
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study of the catalytic hydrodeoxygenation of furan, methylfuran and benzofurane on MoS2†
Wilfried G. Kanhounnon *a,
Saber Gueddida*b,
Simplice Koudjinaa,
Frédéric Richardc,
Guy Y. S. Atohouna,
Jean-François Pauld,
Sébastien Lebègueb and
Michael Badawi*e
*a,
Saber Gueddida*b,
Simplice Koudjinaa,
Frédéric Richardc,
Guy Y. S. Atohouna,
Jean-François Pauld,
Sébastien Lebègueb and
Michael Badawi*e
aLaboratoire de Chimie Physique – Matériaux et Modélisation Moléculaire (LCP3M)/Unité de Chimie Théorique et de Modélisation Moléculaire (UCT2M), Université d’Abomey-Calavi, Cotonou, Benin. E-mail: gbedode.kanhounnon@uac.bj
bUniversité de Lorraine, Laboratoire de Physique et Chimie Théoriques, Vandoeuvre-Les-Nancy F-54506, France. E-mail: saber.gueddida@univ-lorraine.fr
cUniversité de Poitiers, CNRS, Institut de Chimie des Milieux et Matériaux de Poitiers, UMR 7285, rue Michel Brunet, BP633, 86022 Poitiers, France
dUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
eUniversité de Lorraine, CNRS, L2CM, F-57000 Metz, France. E-mail: michael.badawi@univ-lorraine.fr
First published on 16th July 2024
Abstract
Herein, we have studied the direct deoxygenation (DDO) (without prior hydrogenation) of furan, 2-methylfuran and benzofuran on the metal edge of MoS2 with a vacancy created under pressure of dihydrogen. For the three molecules, we found that the desorption of the water molecule for the regeneration of the vacancy is the most endothermic. Based on the thermodynamic and kinetic aspects, the reactivity order of the oxygenated compounds is furan ≈ 2-methylfuran > benzofuran, which is in agreement with literature. We present the key stages of the mechanisms and highlight the effects of substituents.
1 Introduction
Energy consumption is increasing every year and could see an increase of 60% in 2030 worldwide1,2 compared to the present days. The growing demand for energy, the progressive depletion of world oil reserves, resulting in the high cost of fossil fuels, have motivated the search for new sources of energy other than traditional oil such as renewable sources3–6 like solar power, hydropower, wind power, fuel cells, and biofuels.Indeed, biofuels are attractive in view of the renewable nature of the energy produced,7 and constitute an interesting alternative to crude oil. In particular, bio-oils derived from biomass pyrolysis are of high interest. However, wood derived bio-oil fractions contain high amounts of oxygenated compounds that could reach 45 wt%,8 including aldehydes, ketones, carboxylic acids, ethers, alcohols, furanics and phenolics. This high oxygen content gives bio-oils a low energetic power, a high viscosity and corrosive properties together with a chemical and thermal unstability.9,10 Therefore, an important challenge to be met for the valorization of bio-oils as fuels is the reduction of the oxygen compounds to a level allowing their co-treatment with fossil fuels in the already existing hydrotreatment units in petroleum refinery using conventional hydrotreatment catalysts.11 This is compatible with the objective of the European Union which, relying on the tendency of fossil fuels to become scarce in the years to come, will require oil companies to incorporate at least 10% of biomass products in fuels.12 Catalytic hydrodeoxygenation (HDO) is becoming increasingly important in the energy industry and a precise understanding of the mechanisms of deoxygenation reactions would allow an improvement in the activity and selectivity of catalysts through the modifications of their composition and structure. Furanic compounds, resulting mainly from the decomposition of hemicellulosic biomass, are strongly present in bio-oils. Among the oxygenated compounds of bio-oils, they are the most refractory to deoxygenation with this order of reactivity: furan > benzofuran > dibenzofuran.13–15 Therefore, they are also used as model compounds in the study of the deoxygenation of bio-oils.16,17
However, so far much work has been conducted on the search for efficient, available and inexpensive hydrotreatment catalysts, or on techniques for separating oxygen and sulfur compounds from hydrocarbons for their subsequent treatment.9,18,19
Conventional molybdenum sulfide (MoS2) with a 50% sulfur coverage rate, is stable under hydrotreatment conditions, and appears as good candidate for HDO processing.20–22 It is well admitted that the active sites are molybdenum atoms exhibiting coordinated unsaturated sites (CUS) located on the edges and also on the corners, which accurate descriptions have been provided by a number of theoretical studies.23–27 Such active sites can be done by removing H2S under hydrogen pressure and therefore molecules will be able to adsorb on vacancies before oxygen elimination.12,28–35 The stability of the sulfided catalysts in the presence of large amounts of oxygenated compounds is critical since modifications of the structure of the active edges of the sulfide phase can be involved.9,36 It has been shown by DFT calculations that the stability of the sulfide phase under typical HDO conditions depends on the H2O/H2S pressure ratio. Taking into account the fact that model compounds for HDO study are just thiophen counterpart and targeting to achieve process that can help obtaining bio-fuels with good octane index. We retain for our study to use CUS on the Mo-edge to elucidate the DDO mechanisms of model compounds.37 Indeed, vacancy created on this edge is not large enough to allow flat adsorption which will make HYD process prevail on that of DDO.
