
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise and stereoselective synthesis of the BCDF tetracyclic ring system of C19-diterpenoid alkaloids†
Fangzhou Yu and
Liang Xu *
*
Key Laboratory of Drug Targeting and Drug Delivery Systems of the Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, P. R. China. E-mail: liangxu@scu.edu.cn
First published on 4th July 2024
Abstract
A new synthetic route for the BCDF tetracyclic ring system of C19-diterpenoid alkaloids (C19-DTAs) has been developed. The key step is a Pd-catalyzed transannular alkenylation that installs a functionalized bridged F ring. The overall strategy is concise and stereoselective, and it provides a valuable new tool for the synthesis of C19-DTAs. The synthesis begins with a bridged [3.2.1] ring system, which is converted to a key intermediate through a series of highly regio- and stereoselective processes. The introduction of an allylic side chain with high precision is accomplished, culminating in a Pd-catalyzed transannular alkenylation that installs a functionalized bridged F ring to yield the BCDF tetracyclic analog of C19-DTAs.
Introduction
Aconitine (1), first isolated by Manske in 1833 and its structure elucidated in 1959,1,2 is a prototypical C19 diterpenoid alkaloid (C19-DTAs). It is the major toxic component of over 250 species of Aconitum plants, commonly referred as monkshood or wolf's bane, which have long been used in traditional medicine.3 Aconitine's primary mode of action involves potent activation of sodium ion channels, preventing repolarization of muscles and neurons. As a result, it can induce ventricular arrhythmias, potentially leading to death.3 While aconitine's high toxicity has limited its clinical use, less toxic analogs and derivatives are emerging as promising candidates for the treatment of pain, inflammation, or arrhythmias.4 They share a common hexacyclic ring system (ABCDEF ring) and typically possess three to nine oxygen substituents such as hydroxy, methoxy, and acyloxy groups (Fig. 1). Their intricate structures and diverse biological activities have attracted considerable attention from synthetic chemists over the past five decades.5 As pioneers in the field, Wiesner et al. developed four generations of fundamental synthetic strategies for the practical synthesis of talatisamine, chasmanine, and 13-desoxydelphonine in the 1970s.6 In the last decade, research groups led by Gin,7 Sarpong,8 Fukuyama,9 Inoue10 and Reisman11 have made significant contributions, culminating in the elegant total syntheses of weisaconitine D, liljestrandinine, cardiopetaline, talatisamine and puberuline C. Recently, Qin and co-workers reported an ingenious total synthesis of vilmoraconitine by using an efficient hydrodealkenylative fragmentation/Mannich strategy.12 While other synthetic efforts exist,13 these remain the only eight natural C19 (C18) DTAs with successful total syntheses to date. Notably, all synthesized C19-DTAs or C18-DTAs possess only three to six oxygen functionalities. The total synthesis of C19-DTAs with a more oxygenated ring system, such as aconitine (1) with its unprecedented nine oxygen functionalities, remains a significant challenge.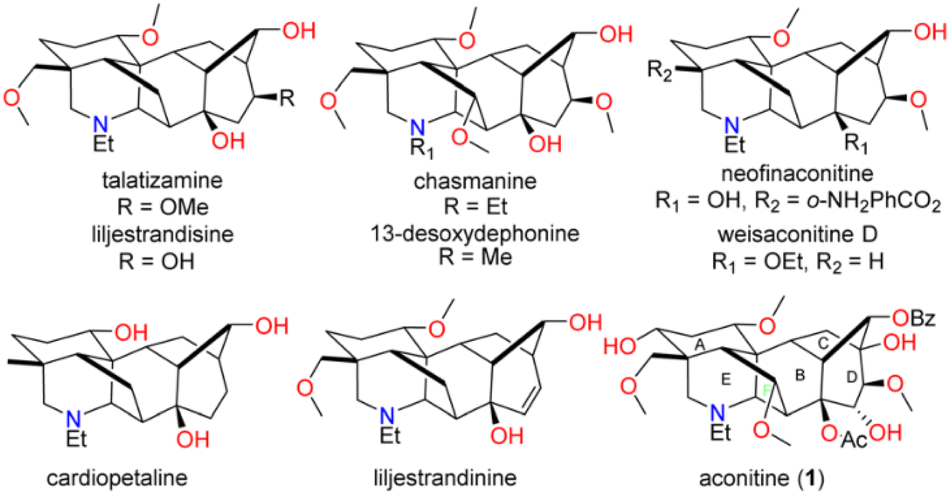 | ||
| Fig. 1 Structures of C18- or C19-diterpenoid alkaloids. | ||
Due to our long-standing research interest in the chemistry of C19-DTAs, we embarked on synthetic studies of aconitine and other bioactive C19-DTAs. In previous synthetic efforts, we have constructed several important ring systems, such as the AEF, ABEF, ABF, BCD and BCDE rings of C19-DTAs.14 In particular, we have previously utilized a cascade oxidative dearomatization/dimerization/retro-DA/IMDA reaction and the Wagner–Meerwein rearrangement as key steps to successfully construct the tricyclic [6.2.1.0] carbocyclic BCD ring analog, providing a potential approach for the construction of the bridged A, E, and F rings in aconitine.
