
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew diterpene lactone derivatives from Aphanamixis polystachya leaves inhibit nitric oxide production in RAW 264.7 cells†
Ngo Anh Bangab,
Duong Thi Hai Yena,
Dan Thi Thuy Hanga,
Pham Hai Yena,
Nguyen Huy Hoanga,
Do Thi Tranga,
Duong Thi Dunga,
Nguyen Thi Cuca,
Nguyen The Cuongc,
Nguyen Xuan Nhiemab,
Bui Huu Taiab and
Phan Van Kiem *ab
*ab
aInstitute of Marine Biochemistry, Vietnam Academy of Science and Technology (VAST), 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: phankiem@yahoo.com
bGraduate University of Science and Technology, VAST, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
cInstitute of Ecology and Biological Resources, VAST, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
First published on 1st July 2024
Abstract
Phytochemical studies on Aphanamixis plants have attracted considerable attention over the past few decades due to the structural diversities and significant biological activities of terpenoids produced by these plants. In the present study, five new acyclic diterpene lactone derivatives, aphanamixionolides A–E (1–5), and three known tirucallane-type triterpenes, namely, piscidinol A (6), hispidone (7), and bourjotinolone A (8), were isolated from the leaves of Aphanamixis polystachya. Their structures were elucidated by comprehensive analyses of HR-ESI-MS and NMR spectroscopic data and by comparison with those reported in the literature. Absolute configurations of the new compounds were determined by experimental and TD-DFT calculated ECD spectra. Compounds 1–8 inhibited NO production with IC50 values of 10.2–37.7 μM, which are comparable to positive control L-NMMA (IC50: 31.5 μM).
Introduction
The genus Aphanamixis (Meliaceae family) is widely distributed in tropical regions of Asia, such as southern China, India, and Southeast Asian countries.1 The leaves, fruits, stem barks, and roots of A. grandifolia and A. polystachya are traditionally used for curing various cancers and inflammations as well as rheumatism and pain.2,3 Phytochemical studies on Aphanamixis plants have attracted considerable attention over the past few decades and mostly focused on the two above-mentioned Aphanamixis species based on their traditional uses.1 The Aphanamixis plants produced terpenoids including triterpenes, diterpenes, and limonoids, which have gained interest due to their structures with high degrees of oxidation, rearrangement and/or dimerization of original backbones.2–6 Additionally, terpenoids produced by Aphanamixis exhibited potent cytotoxic, insecticidal, and anti-inflammatory activities.1 The structural diversities and significant anti-inflammatory activities inspired our phytochemical investigations of Aphanamixis plants. Herein, we describe the isolation and structure elucidation of five new acyclic diterpene lactone derivatives and three known tirucallane-type triterpenes from the leaves of A. polystachya. These compounds were also evaluated for their anti-inflammatory activities by inhibiting NO production in LPS-activated RAW 264.7 cells.Experimental
General experimental procedures