Previously, our research investigated the hydrodeoxygenation (HDO) of furan and methylfuran on MoS2 and WS2,38,39 employing models represented as MoS3H3+ and WS3H+, respectively. This study successfully identified reaction intermediates involved in the hydrodeoxygenation process. However, the limitations of employing catalyst models containing only one Mo or W atom restricted the exploration of certain aspects of the HDO mechanism, such as H2 dissociation. This current work addresses these limitations by utilizing more realistic MoS2 models. These models not only enable the examination of the direct deoxygenation (DDO) mechanism, which is generally more complex than HDO, but also facilitate the exploration of Co or Ni promoting effects. By considering these factors, our study aims to provide deeper insights into the catalytic processes involved, thereby contributing to the advancement of knowledge in this field.
The aim of the present work is to shed insights on the direct deoxygenation of furan and two of its derivatives on the Mo-edge's CUS of MoS2 nanocrystalite, a really determinant fact in the process enhancing.
2 Computational details
Periodic density functional theory (DFT) calculations were carried out by means of the Vienna ab initio simulation package (VASP) code40 and the generalized gradient approximation (GGA) for the exchange–correlation potential as parametrized by Perdew, Burke and Ernzerhof (PBE).41 The projector augmented wave (PAW) method42 was used for the basis functions as provided by VASP. The weak van der Waals interactions were included within the Grimme approximation (PBE + D2).43,44The geometrical structures of the considered molecules are given in Fig. 1. Adsorption sites are generated by creating vacancies at the edges of the MoS2 structure. This is achieved by removing sulfur atoms under hydrogen pressure, resulting in the formation of H2S. Consequently, the MoS2 surface used in our model contains 32 Mo atoms and 63 S atoms instead of the usual 64 S atoms. The MoS2 nanocrystalite exhibits two edges, the (1 0 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) plan called hereafter the M-edge and the ( 0 1 0) plan known as the sulfur edge (S-edge). The lattice parameters for these periodic structures are a = 12.64 Å, b = 12.29 Å, c = 25.00 Å, and α = β = γ = 90°. In all cases, we used a periodic supercell separated by 15 Å of vacuum region in the z direction.
0) plan called hereafter the M-edge and the ( 0 1 0) plan known as the sulfur edge (S-edge). The lattice parameters for these periodic structures are a = 12.64 Å, b = 12.29 Å, c = 25.00 Å, and α = β = γ = 90°. In all cases, we used a periodic supercell separated by 15 Å of vacuum region in the z direction.
 | ||
| Fig. 1 Molecular structure of the: (a) furan, (b) 2-methylfuran, (c) benzofuran molecules. | ||
The energy cutoff for the plane-wave expansion was set at 450 eV to ensure the convergence of our calculations. The integration over the Brillouin zone was sampled by 3 × 3 × 1 k-points, using the Monkhorst–Pack scheme. The Kohn–Sham self-consistent total energy differences were converged within 10−6 eV. For the atomic relaxation, the position of all the atoms of the considered molecule and the atoms of the first two layers of the MoS2 surface were relaxed by nullifying the forces on the relaxed atoms with a precision of 0.03 eV Å−1 while all other atoms were kept fixed.
3 Results and discussion
3.1 DDO route of furan
Direct deoxygenation of furan leads to butadiene that would only be present in the form of traces under the typical conditions of the hydrodeoxygenation process. It therefore hydrogenates very quickly and leads to the formation of butenes and butane molecules.9 Here the formation of the butadiene molecule was only studied, which represents the first part of the reaction mechanism. In this work, the DDO mechanism utilized is based on the well-established experimental and theoretical studies of the hydrodesulfurization mechanism of thiophene, with furan serving as its oxygen counterpart where the sulfur atom is substituted by an oxygen atom.45,46 The starting point of the catalytic cycle, as shown in Fig. S1 in the ESI,† involves a metal edge surface with a vacancy. At this point, furan (or its derivatives) is adsorbed, and a dihydrogen molecule is positioned sufficiently far from the surface. It is admitted that the dihydrogen molecule dissociates heterolytically.32,47,48 The initial position of the hydrogen atom, placed far from the surface, serves as the starting point to study the dissociation of H2. This dissociation is a crucial step in the mechanism. It can occur with a proton on a sulfur atom and a hydride on a neighboring molybdenum one, this step being slightly exothermic. As this dissociation has a low activation energy (19.2 kJ mol−1), such a step can occur at CUS sites present on sulfide catalysts, consistent with previous results.47,49–52Fig. 2 shows the energetic profile for the DDO route of furan using the calculated adsorption energies of each reaction step. The calculated adsorption energies and the activation energies of the various reaction stages for the three molecules are summarized in Table 1 and 2, respectively. The addition of the first hydrogen atom on the carbon in the α position of oxygen is endothermic with an energy value of 41 kJ mol−1. The interatomic distance C–O changes from 1.4 Å in the adsorbed state to 1.5 Å in the formed complex. The addition of the first H atom hence allows an activation of the C–O bond, favoriting its scission. This step is highly exothermic with an energy value of −128 kJ mol−1 since it allows to restor the conjugation in the butadienolate formed. Furthermore, this step is kinetically more favored than the previous as its activation energy is weaker (by 25 kJ mol−1) compared to hydrogen addition.
