
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel diamine-scaffold based N-acetylgalactosamine (GalNAc)–siRNA conjugate: synthesis and in vivo activities†
Qiang Li *ab,
Mingxin Dong*a and
Pu Chen*bc
*ab,
Mingxin Dong*a and
Pu Chen*bc
aDepartment of Medicinal Chemistry, School of Pharmacy, Qingdao University, Qingdao 266021, China. E-mail: sdhyliqiang@163.com; Mxdong64@qdu.edu.cn
bResearch and Development Department, NanoPeptide (Qingdao) Biotechnology Ltd, Qingdao, China
cDepartment of Chemical Engineering, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, ON, Canada. E-mail: p4chen@uwaterloo.ca
First published on 30th May 2024
Abstract
GalNAc-conjugated siRNA has shown remarkable potential in liver-targeted delivery in recent years. In general, tetrahydroxymethylmethane or other branching clusters constitute the basis of GalNAc's structure, which yields trivalent or tetravalent ligands. A novel diamine-scaffold GalNAc conjugate was synthesized and evaluated for its efficiency in siRNA administration. It exhibits comparable siRNA delivery effectiveness to a GalNAc NAG37 phase II clinical drug candidate targeting ANGPTL3. In addition, it exhibits more powerful silencing activity when connected to the 3′-end of the sense strand with an additional PS-linkage instead of a PO linkage between the ligand and the oligomer compared to a GalNAc L96 standard targeting TTR. Taken together, the incorporation of a diamine-scaffold into the GalNAc conjugate structure has potential in the field of gene therapy.
Introduction
Due to their particular physiological and physicochemical qualities, RNA-based therapies, including antisense oligonucleotides (ASOs), small interfering RNA (siRNA), and other RNA molecules, offer considerable potential.1–5 In fact, more than 80% of known disease-causing protein targets in the human genome are tricky to target with appropriate binding sites utilizing small molecules or antibodies.6 RNA-based therapeutics, especially ASO and siRNA which target mRNA translation or the function of long non-coding RNA (lncRNA), are not constrained by protein structure. They are increasingly playing crucial roles in treating “undruggable” diseases, such as rare diseases and metabolic diseases.7,8However, these oligonucleotides contain multiple negative charges, resulting in low cell uptake efficiency and limiting their potential for therapeutic efficacy.9 The development of RNA-based therapies relies greatly on developments in delivery technology.10–12 Currently, conjugated connection delivery systems (small molecule ligands, antibodies, and other compounds) are the most extensively employed delivery strategies for siRNA.13–16 Among them, N-acetylgalactosamine (GalNAc) conjugates are successful and widely used for liver-targeted siRNA delivery due to their strong interactions with the asialoglycoprotein receptor (ASGPR).13,17–19 Therefore, a number of siRNA medicines conjugated with different GalNAc ligands have been approved by the U.S. Food and Drug Administration (FDA) or have shown positive results in recent clinical investigations.20 For example, inclisiran (ALN-PCSsc), an siRNA conjugated to the GalNAc ligand L96, reduces low-density lipoprotein (LDL) cholesterol by directly inhibiting the synthesis of proprotein convertase subtilisin 9 (PCSK9) in hepatocytes.21,22
Anlylam, Arrowhead, Silence, Dicerna, and other siRNA pharmaceutical companies have developed a series of GalNAcylated oligonucleotides at the 5′-end, 3′-end, or middle of the sequence.19,23 On the one hand, the modified nucleoside phosphoramidite with an individual GalNAc residue connected to either the nucleobase or the 2′ or 3′ position on the ribose may be inserted at any position of the strand during solid phase synthesis.24 On the other hand, the trivalent or tetravalent GalNAc ligands might alternatively attached to either the 5′-end or 3′-end of the sense strand.19 The latter relies on core structures based on tetrahydroxymethylmethane, tris, γ-glutamyl dipeptide, or other branched clusters.18,19
Herein, we report the synthesis of novel diamine-scaffold-based GalNAc ligands and introduce them at the terminal end of the sense strand. Specifically, TG1 or TG2 was attached to the 5′-end or 3′-end of oligonucleotides, respectively (Fig. 1). Subsequent biological investigations revealed that TG1 had delivery capabilities comparable to those of NAG37. Furthermore, the silencing effect of TG2-modified siRNA is equivalent to that of L96-modified siRNA.
