
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, molecular docking and in vitro anticancer activities of 1-(4-(benzamido)phenyl)-3-arylurea derivatives†
Prafulla Sabale *a,
Nusrat Sayyada,
Abuzer Alib,
Vidya Sabalec,
Mohammed Kaleem*d,
Turky Omar Asare,
Amena Alif,
Md. Ali Mujtabag and
Md. Khalid Anwerh
*a,
Nusrat Sayyada,
Abuzer Alib,
Vidya Sabalec,
Mohammed Kaleem*d,
Turky Omar Asare,
Amena Alif,
Md. Ali Mujtabag and
Md. Khalid Anwerh
aDepartment of Pharmaceutical Sciences, Rashtrasant Tukadoji Maharaj Nagpur University, Mahatma Jyotiba Fuley Shaikshanik Parisar, Nagpur-440033, India. E-mail: prafullasable@yahoo.com; nusratsayyad11@gmail.com; Tel: +919158537050
bDepartment of Pharmacognosy, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia. E-mail: abuali@tu.edu.sa
cDepartment of Pharmaceutics, Dadasaheb Balpande College of Pharmacy, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur, Maharashtra 440037, India. E-mail: vidyasabale@gmail.com
dDepartment of Pharmacology, Dadasaheb Balpande College of Pharmacy, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur, Maharashtra 440037, India. E-mail: kaleemmubin88@gmail.com
eDepartment of Biology, College of Science and Arts at Alkamil, University of Jeddah, Saudi Arabia. E-mail: tasar@uj.edu.sa
fDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia. E-mail: amrathore@tu.edu.sa
gDepartment of Pharmaceutics, Faculty of Pharmacy, Northern Border University, Arar, Saudi Arabia. E-mail: M.Mujtaba@nbu.edu.sa
hDepartment of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, P.O. Box 173, Al-Kharj 11942, Saudi Arabia. E-mail: m.anwer@psau.edu.sa
First published on 29th July 2024
Abstract
In both premenopausal and postmenopausal women, oestrogens play a critical role in the development of breast cancer. Aromatase is an enzyme that catalyses the final step in the biosynthesis of estrogen and has emerged as a promising target for therapeutic intervention. This study aimed to design and evaluate novel 1-(4-(benzamido)phenyl)-3-arylurea derivatives as potential aromatase inhibitors. Through molecular docking, promising leads were identified and synthesized. Spectroscopic techniques confirmed their structural integrity. Cytotoxicity against various cancer cell lines was assessed using MTT assay. Docking investigations against the aromatase enzyme (3s7s) elucidated binding interactions and energies. Compound 6g, exhibiting a binding energy of −8.6 kcal mol−1 and interacting with ALA306 and THR310 residues, showed the most promising activity. It demonstrated GI50 values ranging from 14.46 μM, 13.97 μM, 11.35 μM, 11.58 μM, and 15.77 μM against A-498, NCI-H23, MDAMB-231, MCF-7, and A-549 respectively. Lastly, the physicochemical, and ADMET properties of the compound were predicted. These findings highlight the potential of 1-(4-(benzamido)phenyl)-3-arylureas as a new class of antitumor agents targeting aromatase. Their versatility and superior activity compared to standard chemotherapeutic agents, like doxorubicin, warrant further investigation for the development of broader-spectrum anticancer drugs.
1. Introduction
Cancer is a complicated disease with some distinct characteristics, one of them is dysregulated cell production.1–3 It is caused by an imbalance between the expression of oncogenes and proto-oncogenes, which is caused by genetic and epigenetic abnormalities.4,5 Globally, the number of cancer diagnoses has climbed to 19.3 million, and each year, 10 million deaths related to cancer are reported. The most common type of cancer among women is breast cancer (BC), globally.6 BC is a highly complex disease with distinct biological subtypes and several targeted prognostic markers of therapeutic significance.7 Approximately 2.3 million (11.7%) new instances of breast cancer were reported in 2020, and 684![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 996 (6.9%) fatalities were linked to the disease.1 Furthermore, It is predicted that by 2040, there will be around 3 million new cases of breast cancer and one million more deaths from the disease.8 The prognosis and clinical manifestations vary considerably amongst patients. BC develops through multiple pathways, including aromatase enzyme-regulated estrogen synthesis that primarily affects the expression of aromatase enzyme is highest in or near breast tumor sites and is known to catalyze the last step of conversion of androgen to estrogen.9 Elevated expressions of estrogen and aromatase in breast cancer tissues than non-cancerous have been reported by several studies.10 Tamoxifen has been used as standard endocrine therapy for a decade, however, the development of third-generation aromatase inhibitors (Anastrozole, Letrozole, Exemestane) has greater efficacy and improved disease-free survival compared to tamoxifen.11 Although many potential aromatase inhibitors have been published. A few numbers are undergoing clinical studies to treat breast cancer but there are possible serious side effects such as loss of bone density, cardiac events, joint and muscle pain, and hot flashes. Many novel series of compounds have been synthesized and their anticancer efficacy was assessed using sorafenib as parent structure.12 The Diaryl urea is a prominent pharmacophore in building anticancer moiety.13 This activity due to pharmacophore screening shows its near-perfect binding with certain acceptors.14 The protons on two nitrogen and oxygen atoms present in urea moiety act as hydrogen bond donors and hydrogen bond acceptors respectively.15 For drug design and development molecular hybridization is an attractive area in medicinal chemistry that aims to combine two or more pharmacophores for the development of a single compound with improved efficacy.16 From a synthetic view, it was of interest to combine heterocycles to benzamide through a urea linker bridge to develop more potent antiproliferative agents.17,18 Concurrently sorafenib and imatinib, as a potent anti-proliferative drug, have structural similarities.19 Dasatinib, a USFDA-approved drug has been accepted as a chemotherapeutic anticancer agent.20 They all include a diphenyl skeleton linked by an amide or urea, and aromatic heterocyclic rings.21 In medicinal chemistry, the amide group is immensely essential for biological molecules due to their improved solubility in the biological environment and bioavailability.22–24 Amide derivatives were associated with a variety of biological activities like anticonvulsant, anti-tuberculosis, insecticidal, antifungal, anti-microbial, anti-HCV, and antiproliferative properties.25–27 The intermolecular forces have increased importance in medicinal chemistry and allow them to interact with a variety of enzymes and receptors due to their high solubility, ease, and efficiency in reaching target sites.28–30
996 (6.9%) fatalities were linked to the disease.1 Furthermore, It is predicted that by 2040, there will be around 3 million new cases of breast cancer and one million more deaths from the disease.8 The prognosis and clinical manifestations vary considerably amongst patients. BC develops through multiple pathways, including aromatase enzyme-regulated estrogen synthesis that primarily affects the expression of aromatase enzyme is highest in or near breast tumor sites and is known to catalyze the last step of conversion of androgen to estrogen.9 Elevated expressions of estrogen and aromatase in breast cancer tissues than non-cancerous have been reported by several studies.10 Tamoxifen has been used as standard endocrine therapy for a decade, however, the development of third-generation aromatase inhibitors (Anastrozole, Letrozole, Exemestane) has greater efficacy and improved disease-free survival compared to tamoxifen.11 Although many potential aromatase inhibitors have been published. A few numbers are undergoing clinical studies to treat breast cancer but there are possible serious side effects such as loss of bone density, cardiac events, joint and muscle pain, and hot flashes. Many novel series of compounds have been synthesized and their anticancer efficacy was assessed using sorafenib as parent structure.12 The Diaryl urea is a prominent pharmacophore in building anticancer moiety.13 This activity due to pharmacophore screening shows its near-perfect binding with certain acceptors.14 The protons on two nitrogen and oxygen atoms present in urea moiety act as hydrogen bond donors and hydrogen bond acceptors respectively.15 For drug design and development molecular hybridization is an attractive area in medicinal chemistry that aims to combine two or more pharmacophores for the development of a single compound with improved efficacy.16 From a synthetic view, it was of interest to combine heterocycles to benzamide through a urea linker bridge to develop more potent antiproliferative agents.17,18 Concurrently sorafenib and imatinib, as a potent anti-proliferative drug, have structural similarities.19 Dasatinib, a USFDA-approved drug has been accepted as a chemotherapeutic anticancer agent.20 They all include a diphenyl skeleton linked by an amide or urea, and aromatic heterocyclic rings.21 In medicinal chemistry, the amide group is immensely essential for biological molecules due to their improved solubility in the biological environment and bioavailability.22–24 Amide derivatives were associated with a variety of biological activities like anticonvulsant, anti-tuberculosis, insecticidal, antifungal, anti-microbial, anti-HCV, and antiproliferative properties.25–27 The intermolecular forces have increased importance in medicinal chemistry and allow them to interact with a variety of enzymes and receptors due to their high solubility, ease, and efficiency in reaching target sites.28–30
2. Material and methods
2.1. Molecular docking
The structures of each designed novel derivative and native ligand have been drawn using ChemDraw Ultra 16.0. (mole. File format). Universal Force Field (UFF) was utilized to carry out the energy minimization or optimization. All the compounds were docked to the crystal structure of human placental aromatase complexed with the breast cancer medication exemestane, using autodock vina 1.1.2 in PyRx 0.8. The crystal structure of the exemestane was procured from the RCSB Protein Data Bank (PDB) as entry 3s7s (https://www.rcsb.org/structure/3S7S).31With the aid of Discovery Studio Visualizer 2019,32 The protein structure of human placental aromatase complexed with the breast cancer drug exemestane was cleaned up, optimized, and made ready for docking. This involved removing unnecessary water molecules and associated ligands. The Vina Wizard Tool in PyRx 0.8 was used to carry out the binding energy investigations. The docking investigation included all ligand molecules (PDBQT Files) and target molecules (aromatase enzymes). To specify the area for interactions around the RBD (to occupy), a three-dimensional grid box of known size (size_x = 51.602628352Ao; size_y = 62.2935613655Ao; size_z = 41.7297385081Ao) was calculated for MD simulation using Autodock tool 1.5.6 with an exhaustiveness value of 8. BIOVIA Discovery Studio Visualizer (version 19.1.0.18287) was employed to locate and record the active amino acid residues in the protein. The entire process of molecular docking has been described in our previous publication by Sayyad et al.33
2.2. Chemistry
2.2.4.1. 1-(4-(Benzamido)phenyl)-3-(pyridin-3-yl)urea (6a). Obtained as off white solid, yield: 69%; molecular formula: C19H16N4O2; molecular weight: 332 g mol−1; m.p.: 258–261 °C; Rf value: 0.76; IR (cm−1): 3324.8 (N–H stretching), 1241.2 (C–N stretching (Ar)) 1520.8 (N–H bending) 1672.9 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching) 1625.1 (CO stretching (amide)), 823.7, 689.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 6.089 (s, –NH, 2H), 7.336, 7.353, 7.491, 7.504, 7.521, 7.558, 7.572, 7.587, 7.601, 7.619, 7.800, 7.860 (m, Ar–CH)., 7.875(s, –NH, 1H); MS: m/z exact mass: 332.13, mass found: 333.73 (M + 1).