However, the stereoselectivity for the preparation of the key rearrangement precursor was still undesirable and needed further improvement. Herein, we report further synthetic studies on the highly functionalized BCD ring analog 4 via an improved synthetic route that further introduced a functionalized bridged F ring via a Pd-catalyzed transannular alkenylation reaction to successfully afford the tetracyclic BCDF ring compound 2, which is an important model substrate intermediate for the total synthesis of aconitine.
We envisioned the synthesis of the tetracyclic aconitine BCDF ring analog 2 (Scheme 1). Initially, compound 3, featuring a [3.2.1] bridged ring structure, could be transformed from compound 2 through a transannular alkenylation reaction.15 Compound 2 could be obtained from ketone 4 via regioselective acylation and stereoselective alkylation reactions. Ketone 4, in turn, could be derived from the 5/6/6 tricyclic ketone 5 via a Wagner–Meerwein rearrangement.16 Finally, compound 5 could be synthesized in a single step through an intramolecular oxidative dearomatization/IMDA cascade reaction of phenol 6 with a dienophile side chain. Although we have previously synthesized analogous tricyclic compounds to 4, developing a rapid, highly stereoselective, and gram-scalable method for the synthesis of the BCD tricyclic compound 4 is still necessary to further expand our synthetic work.
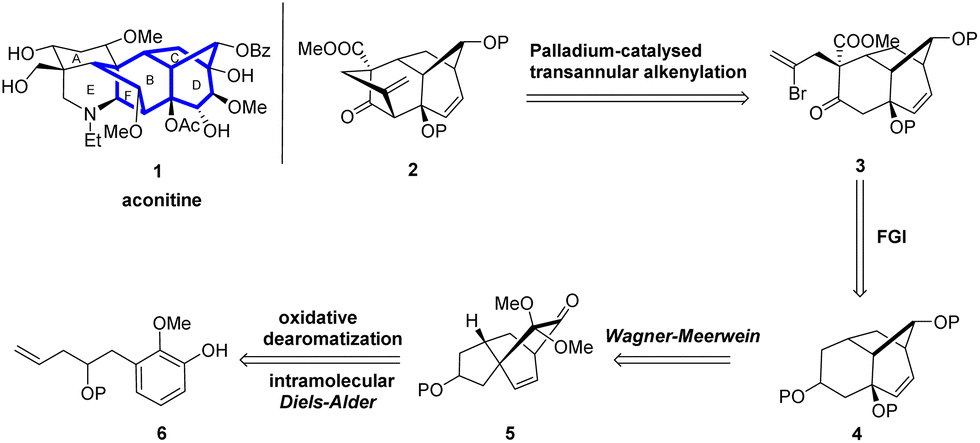 | ||
| Scheme 1 Synthetic plan for the BCDF ring system 2. | ||
Results and discussion
Our synthetic studies began with the preparation of vinyl arene 13 as shown in Scheme 2. First, commercially available 2,3-dihydroxybenzaldehyde 7 was converted on a 10 gram scale to 3-hydroxy-2-methoxybenzaldehyde 8 via a known regioselective phenolic hydroxyl–methylation reaction.17 Compound 8 was then converted to the TBS phenol ether 9 under standard conditions. Next, the Wittig reaction of compound 9 with a phosphonium salt generated the enol ether intermediate 10, which was subsequently subjected to in situ acid-promoted hydrolysis, affording the one-carbon homologated aldehyde 11 with an overall yield of 71% across two steps.18 Finally, aldehyde 11 was subjected to a Barbier allylation reaction19 with allyl bromide in the presence of Zn/NH4Cl to introduce the terminal olefin side chain, yielding highly functionalized phenol 12. Selective protection of the resulting homoallylic alcohol with TBS then afforded phenol 13.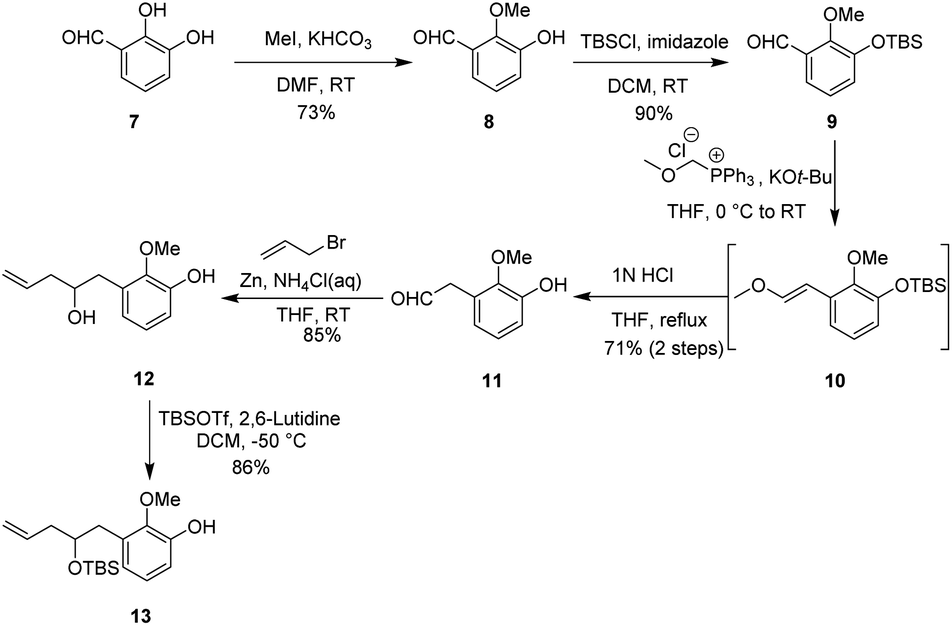 | ||
| Scheme 2 Synthesis of vinyl arene 13. | ||
After successfully obtaining vinyl phenol 13, we proceeded with the oxidative dearomatization/intramolecular Diels–Alder reaction (IMDA) and the subsequent planned Wagner–Meerwein rearrangement (Scheme 3). Fortunately, phenol 13 with bis(acetoxy)iodobenzene (PIDA) under standard oxidative conditions, uniquely produced the relatively stable o-quinone monoacetal 14 at 0 °C in methanol and, notably, the typically observed o-quinone monoacetal dimerization at room temperature20 did not occur. Rapid column chromatographic purification of compound 14, followed by reflux in toluene, easily afforded a separable mixture of endo diastereoisomers 15a and 15b, with a small amount of the undesired exo addition product (15%),21 achieving the expected endo stereoselectivity with a combined yield of 72% for the two steps. The α-stereochemistry of the OTBS substituent was determined in 15a by observing the NOE correlation between the vinyl proton signal (δ = 6.24, m) and the tert-butyl proton signal (δ = 0.88, s) of the OTBS substituent. Notably, 15a or 15b showed a resemblance in the position of the carbonyl and acetal substituents adjacent to the [2.2.2] bicyclooctane bridge portion with our previously synthesized tricyclic analog 15x,14 which could potentially enhance the stereoselectivity of the key Wagner–Meerwein rearrangement precursor. It was encouraging to find that the ketone carbonyl reduction of 15a proceeded exclusively from the more hindered β-face. This selectivity may have been influenced by the potential torsional steering effect of the neighboring gem-dimethoxy group.8 By using sodium borohydride (NaBH4) for the attack from the more crowded β-face, we were able to synthesize compound 16 with high stereoselectivity. Subsequent treatment with sodium hydride (NaH) and iodomethane (MeI) converted the newly formed methylene hydroxy group into the methoxy ether compound 17, achieving a 99% yield. After removal of the dimethyl ketal functional group under acidic conditions to give the ketone 18, a stereoselective reduction with NaBH4 occurred from the β-side again (due to the steric hindrance of the α-side OMe substituent), leading to the formation of alcohol 19 with the hydroxy group oriented to the α-position in a combined yield of 82% in two steps. Subsequently, by activating 19 with trifluorometha-nesulfonic anhydride (Tf2O) in DCM and using methanol as nucleophile, the key Wagner–Meerwein rearrangement was performed in a one-pot manner without the need to isolate the potential intermediate triflate, directly yielded the allyl methyl ether isomers 20a and 20b in yields of 72% and 8%, respectively.22 We attribute the predominance of 20a as the major product to the absence of a strained bridgehead double bond. In contrast, 20b has a strained bridgehead olefin, making its formation less favored. When water is used instead of methanol to trap the allyl carbocation, the C8-hydroxy substituted rearranged product is generated, along with a small amount of the allyl isomer, although in lower yield.
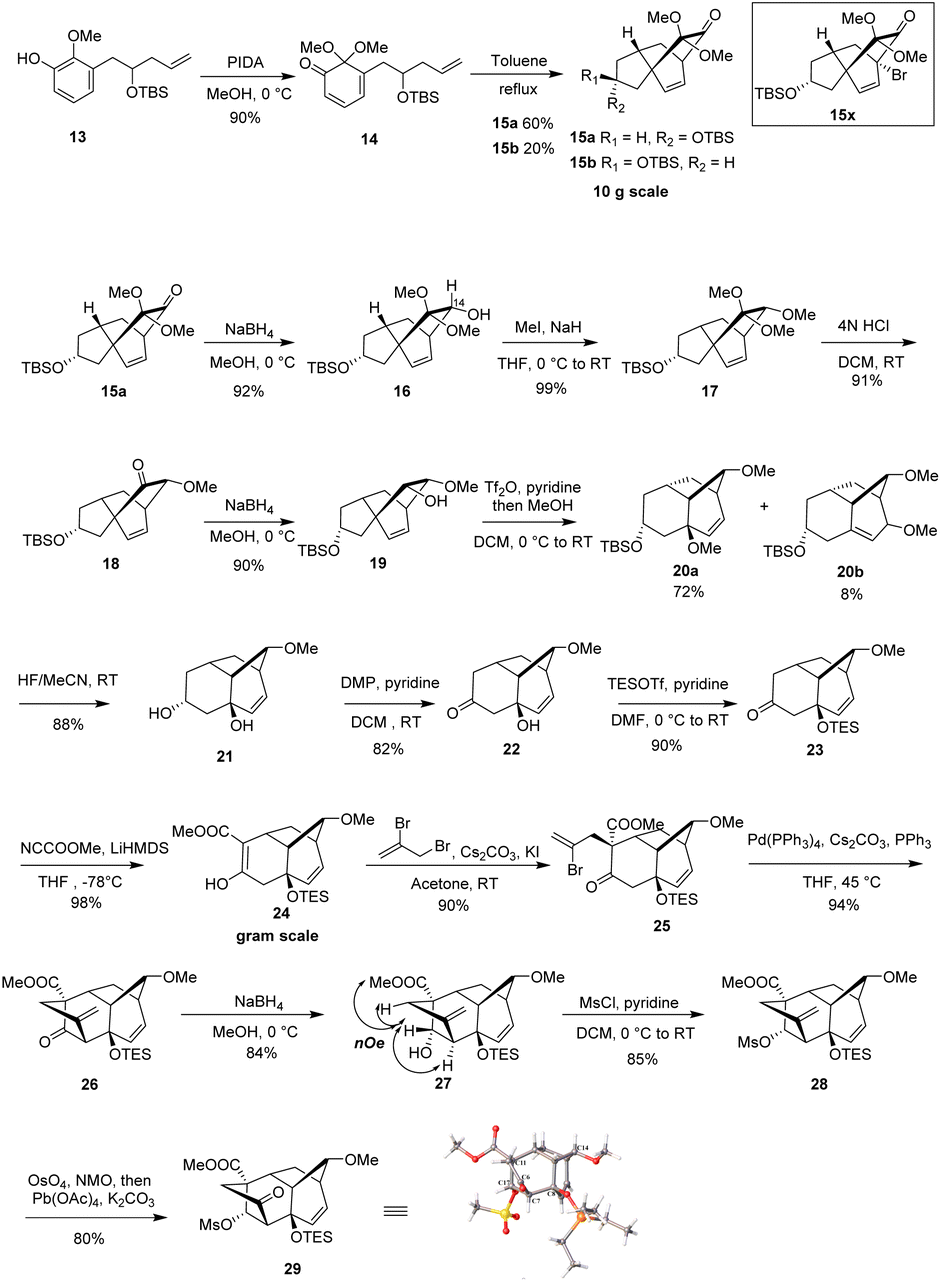 | ||
| Scheme 3 Synthesis of BCDF analogue 29. | ||
After successfully synthesizing bicyclo [3.2.1] octane vinyl ether isomers 20a and 20b, we investigated their further transformation into the BCDF tetracyclic target molecule. Treatment of a mixture of 20a and 20b with 40% aqueous HF resulted in both TBS deprotection and removal of the allylic methyl ether, forming the diol 21. This demonstrates that the vinyl isomer 20b can also generate the same allylic alcohol under acidic conditions, probably due to the formation of a more stable allylic tertiary carbocation. The addition of the hydroxy group is controlled by the relative stability of the olefin isomers. Subsequent oxidation of the C-17 secondary alcohol in 21 with Dess–Martin periodinane (DMP) afforded the alcohol–ketone 22. After TES protection of the exposed C-8 tertiary alcohol, deprotonation of ketone 23 with lithium hexamethyldisilazane (LiHMDS) and subsequent introduction of a methoxycarbonyl group using Mander's reagent23 (methyl cyanoformate) afforded enol ester 24 in high regioselectivity (98% yield) due to the steric hindrance caused by the OTES group at the adjacent C-7 position. C-Alkylation of enol ester 24 at C-11 with 2,3-dibromopropene to introduce the F ring side chain initially yielded significant O-alkylation by-products. However, the addition of KI as an additive24 facilitated the regioselective formation of the C-alkylated product 25 in 90% yield. Based on HSAB25 theory, this improved regioselectivity is attributed to the in situ replacement of the allyl bromide atom with a more nucleophilic iodide ion, forming a softer allyl iodine intermediate that preferentially reacts with the carbon nucleophile. Although the stereochemistry of the newly installed allylic side chain was initially unclear, subsequent experiments revealed an 11β configuration, likely due to the greater accessibility of the exo face of the ester enolate intermediate. Transannular alkenylation26 with Pd(PPh3)4 efficiently completed the F ring construction, affording the BCDF tetracyclic product 26. Initially, low yields were observed, we speculated this may attributed to the degradation of catalyst Pd(PPh3)4 during the reaction. Adding PPh3 ligand to the reaction inhibited ligand dissociation from the Pd(0) center, ultimately leading to a successful 94% yield under milder conditions. Finally, reduction of the C-17 ketone in 26 with NaBH4 provided the β-hydroxy compound 27 in a highly stereoselective manner. NOE experiments on 27 confirmed the 11β configuration of the allyl side chain, as evidenced by the spatial correlations between the C-17 hydroxy methine protons and both the C-5 methylene and ester methoxy methylene protons. The whole stereochemical structure of 27 was further unambiguously established on the basis of the X-ray crystallography analysis of its ketone derivative 29, which was prepared by sequential sulfonylation of hydroxyl group to give 28 and oxidative cleavage of the exocyclic olefin by treatment with osmium tetraoxide/NMO followed by lead tetraacetate.
Conclusions
In summary, we have developed an efficient synthetic route that, for the first time, assembles the tetracyclic BCDF ring analog of C19-DTAs in 14 steps with an overall yield of 10.8%. This synthetic route is based on the preparation of the gram-scale tricyclic BCD compound 24, with key steps including the highly stereoselective reduction of the ketone intermediate 15a, followed by a Wagner–Meerwein rearrangement, then introduce an allylic side chain with high regioselectivity and stereoselectivity. Finally, a Pd-catalyzed transannular alkenylation was used to further introduce a functionalized bridged F-ring, efficiently providing the BCDF tetracyclic analog of C19-DTAs. Further experiments are in progress.Data availability
The data supporting this article have been included as part of the ESI. Crystallographic data for compound 29 has been deposited at the CCDC under [accession number 2362943] and can be obtained from https://deposit.ccdc.cam.ac.uk/.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant Numbers 21472129 and 21272163).Notes and references
- M. Przybylska and L. Marion, Can. J. Chem., 1959, 37, 1116 CrossRef CAS; K. Wiesner, D. L. Simmons and L. R. Fowler, Tetrahedron Lett., 1959, 18, 1 CrossRef.
- F. P. Wang and Q. H. Chen, The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, 2010, p. 1 Search PubMed.
- Y. Fujita, K. Terui, M. Fujita, A. Kakizaki, N. Sato, K. Oikawa, H. Aoki, K. Takahashi and S. Endo, J. Anal. Toxicol., 2007, 31, 132 CrossRef CAS PubMed.
- A. Ameri, Prog. Neurobiol., 1998, 56, 211 CrossRef CAS PubMed; M. Stevens, S. Peigneur and J. Tytgat, Front. Pharmacol, 2011, 2, 71 Search PubMed; B. Borcsa, L. Fodor, D. Csupor, P. Forgo, A. t. Molnar and J. Hohmann, Planta Med., 2014, 80, 231 CrossRef PubMed.
- M. Brimble, K. Goodall and D. Barker, Synlett, 2005, 2005, 1809 CrossRef CAS; A. M. Hamlin, J. K. Kisunzu and R. Sarpong, Org. Biomol. Chem., 2014, 12, 1846 RSC; B. Liu, G. Zhu and R. Liu, Synthesis, 2015, 47, 2691 CrossRef; X. Y. Liu and Y. Qin, Synlett, 2015, 4, 1010 Search PubMed.
- K. Wiesner, T. Tsai, K. Huber, S. Bolton and R. Vlahov, J. Am. Chem. Soc., 1974, 96, 4990 CrossRef CAS; K. Wiesner, Pure Appl. Chem., 1975, 41, 93 CrossRef; S.-F. Lee, G. M. Sathe, W. W. Sy, P.-T. Ho and K. Wiesner, Can. J. Chem., 1976, 54, 1039 CrossRef; K. Wiesner, T. Y. Tsai and K. P. Nambiar, Can. J. Chem., 1978, 56, 1451 CrossRef; K. Wiesner, Pure Appl. Chem., 1979, 51, 689 CrossRef.
- Y. Shi, J. T. Wilmot, L. U. Nordstrom, D. S. Tan and D. Y. Gin, J. Am. Chem. Soc., 2013, 135, 14313 CrossRef CAS PubMed.
- C. J. Marth, G. M. Gallego, J. C. Lee, T. P. Lebold, S. Kulyk, K. G. Kou, J. Qin, R. Lilien and R. Sarpong, Nature, 2015, 528, 493 CrossRef CAS PubMed; K. G. M. Kou, S. Kulyk, C. J. Marth, J. C. Lee, N. A. Doering, B. X. Li, G. M. Gallego, T. P. Lebold and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 13882 CrossRef PubMed.
- Y. Nishiyama, S. Yokoshima and T. Fukuyama, Org. Lett., 2016, 18, 2359 CrossRef CAS PubMed; Y. Nishiyama, S. Yokoshima and T. Fukuyama, Org. Lett., 2017, 19, 5833 CrossRef PubMed.
- K. Hagiwara, T. Tabuchi, D. Urabe and M. Inoue, Chem. Sci., 2016, 7, 4372 RSC; D. Kamakura, H. Todoroki, D. Urabe, K. Hagiwara and M. Inoue, Angew. Chem., Int. Ed., 2020, 59, 479 CrossRef CAS PubMed; T. Shimakawa, S. Nakamura, H. Asai, K. Hagiwara and M. Inoue, J. Am. Chem. Soc., 2023, 145(1), 600–609 CrossRef PubMed.
- A. R. Wong, N. J. Fastuca, V. W. Mak, J. K. Kerkovius, S. M. Stevenson and S. E. Reisman, ACS Cent. Sci., 2021, 7, 1311 CrossRef CAS PubMed.
- J. Ji, J. Chen, S. Qin, W. Li, J. Zhao, G. Li, H. Song, X. Y. Liu and Y. Qin, J. Am. Chem. Soc., 2023, 145, 3903 CrossRef CAS PubMed.
- R. M. Conrad and J. D. B. RM Conrad, Org. Lett., 2007, 9, 5465 CrossRef CAS PubMed; Z.-K. Yang, Q.-H. Chen and F.-P. Wang, Tetrahedron Lett., 2011, 67, 4192 CrossRef; K. J. Goodall, M. A. Brimble and D. Barker, Tetrahedron Lett., 2012, 68, 5759 CrossRef; T. Tabuchi, D. Urabe and M. Inoue, J. Org. Chem., 2016, 81, 10204 CrossRef PubMed; X.-H. Zhou, Y. Liu, R.-J. Zhou, H. Song, X.-Y. Liu and Y. Qin, Chem. Commun., 2018, 54, 12258 RSC; R.-J. Zhou, G.-Y. Dai, X.-H. Zhou, M.-J. Zhang, P.-Z. Wu, D. Zhang, H. Song, X.-Y. Liu and Y. Qin, Org. Chem. Front., 2019, 6, 377 RSC; Z. Meng, F. Mi, F. Lu, W. Tan, X.-Y. Liu and Y. Qin, Chin. Chem. Lett., 2020, 31, 1903 CrossRef.
- H. Cheng, F.-H. Zeng, D. Ma, M.-L. Jiang, L. Xu and F.-P. Wang, Org. Lett., 2014, 16, 2299 CrossRef CAS PubMed; X. Yang, B. Cheng, H. Cheng, L. Xu and J.-L. Wang, Chin. Chem. Lett., 2017, 28, 1788 CrossRef; M. Liu, C. Cheng, W. Xiong, H. Cheng, J.-L. Wang and L. Xu, Org. Chem. Front., 2018, 5, 1502 RSC; Z. Lv, L. Gao, C. Cheng, W. Niu, J. L. Wang and L. Xu, Chem.–Asian J., 2018, 13, 955 CrossRef PubMed; S.-Y. Cao, H.-J. Yue, M.-Q. Zhu and L. Xu, Org. Chem. Front., 2020, 7, 933 RSC; T. Guo, F. Peng, X. Song, J. Lei, F. Yu, H. Chu, K. Yang and L. Xu, Org. Chem. Front., 2023, 10, 675 RSC.
- D. Bankston, F. Fang, E. Huie and S. Xie, J. Org. Chem., 1999, 64, 3461 CrossRef CAS PubMed; M. Oestreich, P. R. Dennison, J. J. Kodanko and L. E. Overman, Angew. Chem., Int. Ed., 2001, 40, 1439 CrossRef PubMed; A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef PubMed; L. A. Arnold, W. Luo and R. K. Guy, Org. Lett., 2004, 6, 3005 CrossRef PubMed.
- For examples of the transformation of the bicyclo[2.2.2] to [3.2.1] skeleton through the Wagner–Meerwein rearrangement, see: S. P. Sethi, K. S. Atwal, R. M. Marini-Bettolo, T. Y. R. Tsai and K. Wiesner, Can. J. Chem., 1980, 58, 1889–1891 CrossRef CAS; M. Asaoka and H. Takei, Tetrahedron Lett., 1987, 28, 6343–6346 CrossRef; W. G. Earley, J. E. Jacobsen, A. Madin, G. P. Meier, C. J. O’donnell, T. Oh, D. W. Old, L. E. Overman and M. J. Sharp, J. Am. Chem. Soc., 2005, 127, 18046–18053 CrossRef PubMed; F. Leonelli, F. Blesi, P. Dirito, A. Trombetta, F. Ceccacci, A. L. Bella, L. M. Migneco and R. M. Bettolo, J. Org. Chem., 2011, 76, 6871–6876 CrossRef PubMed; L. Q. Song, G. L. Zhu, Y. J. Liu, B. Liu and S. Qin, J. Am. Chem. Soc., 2015, 137, 13706–13714 CrossRef PubMed; C. J. Marth, G. M. Gallego, J. C. Lee, T. P. Lebold, S. Kulyk, K. G. Kou, J. Qin, R. Lilien and R. Sarpong, Nature, 2015, 528, 493 CrossRef PubMed; X. Y. Liu, F. P. Wang and Y. Qin, Acc. Chem. Res., 2021, 54, 22–34 CrossRef PubMed.