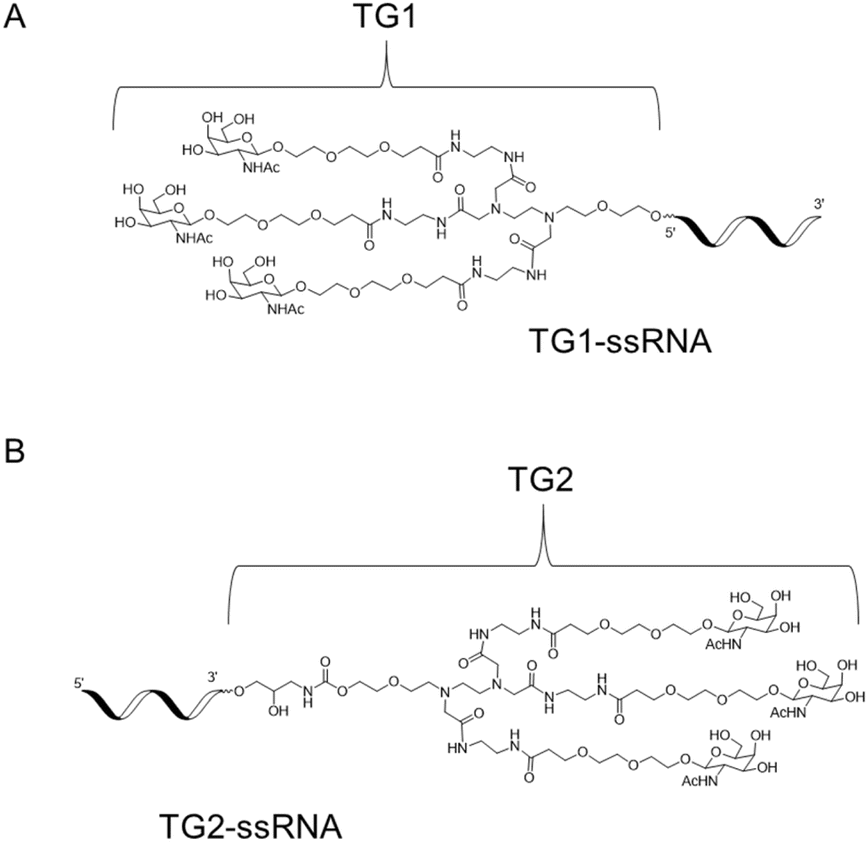 | ||
| Fig. 1 The structure of 5′-end GalNAc TG1 (A) and 3′-end GalNAc TG2 (B). | ||
Results
First, as indicated in Scheme 1, we created a trivalent GalNAc precursor 7. This precursor had a diamine core structure. According to the reported literature, starting materials 1 and 4 were obtained.25,26 In brief, 1 was coupled with amino 2 in the presence of sodium iodide and K2CO3, followed by deprotection with HCl to produce the key intermediate 3. Subsequently, using TMSOTf as a catalyst, a traditional glycosidation reaction between tert-butyl 3-(2-(2-hydroxyethoxy)ethoxy)propanoate 4 and completely acetylated galactosamine produced 5.27 Trifluoroacetic acid (TFA) was used to cleave compound 5's tert-butyl ester, yielding another essential intermediate, compound 6. In order to create the core diamine scaffold, the acid 6 was activated by EDCI and NHS, then coupled with compound 3 in the presence of DIPEA to build the core diamine scaffold, and deprotected to generate the OH-naked precursor 7. The structures of key compounds 3 and 6 were characterized using NMR methods (Fig. S1 and S2†).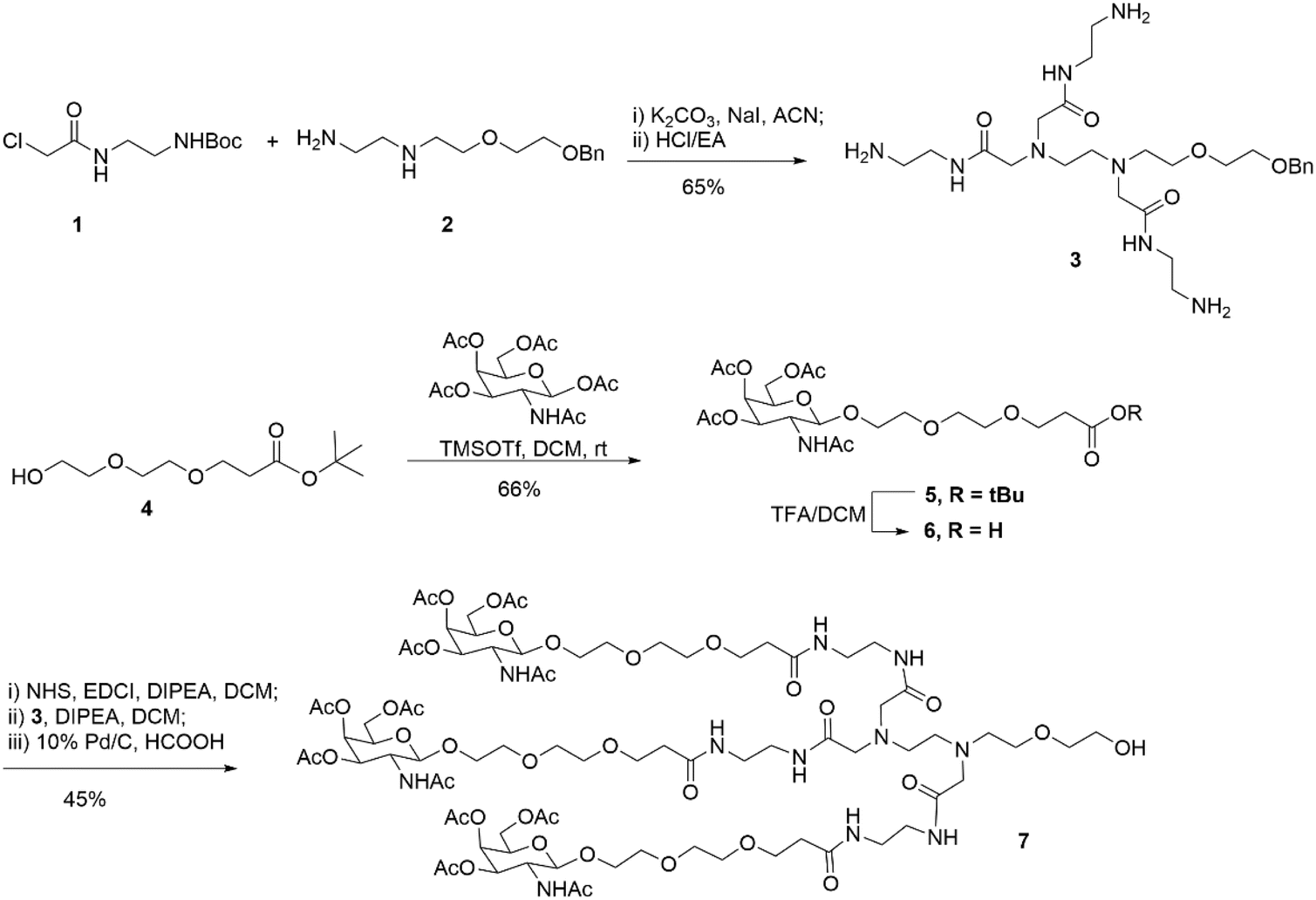 | ||
