DOI:
10.1039/D4RA03387F
(Paper)
RSC Adv., 2024,
14, 22665-22675
Effect of a solvothermal method using DMF on the dispersibility of rGO, application of rGO as a CDI electrode material, and recovery of sp2-hybridized carbon†
Received
8th May 2024
, Accepted 4th July 2024
First published on 18th July 2024
Abstract
Graphene is prized for its large surface area and superior electrical properties. Efforts to maximize the electrical conductivity of graphene commonly result in the recovery of sp2-hybridized carbon in the form of reduced graphene oxide (rGO). However, rGO shows poor dispersibility and aggregation when mixed with other materials without hydrophilic functional groups, This could lead to electrode delamination, agglomeration, and reduced efficiency. This study focuses on the impact of solvothermal reduction on the dispersibility and capacitance of rGO compared with chemical reduction. The results show that the dispersibility of rGO-D obtained through solvothermal reduction using N,N-dimethylformamide improved compared to that obtained through chemical reduction (rGO-H). Furthermore, when utilized as a material for CDI, an improvement in deionization efficiency was observed in the AC@rGO-D-based CDI system compared to AC@rGO-H and AC. However, the specific surface area, a key factor affecting CDI efficiency, was higher in rGO-H (249.572 m2 g−1) than in rGO-D (150.661 m2 g−1). While AC@rGO-H is expected to exhibit higher deionization efficiency due to its greater specific surface area, the opposite was observed. This highlights the effect of the improved dispersibility of rGO-D and underscores its potential as a valuable material for CDI applications.
Introduction
The increasing energy demand has triggered increasing research focused on sustainable and renewable power sources and emphasizing technological advancements in energy storage and conversion devices. Fossil fuels, coal, and oil, the most widely used energy sources, result in the complex and continuous deterioration of the global environment through the emission of greenhouse gases, such as carbon dioxide. As a result, the need for eco-friendly and renewable energy sources has become important. Among these, supercapacitors have attracted attention as modern energy storage devices owing to their high current density, fast charging–discharging, durability, and low maintenance.1,2 While various electrode materials are being researched, carbon nanomaterials stand out for their unique physicochemical and electrochemical properties. Moreover, owing to their porous characteristics, they are utilized in environmental engineering applications such as adsorption and filtration for pollutant treatment with electrical sources.3–6 One of these materials is graphene—characterized by a single layer of carbon atoms arranged in a two-dimensional honeycomb structure and extracted from graphite, which comprises numerous carbon layers bound by weak van der Waals forces.7,8 Celebrated as the “new dream material,” graphene boasts an extensive surface area, high electrical conductivity, and distinctive properties. It is utilized across diverse industries, including electronics, sensors, supercapacitors, and composite materials.7,9–13 Consequently, leveraging graphene and other electrode materials with high specific surface area (SSA) and electrical characteristics in capacitors has become a focal point of research.4,14–17 Graphene is produced using several methodologies, such as mechanical methods, epitaxial growth, chemical vapor deposition, and chemical exfoliation.18 Chemical exfoliation, which includes techniques such as Hummers' method and advanced processes such as the Couette–Taylor flow method, is particularly favored for its high production and cost efficiency in generating graphene flakes.19–21 Unfortunately, the electrical properties of graphene flakes derived from chemical exfoliation, known as graphene oxide (GO), are compromised by the presence of oxygen functional groups that impede electron mobility.22 The reduction of GO is necessary to enhance its electrical properties, with chemical reduction methods employing agents such as hydrazine monohydrate being widely used.18,23–26 The resulting reduced graphene oxide (rGO) shows improved electrical properties, but it suffers from lower polarity and dispersibility and tends to undergo re-stacking itself.27,28 Additionally, it can interrupt homogeneous mixing with other materials, which can cause aggregation, resulting in detachment and cracking. Also, this could potentially result in decreased efficiency. Therefore, research is needed to address the low dispersibility of rGO. This study aims to investigate the dispersibility and aggregation tendencies of rGO produced via the prevalent chemical reduction method. Additionally, it examines the dispersibility of rGO produced through another reduction method known as solvothermal reduction. A comparison of the dispersibility of rGO produced by these two different methods will be conducted, and surface analysis and functional group analysis will be performed to identify the underlying causes. Finally, we aim to compare and validate the impact of differences in the dispersibility, electrochemical properties of rGO and deionization efficiency of the CDI system, resulting from two different reduction processes. This study aims to identify the reduction processes of rGO that exhibit relatively higher dispersion stability.
