
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen advancements towards the electrochemical synthesis of heterocycles
Sonia Zeba Hashmi *a,
Diksha Baretha,
Jaya Dwivedia,
Dharma Kishorea and
P. A. Alvi*b
*a,
Diksha Baretha,
Jaya Dwivedia,
Dharma Kishorea and
P. A. Alvi*b
aDepartment of Chemistry, Banasthali Vidyapith, Banasthali-304022, Rajasthan, India. E-mail: zebahashmi@gmail.com
bDepartment of Physical Sciences, Banasthali Vidyapith, Banasthali-304022, Rajasthan, India. E-mail: drpaalvi@gmail.com
First published on 7th June 2024
Abstract
Heterocyclic chemistry is a large field with diverse applications in the areas of biological research and pharmaceutical advancement. Numerous initiatives have been proposed to further enhance the reaction conditions to reach these compounds without using harmful compounds. This paper focuses on the recent advances in the eco-friendly and green synthetic procedures to synthesize N-, S-, and O-heterocycles. This approach demonstrates considerable potential in accessing such compounds while circumventing the need for stoichiometric quantities of oxidizing/reducing agents or catalysts containing precious metals. Merely employing catalytic quantities of these substances proves sufficient, thereby offering an optimal means of contributing to resource efficiency. Renewable electricity plays a crucial role in generating environmentally friendly electrons (oxidant/reductant) that serve as catalysts for a series of reactions. These reactions involve the production of reactive intermediates, which in turn allow the synthesis of new chemical bonds, enabling beneficial transformations to occur. Furthermore, the utilization of metals as active catalysts in electrochemical activation has been recognized as an effective approach for achieving selective functionalization. The aim of this review was to summarize the electrochemical synthetic procedures so that the undesirable side reactions can be considerably reduced and the practical potential range of the chemical reactions can be expanded significantly.
Introduction
Significant progress in electrochemical engineering technology in the past thirty years has facilitated the creation of alternative approaches to mitigate and avoid the production of harmful pollutants that negatively impact the environment. Implementing this method in reality poses challenges, and it is more practical and achievable to utilize technologies that can reduce and transform pollutants into environmentally benign by products. This article explores different approaches to the electrosynthesis of heterocyclic molecules. Electrochemical technologies have played a vital role in developing methods to remove harmful gasses such as carbon dioxide, nitrous oxides, and sulfur dioxide. This strategy effectively prevents energy waste and minimizes the release of greenhouse gas emissions.The chemical sector makes a significant contribution to global pollution. In response to increasingly strict and strictly enforced environmental regulations, the chemical industry should adopt the following guidelines in the 21st century and beyond: (1) prioritize waste avoidance and minimization, (2) eliminate, recover, and recycle waste, (3) achieve environmentally acceptable and harmless waste disposal, (4) significantly reduce the generation of pollutants and energy consumption in the chemical process industry, without involving the combustion of fossil fuels, and (5) promote the generation and storage of clean energy.
Due to the immense applicability of the heterocyclic compounds, the synthesis and alteration of heterocyclic structures often takes place in conditions that may require the use of toxic substances such as acids or bases the requirement of high temperatures to generate the desired heterocyclic system. By utilizing the electron as a reactant, the need for further chemicals is eliminated, making the process of disposing, recycling, and recovering of treated solutions more straightforward. The broad versatility of electrochemical processes in both their electrosynthesis and elimination, recovery, recycling within the process industry stems from the fact that electrons can be easily removed by oxidation or reduction, unlike conventional chemicals. This can be accomplished by utilizing electrode materials that possess high resistance to oxidation or reduction and employing inert electrolytes that can tolerate electrode potentials ranging from +3 V to −3 V compared to the standard calomel electrode. Reduction may be achieved with a power that is equivalent to or superior to that of Na or Li-metals in amine solvents, and oxidations can be carried out at the chemical fluorine level. The electron is an inexpensive, versatile, and highly pure reagent. The use of electrons as reagents to induce chemical changes offers several advantages. Firstly, it is cost-competitive compared to alternative processes. Secondly, it allows for selective chemistry, meaning that specific reactions can be targeted. Thirdly, it has broad applicability, making it suitable for a wide range of reactions. Fourthly, it results in less pollution compared to most competing processes. Fifthly, it requires lower temperature conditions compared to equivalent techniques, especially those involving high temperatures. Sixthly, the involved equipment and operations are mainly simple and, if properly inexpensive or designed compared to conventional technologies. Lastly, the foremost electrochemical parameters, potential and current, are particularly compatible for data acquisition, process automation, and control.
Scientists have mostly focused their efforts on developing sustainable and environment friendly technologies that adhere to both economic and environmental constraints, by avoiding the use of harmful substances.1 In the years to come, there shall be a growing interest in the utilization of organic electrochemical synthesis as a versatile and potent approach for constructing heterocyclic structures.2–5 This method has demonstrated superior efficiency and selectivity compared to traditional synthetic methods. Moreover, it is worth to notice that the green methodologies involved in the electrosynthesis complies with nine out of the 12 principles of green chemistry, as depicted in Fig. 1. Agricultural chemicals, physiologically active natural organic compounds, and pharmaceuticals contain at least one heterocyclic ring.6,7 Electricity-initiated biological transformations possess intrinsic sustainability and environmental benefits, as they only necessitate mild operating conditions.8,9
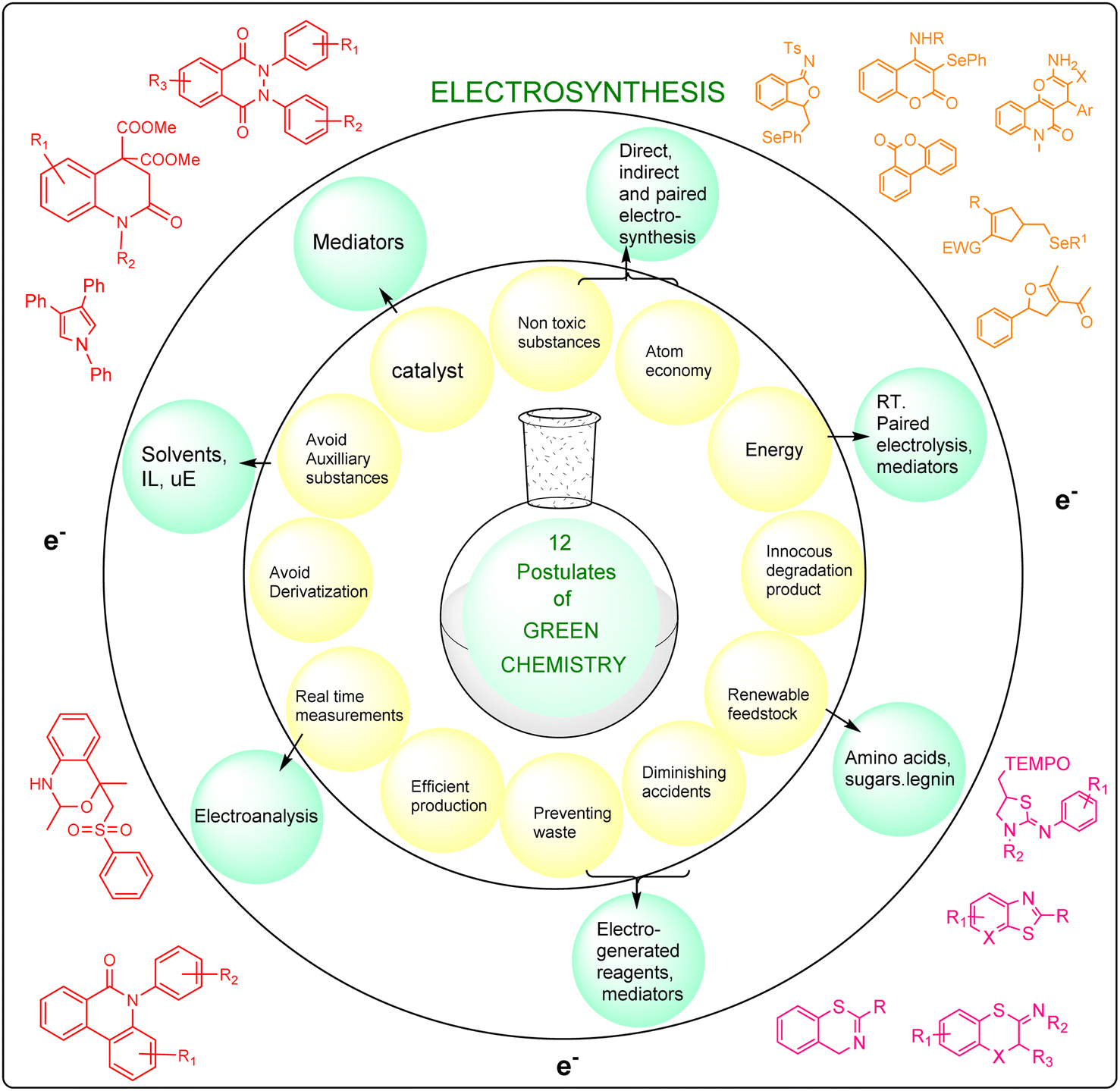 | ||
| Fig. 1 Relationship between 12 postulates of green chemistry and 9 principles of electrosynthesis. | ||
Nevertheless, there are certain apprehensions that arise when employing an electrochemical process in chemical transformations. One notable concern pertains to the necessity of possessing a comprehensive electrochemical apparatus, which is commonly associated with substantial expenses for both acquisition and operation. Moreover, the utilization of a supporting electrolyte is commonly employed to facilitate e-transfer in a solution. The selection of a suitable solvent for electrosynthesis can be a complex task due to the usage of poorly conductive solvents such as tetrahydrofuran and toluene. The electrochemical reactions involving the use of metal catalysts within easily accessible undivided cells are somewhat restricted because of its tendency to get readily reduced to zerovalent metals at the cathode. Additionally, when chemical and electrical reactions are conducted in split cells, the use of expensive ion exchange membranes becomes necessary in order to separate the negative and positive electrodes.10 The use of an electrochemical process in chemical transformations raises a number of apparent issues, such as the need for extensive electrochemical equipment that is often expensive to purchase and maintain. The selection of solvent can be difficult in electrosynthesis since toluene, tetrahydrofuran, and other less conductive solvents are utilized. Furthermore, a supporting electrolyte is typically employed to aid electron transit in solution. The majority of metallic ions are easily reduced at the cathode to zero-valent metals, which restricts the use of metal catalysts in electrochemical reactions occurring in easily accessible undivided cells. Additionally, while customizing the electronic configuration to achieve the best binding, thermodynamics, and even predictive manipulations are also challenging. Another obstacle that lies in the occurrence of side reactions, is the reduction in the current efficiency which can lead to deterioration of the electrode.
Electrochemical synthesis has experienced rapid development in recent years, contrasting with the previously steady evolution. The concept of utilizing electricity in organic synthesis has emerged as a new era with expanding opportunities and advantages. The drive to create cleaner and more cost-effective synthetic methods has insisted chemists to explore novel approaches for activating organic molecules. Electrosynthesis stands out as one of the most cost-effective and environmentally friendly techniques, as it enables oxidations and/or reductions to be conducted without the requirement for additional chemical reagents. Minimizing the number of stages results in energy conservation, contributing positively to both environmental sustainability and the economic evaluation of manufacturing processes.
Electrochemical processes have inherent advantages over conventional reagent-based methods, while transforming renewable biogenic compounds and natural raw materials into complex compounds with high added value, such as pharmaceuticals or other chemicals and natural products. Now a days electrocatalysis integrated with electrosynthesis is becoming an emerging trend. This includes utilization of flow chemistry for scaling up the processes to enhance efficiency and sustainability. Moreover, there is a focus on incorporating new synthetic feedstocks to further diversify the scope of electrochemical reactions.11
In this article, we aim to provide a comprehensive overview of the advancements achieved in the electrochemical synthesis of N-, S-, and O-heterocyclic compounds using intermolecular and intramolecular cyclization reactions over the past few years.
Electrosynthesis of N-heterocycles
The significance of N-containing heterocyclic compounds in the field of biomedicine12 has led to the advancement of synthetic techniques employed in their synthesis. In the year 2018, Wu and colleagues published a study documenting the electrochemical reaction employed for the intramolecular cross-coupling of C(sp3)–H and C(sp2)–H bonds. The catalyst used in this reaction was Cp2Fe, as illustrated in Scheme 1. A family of oxindole cycle of C8H12O3 compounds 2 and 4 was synthesized with high efficiency.13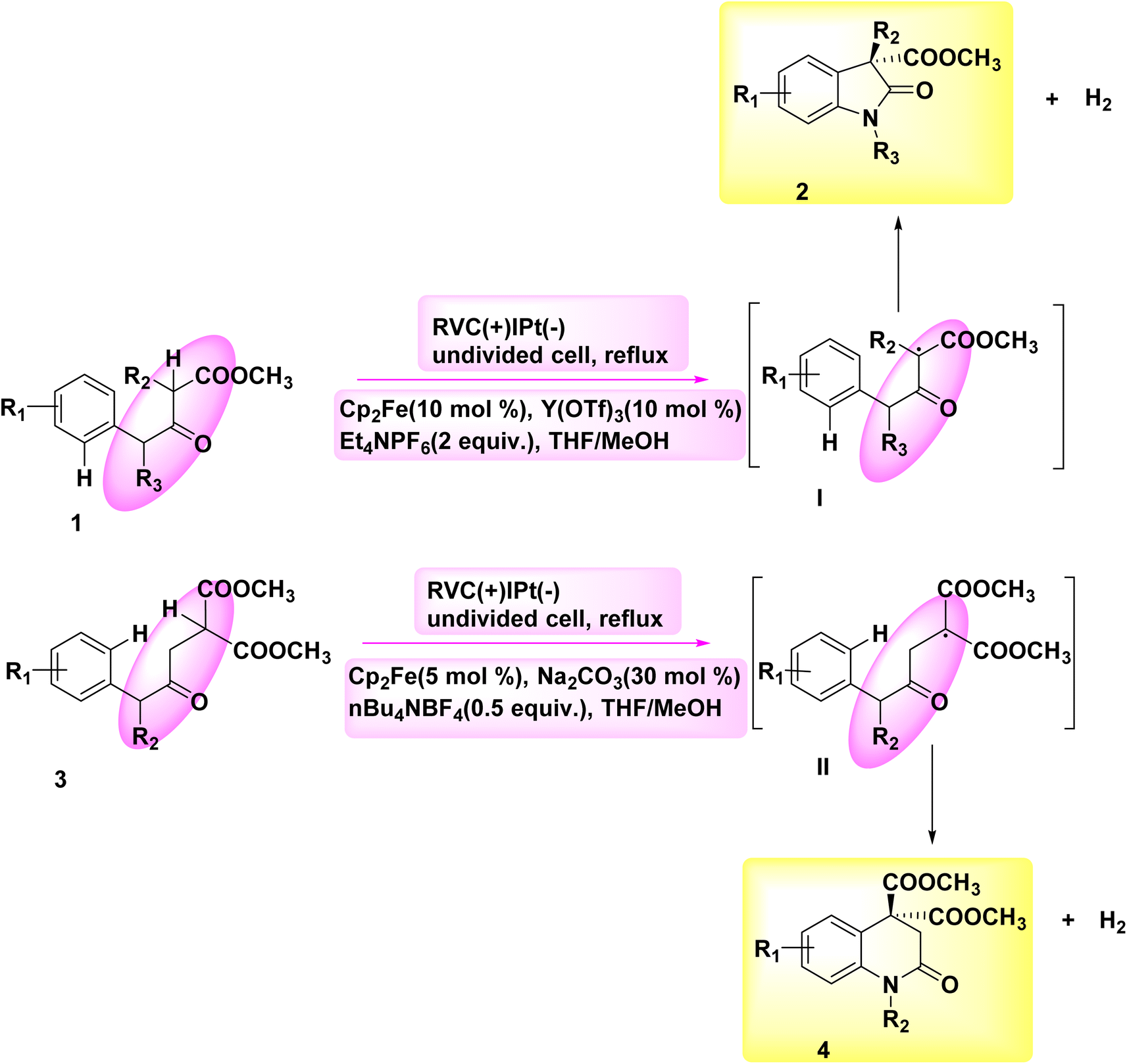 | ||
| Scheme 1 Electrochemical synthesis of oxindole ring compounds 2 and 4. | ||
In 2018, Hu et al. introduced an alternative technique for the electrochemical synthesis of N-heterocycles, Scheme 2. One of the techniques employed in the synthesis of nitrogen-containing compounds with high saturation involves the cross-coupling of C(sp3)–H and N–H bonds. Typically, supplementary oxidizing agents or halogenated compounds are required in such processes. The authors have presented a commendable study on the electrochemical synthesis of a five-membered ring, demonstrating the absence of additional oxidants or hazardous chemicals. A significant yield of pyrrolidine 6 was achieved from sulfonamide 5, resulting in the generation of a substantial family of pyrrolidine compounds. The process of electrolysis was conducted in an undivided cell utilizing a cathode made of Pt plate and an anode composed of a carbon rod. Tetrabutylammonium acetate has the ability to form intermolecular H2-bonds with amide molecules, hence aiding in the breaking of the nitrogen–hydrogen bond. Furthermore, it is employed as an electrolyte in several applications. In this approach, the inclusion of additional oxidants and the N-halogenation step can be omitted. The production of benzylic and non-activated primary, secondary, and tertiary C(sp3)–H amination can be achieved with favorable yields.14–16
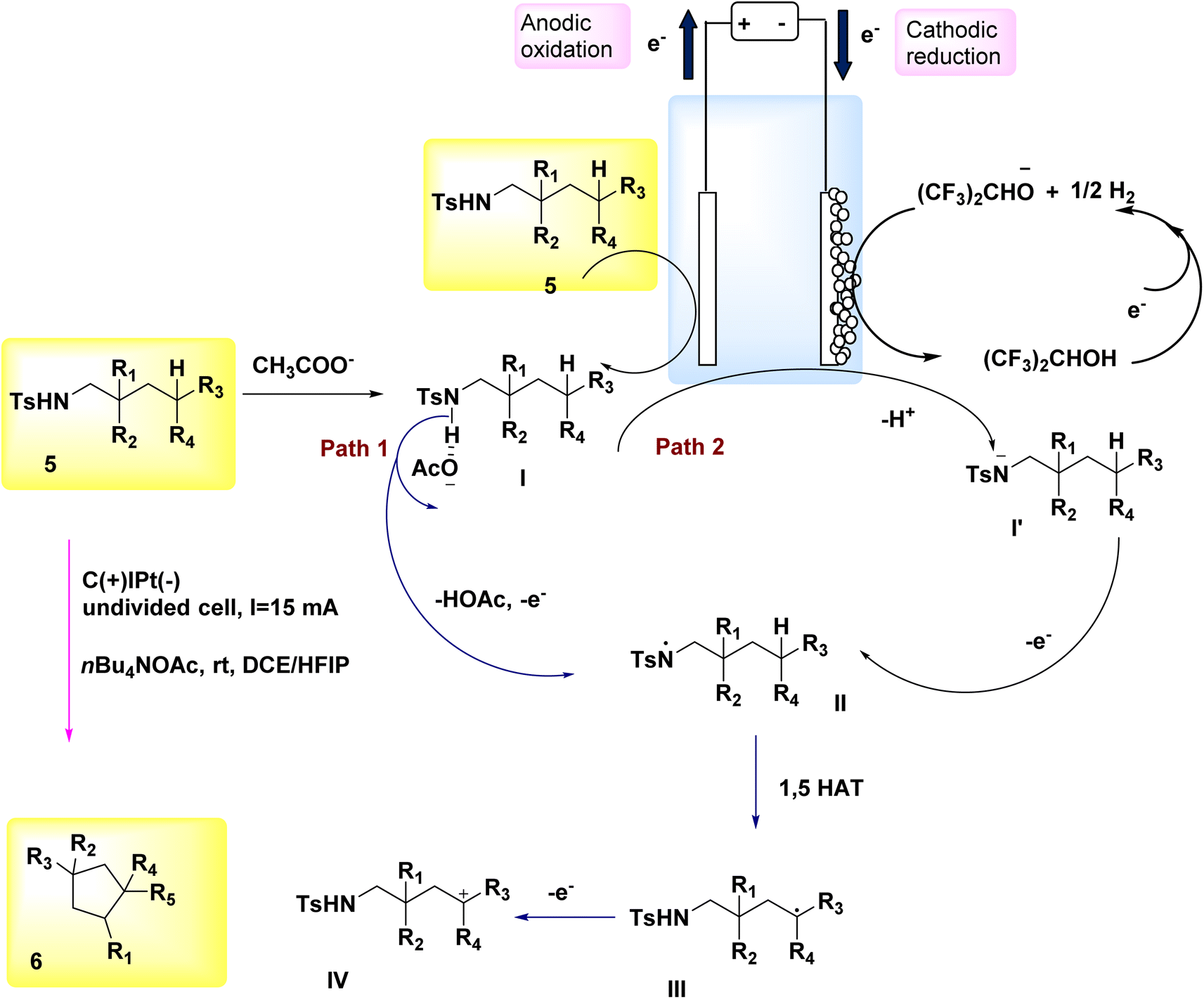 | ||
| Scheme 2 Electrochemical synthesis of substituted pyrrolidine 6 with its proposed mechanism. | ||
The process was initiated by the formation of bonding complexes between sulfonamide 5 and acetate. The formation of the N-centered radical intermediate II was initiated by a process of single electron oxidation occurring at the anode (path 1). A carbon-centered radical III was formed as a result of the 1,5-HAT reaction, where a carbon–hydrogen bond was cleaved by an aminyl radical. Subsequently, the radical species underwent oxidation to yield the C-cation intermediate IV. The cyclization product 6 is formed as a result of the nucleophilic attack and subsequent proton eliminations of the sulfonamide. In the framework of the reaction phase, the concurrent reduction of protons at the cathode would result in the production of molecular H2. This process eliminates the need for a stoichiometric external oxidant. The alkoxide that is generated has the ability to deprotonate the substrate, leading to the creation of N anion I′. This intermediate can then undergo rapid oxidation on the (+), culminating in the synthesis of the N-centered radical intermediate II (path 2)
In the year 2018, a novel approach was employed for the generation of N-acyloxy NH2s electrochemically, marking the first instance of utilizing an inner-sphere electron-transfer mechanism for this purpose (Scheme 3). A cellular system consisting of a cathode made of graphite (C) and an anode made of Pt (Pt) was utilized. The NH2s that are generated in situ undergo intramolecular C(sp2/sp3)–H aminations in the presence of NaBr, which serves as both an electrolyte and catalyst. This reaction leads to the formation of quinolinone 8 and indolinone 9 products, exhibiting remarkable regio- and chemoselectivities.17,18
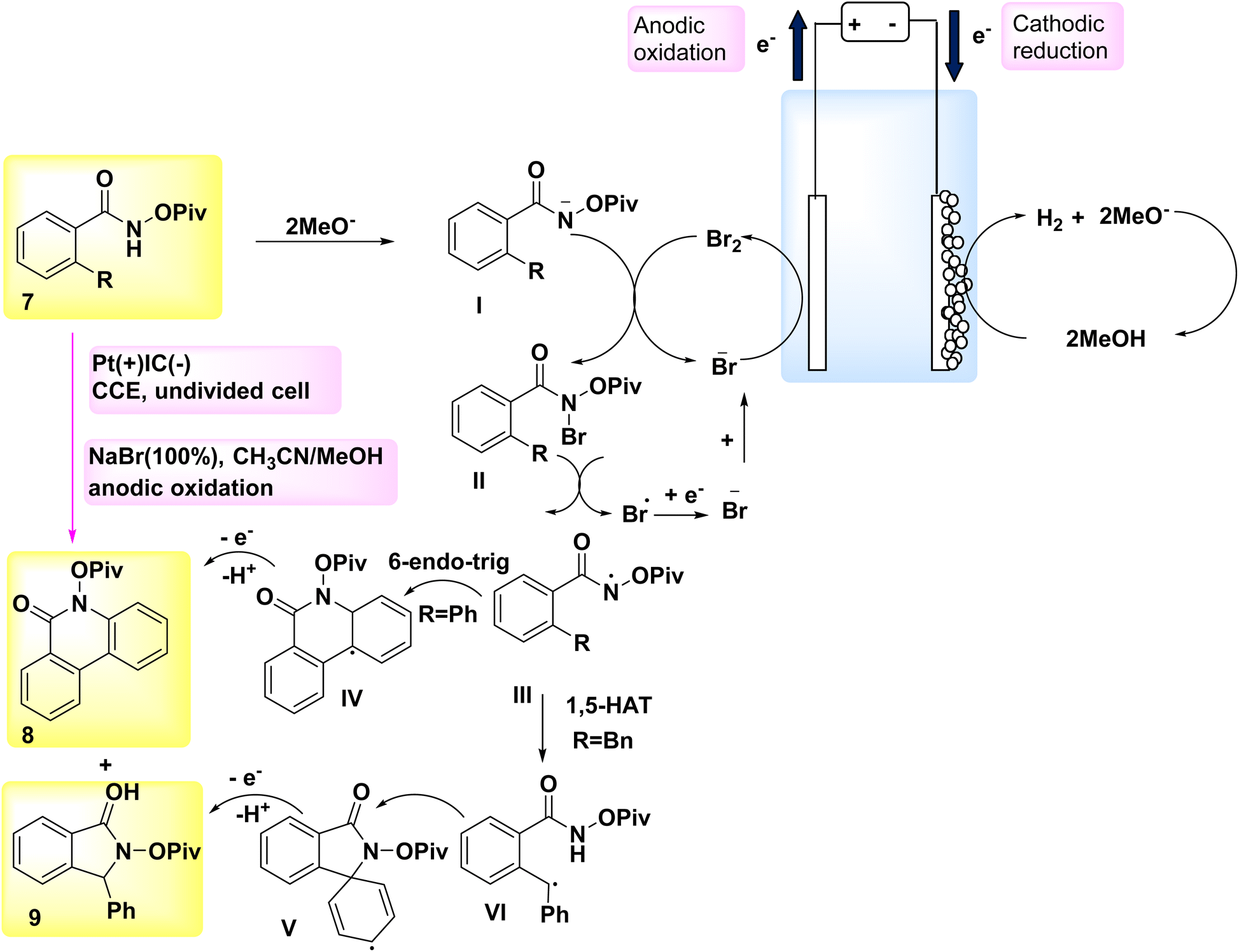 | ||
| Scheme 3 Electrochemical synthesis of quinolinone 8 and indolinone 9 with its proposed mechanism. | ||
A hypothetical method was built based on the findings from these results and relevant studies (Scheme 3). Intermediate II is formed when a methoxide ion (MeO−), which acts as an electrogenerated base, is present, and it facilitates the collection of anodically produced Br2 by the substrate 7.
After the cleavage of the nitrogen and bromine bonds, the formation of N-acyloxy amidyl III occurs, which then undergoes cyclization by a 6-endo-trig mechanism to produce intermediate IV. The generation of Br-radicals was seen in the cyclic voltammetry tests, providing evidence for the breaking of the nitrogen-bromine (N–Br) bond. The presence of steric hindrance caused by the –OPiv group effectively mitigates the formation of an undesired intermediate.
The aromatization process ultimately converts intermediate IV into the sp2 carbon–hydrogen amination product 8. The postulated mechanism for the development of benzylic radical VI in the sp3 carbon–hydrogen amination process involves a 1,5-H2 atom transfer. The benzylic radical VI undergoes further oxidation and then undergoes intramolecular cyclization, resulting of product 9 formation.
In the year 2018, an alternative technique for electro-anodic oxidation was employed in order to establish a novel and enduring supply of N arylphenanthredin-6-one derivatives. The conversion of C19H15NO 10 to N, C coupled product 11 (Scheme 4) is achieved using an undivided Teflon cell equipped with cathodic graphite and anodic nickel electrodes. The solvent used in this study is 1,1,1,3,3,3-hexafluoroisopropanol. The utilization of a moderator is not obligatory in conjunction with this system owing to its exceptional electrical efficiency. An accessible or cost-effective method for synthesizing this category of chemicals is facilitated by readily obtainable and economical initial reactants. The production of a diverse array of derivatives is achievable, and the identification of valuable functionalities that facilitate subsequent reactions is acknowledged. The scalability of this technology allows for effortless adjustments in both larger and smaller scales.19
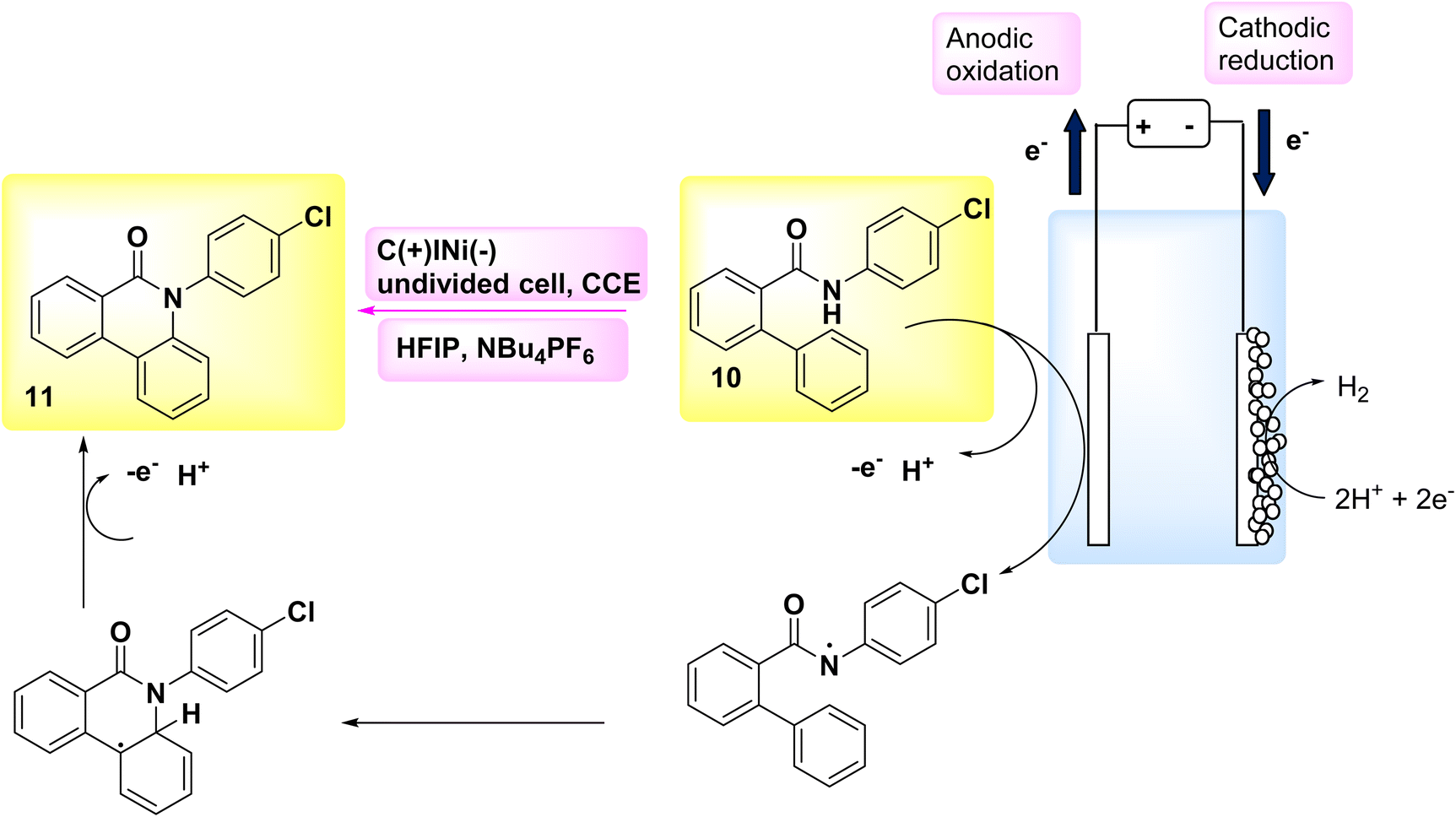 | ||
| Scheme 4 Electrochemical synthesis of N-arylphenanthredin-6-one 11 with its proposed mechanism. | ||
Scheme 4 illustrates the proposed procedure for the synthesis of compound 11. The initiation of cyclization occurs through the oxidation of the substrate at the anode electrode, at which the generation of an NH2 I is seen. The anilide could be deprotonated by an 1,1,1,3,3,3-hexafluoroisopropanolanion that is generated in situ. The N-aryl system has the ability to provide stabilization to the NH2. In this particular situation, the NH2 establishes a nitrogen, carbon link with the second phenyl moiety of the biphenyl scaffold, which is unsubstituted. Consequently, a radical is formed within the lactam system. The product 11 undergoes a secondary oxidation phase, subsequent to which a proton extrusion occurs.
According to a paper in 2018, the oxidative [4 + 2] annulation method, specifically Scheme 5, was identified as an efficient method for the rapid synthesis of six-membered heterocycles. In this methodology, tertiary anilines and alkenes engage in an [4 + 2] electrochemical annulation process, resulting in the formation of tetrahydroquinolines in a homogeneous manner, without the requirement of metallic catalysts or external oxidizing agents. In order to assess the reaction conditions, model substrates DMA 13 and C10H11NO 12 were employed. C18H20N2O 14 may be synthesized with a 72% yield in an undivided cell by employing nBu4NBF4 as the CH3CN/AcOH and electrolyte as cosolvents for a duration of 6 hours.20,21
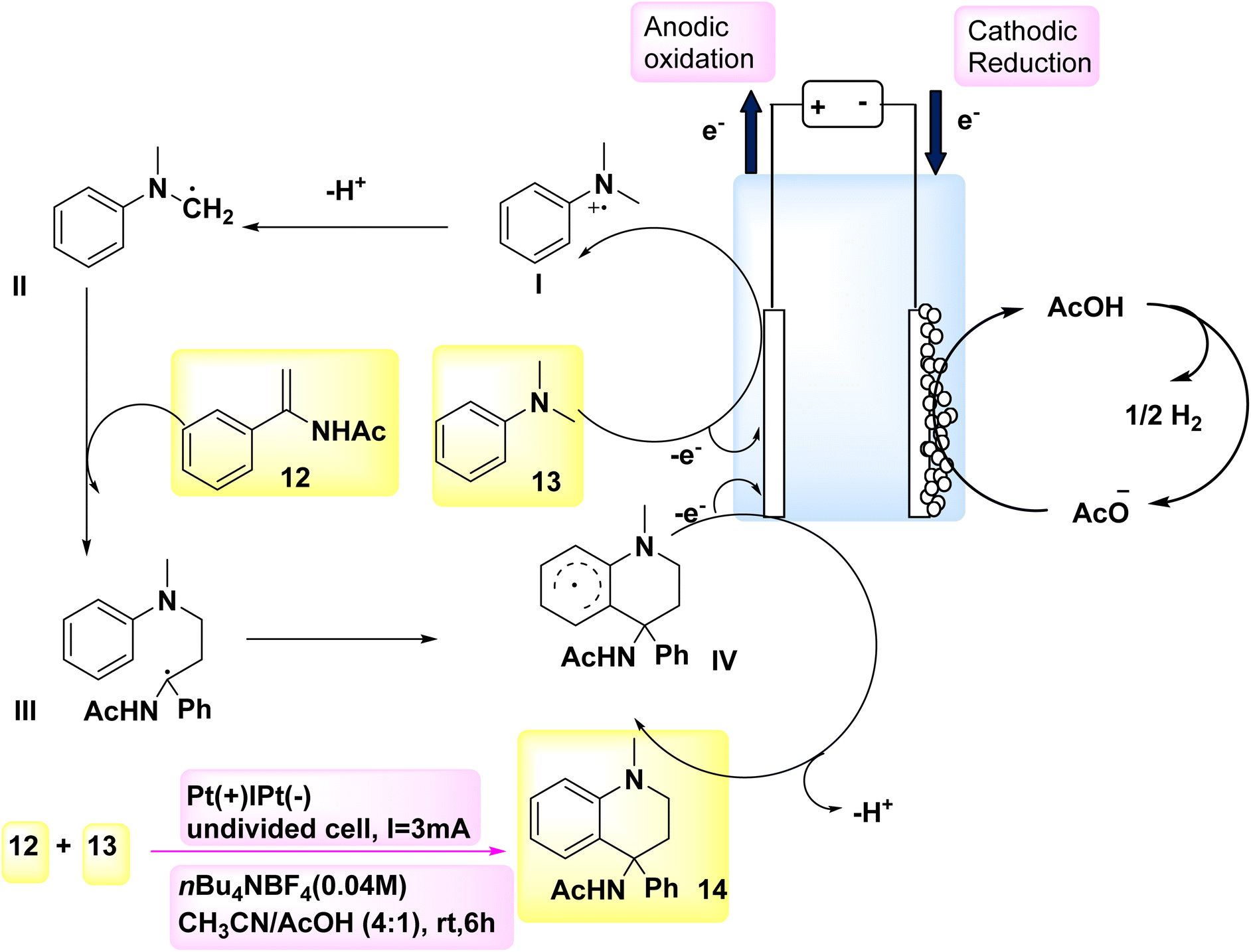 | ||
| Scheme 5 Electrochemical synthesis of N-(1-methyl-4-phenyl-1,2,3,4-tetrahydro-quinolin-4-yl)acetamide 14 with its proposed mechanism. | ||
Scheme 5 presents a possible mechanism along with its synthesis for the reaction involving compound 12 and compound 13. Initially, the oxidation of 13 occurs, resulting in the generation of a radical cation. This radical cation can be stabilized by acetic acid, thereby facilitating the anodic reaction. The tertiary-amino carbon radical is formed through a process involving resonance of the radical cation followed by deprotonation. The radical II has the potential to engage in a radical addition reaction with 12. The intended product 14 will be generated during the subsequent anodic oxidation, wherein an intramolecular cyclization process is taken part in by the resulting radical species III. The production of H2 gas occurs by the concurrent (−) reduction of acetic acid.
In the year 2018, two interrelated electrochemical techniques utilizing Co catalysts were employed for the synthesis of substituted oxindoles through radical pathways. The effectiveness and environmental friendliness of the electrochemical Co-catalyzed system were described, as it achieved high yields of arylation 18 or alkylation 19 compounds at ambient temperature without the need for stoichiometric oxidants. Several substituted oxindoles were synthesized using electrochemical reactions of N-arylacrylamides 15 with either potassium alkyltrifluoroborates 17 or arylhydrazines 16 in environmentally friendly conditions. The reaction is carried out using platinum as the cathode and reticulated vitreous carbon as the anode. The absence of oxidants in these two reactions has support a groundbreaking approach for the synthesis of novel radical oxidative couplings. Furthermore, the synthesis of molecules possessing an all-carbon chiral core can potentially be achieved using a Co-catalyzed radical reaction involving the reaction between N-arylacrylamides and potassium alkyltrifluoroborates. These two unique approaches for acquiring substituted oxindoles are anticipated to yield advantageous outcomes in the field of organic synthesis.22
The anodic oxidations of phenylhydrazine 16 or C7H7BF3K 17 were facilitated by Co salts. This resulted in the formation of benzyl or phenyl free radicals, which subsequently acted as catalysts for the reaction (Scheme 6-I). The process of cathodic reduction is responsible for the production of molecular H2 as depicted in Scheme 6-II. The compound Ph˙ or 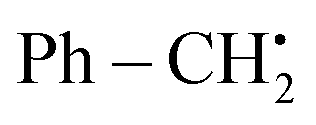 was subjected to a direct assault by 17 throughout the reaction, resulting in the formation of radical intermediate I or I′. Subsequently, an intramolecular cyclization process was employed to generate radical II or II′. After the transfer of a solitary electron from cobalt III to cobalt II, the anode undergoes oxidation, resulting in the fabrication of the oxidized radical II or II′ and the formation of the cationic intermediate III or III′. In the final step, deprotonation was employed to facilitate the conversion of intermediate III or III′ into the desired products 18 and 19, respectively (as seen in Scheme 6-III).
was subjected to a direct assault by 17 throughout the reaction, resulting in the formation of radical intermediate I or I′. Subsequently, an intramolecular cyclization process was employed to generate radical II or II′. After the transfer of a solitary electron from cobalt III to cobalt II, the anode undergoes oxidation, resulting in the fabrication of the oxidized radical II or II′ and the formation of the cationic intermediate III or III′. In the final step, deprotonation was employed to facilitate the conversion of intermediate III or III′ into the desired products 18 and 19, respectively (as seen in Scheme 6-III).
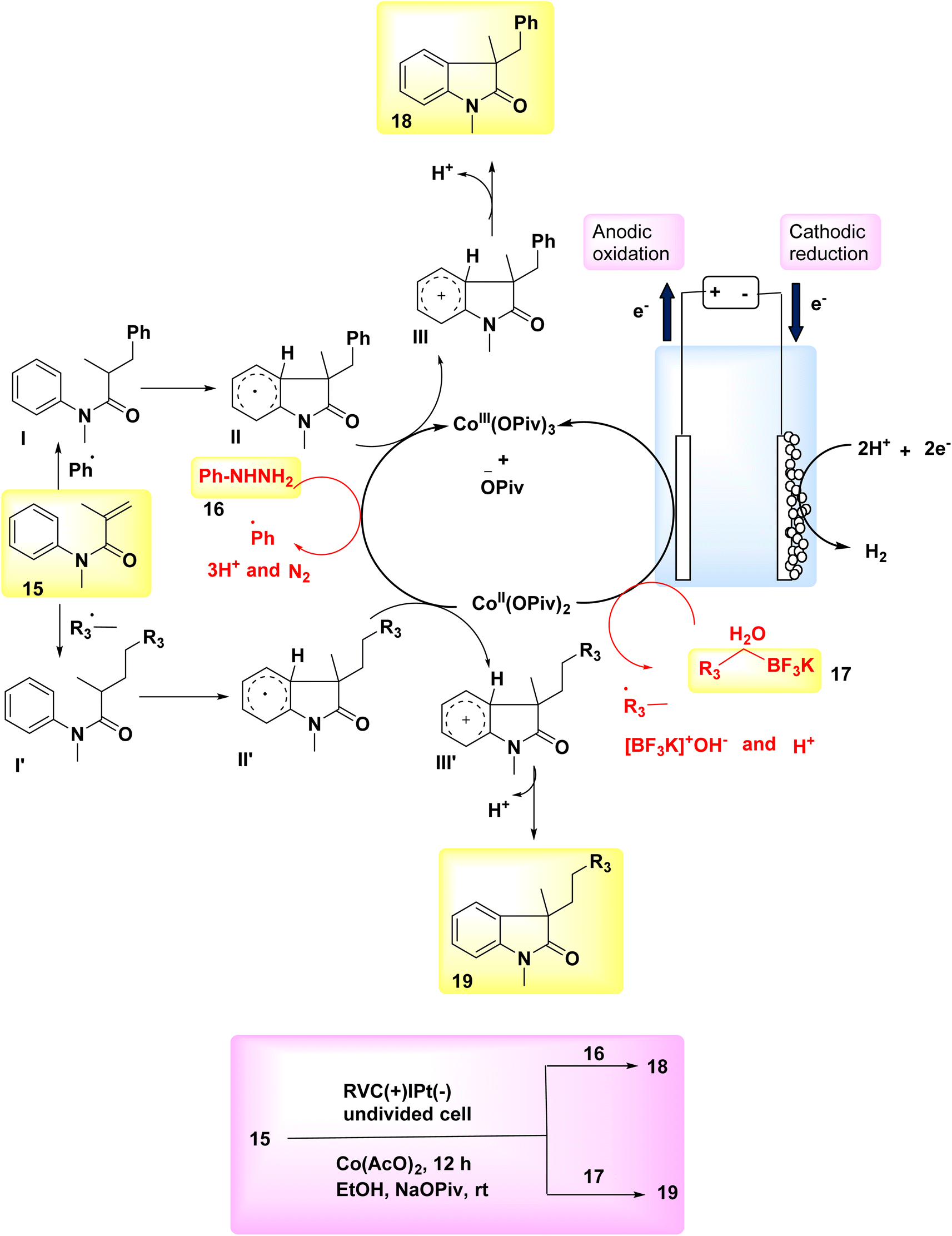 | ||
| Scheme 6 Electrochemical arylation 18 and alkylation 19 products with its proposed mechanism. | ||
In the year 2018, researchers employed electrochemical synthesis as a means to generate a novel and environmentally friendly method for obtaining phthalazin-1,4-diones 21. This approach can effectively circumvent the use of hydrazine chemicals, as it is used in many of the synthetic procedures for the incorporation of pyrazole ring in the hybrid multi-component drug synthesis.23–25 They have been shown to have substantial toxicity or carcinogenic qualities.