Melting points were measured using a Mel-Temp 3.0 apparatus. ECD spectra were recorded using a ChiraScan circular dichroism spectrometer. Optical rotation was measured using a Jasco P2000 polarimeter. HR-ESI-MS spectra were recorded using an Agilent 6530 Accurate-Mass QTOF. NMR spectra were recorded using a Bruker AvanceNEO 600 MHz FT-NMR spectrometer. Semi-preparative HPLC was performed using an Agilent 1260 infinity II system equipped with a YMC J'sphere ODS-H80 (20 × 250 mm, 4 μm) HPLC column, running at flow rate of 3 mL min−1. Thin-layer chromatography was carried out on silica gel or reversed phase C18 (RP-18) pre-coated plates. Gravity column chromatography was performed using silica gel (40–63 μm) or RP-18 (150 μm) as the stationary phase.Plant material
The leaves of Aphanamixis polystachya (Wall.) R. Parker were collected in September 2022 at Tamdao National Park, Vinh Phuc province, Vietnam, and taxonomically identified by Dr Nguyen The Cuong, Institute of Ecology and Biological Resources, VAST. Voucher specimens (No. NCCT-P125L) were kept at the Institute of Marine Biochemistry, VAST.Extraction and isolation
Dried powdered A. polystachya leaves (5 kg) were extracted with methanol in an ultrasonic bath at room temperature for three times (15 L MeOH and 1 h for each time). After the removal of methanol, the residue (500 g) was suspended with water (3.0 L) and successively separated with n-hexane and dichloromethane. The dichloromethane soluble fraction (100 g) was separated on a silica gel column and eluted with a gradient system of dichloromethane/methanol (40/1–5/1, v/v) to give four fractions, APLD1–APLD4. Fraction APLD3 was separated on a RP-18 column and eluted with acetone/water (1/2, v/v) to give five fractions, APLD3A–APLD3E. Fraction APLD3C was purified by semi-preparative HPLC using acetonitrile/water (7/3, v/v) to give compound 8 (4.0 mg, tR 52.7 min). Fraction APLD3E was separated on a silica gel column and eluted with n-hexane/acetone (5/1, v/v) to give three fractions, APLD3E1–APLD3E3. Fraction APLD3E1 was purified by semi-preparative HPLC using acetonitrile/water (9/1, v/v) to give compound 7 (6.5 mg, tR 61.2 min). Fraction APLD3E2 was also purified by semi-preparative HPLC using acetonitrile/water (9/1, v/v) to give compound 6 (5.0 mg, tR 53.3 min). Fraction APLD4 was chromatographed using a silica gel column and eluted with n-hexane/acetone (6/1, v/v) to give three fractions, APLD4A–APLD4C. Fraction APLD4B was purified by semi-preparative HPLC using acetonitrile/water (6/4, v/v) to give three compounds, 1 (4.0 mg, tR 42.2 min), 5 (24.2 mg, tR 45.2 min), and 2 (5.0 mg, tR 48.9 min). Fraction APLD4C was purified by semi-preparative HPLC using acetonitrile/water (45/55, v/v) to give two compounds, 3 (6.2 mg, tR 63.2 min) and 4 (7.9 mg, tR 68.9 min) (Fig. 1).| (5S,11R)-Aphanamixionolide A | (1) |
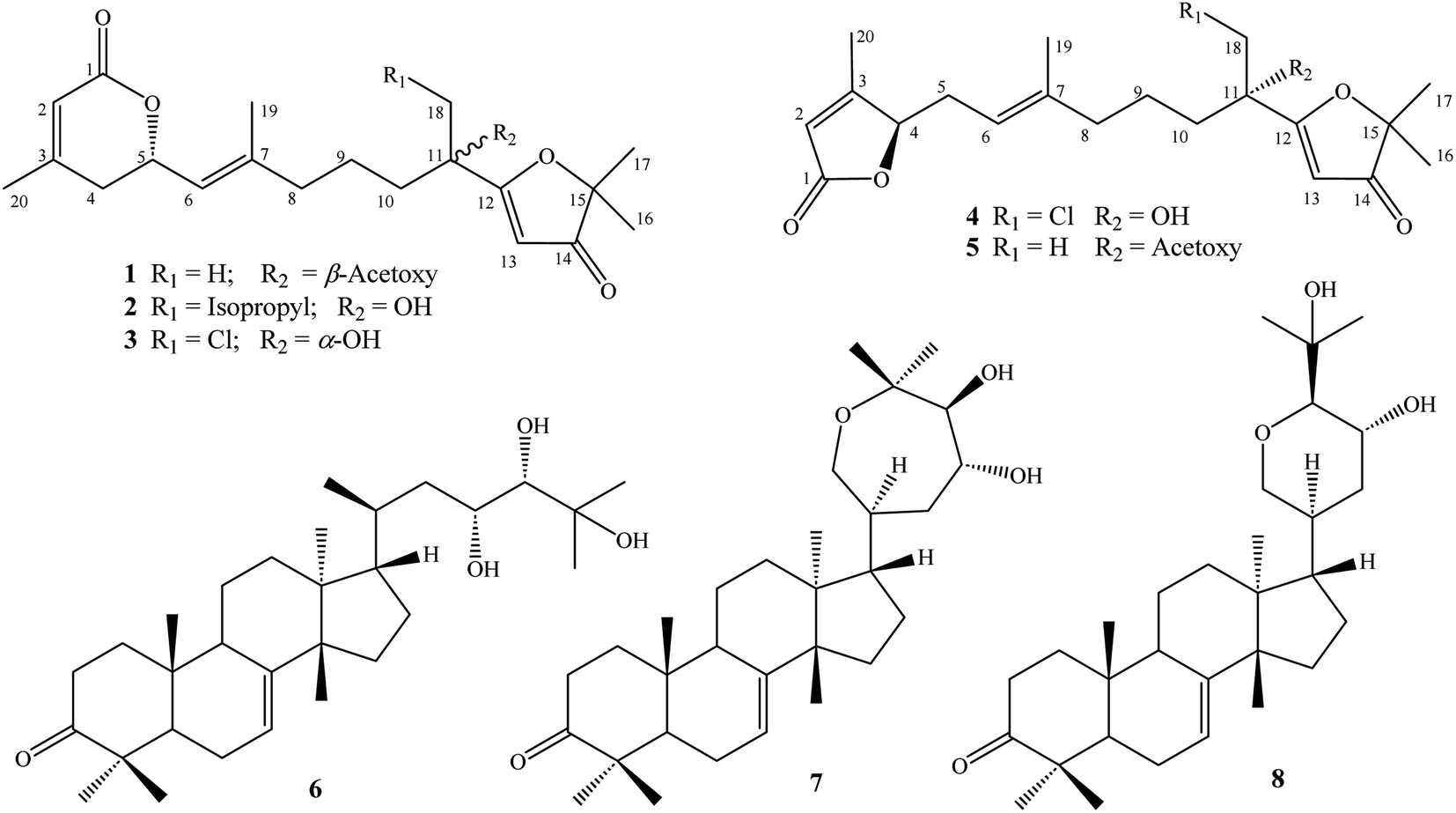 | ||
| Fig. 1 Structures of compounds 1–8 isolated from the leaves of Aphanamixis polystachya. | ||
White amorphous powder; mp 57.2–57.6; [α]28D: +51.6 (c 0.1, MeOH); ECD (MeOH) θ(λ nm): −5.4(215), −6.3(249) mdeg; HR-ESI-MS: m/z 391.2122 [M + H]+ (calcd for [C22H31O6]+, 391.2115); 1H- and 13C-NMR data are given in Table 1.