N stretching) 1625.1 (CO stretching (amide)), 823.7, 689.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 6.089 (s, –NH, 2H), 7.336, 7.353, 7.491, 7.504, 7.521, 7.558, 7.572, 7.587, 7.601, 7.619, 7.800, 7.860 (m, Ar–CH)., 7.875(s, –NH, 1H); MS: m/z exact mass: 332.13, mass found: 333.73 (M + 1).
2.2.4.2. 1-(4-(Benzamido)phenyl)-3-(thiazol-2-yl)urea (6b). Yielded as a solid off-white, yield: 71%; molecular formula: C17H14N4O2S; molecular weight: 338 g mol−1; m.p.: 371–374 °C; Rf value: 0.53; IR (neat cm−1): 3324.8 (N–H stretching), 1312.0 (C–N stretching (Ar)) 1576.7 (N–H bending) 1672.9 (C
N stretching) 1625.1 (CO stretching (amide)), 820.0, 715.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 6.029 (s, 2H, –NH urea), 7.93(s, –NH, 1H benzamide), 7.88, 7.84, 7.50, 7.39, 7.36, 7.31, 7.28, 7.25, 6.701, 6.691 (m, Ar–CH); MS: m/z exact mass: 338.08, mass found: 339.01 (M + 1).
2.2.4.3. 1-(4-(Benzamido)phenyl)-3-(thiophen-2-yl)urea (6c). Yielded as a solid off-white, yield: 60%; molecular formula: C18H15N3O2S; molecular weight: 337 g mol−1; m.p.: 343–346 °C; Rf value: 0.81; IR (neat cm−1) 3324.8 (N–H stretching), 1241.2 (C–N stretching (Ar)) 1520.8 (N–H bending) 1672.9 (C
N stretching) 1625.1 (CO stretching (amide)), 823.7, 689.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 5.99 (s, 1H, –NH urea), 6.00 (s, 1H, –NH urea), 6.39, 6.41, 6.56, 6.59, 6.6., 6.71, 7.22, 7.25, 7.27, 7.30, 7.32, 7.36, 7.58, 7.90, 7.95(m, Ar–CH), 8.00(s, –NH, 1H benzamide); MS: m/z exact mass: 337.09, mass found: 338.41 (M + 1).
2.2.4.4. 1-(4-(Benzamido)phenyl)-3-(1H-benzo[d]imidazole-2-yl)urea (6d). Obtained as an off white solid, yield:74%; molecular formula: C21H17N5O2; molecular weight: 371 g mol−1; m.p.: 415–417 °C; Rf value: 0.39; IR (neat cm−1): 3324.8 (N–H stretching), 1285.9 (C–N stretching (Ar)) 1580.4 (N–H bending) 1677.3 (C
N stretching) 1625.1 (CO stretching (amide)), 820.0, 708.2, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 5.00 (s, 1H, –NH benzimidazole), 6.18 (s, 2H, –NH urea), 7.17, 7.19, 7.20, 7.39, 7.41, 7.44, 7.48, 7.50, 7.64, 7.70, 7.73, 7.76, 8.02, 8.05(m, Ar–CH), 8.10(s, –NH, 1H benzamide); MS: m/z exact mass: 371.14, mass found: 372.52 (M + 1).
2.2.4.5. 1-(4-(Benzamido)phenyl)-3-(1H-benzo[d]imidazole-5-yl)urea (6e). Yielded as a solid off-white, yield:76%; molecular formula: C21H17N5O2; molecular weight: 371 g mol−1; m.p.: 407–409 °C; Rf value: 0.67; IR (neat cm−1): 3324.8 (N–H stretching), 1312.0 (C–N stretching (Ar)) 1576.7 (N–H bending) 1677.3 (C
N stretching) 1625.1 (CO stretching (amide)), 820.0, 704.5, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 4.92 (s, 1H, –NH benzimidazole), 6.03 (s, 2H, –NH urea), 7.21, 7.31, 7.32, 7.38, 7.40, 7.44, 7.58, 7.60, 7.62, 7.65, 7.92, 7.96, 8.01(m, Ar–CH), 8.10(s, –NH, 1H benzamide); MS: m/z exact mass: 371.14, mass found: 372.02 (M + 1).
2.2.4.6. 1-(4-(Benzamido)phenyl)-3-methylurea (6f). Yielded as a solid off-white, yield: 69%; molecular formula: C15H15N3O2; molecular weight: 269 g mol−1; m.p.: 342–344; Rf value: 0.68; IR (neat cm−1): 3324.8 (N–H stretching), 1312.0 (C–N stretching (Ar)) 1576.7 (N–H bending) 1672.9 (C
N stretching) 1625.1 (CO stretching (amide)), 823.7, 715.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 2.68 (s, 1H, –CH3 methyl), 6.01 (s, 2H, –NH urea), 7.32, 7.35, 7.38, 7.40, 7.44, 7.59, 7.90, 7.93(m, Ar–CH), 8.00(s, –NH, 1H benzamide); MS: m/z exact mass: 269.12, mass found: 270.14 (M + 1).
2.2.4.7. 1-(4-(Benzamido)phenyl)-3-o-tolylurea (6g). Yielded as a solid off-white, yield: 73%; molecular formula: C21H19N3O2; molecular weight: 345 g mol−1; m.p.: 445–447; Rf value: 0.59; IR (neat cm−1): 3324.8 (N–H stretching), 1312.0 (C–N stretching (Ar)) 1576.7 (N–H bending) 1654.9 (C
N stretching) 1625.1 (CO stretching (amide)), 820.0, 715.6, 894.6 (aromatic C–H bending); 1H NMR (500 MHz, CDCl3) (chemical shift δ): 3.15 (s, 1H, –CH3 methyl), 6.00 (s, 1H, –NH urea), 6.03 (s, 1H, –NH urea), 6.72, 6.76, 6.80, 6.92, 6.97, 6.99, 7.30, 7.33, 7.37, 7.40, 7.45, 7.57, 7.92, 7.97(m, Ar–CH), 8.01(s, –NH, 1H benzamide); 13C NMR (125 MHz, DMSO) 21.0(–CH3), 111.56(–CH), 117.54(–CH), 121.77(–CH), 123.97(–CH), 125.18(–CH), 126.39(–CH), 127.17(–CH), 128.33(–CH), 128.42(–CH), 127.58(–CH), 128.65(–CH), 128.83(–CH), 128.97(–CH), 129.20(–CH), 131.61(–CH), 134.0(–CH), 134.78(–CH), 138.06(–CH), 155.36(–CO urea), 165.57(–CONH). MS: m/z exact mass: 345.15, mass found: 346.19 (M + 1).
2.3. Biological evaluation
The biological research has been conducted at the “National Centre for Cell Science” in Pune, India. All cell lines were purchased from the National Centre for Cell Science in Pune, Maharashtra, India. The cancer cell lines viz A-498 (renal carcinoma), NCI-H23 (lung carcinoma), A549 (lung adenocarcinoma), MCF-7 (breast adenocarcinoma), and MDA-MB-231 (breast adenocarcinoma), were used for study at the National Centre for Cell Science (Pune, India).The cell lines were processed in a lab as soon as they were delivered, and the first cells to be processed were cryopreserved in liquid nitrogen for subsequent use. After being meticulously cared for by guidelines, the cultivated cell lines reached maturity in less than eight weeks. Approximately 70–80% of the cells from grown tissues that were sown during the tests were confluent. The cell lines were preserved in complete media containing “Dulbecco's modified Eagle medium (Cell Clone Genetix brand, Catalogue No.: CC3004) along with penicillin-streptomycin (50 U ml−1, 50 mg ml−1; HiMedia, Catalogue No.: A002) and 10% fetal bovine serum by maintaining air (95%) CO2 (5%) at a temperature of 37 °C.