The experimental procedure involved the utilization of a single, non-partitioned cell including graphite (+) and a Pt (−). The solvents employed were 1,1,1,3,3,3-hexafluoroisopropanol and NBu4PF6. The starting material used in this study was phthaldianilide 20, as depicted in Scheme 7. The utilization of this method presents a valuable alternative to the traditional synthetic route due to its utilization of easily accessible and cost-effective starting materials. The utilization of a straightforward configuration, the exclusion of metallic catalysts and organic oxidizers, and the availability of electrode materials that can be scaled up and have a prolonged lifespan, facilitate convenient and enduring accessibility to this particular category of substrates. A N–N connection with an anodic character is formed. This methodology facilitates the synthesis of a diverse array of derivatives, hence enabling the incorporation of valuable functionalities that can support subsequent reactions. Additionally, this approach offers the opportunity to obtain nonsymmetrical compounds.26,27
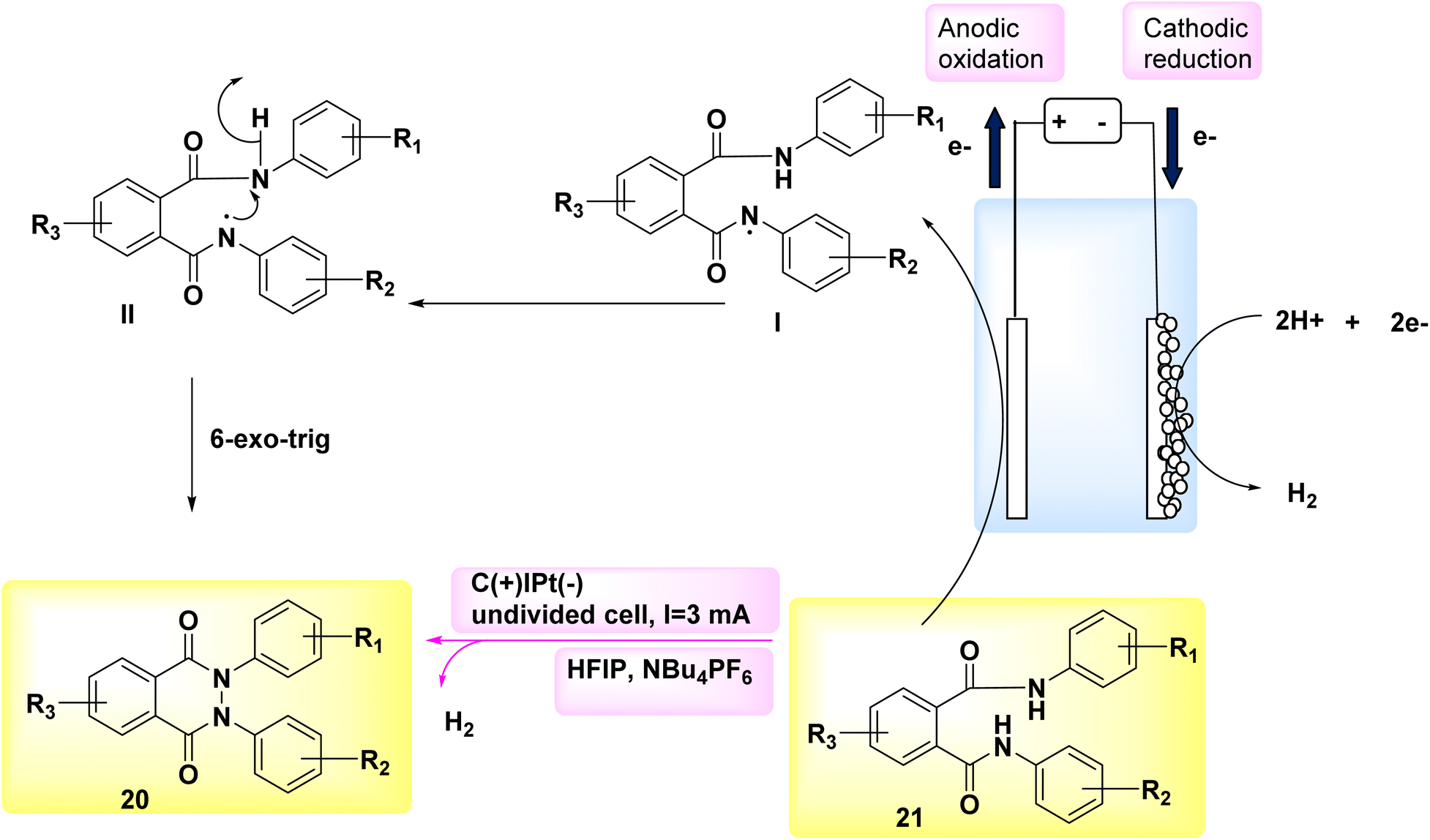 | ||
| Scheme 7 Electrochemical synthesis of phthalazin-1,4-diones 21 with its proposed mechanism. | ||
The year 2018 witnessed the evoluation of a systematic and stereoselective approach for the synthesis of indolines and azaindolines 23. This method used the (3 + 2) annulation of heteroarylaminesintramolecular dehydrogenation 22 with coupled substituted alkenes. The reaction was facilitated by the use of oxidizing agents or metal catalysts Scheme 8. The procedure employs a cascade radical cyclization mechanism to produce C–C and C–N bonds sequentially. The initial phase of the bicyclization process facilitates the production of a 6-membered ring, hence ensuring the effective evoluation of the subsequent C–C bond, which plays a crucial role in the overall effectiveness of the annulation. The electrosynthetic approach enables the complete synthesis of (±)-hinckdentine using commercially available components.28
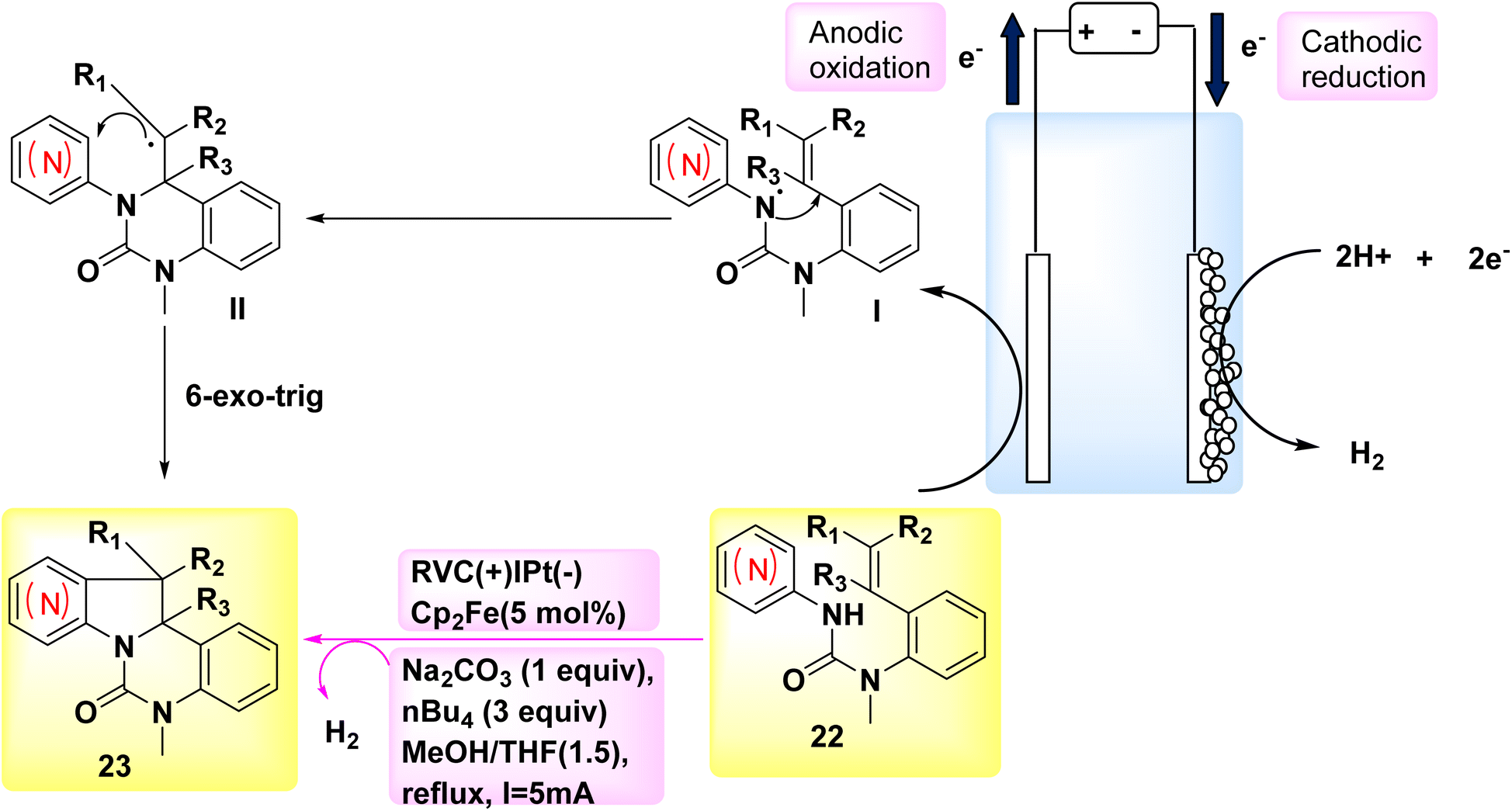 | ||
| Scheme 8 Electrochemical synthesis of azaindolines 23 with its proposed mechanism. | ||
In the year 2018, a radical cyclization cascade, namely a 5-exo-trig or 7-endo-trig mechanism, was utilized for the electrochemical synthesis of 7-membered carbocycles, denoted as compound 25 in Scheme 9. The initial cyclization step of the cascade process results in the formation of a five-membered ring, accompanied by the transposition of the radical center and the remaining alkene. The trans-configuration of the molecule induces a region-selective 7-endo cyclization of the 6-heptenyl radical. The experiment was performed within a single cell, utilizing a solvent mixture of tetrahydrofuran and methanol, with nBu4NBF4 serving as the electrolyte.29
 | ||
| Scheme 9 Electrosynthesis of 7 membered functionalized carbocycles 25. | ||
The synthesis of chiralaxial imidazopyridine-containing biaryls involves a series of events that proceed through a radical cyclization cascade mechanism.30 A novel and groundbreaking C–N linked cyclization technique, known as regiospecific [3 + 2] annulations, was utilized in the electrochemical synthesis of imidazo-fused N-heteroaromatics.31 The electrosynthesis process was made possible in 2018 due to the evolution of a tetra-arylhydrazine catalyst that generates NH2 and the subsequent discovery of the distinct reactivities exhibited by nitrogen and C-centered radicals in constructing C–N links. In order to achieve this purpose, a readily accessible carbamate 26 was employed as a representative substrate. An undivided cell, equipped with a reticulated vitreous carbon-anode and a Pt-cathode, was utilized to evaluate various electrolysis conditions. The most favorable outcomes were obtained when a steady current was applied to the electrolysis of 26 in a mixed solvent of CH3CN and H2O, while under reflux conditions and in the presence of tetraarylhydrazines-catalyst. Despite the fact that tetraarylhydrazines are readily accessible, they have not been employed as a redox catalyst in any known instances.32,33 The imidazopyridine compound 27 (Scheme 10) was successfully extracted with a yield of 89% under the given conditions.34
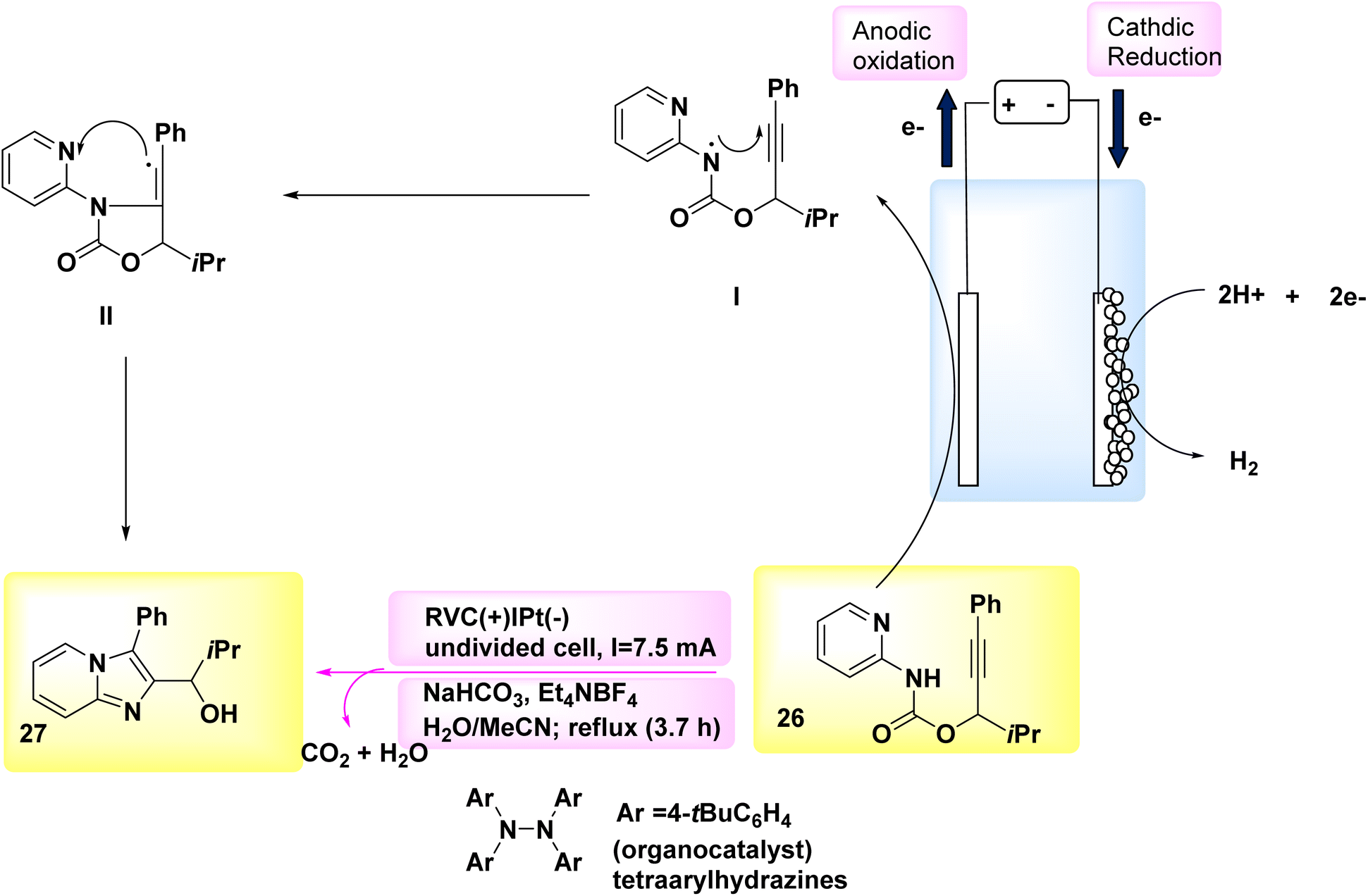 | ||
| Scheme 10 Electrochemical synthesis of fused imidazo–pyridine 27 with its proposed mechanism. | ||
A reagent-free intramolecular dehydrogenative carbon–nitrogen cross-coupling technique for mild electrolysis was discovered in 2018 (Scheme 11). The synthesis of valuable C6H5N3 30 and their derivatives can be achieved by a convenient one-pot technique using readily available aldehydes and 2-hydrazinopyridines 28. This synthetic approach is both atom- and step-economic. The approach described herein, which may be easily implemented on a gram scale in the absence of oxidizing agents or metallic catalysts, applies to a wide range of functional groups. The present study employed a novel methodology for the synthesis of Xanax, a widely prescribed medicine, along with subsequent late-stage functionalization to introduce chemical heterogeneity in pharmacologically significant lead compounds.35
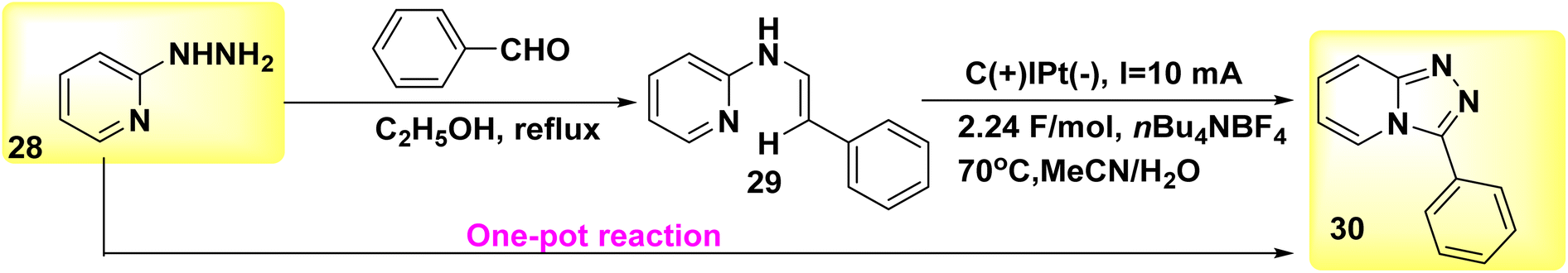 | ||
| Scheme 11 Electrochemical synthesis of 1,2,4-tetrazolo [4,3-α]pyridines 30. | ||
The year 2018 witnessed the evoluation of a method for synthesizing 3,5-disubstituted-1,2,4-thiadiazoles using an electrochemical process involving the NH4I-mediated dimerization of thioamides 31 (Scheme 12).
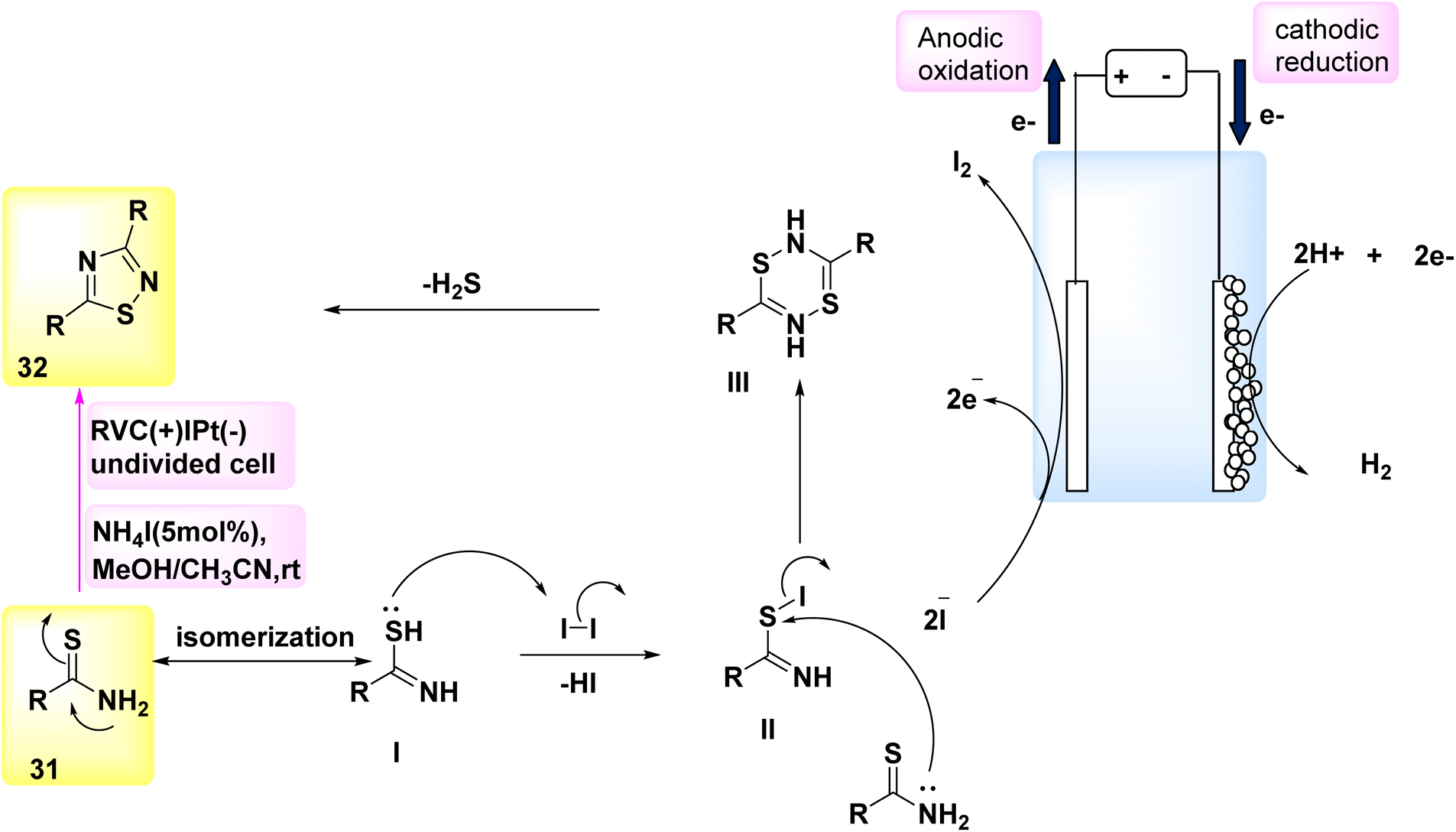 | ||
| Scheme 12 Electrochemical synthesis of 3,5-disubstituted-1,2,4-thiadiazoles 32 with its proposed mechanism. | ||
The electrosynthesis technique described in this study employs NH2I as both a catalyst and electrolyte. Notably, this method eliminates the need for oxidizing agents, hence enabling the synthesis of a diverse array of 1,2,4-thiadiazole molecules. The procedure serves as a demonstration of the electrochemical formation of S–N bonds.32
In the year 2018, significant progress was achieved in the field of electrochemical reactions for the synthesis of functionalized tetrazoles, with a particular emphasis on their ecological compatibility. The simple reaction did not require oxidants or metal catalysts, and a range of chemicals were deemed suitable under the favorable conditions.
Significantly, this reaction can be conveniently conducted in a singular vessel or on a scale of grams. Additional applications of this inovationare currently being evaluated.33 The electrochemical reaction was conducted in a basic undivided cell under conditions of continuous current.
The anode is composed of reticulated vitreous carbon, whilst the (−) consists of a Pt-plate. Tetrazole 35 was successfully combined with a high yield of 90% using the process of electrolyzing hydrazone 33 and TMSN3 34 at a temperature of 0 °C. This reaction took place in a solvent mixture consisting of CH3CN and CH3OH.
In this experiment, the electrolyte employed was lithium perchlorate (LiClO4) as described in Scheme 13.
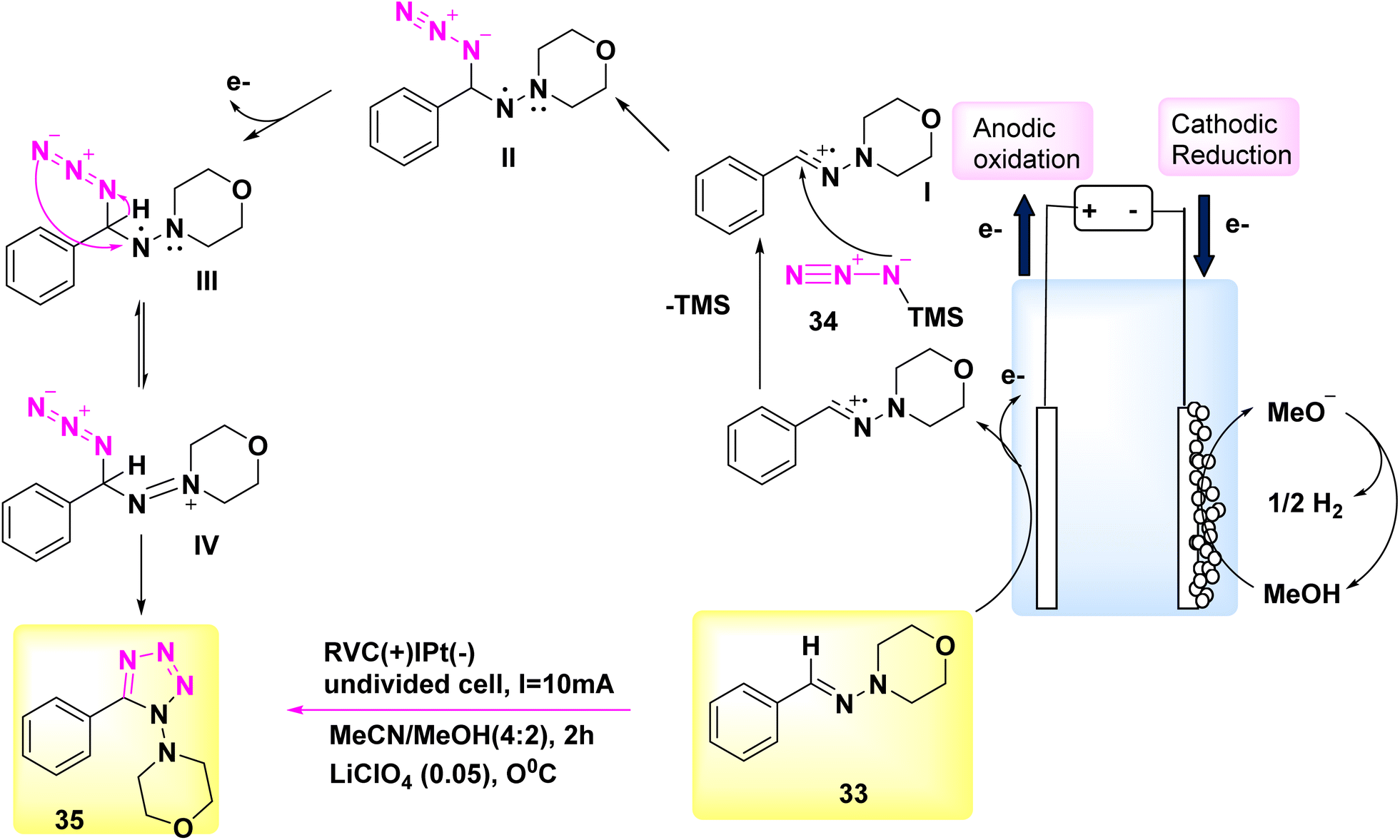 | ||
| Scheme 13 Electrochemical synthesis of 1,5-disubstituted tetrazole 35 with its proposed mechanism. | ||
The author presents a viable technique in Scheme 13. The formation of carbocation intermediate I can occur through anodic oxidation of 33. Subsequently, this intermediate can react with TMSN3 to establish a carbon–nitrogen bond, resulting in the generation of intermediate II. Afterwards, the anode has the potential to undergo oxidation of the N-centered radical, resulting in the development of intermediate III. This intermediate exhibits resonance with IV. After intramolecular deprotonation and cyclization, facilitated by the methoxide ion generated during the cathodic reduction of CH3OH, compound III/IV undergoes a conversion process resulting in the production of tetrazole product 35.
In the year 2018, a novel electrochemical methodology was employed to achieve the initial direct aziridination of alkenes, with potential applicability to styrenes bearing multiple substituents. C3H2F6O sulfamate 37 was employed as a nucleophilic N-source. The mechanistic experiments suggest that the electrochemical process involves the sequential production of two carbon-nitrogen bonds, facilitated by the interaction between cationic carbon species or sulfamate.
In order to assess the feasibility of this approach, a group of 36 subjects were subjected to electrochemical methods at a voltage of 5 V, utilizing LiClO4 as the electrolyte. Graphite felt electrodes were employed, and the experiment was conducted in the presence of sulfamate 37 dissolved in C2H3N. The aziridine 38 was obtained with a yield of 87% using 2,6-lutidine as a base, as described in Scheme 14.36
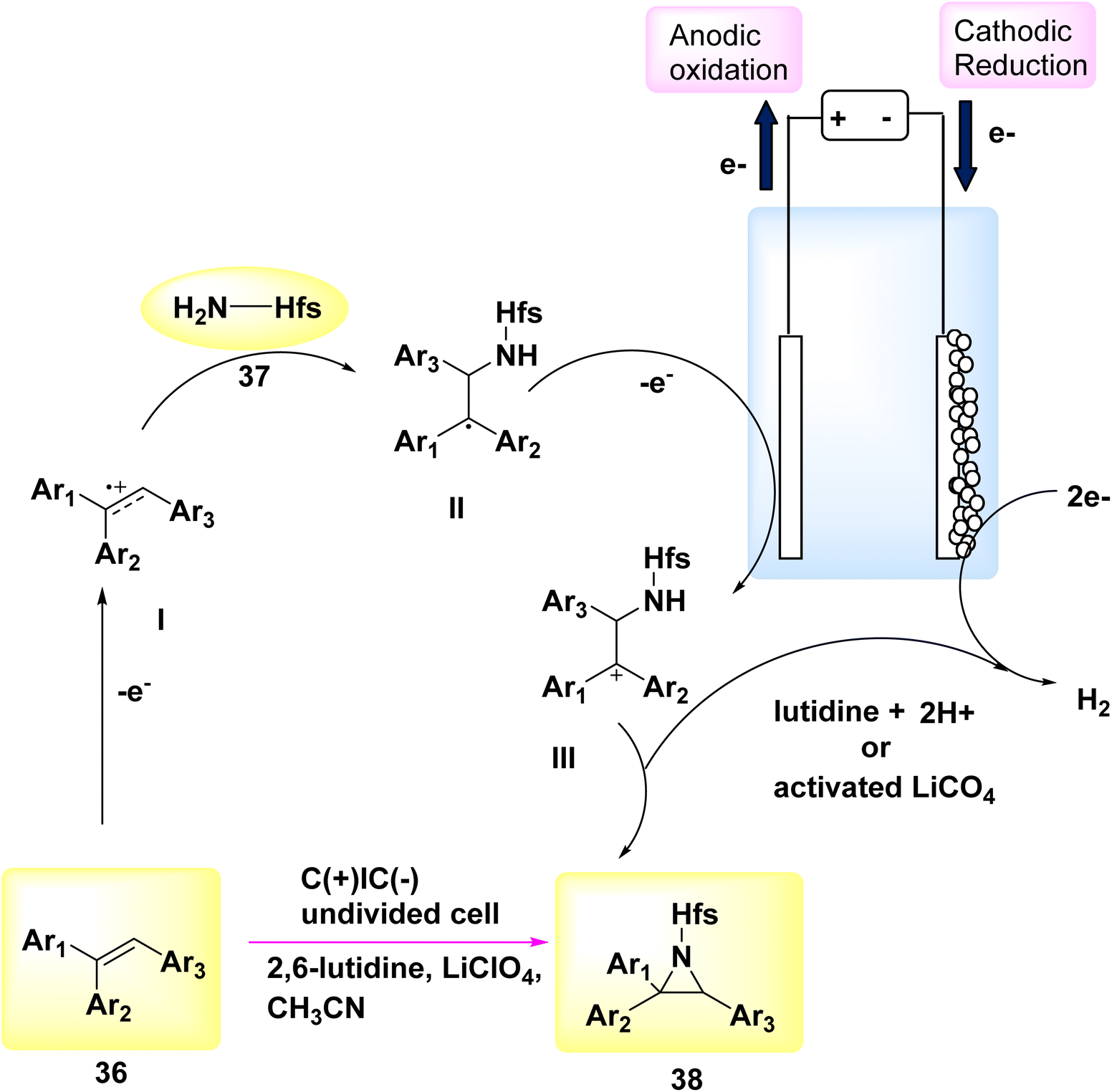 | ||
| Scheme 14 Electrochemical synthesis of aziridine 38 with its proposed mechanism. | ||
A proposal was made to establish a reaction route, as seen in Scheme 14. The alkene undergoes an anodization process, leading to the generation of carbocation radical I. The sulfamate nucleophile is introduced, and lutidine is employed as a deprotonating agent, leading to the formation of a neutral radical species II. This species is subsequently oxidized on the anode, resulting in the production of carbocation III. The aziridine product 38 is ultimately achieved by the process of ring closure. Lutidine is regenerated at the cathode through the process of H2 release. Furthermore, the cathode discharge process can involve the utilization of LiClO4, activated potassium benzyl trifluoroborate, or CH3CN.
In the year 2018, a method based on electrochemical reactions was employed to illustrate the dehydrogenative carbon–hydrogen/nitrogen–hydrogen [4 + 2] annulation of amides 39 utilizing either ethylene 40 or ethyne 41. Nevertheless, there are only a limited number of methodologies that can be employed to introduce ethylene or ethyne into intricate chemical structures. The compound Co(acac)2 was effectively employed as a catalyst in the existence of NaOPiv and undivided electrolytic conditions for the reaction involving ethylene 40. Under the conditions of a constant current electrolysis of 4.0 mA, a significant yield of 89% for the cyclization product 42 was obtained after 4 hours. The electrochemical procedure required the use of the aminoquinoline leading group, as the presence of pyridine or pyridine-N-oxide as directing groups did not yield any favorable result. The selection of a Co catalyst precursor had a pivotal role in achieving a high reaction yield. An experiment was conducted using a 5 mmol scale reaction in a bigger divided cell with a constant current of 30 mA (Scheme 15). Both the (+) and (−) electrodes were constructed using carbon fabric. Fortunately, a significant amount of 0.90 grams, which accounts for 66% of the initial 56 grams, was successfully removed following a 13 hours electrolysis process.37
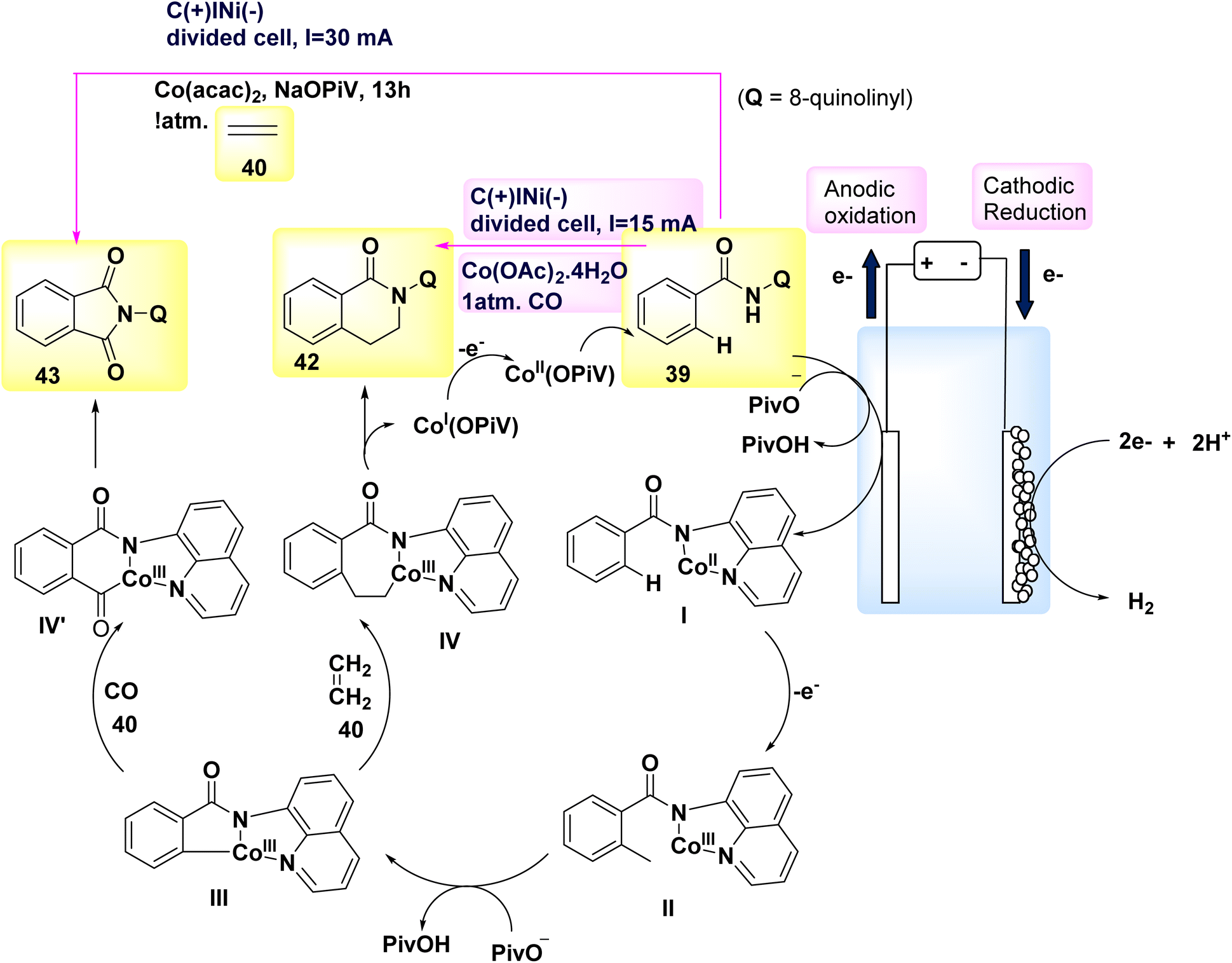 | ||
| Scheme 15 Electrochemical synthesis of 2-(quinolin-2-yl)-3,4-dihydroisoquinolin-1(2H)-one 42 and 2-(quinolin-2-yl)isoindoline-1,3-dione 43 with its proposed mechanism. | ||
One of the most straightforward methods for synthesizing CO compounds involves the utilization of oxidative carbonylation with CO (Scheme 15), a process that is facilitated by transition metals as catalysts. In the year 2018, the intramolecular CH/NH carbonylation process was successfully conducted using anodic oxidation, with C17H14N2O 39 as the chosen substrate. At a constant current of 15 mA, an isolated yield of 43 with an efficiency of 85% could be achieved using electrolysis. The catalyst Co(OAc)2·4H2O exhibited the most superior performance.38
In light of the mentioned results, a putative mechanism is proposed for Scheme 15. By utilizing NaO–Piv·H2O, it is possible to synthesize Co(II) complex I as a bidentate N-coordinated Co(II) complex, which first coordinates with 39. Subsequently, the anode facilitates the direct oxidation of complex I, resulting in the production of Co(III) complex II. Complex II undergoes intramolecular carbon–hydrogen activation facilitated by NaOPiv·H2O, resulting in the formation of cyclic Co(III) complex III. The final products, denoted as 42 and 43, are synthesized through the processes of ethylene 40 or CO-insertion and the subsequent reductive elimination of the Co(III) species. To restore the Co(II) catalyst, the Co(I) species generated during reductive exclusion undergo oxidation at the C-anode. The presence of a considerable quantity of H2 gas in the reaction system, as detected by gas chromatography following the cessation of the reaction, suggests that the cathodic reaction most likely involves proton reduction.