| (5S,11R/11S)-Aphanamixionolide B | (2) |
| No. | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| a Observed in a pair of signals: 74.12/74.08.b Observed in a pair of signals: 122.54/122.51.c Observed in a pair of signals: 142.01/141.77.d Observed in a pair of signals: 39.35/39.27.e Observed in a pair of signals: 20.97/20.90.f Observed in a pair of signals: 40.34/40.30.g Observed in a pair of signals: 76.11/76.05.h Observed in a pair of signals: 89.64/89.61.i Observed in a pair of signals: 48.16/48.13.k Observed in a pair of signals: δC 24.36/δH 0.99 and δC 24.12/δH 0.89. | ||||||||||
| 1 | 165.2 | — | 165.2 | — | 165.3 | — | 173.2 | — | 173.0 | — |
| 2 | 116.7 | 5.82 (s) | 116.7 | 5.82 (s) | 116.7 | 5.83 (s) | 117.5 | 5.81 (s) | 117.4 | 5.81 (s) |
| 3 | 157.0 | — | 157.0 | — | 157.1 | — | 168.1 | — | 168.1 | — |
| 4 | 35.1 | 2.39 (dd, 18.0, 10.8) | 35.1 | 2.39 (dd, 18.0, 10.8) | 35.1 | 2.39 (dd, 18.0, 10.8) | 84.1 | 4.89 (t, 4.8) | 84.2 | 4.88 (t, 5.4) |
| 2.22 (dd, 18.0, 3.6) | 2.22 (dd, 18.0, 3.6) | 2.20 (dd, 18.0, 3.6) | ||||||||
| 5 | 74.1 | 5.11 (m) | 74.1a | 5.10 (m) | 74.1 | 5.10 (m) | 29.9 | 2.71 (m) | 30.2 | 2.68 (m) |
| 2.32 (m) | 2.32 (m) | |||||||||
| 6 | 122.6 | 5.33 (d, 9.0) | 122.5b | 5.32 (d, 8.4) | 122.8 | 5.33 (d, 9.0) | 116.7 | 4.98 (t, 7.8) | 116.9 | 5.05 (t, 7.2) |
| 7 | 141.6 | — | 142.0c | — | 141.4 | — | 139.2 | — | 138.9 | — |
| 8 | 39.1 | 2.04 (m) | 39.3d | 2.04 (m) | 39.1 | 2.03 (m) | 39.4 | 2.00 (m) | 39.4 | 2.00 (m) |
| 2.08 (m) | ||||||||||
| 9 | 21.1 | 1.46 (m) | 21.0e | 1.30 (m) | 21.0 | 1.40 (m) | 21.3 | 1.32 (m) | 21.3 | 1.40 (m) |
| 1.57 (m) | 1.66 (m) | 1.58 (m) | ||||||||
| 10 | 38.1 | 1.90 (m) | 40.3f | 1.62 (m) | 35.9 | 1.68 (m) | 35.9 | 1.61 (m) | 38.0 | 1.84 (m) |
| 1.73 (m) | 1.75 (m) | 1.68 (m) | ||||||||
| 11 | 79.2 | — | 76.0g | — | 75.3 | — | 75.3 | — | 79.1 | — |
| 12 | 190.9 | — | 194.5 | — | 190.4 | — | 190.7 | — | 191.0 | — |
| 13 | 100.1 | 5.45 (s) | 101.4 | 5.64 (s) | 103.1 | 5.45 (s) | 102.9 | 5.72 (s) | 100.0 | 5.45 (s) |
| 14 | 206.7 | — | 206.8 | — | 206.6 | — | 206.7 | — | 206.7 | — |
| 15 | 89.3 | — | 89.6h | — | 90.2 | — | 90.1 | — | 89.3 | — |
| 16 | 22.6 | 1.38 (s) | 23.0 | 1.39 (s) | 22.9 | 1.40 (s) | 22.9 | 1.40 (s) | 22.6 | 1.36 (s) |
| 17 | 22.6 | 1.36 (s) | 22.6 | 1.38 (s) | 22.7 | 1.39 (s) | 22.7 | 1.39 (s) | 22.6 | 1.35 (s) |
| 18 | 22.3 | 1.68 (s) | 48.1i | 1.60 (m) | 51.0 | 3.92 (d, 11.4) | 50.9 | 3.91 (d, 11.4) | 22.3 | 1.67 (s) |
| 1.77 (m) | 3.61 (d, 11.4) | 3.62 (d, 11.4) | ||||||||
| 19 | 16.6 | 1.69 (s) | 16.6 | 1.67 (s) | 16.4 | 1.67 (s) | 16.0 | 1.60 (s) | 16.1 | 1.60 (s) |
| 20 | 23.0 | 2.00 (s) | 23.0 | 1.99 (s) | 23.0 | 1.98 (s) | 13.9 | 2.05 (s) | 13.9 | 2.05 (s) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| 11-Acetoxy | 18-Isopropyl | 11-Acetoxy | ||||||||
| 1′ | 169.3 | — | 24.3 | 1.75 (m) | 169.3 | — | ||||
| 2′ | 21.4 | 2.07 (s) | 24.4k | 0.99 (d, 7.0) | 21.4 | 2.07 (s) | ||||
| 3′ | 24.4k | 0.99 (d, 7.0) | ||||||||
White amorphous powder; mp 73.1–73.9; [α]28D: +19.3 (c 0.1, MeOH); ECD (MeOH) θ(λ nm): −6.3(215), −6.6(250), +0.9(299), −0.5(338) mdeg; HR-ESI-MS: m/z 391.2492 [M + H]+ (calcd for [C23H35O5]+, 391.2479); 1H- and 13C-NMR data are given in Table 1.
| (5S,11S)-Aphanamixionolide C | (3) |
White amorphous powder; mp 78.1–79.0; [α]28D: −26.5 (c 0.1, MeOH); ECD (MeOH) θ(λ nm): −4.1(215), −6.2(252) mdeg; HR-ESI-MS: m/z 383.1627 [M + H]+ (calcd for [C20H2835ClO5]+, 383.1620) and m/z 385.1605 [M + 2amu + H]+ (calcd for [C20H2837ClO5]+, 385.1605); 1H- and 13C-NMR data are given in Table 1.
| (4R,11S)-Aphanamixionolide D | (4) |
White amorphous powder; mp 76.5–77.3; [α]28D: −40.2 (c 0.1, MeOH); ECD (MeOH) θ(λ nm): −11.4(211), −2.7(241) mdeg; HR-ESI-MS: m/z 383.1625 [M + H]+ (calcd for [C20H2835ClO5]+, 383.1620) and m/z 385.1600 [M + 2amu + H]+ (calcd for [C20H2837ClO5]+, 385.1605); 1H- and 13C-NMR data are given in Table 1.
| (4R,11S)-Aphanamixionolide E | (5) |
White amorphous powder; mp 55.4–56.1; [α]28D: −33.1 (c 0.1, MeOH); ECD (MeOH) θ(λ nm): −22.0(210) mdeg; HR-ESI-MS: m/z 391.2118 [M + H]+ (calcd for [C22H31O6]+, 391.2115); 1H- and 13C-NMR data are given in Table 1.