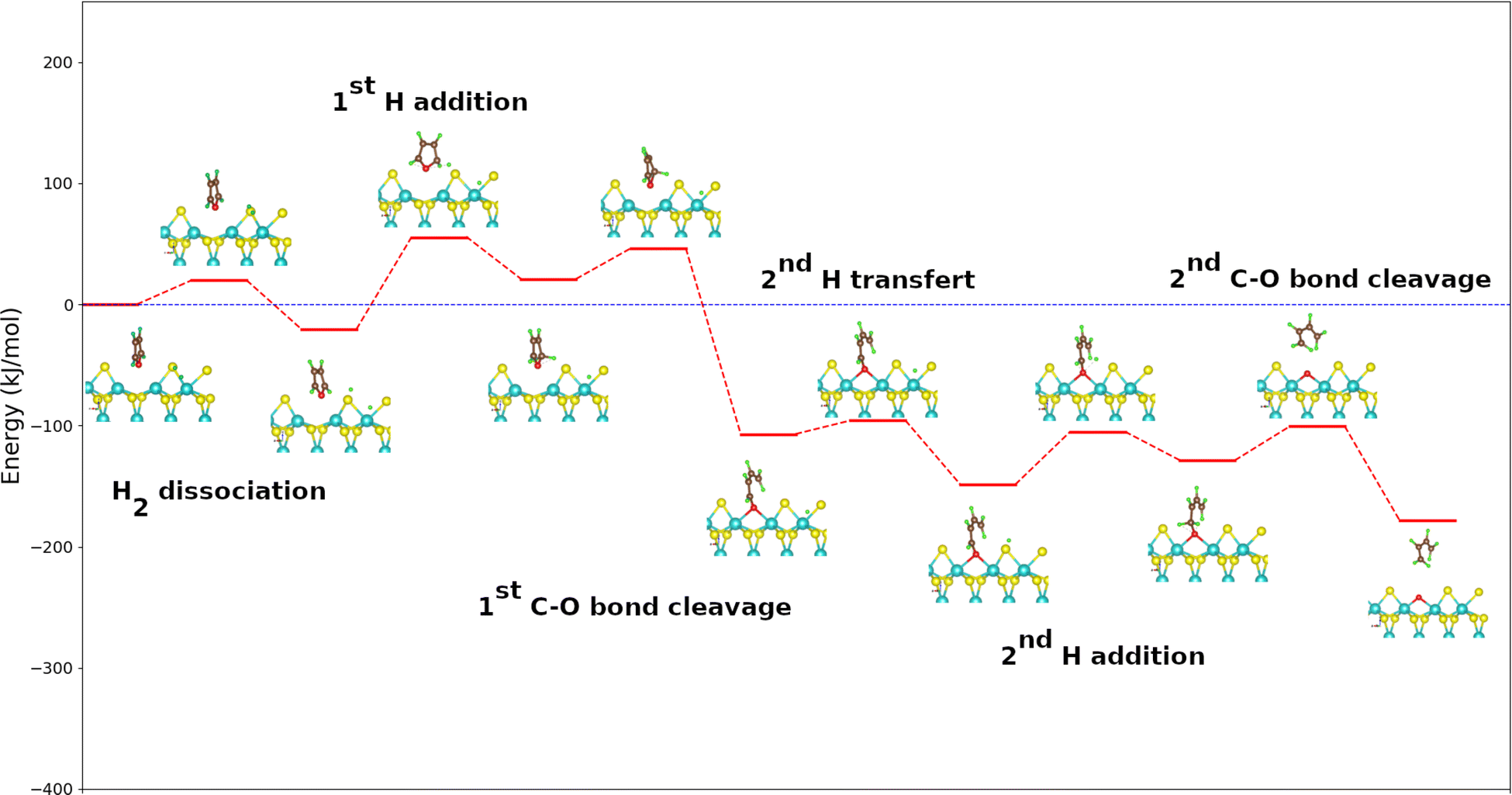 | ||
| Fig. 2 Energy diagram of the DDO route of furan on the metal edge of MoS2 surface using the PBE + D2 approximation. For clearer images, please refer to Fig. S6 in the ESI.† | ||
| Activation energies of the various stages of the reaction | |||||
|---|---|---|---|---|---|
| Molecule/reaction step | Hydrogen addition (kJ mol−1) | Broken C–O bonds (kJ mol−1) | |||
| Furan | 1st | 76 | 1st | 25.38 | |
| 2nd | 44 | 2nd | — | ||
| Methylfuran | 2H | 1st | 74 | 1st | 55 |
| 2nd | 39 | 2nd | 25 | ||
| 5H | 1st | 51 | 1st | 83 | |
| 2nd | 56 | 2nd | — | ||
| Benzofuran | 98 | 1st | 24 | ||
| Addition of the 2nd H and cleavage | 125 | ||||
| Energies of the various reaction stages for molecules | ||||||||
|---|---|---|---|---|---|---|---|---|
| Molecule/reaction step | H Added | Transfer of H from Mo to S | Broken C–O bonds | Desorption | ||||
| Furan | 1st | 41 | −42 | 1st | −128 | Addition of H2 | −68 | |
| 2nd | 20 | 2nd | −79 | Desorption of H2O | 64 | |||
| Methylfuran | 2H | 1st | 38 | −44 | 1st | −121 | Addition of H2 | −68 |
| 2nd | 19 | 2nd | −55 | Desorption of H2O | 64 | |||
| 5H | 1st | 10 | −36 | 1st | −97 | Addition of H2 | −68 | |
| 2nd | 48 | 2nd | −84 | Desorption of H2O | 64 | |||
| Benzofuran | 1st | 46 | 1 | 1st | −193 | Addition of H2 | −64 | |
| Addition of the 2nd H and cleavage | −25 | Desorption of H2O | 64 | |||||
The displacement of the second hydrogen atom to the sulfur one closest to the butadienolate is slightly exothermic. A transfer of an H from a Mo atom to a neighboring S is almost athermal and little active.33 The activation energy of this step, which does not depend on the molecule model but on the catalytic surface, should not change significantly from one molecule to another. We then admit its calculated value for the case of the benzofuran molecule which is 27 kJ mol−1 for all the calculated mechanisms. The activation energy involved in the second hydrogen addition is much lower than the one involved in the first H addition (76 kJ mol−1). The addition of 2nd H atom is slightly endothermic (20 kJ mol−1) and leads to the formation of an intermediate reaction. The activation energy (44 kJ mol−1) is lower than that of the first addition, which can be attributed to the reduction of steric hindrance on the catalyst surface following the opening of the cycle after the first break. The release of the butadiene molecule by breaking the second C–O bond is exothermic with an energy value of −50 kJ mol−1 and leaves an oxygen atom on the metal edge of the surface. The calculation of the transition state for this phase was not conclusive. Nevertheless, we estimate that this step will be significantly more kinetically favored than the addition of the first hydrogen atom due to the much greater stability of the product relative to the reactant. The vacancy regeneration which is common to all the studied molecules, occcurs in three steps. The first is the dissociation of H2 molecule on the surface with the removed oxygen of the molecule still remainded adsorbed on it. It is remarked that one proton is