In the year 2018, a series of experiments involving competition, KIE measurements, and cyclic voltammetry investigations were conducted in order to gain a better understanding of the carbon–hydrogen/nitrogen–hydrogen annulation process. These investigations resulted in the identification of key mechanistic insights, which in turn led to the evoluation of a schematic representation (Scheme 16). This illustration depicted the formation of an intermediate molecule, denoted as I, followed by the alkyne addition 46 to generate the crucial intermediate II. Through the process of reductive elimination, the required product 47 or 48 is respectively synthesized.39
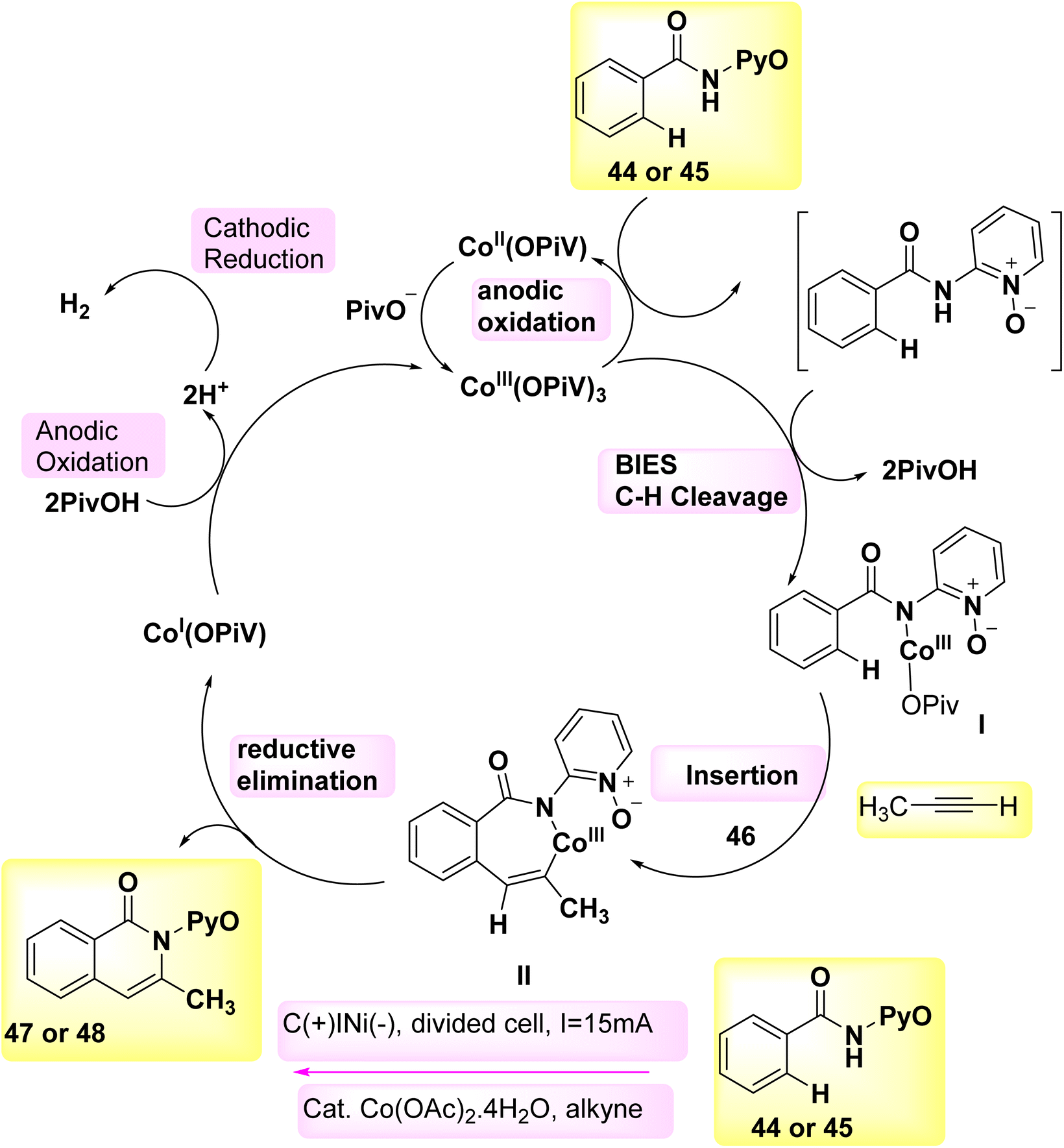 | ||
| Scheme 16 Electrochemical synthesis of isoquinolones 47 and pyridones 48 with its proposed mechanism. | ||
In the year 2018, allenes were employed for the purpose of electrochemically activating C–H bonds, which were afterwards connected to Co catalysis with a high degree of versatility. Therefore, under mild circumstances, the process of allene 50 annulations was successfully achieved, specifically targeting carbon–hydrogen/nitrogen–hydrogen functionalizations. The reactions exhibited favorable levels of chemo-, site-, and regioselectivity. The conversion of Substrate 49 into product 51 occurs. The discovery was made that employing a reduced vanadium compound (RVC) as an anode yielded favorable results. Furthermore, it was shown that various Co-salts might potentially serve as the precatalyst. The electrochemical activation of C–H bonds demonstrated efficacy when employing various solvents, such as polar protic alcohols, tetrahydrofuran (THF), and dichloromethane (CH2Cl2). Hence, the utilization of MeOH as the solvent and sodium pivalate as the additive yielded the most optimal reaction conditions, as depicted in Scheme 17.40
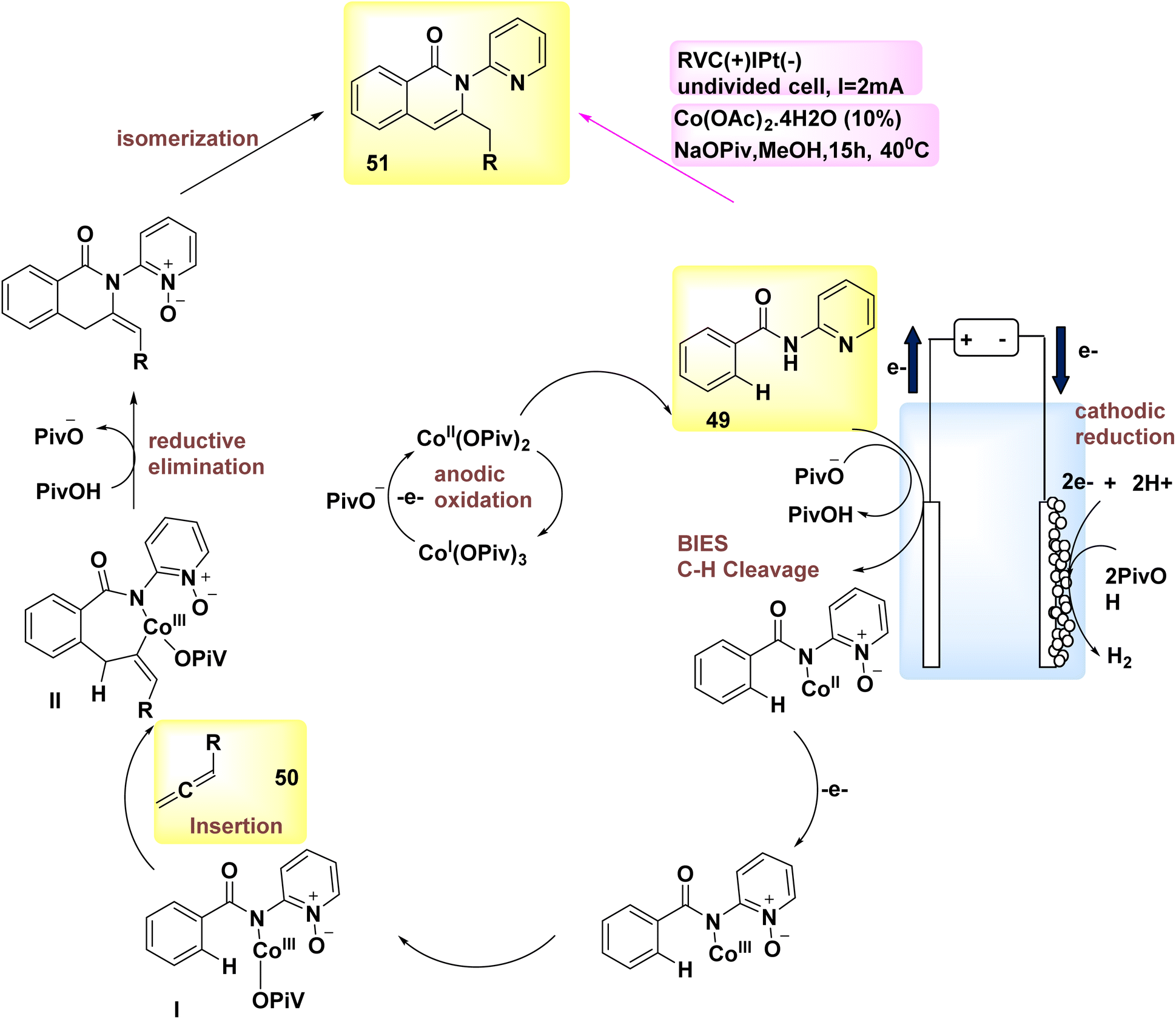 | ||
| Scheme 17 Electrochemical synthesis of carbon–hydrogen annulation product 51 with its proposed mechanism. | ||
The described mechanism is mentioned in Scheme 17, depending on both experimental and theoretical mechanistic investigations. It facilitates the process of achieving a successful BIES–carbon–H2 scission by utilizing carboxylate assistance. The exo-methylene isoquinolone III is synthesized through the reaction of allene 50 and subsequent reductive elimination, resulting in the isomerization and formation of product 51.
Subsequently, the pivotal process of anodic oxidation effectively restores the active Co catalyst, resulting in the sole byproduct of H2 gas.
The year 2018 witnessed the demonstration of the utilization of flexible Co-catalyzed activation and annulation of internal alkynes 53 and 52 for the purpose of achieving substituted isoquinolone product 54. The electro-oxidative carbon–hydrogen activation array was demonstrated using earth abundant Co catalysts in an undivided cell arrangement. The reaction parameters were kept at a very low level, and the experiments were conducted at room temperature. The exclusive production of H2 gas as a byproduct is achieved through the utilization of electrochemical Co catalysis, which involves the prevention of metallic oxidizing agents.41
Scheme 18 has presented a plausible mechanism, drawing upon the fundamental research conducted. Initially, the process of anodic oxidation is employed to generate the Co(III) salt with catalytic properties. Afterwards, the process of carbon–hydrogen coation, facilitated by carboxylate, results in the formation of Co(III) species I. After the process of migratory insertion, the formation of Co(III) complex II occurs. The process involves the concurrent release of isoquinolone 54 and Co(I) intermediate, which is then followed by reductive elimination. The process of anodic oxidation was employed for the purpose of regenerating Co(III) carboxylate, a compound known for its catalytic properties.
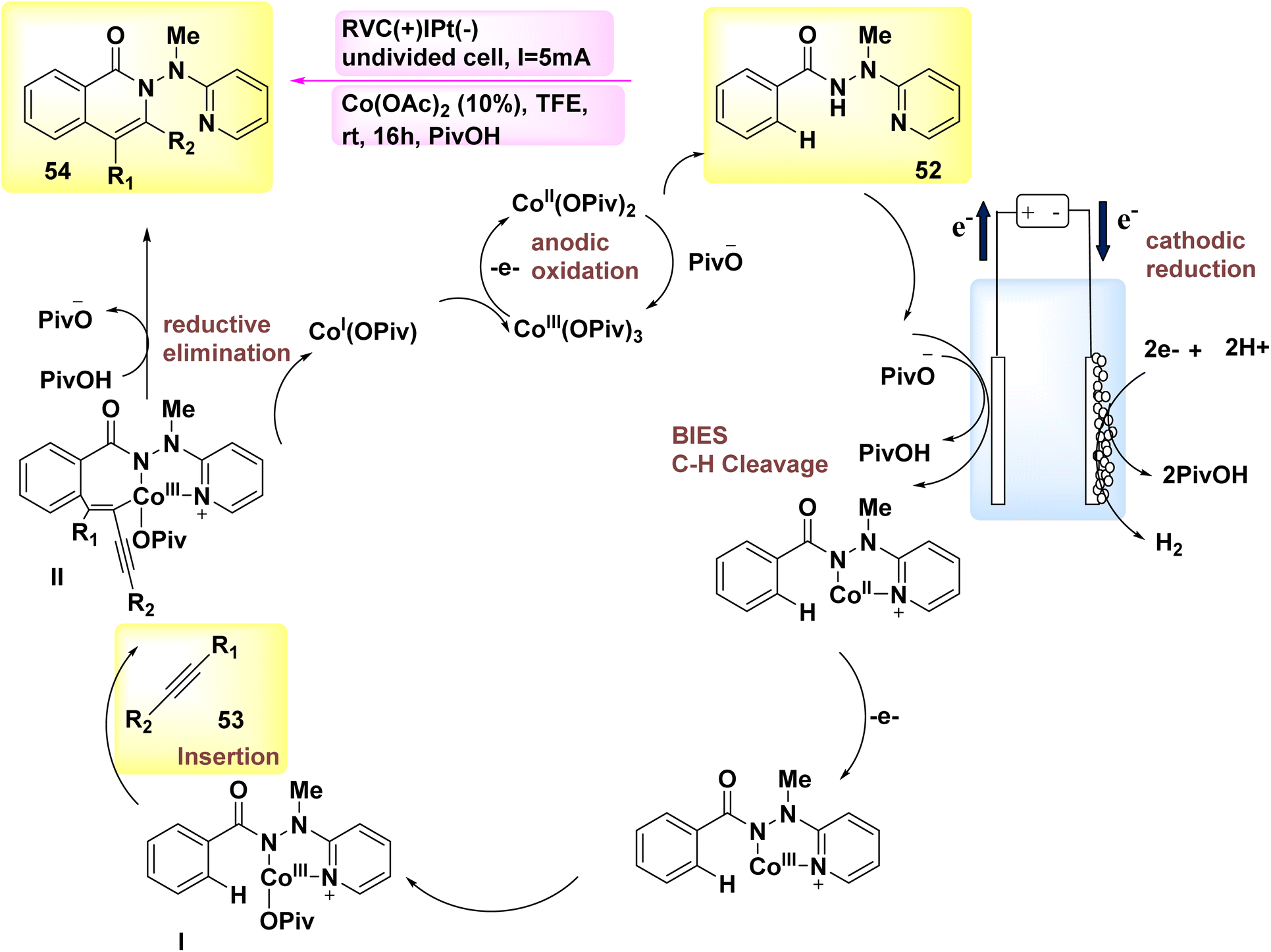 | ||
| Scheme 18 Electrochemical synthesis of substituted isoquinolone 54 with its proposed mechanism. | ||
In general, Co-electrocatalysis circumvents the need for employing costly and hazardous oxidizing agents in stoichiometric quantities, hence producing H2.
In the year 2018, researchers discovered that ethyl naphthalen-1-ylcarbamate 55 can be simply used to electrooxidatively annulate alkyne 53. The rhodium(III) catalysts commonly utilized shown a lack of efficacy in generating the desired product, resulting in a mere 70% yield. DMF and t-AmOH were identified as solvents that yielded favorable outcomes among a set of conventional solvents. However, it was found that the inclusion of C2H3KO2 in a solvent blend consisting of t-ammonium hydroxide and H2O proved to be the most efficacious.42
According to mechanistic study, it has been proposed that the initiation of a viable catalytic cycle can be achieved through the facile activation of an organometallic CH bond (as seen in Scheme 19).
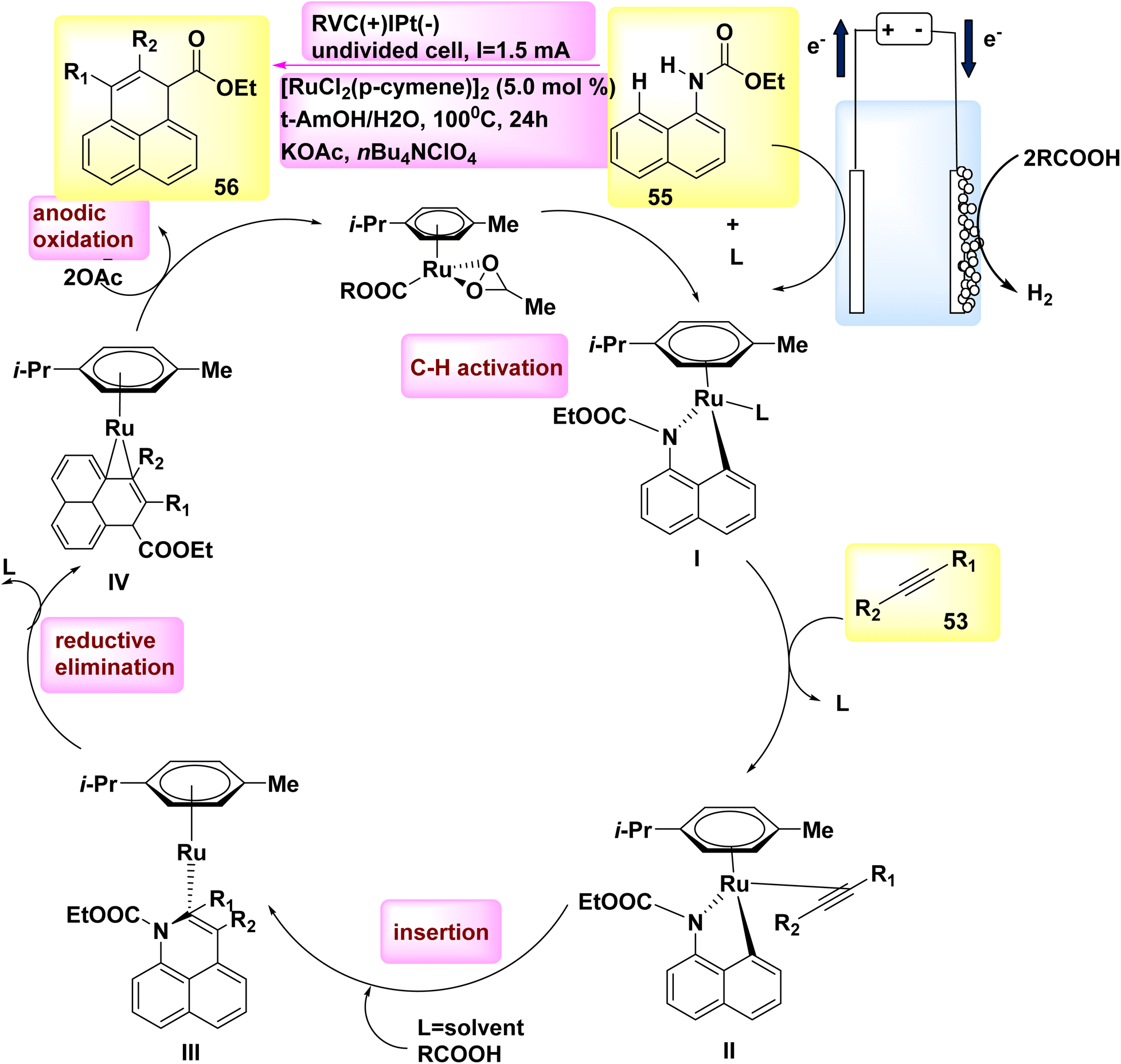 | ||
| Scheme 19 Electrochemical synthesis of rhodium(III) catalysed annulated product 56 with its proposed mechanism. | ||
Therefore, the reaction yields ruthena(II) cycle I and two equivalents of COOH. The ruthena(II) cycle III, consisting of seven members, is then obtained through alkyne inclusion. This cycle is rapidly converted into Ru(0) sandwich complex IV via reductive elimination.
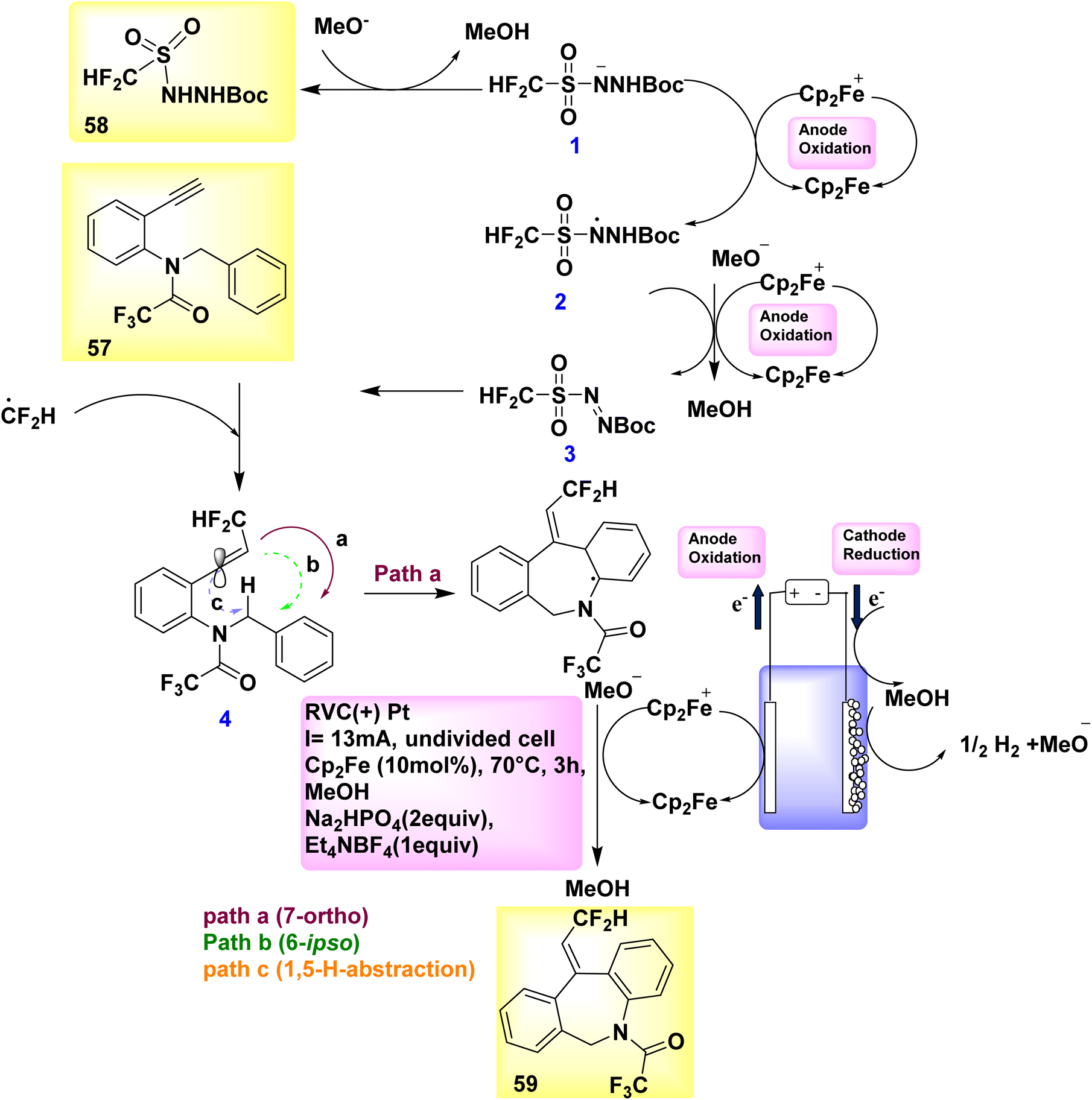
Catalytic reduction produces just molecular H2 as a byproduct in a stoichiometric way, whereas anodic oxidation is principally responsible for the crucial reoxidation of the resulting Ru(0) complex IV. In the year 2018, a novel approach was developed to synthesize fluorinated dibenzazepines 59 by the discovery of a previously unidentified difluoromethylation reagent, CF2HSO2NHNHBoc 58 (Scheme 20).
 | ||
| Scheme 20 Electrochemical synthesis of fluorinated dibenzazepine 59 with its proposed mechanism. | ||
In order to generate the compound with a molecular weight of 59, the CF2H radical, which is generated in the presence of ferrocene, engages in a unique alkyne reaction at 57. This reaction is followed by homolytic aromatic substitution process, resulting in the formation of a seven-membered ring. The stereoselective synthesis of fluorinated dibenzazepine 59 (yield = 70%) was successfully accomplished through the electrolysis of amide 57, which served as the radical acceptor and carried a terminal alkynyl group. The aforementioned reaction takes place in the presence of methanol at 70 °C temperature, employing ferrocene (Cp2Fe) as the mediator.43
Scheme 20 provides a viable explanation for the formation of fluorinated dibenzazepine 59. The initial stage of the electrolytic reaction involves the oxidation of Cp2Fe to Cp2Fe+. The process of cathodic reduction of CH3OH results in the formation of H2 gas (H2) and methoxide ions (MeO−). Consequently, the oxidation of the conjugate base of 58 by Cp2Fe+ likely facilitates the production of diazene III, maybe in conjunction with N-radical II. The occurrence of current efficiency can be attributed to the reduction of Cp2Fe+ to Cp2Fe at the cathode within an undivided cell. Upon the decomposition of III, the CF2H radical is generated, which subsequently engages with 57 to yield vinyl radical IV. Based on computational simulations (Scheme 20), it was determined that the 7-ortho cyclization (path-a) exhibited a higher kinetic preference in the present process compared to the 6-ipso cyclization (path-b) or 1,5-H abstraction (path-c). To generate the radical intermediate V, the C-radical IV proceeds along a regio- and stereoselective pathway, designated as path-a. The dibenzazepine product 59 is ultimately formed through the process of rearomatization of V, which occurs as a result of the removal of an electron (e−) and a proton (H+).
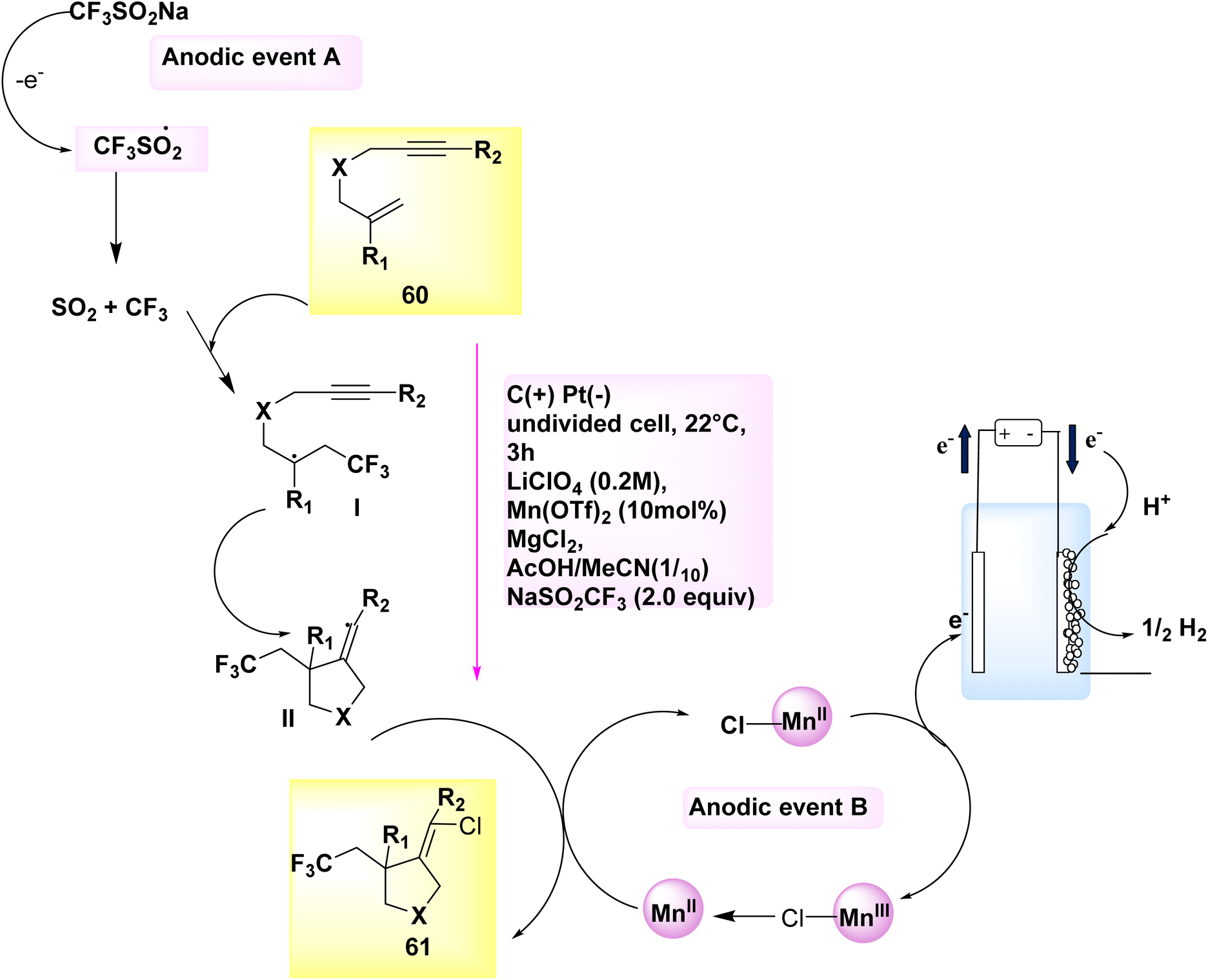
The electrocatalytic synthesis of chlorotrifluoromethylated pyrrolidine derivatives has been documented in the years 2018 and 2020. The process of anodically linked electrolysis involves the simultaneous generation of two reactive radical species at the anode, followed by their subsequent convergence and beneficial reaction. This procedure enables the desired outcome to be achieved. The addition of these intermediates to the alkene is regulated by a redox-active Mn catalyst. The eneyne cyclization products can be synthesized with a high degree of stereoselectivity in relation to the alkene geometry by employing 2,2′-bipyridine as the ligand. Remarkably, when subjected to only slightly modified reaction conditions, the 1,6-enyne substrates 60 underwent difunctionalization, resulting in the formation of chlorotrifluoromethylated pyrrolidines 61. In this experimental protocol, lithium perchlorate (LiClO4) was employed as the electrolyte, while a combination of CH3COOH and CH3CN served as the solvent within an undivided cell.
The reaction is conducted at a temperature of 22 °C for a duration of 3 hours. In this particular instance, the utilization of the (bpy)-bidentate ligand significantly enhanced the stereochemical properties of the products 61, as depicted in Scheme 21.44,45
 | ||
| Scheme 21 Electrochemical synthesis of chlorotrifluoro methylated pyrrolidines 61 with its proposed mechanism. | ||
A cyclic system was developed Scheme 21 for the purpose of facilitating the electrochemical ene-yne cyclization. Anodically connected electrolysis facilitates the participation of catalysts and functional group donors in promoting the anodic events A and B. A C-centered radical with sp3 hybridization is formed through the addition of the transitory and highly reactive CF3 radical to the trisubstituted alkene 60. The aforementioned intermediate is subsequently subjected to an intramolecular addition reaction with the alkyne, resulting in the formation of an intermediate containing an alkenyl radical, denoted as II. The highly reactive C-centered radical is employed in close proximity to an open-shell metal complex ([MnIII]–Cl), where it undergoes a conversion into an alkenyl chloride 61 through a radical atom transfer process. In the present experimental protocol, the catalyst experiences a process of single-electron oxidation at the electrode, leading to a subsequent transition back to the MnII oxidation state.
In 2019, Gao and colleagues published an exceptional method for synthesizing pyrrole derivatives. Pyrroles 64 were synthesized in an undivided cell utilizing a carbon plate as the anode and a Pt cathode. By subjecting easily obtainable aryl-acetaldehydes 62 and primary amines 63 to electrolysis at a constant current of 10 mA, a wide range of C4H5N derivatives were produced in high yields (Scheme 22). In addition, in humble settings, this reaction may exhibit robust resistance to functional groups and consistent reproducibility.46
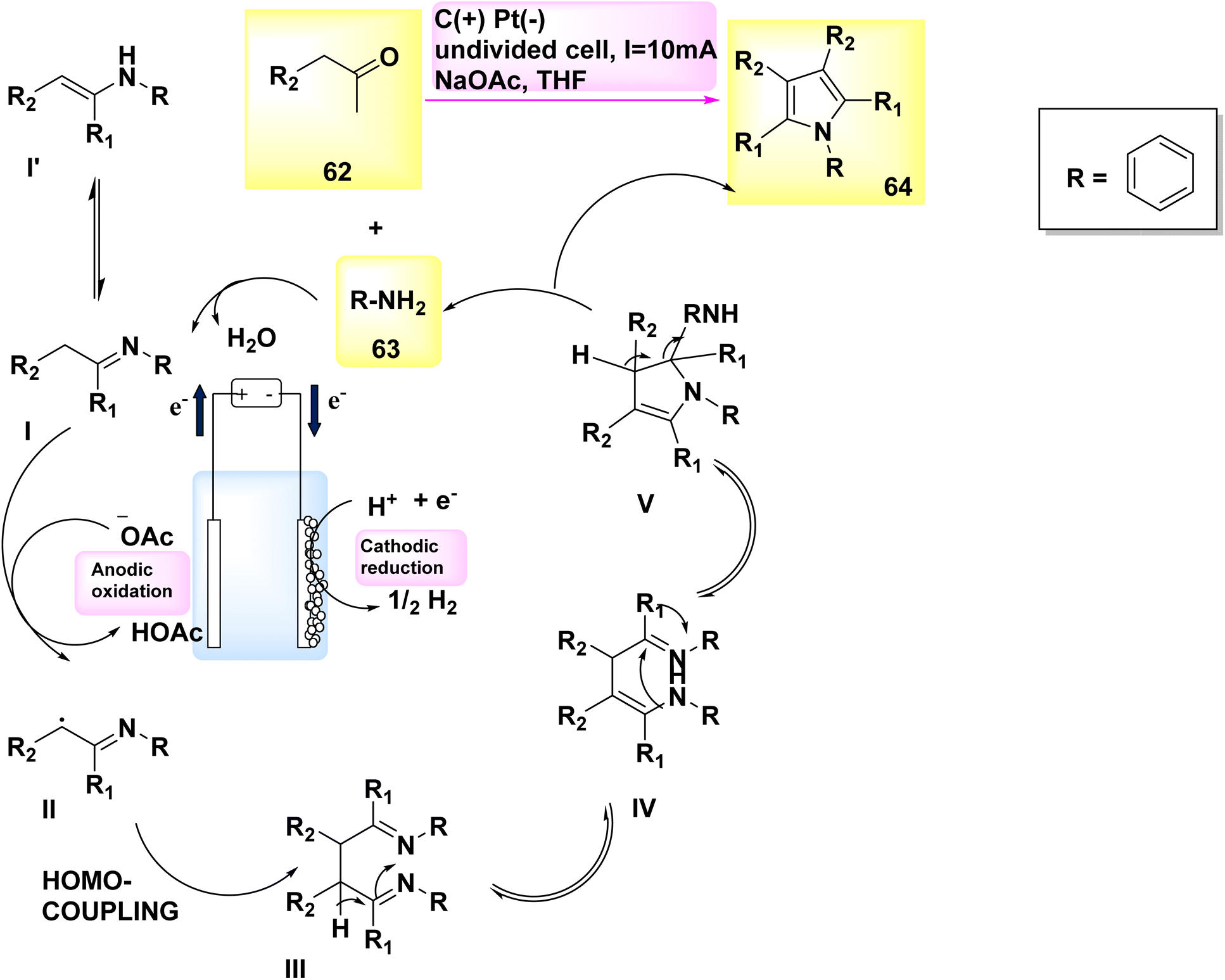 | ||
| Scheme 22 Electrochemical synthesis of 1,3,4-triphenyl pyrrole 64 with its proposed mechanism. | ||
In the year 2019, an electrolytic operation was successfully conducted without the presence of an external oxidant and the utilization of a catalyst. The approach involves a controlled and straightforward electrolysis process of sodium sulfinates 31. This is carried out in an undivided cell, utilizing a solvent mixture of methyl cyanideor water, along with the addition of Et4NClO4 salt as an electrolyte. The electrochemical trifluoromethylation and cycling of N-arylacrylamide 65 were conducted under conditions of constant current, utilizing CF3SO2Na 66. This notion has the potential to be applied to a diverse array of functional categories. A yield of 74% was achieved in the synthesis of the desired compound, C12H11F3INO 67 (Scheme 23).47
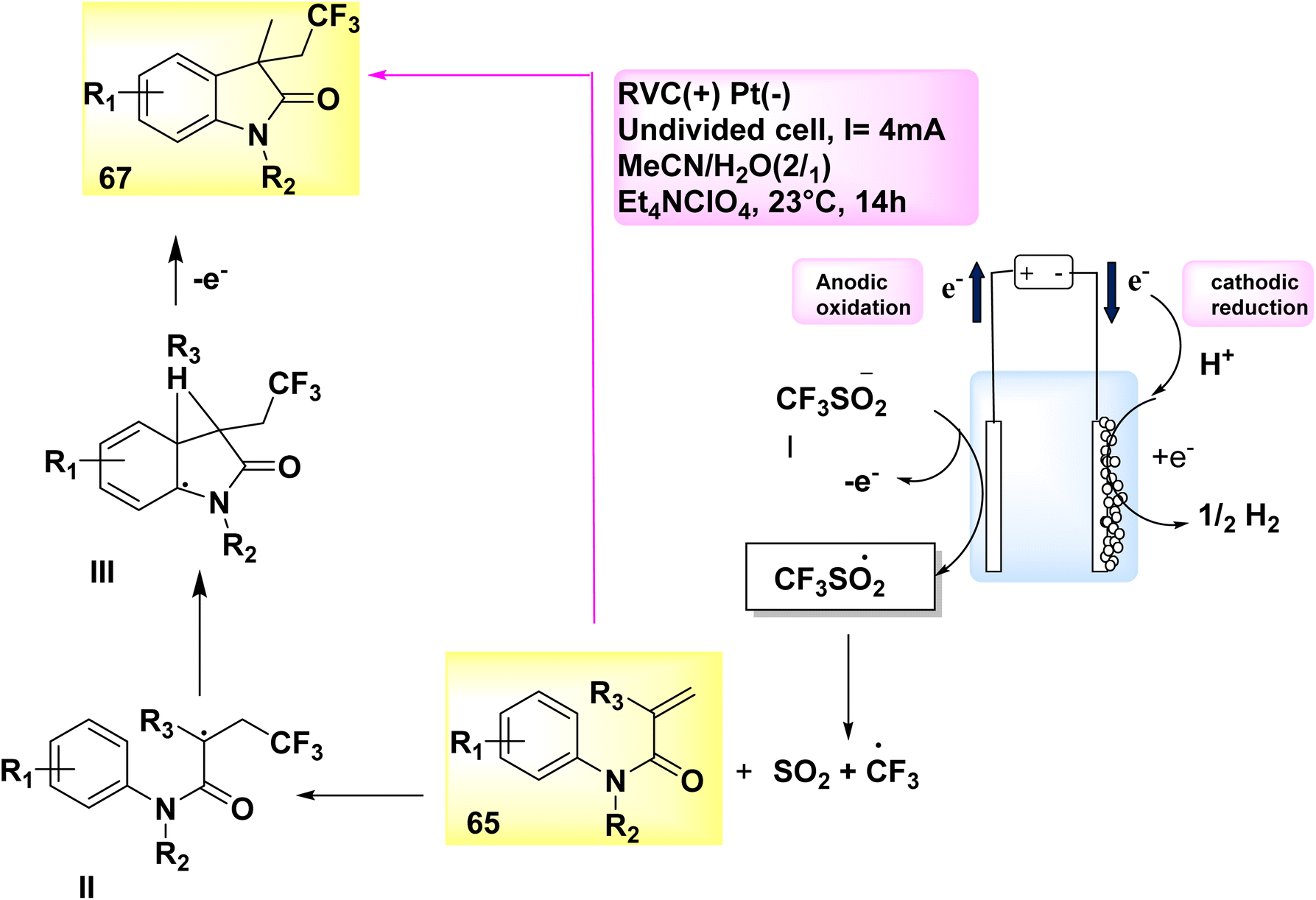 | ||
| Scheme 23 Electrochemical synthesis of 1,3-dimethyl-3-(2,2,2-trifluoroethyl)indolin-2-one 67 with its proposed mechanism. | ||
A corresponding radical is generated in a desulfurative process, leading to the conversion of sulfinate anion I into CF3 radical. Within the specified location, the CF3 radical initiates an assault on the alkene 65, resulting in the formation of intermediate II. Subsequently, intermediate II undergoes a transformation process, leading to the formation of intermediate III. Under conditions of anodic oxidation, a higher degree of aromatization leads to the formation of the same product 67. At the cathode, the process of reduction occurs where H2 cations undergo simultaneous reduction, resulting in the production of molecular H2.
In the year 2019, Hu et al. introduced a novel methodology for synthesizing N–O heterocycles. A Cu-catalyzed electrosynthesis was employed to develop an aza-Wacker cyclization process. The utilization of this tandem methodology enables the occurrence of substrate transformations that yield R – intermediates, so significantly broadening the scope of action. Alkene-substituted oxazolone 69 was synthesized under moderate conditions. The experiment commenced by employing electrochemical oxidative amination on crotyl N-phenylcarbamate 68, utilizing a Cu catalyst. It was found that the target compound 69 could be synthesized under ambient conditions using a partitioned cell setup. The experimental setup included CH3OH as the solvent, C-fiber as the electrode, LiClO4 as the electrolyte, diacetoxycopper as the catalyst, sodium acetate as the base, and a constant current of 3 mA.48
The aforementioned observations described as the basis for the evoluation of the electrochemical formal called aza-Wacker cyclization mechanism, as depicted in Scheme 24. Initially, substrate 68 undergoes a reaction with the base, I, resulting in the formation of an adduct. Subsequently, this adduct is subjected to oxidation at the anode, leading to the generation of an NH2, denoted as II. The radical undergoes cyclization to yield radical III, which is subsequently captured by copper(II) to form copper(III) alkyl intermediate IV. After undergoing a base-catalyzed elimination reaction, the resulting product is identified as product 69, which leads to the formation of a copper(I) species. In order to reinitiate the catalytic cycle, the latter undergoes oxidation at the electrode, resulting in the constitution of copper(II).
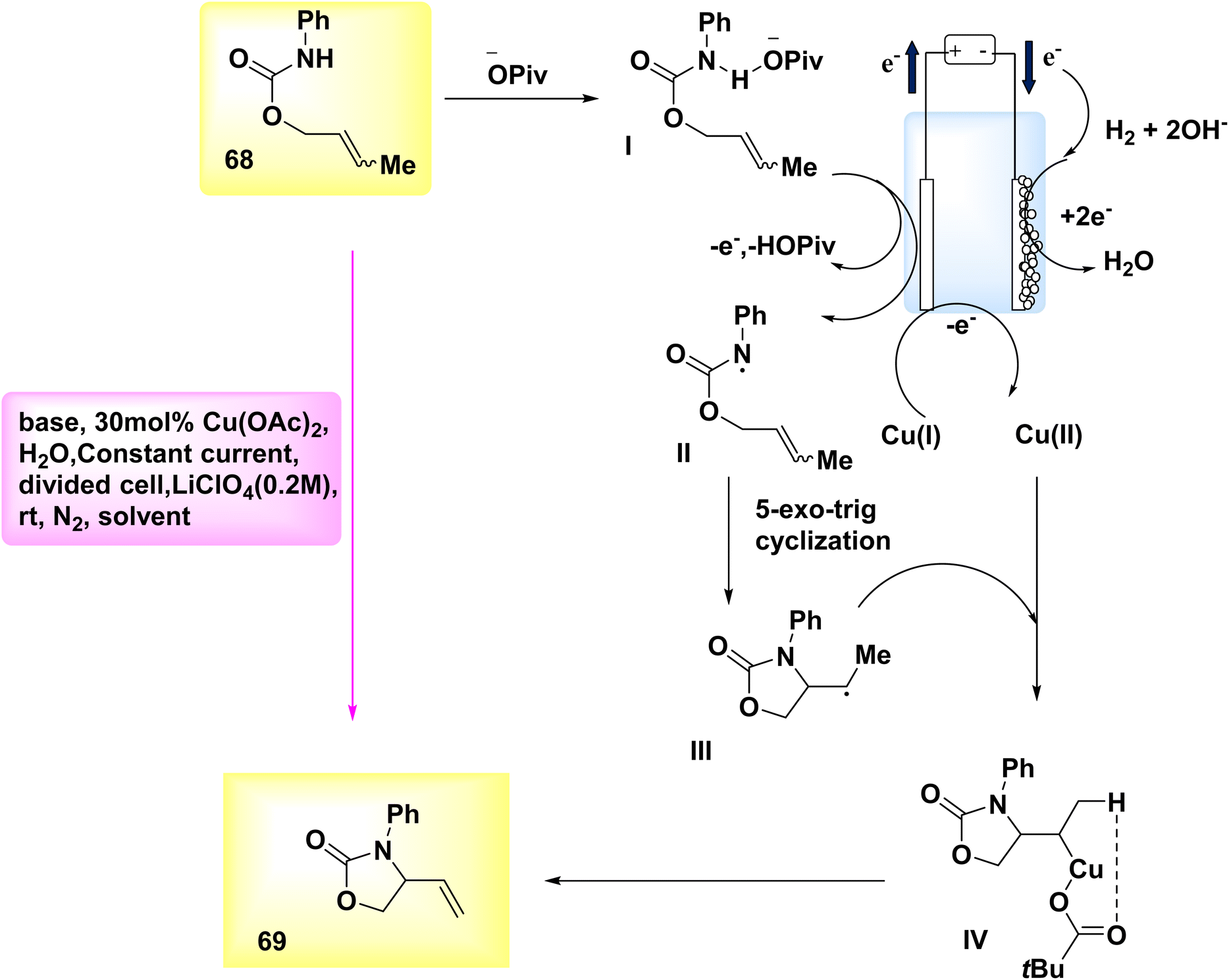 | ||
| Scheme 24 Electrochemical synthesis of alkene-substituted oxazolone 69 with its proposed mechanism. | ||
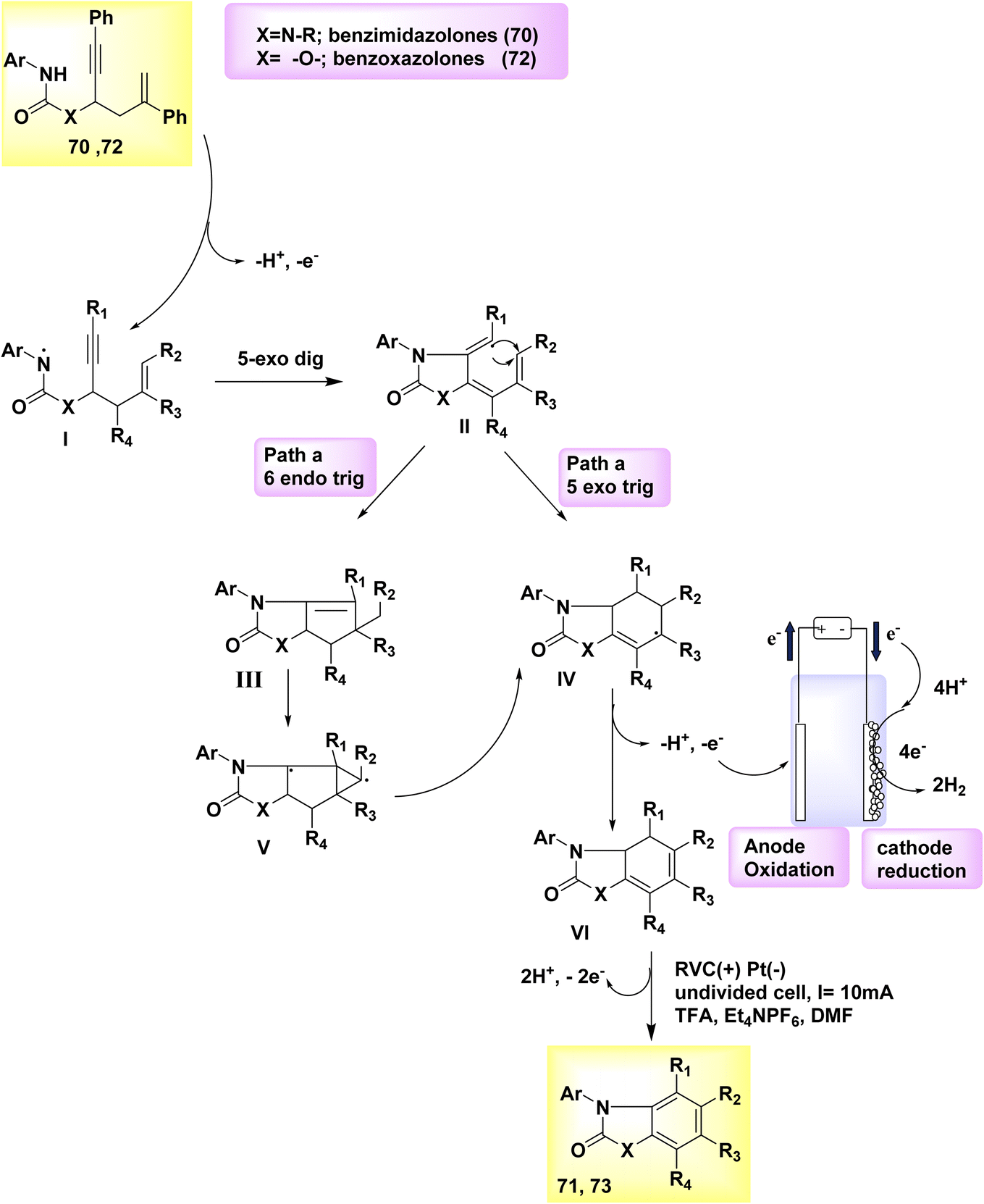
In the year 2019, there was a proposal to utilize a dehydrogenative cyclization cascade for the synthesis of benzimidazolone and benzoxazolone derivatives with high levels of substitution. This approach involves the generation of homo- and heterocyclic ringed structures. The benzimidazolone 71 and benzoxazolone 73 are fundamental structures found in various pharmaceutical drugs. The process of biscyclization/dehydrogenation involves the conversion of arylamine-tethered 1,5-enynes into functionalized benzanellated heterocycles, exhibiting precise control over regioselectivity. The mechanism of H2 evolution is responsible for driving electricity-powered oxidative reactions, hence eliminating the need for metal catalysts and oxidants altogether. The electrochemical dehydrogenative interconversion of an easily accessible urea substrate 70 was investigated in Scheme 25. In order to mitigate the susceptibility of compound 72 to base-promoted ionic hydroamidation, deliberate efforts were made to exclude any base additions.
 | ||
| Scheme 25 Electrochemical synthesis of benzimidazolone 71 and benzoxazolone 73 with its proposed mechanism. | ||
In contrast, the substrate exhibits stability even in an acidic milieu. Researchers successfully synthesized the benzimidazolone product 71 with a yield of 83% by performing an electrolytic method at a temperature of 100 °C. In this experimental procedure, a consistent current of 10 mA was utilized, along with C2HF3O2 as an additive and dimethylformamide as the solvent. The anode used was made of reticulated vitreous C, while the (−) was composed of Pt. One possible approach for synthesizing functionalized benzoxazolones 73 from propargylic carbamates 72 involves employing an electrochemical cyclization cascade, Scheme 25. The yield was notably enhanced by conducting the electrochemical process in a solvent of TFE and including C2H4O2 as an addition while maintaining a temperature of 80 °C.49
To generate an NH2, the arylamine moiety 70 of the starting material is initially subjected to anodic oxidation, followed by deprotonation, and subsequently undergoes a 5-exo-dig cyclization to form a vinyl radical II. The intramolecular 6-endo-trig cyclization of II leads to the formation of III, which possesses an acyclic carbon structure. As an alternative, compound II can undergo a 5-exo-trig cyclization reaction to yield compound IV. Subsequently, compound IV can be transformed into compound III through the involvement of an intermediate a tricyclic radical V. The vinyl radical cyclization typically results in the formation of both the 6-endo and 5-exo products, namely 71 and 73, in combination.
