DOI:
10.1039/D4RA03208J
(Paper)
RSC Adv., 2024,
14, 24141-24151
Bio-based sunflower carbon/polyethylene glycol shape-stabilized phase change materials for thermal energy storage†
Received
30th April 2024
, Accepted 13th June 2024
First published on 2nd August 2024
Abstract
The exploitation of shape-stabilized phase change materials with high thermal conductivity and energy storage capacity is an effective strategy for improving energy efficiency. In this work, sunflower stem carbon/polyethylene glycol (SS-PEG) and sunflower receptacle carbon/polyethylene glycol (SR-PEG) shape-stabilized phase change materials, utilizing sunflower stem and receptacle biomass carbon with high specific surface area and pore volume obtained by carbonization as frameworks and polyethylene glycol as an energy storage material, were prepared by the vacuum impregnation method. The ability to load polyethylene glycol into the pore structure of carbon materials in different sunflower parts was mainly investigated, and the micro-morphology, compositional structure and thermal properties were characterized and analyzed using SEM, IR spectroscopy, XRD, DSC and TG techniques. The results showed that the carbonized sunflower stems maintained the sieve pore structure, and the carbonized sunflower receptacle was a macroporous structure containing a large number of three-dimensional interconnections. At the same time, the interaction between polyethylene glycol and each carbon material occurred through physisorption. The melting enthalpies of SS-PEG and SR-PEG shape-stabilized phase change materials were 153.4 J g−1 and 171.5 J g−1, respectively, and the loading rates reached 81.9% and 91.5%, with initial thermal decomposition temperatures (T5%) of 344 °C and 368 °C.
1. Introduction
With the rapid development of the global society and economy, energy consumption and environmental pollution from fossil energy sources such as coal have been increasing.1–3 To meet human needs and achieve the goal of energy conservation, emission reduction, and improvement of environmental pollution, the future global energy trend is to reduce the use of disposable traditional energy sources such as coal, oil, and natural gas, develop new energy sources and promote the development of renewable energy sources. Thermal energy storage (TES) technology is an effective strategy to improve energy utilization, which mainly consists of four types: sensible heat, latent heat, thermochemical, and sorption heat storage.4,5 Among the various thermal energy storage methods, latent heat storage (LHS) based on phase change materials (PCMs) is an efficient energy storage technology with high energy storage density within the phase change temperature interval of the material, which utilizes the change of physical state of phase change materials to absorb or release energy and realizes high-efficiency energy storage.6,7
At present, phase change materials with considerable energy storage density have become a research hotspot in the industry as energy-saving materials and have been widely used in the fields of solar energy systems,8 the automobile industry,9 astronautics and aeronautics,10 energy-saving buildings,11 and intelligent textiles.12 These are mainly divided into solid–solid phase change materials and solid–liquid phase change materials according to the classification of the phase transition form of PCMs. Solid–solid phase change materials are primarily organic materials with good application prospects in thermal energy storage due to their advantages of no-phase separation, small volume change, anti-leakage, and excellent mechanical properties,13,14 mainly including polyol phase change materials, layered chalcogenides and polymers, and their latent heat of phase transition is smaller than that of solid–liquid phase change materials while their phase transition temperature is relatively high. Solid–liquid phase change materials can be divided into organic phase change materials, inorganic phase change materials, and eutectic phase change materials according to their chemical compositions, including paraffin, fatty acids, alcohols, crystalline hydrated salts, molten salts, and eutectic hydrated salts, which have been widely used due to their sizeable latent heat values, many types and small volume changes.15–17 Among them, polyethylene glycol (PEG) is an organic phase change material with the advantages of non-toxicity, favorable biocompatibility, and excellent thermal stability.14 However, the fluidity and the susceptibility to leakage of the liquid phase of polyethylene glycol during the phase change process and low thermal conductivity limit their applications.1,18,19 Confining polyethylene glycol to porous-based supporting materials such as polymers,20,21 foam metal,22,23 and porous carbon materials19,24 to prepare shape-stabilized phase change materials is an effective strategy to address the above drawbacks.
Biomass porous carbon materials have attracted widespread attention and are widely used in many fields, such as energy storage,25,26 supercapacitors,27,28 wastewater treatment,29,30 and electromagnetic shielding,31,32 because of their vast source, low cost, and renewability, and are synthesized from biomass through thermochemical degradation processes such as gasification, hydrothermal carbonization, and high-temperature pyrolysis under anoxic or micro-oxygen conditions.33 Biomass is a plant resource composed of organic carbon,34 which primarily includes agricultural by-products such as straw, rice husk, corn kernel, fruit shells, and bamboo wood, and its chemical composition is mainly cellulose, hemicellulose and lignin, some of which contain a small amount of silica.35 Compared with ordinary carbon materials, biomass porous carbon materials have apparent advantages: cross-linked three-dimensional porous skeleton structure, high specific surface area, strong corrosion and heat resistance, and high thermal and electrical conductivity. Currently, biomass porous carbon materials are prepared using carbonization, activation and template methods. Among them, the direct carbonization method has the advantages of simple operation and low cost, which involves thermal decomposition at a high temperature in an inert gas (N2 or He) atmosphere, and the non-carbon elements and volatile components are removed through various physical and chemical reactions, resulting in the formation of porous carbon.36 Wei et al.37 prepared succulent-based carbon aerogel (SCA) by carbonizing the leaves of succulent plants and introducing molten paraffin into the SCA to obtain a shape-stabilized phase change material. The results showed that the maximum loading of SCA to paraffin was up to 95%, and the melting enthalpy of the composite phase change material was 130.1 J g−1, which was only 1.8% lower than that of pure paraffin. Ji et al.38 prepared a 3D porous network carbon aerogel with “soft–rigid” network structures by direct carbonization of pomelo peels as a support matrix loading paraffin wax to acquire a composite phase change material, which showed a significant latent heat storage capacity of 159.9 J g−1 and exhibited excellent thermal reliability after 25 frequent heating/cooling cycles. The composite phase change materials exhibit ultra-low density, excellent thermal stability, enhanced thermal and electrical conductivity, shape stability, and leakage resistance.