| Scheme 1 Synthesis of diamine-scaffold-based GalNAc precursor 7. | ||
Subsequently, two strategies for conjugation at either the 5′-end or the 3′-end of the sense strand were considered (Fig. 1). Therefore, on the oner hand, the phosphoramidite 8 supporting TG1–ssRNA was obtained from precursor 7 using a conventional method (Scheme 2A). On the other hand, the diamine-scaffold-based GalNAc CPG, which can facilitate the synthesis of TG2–ssRNA, was synthesized (Scheme 2B).24 In short, precursor 7 was activated by CDI, followed by reaction with 1-amino-3-O-(4,4′-dimethoxytrityl)-2,3-propanediol 9 (ref. 28) to yield carbamate 10. In the presence of Et3N and a catalytic amount of DMAP, the reaction of 10 with succinic acid anhydride yields intermediate 11 with an extended carboxyl group.29 The –COOH group of compound 11 was then conjugated with the –NH2 group of 500 Å CPG through condensation, followed by AC2O capping to produce GalNAc CPG 12.
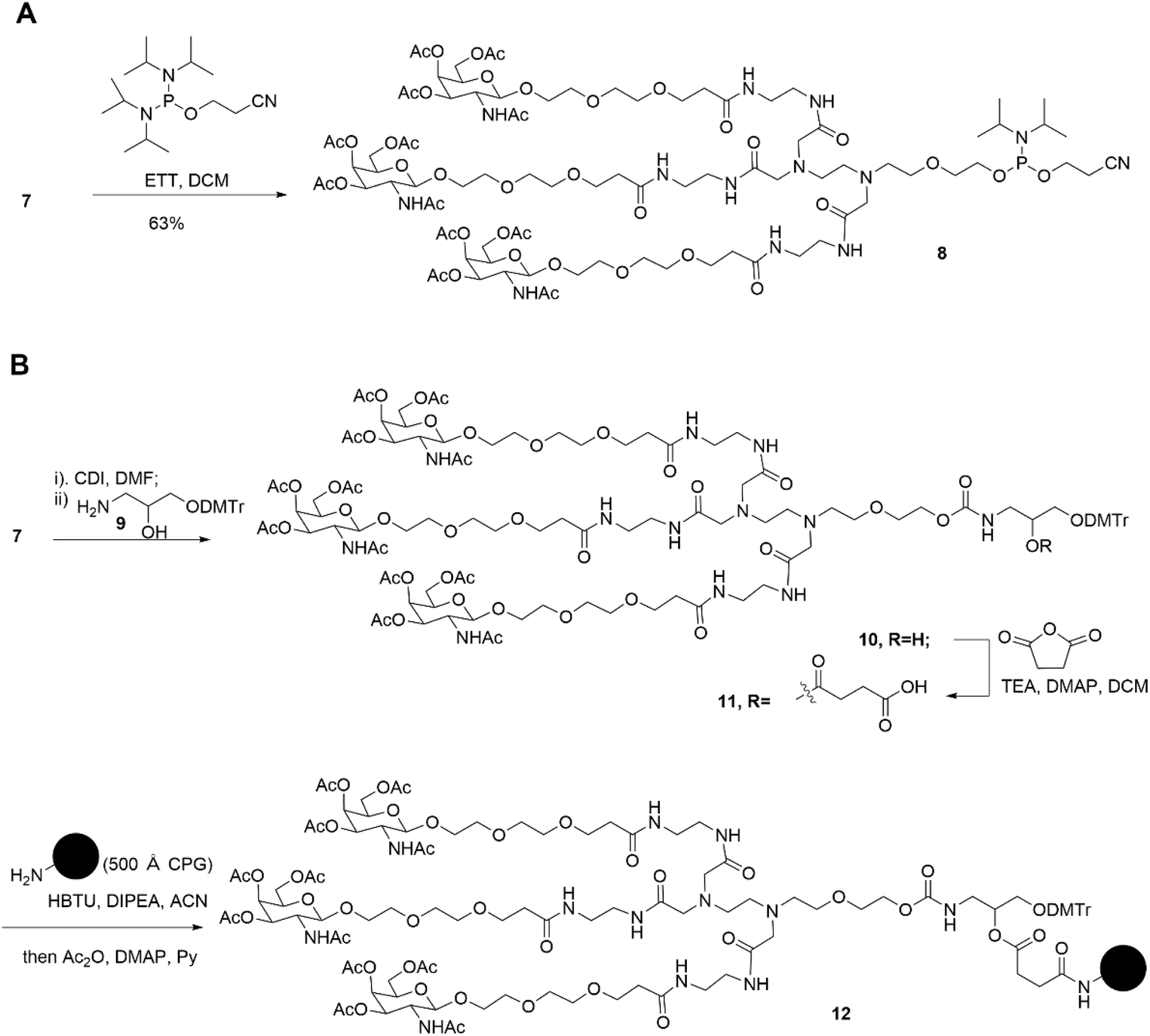 | ||
| Scheme 2 Synthesis of diamine-scaffold-based phosphoramidite (A) and diamine-scaffold-based GalNAc CPG (B). | ||
All siRNA (24–32, as shown in Tables 1, 2 and Fig. S3, S14†), including ligand-conjugated siRNA, were synthesized using standard solid-phase oligonucleotide synthesis methods. Specifically, GalNAc phosphoramidite 8 or NAG37 (synthesized with reference to Arrowhead Pharmaceuticals) is used as the final linking unit to obtain the 5′-end GalNAc-conjugated sense strands 14, 15, and 19. The 3′-end GalNAc-modified sense strands 18, 16, 20, 21, and 22 were collected from solid support L96-CPG or 12, respectively. The phosphoramidite with an inverted abasic site used in strand 15 was synthesized according to Arrowhead Pharmaceuticals. For the fluorescent-labeled antisense strand 17, we utilized commercially available NH2-protected C7-CPG and AF647 NHS ester (Fig. S3†). The overall yields of these modified strands were comparable to those of the natural strands 13 or 23 after standard deprotection and HPLC purification. All siRNA duplexes were obtained by annealing equimolar amounts of antisense strands with the corresponding sense strands.