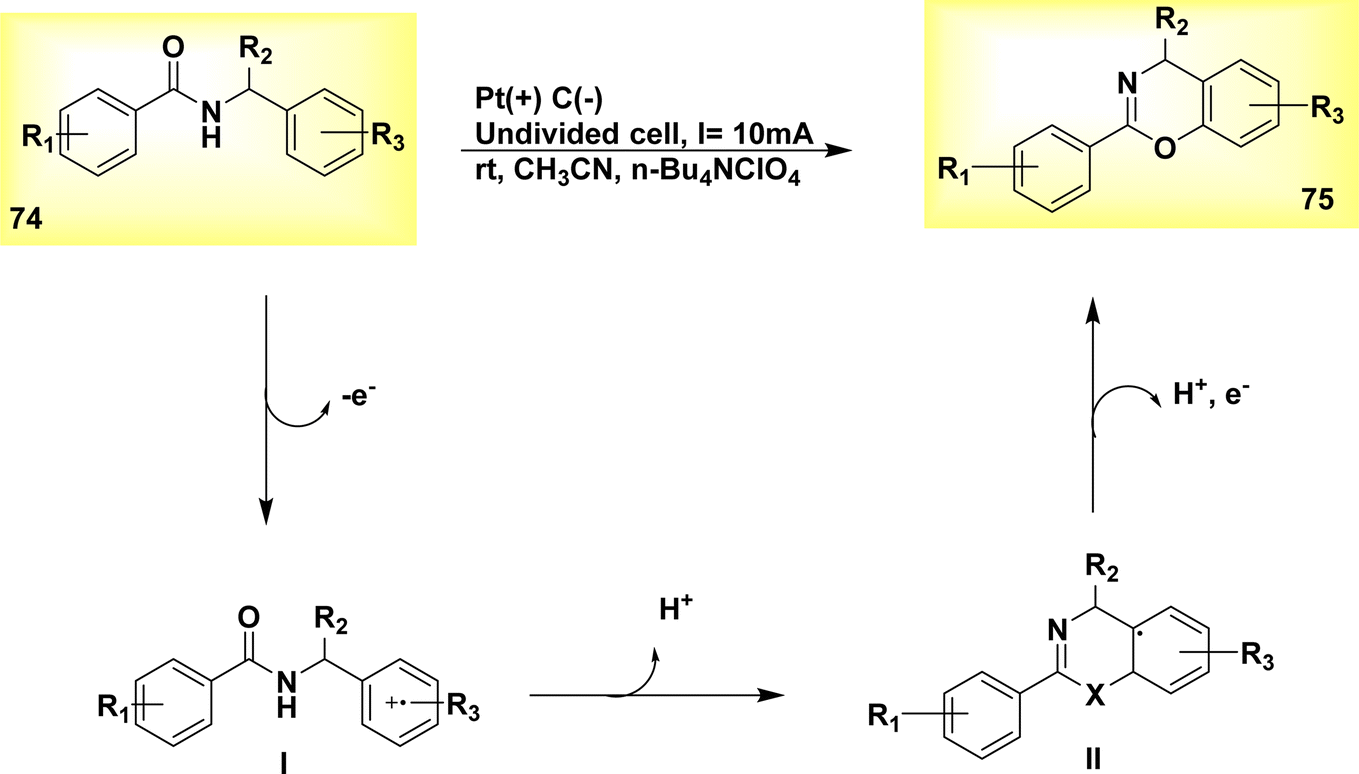
The electrochemical dehydrogenative cyclization of N-benzylamides 74 was observed in the year 2019. In an undivided cell at room temperature, a Pt-plate anode and a graphite rod cathode were utilized. A steady current of 10 mA was supplied for duration of 2 hours. The inhibition of oxidative degradation of the products was effective, resulting in the production of 4H-1,3-benzoxazines 75, Scheme 26, irrespective of the substituents at the benzylic position. The proposed methodology has the ability to synthesize 4H-1,3-benzothiazines 75.50
 | ||
| Scheme 26 Electrochemical synthesis of benzimidazolone 71 and benzoxazolone 73 with its proposed mechanism. | ||
The substrate 74 under went oxidation of its benzylic moiety by electrolysis conditions, resulting in the radical cation I formation. Subsequently, this radical cation underwent cyclization and deprotonation, leading to the formation of the intermediate radical II. Ultimately, the compound underwent oxidation followed by rearomatization, resulting in the formation of the cyclic product with a yield of 89%.
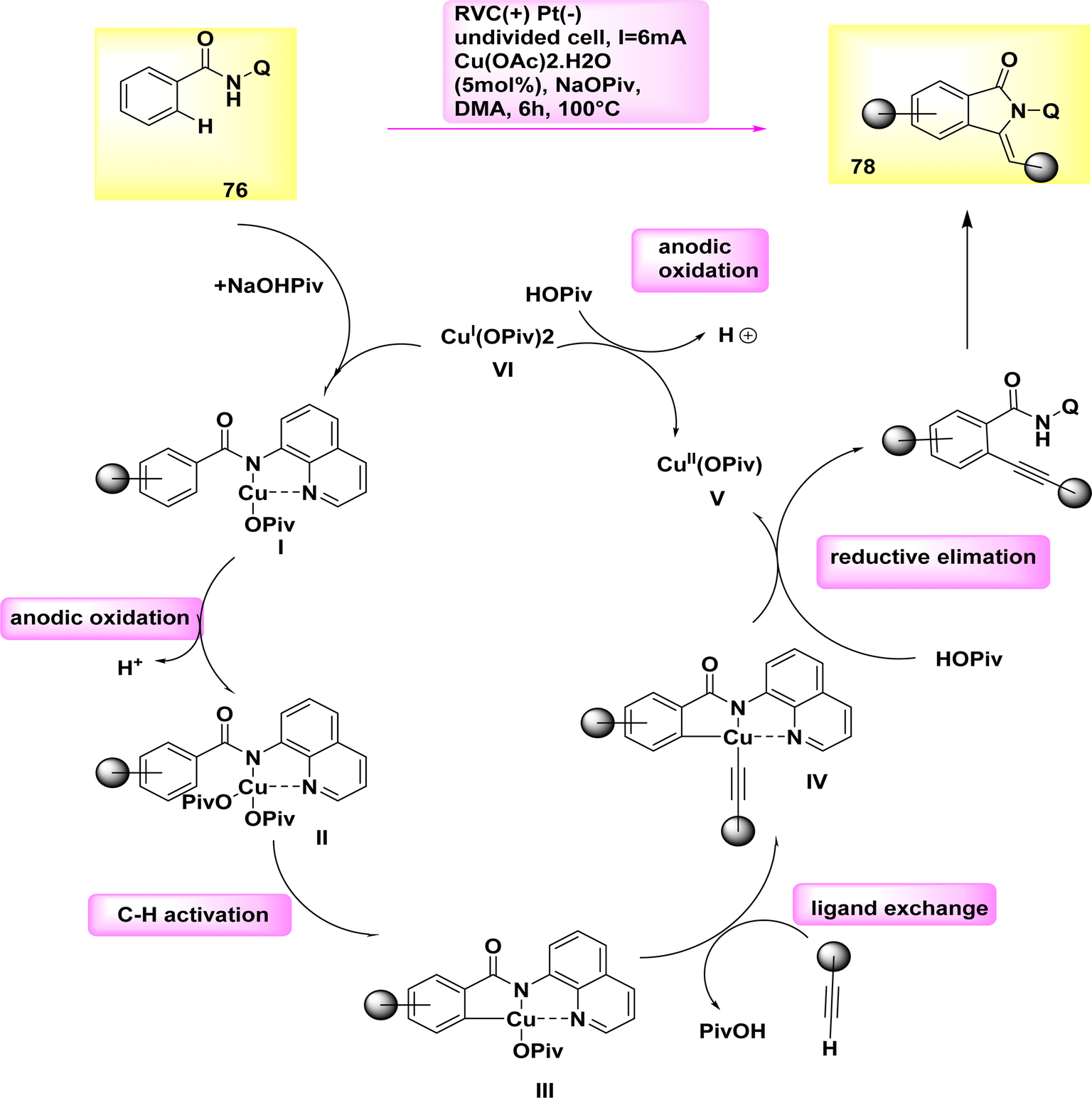
In the year 2019, a cupra electro-catalyzed electrolytic approach was successfully employed to synthesize isoindolones of significant synthetic importance. In a simplified and uncomplicated cellular configuration, Cu work as a catalyst for the electro-oxidative activation of CH or NH bonds in benzamide 76 when combined with terminal alkyne 77.
Cu(II) acetate, which is characterized by its flexibility, affordability, and non-toxicity, is utilized as a catalyst. The optimal conditions for synthesizing isoindolone 78 were determined to be at a temperature of 100 °C in dimethylacetamide, utilizing catalytic amounts of Cu(OAc)2·H2O and NaOPiv as the most effective additive.51
A potential catalytic cycle was proposed, supported by thorough mechanistic research. The cycle initiates with the coupling of substrate 76 and concludes with the oxidation of Cu(II) to generate the active catalytic Cu(III) carboxylate species Scheme 27. The Cu(III) intermediate IV is then formed through a direct CH activation when the electron-deficient benzamide II is present with a carboxylate. The alkynylated arene VII is synthesized through the metallization of the terminal alkyne 77 with the assistance of carboxylate, followed by reductive elimination to create the desired isoindolon 78. The Cu(I) complex is oxidized at the anode to regenerate the catalytically active Cu(III) species.
 | ||
| Scheme 27 Electrochemical synthesis of isoindolone 78 with its proposed mechanism. | ||
The electrocatalytic process of C–H activation using isocyanides is being investigated. At ambient temperature, in the absence of oxidizing agents, the readily accessible Co-catalysts also facilitate efficient electrooxidation carbon–hydrogen/nitrogen–hydrogen functionalization of benzhydrazides 52 employing cost-effective CO. The metallic electrocatalysis is facilitated by the utilization of a detachable pyridyl support in a practical undivided cell configuration. This design allows for a cost-effective method to synthesize physiologically significant imidates 93 and 94. In a broad sense, the utilization of electricity hinders the activation of CH bonds by equimolar amounts of d-and f-block elements, which are both costly and detrimental. However, this hindrance can be overcome with the use of a lasting Co catalyst that is abundant in the Earth's aqueous environment. The observation that only benzhydrazides 52 shown the capability to facilitate the insertion of isocyanide highlights the inherent challenges associated with the Co-electrooxidative C–H activation mechanism (Scheme 28).52
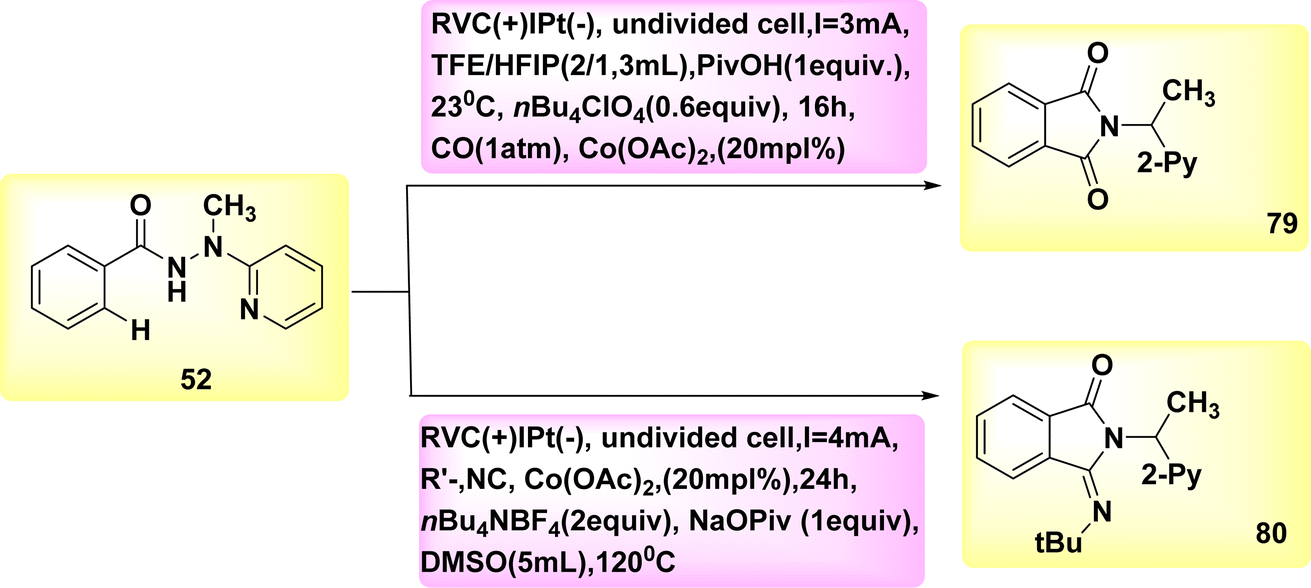 | ||
| Scheme 28 Electrochemical synthesis of substituted cyclic imidates 79 and 80. | ||
A possible catalytic cycle was described based on mechanistic discoveries Scheme 29. An intermediate, referred to as II or II′, is generated through the process of simple carboxylate-assisted CH activation. Additionally, a six-membered Co(III) cycle, known as III or III′, is also synthesized. The formation of III/III′ is achieved through consecutive migrating insertion. The production of the necessary products, specifically 79 or 80, is achieved by the process of reductive elimination. This process also results in the formation of Co(I) species.
 | ||
| Scheme 29 Electrochemical synthesis of substituted cyclic imidates 79 and 80 with its proposed mechanism. | ||
In conclusion, the utilization of anodic oxidation serves as a means to regenerate the catalytically active Co(III) carboxylate complex. This approach eliminates the requirement for costly and dangerous metal oxidants, while generating molecular H2 as the individual byproduct.
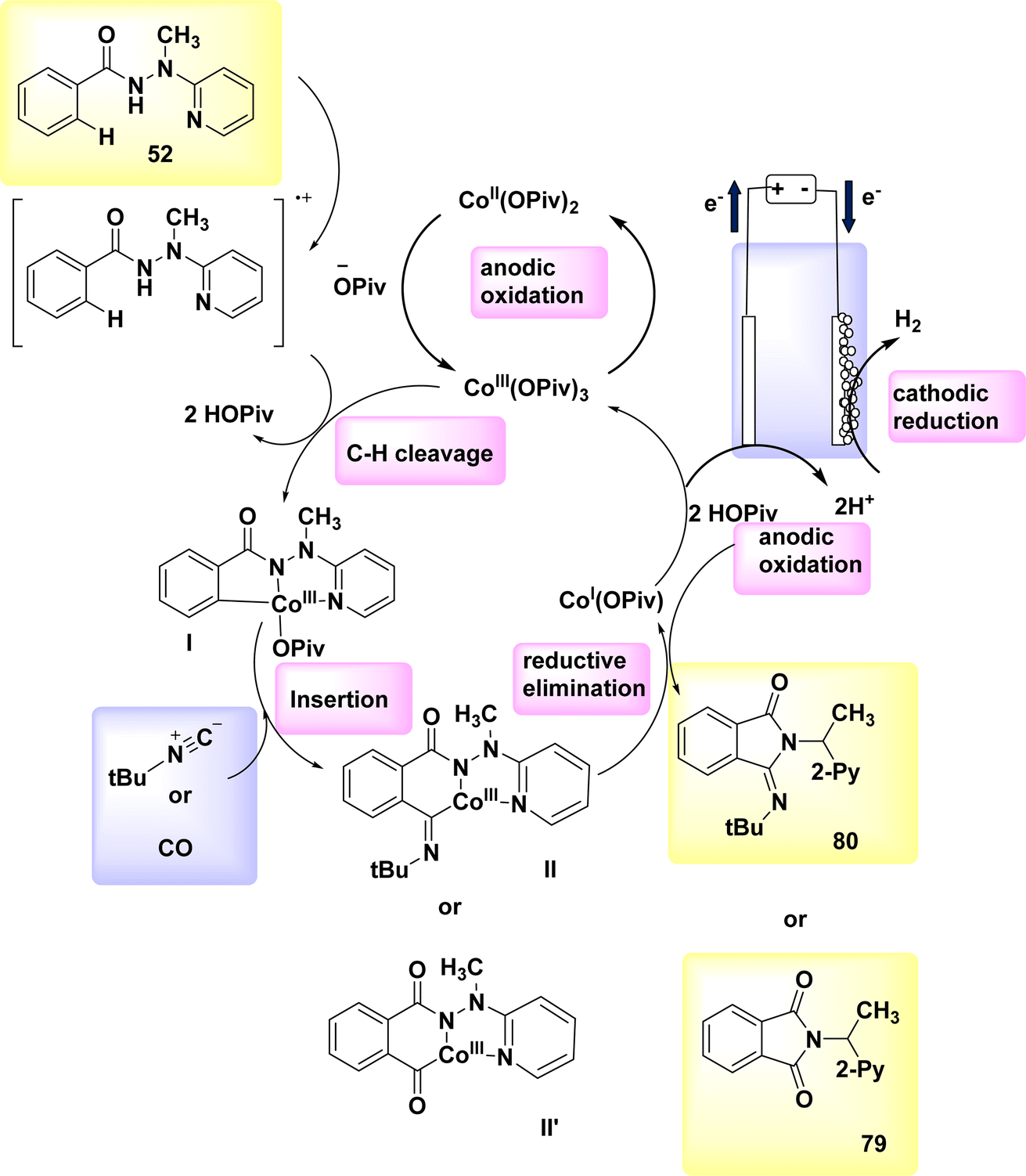
In the year 2019, a comprehensive electrocatalytic strategy (Scheme 30) was formed for the CH/NH annulation process. This technique utilized economical and easily accessible Co salts.
 | ||
| Scheme 30 Electrochemical synthesis of isoquinolone 82 with its proposed mechanism. | ||
The discovery of diynes 81 has been unveiled. The electro-oxidative Co-catalysis occurred in a elementary undivided cell at benign reaction conditions, demonstrating outstanding compatibility with various functional groups. The utilization of electricity obviates the need for potentially dangerous and/or costly chemical substances. The experiment involved the utilization of a reticulated vitreous C(+) and a Pt(−). The reaction was carried out in a solvent known as TFE at a temperature of 60 °C.53,54
The initial step in the production of the catalytically efficient Co(III) salt involves the utilization of anodic oxidation. The subsequent step involves the generation of Co(III) species through a direct C–H Coation process with carboxylate support. The synthesis of Co(III) complex II proceeds through migratory insertion, wherein the Co(I) intermediate and isoquinolone 82 compound are simultaneously released, developed by reductive elimination. The Co(III) carboxylate, which exhibited catalytic activity, was restored through the process of anodic oxidation. In general, Co-electrocatalysis is capable of generating H2 without the need for stoichiometric quantities of expensive and hazardous oxidizing agents.
Aza-PAHs, nitrogen-doped polycyclic aromatic hydrocarbons, find extensive applications within the realm of materials research. In the year 2019, a method including the utilization of annulation or Rh-catalyzed activation was employed to achieve the electrochemical synthesis of polycyclic aromatic hydrocarbons. The regio and chemo selectivity observed in this study was attributed to the functional properties of o-methyl amidoxime. Amidoxime 83 and diphenylacetylene 84 were employed as initiators for the reaction to achieve the intended objective. The target product 85 was isolated through the utilization of KOAc as a base, methanol as a solvent, and a current of 2 mA. The results demonstrated that the inclusion of a little amount of R-carboxylic yielded favorable results, with (CH2)6(CH)3(CCO2H) acid exhibiting the most significant effects.
The effectiveness of the process was further improved by the utilization of a rhodium catalyst with a positive charge. This catalyst facilitated the synthesis of product 85 with a yield of 90% at a temperature of 35 °C (Scheme 31).55
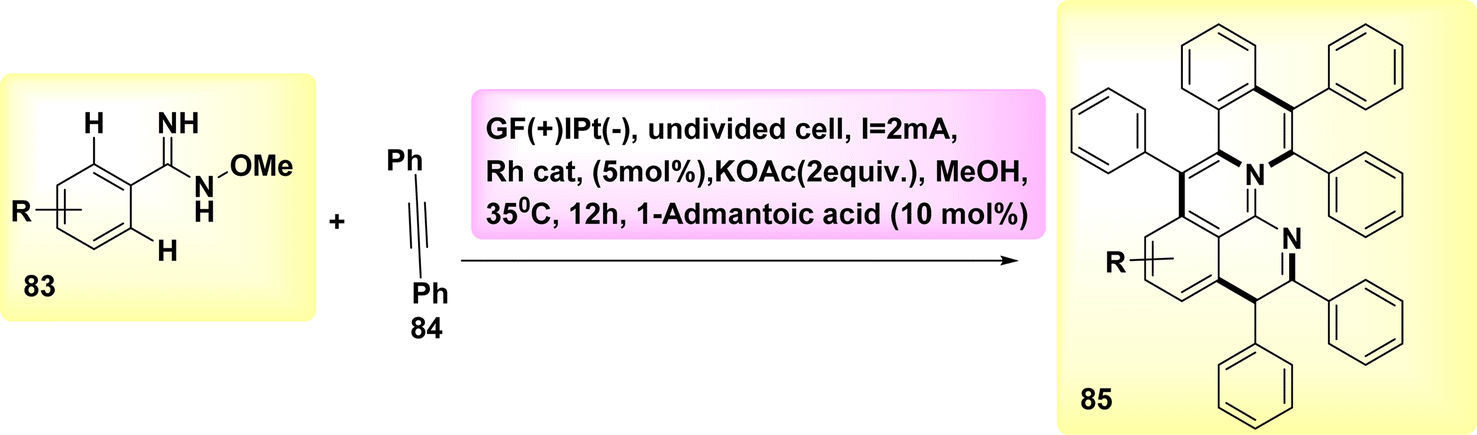 | ||
| Scheme 31 Electrochemical synthesis of nitrogen-doped polycyclic aromatic hydrocarbons (aza-PAHs) 85. | ||
In 2019, significant advancements were made in the field of metallaelectro-catalyzed annulations and activation. Consequently, a variety of carbon–hydrogen/nitrogen–hydrogen functionalizations were found to be feasible, both intramolecularly and intermolecularly, for alkyne 87 annulations. These functionalizations demonstrated significant levels of durability, sensitivity, and selectivity towards functional groups. The electrochemical reaction between imidate 86 and asymmetrical alkyne 87 resulted in the formation of the desired isoquinoline 88 with a yield of 90% (Scheme 32). In the experimental setup, methanol (CH3OH) is employed as the solvent, catalyst, while NaOPiv and PivOH are introduced. Additionally, Pt-plate and GF are utilized as the negatively and positively charged electrodes, respectively, in an undivided cell configuration.56
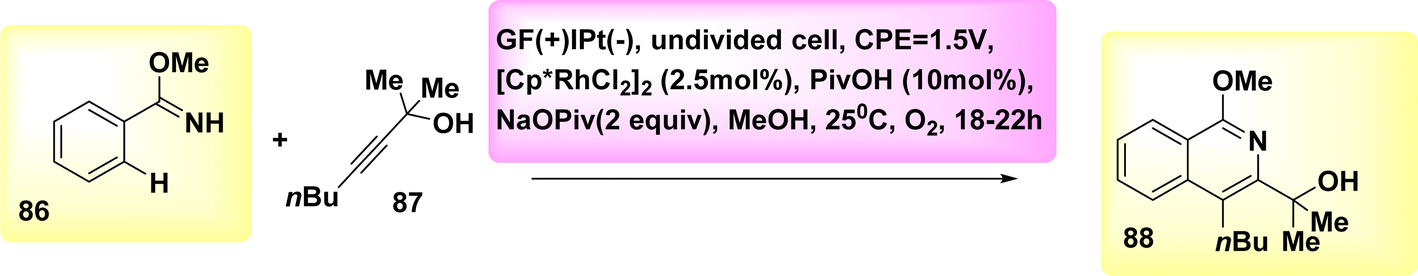 | ||
| Scheme 32 Electrochemical synthesis of isoquinoline 88. | ||
Scheme 33 proposes a plausible mechanism. The compound Cp*Rh(OPiv)2 I is synthesized through the interaction of NaOPiv with a catalyst precursor. This resulting compound then reacts with substrate 86, leading to the formation of the cyclometalated complex II via a straightforward CH-activation process. The rhoda(III) cycle IV is then generated by the processes of alkyne coordination III and migrating insertion.
 | ||
| Scheme 33 Electrochemical synthesis of isoquinoline 88 with its proposed mechanism. | ||
The rhoda(IV) cycle V efficiently generates intermediate VI by the process of anodic oxidation. The regeneration of catalyst I is subsequently facilitated through anodic oxidation, a process that can be accelerated by the presence of oxygen. Aerobic and anodic oxidations have been identified as potential mechanisms for the reoxidation of Rh II–Rh III. The rhoda(III) cycle IV has the capability to directly provide product 88, along with a diminished amount of Rh (rhodium) that has the potential to undergo reoxidation.
The process of cathodic proton reduction exclusively yields molecular H2 as a stoichiometric byproduct, a conclusion that has been substantiated through meticulous headspace gas chromatography examination.
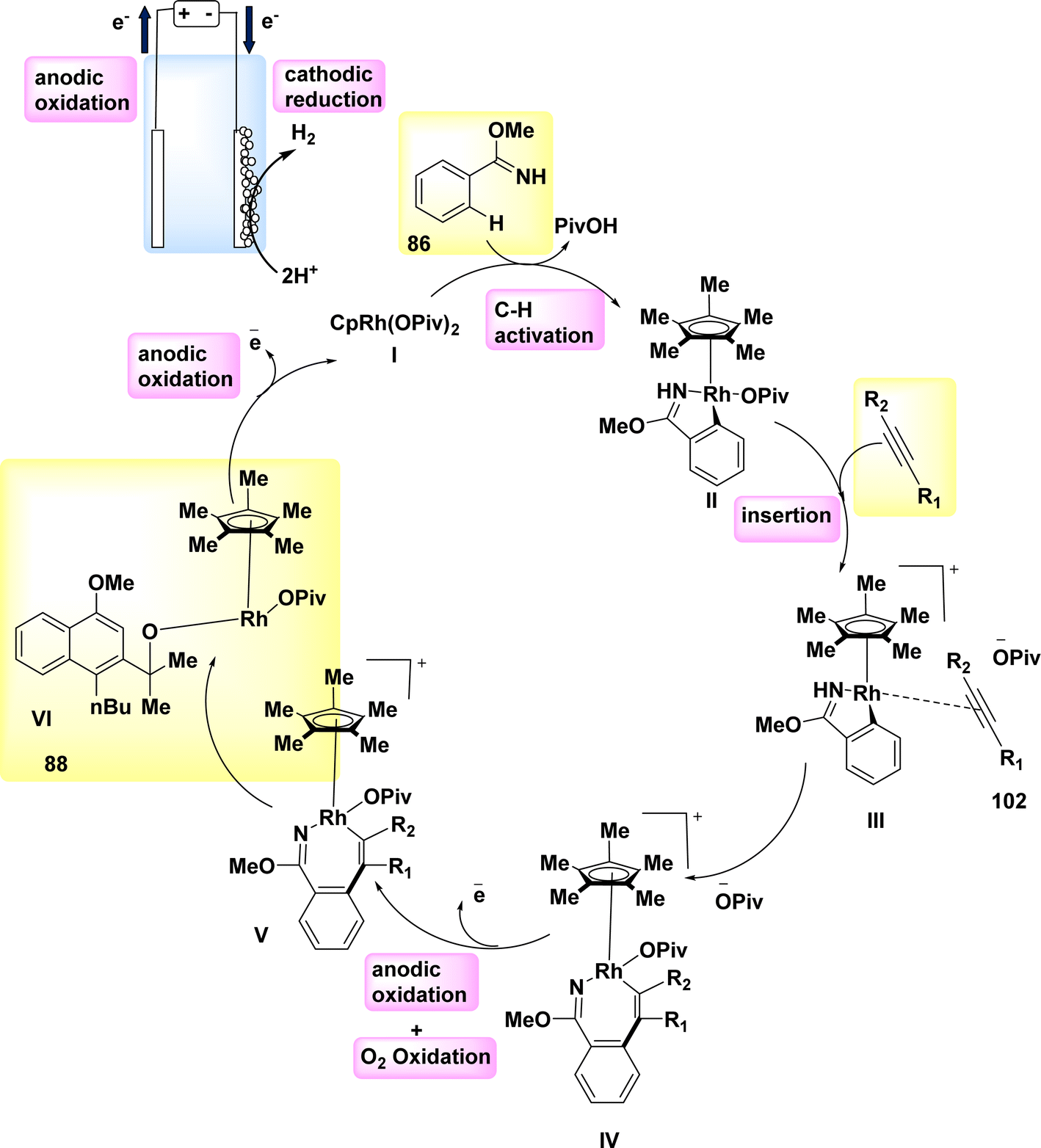
In the year 2019, a novel method for direct electrochemical synthesis was developed, which involved the breakage of carbon–carbon bonds without the need for a catalyst (Scheme 34). The most favorable outcomes were achieved with the direct electrolysis of substrate 89 under a consistent current of 8 mA, utilizing a blended electrolyte solution composed of MeCN/H2O and nBu4NBF4.
 | ||
| Scheme 34 Electrochemical synthesis of 9-membered lactam 90 with its proposed mechanism. | ||
The 9-membered lactam 90 was obtained with a yield of 98% without the use of any extra bases or catalysts.57
A mechanism that is considered feasible is suggested, drawing upon our mechanistic discoveries (Scheme 34). The NH bond in compound 89 undergoes anodic oxidation, resulting in the formation of intermediate I. The intermediate undergoes cyclization, resulting in the formation of radical II. This radical then undergoes selective cleavage of a C–C bond, leading to the production of radical III. Ultimately, the medium-sized lactam 90 can be synthesized through the oxidation of the ketyl radical III via the transfer of a single electron, followed by proton loss.
Nevertheless, the possibility of the cationic pathway cannot be disregarded due to the presence of two closely spaced oxidative waves at 89 in the CV.
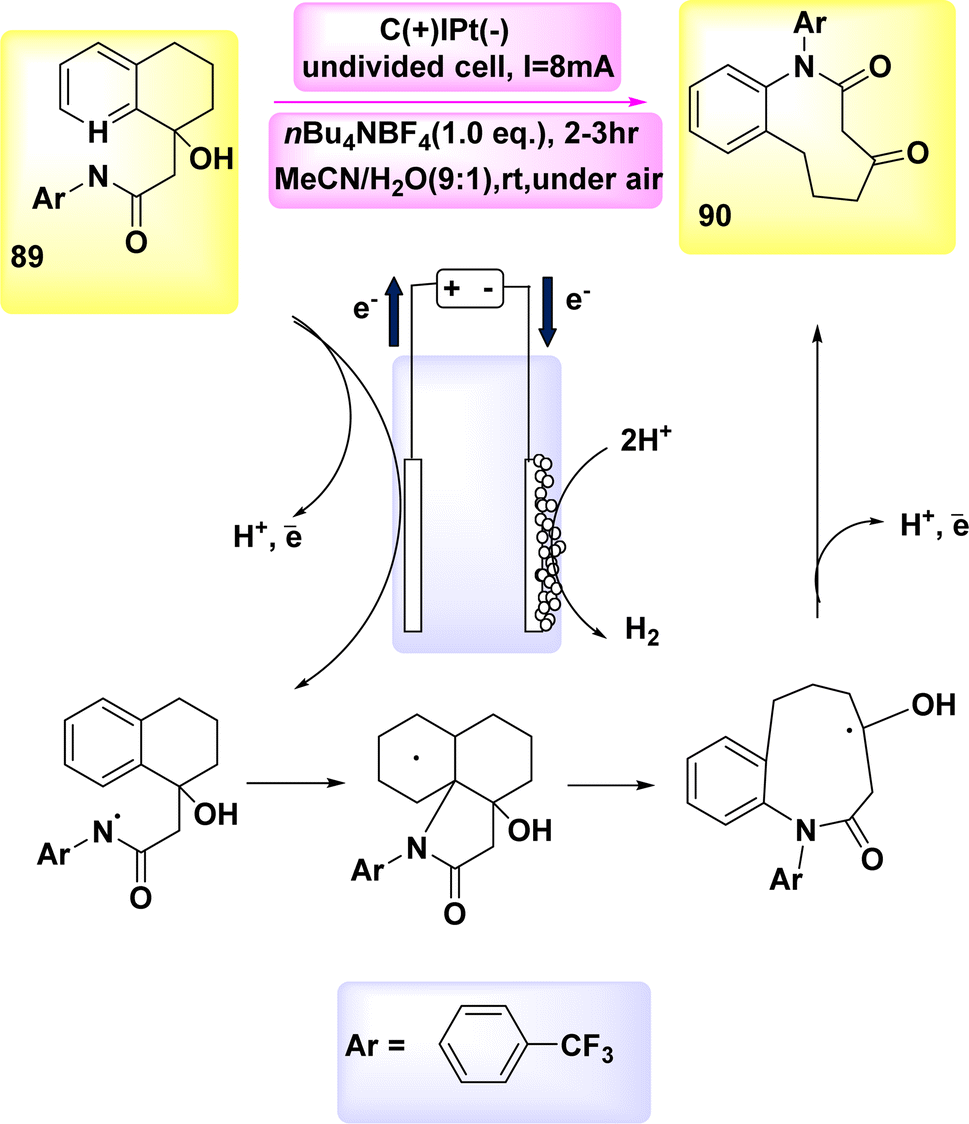
In the year 2020, the author and their colleagues presented a viable approach for the electrochemical synthesis of sulfonated 4H-3,1-benzoxazines. N-(2-(prop-1-en-2-yl)phenyl)benzamide was reacted with p-toluenesulfonylhydrazine to produce sulfonated 4H-3,1-benzoxazine in a yield of 78% (Scheme 35).
 | ||
| Scheme 35 Electrochemical synthesis of sulfonated 4H-3,1-benzoxazines 93 with its proposed mechanism. | ||
The most favorable outcome was achieved by performing the reactions in an undivided cell with a carbon rod as the anode and a Pt foil as the cathode. The cell was filled with anhydrous MeCN and nBu4NBF4 was used as the electrolyte. The reactions were carried out under galvanostatic conditions at ambient temperature.
The approach exhibits a larger range of substrate compatibility, accommodating diverse functional groups, and operates at ambient temperatures without the need for metal catalysts or external oxidizing agents.58
The electro-oxidation and deprotonation of CH3C6H4SO2NHNH2 92 led to the generation of the corresponding sulfonyl radical II, which subsequently resulted in the liberation of N2. The sulfonyl radical II was subsequently reacted with alkene 91 to generate the radical intermediate III. The radical intermediate III, might subsequently undergo quick oxidation at the anode, resulting in the generation of the corresponding carbon cation, referred to as IV. The sulfonated 4H-3,1-benzoxazine 93 was synthesized through a series of reactions involving nucleophilic attack and subsequent deprotonation. The protons underwent synchronous transformation into H2 through cathodic reduction, Scheme 35.
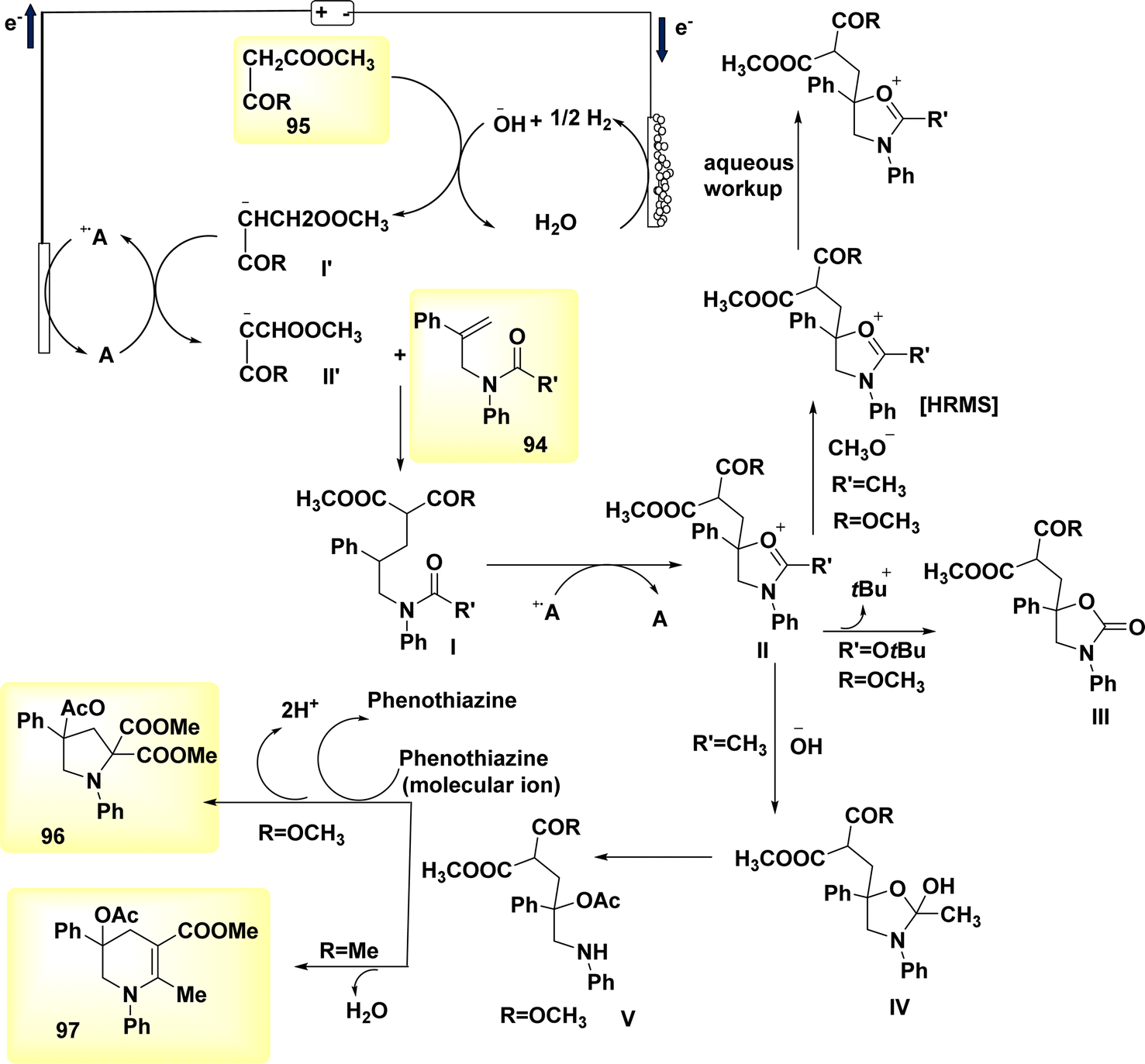
In a study conducted by He et al. in 2018 and 2020, the authors documented the synthesis of pyrrolidines 96 and tetrahydropyridines 97 derivatives using a dehydrogenative annulations technique. This method utilized readily available chemicals, Scheme 36. In this methodology, the utilization of a catalytic quantity of a redox catalyst based on phenothiazine facilitates efficient and selective intermolecular radical reactions of C8H12O3 compounds. The utilization of this approach is currently under consideration as a means to promote diverse oxidative radical processes of 1,3-dicarbonyl molecules.59,60 In this study, the electrolysis of N-allyl amide 94 with dimethyl malonate 95 was conducted. The optimal reaction system was identified as HCO2Na (0.3 equivalents) serving as the base additive, along with phenothiazine (20 mole%) acting as the redox catalyst. This system was employed in a refluxed mixture of tBuOMe, MeCN, and H2O, with a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1. The pyrrolidine derivative 96 was synthesized, resulting in a yield of 70%, using the given conditions. A reverse voltage converter (RVC) anode and a Pt (Pt) cathode were employed.
:3:1. The pyrrolidine derivative 96 was synthesized, resulting in a yield of 70%, using the given conditions. A reverse voltage converter (RVC) anode and a Pt (Pt) cathode were employed.
 | ||
| Scheme 36 Electrochemical synthesis of pyrrolidine 96 and tetrahydropyridine 97 with its proposed mechanism. | ||
The utilization of a β-ketoester as the coupling partner in the reaction led to the production of a tetrahydropyridine product 97 by a (4 + 2) annulation process, without the generation of a pyrrolidine derivative 96. The reaction involving the annulation of methyl acetoacetate (a β-ketoester) with an enyne exhibited a unique outcome, resulting in a 13% yield of pyrrolidine and a 53% yield of C5H9N product. It is noteworthy to mention that these annulations were not dependent on the use of sodium formate and were advantageous in terms of reduced current densities.