TD-DFT calculated ECD spectra
Conformational searches and geometric equilibrium were carried out using the Spartan 18 program (Wavefunction Inc., Irvine, CA, USA). Possible conformations were optimized and subjected to TDDFT calculations using the Gaussian 16 program (Gaussian Inc., Wallingford, CT, USA). The calculated ECD spectra were composed based on the Boltzmann distribution of the stable conformers using the SpecDis v1.71 software (University of Wuerzburg, Wuerzburg, Germany). The calculations were performed as described previously.7,8 Briefly, five pairs of dia-stereoisomers, namely, 1a-(5S,11S)/1b-(5S,11R), 2a-(5S,11S)/2b-(5S,11R), 3a-(5S,11S)/3b-(5S,11R), 4a-(4R,11S)/4b-(4S,11S), and 5a-(4R,11R)/5b-(4R,11S) were submitted to conformational searches at the Molecular Mechanics MMFF and performed equilibrium geometry with semi-empirical PM3 set. Conformers were collected with their relative energy lower than 40 kJ mol−1. The top 20 stable conformers were selected and optimized by DFT calculations at the B3LYP/6-31G(d,p) basis set. The solvent effects were determined by integral equation formalism polarizable continuum model (IEFPCM) calculations with methanol as the solvent. The optimized conformers were then subjected to TD-DFT calculations using B3LYP/6-31G(d,p) or wB97XD/6-31G(d,p) functions and methanol as solvent effects in an IEFPCM. The ECD spectra at 30 excited states for each conformer were recorded and summed to obtain theoretical ECD spectra of each stereoisomer. The ECD spectra of corresponding enantiomers 1c/1d, 2c/2d, 3c/3d, 4c/4d, and 5c/5d were composed by mirror imaging those of 1a/1b, 2a/2b, 3a/3b, 4a/4b, and 5a/5b, respectively.Nitric oxide assay
Experiments were conducted following the same procedure as described in previous reports and referred to ESI.†Results and discussion
The methanol extract of A. polystachya was well mixed with water and successively separated with n-hexane and dichloromethane. The dichloromethane soluble fraction was subjected to gravity column chromatography using silica gel or RP-18 resin as the stationary phase, and further purified by semipreparative HPLC to give five new diterpenoids (1–5) and three known triterpenoids (6–8).Compound 1 was obtained as a white amorphous powder. Its molecular formula, C22H30O6, was determined by protonated ion [M + H]+ at m/z 391.2122 in the HR-ESI-MS (calcd for [C22H31O6]+, 391.2115), indicating eight degrees of unsaturation. The 1H-NMR spectrum of 1 contained six singlet methyl groups [δH 2.07, 2.00, 1.69, 1.68, 1.38, and 1.36 (each, 3H)], three olefinic protons [δH 5.45 (1H, s), 5.33 (1H, d, J = 9.0 Hz), and 5.82 (1H, s)], and one oxygenated methine group [δH 5.11 (1H, m)]. The 13C-NMR and HSQC spectra of 1 showed 22 carbons including six methyl groups, four methylenes, four methines, and eight non-protonated carbons. The methyl [δC 21.4/δH 2.07 (3H, s)] and carbonyl signals (δC 169.3) were assigned to an acetoxy group. Other 20 carbons suggested for a diterpenoid backbone. The COSY spectrum of 1 revealed two coupled systems, including H2-4 (δH 2.39 and 2.22)/H-5 (δH 5.11)/H-6 (δH 5.33) and H2-8 (δH 2.04)/H2-9 (δH 1.46)/H2-10 (δH 1.90). The HMBC correlations between H3-20 (δH 2.00) and C-2 (δC 116.7)/C-3 (δC 157.0)/C-4 (δC 35.1), and H-2 (δH 5.82) and C-1 (δC 165.2) indicated a terminal carboxylate functional group at C-1 and a double bond at C-2/C-3. The HMBC correlations between H3-19 (δH 1.69) and C-6 (δC 122.6)/C-7 (δC 141.6)/C-8 (δC 39.1) indicated another double bond at C-6/C-7. Furthermore, a carbon chemical shift of C-14 (δC 206.7) and the HMBC correlations between H3-16 (δH 1.38)/H3-17 (δH 1.36) and C-15 (δC 89.3)/C-14 (δC 206.7) confirmed a ketone functional group at C-14. Then, the HMBC correlations between H-13 (δH 5.45) and C-14 (δC 206.7)/C-12 (δC 190.9), and H3-18 (δH 1.68) and C-10 (δC 38.1)/C-11 (δC 79.2)/C-13 (δC 100.1) suggested the last double bond at C-12/C-13 and fully established an acyclic diterpene backbone. Carbon chemical shifts of C-5 (δC 74.1), C-11 (δC 79.2), C-12 (δC 190.9), and C-15 (δC 89.3) indicated oxygen-bearing carbons. Additionally, de-shielded signals of C-12 (δC 190.9) and C-15 (δC 89.3) suggested the existence of an ether bridge between these carbons, forming a furan-3-one moiety. The strong downfield shifted signals of oxygenated olefinic carbon (C-12: δC 190.9) and carbinol carbon (C-15: δC 89.3) in the furan-3-one moiety are consistent with those previously reported in novel nemoralisin-type diterpenoids from Aphanamixis plants.9–12 Although interactions between H-5 (δH 5.11) and C-1 (δC 165.2) are not clearly observed in the HMBC spectrum, the chemical shift values at those positions (δH-5 5.11, δC-5 74.1, δC-1 165.2) are consistent with the presence of lactone bridge between C-1 and C-5 as compared to compound 2 (showing weak HMBC correlation between H-5 and C-1). The acetoxy group, therefore, was assigned at the last oxygen bearing carbon C-11. The geometry of double bond at C-6/C-7 was determined to be E-configuration based on NOESY correlations of H3-19 (δH 1.69)/H-5 (δH 5.11) and H-6 (δH 5.33)/H2-8 (δH 2.04). Later, absolute configurations at C-5 and C-11 were elucidated by experimental and TD-DFT calculated ECD spectra. The theoretical ECD spectra of the four possible stereoisomers of 1, namely, (5S,11S)-1a, (5S,11R)-1b, (5R,11R)-1c, and (5R,11S)-1d were computed. The experimental ECD spectrum of 1 displayed negative Cotton effects at wavelengths of 215 nm (−5.4 mdeg) and 249 nm (−6.3 mdeg), which is consistent with those calculated ECD spectrum for (5S,11R)-1b isomers. Consequently, the structure of compound 1 was completely determined and it was named (5S,11R)-aphanamixionolide A.