adsorbed on the oxygen atom and one hydride on the neighbouring molybden one. This step slightly exothermic is kinetically limited than all the steps of the oxygen atom removing from the molecule (71 kJ mol−1). The second step was the transfert of H atom from the molybdenum atom to the formed hydroxide on the surface to form water molecule. It exhibits strong exothermicity, coupled with a relatively low activation energy of 47 kJ mol−1. The last step is the desorption of water molecule from the surface. It is kinetically favored but highly endothermic (73 kJ mol−1). By desorption of water, the vacancy is regenerated because it is less endothermic (73 kJ mol−1) than the creation of a vacancy by extraction of a sulfur atom (93 kJ mol−1) as highlighted in Fig. 3.
 | ||
| Fig. 3 Energy diagram for vacancy regeneration on the metal edge of MoS2 surface after oxygen atom removal from the studied molecules. For clearer images, please refer to Fig. S3 in the ESI.† | ||
As expected, the cleavages of the C–O bond and the transfer of a hydrogen atom from a molybdenum to a sulfur atom as proposed in the mechanism are exothermic, while the additions of hydrogen and the vacancy regeneration by water desorption are endothermic. It can be assume that the addition of the first hydrogen atom would be the limiting step of the whole process.
3.2 DDO of 2-methylfuran
A similar reaction mechanism for the DDO route of 2-methylfuran molecule as that of furan is proposed in one. The fact that the molecule has a non-symmetrical geometry offers two possible routes of deoxygenation: (1) the attack by the methylated carbon atom adjacent to the oxygen atom (denoted hereafter 2H route) or (2) the attack by the second unmethylated carbon atom adjacent to the oxygen atom (denoted hereafter 5H pathway). Fig. 4 shows the PBE + D2 calculated energy diagram for the DDO route of 2-methylfuran on the metal edge of MoS2 surface. The dissociation of H2 follows the same tendency as for the case of furan. The addition of the first H atom on the carbon in α position of the oxygen for both pathways is endothermic (44 kJ mol−1 for the 2H route and 16 kJ mol−1 for the 5H route) but less endothermic than the homologous step calculated from furan. This fact can be explained by the donor inducing effect of the methyl group on the adjacent carbons of the oxygen atom which would make the new C–H bond formed in 2-methylfuran stronger. This reaction step is more exothermic on the 5H route than on the 2H one. The steric hindrance created by the methyl group may be responsible of this expected difference in reactivity. It is on the 2H path slower than for the reaction of furan and faster on the 5H path. The activation energies are 80 kJ mol−1 on the 2H route and 57 kJ mol−1 on the 5H route against 76 kJ mol−1 for the furan molecule (Table 2). The reaction following the 2H route leads to 2-hydro-2-methylfuran while following the 5H route 5-hydro-2-methylfuran can be formed. These intermediates thus formed can undergo a cleavage of their C–O bonds. The cleavage of the C–O bonds is strongly exothermic with a reaction energy