The study's findings have presented a beneficial mechanistic approach in Scheme 36. The initial stage of the process involves the anodic oxidation of A, resulting in the formation of the radical cation A˙+. Simultaneously, at the cathode, a process takes place where water is reduced to OH− ion and H2 gas. The H2 gas then deprotonates the 1,3-dicarbonyl molecule 95, resulting in the formation of the anion I′, which possesses a higher oxidizability. The process of single-electron transfer results in the development of a radical II′. This radical then combines with the alkenyl group of the N-allyl amide 94, leading to the generation of a tertiary C-radical I. Intermediate I undergoes intramolecular oxidation and subsequent entrapment of its carbonyl group, rather than the Co of the 1,3-dicarbonyl moiety, resulting in the formation of compound II. The reaction involving the removal of the tertiary butyl group yields a cyclic carbamate III.
Amides can undergo a reaction with water or hydroxide ion, resulting in the formation of a secondary amine. This secondary amine can then participate in a C(sp3)–H/N–H cross coupling reaction, leading to the formation of the depicted product 96 (where R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH3). Alternatively, in the presence of intramolecular dehydration, the secondary amine can generate tetrahydropyridine 97 (where RCH3).
OCH3). Alternatively, in the presence of intramolecular dehydration, the secondary amine can generate tetrahydropyridine 97 (where RCH3).
The 5-exo-dig cyclization of NH2s was employed in the year 2020 to successfully synthesize oxazol-2-ones 99 and imidazole-2-ones 99′, Scheme 37. The electrosynthesis process relies on the dual functionality of TEMPO as both an O2-atom donor and a redox facilitator in the formation of NH2I. The reactions are conducted in a straightforward experimental arrangement under gentle operating conditions.61
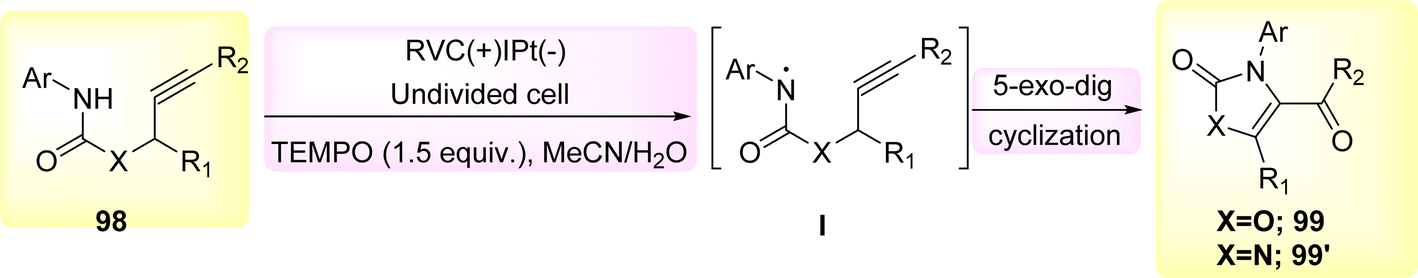 | ||
| Scheme 37 Electrosynthesis of oxazol-2-ones 99 and imidazole-2-ones 99′. | ||
The broad and useful procedure of electrochemically forming isoxazolidine-fused isoquinolin-1(2H)-ones 101 through the employment of NH2s has been documented in 2020 Scheme 37. In the optimization experiment, the compound C17H15NO2 100 was chosen as the outline molecule. The intended oxidation-induced intramolecular annulation product 101 was successfully synthesized with a yield of 93% under a constant current of less than 2 mA and using nBu4NBF4 as the electrolyte.62
Scheme 38 presents a viable technique. Initially, the process involves the cathodic reduction of ethanol, resulting in the formation of ethoxide ions. Subsequently, these ethoxide ions undergo deprotonation, leading to the generation of anion I. Subsequently, the anion I undergoes selective oxidation in order to produce radical II. The compound II participates in the 5-exo-dig annulation reaction, resulting in the formation of radical III. Subsequently, a second annulation reaction occurs, leading to the generation of delocalized radical IV. Ultimately, the process of electron-oxidation leads to the rearomatization of IV, resulting in the formation of compound 101.
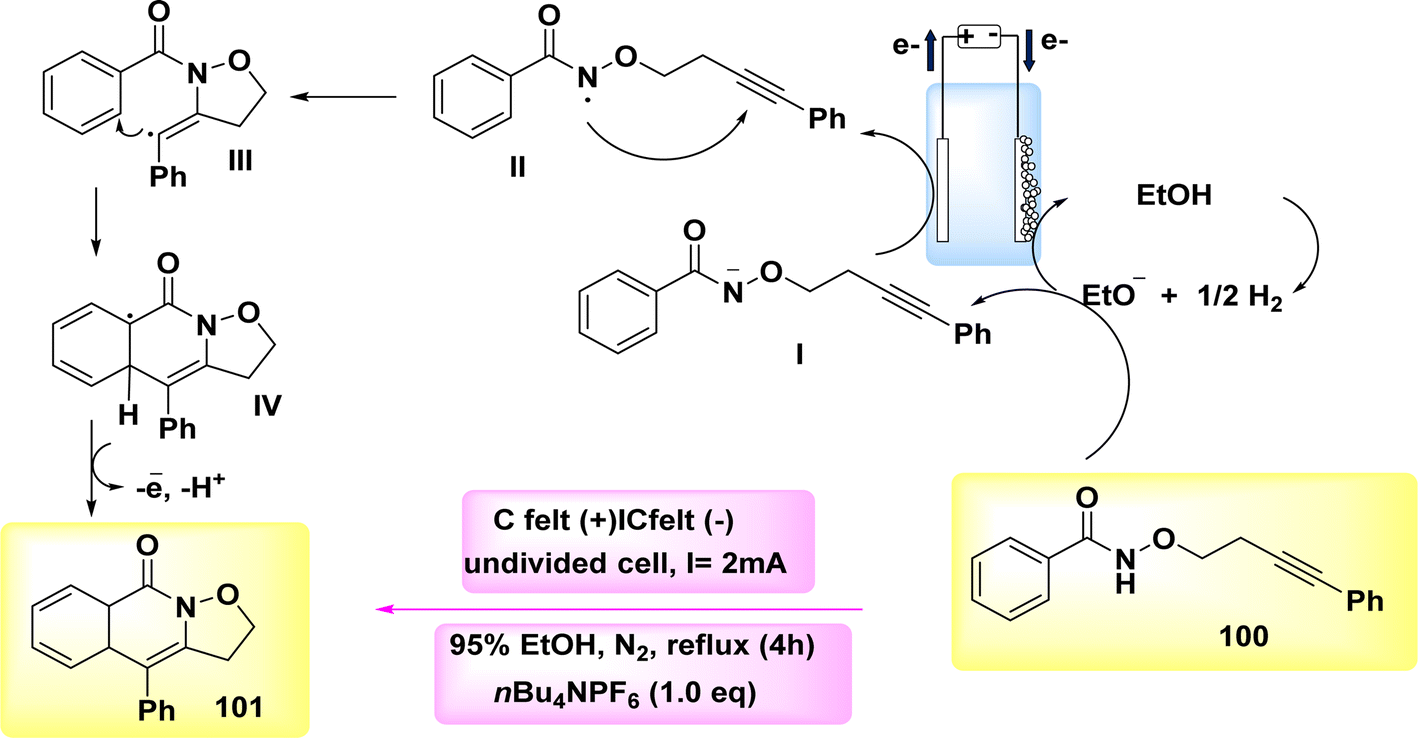 | ||
| Scheme 38 Electrochemical synthesis of isoxazolidine-fused isoquinolin-1(2H)-ones 101 with its proposed mechanism. | ||
The year 2020 witnessed the presentation of a very adaptable and potent method for the electrochemical production of Zincke intermediate I, which carries a double positive charge. The compound known as phenoxy-acetate 102 exhibited notable reactivity in the context of 2-fold amination reactions. The pyridinium intermediates I release amines that induce the development of an intramolecular heterocycle 103. A two-step amination process, which demonstrated a yield of 90%, was discovered, Scheme 38. This process is characterized by its lack of reliance on metal catalysts or leaving groups, rendering it a unique and robust approach. The best settings for electrolysis involve a divided cell arrangement with an isostatic graphite-(+) and Pt-cathode, a Thomapor-separator, and a two-step electrolysis process. The first step involves a current density of 5 mA cm−2 and a total charge of 3.4 F, while the second step involves a current density of 10 mA cm−2 and a total charge of 2.1 F. These electrolysis conditions are conducted at a temperature of 25 °C.63
When pyridine is present, the process of anodic oxidation of compound 102 typically leads to the formation of pyridinium intermediates I, which are positively charged and exhibit a relatively high level of stability. In the subsequent stage, species I undergo a treatment with pyridine, followed by the extraction of an aromatic primary amine group. The ester functionality of phenoxy acetate derivative 102 has the potential to stabilize compound I through various interactions, such as π–π, π-non-bonding, or nonbonding cationic connections. Enhanced oxidation reactions can potentially be facilitated by the stability of the e−-deficient substituent. The aforementioned contacts serve the purpose of concealing the positive charge, mitigating shielding effects, diminishing the influence of electron withdrawal, and facilitating increased oxidation of I, as illustrated in Scheme 39.
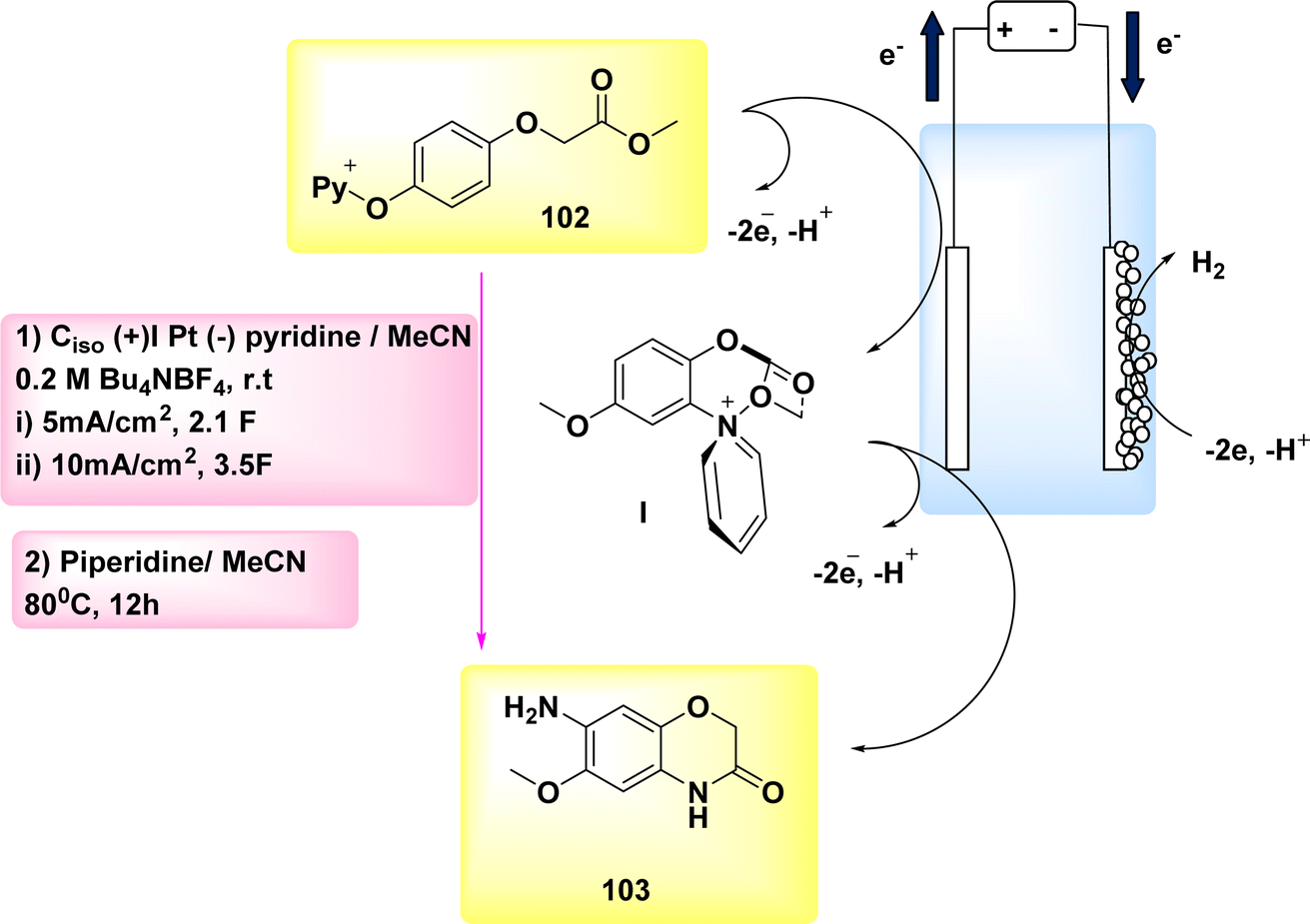 | ||
| Scheme 39 Electrochemical synthesis of 2H-benzo[b][1,4]oxazine-3(4H)-one 103 with its proposed mechanism. | ||
The catalysis of C–H functionalization by Co has occurred as a groundbreaking approach for chemical synthesis in the year 2020. In this study, the synthesis of biologically significant isoquinolones 107 and pyridines 108 was achieved by the utilization of aryl and alkenyl amides 104 or 105, which were derived from pyridine N-oxide. This synthesis method involved C–H or N–H activation, and the use of Co-electrocatalysis. The results of this study demonstrated the potential of Co-electrocatalysis in the synthesis of these important compounds, Scheme 40. The transition was observed at ambient temperature under highly mild reaction conditions and resulted in high yields in a solvent mixture containing H2O. A cellular system consisting of a single cell is utilized, wherein the cathode is composed of Ptand the anode is composed of RVC. This system is employed to facilitate the operation for duration of 16 hours at a temperature of 23 °C. Consequently, a novel approach has been developed to establish a comprehensive resource economy. This approach utilizes renewable green power as a redox agent, which operates in redox mediator-free conditions and only generates valuable H2.64–66
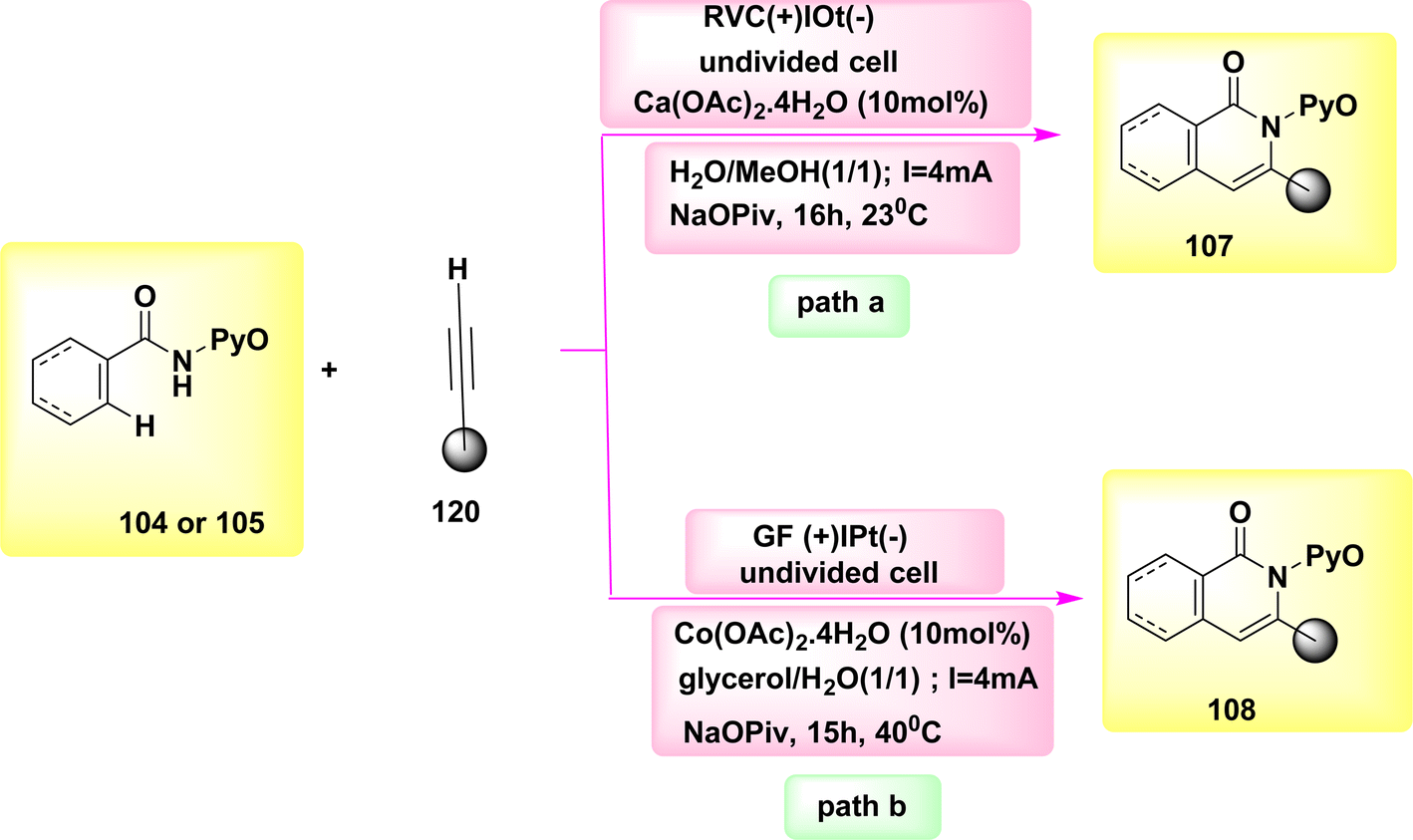 | ||
| Scheme 40 Electrochemical synthesis of isoquinolones 107 and pyridines 108 with its proposed mechanism. | ||
The experimental conditions for the reaction (Scheme 40-path a) were kept constant, except for the substitution of an RVC anode with a GF anode. This modification resulted in product yields ranging from 70%–76%.67
The utilization of glycerol derived from biomass as a medium for electrochemically facilitated CH activation pathways has not been previously recorded in academic literature. The utilization of renewable energies for the formation of C–C or NC bonds, without the production of molecular H2 as a byproduct, has contributed to the advancement of resource efficiency. The utilization of a Co catalyst devoid of Cp* has facilitated the achievement of sustainable C–H activation of amides, eliminating the need for the use of detrimental metal oxidants. The carbon–hydrogen/nitrogen–hydrogen functionalization process was successfully carried out in an aqueous glycerol solution at a temperature of 40 °C. Significantly, previous studies have demonstrated the feasibility of conducting electrocatalytic C–H activations through the utilization of sustainable solar and wind energy sources. The integration of renewable solvents and alternative energy sources in molecular catalysis, specifically through Scheme 40-path b, holds promise for advancing the evoluation of more sustainable future energy economies.68
In 2020, the feasibility of electro-oxidative Co catalysis was achieved (Scheme 41). The electrochemical activation of carbon-hydrogen bonds in compound 52 using allene 50 was conducted in an undivided cell configuration, resulting in significant levels of chemoselectivity and regioselectivity. The intended regioselective carbon–hydrogen annulation product 109 was obtained in a yield of 91% when either C2H3F3O or CH3OH was employed as the solvent. The yield of the synthesis was significantly enhanced when conducted at a temperature of 40 °C. Upon analysis of substitute additives, it was shown that sodium acetate NaOAc had a slightly superior performance compared to NaOPiv and PivOH.69
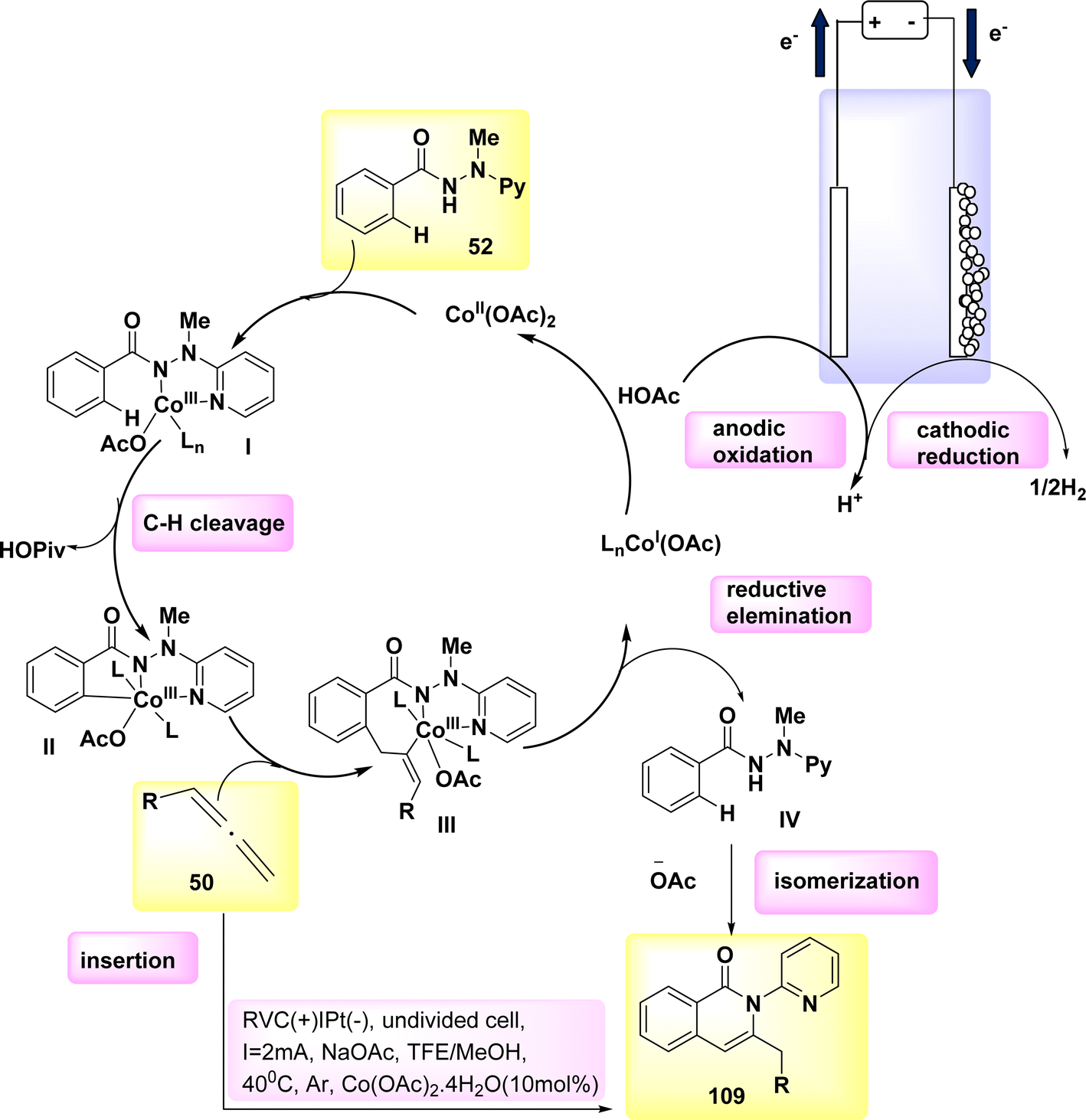 | ||
| Scheme 41 Electrochemical synthesis of carbon–H2 annulation product 109 with its proposed mechanism. | ||
A plausible catalytic cycle was suggested and is depicted in Scheme 41, building upon the previously discussed mechanistic findings. The electrooxidative CH activation was catalyzed by anodic oxidation, subsequently leading to a carboxylated-assisted BIES CH cobaltation process, resulting in the formation of Co(III) complex II. The Co(I) complex and exomethylene isoquinolone IV were synthesized using a subsequent reductive elimination process and regioselective allene insertion. This reaction pathway allowed for the isomerization of the resulting compounds, ultimately yielding the desired product 109. The process of anode oxidation was employed to reconstruct the Co(III) complex I, which is essential for catalytic activity. This step completes the overall catalytic cycle. In general, the cobalt-electrocatalysis approach effectively circumvented the utilization of chemical oxidants, resulting in the production of just molecular H2 as waste.
In 2020, observed the discovery that, in the absence of metallic oxidizing agents, imidazoles 110 and alkynes 84 can go through a Ru(Ru)-catalyzed electrosynthesis process to produce a range of N-fused[5,6]-bicyclic heteroarenes 111 through electrochemical carbon–hydrogen/nitrogen–hydrogen annulation that is selective and sensitive. To get the intended result, an indivisible cell configuration with a Pt-(−) and GF-(+) is employed 111. But when compared to other solvents, DMF yielded the greatest results.70
Based on the mechanistic findings (Scheme 42), it is proposed that the catalytic cycle commences with a rapid organometallic CH activation. Consequently, the production of II occurs. The C12H16N2Ru2 IV is subsequently synthesized using alkyne coordination and migratory insertion processes, followed by anodization to yield the Ru(III) complex V. The formation of VI occurs through a process known as ring opening of V. The formation of Ru(I) complex VII occurs through a process of oxidation-induced reductive elimination, followed by subsequent anodic reoxidation.
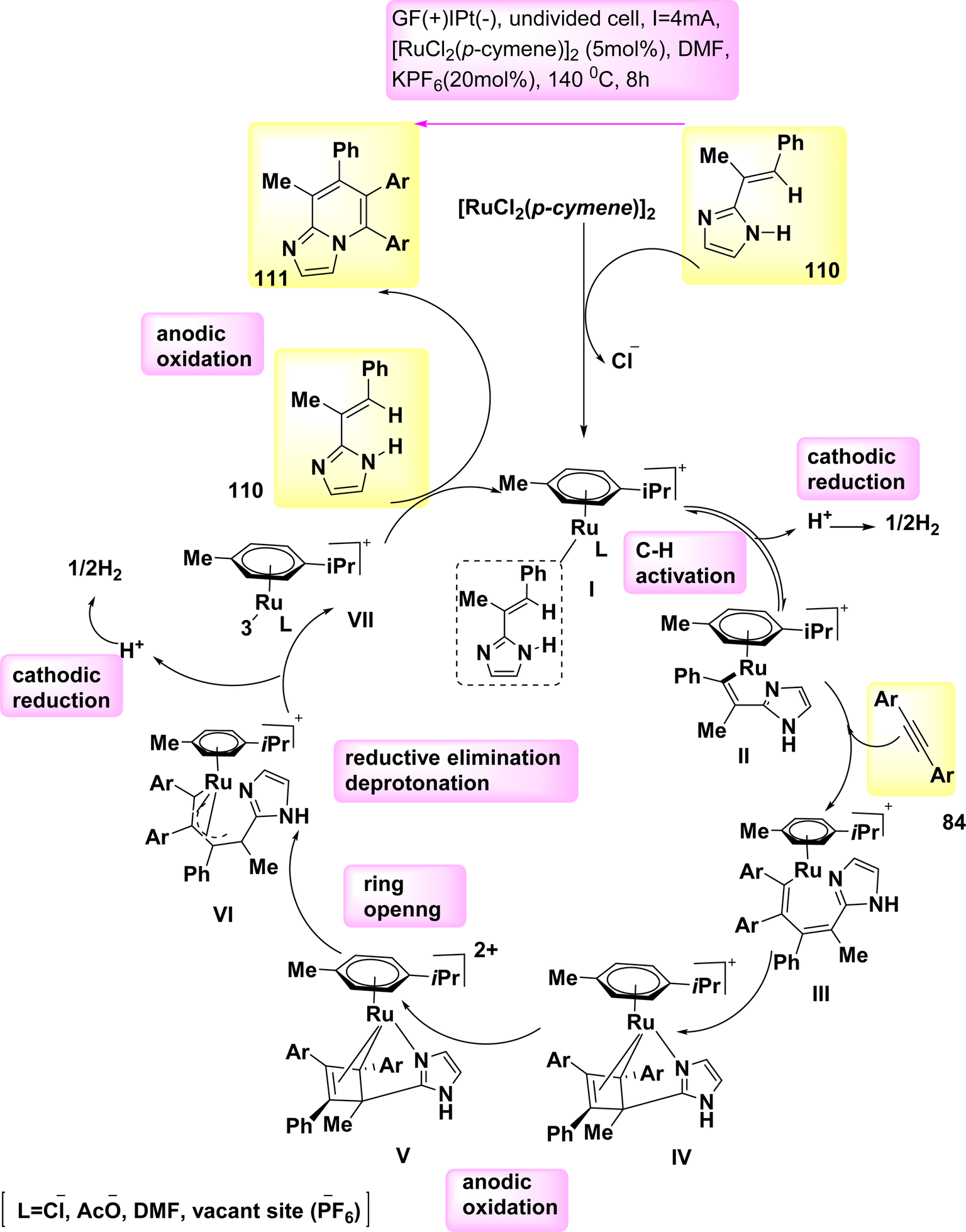 | ||
| Scheme 42 Electrochemical synthesis of bridge head N-fused[5,6]-bicyclic heteroarenes 111 with its proposed mechanism. | ||
In the year 2021, a transformation involving the conversion of a linear molecule known as BrHN into a cyclic structure, 112, Scheme 43, was successfully achieved. The β-lactams, also referred to as azetidin-2-ones, are a class of heterocyclic compounds characterized by a four-membered ring structure and the presence of an amide. Elaborating on the significance of this category of compounds in the realm of antibiotics is deemed unnecessary due to their widespread recognition within the biomedical sciences.71
 | ||
| Scheme 43 Synthesis of azetidin-2-ones 112. | ||
A facile electrochemical synthesis of β-lactams has been successfully accomplished using constant current electrolysis in a suitable solvent with a volatile organic compound (VOC)-supporting electrolyte solution containing C4H12+ salt. This method involves the addition of bromoamides 113 and results in the production of an electrogenerated base. By employing this approach, the necessity for bases and probases is circumvented, leading to the attainment of high yields of β-lactams 112 (Scheme 44). Typically, 113, which possess a leaving group in the β-position, can undergo deprotonation at the N atom under normal circumstances. This deprotonation generates a (−)ion that facilitates internal nucleophilic displacement, ultimately yielding the corresponding β-lactams.
 | ||
| Scheme 44 Electrochemical synthesis of azetidin-2-ones 112. | ||
The cyclization of a linear bromoamide 113 can be achieved by deprotonating a carbon atom. In this particular situation, the attainment of suitable acidity necessitates the utilization of ethoxy-carbonyl, which functions as an electron-withdrawing group. The generation of N-heterocyclic carbene (Scheme 45) occurs by the reduction of BMIm+ at the negative electrode. The NHC can function as both a nucleophile and a base. In this context, it acts as a deprotonating agent for the bromoamide 113, leading to the evoluation of a ring structure. Carbenes have demonstrated efficacy and environmental compatibility as valuable instruments for conducting chemical synthesis. The electrochemical technique described in this study offers a simplified process as it does not necessitate the use of any probase other than the IL solvent. The initial IL cation is generated by the NHC when it acts as a base in practice. The yields of β-lactams 112 are also satisfactory when the internal displacement occurs at a CBr site that is disubstituted, regardless of the nitrogen terminus.
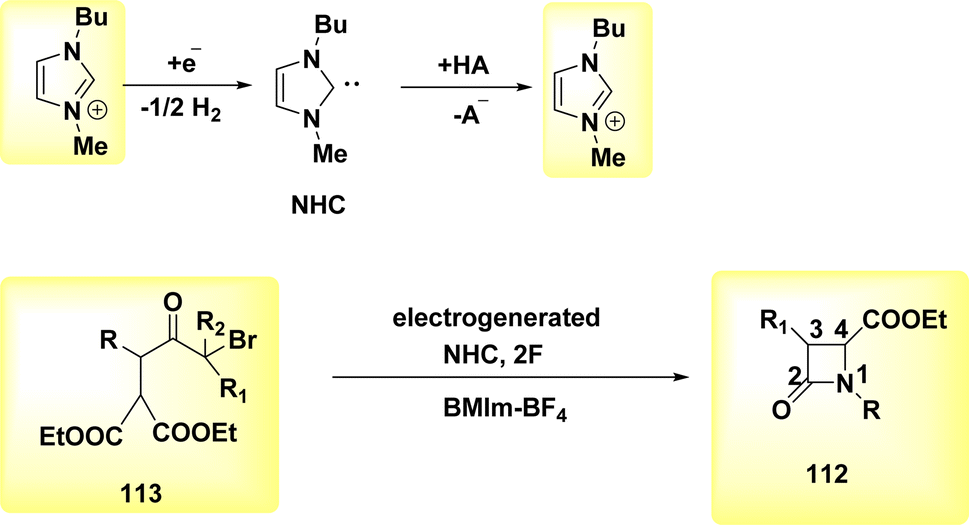 | ||
| Scheme 45 Electrochemical synthesis of azetidin-2-ones 112. | ||
[2 + 2], the most widely recognized method for synthesizing β-lactams 112 is through the cycloaddition reaction, specifically the Staudinger reaction, involving imine 114 and ketene, Scheme 46. The ketene, known for its inherent instability, is commonly generated through the in situ dehydrohalogenation of a suitable acyl halide 115.
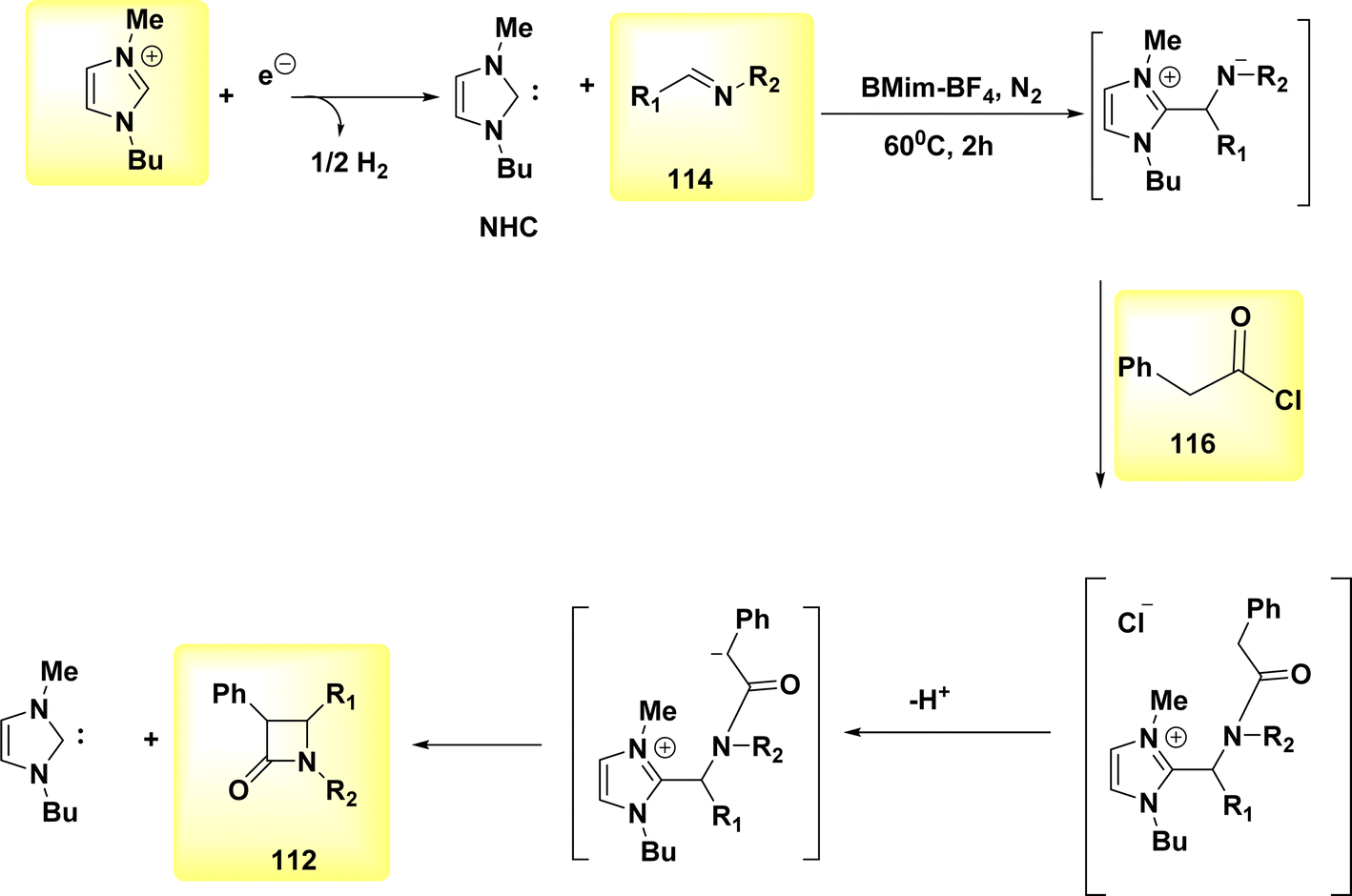 | ||
| Scheme 46 Electrochemical synthesis of azetidin-2-ones 112 with proposed mechanism. | ||
The stereochemical outcome of the occurrence is not neglected due to its unexpected character. Indeed, it is possible to achieve either a concerted or two-step process in the final β-lactam 112, depending on the varying relative configurations. During the electrosynthesis process of the ionic liquid BMIm-BF4, the formation of cis-lactams was shown to occur predominantly, with the preference being determined by the configuration of the nitrogen atom. However, due to the varying amount of NHC, the inclusion of Et3N (an external base) was required to achieve good yields. The imine 114 (Scheme 46) was activated by the electrogenerated NHC, which functioned as botha nucleophile and a base in the reaction. This finding eliminates the possibility of a coordinated mechanism.72
The final electrochemical step employed in the production of β-lactams 112 entails the induction of ring contraction by anodic means. The electrochemical approach anticipated the potential to generate the active “I+” in its original location through anodic oxidation of I− from the supporting electrolyte, while inadvertently forming an electrogenerated base at the cathode. The reaction was conducted in acetonitrile at a temperature of 80 °C in the presence of chiral pyrrolidones 117, resulting in a high yield (Scheme 47).
 | ||
| Scheme 47 Electrochemical synthesis of azetidin-2-ones 112 with proposed mechanism. | ||
The electroactive species in question is the I-anion, which undergoes oxidation to become “I+” and subsequently reacts with the open form of iodide to generate the corresponding α-I−. The compound known as β-lactam 112 is synthesized through the process of intramolecular iodide translocation and subsequent base deprotonation. It is highly probable that this reaction occurs with equal amounts of the R- and S-forms at the C3 position.
The process of equilibration in alkaline solutions results in the formation of the S-diastereoisomer as the predominant product. This particular isomer can be obtained in a pure state by the process of crystallization, as it exhibits the highest level of thermodynamic stability.
In the year 2021, Zhou73 and colleagues presented a novel electro-oxidative method for the formation of C–N bonds. This strategy was shown to be suitable for large-scale synthesis, enabling the synthesis of several compounds (Scheme 48). The initial step was the coupling of the model substrate C16H13NO 118 with a substituted pyrazole 119. This reaction was carried out using an undivided cell, where Pt served as both the cathode and anode.
 | ||
| Scheme 48 Electrochemical synthesis of 2-heteroaryl-C16H13NO 120 with proposed mechanism. | ||
The best condition was eventually produced by employing a solvent mixture of dichloromethane/tetrafluoroethylene, nBu4NBF4 as the electrolyte, and a reaction mixture containing 0.5 mmol of compound 118 and 0.5 mmol of compound 119. The reaction was conducted at room temperature under atmospheric conditions. The product with the designation 120, which exhibited a yield of 69%, was subjected to separation.73
This study presents a feasible mechanistic approach for the electrochemical oxidative azolation of indoles 118, which is based on thorough mechanistic investigations and pertinent literature sources (Scheme 48). Due to its comparatively lower oxidative potential in comparison to pyrazole 119, indole 118 exhibits anodic oxidation as its initial reaction. This leads to the intermediate I formation, which subsequently engages in nucleophilic addition with 118, leading in the elimination of a proton and the formation of radical intermediate II. In order to obtain the desired isolated product 120, it is necessary to subject the radical intermediate to subsequent oxidation and tautomerization processes.
Electrosynthesis of S-heterocycles
The S-heterocycles, including derivatives of thiazole-2-imine 136, has exhibited a significant rise in their pharmacological utilization owing to their noteworthy therapeutic capabilities, such as analgesic properties, anti-inflammatory effects, kinase inhibition activities, antibacterial attributes,79 antifungal properties,74 and melanin reduction activity.75M. Islam et al. described the electrochemical synthesis of C3H6N2S 136 from thiourea-tethered terminal alkenes 135 in 2019.76 The electrolysis was carried out in a flow-reactor system, employing, C9H18NO˙ as a redox catalyst. The catalyst acts as a nucleophilic reagent according to the mechanism proposed (Scheme 49).
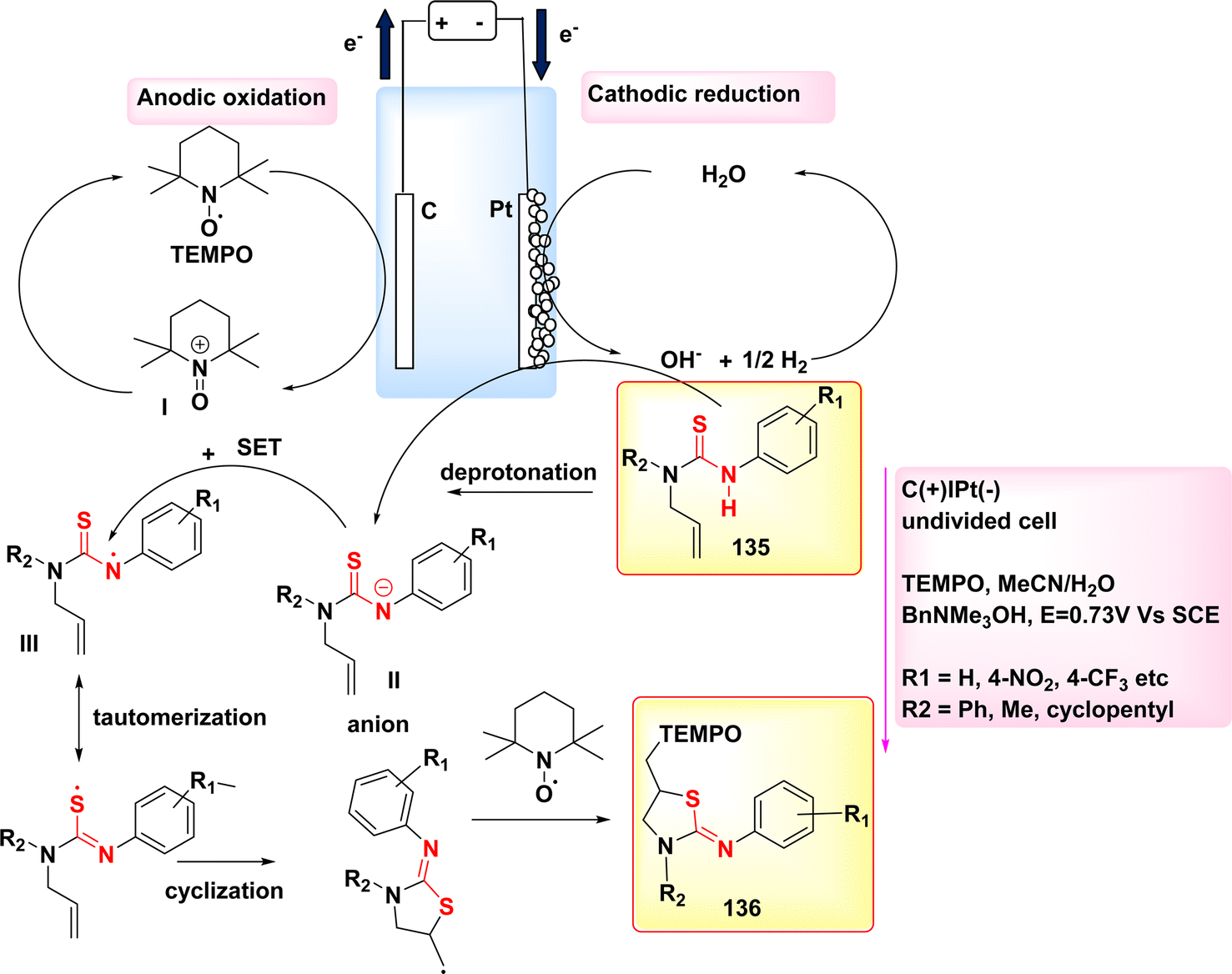 | ||
| Scheme 49 Electrochemical synthesis with the proposed mechanism of thiazolidin-2-imines 136. | ||
The process of cathodic reduction involves the conversion of water into hydroxide ions (OH−) and H2 gas (H2), while the subsequent anodic oxidation involves the conversion of tropospheric emission: monitoring of pollution into oxo-ammonium ion I. Phenylurea undergoes deprotonation through the action of the hydroxide ion, leading to the formation of an anion II centered on the nitrogen atom. A nitrogen-centered radical III, which is lacking in electrons, is formed through a subsequent SET reaction between an anion II and an intermediate I. This process serves to regenerate the TEMPO radical molecule. The compound known as thiazolidin-2-imine 136 exhibits a N-radical tautomerization process in the presence of the thiocarbonyl group, resulting in the formation of a sulfur radical. The aforementioned radical undergoes cyclization, resulting in the formation of another radical at the terminal carbon. This radical then interacts with the tropospheric emission: monitoring of pollution radical molecule, leading to the production of the difunctionalized oxysulfurization product 136.