Results and discussion
Characteristics of rGOs
The distinctions between rGOs synthesized via two reduction techniques designed to eliminate oxygen functional groups were analyzed by comparing the oxygen content of the two rGO samples to determine the extent to which they were reduced. Fig. 1 displays the FE-SEM images of GO and rGO. Many single layers of GO and rGOs were identified in each sample. In Fig. S4,† EDS mapping confirmed that the carbon single layers in rGO corresponded to those observed in the images. Table 1, showing the XPS analysis results, shows the proportions of carbon, oxygen, sulfur, and nitrogen elements in GO and the rGO variants. The oxygen content in rGO-D and rGO-H was reduced to 12.8% and 7.8%, respectively, which was considerably lower than that of GO (45.5%). This reduction process decreased the oxygen content in both rGO variants by approximately 33–37%. Furthermore, the carbon-to-oxygen (C/O) atomic ratio increased from 1.46 in GO to 6.59 in rGO-D and 11.48 in rGO-H owing to oxygen reduction. The C/O ratio of rGO obtained via solvothermal reduction typically ranges from 2.50 to 3.84.29 Comparing these values highlights the superior reduction efficacy of the rGOs produced in this study. Nitrogen was also detected in both the rGO-D and rGO-H samples, which was attributed to the reducing agents DMF and hydrazine. Additionally, the purification process entails the removal of sulphur via washing, further enhancing the purity of GO. The reduction and subsequent washing processes further effectively diminished the sulfur content of the rGO samples. XPS explains the relative elemental composition ratios within a substance through the peak values. Fig. S5† depicts the XPS spectra of GO and the two rGO variants. The C 1s and O 1s spectra of GO show peaks at 284.58 eV and 532.03 eV, respectively.
 |
| | Fig. 1 FE-SEM image of the prepared graphene materials. (a) GO; (b) rGO-D; (c) rGO-H. | |
Table 1 Atomic ratio (%) of graphene materials and carbon/oxygen (C/O) ratio and carbon/nitrogen (C/N) ratio. (a) GO; (b) rGO-D; (c) rGO-H
| Atomic (%) |
C (carbon) |
O (oxygen) |
S (sulphur) |
N (nitrogen) |
C/O |
C/N |
| (a) GO |
66.54 |
45.54 |
1.01 |
— |
1.46 |
— |
| (b) rGO-D |
84.51 |
12.83 |
0.08 |
2.59 |
6.59 |
32.63 |
| (c) rGO-H |
89.91 |
7.83 |
0.10 |
2.17 |
11.48 |
41.43 |