of −121 kJ mol−1 for the 2H pathway and −97 kJ mol−1 for the 5H pathway (Table 3). As for furan, the conjugation found in the intermediates formed could explain this exothermicity. Here, the reaction following the 2H pathway is more exothermic in contrast to the addition of the H atom, but less exothermic than the corresponding calculated step of furan. The calculated activation energies are 55 kJ mol−1 for the 2H route and 83 kJ mol−1 for the 5H one. The 2H route is more favored kinetically than the 5H route. The cleavage along the 2H pathway occurs with greater kinetic ease than adding the first H atom, yet it remains kinetically less favored than the analogous step in the deoxygenation of the furan molecule. For the 5H pathway, cleavage is more kinetically challenging compared to the initial hydrogen addition step.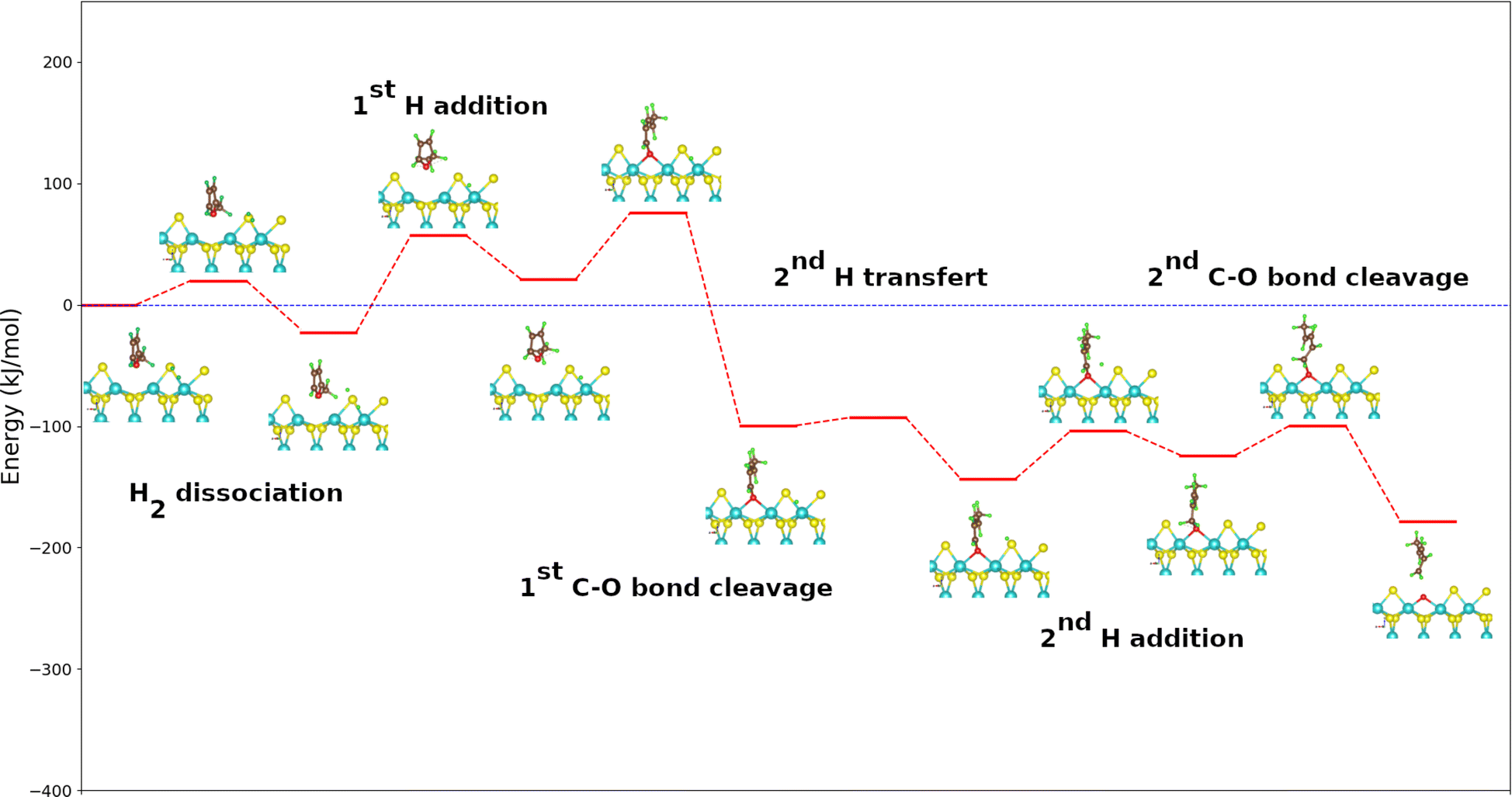 | ||
| Fig. 4 Energy diagram of DDO (5H) route of the 2-methylfuran molecule on the metal edge of MoS2 surface using the PBE + D2 approximation. For clearer images, please refer to Fig. S4 in the ESI.† | ||
| Molecules | Rate determining steps | Activation energy (kJ mol−1) | |
|---|---|---|---|
| Furan | Addition of the 1st H | 76 | |
| Methylfuran | 2H route | Addition of the 1st H | 74 |
| 5H route | Rupture of the first C–O bond | 83 | |
| Benzofuran | Addition of the 2nd H and breaking of 2nd C–O bond | 125 | |
The displacement of the second hydrogen atom to the sulfur atom closest to 2-methylbutadienolate and pentadienolate is exothermic (−44 kJ mol−1 for the 2H pathway and −36 kJ mol−1 for the 5H pathway). This step depends mainly on the surface rather than on the molecule, the activation energy being the same with the one calculted for furan deoxygenation. The addition of the second H is endothermic (19 kJ mol−1 for the 2H pathway and 48 kJ mol−1 for the 5H pathway), the former being less endothermic. It proceeds more rapidly than the first addition of H, with an activation energy of 39 kJ mol−1 compared to 80 kJ mol−1, while for the 5H pathway, this step is as rapid as the first addition given the almost equal activation energies (56 and 57 kJ mol−1 respectively). The methyl group in substitution on the product of the cleavage of the first C–O bond for the 5H pathway constitutes a hindrance for the accession of the second hydrogen atom added to the targeted carbon atom. This explains why the second addition of H along the 5H pathway is kinetically less favored than the first. The release of pentadiene by breaking the second C–O bond is exothermic in both ways. However, the relative instability of the initial system compared to the final one created by the methyl group makes the reaction along the 5H route more exothermic (−83 kJ mol−1 against −54 kJ mol−1 for the 2H route). The calculated activation energy (24 kJ mol−1) following the 2H path is lower than all the other activation energies of the whole process. The deficiency regeneration steps are identical according to the two routes and to those of furan. Desorption of water is also the most endothermic stage of all the stages here (see Table 3).