Folgueiras-Amador et al. in 2018, devised a novel method to enable continuous flow of electrochemical dehydrogenative C–S bond formation, without the need of a catalyst or a supporting electrolyte. The conversion of a diverse array of N-arylthioamides 137 to their corresponding benzothiazoles 138 has been achieved with excellent yields and great current efficiency (Scheme 50). The experiment involved utilizing a Pt plate as the cathode and carbon as the anode. The reaction was conducted within an undivided cell under conditions of a consistent current. The authors have shown that this transformation is achieved exclusively by the utilization of a solvent of high laboratory quality and electricity, without the need of a degassing or a flux environment. It examines three advantages of employing electrochemistry in flow systems: (i) the ability to scale up the reaction without necessitating a larger reactor; (ii) the feasibility of conducting a reaction without the need for a supporting electrolyte; and (iii) the substantial influence of utilizing a well-designed reaction solution, which can be achieved through the implementation of flow systems.77
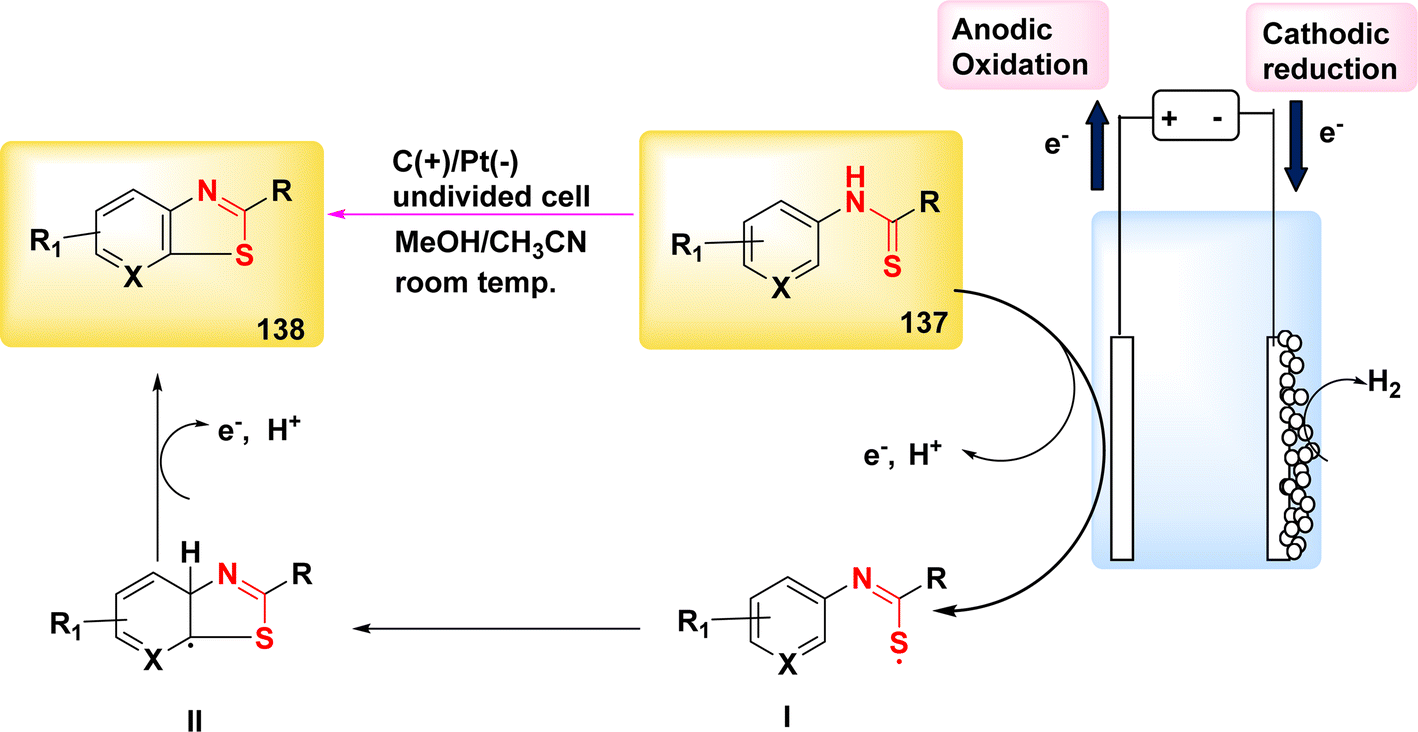 | ||
| Scheme 50 Electrochemical synthesis of benzothiazoles 138 with its proposed mechanism. | ||
It was proposed that the anodic oxidation served as the initial step in the sequence of reactions involving thioamide derivatives 137, leading to the formation of radical intermediates I and II. The final compound 138 is obtained through cyclization and subsequent deprotonation (Scheme 50).
C. Huang et al. in 2019 synthesized the six-membered heterocyclic ring in C8H6OS and C8H7NS through the utilization of intramolecular dehydrogenative C–S coupling.78 The avoidance of oxidative desulfurization, which is a common side reaction for thioamides 139, is achieved by carrying out the reactions within an acidic flow cell. The selection of the model substrate, thioamide 139, was made in order to determine the optimal reaction conditions. The electrolysis was conducted using a flow electrolytic cell equipped with a polyvinylidene fluoride (PVDF) anode loaded with carbon and a Pt-(cathode). Extensive experimentation revealed that the implementation of a continuous-flow electrolytic technique, utilizing a mixture of solvents including MeCN and TFA in a ratio of 9:1, in the existence of Sc(OTf)3, resulted in the successful extraction of C8H6OS 140 with a yield of 73%.
The generation of SNH2 cation II, illustrating the observed concordance between the neutral SNH2 III and the thioamide substrate I which is attributed to the outcomes of a one-electron anodic oxidation process. The ultimate outcome, denoted as V, is achieved through the process of radical cyclization, which is subsequently followed by oxidative rearomatization. The expectation is that cyclization via the protonated radical II will exhibit more efficacy compared to cyclization via the neutral species III. This is due to the higher electrophilicity of the S-radical in intermediates II, rendering it more reactive towards the phenyl ring than the S-radical in intermediates III. It is probable that TFA undergoes ligand exchange with Sc(OTf)3 inside the reaction mixture, resulting in the formation of the more potent acid, TfOH. This enhances the level of protonated form II. The radical III has the ability to undergo dimerization and hydrolysis reactions, resulting in the formation of desulfurization product 140 by a slow cyclization process (Scheme 51).
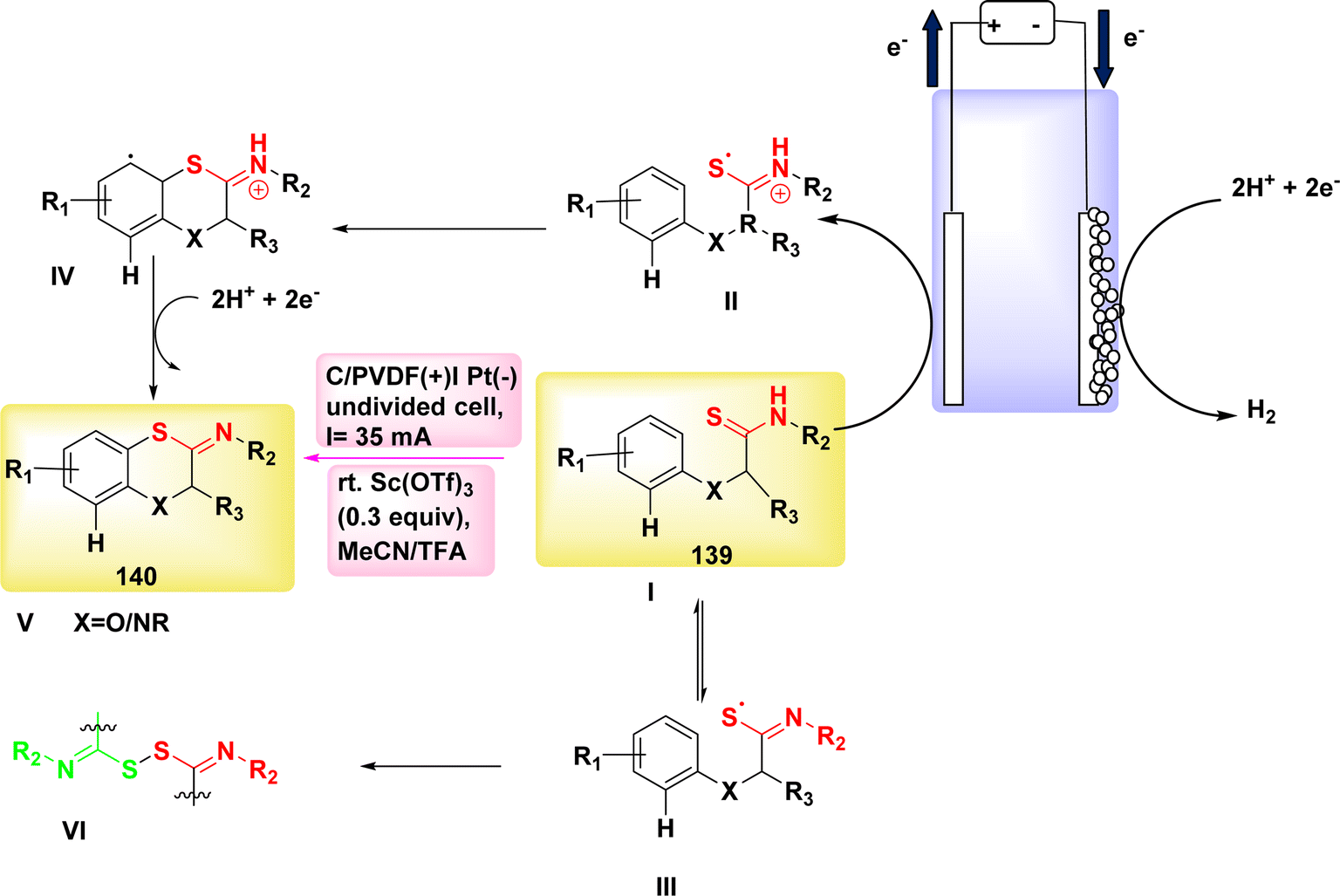 | ||
| Scheme 51 Electrochemical synthesis of 1,4-benzoxathiin 140 with its proposed mechanism. | ||
In a study conducted in 2019, a range of functionalized 1,3-benzothiazines 142 were synthesized using an acidic flow cell (Scheme 52). This method effectively prevents oxidative desulfurization, a common undesired side reaction observed in thioamide processes. The SNH2 cation is widely believed to be the pivotal intermediate. The electrolytic process was conducted within a flow cell, utilizing a Pt cathode and a carbon anode that was impregnated with –(C2H2F2)n−. An assessment of the Rxn conditions was conducted, encompassing several parameters such as current (42 mA), flow rate (0.3 mL min−1), additives, and solvent (TfOH, 0.06 M) in MeCN. It was determined that this particular set of conditions yielded the most favorable outcome.
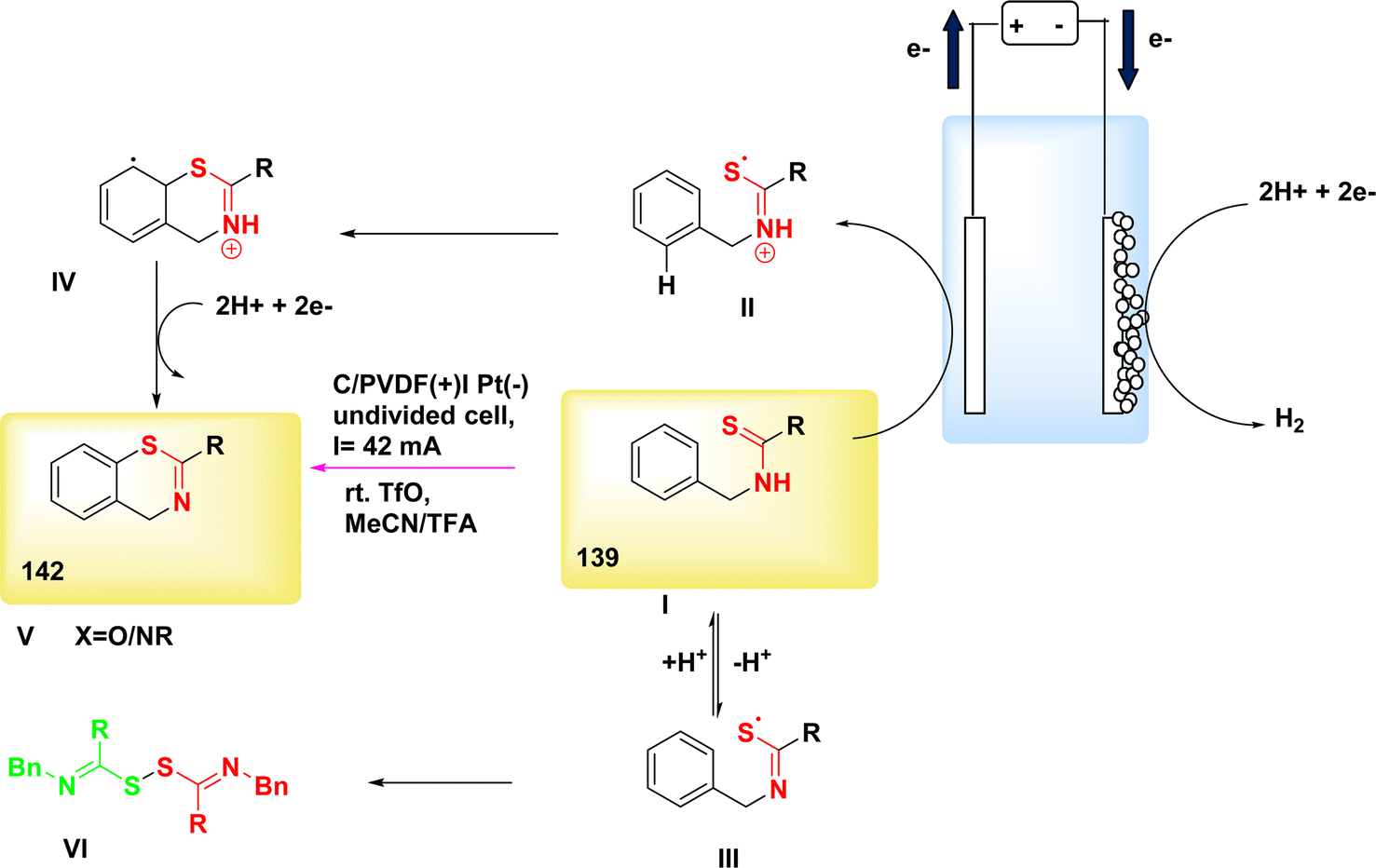 | ||
| Scheme 52 Electrochemical synthesis of 1,3-benzothiazines 142 with its proposed mechanism. | ||
The inclusion of trifluoromethanesulfonic acid (TfOH) resulted in an increase in the conductivity of the reaction solution, eliminating the need for any additional supporting salt. Under the given conditions, the reaction of 139 resulted in the formation of the intended product 142 with a satisfactory yield.80
At the anode, the thioamide I undergoes oxidation through SET to generate radical cation II. Subsequently, radical cation II undergoes cyclization and oxidative aromatization, resulting in the formation of the ultimate heterocycle V. The removal of a proton from the intermediate II results in the formation of III, which exhibits a lower susceptibility to cyclization compared to II. The process of dimerization of radical III can lead to the development of radical VI, which subsequently undergoes hydrolysis to yield desulfurized material. The introduction of trifluoromethanesulfonic acid (TfOH) leads to a more advantageous equilibrium on compound II, resulting in a decrease in the process of desulfurization.
According to a report in 2019, the introduction of sulfur element 144, to N-tosylhydrazones 143 leads to the formation of C2H2N2S through the utilization of an electrochemical method that does not require the presence of metals or oxidants (Scheme 53).
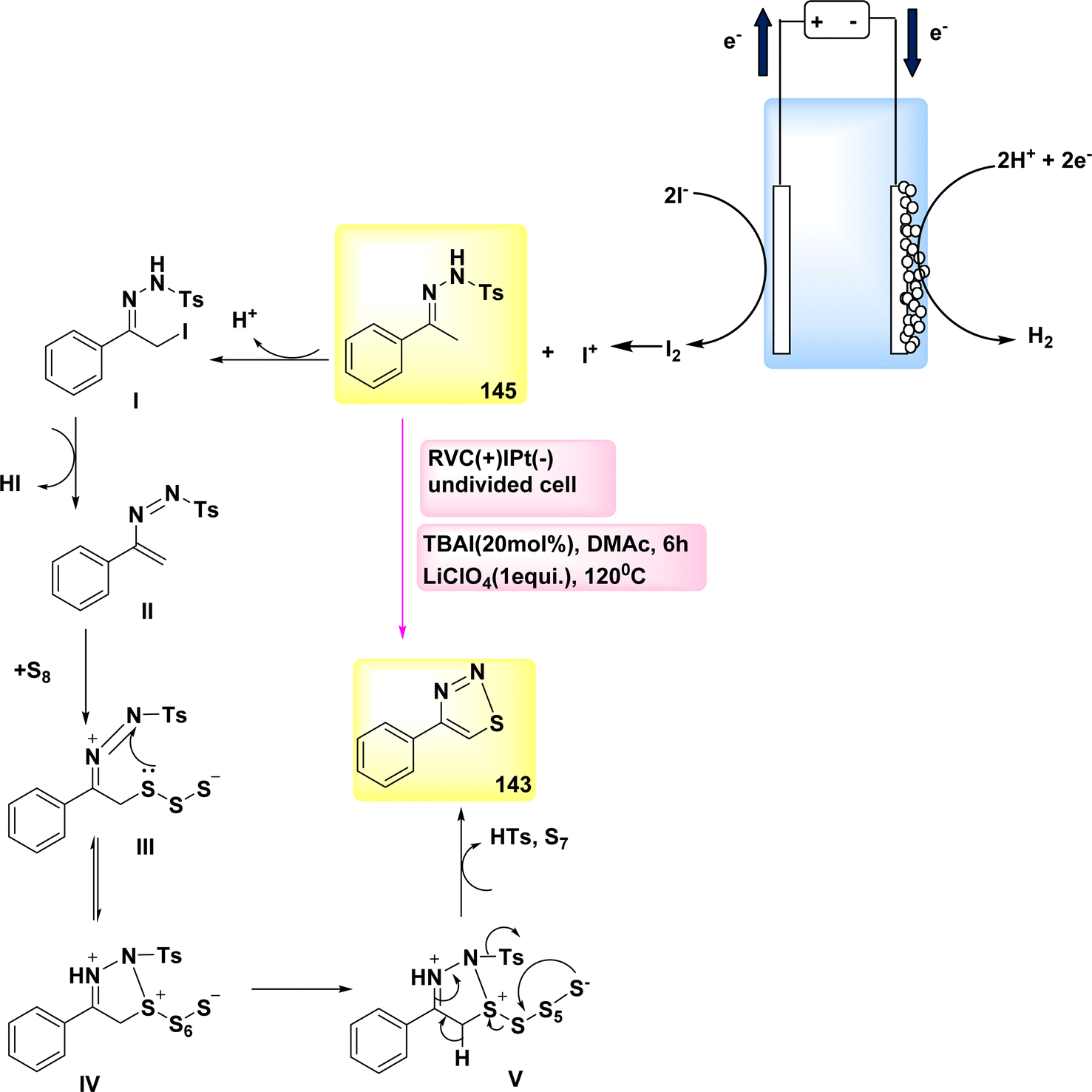 | ||
| Scheme 53 Electrochemical synthesis of 1,2,3-thiadiazoles 145 with its proposed mechanism. | ||
The coupling reaction involved the combination of tosylhydrazone 143, derived from acetophenone, with sulfur 144. The annulation product 145 was synthesized with an 80% yield by employing a catalyst of TBAI (tetrabutylNH2I) at a concentration of 20 mol%, along with LiClO4 as the electrolyte, in a DMAc (dimethylacetamide) solvent. The reaction was carried out at a temperature of 120 °C for duration of 6 hours.86
The process of electrochemical oxidation at the anode resulted in the production of I2 through the oxidation of 2I−. This I2 was then effectively transformed into I− and I+ ions. The conversion of acetophenone tosylhydrazone 143 into intermediate I was achieved through α-iodation, whilst the formation of azoalkene II occurred via HI elimination.
The completion of the reaction cycle can be achieved through the utilization of various methods, which involve the continued oxidation of the generated iodine anions. Zwitterion III was synthesized through the addition of sulfur (S8) to azoalkene II. The aforementioned compoundundergoes a process of cyclization, resulting in the development of intermediate V. The intended product 145 was ultimately synthesized through the elimination of S7 and TsH.
H2 production was achieved through reduction at the cathode, so completing the electrochemical cycle.
In the year 2021, a viable and ecologically sustainable electrochemical method was developed for the synthesis of C-3-sulfonated benzothiophenes 148. This method was conducted under conditions that did not require the use of oxidants or catalysts. The reaction involved the combination of 2-alkynylthio-anisoles 146 with sodium sulfinates 147. Sulfonated benzothiophenes with notable and useful functional groups have been successfully synthesized under conditions of consistent current, resulting in moderate to high yields. A single, unpartitioned cell was utilized for the reaction, with a graphite rod employed as the anode and Pt utilized as the cathode (Scheme 54).
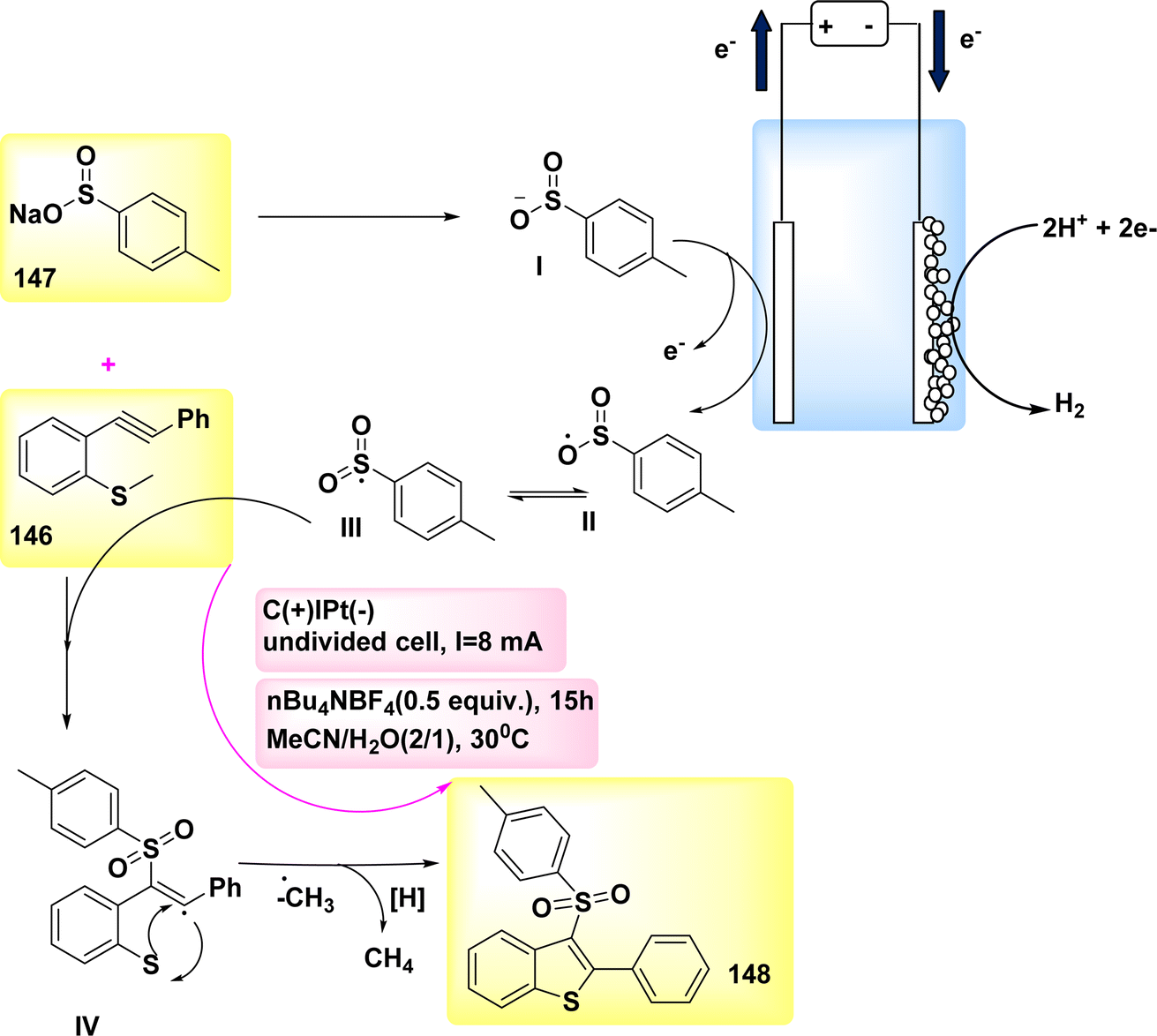 | ||
| Scheme 54 Electrochemical synthesis of C-3-sulfonated benzothiophenes 148 with its proposed mechanism. | ||
The electrolyte employed in this study was nBu4BF4 at a concentration of 2.5 mmol. The solvent system consisted of a mixture of CH3CN and H2O in a ratio of 2:1. To achieve a yield of 68% for product 148, the mixture was subjected to agitation at a temperature of 30 °C and electrolyzed for 15 hours using a current of 8 mA within an oil bath.81
Scheme 54 depicts a possible mechanistic route for the electrocatalytic sulfonylation process. The reaction phase was initiated by the oxidation of sodium p-tolylsulfinate 147 at the positive electrode, resulting in the formation of radical intermediate II or III through the loss of an electron. Resonance structures can be observed between the arylsulfonyl radicals III and II. The formation of the intermediate vinyl radical IV occurs through the intermolecular radical addition of sulfonyl radical III to the alkynyl molecule of 146. Subsequently, the methylthio-moiety initiates an assault on intermediate IV, resulting in the liberation of the desired product 148 and the emission of a methyl radical. The H2 present in the reaction mixture then undergoes a reaction with the methyl radical, resulting in the formation of methane.
Electrosynthesis of O-heterocycles
O-Heterocycles constitute a significant class of biologically active compounds. One possible explanation for this phenomenon could be the inherent prevalence and wide-ranging physiological roles of these entities, which are harnessed in the advancement of novel pharmaceuticals.82In 2018, Li and colleagues published a series of O-heterocycles (Scheme 56). The electro-synthesis described in this study involves the utilization of C8Cl2N2O2 as a redox-mediator and low-cost glassy carbon electrodes. This setup facilitates the intramolecular lactonization process of C13H10O2 derivatives 149, resulting in the production of a diverse range of 6H-benzo[c]chromen-6-ones 150 with high efficiency.83 The experiment was conducted at ambient temperature using C13H10O2 149 as a representative substrate in an undivided cell containing a MeCN/LiClO4 electrolyte solution. This resulted in the production of the desired chemical 150.
The process commences with the compound C8Cl2N2O2 facilitating a homogenous electron transfer from 149 to radical I. This subsequently enables the regeneration of DDQ through anodic oxidation. The addition of radical I to an arene ring results in the development of intermediate II. The aryl radical II underwent anodic oxidation once again, resulting in a yield of 150. The inclusion of 2,6-lutidine in the system will enhance the process of linked electron-transfer by promoting the deprotonation of both 149 and intermediate II.
In a similar vein, Shao et al.84 (2018) have shown the electrochemical oxidative coupling within the same framework (Scheme 55 c).
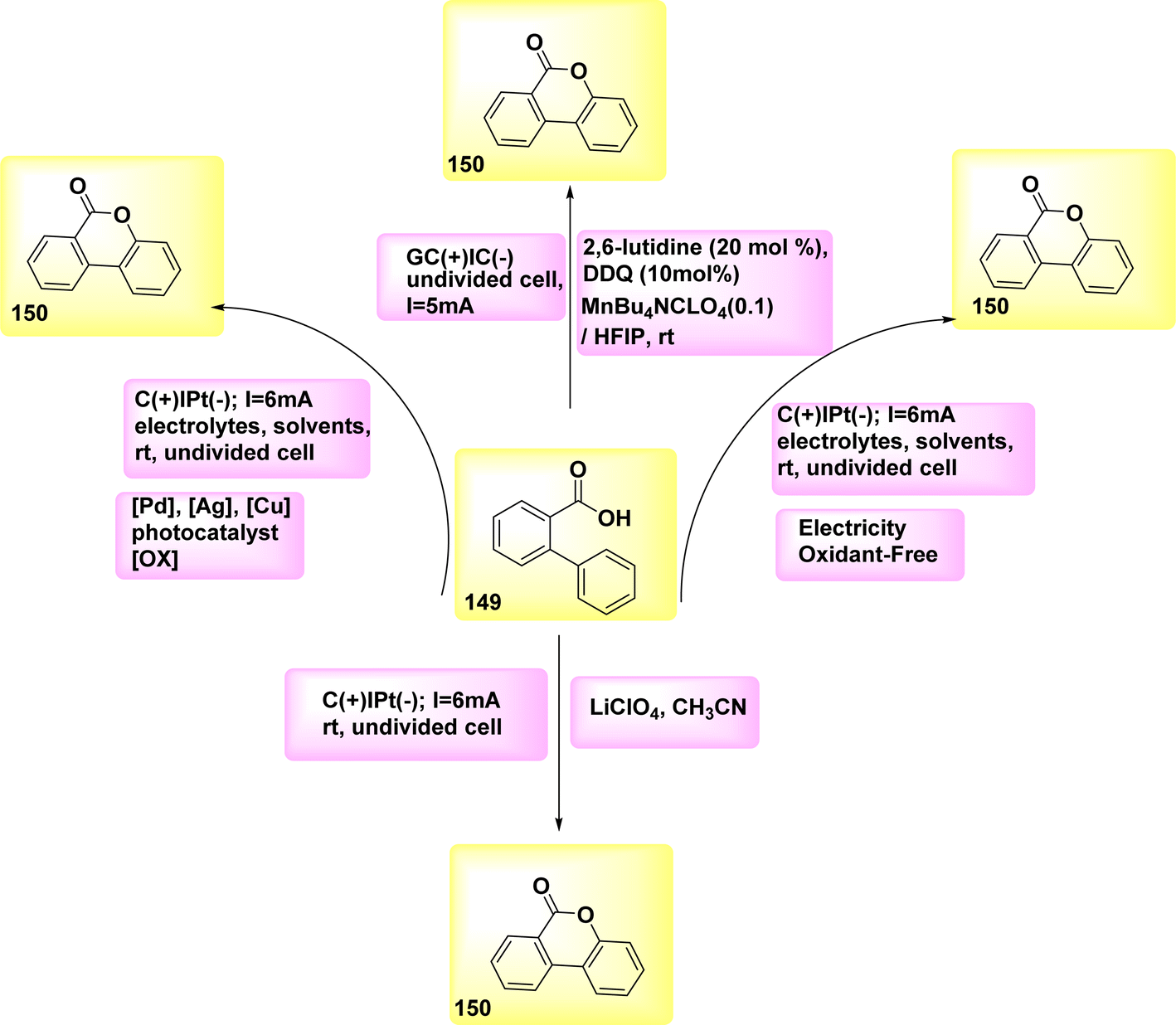 | ||
| Scheme 55 Electrosynthesis of 6H-benzo[c]chromen-6-ones 150. | ||
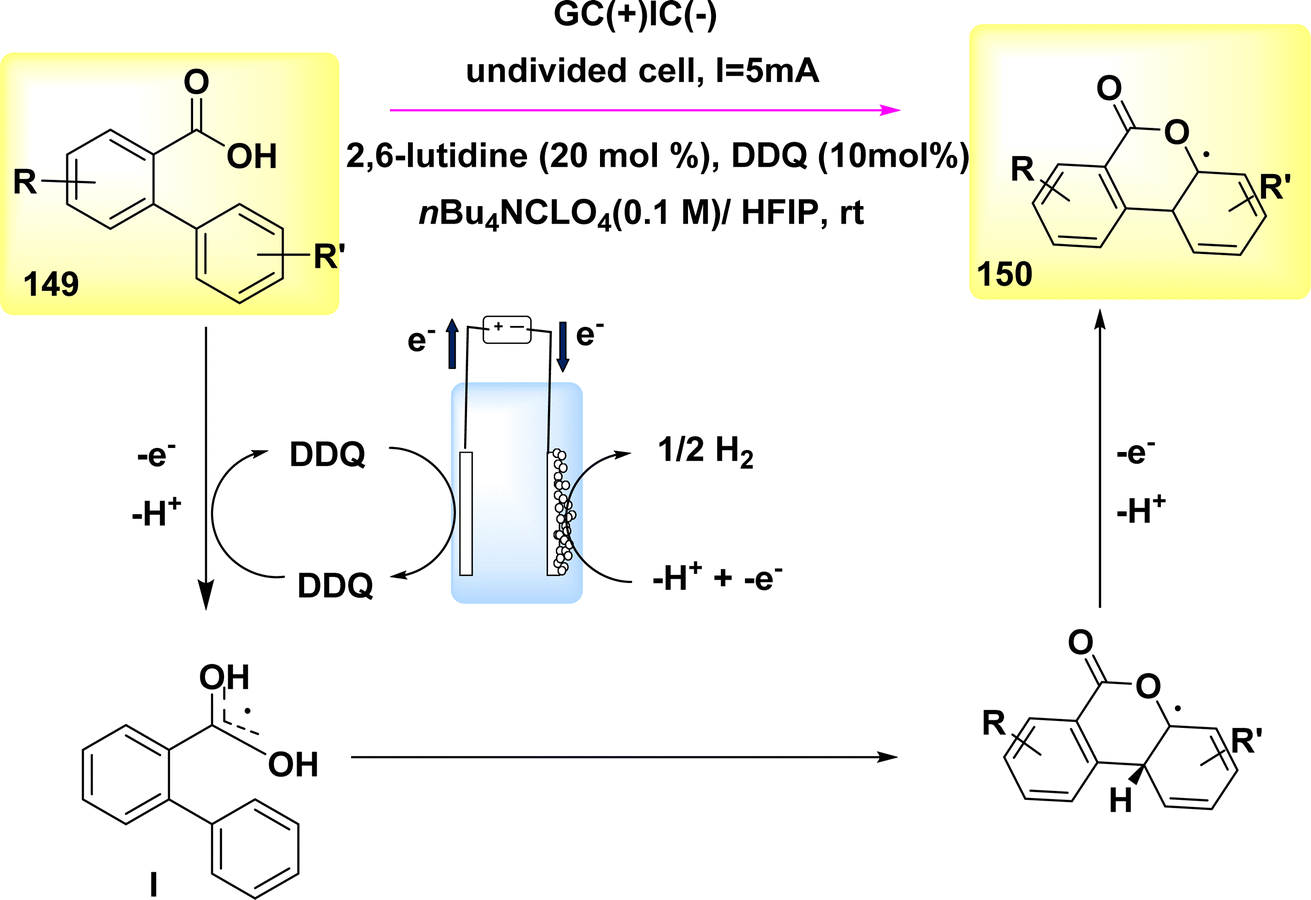 | ||
| Scheme 56 Electrosynthesis of 6H-benzo[c]chromen-6-ones 150 with its proposed mechanism. | ||
The electrochemical synthesis of lactones 150 has been achieved using 2-arylbenzoic acids 149. The C–H or O–H cross-coupling reaction of C13H10O2 acid 149 can be successfully conducted under mild conditions, both with and without the need for excess oxidants and metallic species. This can be performed by utilizing anodic oxidation and cathodic H2 development in a straightforward undivided cell.
Various C12H16O2 acids have been found to generate the corresponding lactones with yields ranging from 30% to 90% when utilizing an inexpensive and environmentally friendly Na2SO4 aqueous solution as a supporting electrolyte. The aforementioned reaction has a considerable degree of utility in natural product synthesis, demonstrating exceptional efficiency when performed on a gram scale.84
Path B exhibits selectivity towards substrates that possess a methoxy group, namely those with a 149′ configuration. The formation of the radical cation I occurs when an anode, which is a positively charged electrode, undergoes single electron transfer oxidation of 149′. The aromatic ring radical cation that is generated is subsequently subjected to assault by the carboxyl group, leading to the formation of intermediate II. The radical adducts II undergo rapid aromatization and deprotonation through anode oxidation, resulting in the formation of the desired product 150′. In contrast, in path A, the process involves the elimination of H2 from the R–COOH at the cathode or the exclusion of a proton through the electro-generated CH3COO–. This leads to the formation of compound III, which is subsequently oxidized to carboxyl radical IV through anode oxidation. The intermediate V, which undergoes cyclization, is rapidly converted into an aromatic compound through a single electron transfer process. Subsequently, the compound is deprotonated through anode oxidation, resulting in the formation of the desired product 150. This product is then subjected to intramolecular assault by a carboxyl radical. The simultaneous cathodic process in both processes A and B involved the reduction of H+ ions to produce H2 gas (Scheme 57).
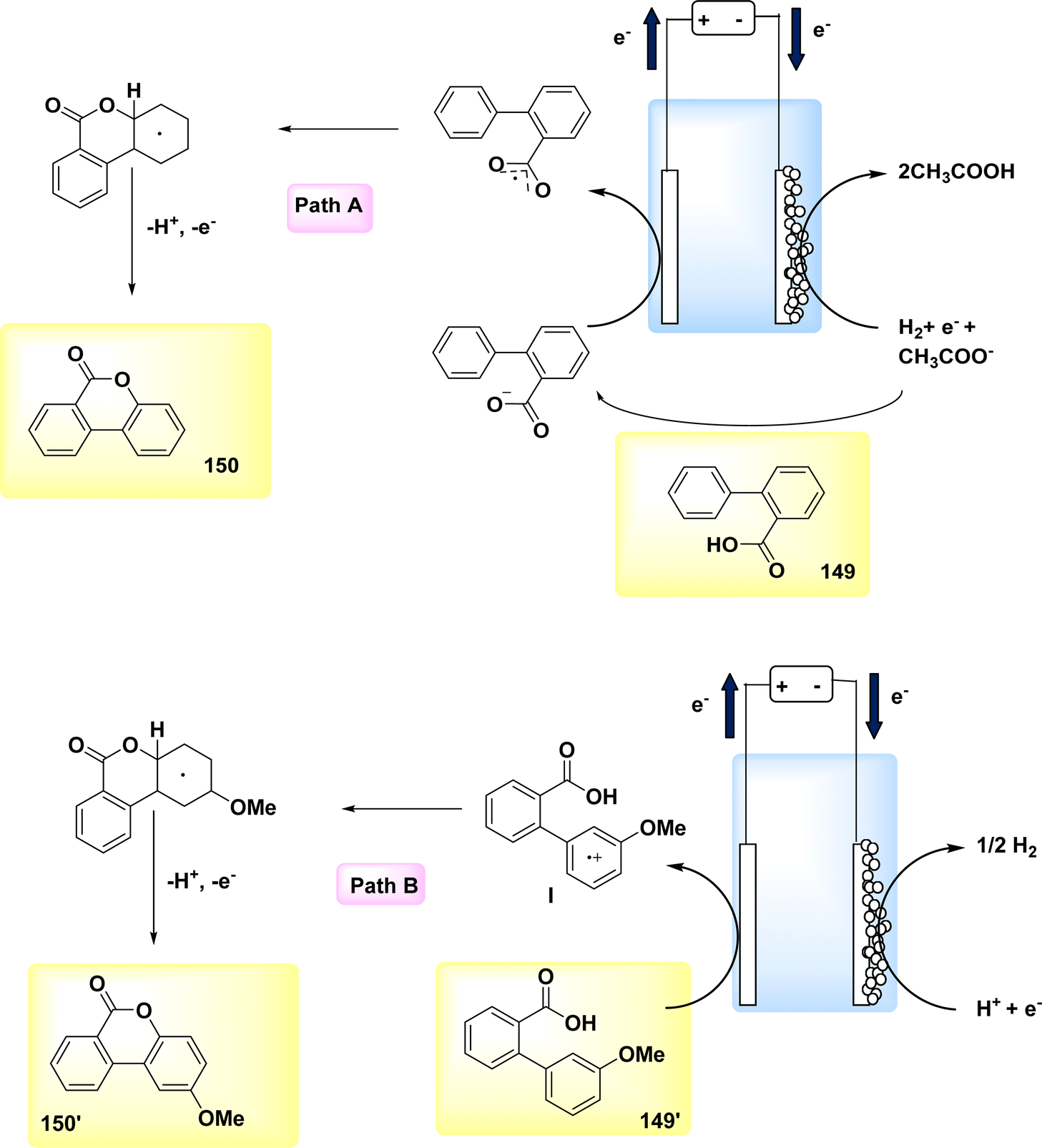 | ||
| Scheme 57 The electrosynthesis of 6H-benzo[c]chromen-6-one 150 and its CH3O− derivative 150′ with its proposed mechanism. | ||
The year 2018 witnessed the demonstration of the electrosynthesis of aromatic lactones 150 by the process of dehydrogenative cyclization. This methodology, which relies on electrochemical techniques, was shown to be highly effective and dependable, as depicted in Scheme 55. The electrochemical technique described in this study is both innovative and advantageous, since it has the potential to be scaled up to 100 grams while operating within reasonable constraints. Interestingly, the utilization of substrates that incorporate heterocycles has the potential to enhance the efficiency of this particular process. Graphite or Pt was employed as the cathode and anode, respectively, in the electrochemical carbon–oxygen cyclization reaction of C13H10O2 acid 149. By subjecting the system to a constant current of 6 mA, the desired product with a yield of 91% was successfully obtained at ambient temperature. A comprehensive depiction of this particular approach has already been elucidated in path A of Scheme 57.85
In the year 2018, a highly effective electrochemical method was developed for the direct lactonization process, demonstrating exceptional regioselectivity, without the need for external oxidants or metals. Under conditions of steady current, the cyclization of 2-biphenylcarboxylic acid 151 occurs readily. In this particular situation, the electrolyte used was tetrabutylammonium tetrafluoroborate (nBu4NBF4), while the solvent system consisted of a combination of methanol and methyl cyanide. The experimental setup involved an undivided cell furnished with a Pt/C-anode and cathode. The scalability of the newly developed methodology was demonstrated utilizing a simple method (Scheme 58) on a 40 g scale.86
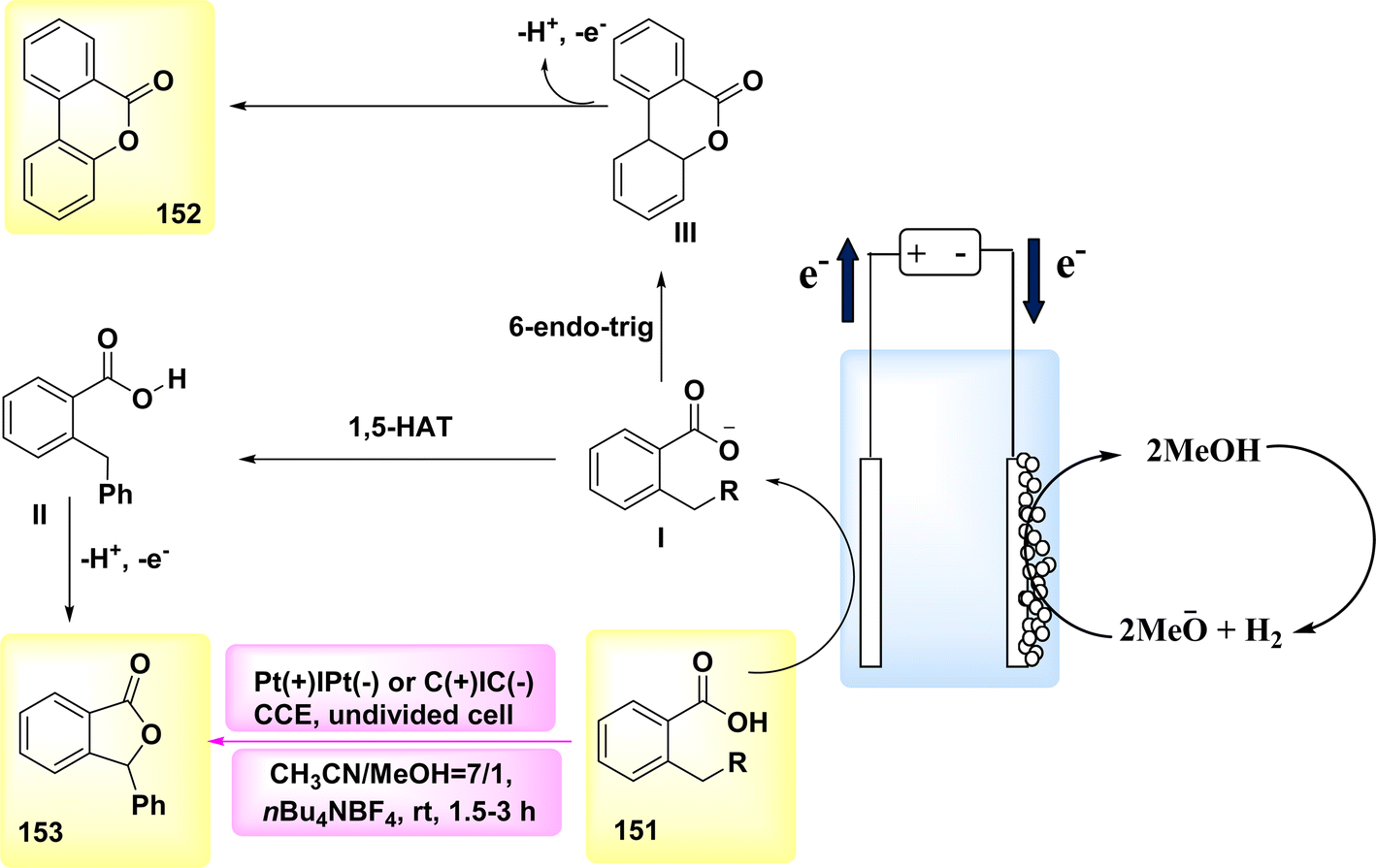 | ||
| Scheme 58 Electrosynthesis of substituted coumarin 152 and tetrahydrofuranone 153 with its proposed mechanism. | ||
The carboxylate radical I exhibits divergent pathways, which are contingent upon the structural characteristics of the substrate. The evoluation of intermediate III is achieved through a 6-endo-trig cyclization. Upon lactonization of the C(sp3)–H link, a H2-atom transfer takes place, resulting in the formation of a stable benzyl radical II. In due course, radicals III and II undergo a process where they undergo the loss of an e− and a proton, resulting in the evoluation of products 152and 153, respectively.
