed.
- P. Kamsri, N. Koohatammakun, A. Srisupan, P. Meewong, A. Punkvang, P. Saparpakorn, S. Hannongbua, P. Wolschann, S. Prueksaaroon, U. Leartsakulpanich and P. Pungpo, Rational design of InhA inhibitors in the class of diphenyl ether derivatives as potential anti-tubercular agents using molecular dynamics simulations, SAR QSAR Environ. Res., 2014, 25, 473–488 CrossRef CAS PubMed.
- M. E. Boyne, T. J. Sullivan, C. W. amEnde, H. Lu, V. Gruppo, D. Heaslip, A. G. Amin, D. Chatterjee, A. Lenaerts, P. J. Tonge and R. A. Slayden, Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors, Antimicrob. Agents Chemother., 2007, 51, 3562–3567 CrossRef CAS PubMed.
- U. H. Manjunatha, S. P. S Rao, R. R. Kondreddi, C. G. Noble, L. R. Camacho, B. H. Tan, S. H. Ng, P. S. Ng, N. L. Ma, S. B. Lakshminarayana, M. Herve, S. W. Barnes, W. Yu, K. Kuhen, F. Blasco, D. Beer, J. R. Walker, P. J. Tonge, R. Glynne, P. W. Smith and T. T. Diagana, Direct inhibitors of InhA are active against Mycobacterium tuberculosis, Sci. Transl. Med., 2015, 7, 269ra3 Search PubMed.
- X. He, A. Alian, R. Stroud and P. R. Ortiz de Montellano, Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis, J. Med. Chem., 2006, 49, 6308–6323 CrossRef CAS PubMed.
- A. Khalifa, A. Khalil, M. M. Abdel-Aziz, A. Albohy and S. Mohamady, Isatin-pyrimidine hybrid derivatives as enoyl acyl carrier protein reductase (InhA) inhibitors against Mycobacterium tuberculosis, Bioorg. Chem., 2023, 138, 106591 CrossRef CAS PubMed.
- P. S. Shirude, P. Madhavapeddi, M. Naik, K. Murugan, V. Shinde, R. Nandishaiah, J. Bhat, A. Kumar, S. Hameed, G. Holdgate, G. Davies, H. McMiken, N. Hegde, A. Ambady, J. Venkatraman, M. Panda, B. Bandodkar, V. K. Sambandamurthy and J. A. Read, Methyl-thiazoles: a novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis, J. Med. Chem., 2013, 56, 8533–8542 CrossRef CAS PubMed.
- A. Campaniço, R. Moreira and F. Lopes, Drug discovery in tuberculosis. New drug targets and antimycobacterial agents, Eur. J. Med. Chem., 2018, 150, 525–545 CrossRef PubMed.
- A. Sabt, M. A. Khedr, W. M. Eldehna, A. I. Elshamy, M. F. Abdelhameed, R. M. Allam and R. Z. Batran, New pyrazolylindolin-2-one based coumarin derivatives as anti-melanoma agents: design, synthesis, dual BRAFV600E/VEGFR-2 inhibition, and computational studies, RSC Adv., 2024, 14, 5907–5925 RSC.
- T. K. Mohamed, R. Z. Batran, S. A. Elseginy, M. M. Ali and A. E. Mahmoud, Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis, Bioorg. Chem., 2019, 85, 253–273 CrossRef CAS PubMed.
- S. J. Hamid and T. Salih, Design, Synthesis, and Anti-Inflammatory Activity of Some Coumarin Schiff Base Derivatives: In silico and in vitro Study, Drug Des., Dev. Ther., 2022, 16, 2275–2288 CrossRef PubMed.
- D. H. Dawood, R. Z. Batran, T. A. Farghaly, M. A. Khedr and M. M. Abdulla, New Coumarin Derivatives as Potent Selective COX-2 Inhibitors: Synthesis, Anti-Inflammatory, QSAR, and Molecular Modeling Studies, Arch. Pharm., 2015, 348, 875–888 CrossRef CAS PubMed.