| siRNA | S/ASa | Sequence (5′–3′)b | Targetc |
|---|---|---|---|
| a S indicates sense strands and AS indicates antisense strands.b Upper-case and lower-case letters represent 2′-deoxy-2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) sugar-modified uridine (U), adenosine (A), guanosine (G), and cytidine (C), respectively. * Indicates a phosphorothioate (PS) linkage. C7-NH2, 3′-amino-modifier C7. AF647: Alexa Fluor 647 fluorophore. InvAb: inverted abasic site. TG1, TG2, and NAG37 represent different GalNAc ligands. Their structures are shown in Fig. 1 and the ESI.c ANG3, angiopoietin-like protein 3. | |||
| 24 | 13 | gcucaacaUAUuugaucagu*a | ANG3 |
| 17 | u*A*c*ugAuCaAaUaUgUuGaGc*C7-NH2-AF647 | ||
| 25 | 14 | TG1*gcucaacaUAUuugaucagu*a | ANG3 |
| 17 | u*A*c*ugAuCaAaUaUgUuGaGc*C7-NH2-AF647 | ||
| 26 | 15 | NAG37*invAb*gcucaacaUAUuugaucagua*invAb | ANG3 |
| 17 | u*A*c*ugAuCaAaUaUgUuGaGc*C7-NH2-AF647 | ||
| 27 | 16 | g*cucaacaUAUuugaucagua*TG2 | ANG3 |
| 17 | u*A*c*ugAuCaAaUaUgUuGaGc*C7-NH2-AF647 | ||
| siRNA | S/ASa | Sequence (5′–3′)b | Targetc | mRNA (%) rel. to PBS | |
|---|---|---|---|---|---|
| Day 14 | Day 39 | ||||
| a S indicates sense strands and AS indicates antisense strands.b Upper-case and lower-case letters represent 2′-deoxy-2′-fluoro (2′-F) and 2′-O-methyl (2′-OMe) sugar-modified uridine (U), adenosine (A), guanosine (G), and cytidine (C), respectively. * Indicates a phosphorothioate (PS) linkage. TG1, TG2, and L96 represent different GalNAc ligands. Their structures are shown in Fig. 1 and the ESI.c TTR, transthyretin. | |||||
| 28 | 18 | c*a*guguUCUugcucuauaaL96 | TTR | 8.94 | 30.2 |
| 23 | u*U*auaGagcaagaAcAcug*u*u | ||||
| 29 | 19 | TG1*caguguUCUugcucuau*a*a | TTR | 9.39 | 53.3 |
| 23 | u*U*auaGagcaagaAcAcug*u*u | ||||
| 30 | 20 | c*a*guguUCUugcucuauaaTG2 | TTR | 7.37 | 44.2 |
| 23 | u*U*auaGagcaagaAcAcug*u*u | ||||
| 31 | 21 | c*a*guguUCUugcucuauaa*TG2 | TTR | 8.89 | 22.0 |
| 23 | u*U*auaGagcaagaAcAcug*u*u | ||||
| 32 | 22 | c*a*guguUCUugcucuaua*a*TG2 | TTR | 7.46 | 22.1 |
| 23 | u*U*auaGagcaagaAcAcug*u*u | ||||
With assembled duplexes on hand, we next investigated the target-delivery capabilities of our 5′-end modified GalNAc ligand TG1. AF647-labeled duplex 24–26 were subcutaneously (SC) administered into BALB/c nude mice. Fluorescence signals were observed in the livers of mice injected with both our GalNAc (TG1)-modified 25 and GalNAc–NAG37-modified 26 thirty minutes after injection, according to bioluminescence imaging. Moreover, 1–2 hours after delivery, there was a noticeable increase in the liver fluorescence signals (Fig. S15†). However, no obvious accumulation of fluorescence signal was observed in the liver of animals injected with siRNA 24. The image of mice injected with duplex 25 shows comparatively greater fluorescent signals in the liver as compared to the image of mice injected with siRNA 26. It might suggest that the ligand TG1 enhances siRNA distribution to the liver at a faster rate than NAG37 ligand. In addition, it was noted that within 3–4 hours of drug injection, the fluorescence signal in the bladder of all three test mice groups was stronger than that of other organs, suggesting that the majority of the siRNA medicines were eliminated through urination. After nine and twenty-four hours, ex vivo pictures of normal organs revealed that siRNAs 25 and 26 were selectively targeted and deposited in the liver, while little to no accumulation was seen in other organs (Fig. 2 and S16†). The small amount of GalNAc-modified siRNA that was discovered in the kidney implies that the kidneys may be able to metabolize certain conjugates, just like they do unconjugated siRNA medications.30
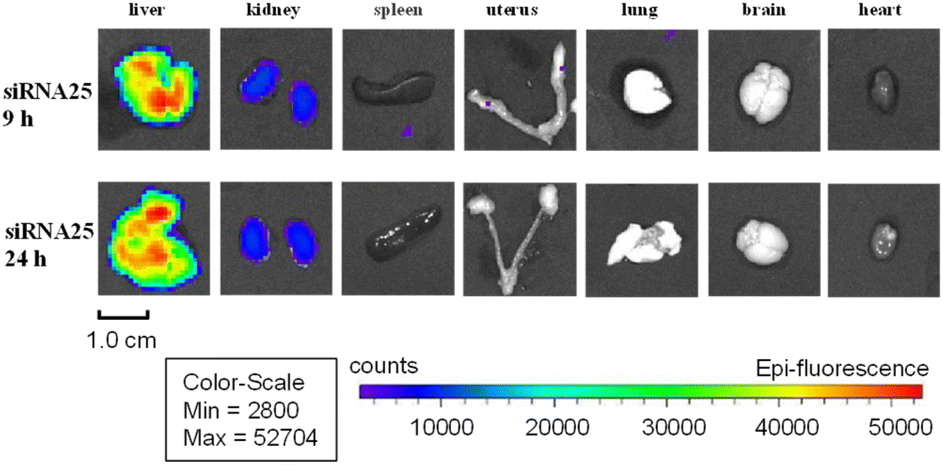 | ||
| Fig. 2 Representative organ images showing the target-delivery capabilities of TG1-conjugated siRNA 24 (9 h or 24 h post-injection into mice). The organ type, fluorescence signal intensity and scale bar are located above and below the graph, respectively. | ||
Apparently, Mice's kidneys appear to have a greater concentration of 24 than their livers. Interestingly, ex vivo images of mice given 24 or 26 treatment showed a higher distribution in extrahepatic organs, such as the spleen and uterus. In contrast, only the liver and kidneys of animals treated with 25 showed obvious fluorescence signals (Fig. 2). Since there are no analogous scenarios for comparable drugs, these subtle distinctions may arise from structural variances between GalNAc–TG1 and GalNAc–NAG37, or differences in modification patterns of the sense strand. Furthermore, the GalNAc–TG2-modified siRNA 27 exhibited comparable outcomes to siRNA 25, demonstrating a noticeable accumulation of fluorescence signal in the liver twenty-four hours after injection, which was slightly higher than that of siRNA 26 (Fig. S17†). Most importantly, both the GalNAc–TG1 and GalNAc–TG2 do not impact cell viability, even when present at high concentrations (Fig. S18†).