According to Xiong et al. (2019), the electrochemical formation of dihydrofuran derivatives 115 was observed in Scheme 59. A significant quantity of 156 was obtained in high yield by the reaction of styrene derivatives 154 and acetylacetone 155. In the experiment an undivided cell is used, with a Pt plate as the cathode, carbon serving as the anode, and CH3CN/1,1,1,3,3,3-hexafluoroisopropanol as the solvent and electrolyte. A steady current of 10 mA was utilized.87
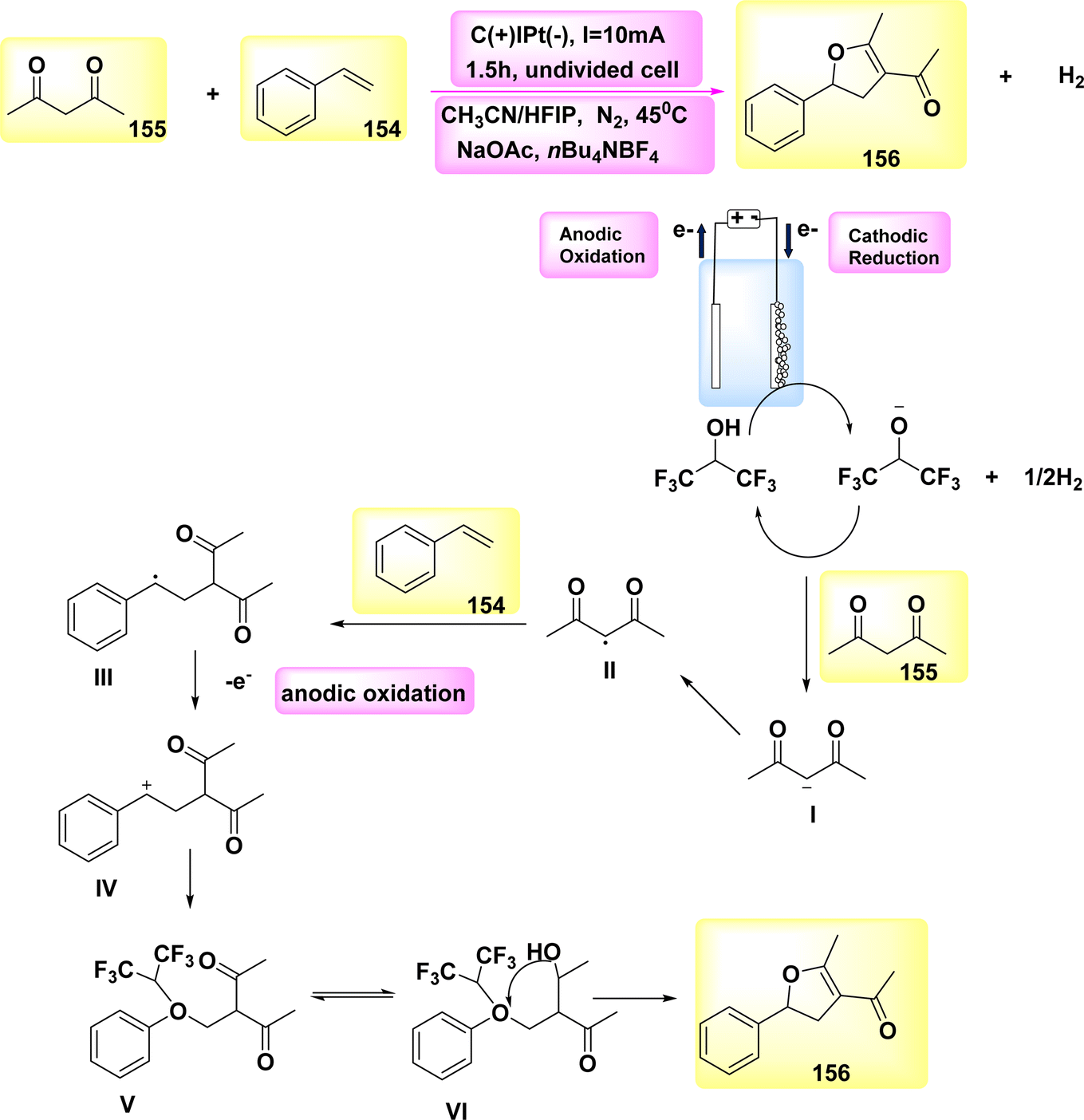 | ||
| Scheme 59 Electrochemical synthesis of substituted dihydrofuran 156 with its mechanism. | ||
By the acetylacetone deprotonation 155 using the base NaOAc or alkoxide, the acetylacetone anion is generated. Subsequently, this anion undergoes oxidation, ensuing the formation of small quantities of the electrophilic C radical II. The stability of this radical can be enhanced by 1,1,1,3,3,3-hexafluoroisopropanol, leading to an anodic event. The C radical II subsequently has a reaction with styrene 154, resulting in the formation of the benzylic radical III. This radical is then subjected to oxidation at the anode, leading to the generation of the benzylic cation IV. The intermediate V, which is likely to undergo tautomerization to VI, initiates an intermolecular nucleophilic attack to release the molecular target 156. This process can be additionally stabilized by the 1,1,1,3,3,3-hexafluoroisopropanolcluster.
In the year 2019, researchers were able to successfully carry out electrochemical oxidation of olefinic carbonyls 158 using readily available diselenides 157. This process enabled theecologically friendly, immediate formation of C–Se and C–O in a configuration. Using substrates 157 and 158, the electrochemical oxidative cyclization reaction was initiated, Scheme 60. The final product was successfully obtained with a Pt-sheet (−) and a graphite-rod(+), utilizing nBu4NBF4 as the electrolyte, acetonitrile as the solvent, andacetic acid as an additive, resulting in a high yield.
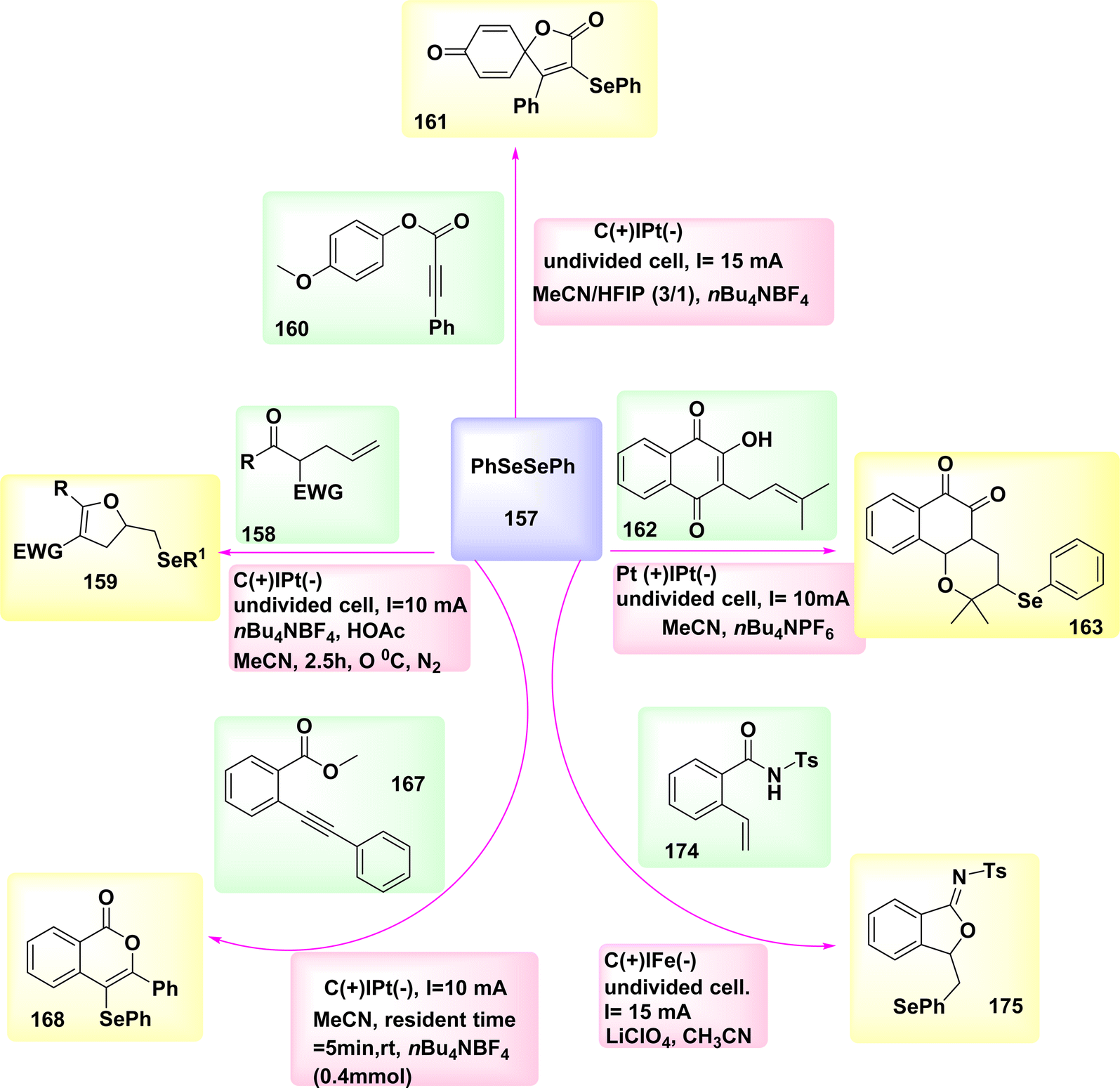 | ||
| Scheme 60 Electrochemical synthesis of substituted selenated dihydrofuran 159, 3-selenated spiro[4.5]trienones 161, selenium-containing naphthoquinones 163, isocoumarin 168, and selenatedisobenzofuran 175. | ||
Using an innovative chelation technique, a diverse range of selenodihydrofurans 159 and selenooxazolines with fragile heterocycles, moderate carbon-iodide bonds, and additional  groups were synthesized. This process occurs by preventing the presence of exogenous metallic and oxidizing substances.88
groups were synthesized. This process occurs by preventing the presence of exogenous metallic and oxidizing substances.88
Scheme 61 illustrates the proposed pathway for the aforementioned reaction. The compound 158 undergoes a reaction with phenyl selenium radical I, resulting in the formation of alkyl radical II. Subsequently, the removal of one electron from alkyl radical II leads to the formation of cation III. The desired product 159 is obtained through a rapid ring closure, subsequent nucleophilic assault on the carbonyl oxygen atom, and deprotonation.
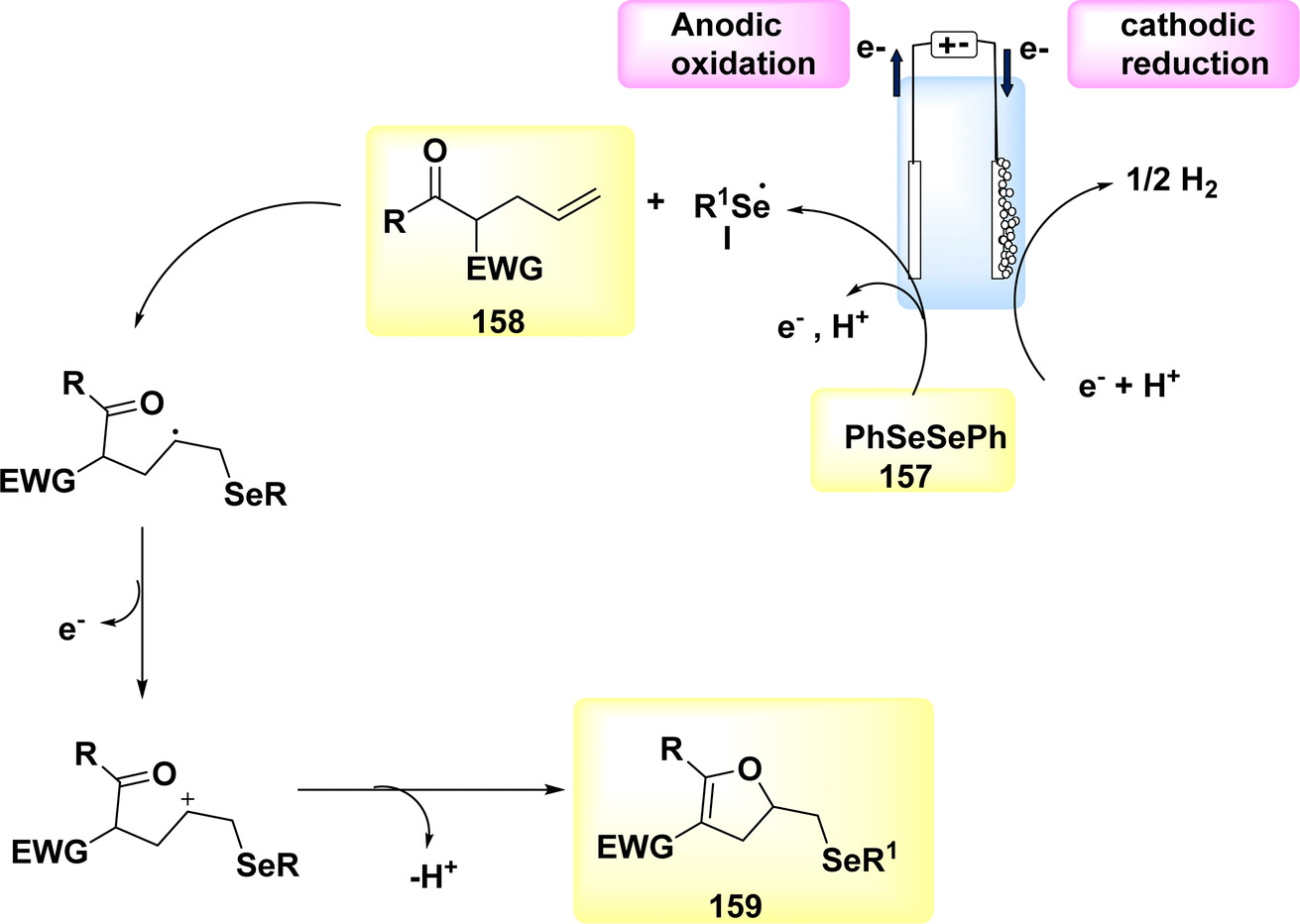 | ||
| Scheme 61 Electrochemical synthesis of substituted selenated dihydrofuran 159 with its mechanism. | ||
For the synthesis of spiro[4.5]trienones, a novel and ecologically sustainable approach has been developed in the year 2020, as outlined in Scheme 60. The process, which exhibited moderate regulation, was conducted within a single, undivided cellular unit. C16H12O3 160 and diphenyl diselenide 157 were chosen as the experimental compounds for the synthesis of 3-selenated spiro[4.5]trienones 161. The reaction was carried outusing a Pt cathode and a graphite anode under constant current conditions of 15 mA. The electrolyte utilized in the CH3CN/1,1,1,3,3,3-hexafluoroisopropanol reaction was nBu4NPF6, resulting in a yield of 84% for the required product 161. Furthermore, substantial quantities of additional analogous compounds were produced.89
A possible reaction mechanism for the electrochemical transition is shown in Scheme 62. The first oxidation of diphenyl diselenide 157 results in the formation of the cationic radical intermediate I. Subsequently, this intermediate undergoes decomposition, giving rise to the generation of radical II and cation III. Subsequently, the carbon–carbon triple bond, the C6H5ClSe radical II reacts with 160 undergo a radical addition process, resulting in the construction of vinyl radical IV. By the presence of a 5-membered ring str. and the inherent stability of the resonant free radical, known as the  IV, the intramolecular spirocyclization to form compound V is typically facilitated. At the (+) further oxidation of intermediate V, resulted in the production of intermediate VI, which is an oxygenium cation. Finally, the formation of product 161 occurs by following path A, by the demethylation of cation VI and the subsequent dearomatization of the aromatic ring. In contrast, it is important to acknowledge that the formation of intermediate VII may potentially occur by the combination of 160 and phenyl selenium cation III. The cyclic selenium cation undergoes intramolecular assault by an aromatic ring possessing a high electron density, resulting in the formation of intermediate VI. Consequently, demethylation of intermediate VI occurs, leading to the production of product 161 through path B.
IV, the intramolecular spirocyclization to form compound V is typically facilitated. At the (+) further oxidation of intermediate V, resulted in the production of intermediate VI, which is an oxygenium cation. Finally, the formation of product 161 occurs by following path A, by the demethylation of cation VI and the subsequent dearomatization of the aromatic ring. In contrast, it is important to acknowledge that the formation of intermediate VII may potentially occur by the combination of 160 and phenyl selenium cation III. The cyclic selenium cation undergoes intramolecular assault by an aromatic ring possessing a high electron density, resulting in the formation of intermediate VI. Consequently, demethylation of intermediate VI occurs, leading to the production of product 161 through path B.
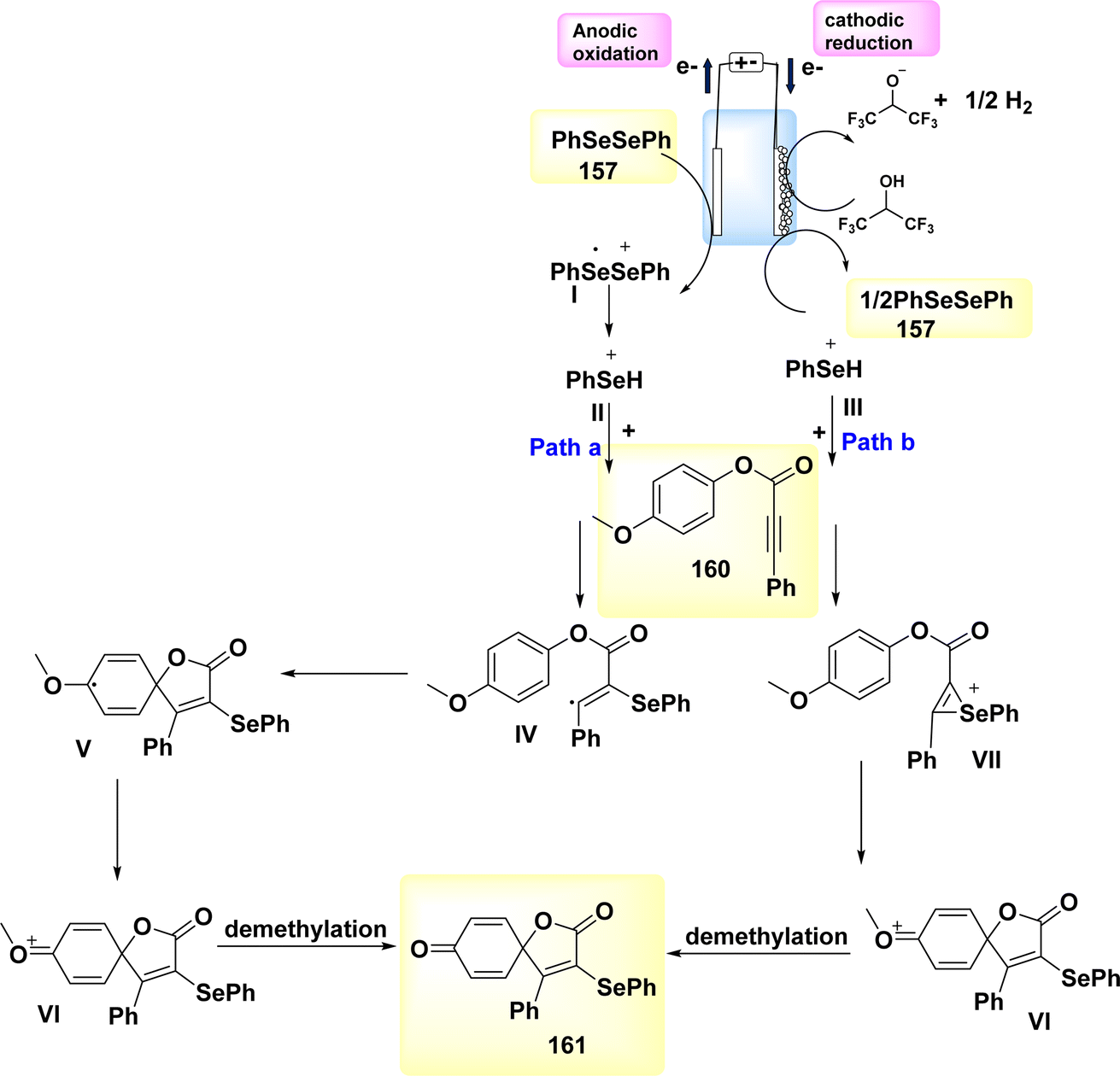 | ||
| Scheme 62 Electrochemical synthesis of 3-selenated spiro[4.5]trienones 161 with its proposed mechanism. | ||
The manufacture of selenium-containing naphthoquinones 163 in the year 2020, is accomplished by the electrochemical selenation process in undivided electrochemical cells (Scheme 60). By employing nBu4NPF6 as the electrolyte and applying a current of 10 mA for a reaction time of 1 hour, this efficient and environmentally friendly technology achieves a yield of 163 (yield = 93%) without the use of oxidizing agents.90
The mechanism for the electrosynthesis of naphthoquinones containing selenium, as hypothesized, is presented in Scheme 63.
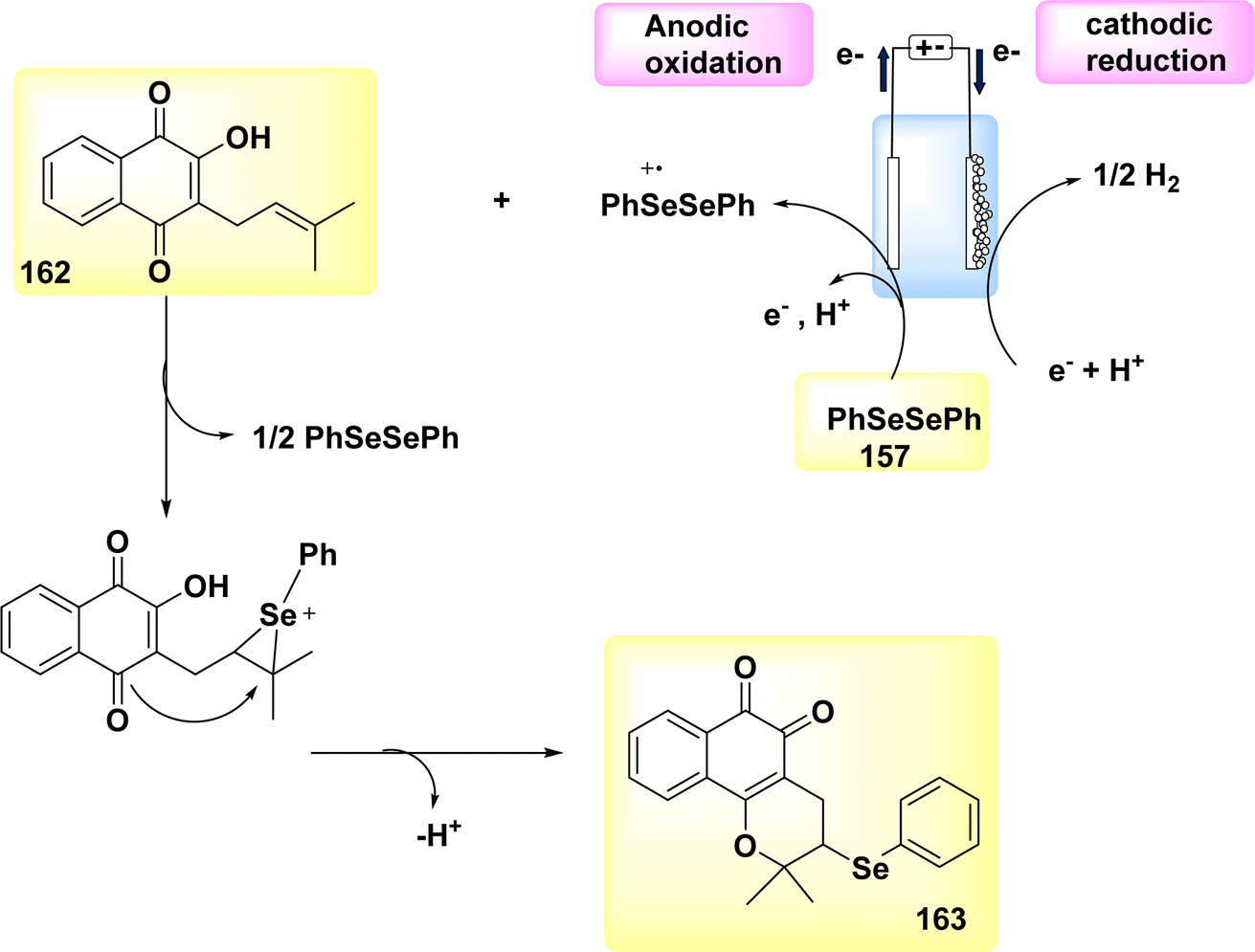 | ||
| Scheme 63 Electrochemical synthesis of selenium-containing naphthoquinones 163 with its proposed mechanism. | ||
The inclusion of lapachol 163 resulted in the observation of a novel anodic reaction occurring at a potential of Ep = 1.55 V vs. the SCE. The oxidation process of product 163 can be observed at the anode, as evidenced by a distinct study conducted on product 163. The presence of both the supplementary anodic incident and the diminishing cathodic incident at higher scan rates suggests that the lapachol 162 and selenium-di-cation II undergo rapid carbophilic reactions to form cationic intermediate III. This intermediate then undergoes nucleophilic cyclization, resulting in the production of product 163 and diselenide 157.
The phenomenon of Ir catalyzed electrocatalysis was observed in the year 2020. This fabrication serves as an example of a user-friendly material that exhibits high stereo- and chemo-selectivity (Scheme 64). The reaction between alkene 165 and 2-hydroxybenzaldehyde 164 results in the formation of substituted isobenzofuran-1(3H)-one 166. The electrolyte employed in the experiment is potassium acetate, and the process takes place within an undivided cell at a temperature of 100 °C for a duration of 18 hours.61,62
 | ||
| Scheme 64 Electrosynthesis of substituted isobenzofuran-1(3H)-one 166. | ||
In the year 2020, to initiate an oxidative cyclization process involving o-(1-alkynyl) benzoate and radicals, a persistent electrochemical microreactor was utilized. The production of isocoumarin 168 (Scheme 60) can be achieved by non oxidant and a metal-free technique. Despite the absence of recent advancements in post processing techniques, the potential for further evoluation in greening this synthesis method remains significant, primarily driven by the improved chemical efficacy. In addition, the scalability of events can be achieved through the implementation of a continuous electrochemical process, resulting in extreme yields of the intended product. This highlights the considerable potential of this approach for future applications in industrial production. 157 and C16H12O2 167 are reacted with two equivalents of nBu4NBF4 in the presence of acetonitrile as the solvent. The amalgamated solution was introduced into the electrochemical microreactor, where a reaction took place under the conditions of a flow rate of 45 L min−1 and a current of 10 mA. A target product yield of 92% can be attained, resulting in the production of 168.91
The intricate mechanism is elucidated by Scheme 65. Initially, diphenyl selenide 157 undergoes oxidation at the anode, thereby producing a cationic radical intermediate I. A phenyl-selenium radical II and a phenyl-selenium cation III is generated by the cleavage of the intermediate I. Subsequently, the C6H5Se cation III under went reduction at the cathode to form diphenyl-selenide. Simultaneously, phenyl-selenium radicals reacted with the carbon–carbon triple bond of o-(1-alkynyl) benzoates 167, resulting in the formation of the remarkably regioselective vinyl radical IV. The intramolecular cyclization of the vinyl group, which is formed in situ, is further facilitated by its contact with the –OMe moiety. This process results in the production of the final product 168 and the liberation of the methyl cation. This cation subsequently undergoes a reaction with the OH− ion present in the solvent, resulting in the evoluation of CH3OH.
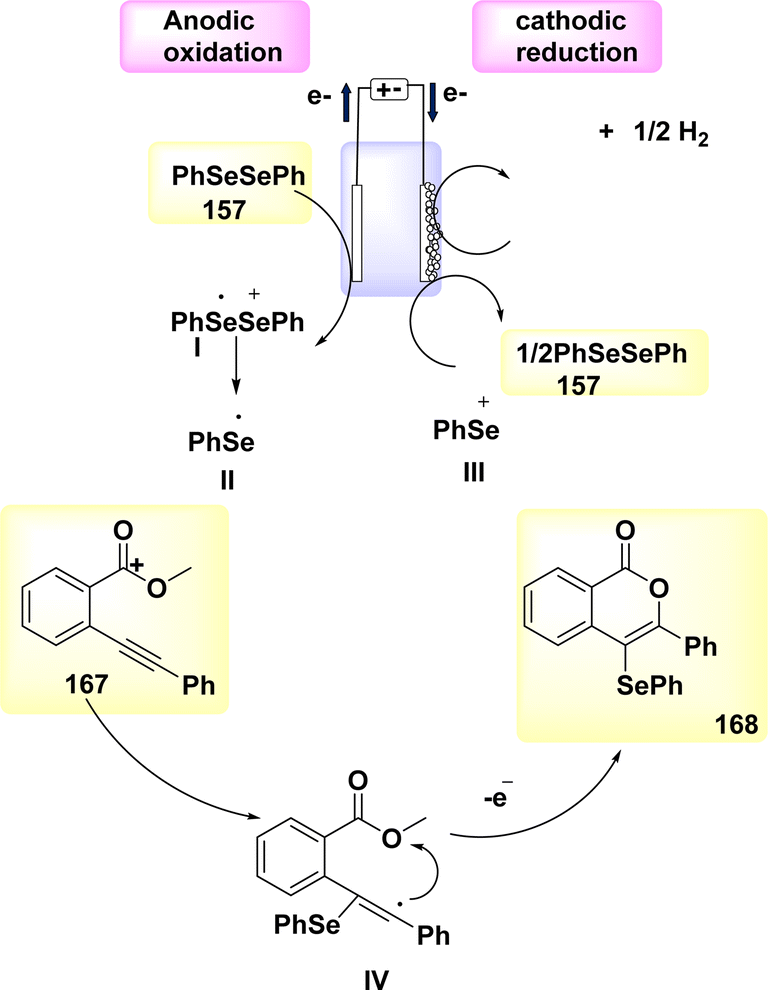 | ||
| Scheme 65 Mechanism of formation of isocoumarin 168. | ||
A C6H5Se radical II and a C6H5Se cation III is created by the cleavage of the intermediate I (Scheme 66).
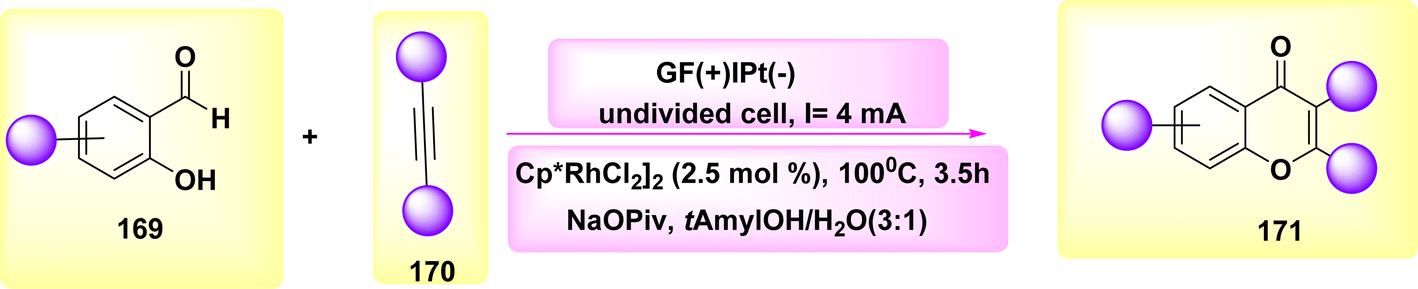 | ||
| Scheme 66 Electrosynthesis of 4H-chromen-4-one 171. | ||
The year 2021 witnessed a proposal on the possibility of synthesizing chromones through the gradual process of electro-formyl C–H activation. Electrocatalysis was conducted using a cell that consisted of an undivided compartment, wherein GF-anode and a Pt-cathode were employed. A 10 mL cell was packed with a mixture containing 2-hydroxybenzaldehyde 169, alkyne 170, sodium pivalate (an electrolyte), [Cp*RhCl2]2 (a catalyst), and t-AmylOH/H2O (4.0 mL, 3:1) as the solvent. In order to synthesize the target compounds 171, an electrocatalytic process was conducted at a temperature of 100 °C using an undivided cell setup. The cell consisted of a GF-anode and a Pt-cathode, and a constant current of 4 mA was applied during the reaction.92
The year 2021 seen the documentation of valuable 7-membered benzoxepine 171 frameworks formed via metal-catalyzed [5 + 2] cyclo additions (Scheme 67). The alkyne annulation method exhibits a broad substrate scope and relies solely on the utilization of electricity as an oxidizing agent. In the electrochemical [5 + 2] cycloadditions, an undivided cell configuration was employed, utilizing graphite as the (+) material and Pt plate as the (−) material. The reactants involved in this process were 2-vinylphenol (172) and diphenylacetylene (84). The target product 173 was obtained with a yield of 88% after subjecting it to rigorous testing using 4-methoxyphenyl-3-phenylpropiolate dimer as the catalyst and sodium pivalate as the electrolyte in a solvent system consisting of tAmOH–H2O at a temperature of 100 °C for a duration of 18 hours.93
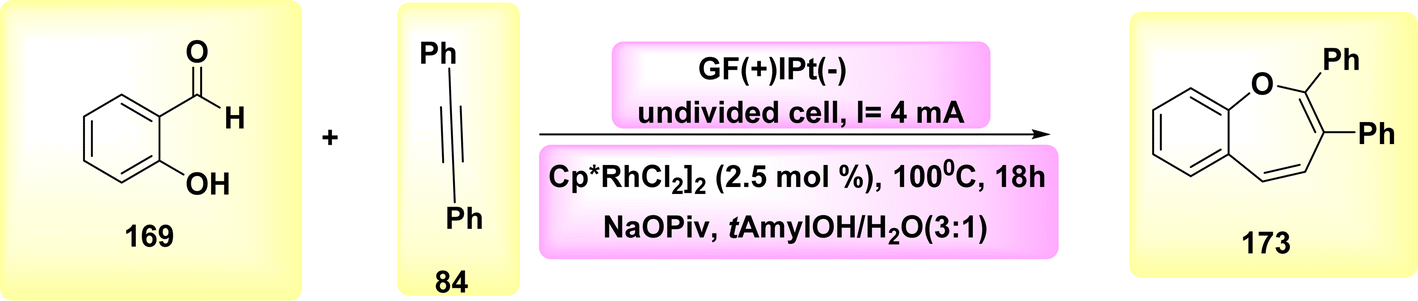 | ||
| Scheme 67 Electrosynthesis of benzoxepine 173. | ||
A potential catalytic cycle is outlined in Scheme 68, whereby the initial step involves the activation of an O2–N2 or H–H bond to generate a rhodacycle I. Complex IV is formed through the process of alkyne coordination-insertion, which subsequently undergoes reductive-elimination, resulting in the production of intermediate III.
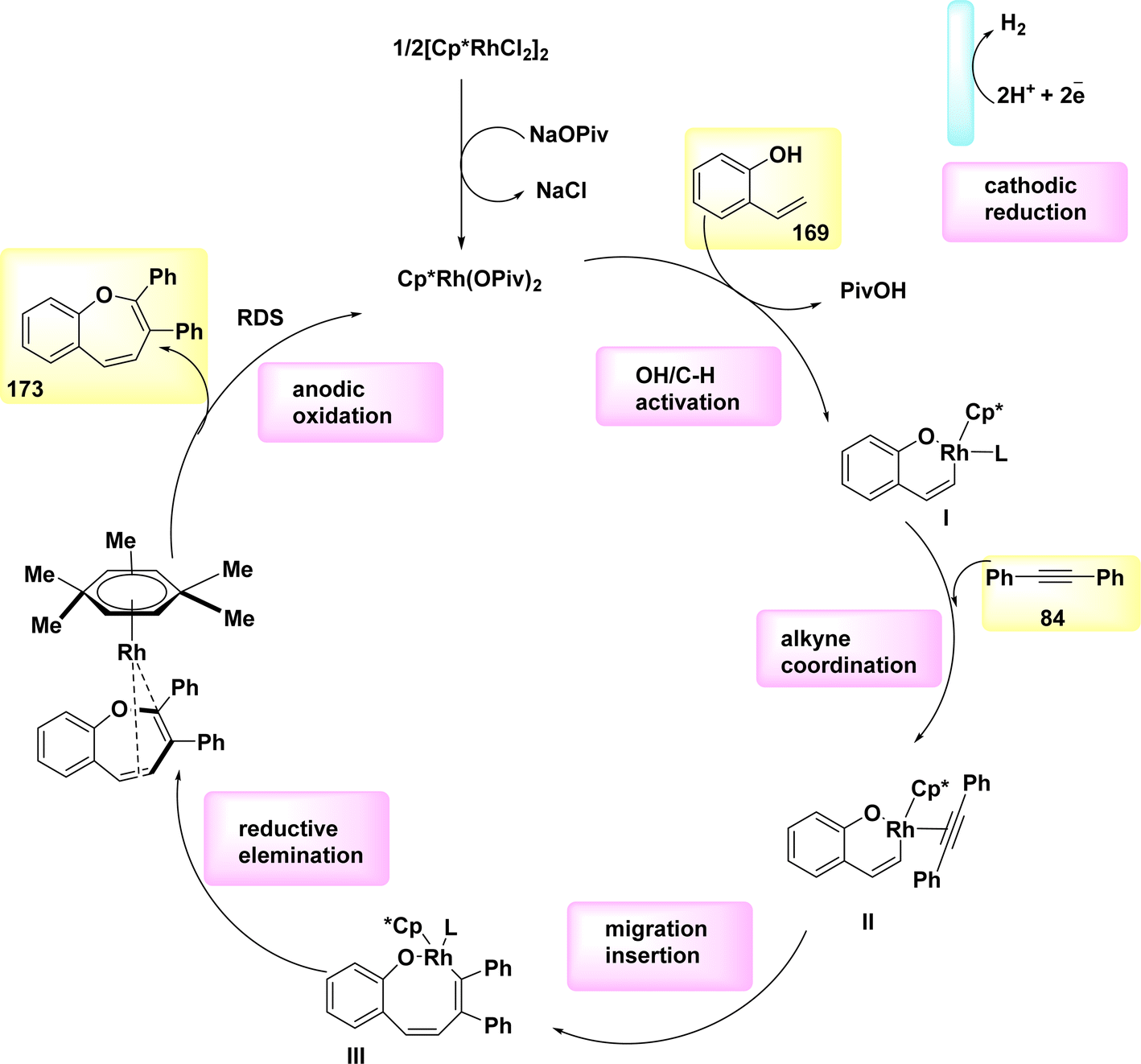 | ||
| Scheme 68 Electrosynthesis of benzoxepine 173 with its proposed mechanism. | ||
In the final analysis, the Rh I species undergoes oxidation to Rh III at the anode, resulting in the liberation of product 173. Simultaneously, molecular H2 is generated as a byproduct at the (−).