- W. B. Li, X. P. Qiao, Z. X. Wang, S. Wang and S. W. Chen, Synthesis and antioxidant activity of conjugates of hydroxytyrosol and coumarin, Bioorg. Chem., 2020, 105, 104427 CrossRef CAS PubMed.
- N. A. A. Latif, R. Z. Batran, S. F. Mohamed, M. A. Khedr, M. I. Kobeasy, S. A. F. Al-Shehri and H. M. Awad, Synthesis, Molecular Docking and Dynamics Simulation Studies of New 7-oxycoumarin Derivatives as Potential Antioxidant Agents, Mini-Rev. Med. Chem., 2018, 18, 1572–1587 CrossRef PubMed.
- S. Dhawan, P. Awolade, P. Kisten, N. Cele, A. S. Pillay, S. Saha, M. Kaur, S. B. Jonnalagadda and P. Singh, Synthesis, Cytotoxicity and Antimicrobial Evaluation of New Coumarin-Tagged beta-Lactam Triazole Hybrid, Chem. Biodiversity, 2020, 17, e1900462 CrossRef CAS PubMed.
- R. Z. Batran, M. A. Khedr, N. A. A. Latif, A. A. Abd El Aty and A. N. Shehata, Synthesis, homology modeling, molecular docking, dynamics, and antifungal screening of new 4-hydroxycoumarin derivatives as potential chitinase inhibitors, J. Mol. Struct., 2019, 1180, 260–271 CrossRef CAS.
- M. Chebaiki, E. Delfourne, R. Tamhaev, S. Danoun, F. Rodriguez, P. Hoffmann, E. Grosjean, F. Goncalves, J. Azéma-Despeyroux, A. Pál, J. Korduláková, N. Preuilh, S. Britton, P. Constant, H. Marrakchi, L. Maveyraud, L. Mourey and C. Lherbet, Discovery of new diaryl ether inhibitors against Mycobacterium tuberculosis targeting the minor portal of InhA, Eur. J. Med. Chem., 2023, 259, 115646 CrossRef CAS PubMed.
- D. S. Reddy, M. Kongot and A. Kumar, Coumarin hybrid derivatives as promising leads to treat tuberculosis: Recent developments and critical aspects of structural design to exhibit anti-tubercular activity, Tuberculosis, 2021, 127, 102050 CrossRef CAS PubMed.
- S. Srivastava, D. Bimal, K. Bohra, B. Singh, P. Ponnan, R. Jain, M. Varma-Basil, J. Maity, M. Thirumal and A. K. Prasad, Synthesis and antimycobacterial activity of 1-(β-d-Ribofuranosyl)-4-coumarinyloxymethyl-/-coumarinyl-1,2,3-triazole, Eur. J. Med. Chem., 2018, 150, 268–281 CrossRef CAS PubMed.
- Z. Q. Xu, W. W. Barrow, W. J. Suling, L. Westbrook, E. Barrow, Y. M. Lin and M. T. Flavin, Anti-HIV natural product (+)-calanolide A is active against both drug-susceptible and drug-resistant strains of Mycobacterium tuberculosis, Bioorg. Med. Chem., 2004, 12, 1199–1207 CrossRef CAS PubMed.
- I. Ahmad, J. P. Thakur, D. Chanda, D. Saikia, F. Khan, S. Dixit, A. Kumar, R. Konwar, A. S. Negi and A. Gupta, Syntheses of lipophilic chalcones and their conformationally restricted analogues as antitubercular agents, Bioorg. Med. Chem. Lett., 2013, 23, 1322–1325 CrossRef CAS PubMed.
- R. Z. Batran, E. Y. Ahmed, H. M. Awad, K. A. Ali and N. A. Abdel Latif, EGFR and PI3K/m-TOR inhibitors: design, microwave assisted synthesis and anticancer activity of thiazole-coumarin hybrids, RSC Adv., 2023, 13, 29070–29085 RSC.
- R. Z. Batran, E. Y. Ahmed, E. S. Nossier, H. M. Awad and N. A. A. Latif, Anticancer activity of new triazolopyrimidine linked coumarin and quinolone hybrids: synthesis, molecular modeling, TrkA, PI3K/AKT and EGFR inhibition, J. Mol. Struct., 2024, 1305, 137790 CrossRef CAS.