Our results show that the C–O bond scission and the transfer of a hydrogen atom from a molybdenum to a sulfur atom are exothermic whereas the hydrogen additions and the regeneration of vacancy by desorption of water are endothermic. The addition of the first hydrogen atom would be the limiting step of the whole process for the 2H pathway as for furan. Following the 5H path, the cleavage of the first C–O bond would be the kinetically determining step. Outside the step of adding the first hydrogen atom, the 2H route is thermodynamically and kinetically more favorable for the direct deoxygenation of 2-methylfuran.
3.3 DDO route of benzofuran
The mechanism adopted for the DDO route of the benzofuran molecule is slightly different from that used for both furan and 2-methylfuran except from dihydrogen dissociation and vacancy regeneration that remain the same. Indeed, the transfer of the hydrogen atom from a molybdenum to the sulfur atom closest to the organic part takes place before the cleavage of the first C–O bond. On the other hand, the addition of the second H atom and the breaking of the second C–O bond occur simultaneously, whereas they are separately for furan and methylfuran used as reactants (see Fig. S2 in the ESI†).Fig. 5 shows the energy diagram for the DDO route of benzofuran onto the MoS2 surface using the PBE + D2 calculated energies. Knowing that the phenylethanol is not observed during the various benzofuran deoxygenation reactions,53,54 the direct deoxygenation reaction is initiated by the addition of the hydrogen atom to the carbon atom on α position of the oxygen atom and not belonging to the benzene cycle. This first step in the process is endothermic and slightly more than the corresponding steps in the deoxygenation of furan and 2-methylfuran. It is also kinetically more challenging to occur than the same steps for these molecules, with an energy value of 98 kJ mol−1. This step is followed by the transfer of the second hydrogen atom to the sulfur atom closest to the organic molecule. It is practically athermic (1 kJ mol−1) unlike the corresponding stages of furan and 2-methylfuran which are all exothermic. This suggests that the transfer of the second hydrogen atom to the sulfur atom closest to the organic part of the reaction system would preferentially take place after the cleavage of the first C–O bond. The cleavage of the first C–O bond after the addition of the H atom is strongly exothermic with an energy value of −193 kJ mol−1, which is more exothermic than the corresponding steps of furan and 2-methylfuran. The calculated activation energy (24 kJ mol−1) is practically equal to the one of the cleavage of the first C–O bond of furan. In comparison to 2-methylfuran, it exhibits a relatively straightforward kinetic pathway, irrespective of the route under consideration. The benzofuran molecule is indeed plan like that of the furan one. This very small difference in activation energy between the furan and the benzofuran on the one hand and the large difference in the same energy between the benzofuran and the 2-methylfuran on the other hand, show that the steric discomfort created by the group methyl would be solely responsible for the difference observed.
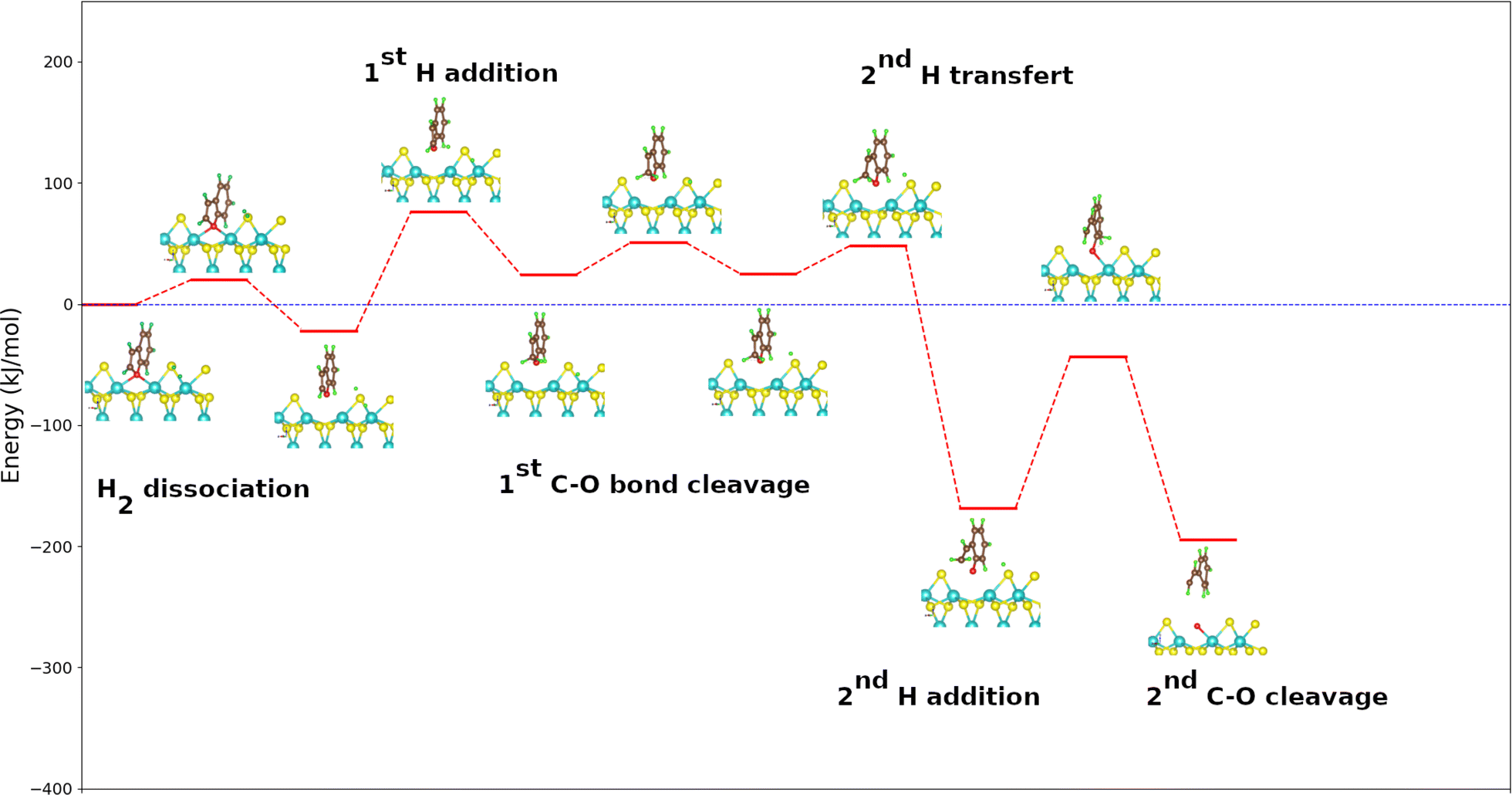 | ||
| Fig. 5 Energy diagram for DDO route of the benzofuran molecule on the metal edge of MoS2 surface. For clearer images, please refer to Fig. S5 in the ESI.† | ||