Compound 2 was obtained as a white amorphous powder. Its molecular formula, C23H35O5, was determined by protonated ion [M + H]+ at m/z 391.2492 in the HR-ESI-MS (calcd for [C23H35O5]+, 391.2479), indicating seven degrees of unsaturation. The 1H and 13C-NMR data of 2 showed close similarity with those of 1, indicating the same acyclic diterpenoid backbone. The NMR data of 2 exhibited signals of an isopropyl group instead of the acetyl group as in compound 1. Additionally, the signals of a methyl group (C-18, δC 22.3/δH 1.68) in 1 were replaced by the signals of a methylene group (C-18, δC 48.1/δH 1.77 and 1.60) in 2, suggesting the position of the isopropyl group at C-18. This deduction was confirmed by COSY correlations of H2-18 (δH 1.77 and 1.60)/H-1′ (δH 1.75)/H3-2′,3′ (δH 0.99) and HMBC correlations between H3-2′,3′ (0.99) and C-1′ (δC 24.3)/C-18 (δC 48.1), H2-18 (δH 1.77 and 1.60) and C-10 (δC 40.3)/C-11 (δC 76.0)/C-12 (δC 194.5). The upfield-shifted signal of C-11 (δC 76.0) in compound 2 compared to that in 1 (C-11: δC 79.2) is consistent with the replacement of an acetoxy group with a hydroxy group at this carbon. The weak HMBC correlation between H-5 (δH 5.10) and C-1 (δC 165.2) further confirmed the lactone bridge between C-1 and C-5. The NOESY correlations of H3-19 (δH 1.67)/H-5 (δH 5.10) and H-6 (δH 5.32)/H2-8 (δH 2.04) confirmed the E-configuration of a double bond at C-6/C-7. Careful examination of the 13C NMR spectra of 2 showed that several carbons significantly divided into a pair of closely chemical shift values (ΔδC < 0.2 ppm, Table 1 and Fig. S16, S17†). This evidence suggested that compound 2 was a pair of dia-stereoisomers at chiral centers C-5 and C-11. The TD-DFT calculation ECD spectra for the four possible stereoisomers (5S,11S)-2a, (5S,11R)-2b, (5R,11R)-2c, and (5R,11S)-2d were performed and compared with the experimental ECD data. The results indicated that compound 2 possessed 5S-configuration (as isomers 2a/2b) and a pair of dia-stereoisomers were formed by difference in absolute configuration at C-11, 11R or 11S. Compound 2, therefore, was determined and it was named (5S,11R/11S)-aphanamixionolide B.
The HR-ESI-MS spectra of 3 showed a pair of isotope protonated molecular ion peaks at m/z 383.1627 [M + H]+ and m/z 385.1605 [M + 2amu + H]+ with an intensity ratio around 3/1, which indicated a molecular formula of C20H27ClO5, containing a chlorine atom (calcd for [C20H2835ClO5]+, 383.1620; and calcd for [C20H2837ClO5]+, 385.1605). The 1H and 13C-NMR data of 3 indicated an acyclic diterpene as compounds 1 and 2. The 1H-NMR spectrum of 3 revealed the presence of four singlet methyl groups [δH 1.98, 1.67, 1.40, and 1.39 (each 3H, s)], suggesting a chlorine atom link to one of the five terminal methyl groups in the diterpene skeleton. Comparison of the NMR data of 3 with those of 1 and 2 showed the difference at signals of methylene group C-18 [δC 51.0/δH 3.92 and 3.61 (each 1H, d, J = 11.4 Hz)], suggesting a chlorine atom link to C-18. This deduction was then confirmed by HMBC correlations between H2-18 (δH 3.92 and 3.61) and C-10 (δC 35.9)/C-11 (δC 75.3)/C-12 (δC 190.4). The NOESY correlations of H3-19 (δH 1.67)/H-5 (δH 5.10) and H-6 (δH 5.33)/H-8 (δH 2.03 and 2.08) confirmed the E-configuration of the double bond at C-6/C-7. Later, absolute configurations 5S and 11S (as isomer 3a) were demonstrated by experimental and TD-DFT calculated ECD spectra, as shown in Fig. 3. Consequently, the structure of 3 was determined and it was named (5S,11S)-aphanamixionolide C.
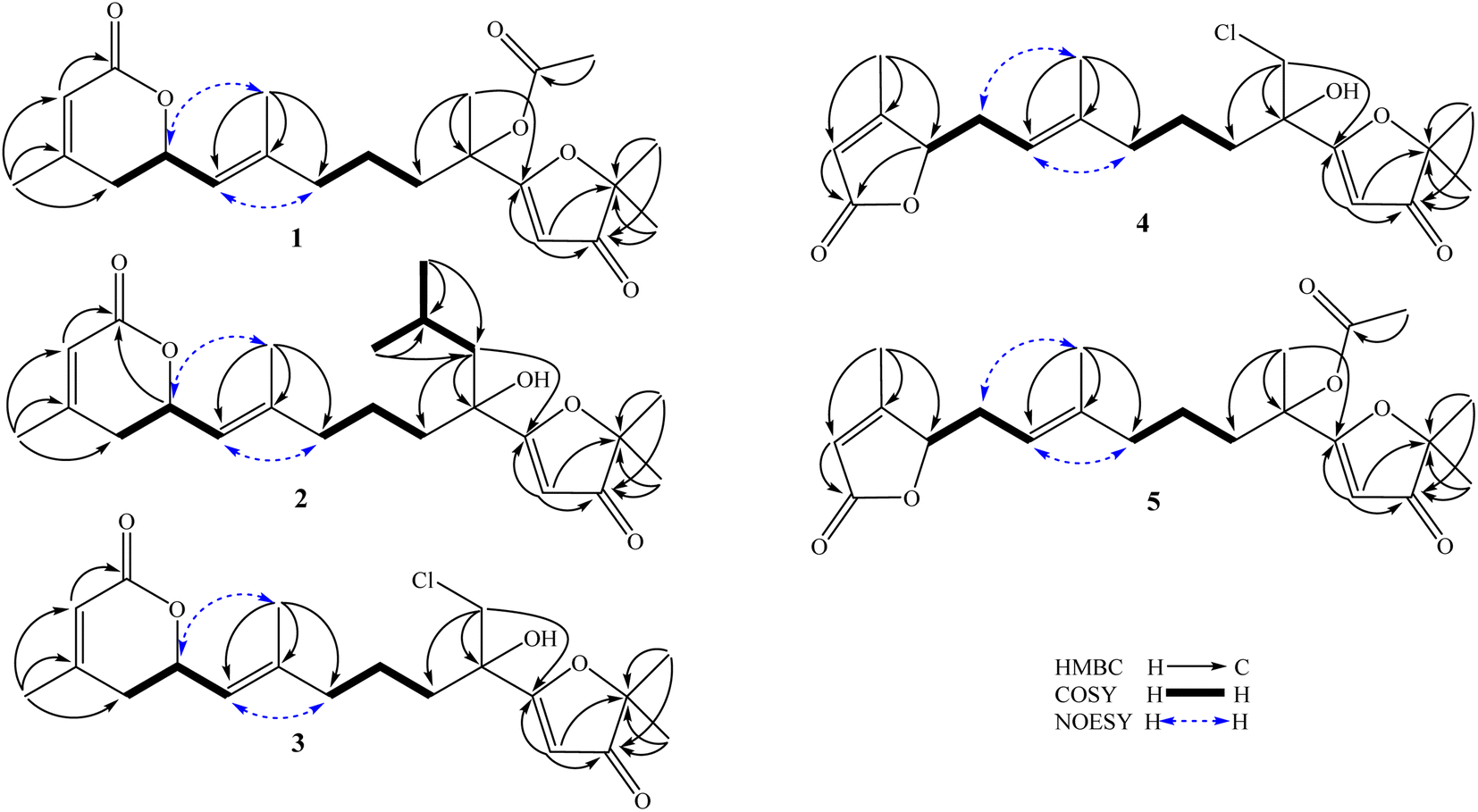 | ||
| Fig. 2 Key HMBC, COSY, and NOESY correlations of compounds 1–5. | ||
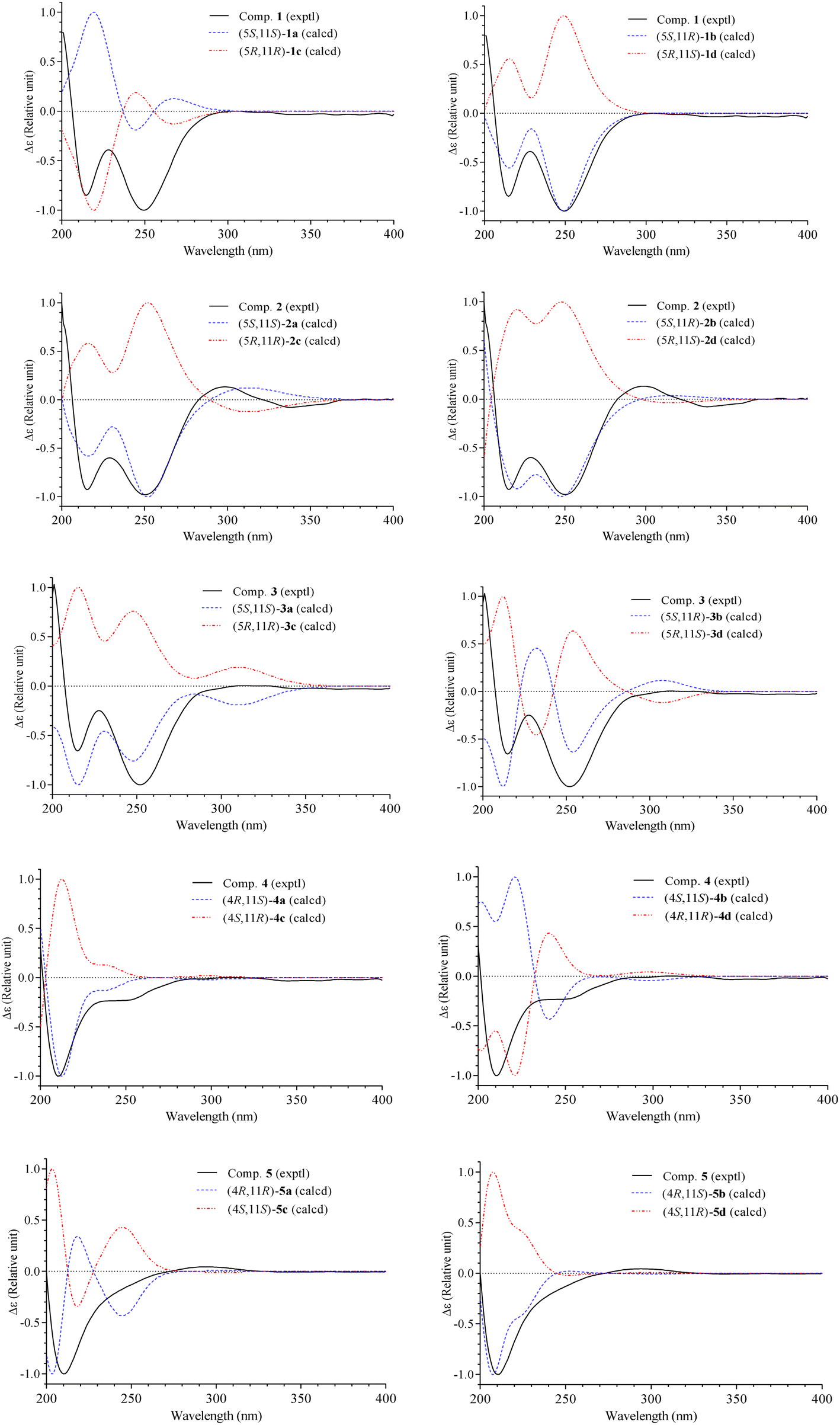 | ||
| Fig. 3 Experimental ECD spectra of 1–5 and TD-DFT calculated ECD spectra for their possible stereoisomers. | ||
Compound 4 was obtained as a white amorphous powder. The HR-ESI-MS spectra of 4 closely resembled those of 3 by a pair of isotope protonated molecular ion peaks at m/z 383.1625 [M + H]+ and m/z 385.1600 [M + 2amu + H]+ with an intensity ratio around 3/1. This evidence indicated that compound 4 had the same molecular formula as that of 3, C20H27ClO5 (calcd for [C20H2835ClO5]+, 383.1620; and calcd for [C20H2837ClO5]+, 385.1605). The 1H and 13C-NMR data of 4 were revealed to be different from those of 3 by signals related to the lactone ring (C-1 to C-7 and C-20, Table 1). The sequencing COSY correlations of H-6 (δH 4.98)/H2-5 (δH 2.71 and 2.32)/H-4 (δH 4.89) and chemical shift values of the methine group C-4 (δC 84.1) indicated that an oxygen-bearing carbon was at C-4 in compound 4 instead of C-5, as shown in compound 3. The HMBC correlations between H3-20 (δH 2.05) and C-2 (δC 117.5)/C-3 (δC 168.1)/C-4 (δC 84.1) confirmed the presence of a double bond at C-2/C-3. The HMBC correlations between H-2 (δH 5.81)/H-4 (δH 4.89) and C-1 (δC 173.2) were demonstrated for a lactone bridge between C-1 and C-4. Compound 4, therefore, contained a γ-lactone instead of a δ-lactone moiety in compounds 1–3. The NOESY correlations of H3-19 (δH 1.60)/H-5 (δH 2.71 and 2.32) and H-6 (δH 4.98)/H-8 (δH 2.00) confirmed the E-configuration of the double bond at C-6/C-7. The absolute configurations 4R and 11S (as isomer 4a) were demonstrated by experimental and TD-DFT calculated ECD spectra, as shown in Fig. 3. Consequently, the structure of 4 was determined and it was named (4R,11S)-aphanamixionolide D.
Compound 5 was obtained as a white amorphous powder. The HR-ESI-MS spectra of 4 closely resembled those of 1 by protonated molecular ion peaks at m/z 391.2118 [M + H]+, indicating the same molecular formula as that of 1, C22H30O6 (calcd for [C22H31O6]+, 391.2115). The NMR spectra data of 5 indicated signals of acetoxy group (δC 169.3 and δC 21.4/δH 2.07) and an acyclic diterpenoid backbone as compounds 1–4 (Table 1). Additionally, the 13C-NMR spectral data of 5 exhibited partially similar carbon chemical shifts with both compounds 1 (from C-8 to C-19 and acetoxy group) and 4 (from C-1 to C-7 and C-20). This evidence indicated that compound 5 possessed two structural fragments, namely, an acetoxylated furan-3-one moiety (as compound 1) and a γ-lactone moiety (as compound 4). The planar structure of 5, including the E-geometric configuration of double bond at C-6/C-7, was then confirmed by analysis of HMBC, COSY, and NOESY correlations, as shown in Fig. 2. Finally, the absolute configurations 4R and 11S (as isomer 5b) were demonstrated by experimental and TD-DFT calculated ECD spectra. Consequently, the structure of 4 was determined and it was named (4R,11S)-aphanamixionolide E.
Other known compounds were determined to be tirucallane-type triterpenes including piscidinol A (6),13,14 hispidone (7),15 and bourjotinolone A15,16 by consistence of their NMR spectral data with those reported in the literature.