- O. M. Abdelhafez, K. M. Amin, H. I. Ali, T. J. Maher and R. Z. Batran, Dopamine release and molecular modeling study of some coumarin derivatives, Neurochem. Int., 2011, 59, 906–912 CrossRef CAS PubMed.
- O. Ebenezer, M. A. Jordaan, G. Carena, T. Bono, M. Shapi and J. A. Tuszynski, An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds, Int. J. Mol. Sci., 2022, 2, 8117 CrossRef PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- M. Zala, J. J. Vora and V. M. Khedkar, Synthesis, Characterization, Antitubercular Activity, and Molecular Docking Studies of Pyrazolylpyrazoline-Clubbed Triazole and Tetrazole Hybrids, ACS Omega, 2023, 8, 20262–20271 CrossRef CAS PubMed.
- A. Foroumadi, F. Soltani, H. Moallemzadeh-Haghighi and A. Shafiee, Synthesis, in vitro-antimycobacterial activity and cytotoxicity of some alkyl alpha-(5-aryl-1, 3, 4-thiadiazole-2-ylthio) acetates, Arch. Pharm., 2005, 338, 112–116 CrossRef CAS PubMed.
- Y. Teneva, R. Simeonova, V. Valcheva and V. T. Angelova, Recent Advances in Anti-Tuberculosis Drug Discovery Based on Hydrazide-Hydrazone and Thiadiazole Derivatives Targeting InhA, Pharmaceuticals, 2023, 16(4), 484 CrossRef CAS PubMed.
- L. Ballell Pages, J. Castro Pichel, R. Fernandez Menendez, E. P. Fernandez Velando, S. Gonzalez Del Valle, A. Mendoza Losana, and M. J. Wolfendale, (Pyrazol-3-yl)-1,3,4-thiadiazole-2-amine and (Pyrazol-3-yl)-1,3,4-thiazole-2-amine Compounds, WO2010/118852A1, PCT publication, 2010.
- Y. Sajja, S. Vanguru, H. R. Vulupala, L. Nagarapu, Y. Perumal, D. Sriram and J. B. Nanubolu, Design, synthesis, and in vitro antituberculosis activity of benzo[6,7] cyclohepta[1,2-b]pyridine-1,3,4-oxadiazole derivatives, Chem. Biol. Drug Des., 2017, 90, 496–500 CrossRef CAS PubMed.
- J. Castro Pichel, R. Fernandez Menendez, E. P. Fernandez Velando, S. Gonzalez Del Valle, and A. Mallo-Rubio, 3-Amino-Pyrazole Derivatives Useful against Tuberculosis, WO2012/049161A1, PCT publication, 2012.
- Z. Xu, C. Gao, Q. C. Ren, X. F. Song, L. S. Feng and Z. S. Lv, Recent advances of pyrazole-containing derivatives as anti-tubercular agents, Eur. J. Med. Chem., 2017, 139, 429–440 CrossRef CAS PubMed.
- H. Zhao, Y. Gao, W. Li, L. Sheng, K. Cui, B. Wang, L. Fu, M. Gao, Z. Lin, X. Zou, M. Jackson, H. Huang, Y. Lu and D. Zhang, Design, Synthesis, and Biological Evaluation of Pyrrole-2-carboxamide Derivatives as Mycobacterial Membrane Protein Large 3 Inhibitors for Treating Drug-Resistant Tuberculosis, J. Med. Chem., 2022, 65, 10534–10553 CrossRef CAS PubMed.
- N. G. Bajad, S. K. Singh, S. K. Singh, T. D. Singh and M. Singh, Indole: A promising scaffold for the discovery and development of potential anti-tubercular agents, Curr. Res. Pharmacol. Drug Discov., 2022, 3, 100119 CrossRef PubMed.
- E. F. Khaleel, A. Sabt, M. Korycka-Machala, R. M. Badi, N. T. Son, N. X. Ha, M. F. Hamissa, A. E. Elsawi, E. B. Elkaeed, B. Dziadek, W. M. Eldehna and J. Dziadek, Identification of new anti-mycobacterial agents based on quinoline-isatin hybrids targeting enoyl acyl carrier protein reductase (InhA), Bioorg. Chem., 2024, 144, 107138 CrossRef CAS PubMed.