Sunflower stems and receptacles have a good pore structure, which provides a circulation path for the carbonized skeleton loaded with phase change materials. Here, we utilize carbonized sunflower stems and receptacles loaded with polyethylene glycol for the first time with a view to preparing two shape-stabilized phase change materials. In this paper, sunflower stem biomass carbon (SSC) and sunflower receptacle biomass carbon (SRC) with high specific surface area and pore volume were prepared from sunflower biomass by direct carbonization, and sunflower stem carbon/polyethylene glycol and sunflower containing carbon/polyethylene glycol shape-stabilized phase change materials were prepared from polyethylene glycol loaded into the two biomass porous carbon materials using vacuum impregnation method. The loading capacity of the porous structure of the two biomass porous carbon materials for polyethylene glycol was investigated, and their thermal storage properties were also explored. Thus, this work opens up new avenues for efficient energy utilization.
2. Experimental methods
2.1. Materials
Sunflowers were collected from the Sichuan Province of China. Polyethylene glycol (PEG, Mn = 4000) was provided by Bayer Co., Ltd (China).
2.2. Preparation of sunflower biomass carbon
The purchased raw materials were washed with deionized water and placed in a sunny environment at room temperature for 24 h. The excess water was wiped off using paper towels, and each part of the sunflower (stem, receptacle, leaves, and petal) was separated appropriately before being placed in a freeze-drying oven at −70 °C for 4 h and then freeze-dried for 48 h. The lyophilized and dehydrated sunflower parts were heated up to 1000 °C under the atmosphere of N2 in a tube furnace at 5 °C min−1 and pyrolyzed for 3 h at this temperature to obtain SSC, SRC, sunflower leaf biomass carbon (SLC) and sunflower petal biomass carbon (SPC), as shown in Fig. 1.
 |
| | Fig. 1 The flowchart for the preparation of shape-stabilized phase change materials. | |
2.3. Preparation of shape-stabilized phase change materials
The PEG was heated to 80 °C in an oven to melt into the liquid state. Then, two samples, SSC and SRC, were immersed into the molten PEG under a vacuum of −0.1 MPa for 4 h to allow the PEG to be adsorbed by the activated charcoal and expel the internal bubbles. Moreover, the samples were removed and placed on filter paper at 80 °C, the excess PEG adsorbed on the surface of the activated charcoal was rolled off until there was no leakage from the filter paper, and the shape-stabilized phase change materials were obtained. The flowchart for preparing shape-stabilized phase change materials is shown in Fig. 1.
2.4. Characterizations
The micro-morphology of samples was investigated by field emission scanning electron microscopy (SEM, JSM-5900LV, Japan). The porous structure of SSC and SRC samples was analyzed using Brunauer–Emmett–Teller (BET, Micromeritics ASAP2460, American). The graphitization degrees of SSC and SRC samples were characterized by Raman spectra (inVia Reflex, American). The chemical structures of samples were studied by Fourier transform infrared spectroscopy (FT-IR, Nicolet 50, American) in the infrared wavenumber from 4000 to 400 cm−1, and X-ray diffraction (XRD, Ultima IV, Japan, Cu Ka radiation at 40 kV and 50 mA). The thermal properties of samples were analyzed by differential scanning calorimetry (DSC, TA Q250, American). A 3–5 mg sample was taken and sealed in an aluminum crucible and placed in the apparatus, and the sample was heated from 10 °C to 90 °C at a heating rate of 10 °C min−1 and then cooled down from 90 °C to 10 °C at a cooling rate of 10 °C min−1 in a nitrogen atmosphere. In order to ensure the reliability of data, three specimens were tested for each sample to obtain an average value. The thermal decomposition behavior of samples was observed by thermogravimetric analysis (TGA, TGA2, Sweden) from 35 to 800 °C at a heating rate of 10 °C min−1 under N2 atmosphere. Thermal conductivity of two shape-stabilized PCMs was conducted by the thermal constant analyzer (Model 2500-OT, Hot disk, Sweden) using a Kapton probe with a radius of 2.001 mm. Solar thermal energy conversion capability was evaluated via an infrared thermal imager (FTLR T420, American). The shape stability of the samples was examined by photographing the leakage of the samples before and after 30 min at 80 °C.
3. Results and discussion
3.1. Structures of SSC and SRC
Fig. 2 shows the SEM images of the raw carbon of each part of the sunflower. Fig. 2a and b shows the morphology of the pore structure of the sunflower stem biomass carbon material. It could be clearly seen that there was a sieve pore structure inside the SSC, which maintained the pore structure of the raw material, while the pith of the stem in the central layer was more easily broken by pressure. The edge of the lignified structure was relatively more potent and retained some of the original sieve pore structure after extrusion and crushing. Fig. 2c and d presented the morphology of the pore structure of the carbon material of the sunflower receptacle, from which it could be obviously observed that the SRC also retained the morphology of the original material, containing a large number of three-dimensional interconnected macroporous structures, with the pore size of 10–50 μm. Fig. 2e–h show the morphology of the pore structures of the sunflower leaf biomass carbon and the sunflower petal biomass carbon materials, respectively. The pore structure of the SLC material is mainly a three-dimensional interpenetrating irregular pore structure, and the pore structure of the SPC material is a unidirectional pore channel structure. Due to the defects of low yield and fragility of the SLC and SPC materials, although they also have excellent pore structures, only SSC and SRC materials were used as the skeleton support materials when impregnating PEG in the later stage.