All isolates (1–8) were evaluated for their anti-inflammatory activities by inhibiting nitric oxide production in LPS-activated RAW 264.7 cells. First, the effects of compounds on the cell viability were examined. At a concentration of 50 μM, compounds 1–8 did not show significant cytotoxicity towards RAW 264.7 cells by MTT tests. The NO inhibitory activity was then evaluated at serial diluted concentrations of 0.4–50 μM. As shown in Table 2, compounds 1–8 inhibited NO production with IC50 values of 10.2–37.7 μM, comparable to positive control L-NMMA (IC50: 31.5 μM). Our results are consistent with previous reports that terpenoid compositions of Aphanamixis plants potentially inhibit LPS-induced NO production activities.2,5,17,18
Conclusions
Phytochemical investigation of the leaves of A. polystachya identified five new acyclic diterpene lactone derivatives (1–5) and three known tirucallane-type triterpenes (6–8). The chemical structures of 1–5 contained two unique structural fragments a γ-lactone/δ-lactone and a furan-3-one moiety, which are rarely found in the nature. Compounds 1–8 exhibited inhibitory effects on NO production in LPS-activated RAW 264.7 cells with IC50 values in the range of 10.2–37.7 μM, comparable to positive control L-NMMA.Author contributions
PV Kiem, NX Nhiem, BH Tai, PH Yen contributed to research idea, structure elucidation, and writing; NT Cuong contributed to sample collection and taxonomical identification. NA Bang, DTH Yen, NH Hoang, DT Trang, DTT Hang, DT Dung, NT Cuc contributed to isolation and NO assay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by Vietnam Academy of Science and Technology (VAST) under grant number NCXS01.02/22-24.Notes and references
- G. W. Wang, H. Z. Jin and W. D. Zhang, Phytochem. Rev., 2013, 12, 915–942 CrossRef CAS.
- X. Yang, S. L. Wu, B. J. Li, Y. P. Li, H. P. He and F. W. Dong, Fitoterapia, 2023, 171, 105709 CrossRef CAS PubMed.
- X. B. Wang, Y. Zhang, J. S. Wang, Y. C. Gu and L. Y. Kong, Tetrahedron Lett., 2013, 54, 6023–6028 CrossRef CAS.
- P. Zhang, S. Xue, W. Huang, C. Wang, Z. Cui, J. Luo and L. Kong, Org. Chem. Front., 2021, 8, 566–571 RSC.
- B. J. Yang, S. R. Fan, J. Y. Cai, Y. T. Wang, C. X. Jing, J. J. Guo, D. Z. Chen and X. J. Hao, J. Org. Chem., 2020, 85, 8597–8602 CrossRef CAS PubMed.
- J. Y. Cai, D. Z. Chen, S. H. Luo, N. C. Kong, Y. Zhang, Y. T. Di, Q. Zhang, J. Hua, S. X. Jing, S. L. Li, S. H. Li, X. J. Hao and H. P. He, J. Nat. Prod., 2014, 77, 472–482 CrossRef CAS PubMed.
- D. T. Trang, D. T. T. Hang, D. T. Dung, N. T. Cuc, P. H. Yen, P. T. T. Huong, L. T. Huyen, N. T. Mai, N. X. Nhiem, B. H. Tai and P. Van Kiem, RSC Adv., 2022, 12, 10646–10652 RSC.
- D. T. T. Hang, D. T. Trang, D. T. Dung, D. T. H. Yen, N. H. Hoang, N. A. Bang, N. T. Cuc, N. X. Nhiem, P. T. T. Huong, B. H. Tai and P. V. Kiem, Phytochemistry, 2021, 190, 112889 CrossRef PubMed.
- F. H. Fang, W. J. Huang, S. Y. Zhou, Z. Z. Han, M. Y. Li, L. F. Liu, X. Z. Wu, X. J. Yao, Y. Li and C. S. Yuan, Eur. J. Org Chem., 2017, 2017, 4429–4433 CrossRef CAS.
- J. Liu, X. F. He, G. H. Wang, E. F. Merino, S. P. Yang, R. X. Zhu, L. S. Gan, H. Zhang, M. B. Cassera, H. Y. Wang, D. G. I. Kingston and J. M. Yue, J. Org. Chem., 2014, 79, 599–607 CrossRef CAS PubMed.
- X. Z. Wu, F. H. Fang, W. J. Huang, Y. Y. Shi, H. Q. Pan, L. Ning and C. S. Yuan, Fitoterapia, 2020, 140, 104431 CrossRef CAS PubMed.
- P. Zhang, L. Cui, Z. Cui, Z. Wang, P. Tang, J. Luo and L. Kong, Fitoterapia, 2022, 159, 105192 CrossRef PubMed.
- J. D. McChesney, J. Dou, R. D. Sindelar, D. K. Goins, L. A. Walker and R. D. Rogers, J. Chem. Crystallogr., 1997, 27, 283–290 CrossRef CAS.
- J. Puripattanavong, S. Weber, V. Brecht and A. W. Frahm, Planta Med., 2000, 66, 740–745 CrossRef CAS PubMed.
- S. D. Jolad, J. J. Hoffmann, J. R. Cole, M. S. Tempesta and R. B. Bates, J. Org. Chem., 1980, 45, 3132–3135 CrossRef CAS.
- W. Bai, H. Y. Yang, X. Z. Jiao, K. N. Feng, J. J. Chen and K. Gao, J. Nat. Prod., 2018, 81, 1777–1785 CrossRef CAS PubMed.
- H. J. Zhang, Y. M. Zhang, J. G. Luo, J. Luo and L. Y. Kong, Org. Biomol. Chem., 2015, 13, 7452–7458 RSC.
- B. H. Tai, N. A. Bang, P. H. Yen, D. T. H. Yen, N. T. Cuc, D. T. Dung, P. T. T. Huong, D. T. Trang, N. X. Nhiem, N. T. Cuong and P. V. Kiem, Phytochemistry, 2024, 220, 113997 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HR-ESI-MS, NMR, and ECD spectra of new compounds of all isolated compounds. See DOI: https://doi.org/10.1039/d4ra02968b |
| This journal is © The Royal Society of Chemistry 2024 |