We next evaluated the ability to silence gene expression in vivo using siRNA 28–32 conjugated to our GalNAc–TG1 and GalNAc–TG2, which were designed to target the rodent transthyretin (TTR) gene (Table 2). GalNAc–L96-conjugated siRNA 28, which is 2 nucleotides shorter than DV22 conjugate from Alnylam, was utilized as a positive control. This siRNA variant features the substitution of a 2′-F modified nucleoside with a 2′-OMe modified nucleoside at position 15 of the sense strand (counted from the 3′-side, Fig. S19†).31 Considering the enhanced effectiveness and prolonged activity of DV22 and DV18 conjugates in mice, the expression of TTR in the liver was assessed using quantitative PCR at 14 and 39 days after administration. After 14 days of SC administration at a dose of 1 mg kg−1, the silencing efficiency of each group was essentially the same, with all groups achieving a silencing efficiency of over 90% (Fig. S20†). Observations in mice 39 days after SC administration of siRNA 29 with GalNAc–TG1 modification at the 5′-end of the sense strand exhibited a that TTR mRNA levels increased to 50% relative to animals treated with PBS controls. In contrast, TTR mRNA expression was still 70% inhibited in the positive control group that received siRNA 26 treatment (Fig. 3). We speculated that the difference in the sense strand connection site (5′-end or 3′-end) between GalNAc–TG1 and GalNAc–L96 may account for the above different result. However, GalNAc–TG2-modified siRNA 30 does not seem to enhance the inhibition of mTTR expression, achieving a 56% inhibition rate which in the range of the values of siRNA 28 and siRNA 29 (Fig. 3). Given the distinct structures of GalNAc–TG2 and GalNAc–L96, their metabolic stability in mice varies significantly. Based on siRNA 30, more in-depth examination of the siRNA chemistry resulted in the creation of siRNA 31, which offers enhanced defense against 3′-exonucleases by adding a PS linkage between GalNAc–TG2 and the oligonucleotide. It should come as no surprise that the additional PS linkage enhanced the inhibition of mTTR expression caused by siRNA 30, resulting in a 78% suppression. On this basis, the siRNA 32 produced by reintroducing an additional PS linkage did not impair its activity. In addition, animals treated with siRNA (35–36) modified with TG3, which contains the trans-4-hydroxyprolinol (tHP) moiety similar to L96, exhibited comparable RNAi efficacy to that of the PS stabilized siRNA 31, either 14 days or 39 days post subcutaneous injection (Fig. S21†). The stabilized chemical moieties, such as PS and tHP, play a crucial role in the receptor-specific targeted delivery of siRNA by protecting the siRNA from nuclease degradation and prolonging the duration of gene silencing. Therefore, the coexistence of the novel diamine-scaffold based GalNAc and stabilized chemical moieties provides a viable approach for RNAi therapeutics.