000 cells per well, MCF-7: 5000 cells per well, and A549: 5000 cells per well. The cells were then kept at 37 °C for 24 h in a humid atmosphere with 5% CO2. The appropriate DMSO dilution was used as the vehicle control before test chemicals were added to wells in triplicate. Following a 72 hours exposure to the test chemical in a humidified environment at 37 °C with 5% CO2, at the end of the incubation time, twenty microliters of MTT reagent (5 mg ml−1) were added to each well. It was then incubated for another two to four hours at 37 °C in a 5% CO2 environment. After the supernatant was removed, the cells were lysed and the formazan needles were dissolved using 200 μl of DMSO. The absorbance was then calculated using an epoch microplate reader at 570 nm. All cell lines were initially screened using test chemical concentrations of 10 and 25 μM. The ones that had an inhibition level of greater than 50% were moved on for GI50 analysis (test chemical concentration inhibiting 50% of the cell population). Nine different concentrations (0.05, 0.1, 5, 1, 5, 10, 30, 50, and 100 M) were tested three times to ascertain the test substances' GI50 value for the respective cell lines. Regression analysis was then used to determine the GI50, which was then expressed in mM using a mean of three replicates.36,372.4. In silico physicochemical, and ADMET predictions
By predicting in silico physicochemical characteristics, adsorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, one can assess a drug candidate's potential. The standard approach suggested in SwissADME (http://swissadme.ch) was used to carry out the ADMET analysis.38 The molecule has been uploaded to the ‘ADMET Evaluation’ option of the ‘services’ section of the ADMETLab 2.0 server. Many of the ADMET attributes of a molecule were calculated and shown by the server after submission.3. Results
3.1. Molecular docking
The crystal coordinates of the aromatase enzyme were taken from http://rcsb.org with PDB ID: 3s7s and the designed molecules (D1–20) were docked into the binding pocket of the enzyme (Fig. 1). The docking scores of the compounds were compared with the co-crystallized ligand where the designed molecules showed good binding scores with good binding affinities. The docking scores of the native ligand and all of its derivatives are listed in Table 1. The comprehensive docking data including residues of active amino acids, type of bond, bond length (Å), and bond class involved in the interactions are illustrated in table D1, D2 and D3 of the (ESI data†).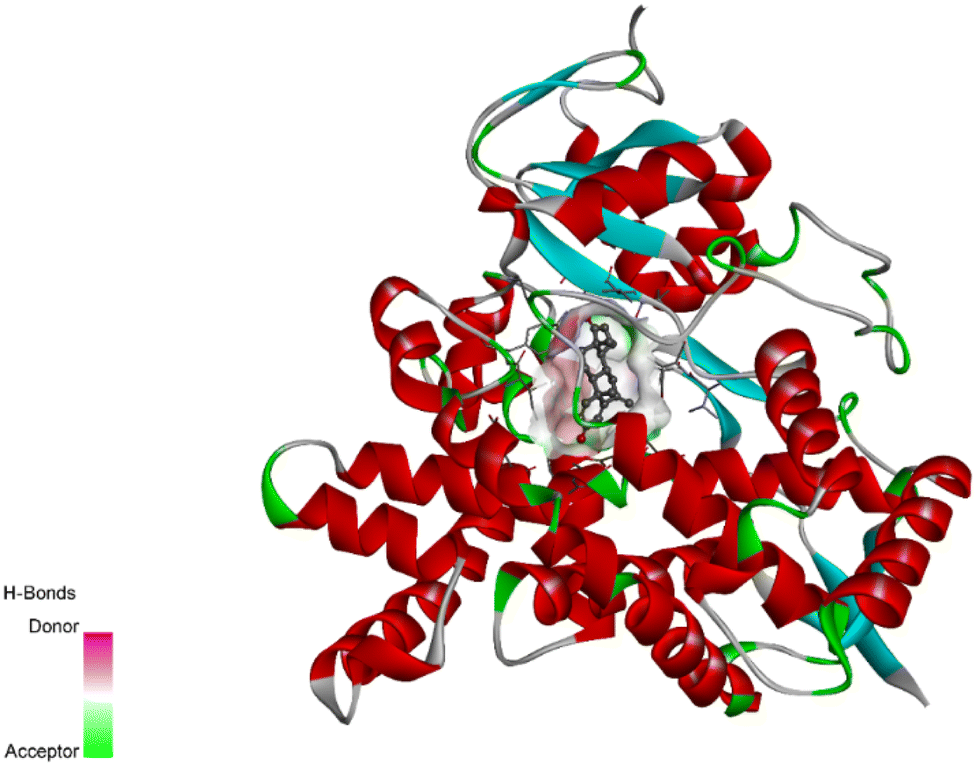 | ||
| Fig. 1 The binding pocket of human aromatase enzyme with the co-crystallized ligand. | ||
| Compound code | Structure | Ligand energy (kcal mol−1) | Binding energy (kcal mol−1) | rmsd/ub | rmsd/lb |
|---|---|---|---|---|---|
| Native ligand | 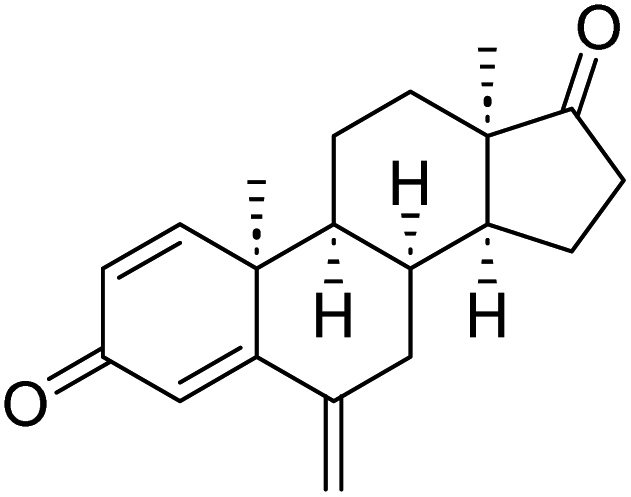 |
1113.62 | −8.9 | 0 | 0 |
| D1 |  |
250.47 | −8.4 | 0 | 0 |
| D2 |  |
277.44 | −8.4 | 0 | 0 |
| D3 (6a) | 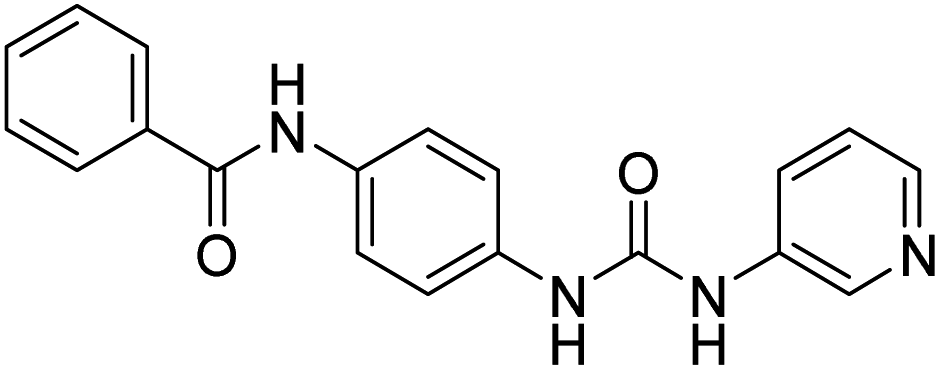 |
287.84 | −8.2 | 0 | 0 |
| D4 | 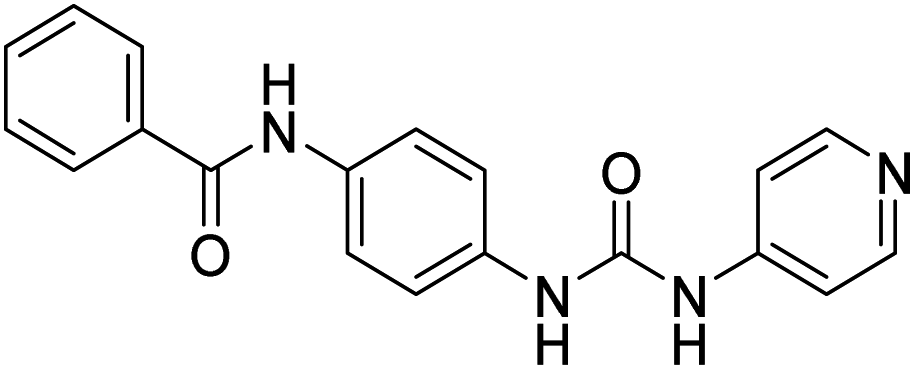 |
275.03 | −8 | 0 | 0 |
| D5 (6b) | 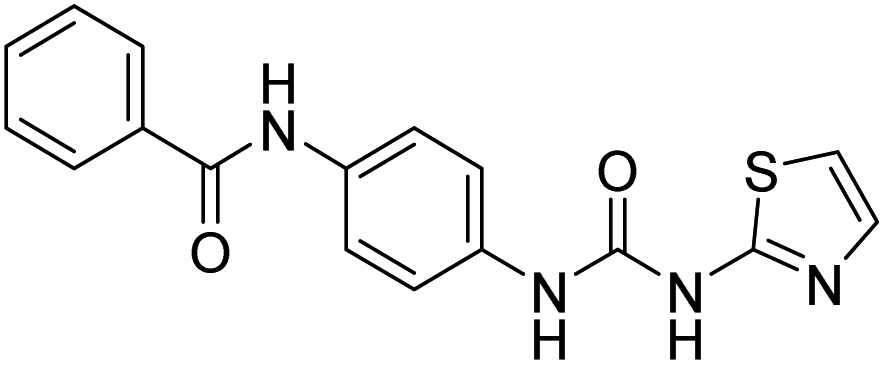 |
458.41 | −8 | 0 | 0 |
| D6 | 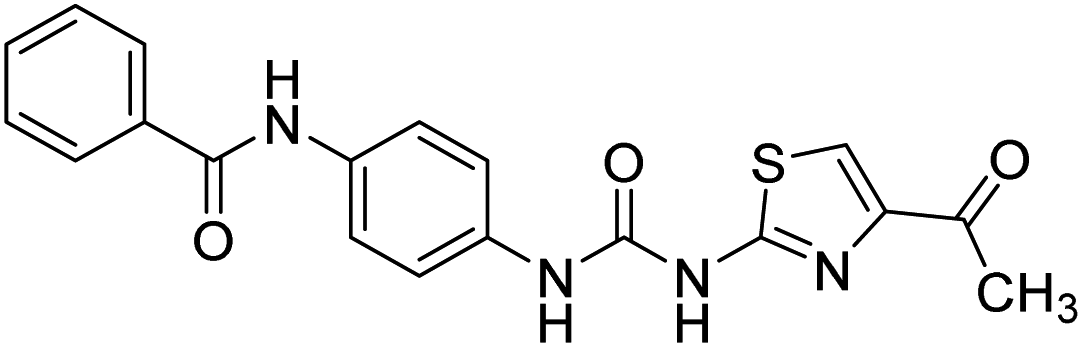 |
575.4 | −7.2 | 0 | 0 |
| D7 (6c) | 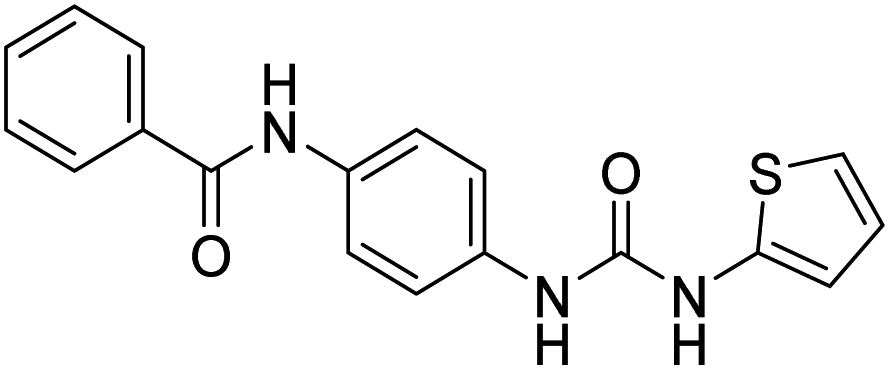 |
402.26 | −8.1 | 0 | 0 |
| D8 | 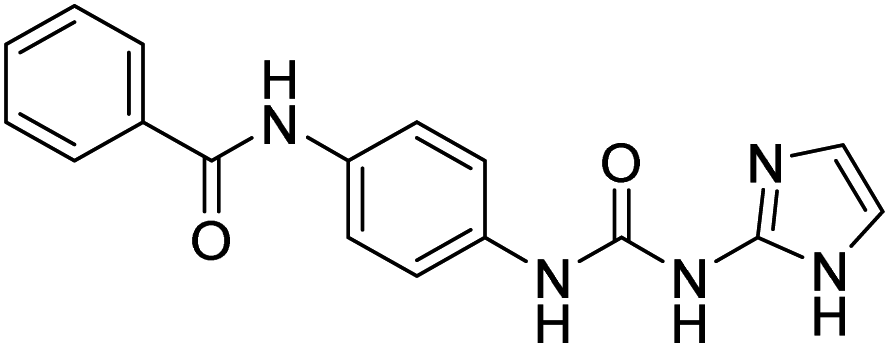 |
420.02 | −8.3 | 0 | 0 |
| D9 (6d) | 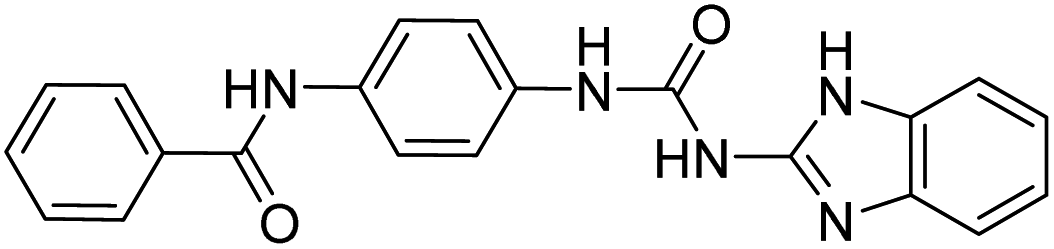 |
514.81 | −7.9 | 0 | 0 |
| D10 (6e) | 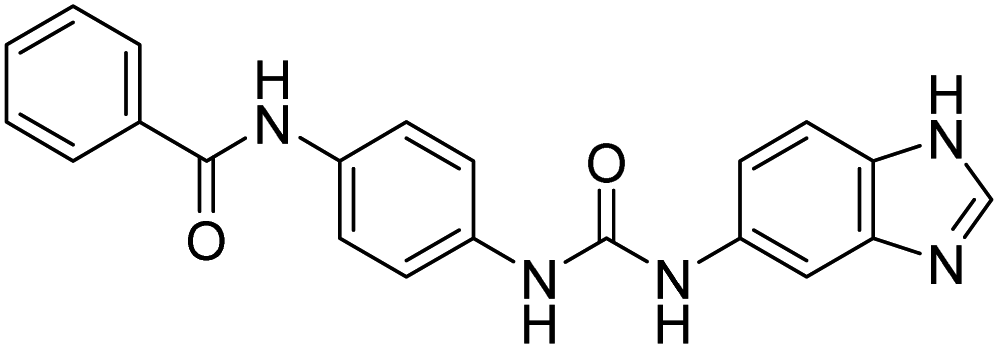 |
528.5 | −7.9 | 0 | 0 |
| D11 (6f) | 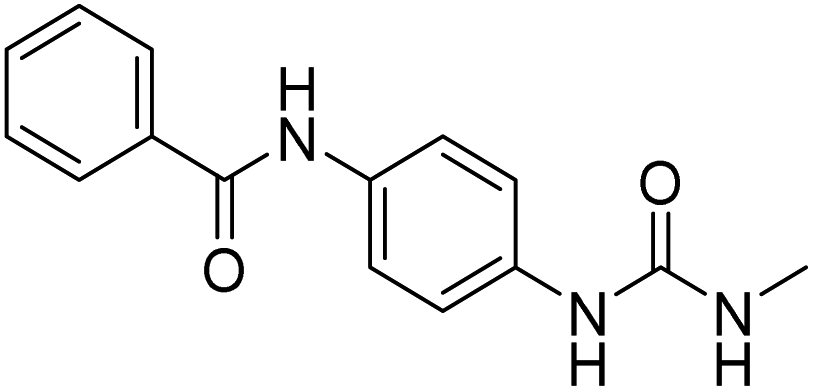 |
175.12 | −7.8 | 0 | 0 |
| D12 | 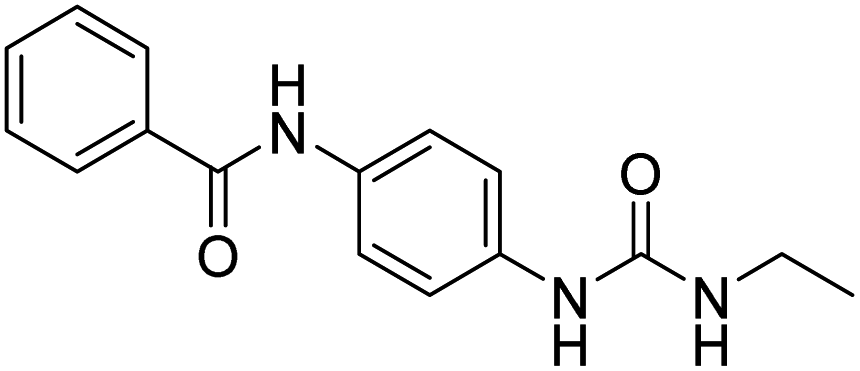 |
174.28 | −7.4 | 0 | 0 |
| D13 | 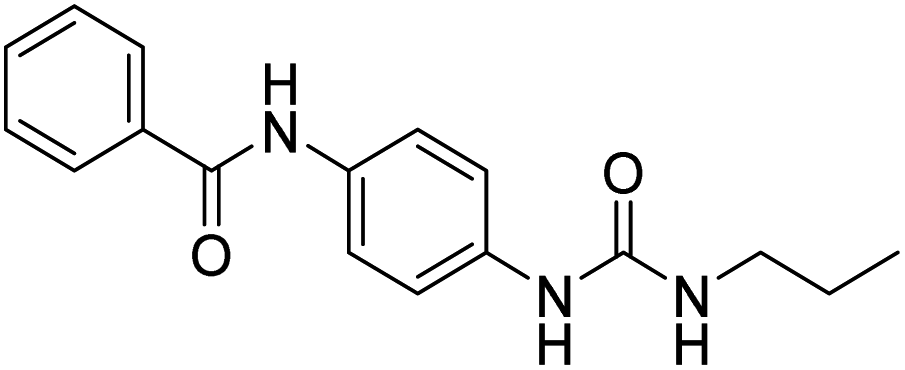 |
172.5 | −6.4 | 0 | 0 |
| D14 | 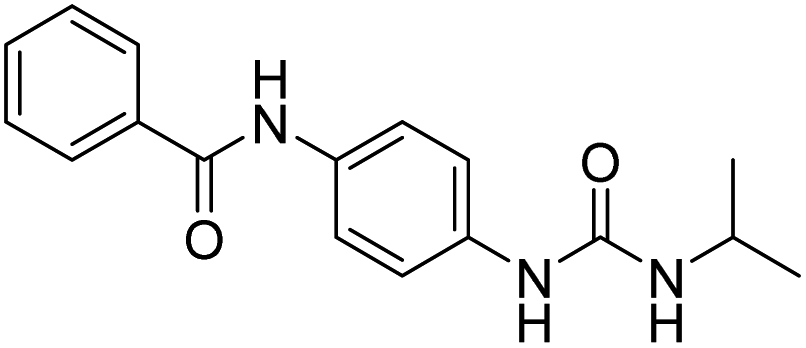 |
177.15 | −7.9 | 0 | 0 |
| D15 | 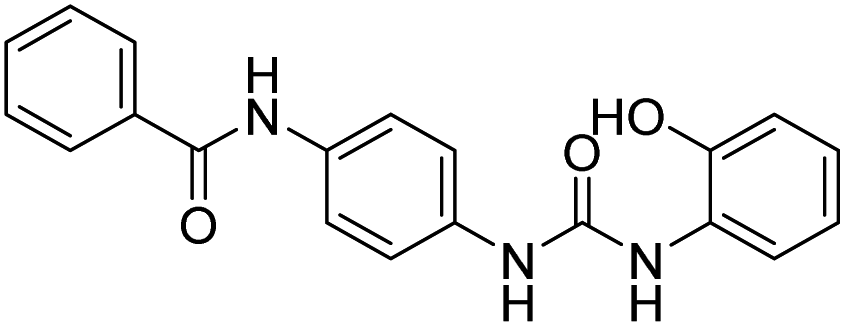 |
274.78 | −7.9 | 0 | 0 |
| D16 | 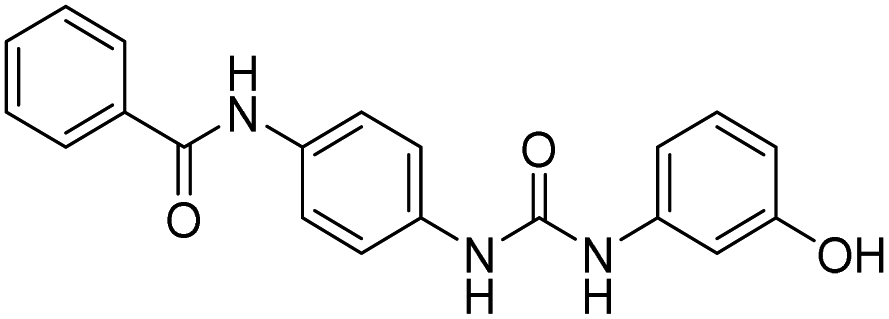 |
284.12 | −8.6 | 0 | 0 |
| D17 | 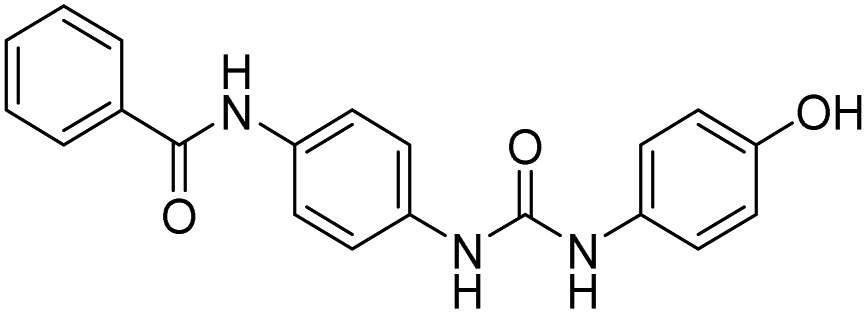 |
300.1 | −8.3 | 0 | 0 |
| D18 | 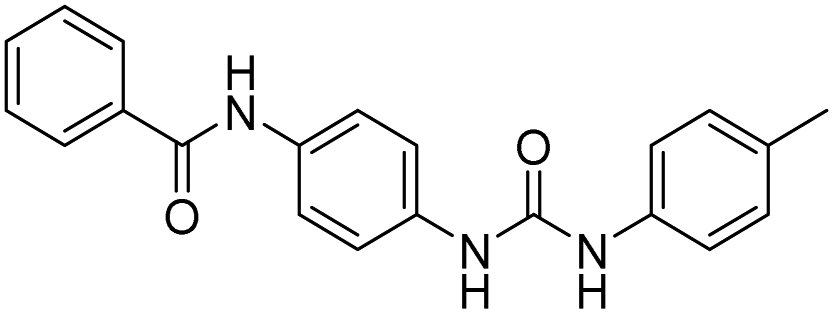 |
276.5 | −8.7 | 0 | 0 |
| D19 (6g) | 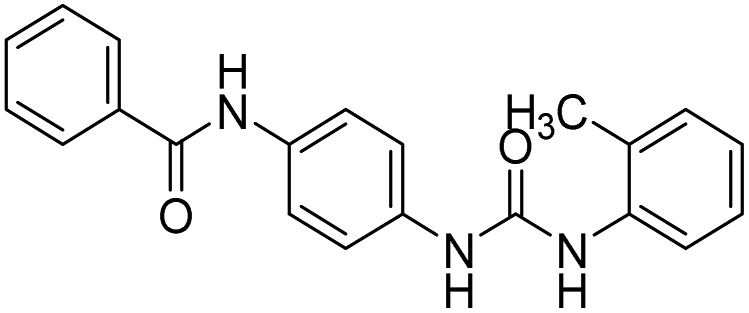 |
275.96 | −8.6 | 0 | 0 |
| D20 |  |
227.72 | −8.6 | 0 | 0 |
3.2. Chemistry
The synthesis of N-(4-(3-phenylureido)phenyl)benzamide (6a–g) derivatives is depicted in Scheme 1. Benzoic acid (1) was coupled with 4-chloroaniline (2) in the presence of DCC resulting in N-(4-chlorophenyl)benzamide (3), which was then refluxed for 12 h with urea in dimethyl formamide in the presence of potassium carbonate to form N-(4-ureidophenyl)benzamide (4). This N-(4-ureidophenyl)benzamide (4) was then refluxed with substituted alkyl/aryl halides (5a–g) in the presence of potassium carbonate to afford the titled N-(4-(3-phenylureido)phenyl)benzamide (6a–g) derivatives.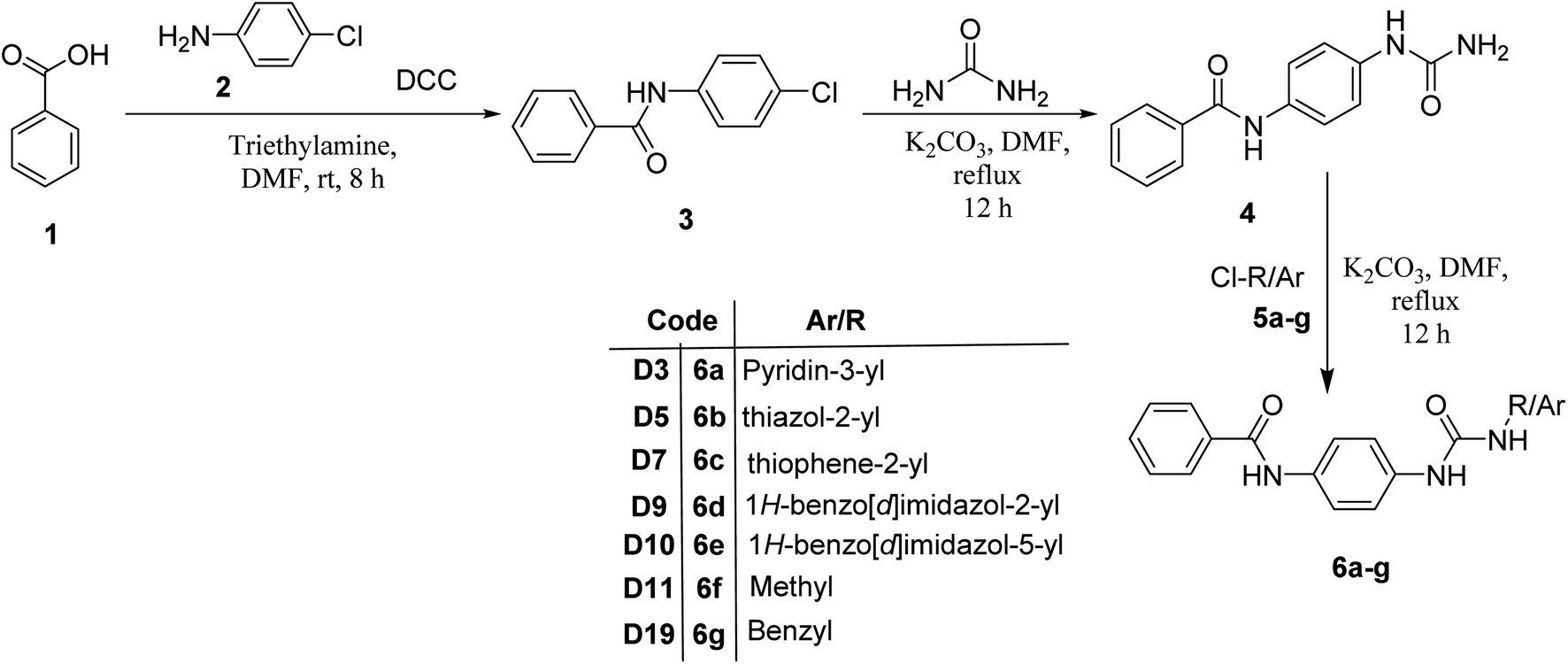 | ||
| Scheme 1 Synthesis of N-(4-(3-phenylureido)phenyl)benzamide derivatives. | ||
3.3. Biological evaluation
| Comp. code | % Viability at 10 μM | ||||
|---|---|---|---|---|---|
| A-498 | NCI-H23 | MDAMB-231 | MCF-7 | A-549 | |
| 6a (D3) | 57.16 | 67.17 | 81.60 | 79.62 | 57.67 |
| 6b (D5) | 59.16 | 81.16 | 91.27 | 90.72 | 71.81 |
| 6c (D7) | 71.12 | 69.19 | 86.95 | 75.15 | 77.13 |
| 6d (D9) | 67.28 | 75.18 | 91.38 | 89.18 | 92.12 |
| 6e (D10) | 81.16 | 92.83 | 90.13 | 89.13 | 79.17 |
| 6f (D11) | 75.15 | 94.07 | 84.42 | 90.17 | 78.16 |
| 6g (D19) | 54.16 | 61.68 | 50.10 | 57.47 | 65.16 |
| Entry | Comp. code | GI50 ± SD | ||||
|---|---|---|---|---|---|---|
| A498 | NCI-H23 | MDAMB-231 | MCF-7 | A-549 | ||
| 1 | 6g (D19) | 14.46 μM ± 0.06079 | 13.97 μM ± 0.07182 | 11.35 μM ± 0.05929 | 11.58 μM ± 0.05363 | 15.77 μM ± 0.05613 |
| 2 | Doxorubicin | 3.45 μM ± 0.09215 | 3.05 μM ± 0.04911 | 3.06 μM ± 0.04619 | 2.70 μM ± 0.02131 | 2.97 μM ± 0.04133 |
3.4. In silico physicochemical and ADMET analysis
The compound's (D19) physicochemical and ADMET properties, which may be found in the ESI,† were predicted using SwissADME (refer to ESI materials†).4. Discussion
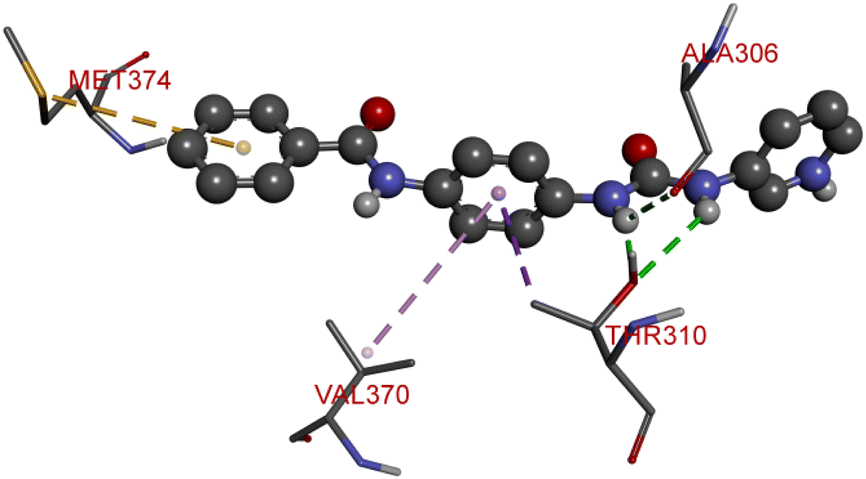
The proliferation of estrogen-sensitive breast cancer cells and the progression of breast cancer can be attributed to the aromatase enzyme.39,40 Therefore, it is postulated that the reduction in aromatase enzyme expression in BC will prevent cell division and growth.41 Even though there are natural and synthetic chemical moieties are available in the market due to various side effects and resistance toward drugs it is necessary to develop new molecules.42 In this work, we have demonstrated that the synthesized molecules show promising percent viability at 10 and 25 μM on an array of five different cell lines. In that MCF7 breast cancer cell lines had shown good inhibitory effects. The downregulation of aromatase enzyme was linked to the inhibitory effects of on panel of cell lines, suggesting that synthesized compounds could be a bright prospect for treating breast cancer. The derivatives were made by applying reaction plans. These novel derivatives were characterized using their mass, 1H NMR, and FTIR spectroscopic data. The 1H NMR, MS, and FTIR spectra of the synthesized compounds were found in the anticipated range. Based on their mass spectra, every synthesized compound has a molecular ion [M + H]+ peak that corresponds to its molecular formula (refer to ESI materials†).The native ligand binding mechanism of the aromatase enzymes crystal structure has been compared to the derivatives binding affinities (PDB ID: 3s7s). Exemestane, the native ligand of the enzyme, has a binding energy of −8.9 kcal mol−1. The enzyme did not produce any hydrogen bonds of any kind. ILE133 (4.78 Å), CYS437 (3.59 Å), VAL370 (3.91 Å), ALA306 (4.12 Å), ILE133 (4.0 Å), ALA306 (5.25 Å), and TRP224 (5.23 Å) are all hydrophobic interaction. It was prevalent for multiple variants to form over two hydrogen bonds with the target. For synthesis and biological activity, only molecules with three or more hydrogen bonds have been chosen. The virtual screening revealed that compounds D3, D5, D7, D9, D10, D11, and D19 were the most efficient molecules as presented in Table 4.
| 3D images | |
|---|---|
| D3 | D5 |
 |
 |
| D7 | D8 |
 |
 |
| D10 | D11 |
 |
 |
| D19 | |
 |
|
Compound D3 exhibited a binding energy of −8.2 kcal mol−1 and established three conventional hydrogen bonds with THR310 (2.99 Å, 2.0 Å), and ALA306 (2.52 Å). Three hydrophobic interactions have been formed with THR310 (3.71 Å), MET374 (5.10 Å), and VAL370 (5.23 Å).
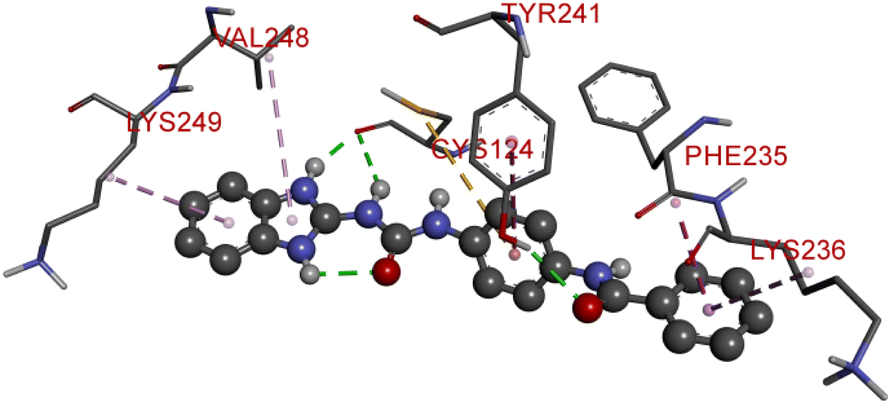
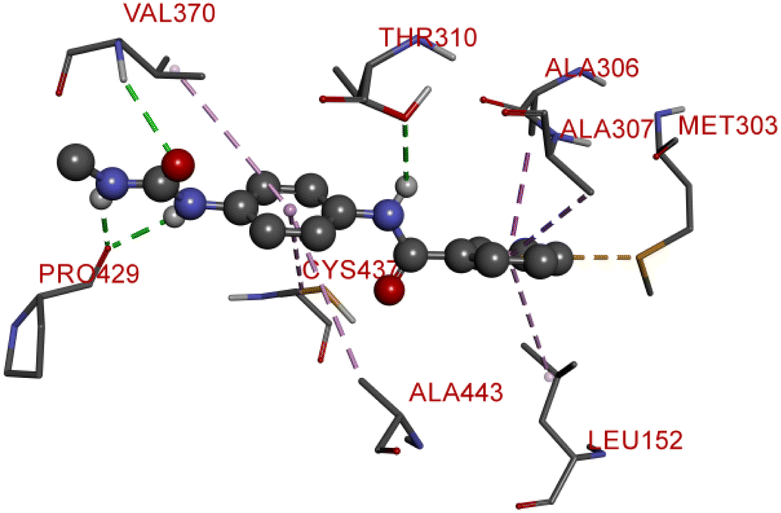
Compound D5 exhibited a binding energy of −8 kcal mol−1 and established three conventional hydrogen bonds with THR310 (2.90 Å, 1.95 Å), and ALA306 (2.73 Å). It has displayed several hydrophobic interactions with THR310 (3.83 Å), MET446 (3.96 Å), MET374 (5.05 Å), VAL370 (5.24 Å), LEU152 (5.32 Å), ALA306 (4.51 Å), and ALA307 (4.24 Å). Compound D7 has shown a binding energy of −8.1 kcal mol−1 and substantiates three conventional hydrogen bonds with THR310 (2.96 Å, 1.87 Å), and ALA306 (2.62 Å). It has resulted in numerous hydrophobic interactions with the same amino acids that compound D5 forms, including Pi-sigma, Pi-sulfur, and Pi-alkyl. Compound D9 demonstrated −7.9 kcal mol−1 binding energy and established three conventional hydrogen bonds with CYS124 (2.62 Å, 1.96 Å), and TYR241 (2.54 Å). Pi-sulfur, amide-Pi stacked, Pi–Pi stacked, and Pi-alkyl bonds have all been produced with CYS124 (5.56 Å), TYR241 (5.34 Å), PHE235; LYS236 (3.85 Å), VAL248 (5.46 Å), LYS249 (4.60 Å), and LYS236 (4.92 Å).
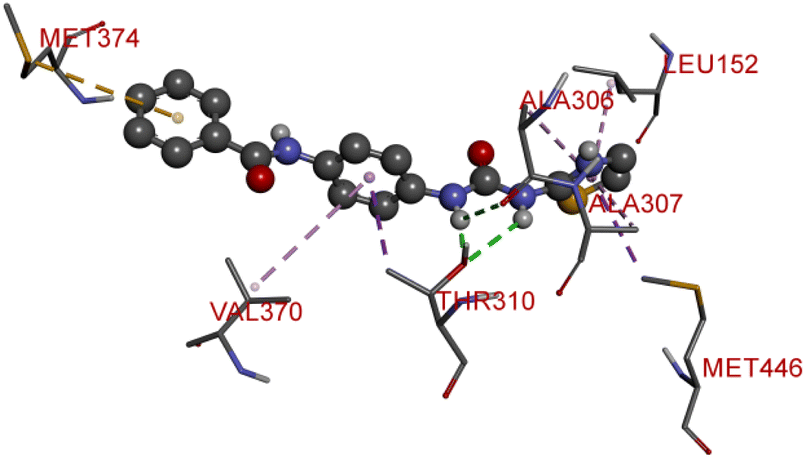
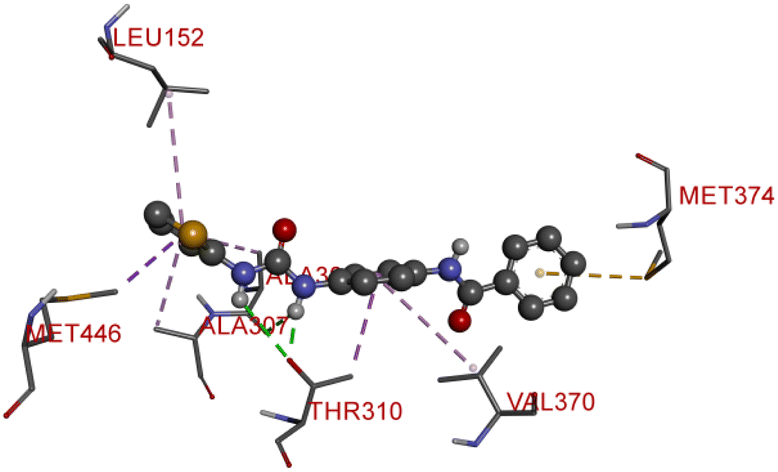
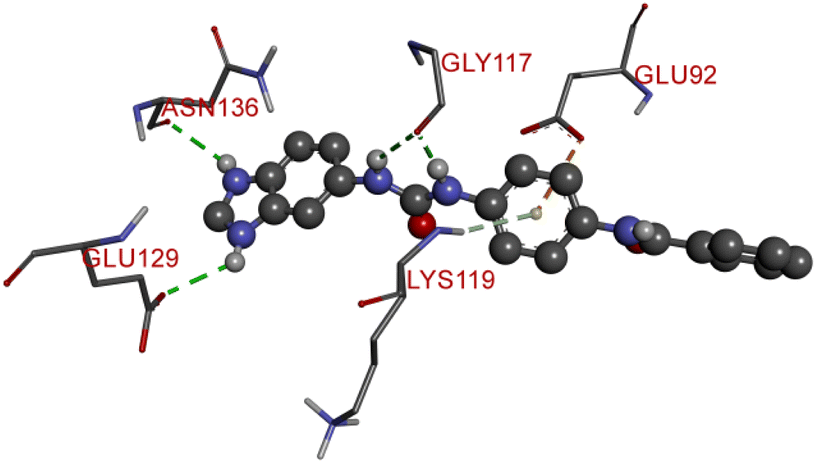
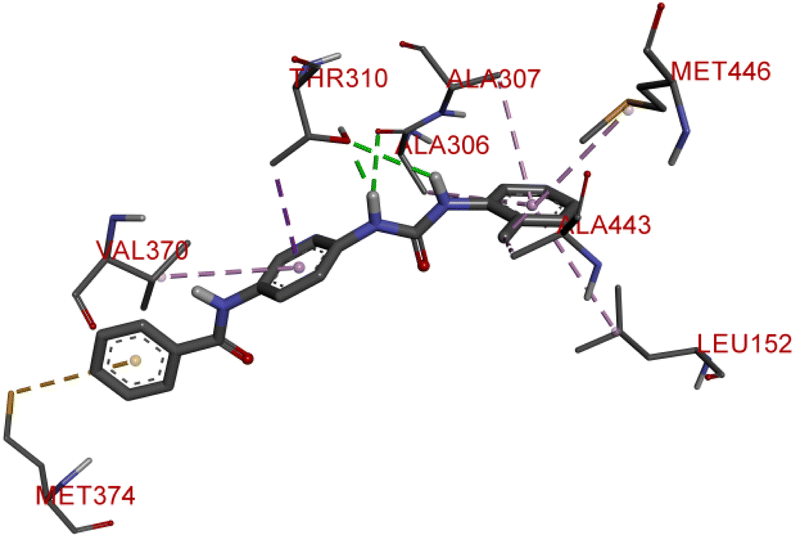
Compound D10 exhibits a binding energy of −7.9 kcal mol−1 and has shown four conventional hydrogen bonds with GLY117 (2.03 Å, 2.67 Å), GLU129 (2.53 Å), and ASN136 (2.55 Å). Additionally, it has produced one Pi-donor and one Pi-anion hydrogen bond with GLU92 (4.05 Å) and LYS119 (3.08 Å), respectively. Compound D11 exhibited a binding energy of −7.8 kcal mol−1 and formed 4 conventional hydrogen bonds with THR310 (2.73 Å), PRO429 (2.16 Å, 2.33 Å), and VAL370 (2.77 Å). It included many Pi-sigma, Pi-sulfur, and Pi-alkyl interactions which contributed to his potency. Compound D19 has shown a binding energy of −8.6 kcal mol−1. Three conventional hydrogen bonds have been established through THR310 (3.02 Å, 1.94 Å), and ALA306 (2.77 Å). Multiple hydrophobic interactions have been developed with THR310 (3.65 Å), MET374 (5.15 Å), ALA443 (3.38 Å), MET446 (5.45 Å), VAL370 (5.18 Å), LEU152 (5.14 Å), ALA306 (4.52 Å), and ALA307 (4.29 Å).
The spectroscopic characterization of data of synthesized compounds from 6a–g can be summarized. The common core of NH stretching 3324.8 cm−1 C–N aryl stretching at 1312 cm−1 NH bending at 1576.7 cm−1 CO stretching at 1625.1 cm−1 and aromatic C–H bending 820.0 cm−1, 715.6 cm−1, 894.6 cm−1. The two NH present in the urea bridge come around 5.99 and 6.00 ppm and amide NH comes around 8.101 ppm. Aromatic region around 6–7 ppm. The 13C NMR spectrum was recorded at 125 MHz in DMSO solution, revealing distinctive chemical shifts for various carbon environments within the molecule. The spectrum features signals at 21.0 ppm, indicative of a methyl group (–CH3), along with multiple signals between 111.56 and 138.06 ppm corresponding to different types of methylene (–CH) present in aromatic rings. Notably, at 155.36 ppm, there is a peak attributed to a carbonyl carbon (–CO) of urea functional group, and at 165.57 ppm, another peak suggests a carbon atom in an amide (–CONH) group. Biological results are shown in the Table 3 along with the concentration–response curve in Fig. 2. From these findings, we believe that further research is necessary to create broader-spectrum anticancer medications because of their adaptability and increased efficacy over conventional chemotherapeutic medicines like doxorubicin. To express the cell-carcinogenic selectivity in vitro tests were conducted to determine the compound's potential to inhibit cell proliferation on a panel of cell lines and compound 6g (D19) shows promising anti-cancer activity with notable potency against MDAMB-231 cells, further optimization may be necessary to enhance its efficacy compared to the positive control, doxorubicin. These results lay the groundwork for future research to explore the therapeutic potential of compound 6g (D19) and optimize its effectiveness as a potential anti-cancer agent.
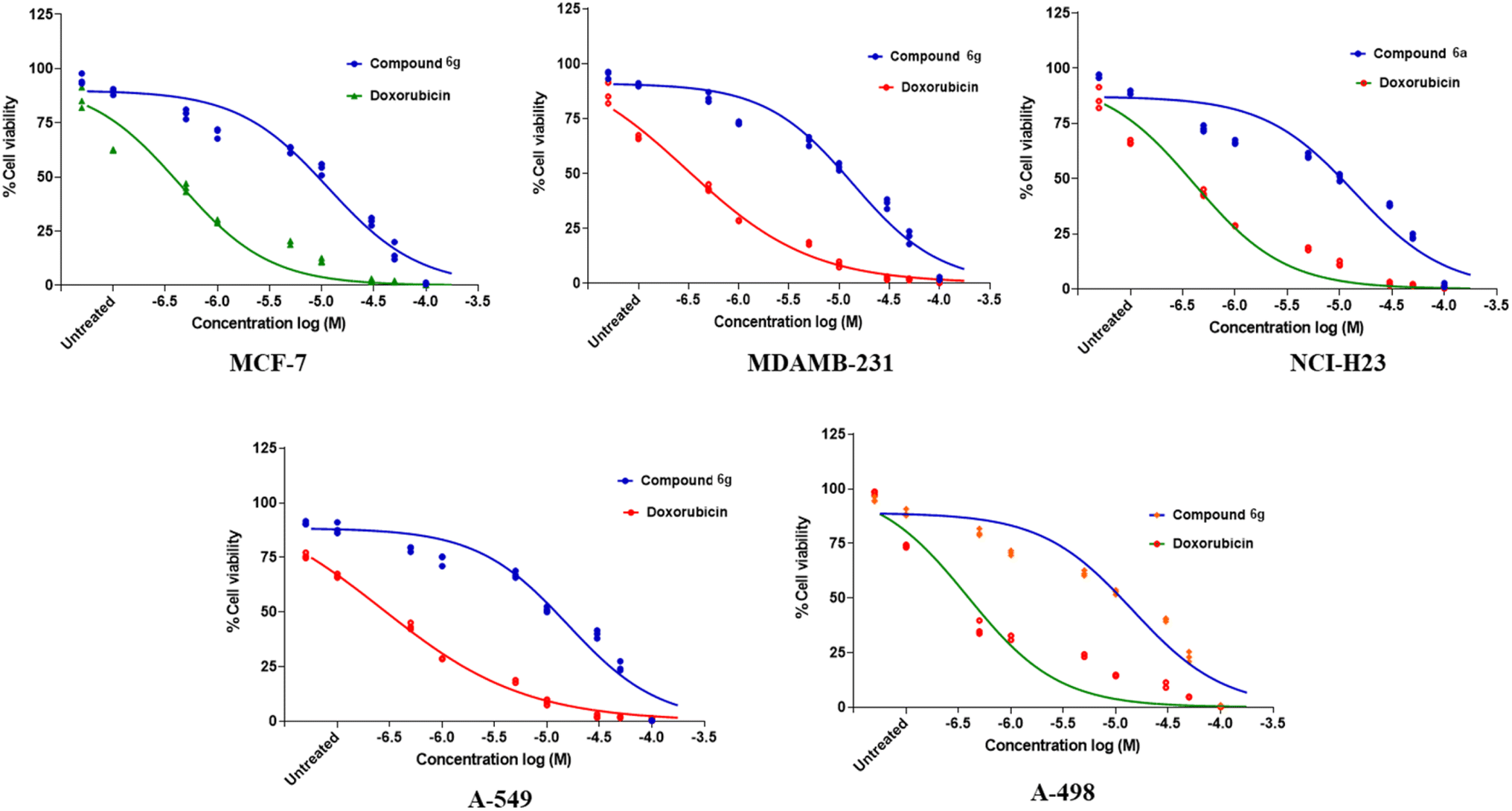 | ||
| Fig. 2 Concentration-response curve for compound 6g. | ||
5. Conclusion
Finally, a novel class of N-(4-(3-phenylureido) phenyl) benzamide (6a–g) derivatives was prepared and tested for antitumor activity against a variety of tumor cell lines. The compound 6g demonstrated good docking scores with noteworthy GI50 value and was found to have potential anti-cancer activity amongst synthesized molecules. Molecular docking analyses indicated the potential molecular interactions that underlie inhibition, which aligns with the findings of experiments. The best compounds revealed in this study have the potential as beginning points and could lead to the development of novel medication candidates for BC treatment. We have evaluated molecules on five different cell lines and in the future could be evaluated for aromatase inhibition activity.Data availability
All the data supporting this article have been presented in the article and the ESI file.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors extend their appreciation to Taif University, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-43). We are thankful to the Department of Pharmaceutical Sciences, RTMNU, Nagpur, India for providing research facility.References
- M. Kaleem, M. H. Dalhat, L. Azmi, T. O. Asar, W. Ahmad, M. Alghanmi, A. Almostadi, T. A. Zughaibi and S. Tabrez, An Insight into Molecular Targets of Breast Cancer Brain Metastasis, Int. J. Mater. Sci., 2022, 23, 11687, DOI:10.3390/ijms231911687.
- M. Kaleem, M. Perwaiz, S. M. Nur, A. O. Abdulrahman, W. Ahmad, F. A. Al-Abbasi, V. Kumar, M. A. Kamal and F. Anwar, Epigenetics of Triple-Negative Breast Cancer via Natural Compounds, Curr. Med. Chem., 2022, 29, 1436–1458, DOI:10.2174/0929867328666210707165530.
- P. Chunarkar-Patil, M. Kaleem, R. Mishra, S. Ray, A. Ahmad, D. Verma, S. Bhayye, R. Dubey, H. Singh and S. Kumar, Anticancer Drug Discovery Based on Natural Products: From Computational Approaches to Clinical Studies, Biomedicines, 2024, 12, 201, DOI:10.3390/biomedicines12010201.
- M. Kaleem, M. Alhosin, K. Khan, W. Ahmad, S. Hosawi, S. M. Nur, H. Choudhry; M. A. Zamzami, F. A. Al-Abbasi and M. N. Javed, Epigenetic Basis of Polyphenols in Cancer Prevention and Therapy, in Polyphenols-based Nanotherapeutics for Cancer Management, ed. S. Tabrez and M. Imran Khan, Springer Singapore, Singapore, 2021, pp. 189–238 ISBN 9789811649349 Search PubMed.