The O 1s peaks in the spectra of rGO-D and rGO-H are less intense than that in the spectrum of GO, highlighting a significant reduction in the oxygen content during the reduction of GO to rGO. The presence of nitrogen, as evidenced by the emergence of the N 1s peak at 398.81 eV, is presumably a result of the use of nitrogen-containing agents such as DMF and hydrazine. These observations suggest that the reduction techniques employing hydrazine and DMF yield a similar extent of reduction. The C/O ratio and along with the oxygen peak indicate that the solvothermal reduction method using DMF is efficacious in reducing GO. Fig. 2 exhibits the C 1s and N 1s spectra of rGO-D and rGO-H, in which carbon adopts various bonding configurations: C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.58 eV), C–O (285.86 eV), C–N (286.87 eV), CO (289.42 eV).30–35 The C–N peak is notably more pronounced in the spectrum of rGO-D than in that of rGO-H. The decreased C/N ratio of rGO-D further demonstrates the synthesis of nitrogen (Table 1). The N 1s spectrum of rGO-D indicates the presence of pyridinic-N (398.6 eV), pyrrolic-N (399.8 eV), and graphitic-N (400.5 eV).36–38 In contrast, rGO-H's N 1s spectrum shows nitrogen in graphitic-N (400.5 eV), NH4+–N (399.4 eV), and N–O (396.5 eV) bonds.38–42 While a similar extent of reduction was achieved using both methods, the C 1s XPS spectra revealed distinct atomic bonding characteristics. rGO-D contained a significant number of C–N bonds, whereas both single and double-bonded nitrogen and oxygen were observed in rGO-H, with the NH4+–N bond being particularly significant. This NH4+–N bond originated from hydrazine monohydrate (N2H4). The existence of N–H bonds in rGO-H is attributed to an intermediary process in which N–H from hydrazine participates in the reduction of GO.23
C (284.58 eV), C–O (285.86 eV), C–N (286.87 eV), CO (289.42 eV).30–35 The C–N peak is notably more pronounced in the spectrum of rGO-D than in that of rGO-H. The decreased C/N ratio of rGO-D further demonstrates the synthesis of nitrogen (Table 1). The N 1s spectrum of rGO-D indicates the presence of pyridinic-N (398.6 eV), pyrrolic-N (399.8 eV), and graphitic-N (400.5 eV).36–38 In contrast, rGO-H's N 1s spectrum shows nitrogen in graphitic-N (400.5 eV), NH4+–N (399.4 eV), and N–O (396.5 eV) bonds.38–42 While a similar extent of reduction was achieved using both methods, the C 1s XPS spectra revealed distinct atomic bonding characteristics. rGO-D contained a significant