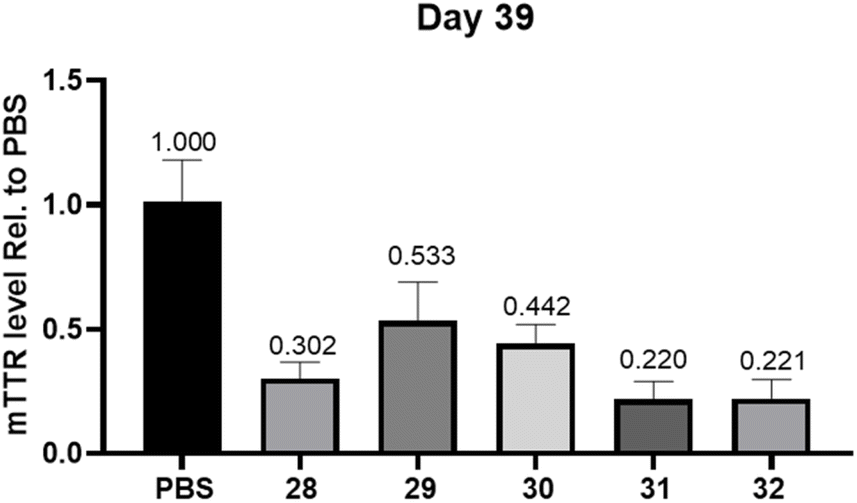 | ||
| Fig. 3 TTR gene silencing results of 39 days after a single subcutaneous dose of 1 mg kg−1 with siRNA 28–32 in wild-type C57BL/6 mice (n = 7). Error bars represent standard errors for TTR mRNA (mTTR) measurements. | ||
Conclusions
We have developed a novel GalNAc ligand precursor 7 based on diamine scaffold, which can be easily synthesized through several coupling steps. Starting from the precursor, the GalNAc phosphoramidite coupled to the 5′-end of the sense strand can be obtained through a one-step reaction, or the GalNAc CPG attached to the 3′-end of the sense strand can be obtained through a simple multi-step conversion. Consequently, several TG1 or TG2 modified siRNAs were then obtained. Importantly, TG1-modified siRNA demonstrates comparable tissue-specific delivery efficiency to a GalNAc NAG37 phase II clinical drug candidate, while TG2-modified siRNA exhibits enhanced inhibition of mTTR expression compared to a GalNAc L96 standard. Subsequently, an examination of the pharmacokinetics (PK) and pharmacodynamics (PD) properties of these GalNAc-conjugated siRNA will be conducted, encompassing an assessment of their in vivo half-lives.32Ethical statement
All the animals were acclimated under standard laboratory conditions (ventilated room, 25 ± 1 °C, 60 ± 5% humidity, 12 h light/dark cycle) and had free access to standard water and food in the Laboratory Animal Center of Marine Biomedical Research Institute of Qingdao (SYXK-2021-0009). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Qingdao” University and approved by the Animal Ethics Committee of “Medical College of Qingdao University” (QDU-AEC-2023187).Author contributions
Qiang Li: methodology, investigation, writing-review & editing, funding acquisition. Mingxin Dong: writing-review & editing; validation. Pu Chen: project administration, writing-review & editing; validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Shandong Postdoctoral Science Foundation [SDCX-ZG-202301015].References
- J. Lieberman, Tapping the RNA world for therapeutics, Nat. Struct. Mol. Biol., 2018, 25, 357–364 CrossRef CAS PubMed.
- S. T. Crooke, B. F. Baker and R. M. Crooke, et al., Antisense technology: an overview and prospectus, Nat. Rev. Drug Discovery, 2021, 20, 427–453 CrossRef CAS PubMed.
- B. Hu, L. Zhong and Y. Weng, et al., Therapeutic siRNA: state of the art, Signal Transduction Targeted Ther., 2020, 5, 101 CrossRef CAS PubMed.
- H. Banoun, mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues, Int. J. Mol. Sci., 2023, 24, 10514 CrossRef CAS PubMed.
- Z. Guizhi and C. Xiao, Aptamer-based targeted therapy, Adv. Drug Delivery Rev., 2018, 134, 65–78 CrossRef PubMed.
- X. Xie, T. Yu and X. Li, et al., Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials, Signal Transduction Targeted Ther., 2023, 8, 335 CrossRef PubMed.
- A. Goga and M. Stoffel, Therapeutic RNA-silencing oligonucleotides in metabolic diseases, Nat. Rev. Drug Discovery, 2022, 21, 417–439 CrossRef CAS PubMed.
- P. G. V. Martini and L. T. Guey, A New Era for Rare Genetic Diseases: Messenger RNA Therapy, Hum. Gene Ther., 2019, 30, 1180–1189 CrossRef CAS PubMed.
- Y. Zhang, C. Sun and C. Wang, et al., Lipids and Lipid Derivatives for RNA Delivery, Chem. Rev., 2021, 121, 12181–12277 CrossRef CAS PubMed.
- K. Paunovska, D. Loughrey and J. E. Dahlman, Drug delivery systems for RNA therapeutics, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed.
- Y. Dong, D. J. Siegwart and D. G. Anderson, Strategies, design, and chemistry in siRNA delivery systems, Adv. Drug Delivery Rev., 2019, 144, 133–147 CrossRef CAS PubMed.