As mentioned above, the addition of the second hydrogen atom and the breaking of the second C–O bond for benzofuran occurred simultaneously. It is exothermic with an energy value of −25 kJ mol−1. The calculated activation energy (125 kJ mol−1) indicates that it is notably less kinetically favored compared to the corresponding stages of furan and methylfuran. This step is therefore the limiting step of the direct deoxygenation route of benzofuran. The desorption of the water molecule for the regeneration of the vacancy is here also the most endothermic for the whole process.
4 Conclusions
The direct hydrodeoxygenation pathway of furanic molecules (furan, 2-methylfuran, and benzofuran) was studied by the DFT/PBE method on a stable surface of MoS2. In reactions involving furan and 2-methylfuran (2H route), the addition of the first hydrogen atom constitutes the rate-determining step. For the 5H route of 2-methylfuran, the cleavage of the first C–O bond is the rate determining step. Our calculations demonstrate that the 2H pathway is expected as the most suitable for the deoxygenation of the 2-meythylfuran. For benzofuran, the concomitant step of adding the second hydrogen atom and breaking the second C–O bond is kinetically determining. For all furanic molecules, the desorption of the water molecule for the regeneration of the vacancy is the most endothermic step. Taking into account the thermodynamic and kinetic aspects, the following order of reactivity for furanic molecules studied can be expected: furan ≈ 2-methylfuran > benzofuran.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was performed using the Lorraine University HPC Mesocenter “Explor” and TGCC under the allocation 2023-A0140810433 by GENCI-EDARI and also the Lille University HPC center. SG, MB and SB also acknowledge financial support through the COMETE project (COnception in silico de Matériaux pour l’EnvironnemenT et l’Energie) cofunded by the European Union under the program “FEDER-FSE Lorraine et Massif des Vosges 2014–2020”. Frederic Richard acknowledges financial support from the European Union (ERDF), “Region Nouvelle Aquitaine”.Notes and references
- A. L. Ahmad, N. M. Yasin, C. Derek and J. Lim, Renewable Sustainable Energy Rev., 2011, 15, 584–593 CrossRef CAS.
- P. Nikulshin, V. Salnikov, A. Varakin and V. Kogan, Catal. Today, 2016, 271, 45–55 CrossRef CAS.
- G. Fogassy, C. Lorentz, G. Toussaint, N. Thegarid, Y. Schuurman and C. Mirodatos, Environ. Prog. Sustainable Energy, 2013, 32, 377–383 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- E. A. Quadrelli, Green Chem., 2016, 18, 328–330 RSC.
- M. N. Nabi, M. M. Rahman, M. A. Islam, F. M. Hossain, P. Brooks, W. N. Rowlands, J. Tulloch, Z. D. Ristovski and R. J. Brown, Energy Convers. Manage., 2015, 96, 588–598 CrossRef CAS.
- H. Von Blottnitz and M. A. Curran, J. Cleaner Prod., 2007, 15, 607–619 CrossRef.
- D. Mohan, C. U. Pittman Jr and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- S. Czernik and A. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- D. Elliott, D. Beckman, A. Bridgwater, J. Diebold, S. Gevert and Y. Solantausta, Energy Fuels, 1991, 5, 399–410 CrossRef CAS.
- M. Daage and R. R. Chianelli, J. Catal., 1994, 149, 414–427 CrossRef CAS.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS.
- Q. Bu, H. Lei, A. H. Zacher, L. Wang, S. Ren, J. Liang, Y. Wei, Y. Liu, J. Tang and Q. Zhang, et al., Bioresour. Technol., 2012, 124, 470–477 CrossRef CAS PubMed.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- A. Y. Bunch and U. S. Ozkan, J. Catal., 2002, 206, 177–187 CrossRef CAS.
- Y. Romero, F. Richard, Y. Renème and S. Brunet, Appl. Catal., A, 2009, 353, 46–53 CrossRef CAS.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1–12 CrossRef CAS.
- E. Furimsky, Catal. Today, 2013, 217, 13–56 CrossRef CAS.
- M. Sun, J. Adjaye and A. E. Nelson, Appl. Catal., A, 2004, 263, 131–143 CrossRef CAS.