 |
| | Fig. 2 SEM images of primary carbon in various parts of sunflower: (a and b) SSC; (c and d) SRC; (e and f) SLC; (g and h) SPC. | |
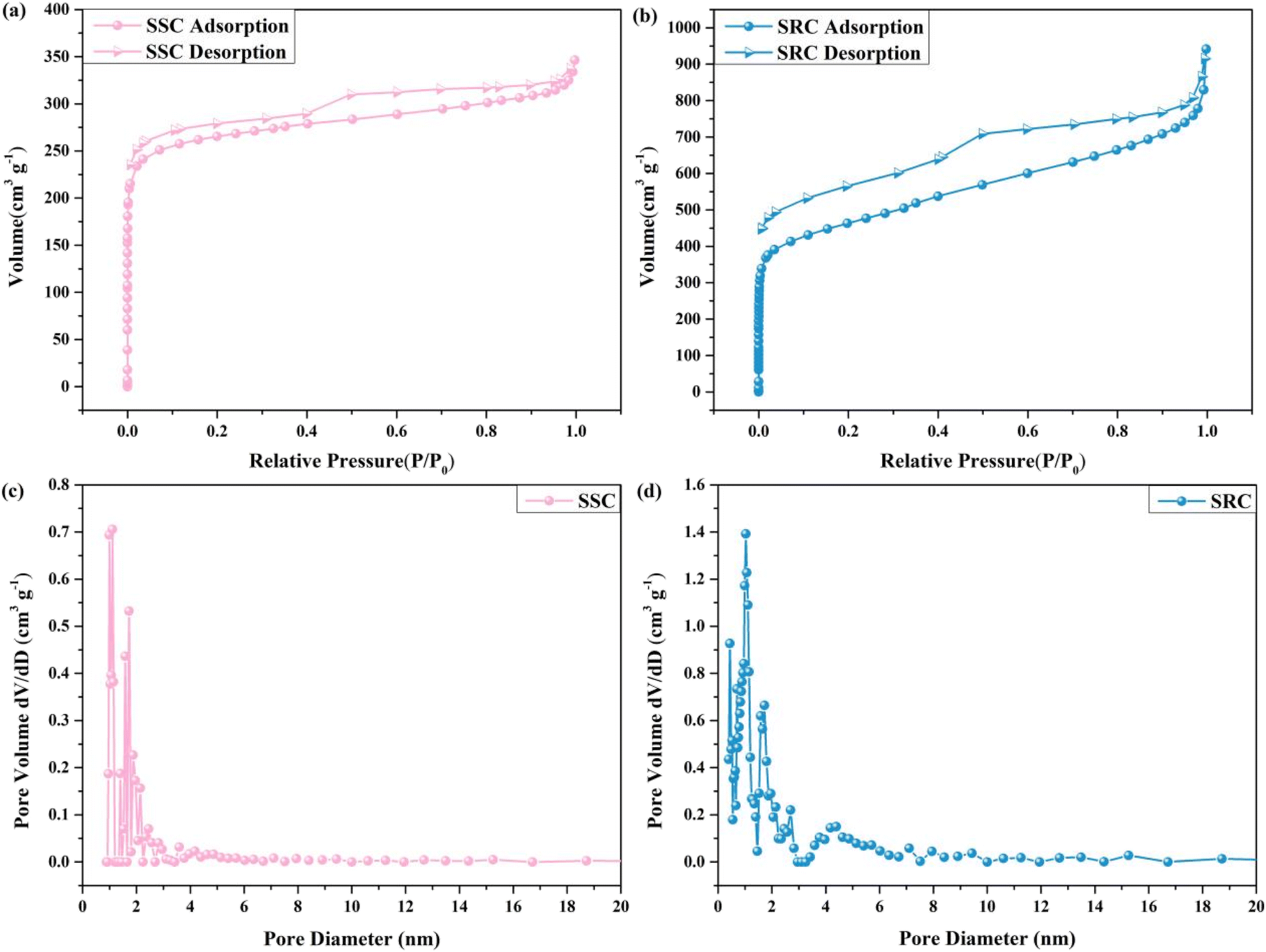
In order to assess the pore structure and adsorption capability of SSC and SRC, the specific surface area and pore size of SSC and SRC were analyzed utilizing N2 adsorption–desorption isotherms and DFT desorption pore size distribution, as shown in Fig. 3 and Table 1. The isotherms of SSC and SRC were both type IV accompanied by an H4 hysteresis loop according to the IUPAC classification.39 Fig. 3a and b showed that the adsorption of SSC and SRC increased rapidly in the relatively low-pressure region, indicating the existence of a strong interaction force between SSC and nitrogen as well as between SRC and nitrogen, along with the existence of a microporous structure. In the higher pressure region (0.5 < P/P0 < 0.99), the adsorption of SSC and SRC again increased steeply due to capillary condensation, demonstrating the presence of meso/macropores. Combining Fig. 3 with Table 1, the specific surface area of SSC and SRC was 1016.60 m2 g−1 and 1637.00 m2 g−1, respectively, and the pore volume of SSC and SRC was 0.51 cm3 g−1 and 1.27 cm3 g−1, which was mainly attributed to the fact that water vapor and carbon dioxide generated from the raw materials can promote the formation of porous structures at high temperatures. Moreover, the average pore size of SSC and SRC was 2.01 nm and 3.10 nm, respectively, showing that SSC and SRC had a multilevel pore structure and their main structure was mesoporous, which contributed to the penetration and loading of organic PCM into SSC and SRC.
 |
| | Fig. 3 Nitrogen adsorption/desorption isotherms: (a) SSC and (b) SRC, and DFT desorption pore size distribution: (c) SSC and (d) SRC. | |
Table 1 BET surface area and pore volume of SSC and SRC
| Samples |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Micro pore volume (cm3 g−1) |
Mean pore size (nm) |
| SSC |
1016.60 |
0.51 |
233.56 |
2.01 |
| SRC |
1637.00 |
1.27 |
376.10 |
3.10 |
The physical properties of the prepared SSC and SRC were investigated by Raman spectroscopy, and the Raman spectra are shown in Fig. 4. The broad peaks at 1348 cm−1 and 1590 cm−1 corresponded to the D and G bands of carbon, respectively, where the relative intensity of the D band represented the degree of defects in the graphite properties, which was known as disordered graphite.40 The G band correlated with the phonon modes of graphite and the formation of graphitic carbon and was used to characterize the sp2 bond structure of the carbon material. The graphite properties of the samples were evaluated using the ratio of the intensity of the G band to the peak intensity of the D band (IG/ID), and the IG/ID values calculated here are 1.10 and 1.06, respectively, indicating that SSC and SRC had high graphitic structures. Meanwhile, the presence of many structural defects in the two bio-carbon materials implied that some of the carbon in the samples was in an amorphous state due to carbonization, and this amorphousness contributed to the enhancement of the pore properties of the carbon materials, which further improved the adsorption capacity of the organic phase-change materials, and thus enhanced their suitability as a support framework for thermal energy storage materials.41
 |
| | Fig. 4 Raman spectra of SSC and SRC. | |
3.2. Structures of shape-stabilized PCMs
Two shape-stabilized phase change materials, SS-PEG and SR-PEG, were obtained by loading PEG into SSC and SRC support skeletons through vacuum impregnation, and the morphology and structure of these two materials are shown in Fig. 5. In Fig. 5a, it was observed that the polyethylene glycol had been completely filled into the sunflower stem carbon material, which suggested that the sieve pore structure of the sunflower stem carbon material facilitated the PEG infiltration and prevented the leakage of PEG through the physisorption force of the pore structure of this material. Fig. 5b found that when the sunflower receptacle carbon material adsorbed polyethylene glycol, the large pores acted as transportation conduits and mesopores and micropores on the pore walls acted as physical adsorption sites to limit the flow of PEG in the liquid state at elevated temperatures. In order to test the shape stabilizing ability of the two stereotyped phase change materials of SS-PEG and SR-PEG, PEG, SS-PEG and SR-PEG at a temperature of 20 °C were used as the comparison samples and heated to 80 °C for 30 min using a temperature higher than the melting point of PEG, which was verified by macroscopically observing the dry and wet state of the filter paper, as shown in Fig. 5c. When the temperature was 20 °C, PEG, SS-PEG and SR-PEG were all in a solid state, and after being heated to 80 °C for 30 min, PEG was transformed from the solid state to the liquid state, SS-PEG and SR-PEG were still in solid state and the filter paper did not show any wetting phenomenon, which implied that SS-PEG and SR-PEG were macroscopically two kinds of shape-stabilized phase change materials. To further study the shape stabilization of the materials, the leakage of the composites was determined by heating the samples cyclically 100 times, and the results are shown in Fig. 5d. After 100 heating cycles, the mass loss of SS-PEG and SR-PEG was 2.49 wt% and 2.64 wt%, respectively, which indicated that the two composites possessed superior leak-proof properties. This excellent leak-proof property can be attributed to the fact that the multistage pore structures of SSC and SRC have sufficient capillary forces to adsorb PCM. During this period, the shape and size of the composites were observed macroscopically without deformation.
 |
| | Fig. 5 SEM images of shape-stabilized phase change materials: (a) SS-PEG and (b) SR-PEG; (c) leakage macroscopic morphology of PEG, SS-PEG and SR-PEG under 80 °C for 30 min; (d) the leakage rate of SS-PEG and SR-PEG after 100 thermal cycles. | |
In order to analyze the component structure of the SS-PEG and SR-PEG shape-stabilized phase change materials, the composition of the samples was investigated utilizing Fourier transform infrared absorption spectroscopy. The infrared spectra of PEG, each carbon material, and the SS-PEG and SR-PEG shape-stabilized phase change materials are shown in Fig. 6. In Fig. 6a, the broad absorption peak of PEG at 3460 cm−1 is attributed to the O–H stretching vibration. The characteristic absorption peak at 2880 cm−1 corresponds to the C–H stretching vibration of methylene, while the absorption peaks at 1470 cm−1 and 1340 cm−1 represent the C–H bending vibration of methylene. The absorption peaks at 1280 cm−1 and 1110 cm−1 refer to the C–O–C stretching vibration, whereas the absorption peaks at 963 cm−1 and 842 cm−1 correspond to the C–O–C bending vibration. As can be observed in Fig. 6b, comparing the infrared spectra of PEG and each carbon material, no new characteristic peaks appeared in the infrared spectra of each of the shape-stabilized phase change materials, which implied that there was no chemical reaction between SSC and PEG as well as SRC and PEG during the preparation of the SS-PEG and SR-PEG shape-stabilized phase change materials. Therefore, it primarily relied on the physical adsorption effect to confine the PEG to the pore structure of each carbon material.42
 |
| | Fig. 6 Infrared spectra of PEG4000, various carbon materials and shape-stabilized phase change materials: (a) PEG4000; (b) SSC, SRC, SS-PEG and SR-PEG. | |
The crystallization behaviors of SS-PEG and SR-PEG were analyzed by XRD, as shown in Fig. 7. The high-intensity diffraction peaks of PEG, SS-PEG, and SR-PEG at 2θ = 19.08° and 23.18° correspond to the characteristic crystal planes (120) and (112), respectively.14,43 Compared with PEG, the SS-PEG and SR-PEG shape-stabilized phase change materials showed a weakening trend in each peak, which implied that SSC and SRC had a limiting effect on the crystallization of polyethylene glycol. Meanwhile, no new diffraction peaks were generated in the SS-PEG and SR-PEG shape-stabilized phase change materials, which indicated that the crystal structures of PEG in SSC and SRC were not altered as well as PEG was adsorbed into the porous structures of SSC and SRC through physical interactions, in agreement with the analytical results in Fig. 6.
 |
| | Fig. 7 XRD patterns of PEG4000, SSC, SRC, SS-PEG and SR-PEG. | |
3.3. Thermal properties and thermal stability of shape-stabilized PCMs
The thermal performance of SS-PEG and SR-PEG shape-stabilized phase change materials were investigated by DSC, and the results are shown in Fig. 8 and Table 2. The enthalpies of melting and crystallization of PEG were 187.4 J g−1 and 174.3 J g−1, respectively, and the melting and crystallization temperatures were 62.5 °C and 28.5 °C. Compared with PEG, the enthalpies decreased for both SS-PEG and SR-PEG shape-stabilized phase change materials. On the one hand, the addition of SSC and SRC reduced the mass ratio of PEG in the composites, which led to a proportional decrease in the enthalpy of melting/crystallization. On the other hand, the physical adsorption effect of SSC and SRC bound the movement of PEG chain segments and decreased the free movement ability of chain segments. Meanwhile, SSC and SRC also destroyed the regularity of PEG molecular chains to decrease the crystallinity and melting/crystallization enthalpy of PEG. Compared to pure PEG, the composites showed a different phase transition process, with the melting point decreasing and the crystallization temperature increasing during the heating/cooling cycle. The strong adsorption of SSC and SRC to PEG led to a decrease in the free movement of the PEG molecular chains. Consequently, the energy required for the free movement of PEG molecular chains increased, and the crystallization temperature heightened.
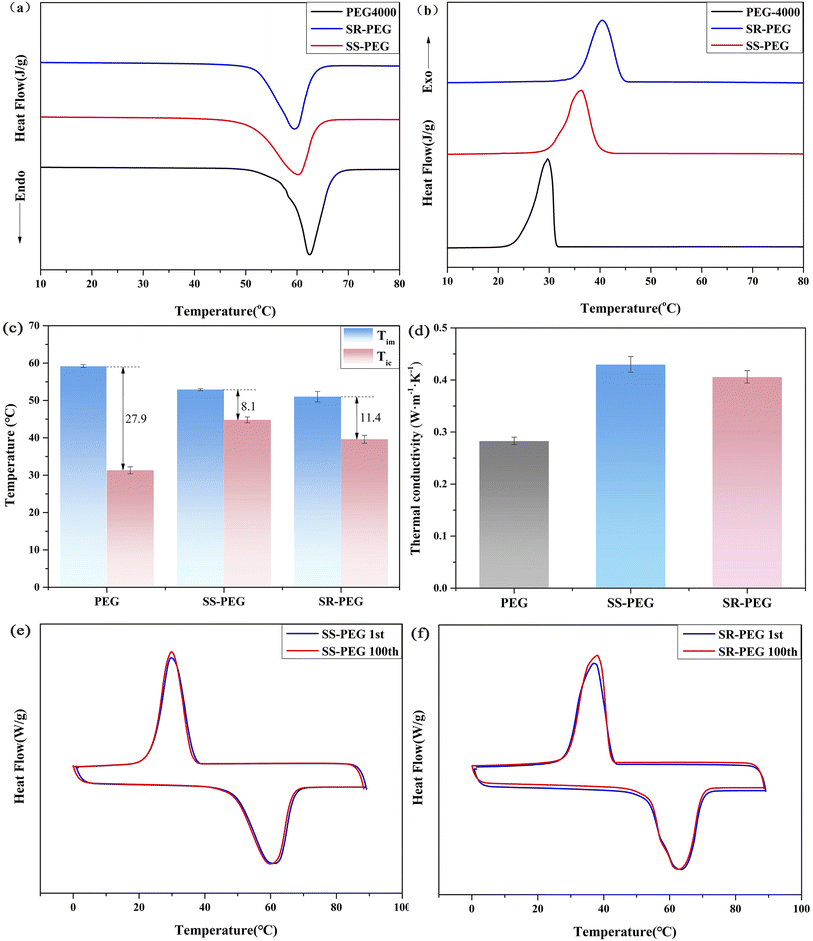 |
| | Fig. 8 DSC curves of PEG4000, SS-PEG and SR-PEG: (a) melting curve, (b) crystallization curve; (c) melting and crystallization temperature of PEG4000, SS-PEG and SR-PEG; (d) the thermal conductivity of PEG, SS-PEG, and SR-PEG; DSC curves of composite PCMs after 100 thermal cycles: (e) SS-PEG, (f) SR-PEG. | |
Table 2 Enthalpy and phase transition temperature of PEG4000 and various shape-stabilized phase change materials
| Samples |
Tim (°C) |
Tm (°C) |
ΔHm (J g−1) |
Tic (°C) |
Tc (°C) |
ΔHc (J g−1) |
R (%) |
| PEG4000 |
59.2 ± 0.4 |
62.5 ± 0.2 |
187.4 ± 0.3 |
31.3 ± 1.1 |
28.5 ± 1.2 |
174.3 ± 0.5 |
— |
| SS-PEG |
52.9 ± 0.3 |
59.7 ± 0.2 |
153.4 ± 0.7 |
44.8 ± 0.9 |
41.4 ± 1.1 |
150.4 ± 0.4 |
81.9 |
| SR-PEG |
51 ± 1.5 |
60 ± 1.1 |
171.5 ± 0.2 |
39.6 ± 1.2 |
36.7 ± 0.6 |
166.5 ± 0.2 |
91.5 |
PEG showed very large hysteresis (initial crystallization temperature much below initial melting temperature), which significantly limited its use as a PCM. In Table 2 and Fig. 8c, it was evident that the thermal hysteresis of the composites was improved with the addition of SSC and SRC. According to some studies,44–46 it is known that the difference in thermal conductivity affected the difference in the hysteresis degree, and under the same conditions, PCM with higher thermal conductivity had a lower melting temperature and higher crystallization temperature, which led to the difference in hysteresis degree. In order to explore the effect of the incorporation of SSC and SRC on the thermal conductivity of the composites, the thermal conductivity of two shape-stabilized PCMs was conducted by the thermal constant analyzer. The thermal conductivity of pure PEG was 0.283 W (m−1 K−1), and the thermal conductivities of SS-PEG and SR-PEG were 0.43 W (m−1 K−1) and 0.406 W (m−1 K−1), respectively. Thermal conductivity of SS-PEG and SR-PEG was significantly enhanced compared to pure polyethylene glycol, which was attributed to the inherently high thermal conductivity of SSC and SRC with higher graphite content, and the three-dimensional interpenetrating porous network structure of SSC and SRC provided a channel for rapid heat transfer. The result is consistent with the literature47,48 that the addition of biomass porous carbon enhances the thermal conductivity of the matrix material. Therefore, the incorporation of SSC and SRC into the composites increased their thermal conductivity and led to a lower hysteresis degree. The thermal hysteresis of SS-PEG was calculated to be lower than that of SR-PEG. Compared with SR-PEG, the percentage of biochar content of SS-PEG was higher, and the thermal hysteresis was lower with relatively less PEG content per unit mass of the composites, which meant that the thermal hysteresis of the composites decreased with the increase in biochar content.
For the sake of further evaluating the energy storage capacity of SS-PEG and SR-PEG shape-stabilized phase change materials, eqn (1) was used to calculate the loading ratio (R)49,50 of sunflower stem and sunflower receptacle biomass carbon (BC) on polyethylene glycol:
| |
 | (1) |
Δ
Hm,PCMs is the melting enthalpy of pure PEG, and Δ
Hm,PCMs/BC is the melting enthalpy of the prepared SS-PEG and SR-PEG shape-stabilized phase change materials. From the calculation, it can be seen that the loading ratios of SSC and SRC to PEG are as high as 81.9% and 91.5%, respectively, indicating that SS-PEG and SR-PEG have excellent heat storage capacity compared with other composite phase change materials in
Table 3.
Table 3 Thermal transition capacity and the loading of recently reported composite PCMs
| Supporting materials |
PCMs |
Latent heat (J g−1) |
Loading (%) |
Ref. |
| Potato derived carbon |
PEG4000 |
91.8 |
50.0 |
51 |
| Potato derived carbon |
PEG4000 |
158.8 |
85.4 |
52 |
| Eggplant-derived porous carbon |
PEG2000 |
149.0 |
90.1 |
53 |
| Hemp-stem-derived biochar |
PEG6000 |
170.44 |
88.62 |
54 |
| Corncob-derived biochar |
PEG6000 |
121.94 |
85.19 |
55 |
| Boron nitride/polypyrrole/wood-based carbon |
PEG6000 |
160 |
78.1 |
56 |
| SSC |
PEG4000 |
153.4 |
81.9 |
This work |
| SRC |
PEG4000 |
171.5 |
91.5 |
This work |
Analysis of 100 melting/freezing cycles was performed using DSC to estimate the cyclic stability of SS-PEG and SR-PEG shape-stabilized phase change materials. The 100-cycle test result, shown in Fig. 8e and f, demonstrated that the DSC curve of SS-PEG and SR-PEG after 100 melting/freezing cycles was almost consistent with the first cycle. Compared with the first cycle, the latent heat of SS-PEG and SR-PEG after 50 melting/freezing cycles lost only 0.59 J g−1 and 0.84 J g−1, respectively, which indicated that both SS-PEG and SR-PEG had excellent thermal cycle stability in thermal energy storage and release.
In the practical application process, the thermal stability of composite phase change materials is a crucial parameter. Fig. 9 shows the thermogravimetric curves of pure PEG, SS-PEG and SR-PEG shape-stabilized phase change materials. As seen in Fig. 9, polyethylene glycol had only one thermogravimetric behavior, and its initial thermal decomposition temperature was 372 °C when the mass loss was 5%, indicating that the polyethylene glycol had good thermal stability. In addition, polyethylene glycol was completely pyrolyzed, and the thermal weight loss rate reached 100% when the temperature increased to 800 °C. Both SS-PEG and SR-PEG also have only one thermal decomposition stage, with initial thermogravimetric loss temperatures (T5%) of 344 °C and 368 °C, respectively. When the temperature reached 418 °C and 436 °C, respectively, the mass loss of SS-PEG and SR-PEG reached the maximum due to the cleavage of PEG molecular chains during the thermal decomposition. Since the weight loss in the whole thermal decomposition process mainly comes from PEG, the load capacity of SSC and SRC to PEG can be further proved by the thermal weight loss rate. When the temperature reached 800 °C, the thermal weight loss rates of SS-PEG and SR-PEG samples were 83.95% and 91.56%, respectively. Combining Table 2 and Fig. 9, the thermal weight loss rate and loading rate of SS-PEG and SR-PEG are roughly the same.
 |
| | Fig. 9 TGA curves of PEG4000, SS-PEG and SR-PEG. | |
In order to investigate the photothermal conversion ability of SS-PEG and SR-PEG shape-stabilized phase change materials, a xenon lamp was used as a light source to irradiate the samples and an infrared thermal camera was used to capture the temperature changes of the samples. Fig. 10a shows the temperature variation curves of PEG, SS-PEG and SR-PEG with time under a xenon lamp, and Fig. 10b shows the infrared thermograms of PEG, SS-PEG and SR-PEG during the warming process. In Fig. 10b, it can be observed that the temperatures of PEG, SS-PEG and SR-PEG increased by 13.1 °C, 19.3 °C and 21.5 °C, respectively, after irradiation under the same xenon lamp irradiation environment for 10 min. The results demonstrated that SS-PEG and SR-PEG had a faster rate of heating compared to PEG, and the temperatures of SS-PEG and SR-PEG reached higher than those of PEG within the same heating time during the heating process in Fig. 10a and b. Therefore, the addition of SSC and SRC improved the heat transfer ability of SS-PEG and SR-PEG, but SR-PEG had better photothermal conversion compared to SS-PEG.
 |
| | Fig. 10 (a) Temperature curves of PEG, SS-PEG and SR-PEG with time under xenon lamp; (b) infrared thermal images of PEG, SS-PEG and SR-PEG during heating. | |
4. Conclusions
Sunflower stem biomass carbon and sunflower receptacle biomass carbon with high specific surface areas and multilayered hierarchical pore structures were prepared by carbonization. The specific surface area of SSC and SRC was 1016.60 m2 g−1 and 1637.00 m2 g−1, respectively, and the average pore size was 2.01 nm and 3.10 nm. Sunflower stem/polyethylene glycol and sunflower receptacle/polyethylene glycol shape-stabilized phase change materials were prepared by vacuum impregnation using polyethylene glycol as a phase change material and biomass carbon materials of sunflower stem and sunflower receptacle as the support skeleton. Due to the excellent pore structure of SSC and SRC, the loading rate of SSC and SRC on polyethylene glycol was as high as 81.9% and 91.5%, and the melting enthalpy of SS-PEG and SR-PEG shape-stabilized phase change materials was 153.4 J g−1 and 171.5 J g−1, respectively. In addition, SS-PEG and SR-PEG had excellent thermal stability and outstanding thermal reliability, and the latent heat remained almost unchanged after 100 cycles. Enhanced heat transfer was observed after the addition of SSC and SRC to PEG. In summary, SS-PEG and SR-PEG shape-stabilized phase change materials have great potential for application in thermal energy storage.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Conceptualization, N. G. and Y. L.; methodology, N. G., J. D., and W. Y.; validation, Y. L. and T. X.; formal analysis, J. D., Y. L., T. X., and C. Y.; investigation, W. Y., B. S., J. L., and B. L.; resources, Y. L. and X. L.; data curation, N. G.; writing – original draft preparation, N. G.; writing – review and editing, Y. L.; supervision, Y. L. and T. X.; project administration, Y. L. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the Chongqing Talent Innovation and Entrepreneurship Team Project of China, Science and Technology Research Project of the Chongqing Education Commission of China (KJZD-M201901101), Chongqing Municipal Education Commission Innovation Group Project of China (CXQT19027), the Chongqing Municipal Graduate Innovation Project of China (CYS22639), the Natural Science Foundation of Chongqing Municipality of China (CSTB2022NSCQ-MSX0275) and the Chongqing Municipal Technology Innovation and Application Development Special Program of China (CSTB2023TIAD-KPX0011). The authors also thank the research group for their cooperation.
References
- Y. Li, B. Wang, W. Zhang, J. Zhao, X. Fang, J. Sun, R. Xia, H. Guo and Y. Liu, Energy, 2022, 254, 124206 CrossRef CAS.
- C. Shao, Y. Zhao and L. Qu, SusMat, 2022, 2, 142–160 CrossRef CAS.
- M. Q. Wu, S. Wu, Y. F. Cai, R. Z. Wang and T. X. Li, Energy Storage Mater., 2021, 42, 380–417 CrossRef.
- M. Wu, T. Li, Q. He, R. Du and R. Wang, Energy, 2022, 239, 121938 CrossRef CAS.
- X. Chen, P. Cheng, Z. Tang, X. Xu, H. Gao and G. Wang, Adv. Sci., 2021, 8, 2001274 CrossRef CAS PubMed.
- W. Aftab, X. Huang, W. Wu, Z. Liang, A. Mahmood and R. Zou, Energy Environ. Sci., 2018, 11, 1392–1424 RSC.
- G. T. Nguyen, RSC Adv., 2023, 13, 7621–7631 RSC.
- S. Khanna, K. S. Reddy and T. K. Mallick, Sol. Energy, 2018, 163, 591–599 CrossRef.
- V. Talele, P. Thorat, Y. P. Gokhale and V. K. Mathew, J. Energy Storage, 2021, 44, 103482 CrossRef.
- E. Oró, A. D. Gracia, A. Castell, M. M. Farid and L. F. Cabeza, Appl. Energy, 2019, 99, 513e533 Search PubMed.
- M. Wu, C. Liu and Z. Rao, Constr. Build. Mater., 2022, 335, 127400 CrossRef CAS.
- Y. Zheng, H. Oguzlu, A. Baldelli, Y. Zhu, M. Song, A. Pratap-Singh and F. Jiang, Carbohydr. Polym., 2022, 291, 119583 CrossRef CAS PubMed.
- C. Chen, W. Liu, Z. Wang, K. Peng, W. Pan and Q. Xie, Sol. Energy Mater. Sol. Cells, 2015, 134, 80–88 CrossRef CAS.
- N. Gao, T. Tang, H. Xiang, W. Zhang, Y. Li, C. Yang, T. Xia and X. Liu, Sol. Energy Mater. Sol. Cells, 2022, 244, 111831 CrossRef CAS.
- B. Xu, P. Li and C. Chan, Appl. Energy, 2015, 160, 286–307 CrossRef.
- P. Sivasamy, A. Devaraju and S. Harikrishnan, Mater. Today, 2018, 5, 14423–14431 CAS.
- B. K. Choure, T. Alam and R. Kumar, J. Energy Storage, 2023, 72, 108161 CrossRef.
- X. Chen, J. Xu, Y. Li, Y. Gao and G. Wang, SusMat, 2023, 3, 510–521 CrossRef CAS.
- K. Zhou, Y. Sheng, W. Guo, L. Wu, H. Wu, X. Hu, Y. Xu, Y. Li, M. Ge, Y. Du, X. Lu and J. Qu, Adv. Compos. Hybrid Mater., 2023, 6, 34 CrossRef CAS.
- X. Fu, Y. Liu, X. Jiang, Q. Wang, Y. Luo and Y. Lyu, Thermochim. Acta, 2018, 665, 111–118 CrossRef CAS.
- J. Tang, M. Yang, F. Yu, X. Chen, L. Tan and G. Wang, Appl. Energy, 2017, 187, 514–522 CrossRef CAS.
- T. Ren, G. Du, Q. Li, Y. Wang, X. Fu, W. Kong, L. Jiang, J. Lei, P. He and Y. Xiao, J. Energy Storage, 2022, 47, 103592 CrossRef.
- H. Zhang, L. Wang, S. Xi, H. Xie and W. Yu, Renewable Energy, 2021, 175, 307–317 CrossRef CAS.
- X. Zhu, Q. Wang, S. Kang, J. Li and X. Jia, Chem. Eng. J., 2020, 395, 125112 CrossRef CAS.
- Z. Zou, W. Wu, Y. Wang and D. Drummer, Mater. Res. Express, 2020, 7, 045601 CrossRef CAS.
- Y. Sun, N. Zhang, X. Pan, W. Zhong, B. Qiu, Y. Cai and Y. Yuan, Int. J. Energy Res., 2021, 45, 20372–20383 CrossRef CAS.
- S. Zhang, Y. Li, Y. Du, X. Ma, J. Lin and S. Chen, Diam. Relat. Mater., 2022, 130, 109507 CrossRef CAS.
- S. Rawat, T. Boobalan, M. Sathish, S. Hotha and B. Thallada, Biomass Bioenergy, 2023, 171, 106747 CrossRef CAS.
- M. Kamali, L. Appels, E. E. Kwon, T. M. Aminabhavi and R. Dewil, Chem. Eng. J., 2021, 420, 129946 CrossRef CAS.
- L. Feng, Z. Gao, T. Hu, S. He, Y. Liu, J. Jiang, Q. Zhao and L. Wei, Chem. Eng. J., 2023, 471, 144772 CrossRef CAS.
- D. Torsello, M. Bartoli, M. Giorcelli, M. Rovere, R. Arrigo, G. Malucelli, A. Tagliaferro and G. Ghigo, Nanomaterials, 2021, 11, 2383 CrossRef CAS PubMed.
- C. Liu, M. Han, J. Lin, W. Liu, J. Liu and Z. Zeng, Carbon, 2023, 208, 255–276 CrossRef CAS.
- Y. Zhang, M. He, L. Wang, J. Yan, B. Ma, X. Zhu, Y. S. Ok, V. Mechtcherine and D. C. W. Tsang, Biochar, 2022, 4, 59 CrossRef CAS.
- L. Ke, Q. Wu, N. Zhou, J. Xiong, Q. Yang, L. Zhang, Y. Wang, L. Dai, R. Zou, Y. Liu, R. Ruan and Y. Wang, Renew. Sustain. Energy Rev., 2022, 165, 112607 CrossRef CAS.
- S. Wang, G. Dai, H. Yang and Z. Luo, Prog. Energy Combust. Sci., 2017, 62, 33–86 CrossRef.
- E. H. Lee, R. S. Park, H. Kim, S. H. Park, S. C. Jung, J. K. Jeon, S. C. Kim and Y. K. Park, J. Ind. Eng. Chem., 2016, 37, 18–21 CrossRef CAS.
- Y. Wei, J. Li, F. Sun, J. Wu and L. Zhao, Green Chem., 2018, 20, 1858–1865 RSC.
- Z. Ji, S. Y. H. Abdalkarim, H. Li, R. A. Asad and H. Y. Yu, Sol. Energy Mater. Sol. Cells, 2023, 259, 112453 CrossRef CAS.
- J. He, G. Zhao, P. Mu, H. Wei, Y. Su, H. Sun, Z. Zhu, W. Liang and A. Li, Sol. Energy Mater. Sol. Cells, 2019, 201, 110111 CrossRef CAS.
- S. Liu, S. Peng, B. Zhang, B. Xue, Z. Yang, S. Wang and G. Xu, RSC Adv., 2022, 12, 9587–9598 RSC.
- D. Sarabandi, G. Roudini and F. Barahuie, J. Energy Storage, 2019, 24, 100795 CrossRef.
- J. S. Hua, C. Yuan, X. Zhao, J. Zhang and J. Du, Energy Sources, Part A, 2019, 41, 86–93 CrossRef CAS.
- L. Jiang, Y. Lei, Q. Liu and J. Lei, Energy, 2020, 193, 116802 CrossRef CAS.
- W. Wu, W. Wu and S. Wang, Appl. Energy, 2019, 236, 10–21 CrossRef CAS.
- Z. Shen, S. Kwon, H. L. Lee, M. Toivakka and K. Oh, Carbohydr. Polym., 2021, 273, 118585 CrossRef CAS PubMed.
- L. Liu, X. Zhang and X. Lin, Renewable Energy, 2022, 187, 572–585 CrossRef CAS.
- D. Sarabandi, G. Roudini and F. Barahuie, J. Energy Storage, 2019, 24, 100795 CrossRef.
- T. Khadiran, M. Z. Hussein, Z. Zainal and R. Rusli, Energy, 2015, 82, 468–478 CrossRef CAS.
- S. Paneliya, S. Khanna, A. P. Singh, Y. K. Patel, A. Vanpariya, N. H. Makani, R. Banerjee and I. Mukhopadhyay, Renewable Energy, 2021, 167, 591–599 CrossRef CAS.
- S. Mandal, S. Ishak, D. E. Lee and T. Park, J. Energy Storage, 2022, 55, 105414 CrossRef.
- B. Tan, Z. Huang, Z. Yin, X. Min, Y. G. Liu, X. Wu and M. Fang, RSC Adv., 2016, 6, 15821–15830 RSC.
- Y. Zhao, X. Min, Z. Huang, Y. g. Liu, X. Wu and M. Fang, Energy Build., 2018, 158, 1049–1062 CrossRef.
- Y. Li, X. Huang, Y. Li, Z. Xi, G. Hai, Z. Tao and G. Wang, Sustainable Energy Fuels, 2020, 4, 1764–1772 RSC.
- R. Yang, X. Guo, H. Wu, W. Kang, K. Song, Y. Li, X. Huang and G. Wang, Biochar, 2022, 4, 38 CrossRef CAS.
- L. Lv, S. Huang, C. Zhou and W. Ma, Ind. Crops Prod., 2023, 204, 117184 CrossRef CAS.
- X. Shi, Y. Meng, R. Bi, Z. Wan, Y. Zhu and O. J. Rojas, Composites, Part B, 2022, 245, 110231 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jiaoli Dua,
Wenbo Yanga,
Bocun Suna,
Juncheng Lia,
Tian Xia
a,
Jiaoli Dua,
Wenbo Yanga,
Bocun Suna,
Juncheng Lia,
Tian Xia