- A. Gupta, J. L. Andresen and R. S. Manan, et al., Nucleic acid delivery for therapeutic applications, Adv. Drug Delivery Rev., 2021, 178, 113834 CrossRef CAS PubMed.
- A. D. Springer and S. F. Dowdy, GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics, Nucleic Acid Ther., 2018, 28, 109–118 CrossRef CAS PubMed.
- S. M. Hammond, A. Aartsma-Rus and S. Alves, et al., Delivery of oligonucleotide-based therapeutics: challenges and opportunities, EMBO Mol. Med., 2021, 13, e13243 CrossRef CAS PubMed.
- B. Malecova, R. S. Burke and M. Cochran, et al., Targeted tissue delivery of RNA therapeutics using antibody-oligonucleotide conjugates (AOCs), Nucleic Acids Res., 2023, 51, 5901–5910 CrossRef CAS PubMed.
- K. Klabenkova, A. Fokina and D. Stetsenko, Chemistry of Peptide-Oligonucleotide Conjugates: A Review, Molecules, 2021, 26, 5420 CrossRef CAS PubMed.
- A. J. Debacker, J. Voutila and M. Catley, et al., Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug, Mol. Ther., 2020, 28, 1759–1771 CrossRef CAS PubMed.
- X. Huang, J. C. Leroux and B. Castagner, Well-Defined Multivalent Ligands for Hepatocytes Targeting via Asialoglycoprotein Receptor, Bioconjugate Chem., 2017, 28, 283–295 CrossRef CAS PubMed.
- V. Kumar and W. B. Turnbull, Targeted delivery of oligonucleotides using multivalent protein-carbohydrate interactions, Chem. Soc. Rev., 2023, 52(4), 1273–1287 RSC.
- Q. Tang and A. Khvorova, RNAi-based drug design: considerations and future directions, Nat. Rev. Drug Discovery, 2024, 23, 341–364 CrossRef CAS PubMed.
- Y. Zhang, H. Chen and L. Hong, et al., Inclisiran: a new generation of lipid-lowering siRNA therapeutic, Front. Pharmacol, 2023, 14, 1260921 CrossRef CAS PubMed.
- Y. N. Lamb, Inclisiran: First Approval, Drugs, 2021, 81, 389–395 CrossRef CAS PubMed.
- J. K. Nair, J. L. Willoughby and A. Chan, et al., Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing, J. Am. Chem. Soc., 2014, 136, 16958–16961 CrossRef CAS PubMed.
- E. A. Ulashchik, Y. V. Martynenko-Makaev and T. P. Akhlamionok, et al., Synthesis of GalNAc-Oligonucleotide Conjugates Using GalNAc Phosphoramidite and Triple-GalNAc CPG Solid Support, Methods Mol. Biol., 2021, 2282, 101–118 CrossRef CAS PubMed.
- H. An, T. Wang and V. Mohan, et al., Solution phase combinatorial chemistry. Discovery of 13- and 15-membered polyazapyridinocyclophane libraries with antibacterial activity, Tetrahedron, 1998, 54, 3999–4012 CrossRef CAS.
- J. Magano, B. Conway and D. Farrand, et al., Scalable and Cost-Effective Synthesis of a Linker for Bioconjugation with a Peptide and a Monoclonal Antibody, Synthesis, 2014, 46, 1399–1406 CrossRef.
- J. H. Kim, H. Yang and J. Park, et al., A general strategy for stereoselective glycosylations, J. Am. Chem. Soc., 2005, 127, 12090–12097 CrossRef CAS PubMed.
- A. V. Azhayev and M. L. Antopolsky, Amide group assisted 3′-dephosphorylation of oligonucleotides synthesized on universal A-supports, Tetrahedron, 2001, 57, 4977–4986 CrossRef CAS.
- V. K. Sharma, M. F. Osborn and M. R. Hassler, et al., Novel Cluster and Monomer-Based GalNAc Structures Induce Effective Uptake of siRNAs in Vitro and in Vivo, Bioconjugate Chem., 2018, 29, 2478–2488 CrossRef CAS PubMed.
- J. K. Nair, H. Attarwala and A. Sehgal, et al., Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates, Nucleic Acids Res., 2017, 45, 10969–10977 CrossRef CAS PubMed.
- D. J. Foster, C. R. Brown and S. Shaikh, et al., Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates, Mol. Ther., 2018, 26, 708–717 CrossRef CAS PubMed.
- R. McDougall, D. Ramsden and S. Agarwal, et al., The Nonclinical Disposition and Pharmacokinetic/Pharmacodynamic Properties of N-Acetylgalactosamine-Conjugated Small Interfering RNA Are Highly Predictable and Build Confidence in Translation to Human, Drug Metab. Dispos., 2022, 50, 781–797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03023k |
| This journal is © The Royal Society of Chemistry 2024 |