In 2021, Bian et al. introduced an alternative methodology for the production of O-heterocycles. In this experiment, the oxidation of C16H15NO3S 174 and diphenyl diselenide 157 in an electrochemical microreactor occurs. The microreactor consisted of a graphite as anode and an iron as cathode. The outcome of the reaction was the production of selenated isobenzofuran 175. Initially, a series of solvents were examined in an experiment conducted using a constant current of 15 mA and LiClO4 as the electrolyte. The desired iminoisobenzofuran product 175 was successfully synthesized with high efficiency when CH3CN was used as the solvent. Subsequently, several electrolytes with supporting properties were subjected to testing. By excluding the use of metals and oxidants, this methodology enabled the synthesis of a diverse range of iminoisobenzofuran derivatives with remarkable yields, as seen in Scheme 69. The utilization of a continuous-flow system in conjunction with electrochemical techniques effectively addresses the challenge of upscaling a batch-type electrolysis process.94
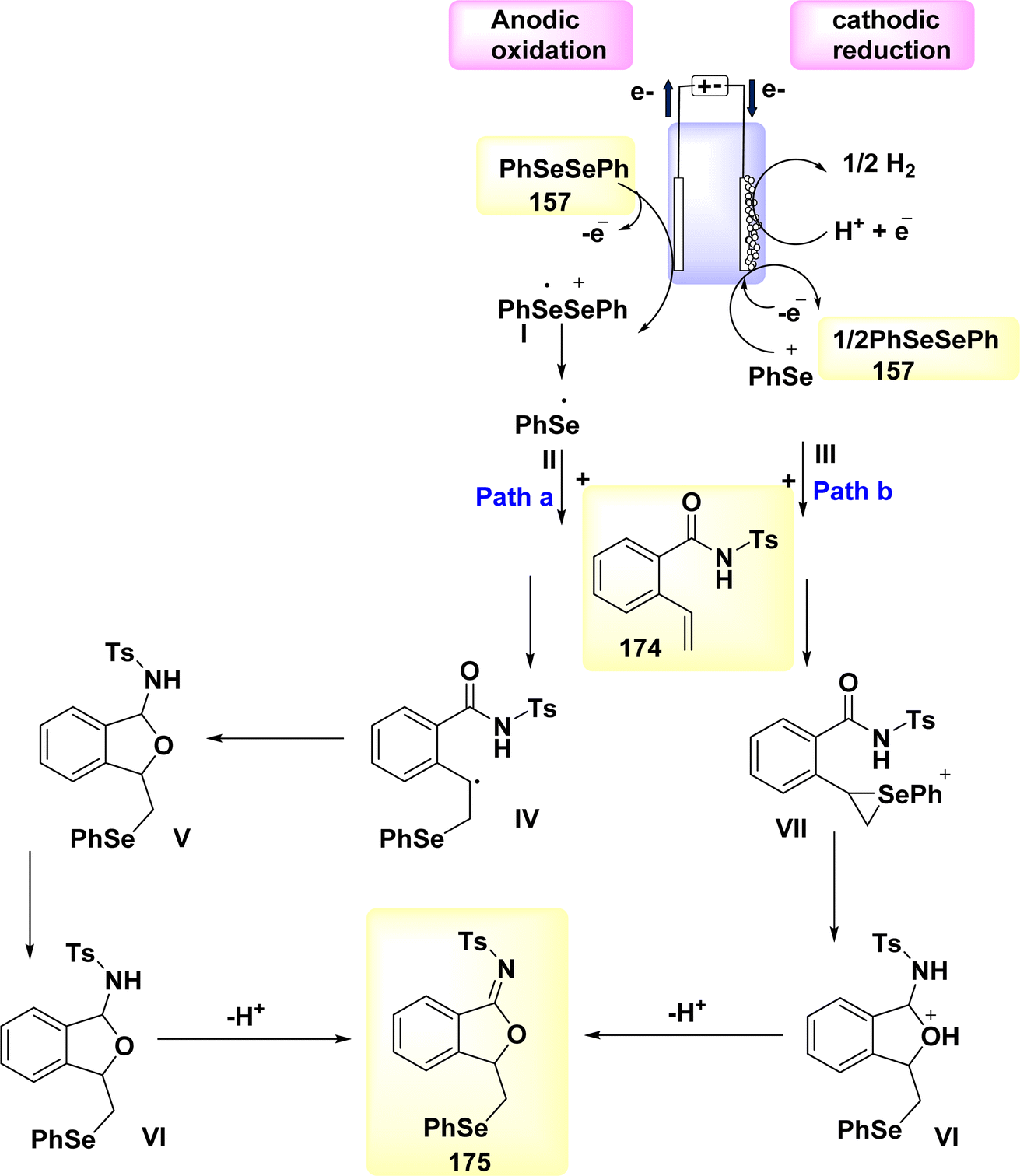 | ||
| Scheme 69 Electrosynthesis of selenated isobenzofuran 175 with its proposed mechanism. | ||
The experiment showcased the occurrence of transition using benzoic acid 176, which exhibited weak coordination. The optimal approach for achieving the desired product 166 was utilizing a solvent mixture of t-AmOH and H2O, with the addition of KOAc, within an undivided cell configuration that was user-friendly. The inclusion of ferrocene/p-benzoquinone (catalytic mediators) in the process may lead to a substantial enhancement in overall efficacy, when compared to the catalytic effects of Rh95 and Ru.96,97
In the years 2018, 2020, and 2021, it is quite probable that an Ir(Ir)-catalyzed cycle was launched to facilitate the process of organometallic carbon–hydrogen functionalization, as depicted in Scheme 70. The construction of a 7-membered Ir(III) cycle IV is subsequently achieved through the process of alkene 168 insertion. Subsequently, this process progresses by reductive elimination and β-hydride elimination to generate intermediate V. The catalytic reoxidation of intermediate V to generate the active molecule I is facilitated by p-benzoquinone. The process of anodic oxidation is occurring on the recently formed hydroquinone.62,66,91,96,98
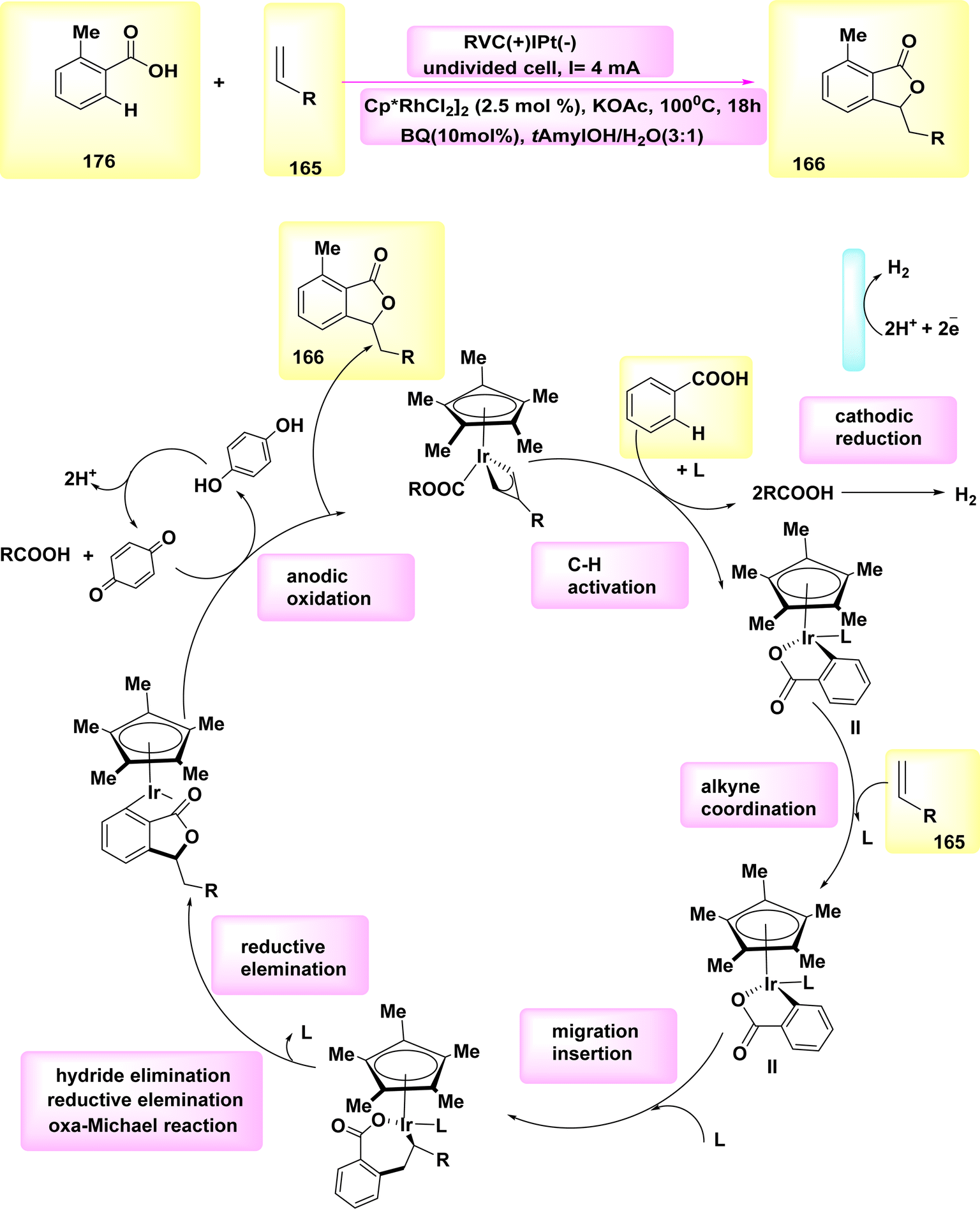 | ||
| Scheme 70 Electrosynthesis of substituted isobenzofuran-1(3H)-one 166 with its proposed mechanism. | ||
The utilization of a Ru catalyst was initially documented in 2018 and subsequently in 2021, as outlined in Scheme 71. The sole oxidizing agent employed in this study was electrical energy, which served to activate the carbon–hydrogen bond. The emission of H2 gas occurred as a by product. The catalyst employed in this procedure had no discernible response to the presence of water and air.
The most favorable outcomes were obtained while doing the experiment in a solvent mixture consisting of tAmOH or H2O, supplemented with NaOPiv. The reaction conditions facilitate the conversion of biphenylacetylene 84 and benzoic acid 176 into substituted 1H-isochromen-1-one 177.97–99
A pragmatic catalytic cycle should begin with a direct organometallic C–H activation, as suggested (Scheme 71). Consequently, the ruthena(II) cycle II is formed along with two equivalents of carboxylic acid. The migration of alkyne insertion subsequently provides the IV. The reductive elimination process is rapidly executed in this cycle, resulting in the formation of complex V. (+) oxidation accomplishes the reoxidation of complex I.
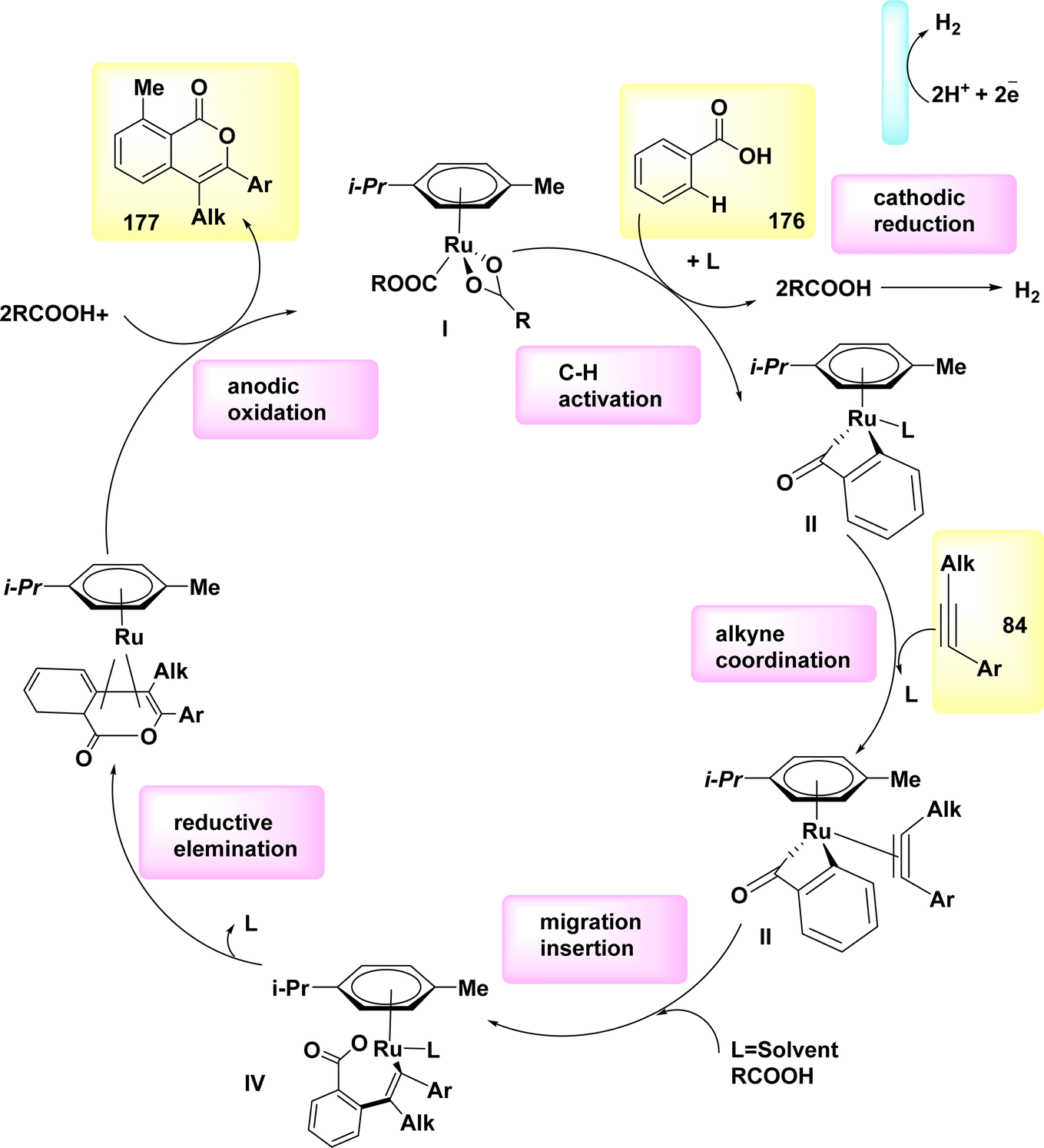 | ||
| Scheme 71 Mechanism for the synthesis of 1H-isochromen-1-one 177. | ||
Conclusion and future perspective
This article primarily examines notable advancements towards the chemical synthesis of heterocycles including nitrogen (N), sulfur (S), and oxygen (O). There is also ongoing discourse over the recognized mechanics of heterocycle evoluation. Organic electrochemistry originated in the 19th century and continues to be a much unexplored area of research. Electrosynthesis is a process that facilitates the selective creation of N-, S-, and O-heterocyclic molecules. This approach presents a less severe situation, decreases the quantity of steps, and gives alternate pathways in comparison to traditional synthesis. Redox reactions are ideal for electrolysis because they involve both reduction and oxidation events happening simultaneously at the (+) and (−), respectively, at the time of the electrolytic process.The utilization of electricity as an alternate method presents a convenient and efficient approach in terms of atom utilization and cost-effectiveness for the formation of molecules.93 Although substantial advancements have been made in this particular area of study, certain challenges persist, notably the need for substantial doses of supporting electrolytes. Under the growing realm of emphasis on environmentally friendly synthesis methods continuous-flow electrochemistry in electrolyte-free conditions is a significant area of concentration.100 Moreover, the recent integration of photochemistry and electrosynthesis has facilitated the evoluation of homogeneous photo electrocatalysis, which exhibits remarkable redox potentials and eliminates the requirement for aggressive chemical redox reagents.101
Nevertheless, the synthesis of cathodic heterocycles seems to be a rare phenomenon, as the majority of recorded electrochemical heterocyclic syntheses are attributed to anodic oxidation. This observation suggests that there exist numerous unexplored domains and opportunities for further investigation within this field. The domain of electro-organic synthesis is experiencing growth, and it is anticipated that further evaluations in electro-reductive methodologies will enhance its scope and provide valuable resolutions to synthetic challenges.102 There are further captivating advancements that are anticipated, like the utilization of sustainable solvents and sustainable energy sources.103 It can be reasonably inferred that the implementation of unique bioderived reaction process medium will advance in parallel with the advancement of innovative heterocyclic reactions.104 There is potential for further advancements in the recycling of transition metals through the utilization of anodic oxidation. The future holds the potential for the advancement of catalyst designs, which will contribute to the expansion of regio- and stereoselective bond functionalizations.105
Electrochemistry is rapidly transitioning from its conventional use to specialized chemical production in a preventive capacity, as well as in recycling and environmental remediation in a curative capacity. Electrochemistry offers a viable alternative to numerous perilous, challenging, and costly procedures across various industries. It is expected to have a significant impact on the advancement of more efficient and cleaner and processes across several industries involved in the production or utilization of chemicals, as it has the potential to address environmental concerns.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Department of Science and Technology-Fund for Improvement of Science and Technology Infrastructure in Universities and Higher Educational Institutions (DST-FIST, India Order Number SR/FST/2022/252) for providing all the necessary facilities.References
- I. T. Horváth, Solvents from nature, Green Chem., 2008, 10(10), 1024–1028 RSC.
- N. Sbei, A. A. Titov, S. Karthikeyan and L. G. Voskressensky, Efficient synthesis of imino-1,3-thiazinan-4-one promoted by acetonitrile electrogenerated base and computational studies with CB1 and 11 βHSD1 molecules, Res. Chem. Intermed., 2020, 46, 5535–5545 CrossRef CAS.
- N. Sbei, G. M. Martins, B. Shirinfar and N. Ahmed, Electrochemical Phosphorylation of Organic Molecules, Chem. Rec., 2020, 20, 1530–1552 CrossRef CAS PubMed.
- N. Sbei, A. A. Titov, E. B. Markova, M. N. Elinson and L. G. Voskressensky, A Facile One-Pot Synthesis of 1,2,3,4-Tetrahydroisoquinoline-1-carbonitriles via the Electrogenerated Cyanide Anions from Acetonitrile, ChemistrySelect, 2020, 5, 4493–4495 CrossRef CAS.
- N. Sbei, R. B. Said, S. Rahali, H. Beya, A. S. Al-Ayed, M. M. Al Mogren, M. L. Benkhoud and M. Seydou, Electrochemical synthesis of hydroxy-thioxo-imidazole carboxylates: an experimental and theoretical study, J. Sulfur Chem., 2020, 41, 117–129 CrossRef CAS.
- N. Sbei, B. Batanero, F. Barba, B. Haouas, L. Fuentes and M. L. Benkhoud, A convenient synthesis of new biological active 5imino-4-thioxo-2-imidazolidinones involving acetonitrile electrogenerated base, Tetrahedron, 2015, 71, 7654–7657 CrossRef CAS.
- Y. Jiang, K. Xu and C. Zeng, Use of Electrochemistry in the Synthesis of Heterocyclic Structures, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS.
- Z.-Y. Mo, X.-Y. Wang, Y.-Z. Zhang, L. Yang, H.-T. Tang and Y. M. Pan, Electrochemically enabled functionalization of indolesoranilines to synthesis of hexafluoro isopropoxyindole and aniline derivatives, Org. Biomol. Chem., 2020, 18, 3832 RSC.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Organic electrosynthesis: A promising green methodology inorganic chemistry, Green Chem., 2010, 12, 2099–2119 RSC.
- G. M. Martins, G. C. Zimmer, S. R. Mendes and N. Ahmed, Electrifying green synthesis: recent advances in electrochemical annulation reactions, Green Chem., 2020, 22(15), 4849–4870 RSC.
- Y. Yuan and A. Lei, Is electrosynthesis always green and advantageous compared to traditional methods?, Nat. Commun., 2020, 11, 2018–2020 CrossRef.
- S. M. Sondhi, N. Singh, A. M. Lahoti, K. Bajaj, A. Kumar, O. Lozach and L. Meijer, Synthesis of acridinyl-thiazolino derivatives and their evaluation for anti-inflammatory, analgesic and kinase inhibition activities, Bioorg. Med. Chem., 2005, 13, 4291–4299 CrossRef CAS.
- Z. Wu, S. Li, H. Long and H. Xu, ElectrochemicaldeH2ative cyclization of 1,3-dicarbonyl compounds, Chem. Commun., 2018, 54, 4601 RSC.
- X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, Electrochemical Oxidation-Induced Site-Selective Intramolecular C(sp3)–H Amination, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS.
- J. Shi, J. Li, W. Zhao, M. Cui, W. Ni, L. Li and S. Zhang, Regioselective intramolecular sp2 C–H amination: direct vs. mediated electrooxidation, Org. Chem. Front., 2021, 8(7), 1581–1586 RSC.
- Q. L. Yang, Y. Y. Li, Y. Liu, T. X. Ren, L. C. Guo, D. C. Wang and H. M. Guo, Electrochemically facilitated oxidative C–H amination enables access to fluorescent N9-fused tricyclic xanthines, Org. Chem. Front., 2021, 8(19), 5410–5417 RSC.
- S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Electrochemical Formation of N-Acyloxy Amidyl Radicals and Their Application: Regioselective Intramolecular Amination of sp2 and sp3CH Bonds, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS.
- P. Qian, L. Xu, W. Wang, L. Zhang, L. Tang, J. Liu and L. Sheng, Electrochemical synthesis of dipyrazolo/dipyrimidine-fused pyridines via oxidative domino cyclization of C (sp3)–H bonds, Org. Chem. Front., 2022, 9(6), 1662–1667 RSC.
- A. Kehl, V. M. Breising, D. Schollmeyer and S. R. Waldvogel, Electrochemical Synthesis of 5-Aryl-phenanthridin-6-one by DeH2ative N, C Bond oxadiazole Formation, Chem.–Eur. J., 2018, 24, 17230–17233 CrossRef CAS PubMed.
- P. Qian, Z. Zhou, L. Wang, Z. Wang, Z. Wang, Z. Zhang and L. Sheng, Electrosynthesis of 2-(1, 3, 4-Oxadiazol-2-yl) aniline Derivatives with Isatins as Amino-Attached C1 Sources, J. Org. Chem., 2020, 85(20), 13029–13036 CrossRef CAS PubMed.
- P. Huang, P. Wang, S. Wang, S. Tang and A. Lei, Electrochemical oxidative [4 + 2] annulation of tertiary anilines and alkenes for the synthesis of tetrahydroquinolines, Green Chem., 2018, 20, 4870–4874 RSC.
- Y. Yu, P. Zheng, Y. Wu and X. Ye, Electrochemical Co-catalyzed CARBON–H2 or N–H oxidation: A facile route to synthesis of substituted oxindoles, Org. Biomol. Chem., 2018, 16, 8917–8921 RSC.
- S. Z. Hashmi, J. Dwivedi, D. Kishore and A. E. Kuznetsov, Synthesis, characterization, and DFT study of the s-triazine analogues of medicinal interest incorporated with five-and six-membered bioactive heterocyclic scaffolds, J. Mol. Struct., 2023, 1278, 134922, DOI:10.1016/j.molstruc.2023.134922.
- D. Bareth, S. Jain, J. Kumawat, D. Kishore, J. Dwivedi and S. Z. Hashmi, Synthetic and pharmacological developments in the hybrid s-triazine moiety: A review, Bioorg. Chem., 2023, 106971 Search PubMed.
- S. Zeba Hashmi and D. Kishore, Novel Synthesis of Imidazo-based and Benzimidazo-based privileged Templates on s-Triazine Nucleus through a Phenoxyl Spacer, J. Heterocycl. Chem., 2017, 54(5), 2912–2915, DOI:10.1002/jhet.2901.
- P. Qian, Z. Yan, Z. Zhou, K. Hu, J. Wang, Z. Li and Z. Wang, Electrocatalytic Intermolecular C (sp3)–H/N–H Coupling of Methyl N-Heteroaromatics with Amines and Amino Acids: Access to Imidazo-Fused N-Heterocycles, Org. Lett., 2018, 20(20), 6359–6363 CrossRef PubMed.
- A. Kehl, T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Electrochemical Conversion of Phthaldianilides to Phthalazin-1,4diones by DeH2ative N–N Bond Formation, Chem.–Eur. J., 2018, 24, 590–593 CrossRef CAS PubMed.
- Z. W. Hou, H. Yan, J. S. Song and H. C. Xu, Electrochemical Synthesis of (Aza)indolines via DeH2ative [3 + 2] Annulation: Application to Total Synthesis of (±)-Hinckdentine A, Chin. J. Chem., 2018, 36, 909–915 CrossRef CAS.
- H. Long, J. Song and H. C. Xu, Electrochemical synthesis of 7 membered carbocycles through cascade 5-: Exo-trig/7- endo-trig radical cyclization, Org. Chem. Front., 2018, 5, 3129–3132 RSC.
- H. Yan, Z. Y. Mao, Z. W. Hou, J. Song and H. C. Xu, A diastereoselectiveapproachtoaxiallychiralbiarylsviaelectrochemically enabledcyclizationcascade, Beilstein J. Org. Chem., 2019, 15, 795–800 CrossRef CAS.
- M. Zhang, Y. Sun, W. Wang, K. Chen, W. Yang and L. Wang, Electrochemical synthesis of sulfonated benzothiophenes using 2 alkynyl thioanisoles and sodium sulfinates, Org. Biomol. Chem., 2021, 19, 3844–3849 RSC.
- Z. Wang, X. Meng, Q. Li, H. Tang, H. Wang and Y. Pan, Electrochemical Synthesis of 3,5-Disubstituted-1,2,4- thiadiazoles through NH4I-Mediated Dimerization of Thioamides, Adv. Synth. Catal., 2018, 360, 4043–4048 CrossRef CAS.
- Z. Ye, F. Wang, Y. Li and F. Zhang, Electrochemical synthesis of tetrazoles: Via metal- and oxidant-free [3 + 2] cycloaddition of azides with hydrazones, Green Chem., 2018, 20, 5271–5275 RSC.
- Z. W. Hou, Z. Y. Mao, Y. Y. Melcamu, X. Lu and H. C. Xu, Electrochemical Synthesis of Imidazo-Fused N-Heteroaromatic Compounds through a C–N Bond-Forming Radical Cascade, Angew. Chem., 2018, 130, 1652–1655 CrossRef.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Electrochemical Synthesis of 1,2,4-Triazole-Fused Heterocycles, Green Chem., 2018, 20, 1732 RSC.
- J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Electrochemical Aziridination by Alkene Activation Using a Sulfamate astheNitrogenSource, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef CAS.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Co-catalyzed electrooxidative CARBON–H2/N–H [4+2] annulations with ethyleneorethyne, Nat. Commun., 2018, 9, 1–7 CrossRef CAS.
- L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C. Pao, J. Chen, J. Lee and A. Lei, Co-catalyzed electrochemical oxidative CH/N–H carbonylation with H2 evolution, ACS Catal., 2018, 8, 5448–5453 CrossRef CAS.
- N. Sauermann, T. H. Meyer and L. Ackermann, Electrochemical Co-Catalyzed CARBON–H2 Activation, Chem.–Eur. J., 2018, 24, 16209–16217 CrossRef CAS.
- T. H. Meyer, J. C. A. Oliveira, S. C. Sau, N. W. J. Ang and L. Ackermann, Electrooxidative Allene Annulations by Mild Co Catalyzed CARBON–H2 Activation, ACS Catal., 2018, 8, 9140–9147 CrossRef CAS.
- R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, Electroremovable Traceless Hydrazides for Co-Catalyzed Electro Oxidative CARBON–H2/N–H Activation with Internal Alkynes, J. Am. Chem. Soc., 2018, 140, 7913–7921 CrossRef CAS PubMed.
- R. Mei, J. Koeller and L. Ackermann, Electrochemical Rucatalyzed alkyne annulations by CARBON–H2/Het–H activation of aryl carbamates or phenols in protic media, Chem. Commun., 2018, 54, 12879–12882 RSC.
- P. Xiong, H. H. Xu, J. Song and H. C. Xu, Electrochemical Difluoromethylarylation of Alkynes, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed.
- P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, 3d metallaelectrocatalysis for resource economical syntheses, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- K. Y. Ye, Z. Song, G. S. Sauer, J. H. Harenberg, N. Fu and S. Lin, Synthesis of Chlorotrifluoromethylated Pyrrolidines by Electrocatalytic Radical Ene-Yne Cyclization, Chem.–Eur. J., 2018, 24, 12274–12279 CrossRef CAS PubMed.
- X. Gao, P. Wang, Q. Wang, J. Chen and A. Lei, Electrochemical oxidative annulations of amines and aldehydesorketones to synthesize polysubstituted pyrroles, Green Chem., 2019, 21, 4941–4945 RSC.
- Z. Ruan, Z. Huang, Z. Xu, G. Mo, X. Tian, X. Yu and L. Ackermann, Catalyst-Free, Direct Electrochemical Tri- and Difluoroalkylation/Cyclization: Access to Functionalized Oxindoles and Quinolinones, Org. Lett., 2019, 21, 1237–1240 CrossRef CAS PubMed.
- X. Yi and X. Hu, Formal Aza-Wacker Cyclization by Tandem Electrochemical Oxidation and Cu Catalysis, Angew. Chem., Int. Ed., 2019, 58, 4700–4704 CrossRef CAS.
- F. Xu, H. Long, J. Song and H. Xu, De Novo Synthesis of Highly Functionalized Benzimidazolones and Benzoxazolones through an Electrochemical DeH2ative Cyclization Cascade, Angew. Chem., 2019, 131, 9115–9119 CrossRef.
- H. Yu, M. Jiao, R. Huang and X. Fang, Electrochemical Intramolecular DeH2ative Coupling of N-Benzyl(thio)amides: A Direct and Facile Synthesis of 4H-1,3-Benzoxazines and 4H-1,3Benzothiazines, Eur. J. Org Chem., 2019, 2019, 2004–2009 CrossRef CAS.
- C. Tian, U. Dhawa, A. Scheremetjew and L. Ackermann, Cupraelectro-Catalyzed Alkyne Annulation: Evidence for Distinct C–H Alkynylation and Decarboxylative CARBON–H2/C–C, ACS Catal., 2019, 9, 7690 CrossRef CAS.
- S. A. Sau, R. Mei, J. Struwe and L. Ackermann, Co electroCatalyzed CARBON-H2 Activation with CO or Isocyanides, ChemSusChem, 2019, 12, 3023 CrossRef CAS.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Electrochemical CARBON-H2/N-H Activation by Water-Tolerant Co CatalysisatRoomTemperature, Angew. Chem., 2018, 130, 2407–2411 CrossRef.
- N. Sauermann, T. H. Meyer and L. Ackermann, Electrochemical Co-Catalyzed CARBON-H2 Activation, Chem.–Eur. J., 2018, 24, 16209–16217 CrossRef CAS.
- W. J. Kong, Z. Shen, L. S. Finger and L. Ackermann, Electrochemical Access to Aza-Polycyclic Aromatic Hydrocarbons: Rhoda-Electrocatalyzed Domino Alkyne Annulations, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS.
- W. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, Flow Rhodaelectro-Catalyzed Alkyne Annulations by Versatile CARBON-H2 Activation: Mechanistic Support for Rhodium (III/IV), J. Am. Chem. Soc., 2019, 141, 17198 CrossRef CAS.
- Z. Xu, Z. Huang, Y. Li, R. Kuniyil, C. Zhang, L. Ackermann and Z. Ruan, Catalyst-free, direct electrochemical synthesis of annulated medium-sized lactams through C-C bond cleavage, Green Chem., 2020, 22, 1099 RSC.
- T. He, W. Zhong and J. Huang, The Synthesis of Sulfonated 4H 3,1-Benzoxazines via an Electro-chemcial Radical Cascade Cyclization, Chem. Commun., 2020, 56, 2735 RSC.
- Z. Wu, S. Li and H. Xu, Synthesis of N-Heterocycles by DeH2ative Annulation of N-Allyl Amides with 1,3-Dicarbonyl Compounds, Angew. Chem., 2018, 130, 14266–14270 CrossRef.
- M. He, Z. Mo, Z. Wang, S. Cheng, R. Xie, H. Tang and Y. Pan, Electrochemical synthesis of 1-naphthols by intermolecular annulation of alkynes with 1,3-dicarbonyl compounds, Org. Lett., 2020, 22, 724–728 CrossRef CAS PubMed.
- Z. Hou and H. Xu, Electrochemically Enabled Intramolecular Amino oxygenation of Alkynes via . NH2 Cyclization, Chin. J. Chem., 2020, 38, 394–398 CrossRef CAS.
- L. B. Zhang, R. S. Geng, Z. C. Wang, G. Y. Ren, L. R. Wen and M. Li, Electrochemical intramolecular CARBON-H2/N-H functionalization for the synthesis of isoxazolidine-fused isoquinolin-1(2H)-ones, Green Chem., 2020, 22, 16–21 RSC.
- L. J. Wesenberg, E. Diehl, T. J. B. Zahringer, C. Dorr, D. Schollmeyer, A. Shimizu, J. Yoshida, U. A. Hellmich and S. R. Waldvogel, Metal-Free Two fold Electrochemical CARBON-H2 Amination of Activated Arenes: Application to Medicinally Relevant Precursor Synthesis, Chem.–Eur. J., 2020, 26, 17574–17580 CrossRef CAS PubMed.
- R. C. Samanta and L. Ackermann, Evolution of Earth-Abundant 3d Metalla electro-Catalyzed CARBON-H2 Activation: From Chelation-Assistance to CARBON-H2 Functionalization without Directing Groups, Chem. Rec., 2021, 21, 2430–2441 CrossRef CAS PubMed.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Electrochemical CARBON-H2/N-H Activation by Water-Tolerant Co Catalysis at Room Temperature, Angew. Chem., 2018, 130, 2407–2411 CrossRef.
- N. Sauermann, T. H. Meyer and L. Ackermann, Electrochemical Co-Catalyzed CARBON-H2 Activation, Chem.–Eur. J., 2018, 24, 16209–16217 CrossRef CAS.
- C. Tian, T. H. Meyer, M. Stangier, U. Dhawa, K. Rauch, L. H. Finger and L. Ackermann, Coaelectro-catalyzed CARBON-H2 activation for resource-economical molecular syntheses, Nat. Protoc., 2020, 15, 1760–1774 CrossRef CAS PubMed.
- T. H. Meyer, G. A. Chesnokov and L. Ackermann, CoaElectrocatalyzed C–H Activation in Biomass- Derived Glycerol:Powered by Renewable Wind and Solar Energy, ChemSusChem, 2020, 13, 668–671 CrossRef CAS.
- R. Mei, X. Fang, L. He, J. Sun, L. Zou, W. Ma and L. Ackermann, Coaelectro-Catalyzed Oxidative Allene Annulation by Electro Removable Hydrazides, Chem. Commun., 2020, 56, 1393 RSC.
- L. Yang, R. Steinbock, A. Scheremetjew, R. Kuniyil, L. H. Finger, A. M. Messinis and L. Ackermann, Azaruthena (II) -bicyclo[3.2.0] heptadiene: Key Intermediate for Ruthenaelectro(II/III/I)-catalyzed Alkyne Annulations, Angew. Chem., Int. Ed., 2020, 59, 11130–11135 CrossRef CAS PubMed.
- M. Bortolami, I. Chiarotto, L. Mattiello, R. Petrucci, D. Rocco, F. Vetica and M. Feroci, Organic electrochemistry: Synthesis and functionalization of β-lactams in the twenty-first century, Heterocycl. Commun., 2021, 27, 32–44 CrossRef CAS.
- M. Feroci, Investigation of the Role of Electrogenerated N-Heterocyclic Carbene in the Staudinger Synthesis in Ionic Liquid, Int. J. Org. Chem., 2011, 1, 191–201 CrossRef CAS.
- N. Zhou, J. Zhao, C. Sun, y. Lai, Z. Ruan and P. Feng, Electro Oxidative C–N Bond Formation through Azolation of Indole Derivatives: An Access to 3 – Substituent-2-(Azol-1-yl)indoles, J. Org. Chem., 2021, 86, 16059–16067 CrossRef CAS.
- S. Bae, H. G. Hahn, K. D. Nam and H. Mah, Solid-phasesynthesis of fungitoxic 2-imino-1,3-thiazolines, J. Comb. Chem., 2005, 7, 7–9 CrossRef CAS PubMed.
- D. S. Kim, Y. M. Jeong, I. K. Park, H. G. Hahn, H. K. Lee, S. B. Kwon, J. H. Jeong, S. J. Yang, U. D. Sohn and K. C. Park, A new 2 imino-1,3-thiazoline derivative, KHG 22394, inhibits melaninsynthesis in mouse B16 melanoma cells, Biol. Pharm. Bull., 2007, 30, 180–183 CrossRef CAS PubMed.
- M. Islam, B. M. Kariuki, Z. Shafiq, T. Wirth and N. Ahmed, Efficient Electrosynthesis of Thiazolidin-2-imines via Oxysulfurization of Thiourea-Tethered Terminal Alkenes using the Flow Microreactor, Eur. J. Org Chem., 2019, 2019, 1371–1376 CrossRef CAS.
- A. A. Folgueiras-Amador, X. Y. Qian, H. C. Xu and T. Wirth, Catalyst- and Supporting-Electrolyte-Free Electrosynthesis of Benzothiazoles and Thiazolopyridines in ContinuousFlow, Chem.–Eur. J., 2018, 24, 487–491 CrossRef CAS PubMed.
- C. Huang, X. Y. Qian and H. C. Xu, Continuous-Flow Electrosynthesis of Benzofused S-Heterocycles by DeH2ative C-S Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6650–6653 CrossRef CAS PubMed.
- C. Huang and H. C. Xu, Synthesis of 1,3-benzothiazines by intramolecular deH2ative C-S cross-coupling in a flow electrolysis cell, Sci. China: Chem., 2019, 62, 1501–1503 CrossRef CAS.
- S. K. Mo, Q. H. Teng, Y. M. Pan and H. T. Tang, Metal- and Oxidant-free Electrosynthesis of 1,2,3-Thiadiazoles from Element Sulfur and N-tosylHydrazones, Adv. Synth. Catal., 2019, 361, 1756 CrossRef CAS.
- D. Zhang, J. Cai, J. Du, X. Wang, W. He, Z. Yang, C. Liu, Z. Fang and K. Guo, Oxidant- and Catalyst-Free Synthesis of Sulfonated Benzothiophenes via Electrooxidative Tandem Cyclization, J. Org. Chem., 2021, 86, 2593–2601 CrossRef CAS PubMed.
- M. Singh, M. Kaur and O. Silakari, Flavones: An important scaffold for medicinal chemistry, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- L. Li, Q. Yang, Z. Jia and S. Luo, Organocatalytic Electrochemical CARBON-H2 Lactonization of Aromatic Carboxylic Acids, Synth, 2018, 50, 2924–2929 CrossRef CAS.
- A. Shao, Y. Gao, N. Li, J. Zhan, C. W. Chiang and A. Lei, Electrochemical Intramolecular C_H/O_H Cross-Coupling of 2-Arylbenzoic Acids, Chin. J. Chem., 2018, 36, 619–624 CrossRef CAS.
- X. Z. Tao, J. Dai, J. Zhou, J. Xu and H. J. Xu, Electrochemical C-O Bond Formation: Facile Access to Aromatic Lactones, Chem.–Eur. J., 2018, 24, 6932–6935 CrossRef CAS PubMed.
- S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Scalable Electrochemical DeH2ative Lactonization of C(sp2/sp3)-H Bonds, Org. Lett., 2018, 20, 252–255 CrossRef CAS PubMed.
- M. Xiong, X. Liang, X. Liang, Y. Pan and A. Lei, Hexafluoro-2-Propanol-Promoted Electro-Oxidative [3 + 2] Annulation of 1,3-Dicarbonyl Compounds and Alkenes, ChemElectroChem, 2019, 6, 3383–3386 CrossRef CAS.
- Z. Guan, Y. Wang, H. Wang, Y. Huang, S. Wang, H. Tang, H. Zhang and A. Lei, Electrochemical oxidative cyclization of olefiniccarbonyls with diselenides, Green Chem., 2019, 21, 4976–4980 RSC.
- J. Hua, Z. Fang, M. Bian, T. Ma, M. Yang, J. Xu, C. K. Liu, W. He, N. Zhu, Z. Yang and K. Guo, Electrochemical Synthesis of Spiro[4.5]trienones through Radical-Initiated Dearomative Spirocyclization, ChemSusChem, 2020, 13, 2053–2059 CrossRef CAS PubMed.
- A. Kharma, C. Jacob, I. A. O. Bozzi, G. A. M. Jardim, A. L. Braga, K. Salomao, C. C. Gatto, M. F. S. Silva, C. Pessoa, M. Stangier, L. Ackermann and E. N. da Silva Junior, Electrochemical Selenation/Cyclization of Quinones: A Rapid, Green and Efficient Access to Functionalized Trypanocidal and Antitumor Compounds, Eur. J. Org. Chem., 2020, 2020, 4474–4486 CrossRef CAS.
- X. Lin, Z. Fang, C. Zeng, C. Zhu, X. Pang, C. Liu, W. He, J. Duan, N. Qin and K. Guo, Continuous Electrochemical Synthesis of Iso-coumarin Derivatives from o-(1-Alkynyl) benzoates under MetalandOxidant-free, Chem.–Eur. J., 2020, 26, 13738–13742 CrossRef CAS PubMed.
- M. Stangier, A. M. Messinis, J. C. A. Oliveira, H. Yu and L. Ackermann, Rhodaelectro-catalyzed access to chromones via formyl CARBON-H2 activation towards peptide electro-labeling, Nat. Commun., 2021, 12, 4736 CrossRef CAS PubMed.
- Y. Wang, J. C. A. Oliveira, Z. Lin and L. Ackermann, Electrooxidative Rhodium-Catalyzed [5 + 2] Annulations via CARBON-H2/O-H Activations, Angew. Chem., Int. Ed., 2021, 60, 6419–6424 CrossRef CAS PubMed.
- M. Bian, J. Hua, T. Ma, J. Xu, C. Cai, Z. Yang, C. Liu, W. He, Z. Fang and K. Guo, Continuous-flow electrosynthesis of selenium substituted imino isobenzofuran via oxidative cyclization of olefinicamides and diselenides, Org. Biomol. Chem., 2021, 19, 3207–3212 RSC.
- Y. Qiu, W. J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Electrooxidative Rhodium-Catalyzed CARBON-H2/CARBON-H2 Activation: Electricity as Oxidant for Cross-DeH2ative Alkenylation, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- Y. Qiu, J. Struwe and L. Ackermann, Metallaelectro-Catalyzed CARBON-H2 Activation by Weak Coordination, Synlett, 2019, 30, 1164–1173 CrossRef CAS.
- T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Resource Economy by Metalla electrocatalysis: Merging Electrochemistry and CARBON-H2 Activation, Trends Chem., 2019, 1, 63–76 CrossRef CAS.
- Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Electrooxidative Ru-Catalyzed CARBON-H2/O-H Annulation by Weak O-Coordination, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed.
- R. Mei, C. Yang, F. Xiong, M. Mao, H. Li, J. Sun, L. Zou, W. Ma and L. Ackermann, Access to 10-Phenanthrenols via Electrochemical CARBON-H2/CARBON-H2 Arylation, Adv. Synth. Catal., 2021, 363, 1120–1125 CrossRef CAS.
- Y. Zhang, C. Ma, J. Struwe, J. Feng, G. Zhu and L. Ackermann, Electrooxidative dearomatization of biaryls: synthesis of tri- and difluoromethylated spiro[5.5] trienones, Chem. Sci., 2021, 12, 10092–10096 RSC.
- R. C. Samanta, T. H. Meyer, I. Siewert and L. Ackermann, Renewable Resources for Sustainable Metallaelectro-Catalysed C–H Activation, Chem. Sci., 2020, 11, 8657–8670 RSC.
- T. H. Meyer and L. Ackermann, Chemistry with Potential: PresentChallenges and Emerging Trends in Organic Electrocatalysis, Aldrichimica Acta, 2021, 54 Search PubMed.
- R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, Co-Catalyzed Oxidative CARBON-H2 Activation: Strategiesand Concepts, ChemSusChem, 2020, 13, 3306 CrossRef CAS PubMed.
- S. Santoro, A. Marrocchi, D. Lanari, L. Ackermann and L. Vaccaro, Towards Sustainable CARBON-H2 Functionalization Reactions: TheEmerging Role of Bio-Based Reaction Media, Chem.–Eur. J., 2018, 24, 13383–13390 CrossRef CAS PubMed.
- R. C. Samanta and L. Ackermann, Evolution of Earth-Abundant 3d-Metallaelectro-Catalyzed CARBON-H2 Activation: From Chelation-Assistanceto CARBON-H2 Functionalization without Directing Groups, Chem. Rec., 2021, 21, 2430–2441 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |