
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ho3+ codoping of GGAG:Ce: a detailed analysis of acceleration of scintillation response and scintillation efficiency loss†
Juraj Páterek *ab,
Pavel Boháčeka,
Bohumil Trundaa,
Vladimir Babina,
Richard Švejkarb,
Karel Jureka,
Jan Rohlíčeka and
Martin Nikl*a
*ab,
Pavel Boháčeka,
Bohumil Trundaa,
Vladimir Babina,
Richard Švejkarb,
Karel Jureka,
Jan Rohlíčeka and
Martin Nikl*a
aInstitute of Physics, Czech Academy of Sciences, Cukrovarnická 10, Prague, Czech Republic. E-mail: paterek@fzu.cz; nikl@fzu.cz
bFaculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Břehová 7, Prague, Czech Republic
First published on 22nd July 2024
Abstract
In this study, we investigate the effects of Ho3+ codoping on the luminescence and scintillation properties of GGAG:Ce, with a particular focus on timing properties and scintillator efficiency. The research reveals that Ho3+ codoping and subsequent resonant energy transfer from Ce3+ to Ho3+ can significantly reduce the 5d1 excited state decay time of Ce3+ and shorten scintillation pulses of GGAG:Ce registered by using photomultipliers, although this reduces scintillator efficiency as well. The study presents a detailed analysis of the loss of scintillator efficiency due to Ho3+ codoping, identifying the most significant loss pathways and estimating their impact. The findings suggest that Ho3+ codoping is an effective method for accelerating the scintillation response of GGAG:Ce. Furthermore, the study presents a high level of consistency of the Ce3+ kinetics with the Inokuti–Hirayama model and with results obtained in the previous studies on similar systems, demonstrating the predictability of the effect of RE3+ codoping on scintillator properties.
Introduction
Codoping with trivalent rare-earth ions (RE3+) was proven to be an effective method for shortening of the activator decay time and scintillation response in Ce3+/Pr3+ activated garnet scintillators. This has been demonstrated in our previous studies on Er3+ and Ho3+ codoping of YAG:Ce (Ce3+ doped yttrium aluminum garnet),1,2 Ho3+ codoping of LuAG:Pr (Pr3+ doped lutetium aluminum garnet)3 and other RE3+ codoping of garnets.4,5The acceleration of the activator decay is enabled by resonant energy transfer (RET). This effect involves transition of one of the centers (donor) to a lower energy state and simultaneous promotion of another distant center (acceptor) to a higher energy state. The mechanism of RET is depicted in Fig. 1.
 | ||
| Fig. 1 Schematic of RET mechanism directed from donor (D, asterisk indicates excited state) to acceptor center (A). Radiative transitions – donor excitation and acceptor emission – drawn with solid line, relaxation to lower energy drawn with dotted line, resonant ET drawn with dashed line. | ||
Unlike reabsorption, where energy emitted in the form of a photon by one center and absorbed by another, the RET does not include the formation of a photon. It is driven by multipole electro–magnetic interaction. RET is enabled between luminescence centers in resonance, i.e. centers whose emission and absorption spectra overlap. Rate of RET is proportional to the overlap of the emission spectra of the donor fD,em and absorption spectra of the acceptor fA,abs and is inversely proportional to the power of the distance between ions R
 | (1) |
| ktot = kinh + krET | (2) |
The downside of the RE3+ codoping is reduction of the donor emission and subsequently scintillator efficiency. Same as shortening of the donor decay time, reduction of the activator emission is caused by RET, hence inevitable. In this sense, the Ho3+ acceptor can be considered a killer center for Ce3+ 5d → 4f emission. Then, using the model for number of UV/visible photons Nph generated per energy of incident radiation E derived in6–8
 | (3) |
Ho3+ ions have been found to be particularly effective acceptor codopants for Ce3+ and Pr3+ activated garnets as they enable acceleration of the Ce3+/Pr3+ decay time due to RET without introducing any additional signal to the detection spectrum or introducing slow components of light, see ref. 1 and 3.
Multiple RE3+ ions other than Ho3+ could be used as acceptor in pair with Ce3+ donor in GGAG, i.e. would reduce the decay time of the 5d state of Ce3+ due to RET. According to ref. 10 and 11, they are Pr3+, Nd3+, Pm3+, Sm3+, Eu3+, Tb3+, Dy3+, Er3+ and Tm3+. However, as shown in our previous studies for Er3+,1,2 Dy3+,3 and Nd3+ (ref. 4) and studies of other authors for Sm3+,12 Eu3+ and Tb3+,13 and Tm3+ (ref. 14) unlike Ho3+ all of them have parity-forbidden 4f → 4f emission positioned in the range of Ce3+ emission which would introduce slow components into detectable emission when using common photomultipliers or even Si-based semiconductor photodetectors. This is counterproductive to the effect of shortening of the scintillation response and would unavoidably lead to impaired timing properties of the scintillator. The situation is specific for Pr3+. This ion is typically used as an activator of garnet scintillators for its fast 5d → 4f emission positioned in UV range but emits also between 480 and 650 nm due to 4f → 4f transitions.15 However in case of Pr3+ codoping of GGAG:Ce, its 5d → 4f emission transition would transfer energy into the Gd sublattice which diminishes fast scintillation response,16 while, the 4f → 4f transitions would remain active and introduce slow light to detectable signal same as the RE3+ ions above. Pm3+ is not considered due to low practical use of this element due to absence of stable isotope.
This study builds on upon these previous findings by examining Ho3+ codoping of GGAG:Ce,Mg (gadolinium aluminum gallium garnet doped with Ce3+ and Mg2+). GGAG:Ce is a representative of multicomponent garnets compounds of general chemical formula of the host (Gd,Lu,Y)3(Al,Ga)5O12. They have been reported firstly in the ceramic form17,18 and their enormously high scintillation light yield up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 phot per MeV and excellent energy resolution of 4.8%@662 keV immediately interested researchers in scintillator field. These materials can be prepared also in single crystal form, most frequently reported by Czochralski technique where even 4 inch diameter large crystals have been achieved.19 Another preparation techniques, e.g. floating zone has also been reported.20 High entropy alloys in multicomponent garnet family were also studied which was fueled by an interest to find unusual stable compositions with unique properties21 and combinatorial research strategy was applied as well.22 Effects of composition and growth parameters on phase formation in multicomponent aluminum garnet crystals was systematically studied.23 Luminescence investigation focused on the interplay between the Ce3+ luminescence center and the host due to decreasing ionization barrier of the Ce3+ 5d1 excited state,24,25 traps states acting in scintillation mechanism were studied by thermoluminescence techniques.26 In the study of scintillation characteristics special attention was paid to stabilization of Ce4+ by stable divalent dopants as Mg2+ or Ca2+ which creates new fast radiative recombination pathway at Cerium centers and accelerates noticeably the scintillation response.27–30 Other codopants were studied for this purpose as well.31 Dependence of the bandgap value on the host composition was also studied32,33 and garnet compounds luminescence and scintillation characteristics were reviewed in ref. 34. The application potential of multicomponent garnets for fast timing application in medical imaging and high energy physics was evaluated in ref. 35 and 36.
000 phot per MeV and excellent energy resolution of 4.8%@662 keV immediately interested researchers in scintillator field. These materials can be prepared also in single crystal form, most frequently reported by Czochralski technique where even 4 inch diameter large crystals have been achieved.19 Another preparation techniques, e.g. floating zone has also been reported.20 High entropy alloys in multicomponent garnet family were also studied which was fueled by an interest to find unusual stable compositions with unique properties21 and combinatorial research strategy was applied as well.22 Effects of composition and growth parameters on phase formation in multicomponent aluminum garnet crystals was systematically studied.23 Luminescence investigation focused on the interplay between the Ce3+ luminescence center and the host due to decreasing ionization barrier of the Ce3+ 5d1 excited state,24,25 traps states acting in scintillation mechanism were studied by thermoluminescence techniques.26 In the study of scintillation characteristics special attention was paid to stabilization of Ce4+ by stable divalent dopants as Mg2+ or Ca2+ which creates new fast radiative recombination pathway at Cerium centers and accelerates noticeably the scintillation response.27–30 Other codopants were studied for this purpose as well.31 Dependence of the bandgap value on the host composition was also studied32,33 and garnet compounds luminescence and scintillation characteristics were reviewed in ref. 34. The application potential of multicomponent garnets for fast timing application in medical imaging and high energy physics was evaluated in ref. 35 and 36.
In this paper, to better understand the mechanisms behind the acceleration of scintillation response due to RE3+ codoping, its benefits and drawbacks, we examine not only direct effects of Ho3+ codoping on scintillation characteristics of GGAG:Ce,Mg like decay time and light yield (LY), but also investigate the effect of Ho3+ codoping on specific stages of scintillation mechanism in detail. Further, findings obtained in this, and previous studies are compared and discussed and build up the picture of the RE3+ codoping for modification of scintillation properties as a method in general.
Experimental methods
Electron probe microanalysis (EPMA) analysis was performed using JEOL JXA-733 microprobe. Crystal structure was examined by powder X-ray diffraction pattern analysis (XRD) measured at powdered small piece of the samples using the Bragg–Brentano focusing configuration on the powder diffractometer Empyrean of PANalytical (λCu, Kα = 1.54184 Å) that was equipped with a fixed divergent slit and PIXcel3D detector. 120 minutes long measurements were made from 4 to 100° 2θ with 0.013° step size and 300 s per step. Absorption spectra were measured with a Shimadzu 3101 PC spectrometer. A Horiba Jobin Yvon 5000M spectrofluorimeter equipped with a TBx-04 photon counting detector was used for the steady-state spectral measurements and measurement of the photoluminescence decay. Excitation was performed with a Seifert tungsten X-ray tube (40 kV, 15 mA) and an Heraus deuterium lamp for radioluminescence and photoluminescence spectroscopy, respectively. All the spectra were corrected for the spectral distortions of the setup. The photoluminescence decay kinetics of the Ce3+ center were measured by a time-correlated single photon counting method37 with a Horiba NanoLED nanosecond excitation source. The scintillation decay curves were obtained with use of 137Cs γ-ray excitation, Tektronix TDS3052C digital phosphor oscilloscope, and a fast photomultiplier Hamamatsu R7207-01 working in current regime. Amplitude spectra for LY measurement38,39 were obtained with a shaping time of 1 μs, 137Cs γ-ray excitation, and a hybrid photomultiplier Photonis PP0475B. All before mentioned measurements were performed at room temperature. Thermally stimulated luminescence (TSL) was measured in range 77–700 K with heating rate 0.1 K s−1. Temperature of the sample was regulated with Janis N2 VPF-800 cryostat. Initially, sample was irradiated with X-ray (40 kV, 15 mA) for 10 minutes at 77 K. Then, spectrally unresolved TSL glow curves were recorded using IBH Scotland TBx-04 photomultiplier in the photon counting mode and 1 s sampling rate. Photoluminescence and scintillation decay kinetics were analyzed using iterative least-square re-convolution method40 and Python packages LMfit41 and SciPy.42Results and discussion
Preparation and composition analysis of the samples
A set of six GGAG crystals was prepared by the Czochralski method43 from melts with starting compositions Gd2.9844−xCe0.015Mg0.0006HoxGa2.7Al2.3O12, with x = 0.00, 0.015, 0.030, 0.045, 0.090 and 0.150. Platelets of the thickness of 1 mm were prepared from the tip parts of the crystal's, see Fig. 2. As the Mg2+ codoping has no effect on the Ce3+–Ho3+ energy transfer process, the materials will be referred only as GGAG:Ce, or Ho3+ codoped GGAG:Ce in the texts below, even though they contain the Mg2+ dopant as well. | ||
| Fig. 2 Photography of GGAG:Ce crystals codoped with various concentrations of Ho3+. The reddish tint is due to Ho3+ codoping of the crystals. | ||
Actual concentrations of the Ce3+ and Ho3+ dopants were determined using the EPMA and absorption spectroscopy. At first, the concentration of Ce3+ and Ho3+ was measured using EPMA for sample x = 0.045 providing 0.18 and 1.97 at% (expressed as a percentage of Gd atoms replaced by the dopant), respectively. Concentration of the dopants in the remaining samples was determined using the integrals of absorption peaks,44 namely the 4f → 5d1 transition for Ce3+ (390–510 nm), 5I8 → 5S2 + 5F4 (520–561 nm) and 5I8 → 5F5 (626–674 nm) transitions for Ho3+. Concentrations of Ce3+ dopant were found at the value of 0.18 ± 0.02 at% for all the samples. For Ho3+ the values of 0.00, 0.13, 0.59, 1.97, 3.56 and 6.21 at% were found. Concentrations of Mg2+ were too low to be measured by EPMA, i.e. lower than 0.01 at%.
XRD analysis confirmed single garnet phase in all the samples (see example in Fig. S2†) with the exception of the highest Ho concentration one, i.e. GGAG:Ce with 6.21 at% of Ho3+, see Fig. S1 in ESI.† In this sample, the secondary phase of the same garnet structure with a little bigger lattice constant was found. Its content (estimated from XRD analysis) is less then 5wt%. EPMA analysis of the secondary phases islands, see Fig. S3,† showed it is most probably due to reduced content of Ho3+ in the secondary phase.
Given the volume of the secondary phase in GGAG:Ce with 6.21 at% of Ho3+ its effect on the studied energy transfer phenomena is considered negligible. Further details on XRD and EPMA analysis are provided in ESI.†
Acceleration of Ce3+ decay time and scintillation properties due to Ho3+ codoping
The effect of Ho3+ codoping of GGAG:Ce was examined using multiple spectroscopic methods. Results of the experiments and discussion of the findings are described in the following paragraphs. First, overlap of the Ce3+ emission and Ho3+ absorption spectra, that is a prerequisite for RET, were studied using photoluminescence steady-state spectroscopy and absorption spectroscopy. Absorption and photoluminescence spectra (excited by 440 nm) of GGAG:Ce and Ho3+ codoped GGAG:Ce crystals are shown in Fig. 3. The non-codoped GGAG:Ce crystal shows typical absorption bands of Ce3+ allowed 4f → 5d1 and 4f → 5d2 transitions at 440 and 340 nm, a wide absorption band below 340 nm induced by charge transfer (CT) absorption band of Ce4+, that is induced by Mg2+ codoping in Ce3+ activated garnets45 and absorption lines of parity forbidden 4f → 4f transition of Gd3+ at around 275 and 310 nm.46 The same absorption patterns are observed in Ho3+ codoped crystals as well. In addition to that, multiple sets of narrow absorption lines of parity forbidden 4f → 4f transitions from Ho3+ ground state 5I8 to 5G6 and 5F1 around 449 nm, 5F3 and 5F2 and 3K8 around 486 nm, 5S2 and 5F4 around 538 nm and 5F5 around 636 nm excited states can be observed. Multiple Ho3+ sets of absorption lines are located below 440 nm as well. For more detailed information on UV/VIS spectrum refer to ref. 47, which reports optical transitions of Ho3+ in structurally similar YAG. Photoluminescence spectra of both non-codoped and Ho3+ codoped GGAG:Ce are dominated by wide Ce3+ 5d → 4f emission band ranging between 450 and 720 nm. In line with,47 that states the emission of Ho3+ in garnet matrix is positioned in the IR spectrum, no Ho3+-related emission is observed in the UV/VIS region. Ce3+ emission bands in Ho3+ codoped GGAG:Ce crystals are deformed due to re-absorption of emitted light by overlapping Ho3+ absorption lines. The same spectral overlap fulfills the prerequisite for RET. | ||
| Fig. 3 Photoluminescence (excitation to 440 nm) and absorption spectra of non-codoped GGAG:Ce and Ho3+ codoped GGAG:Ce shows the spectral overlap. | ||
The time-resolved PL spectroscopy of Ho3+ codoped GGAG:Ce crystals was used to investigate changes of the Ce3+ decay kinetics due to Ho3+ codoping and related RET, see Fig. 4. Time-resolved PL spectra of Ce3+ decays with excitation to 455 nm and emission 530 nm were recorded and reveals substantial acceleration of Ce3+ decay in Ho3+ codoped and the fact the acceleration is proportional to Ho3+ content. The acquired decay curves were fitted to Inokuti–Hirayama (IH) model for donor luminescence kinetics. Assuming homogenous distribution of the donor and acceptors centers through the crystal, decay kinetics of the donor center I(t) will obey the following
 | (4) |
 , where Γ(x) is the gamma function, c is the concentration of the acceptor center and c0 is critical concentration of the acceptor, i.e. the concentration of acceptor that yields rate of RET equal to the rate of the inherent decay rate of the donor, i.e. krET = kinh. The best match was achieved for s = 6, which refers to dipole–dipole interaction between Ce3+ and Ho3+. In line with presumptions of Inokutu–Hirayama model, parameter α is proportional to Ho3+ content, see the inset of Fig. 4. Linearity of the relation between parameter α and Ho3+ content was used to determine the critical concentration of Ho3+ in GGAG:Ce to 4.6 at%. The calculated 1/e decay time of Ce3+ center was shortened from 56 ns for the non-codoped GGAG:Ce to 8 ns for the GGAG:Ce codoped with 6.2 at% of Ho3+. Refer to Table 1 for all calculated 1/e decay times.
, where Γ(x) is the gamma function, c is the concentration of the acceptor center and c0 is critical concentration of the acceptor, i.e. the concentration of acceptor that yields rate of RET equal to the rate of the inherent decay rate of the donor, i.e. krET = kinh. The best match was achieved for s = 6, which refers to dipole–dipole interaction between Ce3+ and Ho3+. In line with presumptions of Inokutu–Hirayama model, parameter α is proportional to Ho3+ content, see the inset of Fig. 4. Linearity of the relation between parameter α and Ho3+ content was used to determine the critical concentration of Ho3+ in GGAG:Ce to 4.6 at%. The calculated 1/e decay time of Ce3+ center was shortened from 56 ns for the non-codoped GGAG:Ce to 8 ns for the GGAG:Ce codoped with 6.2 at% of Ho3+. Refer to Table 1 for all calculated 1/e decay times.
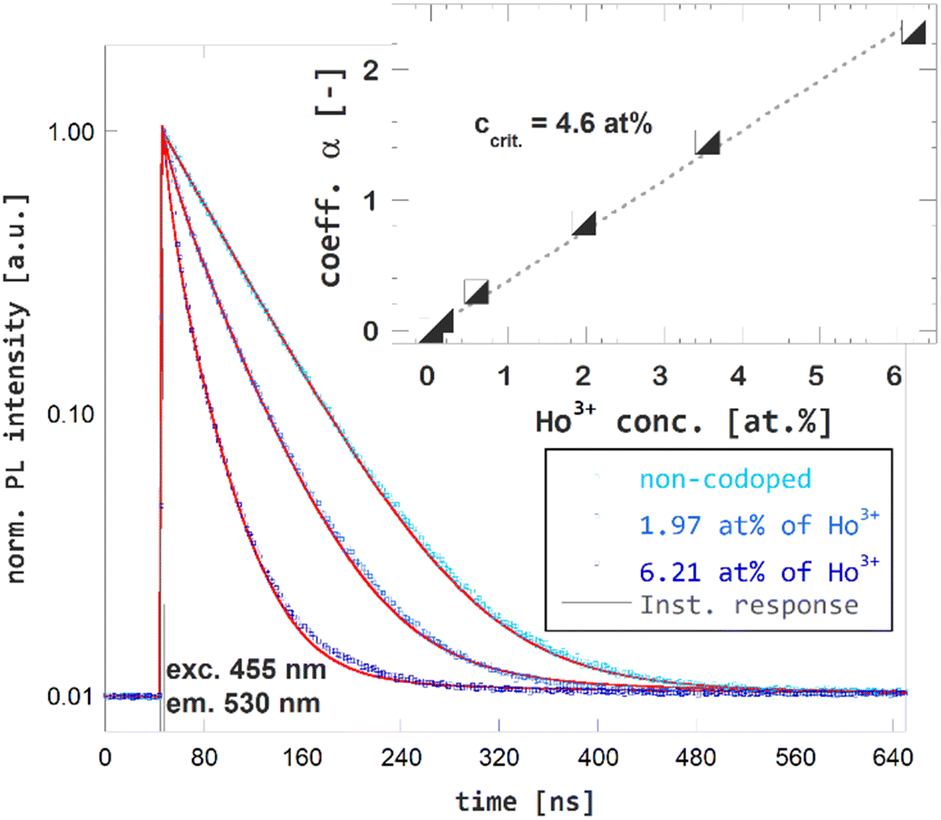 | ||
| Fig. 4 Photoluminescence decay kinetics of Ce3+ (excitation 455 nm, emission 530 nm) in non-codoped GGAG:Ce and Ho3+ codoped GGAG:Ce. The red lines show the IH model fit of the data. | ||
| Ho3+ conc. [at%] | PL τ1/e [ns] | Rel. PL τ1/e [%] | SC τ1/e [ns] | Rel. SC τ1/e [%] | Rel. LY [%] | Rel. eff. [%] |
|---|---|---|---|---|---|---|
| 0.00 | 56.1 | 100 | 90.3 | 100 | 100 | 100 |
| 0.13 | 51.9 | 92 | 105.5 | 117 | 98 | 111 |
| 0.59 | 41.8 | 74 | 82.3 | 91 | 75 | 89 |
| 1.97 | 25.2 | 45 | 53.1 | 59 | 46 | 59 |
| 3.56 | 14.7 | 26 | 36.4 | 40 | 28 | 40 |
| 6.21 | 7.9 | 14 | 25.2 | 28 | 15 | 22 |
In the next paragraph, a comparison of the Ho3+-codoping of GGAG:Ce and YAG:Ce grown by edge-defined growth method, that was investigated in our previous study,1 will be discussed. As the both matrices (GGAG and YAG) are structurally very similar and the same donor–acceptor pair was used in the studies analogical effects of the Ho3+-codoping are expected. In both cases, RET is enabled by overlap of the wide Ce3+ 5d → 4f emission band and Ho3+ absorption lines related to parity-forbidden 4f → 4f transitions and a good match of Ce3+ PL kinetics with IH model was achieved. The Ce3+–Ho3+ interaction is of dipole–dipole type, in both systems. The critical concentration of Ho3+ in GGAG:Ce was found just slightly higher when compared to 4.4 at% found for Ho3+ in YAG:Ce. The difference in critical concentration can be accounted by either of two following explanations or their combination. First, the lattice parameters increase when Y and Al are substituted by Gd and Ga, respectively – lattice parameter increase from 12 to 12.21 and 12.55 Å for Y3Al5O12, Gd3Al5O12 and Gd3Ga5O12, respectively.48 Hence, higher content of Ho3+ acceptor is required to achieve the mean distance between donor–acceptor pairs to be critical distance in GGAG:Ce. Second, the actual and nominal values of Ho3+ content in Ho3+ codoped YAG:Ce crystals may vary, as the nominal values refer to content of Ho3+ in melt.
Consistency of results observed in structurally similar, but not identical GGAG and YAG matrices, grown by different methods and a good match of the measured PL decay curves with the IH model in both cases make the Ho3+-codoping well predictable method for tuning of Ce3+ luminescence kinetics. It is necessary to say, the application of the studied method is not restricted to RE3+ codoping of Ce3+ and Pr3+ activated garnets, but can be universally applied to any family of matrices and combination of donor–acceptor pairs. The only condition is the resonance between the donor and acceptor transitions.
The effect of Ho3+-codoping of GGAG:Ce on its scintillation kinetics was studied using time-resolved spectroscopy of scintillation pulses. The scintillation decay curves for non-codoped GGAG:Ce and Ho3+ codoped GGAG:Ce crystals are presented in Fig. 5. The effect of shortening Ce3+ decay time due to Ho3+ codoping is evident in the scintillation response as well. The 1/e decay time dropped from 90 ns for non-codoped GGAG:Ce to 25 ns for the GGAG:Ce with 6.2 at% of Ho3+ codopant. Refer to Table 1 for 1/e scintillation decay times of all examined crystals.
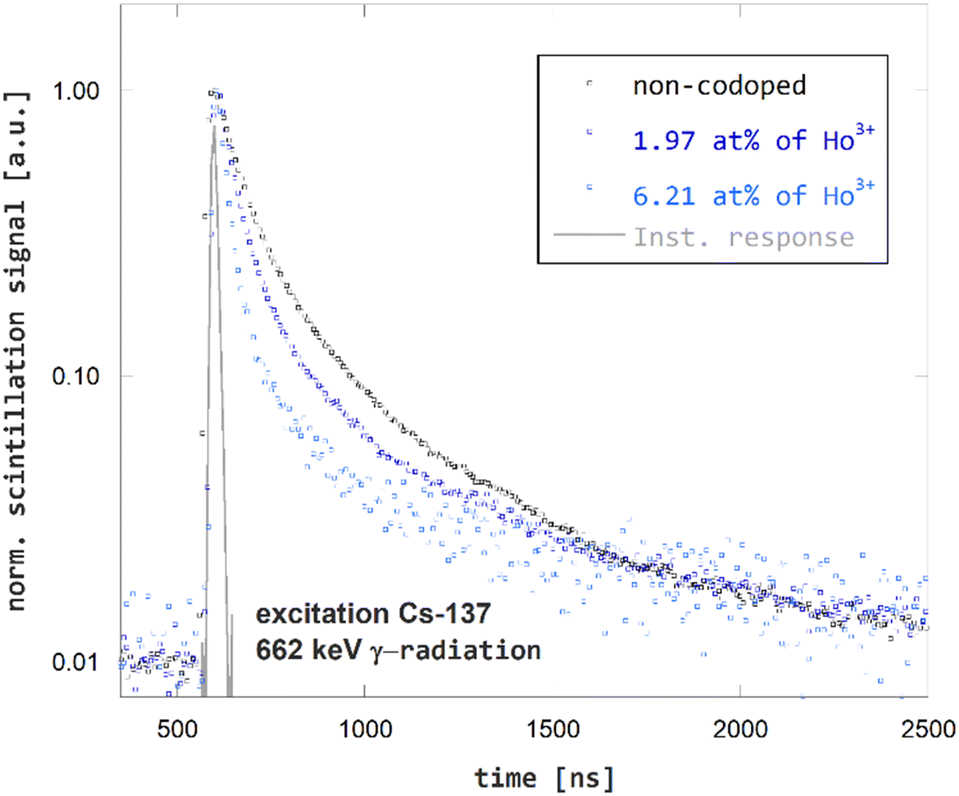 | ||
| Fig. 5 Scintillation decay kinetics of non-codoped GGAG:Ce and Ho3+ codoped GGAG:Ce excited by 137Cs γ-radiation. | ||
RL spectra confirm the expected trend of overall efficiency decrease in Ho3+ codoped GGAG:Ce crystals, see the Fig. 6, the concentration dependence of RL spectra integrals in the inset of this figure and Table 1 for listed values of relative overall efficiency (compared to that non-codoped GGAG:Ce). In general, the overall efficiency decreases with Ho3+ concentration. For the GGAG:Ce with the highest content of Ho3+ codopant the RL spectrum integral drops to 22% of the non-codoped GGAG:Ce. The only deviation from the decreasing trend can be seen for the crystal codoped with 0.13 at% of Ho3+ which shows a bit superior RL intensity than the non-codoped GGAG:Ce.
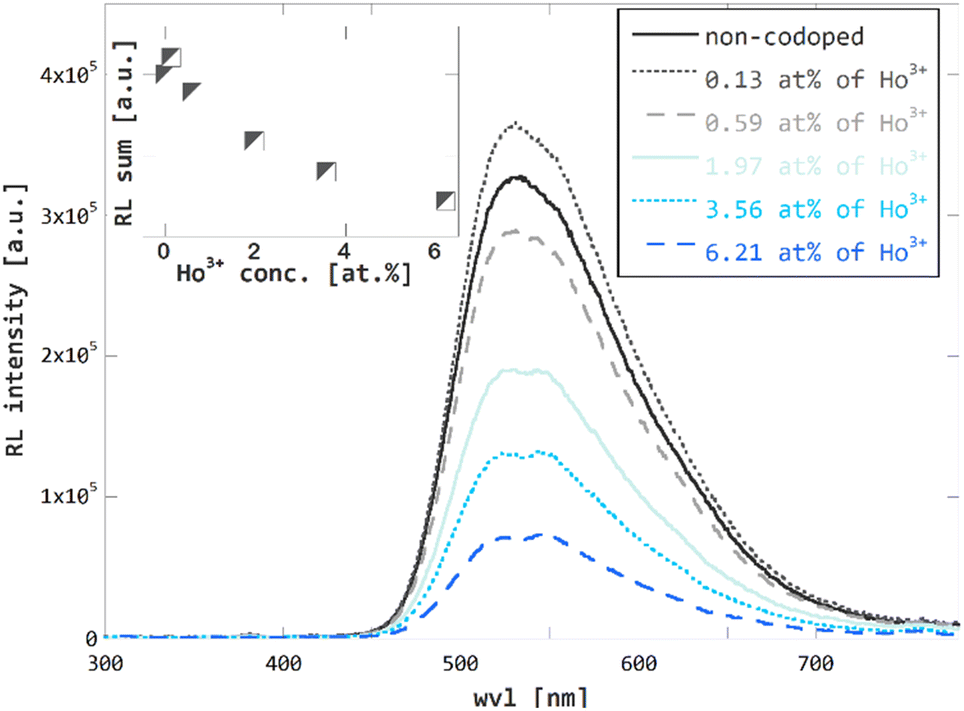 | ||
| Fig. 6 Radioluminescence (40 kV, 15 mA X-rays) spectra of non-codoped GGAG:Ce and Ho3+ codoped GGAG:Ce. Radioluminescence spectra integrals plotted against Ho3+ codopant concentration in the inset. | ||
The same crystal deviates from the decreasing trend of scintillation 1/e scintillation decay time with value greater by 17% than that of the non-codoped crystal. No such pattern is observed in the trend of 1/e photoluminescence decay time or LY measurement, i.e. measurements that are mostly determined by the fast components of the scintillation pulses. Due to these facts, the deviation of RL and 1/e scintillation decay time is attributed to higher contribution of the slow components, probably due to increased content of traps and defects in the crystal.
In line with our observations from previous studies on Ho3+ codoped YAG:Ce1 and LuAG:Pr,3 no or just negligible Ho3+-related emission in the UV-VIS region is observed in RL spectra of Ho3+ codoped GGAG:Ce. According to ref. 47, the emission of Ho3+ centers in YAG host is located in IR region. The same behavior is anticipated for Ho3+ in GGAG. As described above, this makes Ho3+ the ideal codopant as it does not introduce any slow components into the detectable light when usual photomultipliers are used as photodetectors. Another advantage of using Ho3+ codoping, or RE3+ codoping in general, for acceleration of scintillation response using RET in garnet hosts is the expected homogeneous distribution of codopant along the crystal due to very favorable segregation coefficient close to 1.49 This ensures rather homogeneous scintillation characteristics in all the volume of the crystal grown. Another, recently published50 acceleration mechanism in heavily doped GAGG:Ce,Mg crystals which is based on luminescence quenching in the Ce–Mg pairs, is much more problematic in this respect.
Analogously to RL spectroscopy, the amplitude spectroscopy of scintillation pulses confirms the expected decreasing trend of LY in the Ho3+ codoped GGAG:Ce crystals as well. A decrease of LY is proportional to Ho3+ concentration. For the highest content of Ho3+ LY drops to 15% when compared to that of the non-codoped GGAG:Ce crystal. The data for LY are summarized in Table 1.
The above findings show two effects of Ho3+ codoping on scintillation properties of on GGAG:Ce. The first is shortening of the scintillation pulses. In terms of 1/e scintillation decay time, the Ho3+ codoping can reduce this quantity by tens of percent in GGAG:Ce which improves timing properties of the scintillator, enabling e.g. increased detection rate. The second effect of Ho3+ codoping is the decrease of scintillator efficiency. In general, decrease of scintillator efficiency is unfavorable as it leads to impaired performance of the material, e.g. impaired energy resolution. In terms of LY, the rate of decrease is slightly higher than that of 1/e scintillation decay time in GGAG:Ce. One can think of RE3+-codoping as a method that enables trading scintillator efficiency for faster scintillation decay. Both measures are put into perspective in Fig. 7 which compares relative LY and scintillation 1/e decay time τ1/e. The values are listed in Table 1.
 | ||
| Fig. 7 Scintillation 1/e decay time τ1/e and LY of Ho3+ codoped GGAG:Ce plotted against Ho3+ codopant concentration in relative scale. | ||
Energy loss pathways due to Ho3+ codoping and their quantification
As shown in the previous section, the Ho3+ codoping of GGAG:Ce induces simultaneously an acceleration of scintillation pulses and loss of scintillator efficiency. This is due to interference of Ho3+ codopant in multiple stages of the scintillation mechanism. The second part of this study is focused on examination of the origin of losses of scintillator efficiency caused by Ho3+ codoping. The main loss pathways are identified and their contribution in overall loss of scintillator efficiency is estimated. The estimations are further compared to the experimental LY data.We identify following energy loss pathways within the scintillation mechanism of GGAG:Ce caused by Ho3+ codoping interferes:
(a) Degradation of the crystal quality due to high concentration of Ho3+ codoping. Introduction of new element, especially if introduced in high concentrations can make the crystal growth unstable, introduce new type of defects, and cause overall impairment of the crystal quality.
(b) Charge carriers capture on Ho3+ centers during the transport stage of scintillation process. Ho3+, same as Ce3+ and other RE3+ ions, creates recombination centers that capture the electrons and holes during the transport stage of the scintillation conversion mechanism. Once electrons and holes are trapped on Ho3+ center, they will slowly deexcite through the dense structure of Ho3+ excited states producing photons in IR region, outside detection range of used photosensitive elements of scintillation detectors. As a result, the amount of energy delivered to Ce3+ centers and used for generation of detectable scintillation photons is reduced by the part captured on Ho3+ codopant and the scintillator efficiency is impaired.
(c) Resonant energy transfer from Ce3+ to Ho3+. This effect enables shortening of the Ce3+ decay time. At the same time, it consumes part of the energy which would be emitted by Ce3+ in form of scintillation photons in absence of Ho3+ codopant but is resonantly transferred to the Ho3+ and consequently emitted in the IR region, i.e. technically lost, as described above.
(d) Reabsorption of Ce3+ emitted light by Ho3+. RET is enabled via overlap of Ce3+ emission and Ho3+ absorption peaks which inevitably enables not only non-radiative (resonant), but also radiative transfer of energy, i.e., part of the Ce3+ emitted photons is reabsorbed by Ho3+ codopant as shown in Fig. 3.
Assume lx, such as 0 ≤ lx ≤ 1, is an estimated loss of scintillator efficiency induced due to one of the described energy loss pathways due to Ho3+ codoping, and fx = 1 − lX is multiplication factor representing scintillator efficiency after accounting the effect of the specific energy loss pathway. Then, if η0 is the efficiency of the non-codoped crystal, the efficiency of the Ho3+ codoped crystal can be estimated as
| η = fdegfCCfRETfreabsη0 | (5) |
 | (6) |
Experimentally obtained values of LY of the non-codoped and Ho3+ codoped GGAG:Ce crystals can be used as independent reference to validate estimates of the scintillation efficiency η/η0.
To assess the effect of Ho3+ codoping of GGAG:Ce on overall crystal quality, mainly impairment due to introduction of new defects due to Ho3+ codoping, spectrally unresolved TSL measurement was performed. See Fig. 8 for the glow curve of the non-codoped GGAG:Ce and GGAG:Ce codoped with 3.6 at% of Ho3+. Both the glow curves are composed of TSL peaks with the maxima at the same temperatures, although their contribution differs in the non-codoped and Ho3+ codoped crystal. No additional TSL peaks referring to a new type of defects due to Ho3+ codoping are observed in Ho3+ codoped crystal. Therefore, loss of efficiency due to degradation of crystal quality and additional traps is considered negligible and the related multiplication factor fdeg is set 1 for all Ho3+ codoped crystals.
 | ||
| Fig. 8 Spectrally unresolved TSL glow curves of non-codoped and Ho3+ codoped GGAG:Ce measured after 10 minutes of irradiation with 40 kV/15 mA X-rays at 77 K and 0.1 K s−1 heating rate. | ||
The amount of energy resonantly transferred from Ce3+ donor to Ho3+ acceptor, i.e. the loss of efficiency due to RET, is proportional to a difference of integrals of the Ce3+ decay curves in the non-codoped and Ho3+ codoped crystal. The multiplication factor fRET is then estimated as
 | (7) |
| Ho3+ conc. [at%] | fdeg | fCC | fRET | freabs | η/η0 | Rel. LY |
|---|---|---|---|---|---|---|
| 0.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 0.13 | 1.00 | 1.00 | 0.94 | 0.99 | 0.93 | 0.98 |
| 0.59 | 1.00 | 1.00 | 0.77 | 0.98 | 0.76 | 0.75 |
| 1.97 | 1.00 | 1.00 | 0.51 | 0.97 | 0.50 | 0.46 |
| 3.56 | 1.00 | 1.00 | 0.32 | 0.97 | 0.31 | 0.28 |
| 6.21 | 1.00 | 1.00 | 0.19 | 0.79 | 0.15 | 0.15 |
To estimate the loss of efficiency due to reabsorption of Ce3+ emitted light on Ho3+, the obtained photoluminescence spectra, shown in Fig. 3, were used. The loss is proportional to size of the reabsorption dips observed in the photoluminescence spectra of the Ho3+ codoped GGAG:Ce. The multiplication factor freabs is estimated as
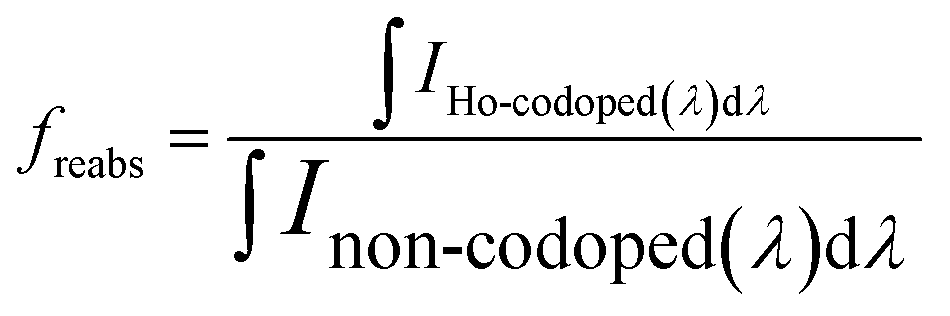 | (8) |
Summarizing the estimates of efficiency loss due to impaired crystal quality, RET, reabsorption on Ho3+ and the relative values of measured LY, we assume the loss of efficiency due to charge carrier capture by Ho3+ are very low or negligible.
In fact, if the multiplicative factor for charge losses due to charge carrier capture fCC is assumed to be 1, we obtain a solid match between the resulting estimate of relative efficiency η/η0 and independently measured relative LY values, see Fig. 9. In case, we assumed the loss of efficiency due to charge carrier capture non-zero, i.e. fCC < 1, the resulting estimate of relative efficiency η/η0 would only deviate from the experimental LY data. Hence the efficiency losses due to charge carrier capture are considered negligible and related multiplicative factor fCC is estimated to 1 for all concentrations of Ho3+.
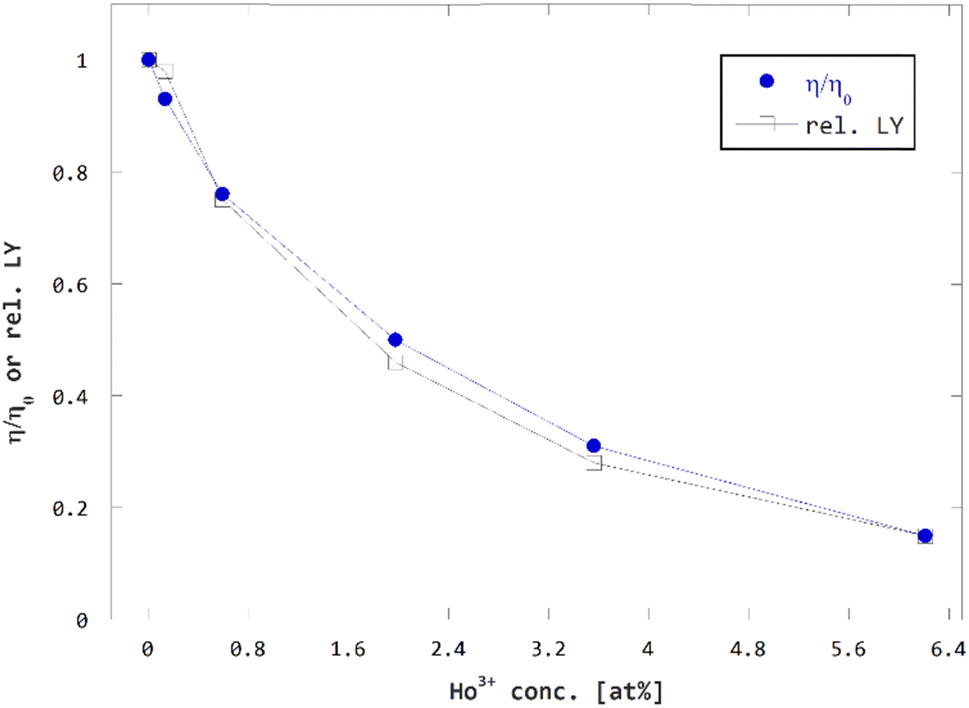 | ||
| Fig. 9 Comparison of concentration dependence of estimated relative efficiency η/η0 and measured relative LY. | ||
The estimations of losses of scintillator efficiency performed in the previous section helped to reveal more on the impact by Ho3+ codoping of GGAG:Ce on specific stages of scintillation mechanism.
The investigation shows by far the greatest part of the losses of scintillator efficiency are due to the RET from Ce3+ to Ho3+, the same process that is causing the acceleration of Ce3+ decay time. A smaller part of losses of scintillation efficiency is due to other means. In other words, most of the lost scintillator efficiency was used for the purpose of the method, i.e. acceleration of the activator decay time. This makes Ho3+ codoping of GGAG:Ce very effective method for modification of timing properties of scintillation response.
Minor losses of efficiency are caused by reabsorption of Ce3+ emitted light by Ho3+ centers. The losses might change for a different geometry of a crystal, however, even for large crystals the losses due to reabsorption should saturate at certain level as Ho3+ absorption lines are overlapping just a part of the broad emission band of Ce3+ and part of the Ce3+ emitted light would remain not absorbed. The losses due to reabsorption of Ce3+ emitted light on Ho3+ do not contribute to shortening of the Ce3+ decay time.
Interestingly, the results also suggest that Ho3+ does not interfere with charge carriers during the transport stage of scintillation conversion in GGAG:Ce, or, in other words, the Ho3+ ability to capture charge carriers is very low when compared to Ce3+, even if the concentration of Ho3+ is an order of magnitude higher. This could be attributed to Mg2+ codoping, that induces formation of Ce4+ centers that are more effective in capturing electrons when compared to Ce3+ capturing holes.
Conclusions
In this study the effect of Ho3+ codoping on GGAG:Ce luminescence and scintillation properties were investigated with the focus on timing properties and scintillator efficiency and the impact of the Ho3+ codoping on different stages of scintillation mechanism of GGAG:Ce. Our results show the Ho3+ codoping and the related resonant energy transfer from Ce3+ to Ho3+ can lead to significant reduction of the Ce3+ 5d1 excited state decay time and shortening scintillation pulses of GGAG:Ce. At the same time scintillator efficiency is reduced as well. We found the 1/e scintillation decay time can be reduced by tens of percent, while light yield decreases by an equivalent amount when using Ho3+ codoping in GAGG:Ce.Moreover, we showed that the Ho3+, unlike other RE3+as Dy3+, Er3+ or Nd3+, is favorable choice of codopant for the examined method due to absence of slow 4f → 4f emission in the UV/VIS region and thus absence of slow components in the detectable light when using usual photomultipliers.
We showed, the emission kinetics of the Ce3+ donor in Ho3+ codoped GGAG:Ce can be consistently described with the Inokuti–Hirayama model. Consistency with this model was observed also in our previous studies on various donor–acceptor pairs and matrices. Furthermore, we showed the method provides consistent results for both GGAG and YAG matrix and crystal growth method when doped by Ce3+–Ho3+ donor–acceptor pairs. Both the consistency with this model and consistency of the results for similar matrices show the effect of RE3+ codoping on scintillator properties is reliably predictable.
Further, losses of the scintillator efficiency due to Ho3+ codoping were analyzed in the detail. The most significant loss-of-efficiency pathways were identified, and their share on total loss of scintillator efficiency was estimated based on the experimental results. The major losses are attributed to the resonant energy transfer from Ce3+ donor to Ho3+ acceptor, i.e. the same mechanism that shortens Ce3+ decay time. Small part of the losses is due to Ho3+ reabsorption of Ce3+ emission and the losses due other pathways are negligible. Thus, we showed the Ho3+ codoping of GAGG:Ce is an effective method for acceleration of its scintillation response, as the largest part of scintillation efficiency losses are due to acceleration itself, not due to effects associated with Ho3+ codoping that do not accelerate the scintillation response.
The unprecedented advantage of the examined method is that it can be applied right away on many existing materials. Its use is not limited to garnet matrices or selection of the Ce3+–Ho3+ donor–acceptor pair, but can be applied to any family of matrices and combination of donor–acceptor pairs that meet the resonance criteria.
Data availability
Data are available upon request from the corresponding authors.Author contributions
Juraj Páterek: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft; Pavel Boháček: crystal growth, resources, writing – review & editing; Bohumil Trunda: crystal growth, resources; Vladimír Babin, Richard Švejkar, Karel Jurek, Jan Rohlíček: investigation; Martin Nikl: funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Czech Science Foundation under Grant No. 21-17731S.References
- J. Páterek, M. Pokorný, S. Sýkorová, A. Stehlík, J. Polák and J. Houžvička, et al., Ho3+ codoping of YAG:Ce: Acceleration of Ce3+ decay kinetics by energy transfer, J. Lumin., 2019, 213, 469–473 CrossRef.
- M. Pokorný, J. Páterek, M. Nikl, S. Sýkorová, A. Stehlík and J. Polák, et al., Concentration dependence of energy transfer Ce3+→Er3+ in YAG host, Opt. Mater., 2018, 86, 338–342 CrossRef.
- J. Páterek, R. Král, J. Pejchal, R. Prokeš and M. Nikl, LuAG:Pr codoped with Ho3+: Acceleration of Pr3+ decay by energy transfer, Radiat. Meas., 2019, 124, 122–126 CrossRef.
- J. Paterek, Acceleration of Scintillation Decay in Single Crystal Y3Al5O12:Ce Scintillators by Codoping, Diploma thesis, Czech Technical University, Prague, Czech Republic, 2017.
- S. Sykorova, J. Páterek, M. Pokorný, R. Kučerková, J. Houžvička, M. Nikl, et al., in Luminescence, Scintillation and Energy Transfer in the Doubly Doped LuAG:Pr,Dy Single Crystal, Chamonix, France, 2017, available from https://indico.cern.ch/event/388511/contributions/2612863/ Search PubMed.
- D. J. Robbins, On Predicting the Maximum Efficiency of Phosphor Systems Excited by Ionizing Radiation, J. Electrochem. Soc., 1980, 127(12), 2694–2702 CrossRef CAS.
- A. Lempicki, A. J. Wojtowicz and E. Berman, Fundamental limits of scintillator performance, Nucl. Instrum. Methods Phys. Res., Sect., 1993, 333(2–3), 304–311 CrossRef CAS.
- P. A. Rodnyi, P. Dorenbos and C. W. E. van Eijk, Energy Loss in Inorganic Scintillators, Phys. Status Solidi B, 1995, 187(1), 15–29 CrossRef CAS.
- K. Han, J. Qiao, S. Zhang, B. Su, B. Lou and C. Ma, et al., Band Alignment Engineering in n s 2 Electrons Doped Metal Halide Perovskites, Laser Photonics Rev., 2023, 17(1), 2200458 CrossRef CAS.
- G. H. Dieke and R. A. Satten, Spectra and Energy Levels of Rare Earth Ions in Crystals, Am. J. Phys., 1970, 38(3), 399–400 CrossRef.
- W. T. Carnall, H. Crosswhite and H. M. Crosswhite, Energy level structure and transition probabilities in the spectra of the trivalent lanthanides in LaF3, Report No.: ANL-78-XX-95, 6417825, 1978, available from: http://www.osti.gov/servlets/purl/6417825/, cited 2018 Oct 1 Search PubMed.
- T. Kunikata, K. Watanabe, P. Kantuptim, T. Kato, D. Nakauchi and N. Kawaguchi, et al., Radioluminescence properties of Sm3+-doped Y3Al5O12 single crystals, Nucl. Instrum. Methods Phys. Res., Sect. B, 2024, 546, 165172 CrossRef CAS.
- J. C. A. Santos, E. P. Silva, D. V. Sampaio, N. R. S. Souza, Y. G. S. Alves and R. S. Silva, Radioluminescence emission of YAG:RE laser-sintered ceramics, Mater. Lett., 2015, 160, 456–458 CrossRef CAS.
- Y. Fujimoto, M. Sugiyama, T. Yanagida, S. Wakahara, S. Suzuki and S. Kurosawa, et al., Comparative study of optical and scintillation properties of Tm3+:YAG, and Tm3+:LuAG single crystals, Opt. Mater., 2013, 35(11), 2023–2026 CrossRef CAS.
- M. Nikl, H. Ogino, A. Krasnikov, A. Beitlerova, A. Yoshikawa and T. Fukuda, Photo- and radioluminescence of Pr-doped Lu3Al5O12 single crystal, Phys. Status Solidi A, 2005, 202(1), R4–R6 CrossRef CAS.
- V. Babin, M. Nikl, K. Kamada, A. Beitlerova and A. Yoshikawa, Effect of the Pr 3+ → Gd 3+ energy transfer in multicomponent garnet single crystal scintillators, J. Phys. D: Appl. Phys., 2013, 46(36), 365303 CrossRef.
- N. J. Cherepy, S. A. Payne, B. W. Sturm, S. P. O'Neal, Z. M. Seeley, O. B. Drury, et al., in Performance of Europium-Doped Strontium Iodide, Transparent Ceramics and Bismuth-Loaded Polymer Scintillators, ed. Franks L. A., James R. B. and Burger A., San Diego, California, USA, 2011, p. 81420W, available from: http://proceedings.spiedigitallibrary.org/proceeding.aspx?doi=10.1117/12.896656, cited 2020 Jan 16 Search PubMed.
- O. B. Drury, N. J. Cherepy, T. A. Hurst and S. A. Payne, Garnet scintillator-based devices for gamma-ray spectroscopy, in 2009 IEEE Nuclear Science Symposium Conference Record (NSS/MIC), IEEE, Orlando, FL, 2009, pp. , pp. 1585–1587, available from: http://ieeexplore.ieee.org/document/5402267/, cited 2024 Jul 6 Search PubMed.
- V. Kochurikhin, K. Kamada, K. Jin Kim, M. Ivanov, L. Gushchina and Y. Shoji, et al., Czochralski growth of 4-inch diameter Ce:Gd3Al2Ga3O12 single crystals for scintillator applications, J. Cryst. Growth, 2020, 531, 125384 CrossRef CAS.
- G. Aad, T. Abajyan, B. Abbott, J. Abdallah, K. S. Abdel and A. A. Abdelalim, et al., Observation of a new particle in the search for the Standard Model Higgs boson with the ATLAS detector at the LHC, Phys. Lett. B, 2012, 716(1), 1–29 CrossRef CAS.
- K. E. Sickafus, C. L. Melcher, M. I. Flynn-Hepford, Y. Wang, G. Jaroslaw and J. P. Smith, et al., Crystal chemistry of rare-earth containing garnets: Prospects for high configurational entropy, J. Solid State Chem., 2022, 310, 122997 CrossRef CAS.
- J. Bárta, K. S. Pestovich, J. A. Valdez, B. W. Wiggins, C. Richards and E. Smith, et al., Compositional screening of Ce-doped (Gd,Lu,Y)3(Al,Ga)5O12 ceramics prepared by quenching from melt and their luminescence properties, J. Alloys Compd., 2021, 889, 161687 CrossRef.
- M. Pianassola, M. Alexander, B. Chakoumakos, M. Koschan, C. Melcher and M. Zhuravleva, Effects of composition and growth parameters on phase formation in multicomponent aluminum garnet crystals, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2022, 78(3), 476–484 CrossRef CAS.
- J. M. Ogiegło, A. Katelnikovas, A. Zych, T. Jüstel, A. Meijerink and C. R. Ronda, Luminescence and Luminescence Quenching in Gd 3 (Ga,Al) 5 O 12 Scintillators Doped with Ce 3+, J. Phys. Chem. A, 2013, 117(12), 2479–2484 CrossRef.
- S. Nargelas, Y. Talochka, A. Vaitkevičius, G. Dosovitskiy, O. Buzanov and A. Vasil’ev, et al., Influence of matrix composition and its fluctuations on excitation relaxation and emission spectrum of Ce ions in (Gd Y1-)3Al2Ga3O12:Ce scintillators, J. Lumin., 2022, 242, 118590 CrossRef CAS.
- W. Drozdowski, K. Brylew, M. E. Witkowski, A. J. Wojtowicz, P. Solarz and K. Kamada, et al., Studies of light yield as a function of temperature and low temperature thermoluminescence of Gd3Al2Ga3O12:Ce scintillator crystals, Opt. Mater., 2014, 36(10), 1665–1669 CrossRef CAS.
- Y. Wu, F. Meng, Q. Li, M. Koschan and C. L. Melcher, Role of Ce 4 + in the Scintillation Mechanism of Codoped Gd 3 Ga 3 Al 2 O 12 : Ce, Phys. Rev. Appl., 2014, 2(4), 044009 CrossRef CAS.
- G. Dantelle, G. Boulon, Y. Guyot, D. Testemale, M. Guzik and S. Kurosawa, et al., Research on Efficient Fast Scintillators: Evidence and X-Ray Absorption Near Edge Spectroscopy Characterization of Ce 4+ in Ce 3+ , Mg 2+ -Co-Doped Gd 3 Al 2 Ga 3 O 12 Garnet Crystal, Phys. Status Solidi B, 2020, 257(8), 1900510 CrossRef CAS.
- K. Bartosiewicz, A. Markovskyi, T. Horiai, D. Szymański, S. Kurosawa and A. Yamaji, et al., A study of Mg2+ ions effect on atoms segregation, defects formation, luminescence and scintillation properties in Ce3+ doped Gd3Al2Ga3O12 single crystals, J. Alloys Compd., 2022, 905, 164154 CrossRef CAS.
- O. Lalinsky, P. Schauer and M. Kucera, Influence of Mg-to-Ce Concentration Ratio on Cathodoluminescence in LuAG and LuGAGG Single-Crystalline Films, Phys. Status Solidi A, 2019, 216(18), 1801016 CrossRef.
- A. Zhang, C. Li, Z. Xue, S. Zhao, P. Qiu and Z. Zhang, et al., Investigation of the Mechanism of Heterovalent Codoping on the Scintillation Properties of GAGG:Ce Crystals, Cryst. Growth Des., 2024, 24(7), 3002–3009 CrossRef CAS.
- D. Spassky, F. Fedyunin, E. Rubtsova, N. Tarabrina, V. Morozov and P. Dzhevakov, et al., Structural, optical and luminescent properties of undoped Gd3AlxGa5-xO12 (x = 0,1,2,3) and Gd2YAl2Ga3O12 single crystals, Opt. Mater., 2022, 125, 112079 CrossRef CAS.
- P. Dorenbos, Electronic structure and optical properties of the lanthanide activated RE3(Al1−xGax)5O12 (RE=Gd, Y, Lu) garnet compounds, J. Lumin., 2013, 134, 310–318 CrossRef CAS.
- Z. Xia and A. Meijerink, Ce 3+ -Doped garnet phosphors: composition modification, luminescence properties and applications, Chem. Soc. Rev., 2017, 46(1), 275–299 RSC.
- T. Kobayashi, S. Yamamoto, S. Okumura, J. Y. Yeom, K. Kamada and A. Yoshikawa, Basic performance of Mg co-doped new scintillator used for TOF-DOI-PET systems, Nucl. Instrum. Methods Phys. Res., Sect., 2017, 842, 14–19 CrossRef CAS.
- L. Martinazzoli, N. Kratochwil, S. Gundacker and E. Auffray, Scintillation properties and timing performance of state-of-the-art Gd 3 Al 2 Ga 3 O12 single crystals, Nucl. Instrum. Methods Phys. Res., Sect., 2021, 1000, 165231 CrossRef CAS.
- D. V. O'Connor and D. Phillips, Time-correlated Single Photon Counting, Academic Press, London, Orlando, 1984, p. 288 Search PubMed.
- J. A. Mares, M. Nikl, N. Solovieva, C. D'Ambrosio, F. de Notaristefani and K. Blazek, et al., Scintillation and spectroscopic properties of Ce3+-doped YAlO3 and Lux(RE)1−xAlO3(RE=Y3+ and Gd3+) scintillators, Nucl. Instrum. Methods Phys. Res., Sect., 2003, 498(1–3), 312–327 CrossRef CAS.
- J. A. Mares, A. Beitlerova, M. Nikl, N. Solovieva, C. D'Ambrosio and K. Blazek, et al., Scintillation response of Ce-doped or intrinsic scintillating crystals in the range up to 1MeV, Radiat. Meas., 2004, 38(4–6), 353–357 CrossRef CAS.
- D. Petschke, dpscience/DLTReconvolution: DLTReconvolution v1.2, Zenodo, 2019, available from: https://zenodo.org/record/3464523, cited 2023 May 3 Search PubMed.
- M. Newville, T. Stensitzki, D. B. Allen and A. Ingargiola, LMFIT: Non-linear Least-Square Minimization and Curve-Fitting for Python, Zenodo, 2014, available from: https://zenodo.org/record/11813, cited 2023 May 3 Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy and D. Cournapeau, et al., SciPy 1.0: fundamental algorithms for scientific computing in Python, Nat. Methods, 2020, 17(3), 261–272 CrossRef CAS.
- J. Czochralski, Ein neues Verfahren zur Messung der Kristallisationsgeschwindigkeit der Metalle, Z. Phys. Chem., 1918, 92U(1), 219–221 CrossRef.
- R. Luther and A. Nikolopulos, Über die Beziehungen zwischen den Absorptionsspektren und der Konstitution der komplexen Kobaltamminsalze, Z. Phys. Chem., 1913, 82U(1), 361–384 CrossRef.
- M. Nikl, K. Kamada, V. Babin, J. Pejchal, K. Pilarova and E. Mihokova, et al., Defect Engineering in Ce-Doped Aluminum Garnet Single Crystal Scintillators, Cryst. Growth Des., 2014, 14(9), 4827–4833 CrossRef CAS.
- K. Kamada, T. Endo, K. Tsutumi, T. Yanagida, Y. Fujimoto and A. Fukabori, et al., Composition Engineering in Cerium-Doped (Lu,Gd) 3 (Ga,Al) 5 O 12 Single-Crystal Scintillators, Cryst. Growth Des., 2011, 11(10), 4484–4490 CrossRef CAS.
- M. Malinowski, Z. Frukacz, M. Szuflińska, A. Wnuk and M. Kaczkan, Optical transitions of Ho3+ in YAG, J. Alloys Compd., 2000, 300–301, 389–394 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards and S. Dacek, et al., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1(1), 011002 CrossRef.
- D. Mateika, E. Völkel and J. Haisma, Lattice-constant-adaptable crystallographics, J. Cryst. Growth, 1990, 102(4), 994–1013 CrossRef CAS.
- L. Martinazzoli, S. Nargelas, P. Boháček, R. Calá, M. Dušek and J. Rohlíček, et al., Compositional engineering of multicomponent garnet scintillators: towards an ultra-accelerated scintillation response, Mater. Adv., 2022, 3(17), 6842–6852 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02866j |
| This journal is © The Royal Society of Chemistry 2024 |