- D. L. Boger, J. Hong, M. Hikota and M. Ishida, J. Am. Chem. Soc., 1999, 121, 2471 CrossRef CAS; A. N. Lowell, P. D. Wall, S. P. Waters and M. C. Kozlowski, Tetrahedron, 2010, 66, 5573 CrossRef.
- L. Fitjer and U. Quabeck, Synth. Commun., 1985, 15, 855 CrossRef CAS.
- C. Petrier and J. L. Luche, J. Org. Chem., 1985, 50, 910 CrossRef CAS.
- Y.-K. Chen, R. K. Peddinti and C.-C. Liao, Chem. Commun., 2001, 15, 1340 RSC; S. K. Chittimalla, S. K. Chittimalla, H.-Y. Shiao, H.-Y. Shiao, C.-C. Liao and C.-C. Liao, Org. Biomol. Chem., 2006, 4, 2267 RSC; H. Cheng, F. H. Zeng, X. Yang, Y. J. Meng, L. Xu and F. P. Wang, Angew. Chem., Int. Ed., 2015, 128, 400 CrossRef CAS.
- A small amount of the undesired exo addition product 15c (ca. 15%, see the ESI†) was isolated in the reaction mixture, and the two desired OTBS substituted diastereomers 15a and 15b could be used as a mixture to transform into the ketone intermediates 22. For clarity of the NMR spectra, the single major isomer 15a was separated from 15b to perform subsequent transformation.
- I. Fernández, F. P. Cossío and M. A. Sierra, Chem. Rev., 2009, 109, 6687 CrossRef CAS PubMed; O. Gutierrez and D. J. Tantillo, J. Org. Chem., 2012, 77, 8845 CrossRef PubMed; C. L. Hugelshofer and T. Magauer, Nat. Prod. Rep., 2017, 34, 228 RSC.
- L. N. Mander and S. P. Sethi, Tetrahedron Lett., 1983, 24, 5425 CrossRef CAS.
- B. Hegedüs and A. F. Krassó, Helv. Chim. Acta, 2004, 53, 959 CrossRef CAS; K. C. Nicolaou, P. K. Sasmal and H. Xu, J. Am. Chem. Soc., 2004, 126, 5493 CrossRef PubMed.
- N. Kornblum, R. A. Smiley, R. K. Blackwood and D. C. Iffland, J. Am. Chem. Soc., 1955, 77, 6269 CrossRef CAS; R. G. Pearson and J. Songstad, J. Am. Chem. Soc., 1967, 89, 1827 CrossRef.
- J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed., 2015, 54, 400 CrossRef CAS PubMed; S. Z. Jiang, X. Y. Zeng, X. Liang, T. Lei, K. Wei and Y. R. Yang, Angew. Chem., Int. Ed., 2016, 55, 4044 CrossRef PubMed; G. Li, X. Xie and L. Zu, Angew. Chem., Int. Ed., 2016, 55, 10483 CrossRef PubMed; W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3500 CrossRef PubMed; M. Jarret, A. Tap, C. Kouklovsky, E. Poupon, L. Evanno and G. Vincent, Angew. Chem., Int. Ed., 2018, 57, 12294 CrossRef PubMed; S. Pan, S. Chen and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 6333 CrossRef PubMed; X. Zhang, B. N. Kakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed., 2019, 58, 6053 CrossRef PubMed; A. Turlik, Y. Chen, A. C. Scruse and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 8088–8092 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. CCDC 2362943. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02821j |
| This journal is © The Royal Society of Chemistry 2024 |