- L. S. Byskov, J. K. Nørskov, B. S. Clausen and H. Topsøe, J. Catal., 1999, 187, 109–122 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, J. Catal., 2000, 189, 129–146 CrossRef CAS.
- E. Hensen, P. v. Kooyman, Y. Van der Meer, A. Van der Kraan, V. De Beer, J. Van Veen and R. Van Santen, J. Catal., 2001, 199, 224–235 CrossRef CAS.
- H. G. Füchtbauer, A. K. Tuxen, Z. Li, H. Topsøe, J. V. Lauritsen and F. Besenbacher, Top. Catal., 2014, 57, 207–214 CrossRef.
- A. K. Tuxen, H. G. Füchtbauer, B. Temel, B. Hinnemann, H. Topsøe, K. G. Knudsen, F. Besenbacher and J. V. Lauritsen, J. Catal., 2012, 295, 146–154 CrossRef CAS.
- M. Šarić, J. Rossmeisl and P. G. Moses, Phys. Chem. Chem. Phys., 2017, 19, 2017–2024 RSC.
- S. Li, Y. Liu, X. Feng, X. Chen and C. Yang, Mol. Catal., 2019, 463, 45–53 CrossRef CAS.
- R. Chianelli, G. Berhault, P. Raybaud, S. Kasztelan, J. Hafner and H. Toulhoat, Appl. Catal., A, 2002, 227, 83–96 CrossRef CAS.
- B. Dupuy, S. Laforge, C. Bachmann, P. Magnoux and F. Richard, J. Mol. Catal. A: Chem., 2012, 363, 273–282 CrossRef.
- H. Toulhoat, P. Raybaud, S. Kasztelan, G. Kresse and J. Hafner, Catal. Today, 1999, 50, 629–636 CrossRef CAS.
- S. Cristol, J. Paul, E. Payen, D. Bougeard, S. Clémendot and F. Hutschka, J. Phys. Chem. B, 2000, 104, 11220–11229 CrossRef CAS.
- S. Cristol, J. Paul, E. Payen, D. Bougeard, S. Clemendot and F. Hutschka, J. Phys. Chem. B, 2002, 106, 5659–5667 CrossRef CAS.
- J.-F. Paul and E. Payen, J. Phys. Chem. B, 2003, 107, 4057–4064 CrossRef CAS.
- M. Sun, A. E. Nelson and J. Adjaye, Catal. Today, 2005, 105, 36–43 CrossRef CAS.
- P. Zheng, A. Duan, K. Chi, L. Zhao, C. Zhang, C. Xu, Z. Zhao, W. Song, X. Wang and J. Fan, Chem. Eng. Sci., 2017, 164, 292–306 CrossRef CAS.
- M. Badawi, J.-F. Paul, S. Cristol and E. Payen, Catal. Commun., 2011, 12, 901–905 CrossRef CAS.
- C. Guo, T. Zhang, M. Niu, S. Cao, S. Wei, Z. Wang, W. Guo, X. Lu and C.-M. L. Wu, Mol. Catal., 2019, 463, 67–76 CrossRef CAS.
- W. G. Kanhounnon, U. A. Kuevi, G. A. Kpotin, S. Koudjina, A. K. Houngue, G. Y. Atohoun, J.-B. Mensah and M. Badawi, J. Mol. Model., 2019, 25, 1–11 CrossRef CAS.
- M. Badawi, S. Cristol, J.-F. Paul and E. Payen, C. R. Chim., 2009, 12, 754–761 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. Bucko, J. Hafner, S. Lebegue and J. G. Angyan, J. Phys. Chem. A, 2010, 114, 11814–11824 CrossRef CAS.
- Q. Jin, B. Chen, Z. Ren, X. Liang, N. Liu and D. Mei, Catal. Today, 2018, 312, 158–167 CrossRef CAS.
- Y. Huang, H. Liu, X. Chen, D. Zhou, C. Wang, J. Du, T. Zhou and S. Wang, J. Phys. Chem. C, 2016, 120, 12012–12021 CrossRef CAS.
- M. Brémaud, L. Vivier, G. Pérot, V. Harlé and C. Bouchy, Appl. Catal., A, 2005, 289, 44–50 CrossRef.
- X.-D. Wen, T. Zeng, B.-T. Teng, F.-Q. Zhang, Y.-W. Li, J. Wang and H. Jiao, J. Mol. Catal. A: Chem., 2006, 249, 191–200 CrossRef CAS.
- P. d'Araujo, C. Thomas, L. Vivier, D. Duprez, G. Pérot and S. Kasztelan, Catal. Lett., 1995, 34, 375–378 CrossRef.
- C. Thomas, L. Vivier, J. Lemberton, S. Kasztelan and G. Pérot, J. Catal., 1997, 167, 1–11 CrossRef CAS.
- C. Thomas, L. Vivier, A. Travert, F. Maugé, S. Kasztelan and G. Pérot, J. Catal., 1998, 179, 495–502 CrossRef CAS.
- C. Thomas, L. Vivier, M. Lescanne, S. Kasztelan and G. Pérot, Catal. Lett., 1999, 58, 33–35 CrossRef CAS.
- C.-L. Lee and D. F. Ollis, J. Catal., 1984, 87, 325–331 CrossRef CAS.
- M. C. Edelman, M. K. Maholland, R. M. Baldwin and S. W. Cowley, J. Catal., 1988, 111, 243–253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03043e |
| This journal is © The Royal Society of Chemistry 2024 |