- M. Kaleem, A. Kayali, R. A. Sheikh, A. Kuerban, M. A. Hassan, N. A. R. Almalki, F. A. Al-Abbasi, F. Anwar, Z. Omran and M. Alhosin, In Vitro and In Vivo Preventive Effects of Thymoquinone against Breast Cancer: Role of DNMT1, Molecules, 2024, 29, 434, DOI:10.3390/molecules29020434.
- E. A. Hobbs, N. Chen, A. Kuriakose, E. Bonefas and B. Lim, Prognostic/Predictive Markers in Systemic Therapy Resistance and Metastasis in Breast Cancer, Ther. Adv. Med. Oncol., 2022, 14, 175883592211126, DOI:10.1177/17588359221112698.
- E. A. Hobbs, N. Chen, A. Kuriakose, E. Bonefas and B. Lim, Prognostic/Predictive Markers in Systemic Therapy Resistance and Metastasis in Breast Cancer, Ther. Adv. Med. Oncol., 2022, 14 DOI:10.1177/17588359221112698.
- M. Arnold, E. Morgan, H. Rumgay, A. Mafra, D. Singh, M. Laversanne, J. Vignat, J. R. Gralow, F. Cardoso and S. Siesling, et al., Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040, Breast, 2022, 66, 15–23 CrossRef PubMed.
- P. M. Sabale, V. P. Sabale and L. C. Potey, Aromatase and Aromatase Inhibitors in Breast Cancer Treatment, J. Curr. Pharma Res., 2018, 9, 2636–2655 CrossRef CAS.
- S. Chen, T. Itoh, K. Wu, D. Zhou and C. Yang, Transcriptional Regulation of Aromatase Expression in Human Breast Tissue, J. Steroid Biochem. Mol. Biol., 2002, 83, 93–99 CrossRef CAS PubMed.
- I. E. van Hellemond, S. M. Geurts and V. C. Tjan-Heijnen, Current Status of Extended Adjuvant Endocrine Therapy in Early Stage Breast Cancer, Curr. Treat. Options Oncol., 2018, 19, 1–18 CrossRef PubMed.
- J. A. Files, M. G. Ko and S. Pruthi, Managing Aromatase Inhibitors in Breast Cancer Survivors: Not Just for Oncologists, Mayo Clin. Proc., 2010, 85, 560–566, DOI:10.4065/mcp.2010.0137.
- L. Garuti, M. Roberti, G. Bottegoni and M. Ferraro, Diaryl Urea: A Privileged Structure in Anticancer Agents, Curr. Med. Chem., 2016, 23, 1528–1548 CrossRef CAS PubMed.
- D. Horvath, Pharmacophore-Based Virtual Screening, Chemoinformatics and Computational Chemical Biology, 2010, pp. 261–298 Search PubMed.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, The Squaramide versus Urea Contest for Anion Recognition, Chem.–Eur. J., 2010, 16, 4368–4380 CrossRef CAS PubMed.
- C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E. J. Barreiro and C. A. M. Fraga, Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- H. Kumar, D. Saini, S. Jain and N. Jain, Pyrazole Scaffold: A Remarkable Tool in the Development of Anticancer Agents, Eur. J. Med. Chem., 2013, 70, 248–258 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Morpholine as Ubiquitous Pharmacophore in Medicinal Chemistry: Deep Insight into the Structure-Activity Relationship (SAR), Bioorg. Chem., 2020, 96, 103578 CrossRef CAS PubMed.
- F. Jin, D. Gao, Q. Wu, F. Liu, Y. Chen, C. Tan and Y. Jiang, Exploration of N-(2-Aminoethyl) Piperidine-4-Carboxamide as a Potential Scaffold for Development of VEGFR-2, ERK-2 and Abl-1 Multikinase Inhibitor, Bioorg. Med. Chem., 2013, 21, 5694–5706 CrossRef CAS PubMed.
- A. Haouala, B. Zanolari, B. Rochat, M. Montemurro, K. Zaman, M. A. Duchosal, H. B. Ris, S. Leyvraz, N. Widmer and L. A. Decosterd, Therapeutic Drug Monitoring of the New Targeted Anticancer Agents Imatinib, Nilotinib, Dasatinib, Sunitinib, Sorafenib and Lapatinib by LC Tandem Mass Spectrometry, J. Chromatogr. B, 2009, 877, 1982–1996 CrossRef CAS PubMed.
- Y.-C. Wu, X.-Y. Ren and G.-W. Rao, Research Progress of Diphenyl Urea Derivatives as Anticancer Agents and Synthetic Methodologies, Mini-Rev. Org. Chem., 2019, 16, 617–630 CrossRef CAS.
- B. Das, A. T. Baidya, A. T. Mathew, A. K. Yadav and R. Kumar, Structural Modification Aimed for Improving Solubility of Lead Compounds in Early Phase Drug Discovery, Bioorg. Med. Chem., 2022, 116614 CrossRef CAS PubMed.
- A. Abelian, M. Dybek, J. Wallach, B. Gaye and A. Adejare, Pharmaceutical Chemistry, in Remington, Elsevier, 2021, pp. 105–128 Search PubMed.
- R. Jana, H. M. Begam and E. Dinda, The Emergence of the C–H Functionalization Strategy in Medicinal Chemistry and Drug Discovery, Chem. Commun., 2021, 57, 10842–10866 RSC.
- N. Kushwaha, R. K. Saini and S. K. Kushwaha, Synthesis of Some Amide Derivatives and Their Biological Activity, Int. J. ChemTech Res., 2011, 3, 203–209 CAS.
- B. S. Sastry, K. S. Babu, T. H. Babu, S. Chandrasekhar, P. V. Srinivas, A. K. Saxena and J. M. Rao, Synthesis and Biological Activity of Amide Derivatives of Nimbolide, Bioorg. Med. Chem. Lett., 2006, 16, 4391–4394 CrossRef CAS PubMed.
- X. Hua, N. Liu, Z. Fan, G. Zong, Y. Ma, K. Lei, H. Yin and G. Wang, Design, Synthesis and Biological Activity Screening of Novel Amide Derivatives Containing Aromatic Thioether Group, Chin. J. Org. Chem., 2019, 39, 2581, DOI:10.6023/cjoc201903004.
- H. Abdel-Halim, The Effectiveness of Using Three-Dimensional Visualization Tools to Improve Students' Understanding of Medicinal Chemistry and Advanced Drug Design Concepts, Int. J. Learn. Teach. Educ. Res., 2020, 19, 170–187 CrossRef.
- A. K. Ghosh and M. Brindisi, Urea Derivatives in Modern Drug Discovery and Medicinal Chemistry, J. Med. Chem., 2019, 63, 2751–2788 CrossRef PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- S. Dallakyan and A. J. Olson, Small-Molecule Library Screening by Docking with PyRx, in Chemical Biology, ed. J. E. Hempel, C. H. Williams and C. C. Hong, Methods in Molecular Biology, Springer New York, New York, NY, 2015, vol. 1263, pp. 243–250 ISBN 978-1-4939-2268-0 Search PubMed.
- Dassault Systèmes, Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, 2017 Search PubMed.
- N. B. Sayyad and P. M. Sabale, Rational Drug Design and In Vitro Cell Line Studies of Some N-(4-(1Hbenzo[d]Imidazole-2-Yl)Phenyl)Arylamine Derivatives as Aromatase Inhibitorsfor the Treatment of Cancer, CEI, Concr. Eng. Int., 2023, 19, 38–48, DOI:10.2174/1573408019666221028142316.
- M. Khattab, F. Ragab, S. Galal and H. El Diwani, synthesis of 4-(1H-benzo(d) imidazole-2-yl) aniline derivatives of expected anti-HCV activity, Int. J. Res. Pharm. Chem., 2012, 2, 937–946 CAS.
- S. Shenvi, K. Kumar, K. S. Hatti, K. Rijesh, L. Diwakar and G. C. Reddy, Synthesis, Anticancer and Antioxidant Activities of 2,4,5-Trimethoxy Chalcones and Analogues from Asaronaldehyde: Structure–Activity Relationship, Eur. J. Med. Chem., 2013, 62, 435–442, DOI:10.1016/j.ejmech.2013.01.018.
- P. Basu and C. Maier, Phytoestrogens and Breast Cancer: In Vitro Anticancer Activities of Isoflavones, Lignans, Coumestans, Stilbenes and Their Analogs and Derivatives, Biomed. Pharmacother., 2018, 107, 1648–1666, DOI:10.1016/j.biopha.2018.08.100.
- K. L. Ameta, N. S. Rathore and B. Kumar, Synthesis and in Vitro Anti Breast Cancer Activity of Some Novel 1,5-Benzothiazepine Derivatives, J. Serb. Chem. Soc., 2012, 77, 725–731 CrossRef CAS.
- S. K. Lee, G. S. Chang, I. H. Lee, J. E. Chung, K. Y. Sung and K. T. No, The Preadme: Pc-Based Program For Batch Prediction of Adme Properties, EuroQSAR 2004, Istanbul, Turkey, 2004, pp. 9.5–10 Search PubMed.
- D. Molehin, F. Rasha, R. L. Rahman and K. Pruitt, Regulation of Aromatase in Cancer, Mol. Cell. Biochem., 2021, 476, 2449–2464, DOI:10.1007/s11010-021-04099-0.
- G. P. Williams and P. D. Darbre, Low-Dose Environmental Endocrine Disruptors, Increase Aromatase Activity, Estradiol Biosynthesis and Cell Proliferation in Human Breast Cells, Mol. Cell. Endocrinol., 2019, 486, 55–64, DOI:10.1016/j.mce.2019.02.016.
- L. Gu, S. T. Saha, J. Thomas and M. Kaur, Targeting Cellular Cholesterol for Anticancer Therapy, FEBS J., 2019, 286, 4192–4208, DOI:10.1111/febs.15018.
- N. Thomford, D. Senthebane, A. Rowe, D. Munro, P. Seele, A. Maroyi and K. Dzobo, Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery, Int. J. Mater. Sci., 2018, 19, 1578, DOI:10.3390/ijms19061578.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02882a |
| This journal is © The Royal Society of Chemistry 2024 |