- A. Bekier, M. Kawka, J. Lach, J. Dziadek, A. Paneth, J. Gatkowska, K. Dzitko and B. Dziadek, Imidazole-Thiosemicarbazide Derivatives as Potent Anti-Mycobacterium Tuberculosis Compounds with Antibiofilm Activity, Cells, 2021, 10, 3476 CrossRef CAS PubMed.
- P. Mugabo and M. Mulubwa, Ethionamide population pharmacokinetics/pharmacodynamics and therapeutic implications in South African adult patients with drug-resistant tuberculosis, Br. J. Clin. Pharmacol., 2021, 87, 3863–3870 CrossRef CAS PubMed.
- A. K. Ojha, A. D. Baughn, D. Sambandan, T. Hsu, X. Trivelli, Y. Guerardel, A. Alahari, L. Kremer, W. R. Jacobs Jr and G. F. Hatfull, Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria, Mol. Microbiol., 2008, 69, 164–174 CrossRef CAS PubMed.
- R. Z. Batran, A. Sabt, M. A. Khedr, A. K. Allayeh, C. Pannecouque and A. F. Kassem, 4-Phenylcoumarin derivatives as new HIV-1 NNRTIs: Design, synthesis, biological activities, and computational studies, Bioorg. Chem., 2023, 141, 106918 CrossRef CAS PubMed.
- S. G. Franzblau, R. S. Witzig, J. C. McLaughlin, P. Torres, G. Madico, A. Hernandez, M. T. Degnan, M. B. Cook, V. K. Quenzer, R. M. Ferguson and R. H. Gilman, Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay, J. Clin. Microbiol., 1998, 36, 362–366 CrossRef CAS PubMed.
- M. Korycka-Machała, M. Kawka, J. Lach, R. Płocińska, A. Bekier, B. Dziadek, A. Brzostek, P. Płociński, D. Strapagiel, M. Szczesio, K. Gobis and J. Dziadek, 2,4-Disubstituted pyridine derivatives are effective against intracellular and biofilm-forming tubercle bacilli, Front. Pharmacol, 2022, 13, 1004632 CrossRef PubMed.
- M. Korycka-Machała, A. Viljoen, J. Pawełczyk, P. Borówka, B. Dziadek, K. Gobis, A. Brzostek, M. Kawka, M. Blaise, D. Strapagiel, L. Kremer and J. Dziadek, 1H-Benzo[d]Imidazole Derivatives Affect MmpL3 in Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2019, 63, e00441 CrossRef PubMed.
- M. Kawka, A. Brzostek, K. Dzitko, J. Kryczka, R. Bednarek, R. Płocińska, P. Płociński, D. Strapagiel, J. Gatkowska, J. Dziadek and B. Dziadek, Mycobacterium tuberculosis Binds Human Serum Amyloid A, and the Interaction Modulates the Colonization of Human Macrophages and the Transcriptional Response of the Pathogen, Cells, 2021, 10, 1264 CrossRef CAS PubMed.
- M. Korycka-Machała, A. Brzostek, B. Dziadek, M. Kawka, T. Popławski, Z. J. Witczak and J. Dziadek, Evaluation of the Mycobactericidal Effect of Thio-functionalized Carbohydrate Derivatives, Molecules, 2017, 22, 812 CrossRef PubMed.
- M. Korycka-Machała, J. Pawełczyk, P. Borówka, B. Dziadek, A. Brzostek, M. Kawka, A. Bekier, S. Rykowski, A. B. Olejniczak, D. Strapagiel, Z. Witczak and J. Dziadek, PPE51 Is Involved in the Uptake of Disaccharides by Mycobacterium tuberculosis, Cells, 2020, 9, 603 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- BIOVIA, and Dassault Systèmes, Discovery Studio Visualizer, V24.0.0, Dassault Systèmes, San Diego, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02746a |
| This journal is © The Royal Society of Chemistry 2024 |