number of C–N bonds, whereas both single and double-bonded nitrogen and oxygen were observed in rGO-H, with the NH4+–N bond being particularly significant. This NH4+–N bond originated from hydrazine monohydrate (N2H4). The existence of N–H bonds in rGO-H is attributed to an intermediary process in which N–H from hydrazine participates in the reduction of GO.23
 |
| | Fig. 2 C 1s and N 1s spectra of rGO-D and rGO-H. (a) C 1s of rGO-D; (b) C 1s of rGO-H; (c) N 1s of rGO-D; (d) N 1s of rGO-H. | |
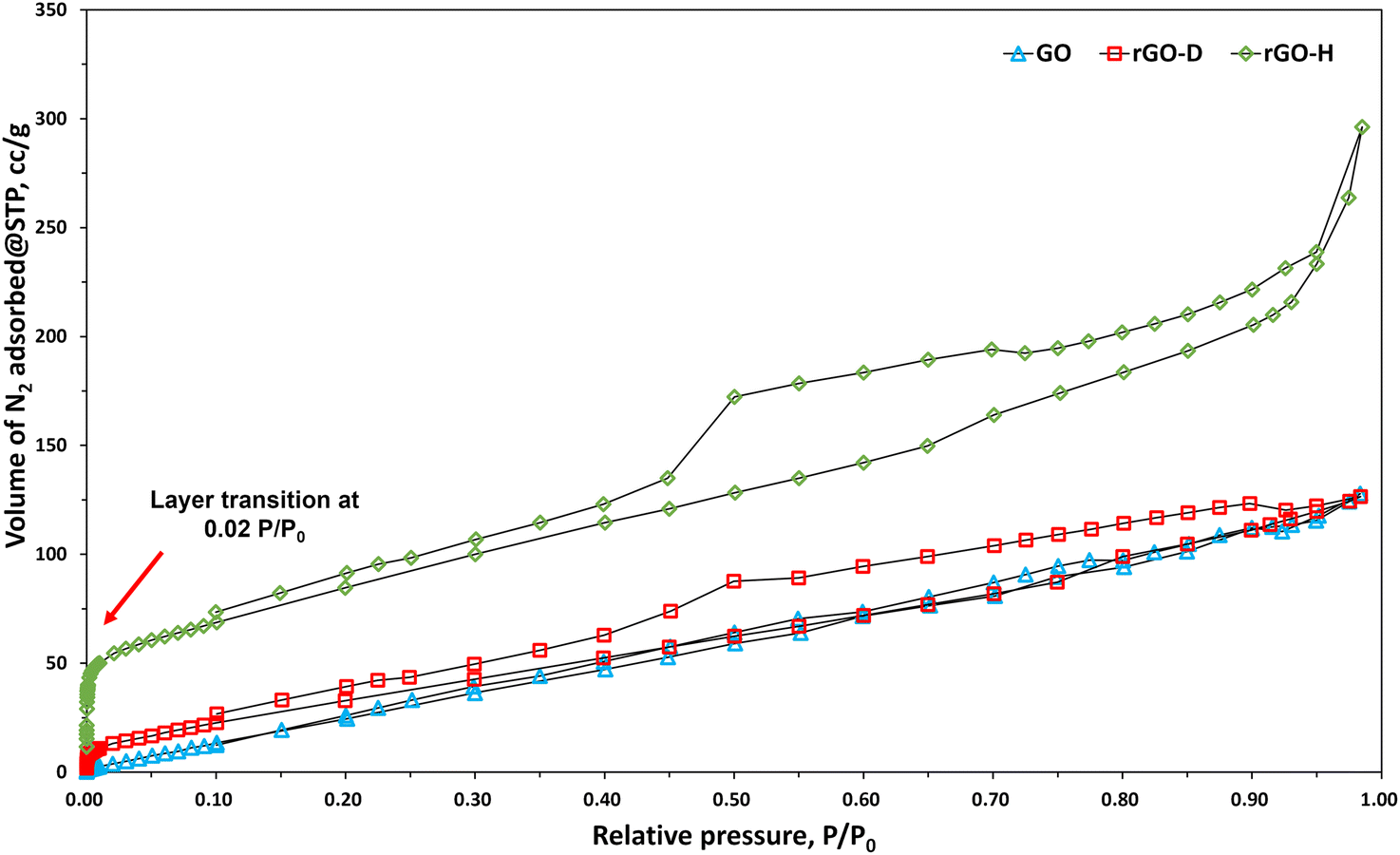
Fig. 3 displays the FT-IR spectra of GO, rGO-D, and rGO-H. The spectrum of GO shows significant peaks attributed to C–O stretching at 1000–1150 cm−1 and 1000–1250 cm−1, along with those arising from C–OH (1400–1450 cm−1), carbonyl CO, (1700–1760 cm−1), and CC aromatic stretching 1614–1635 cm−1.43–45 Further magnification reveals peaks attributed to C–O (1089 cm−1), C–N (1210 cm−1), C–H (1350–1470 cm−1), N–H (1587 cm−1), CC (1595 cm−1), amide CO (1630–1680 cm−1), and carbonyl CO (1700–1760 cm−1) in the FT-IR spectra of rGO-D and rGO-H.36,46–50 Common features between the XPS and FT-IR analysis results of the two types of rGO are observed. In the case of rGO-D, it is the presence of carbon–nitrogen bonds (N–C), synthesized during the reduction process (Fig. 2c, d and 3). Additionally, the observed C–H bonds are indicative of DMF-derived methyl groups (–CH3) in rGO-D. DMF undergoes decarbonylation at 153 °C, resulting in the decomposition of carbon monoxide and the formation of dimethylamine.51–53 Carbon monoxide, a widely used reducing agent, not only reduces GO but also reacts with dimethylamine to form nitrogen-doped heterocyclic graphene and methyl groups in the base and edge. As a result, the use of DMF in the reduction process can also lead to the formation of graphite-N, pyrrolic-N, and pyridine-N.36,48,49,54 The FT-IR spectra of rGO-H reveal the presence of N–H bonds, which is consistent with the N 1s spectra (NH4+–N). Hydrazine readily opens epoxide rings and forms hydrazinoalcohols, which form double bonds via the thermal elimination of diimide. Similarly, carbonyl groups can be reduced by hydrazine to form C–N bonds. The mechanism through which epoxide groups are restored to the sp2-hybridized form in GO by hydrazine is well understood through previous research.23,55 Fig. 4 shows the nitrogen adsorption isotherms of GO, rGO-D, and rGO-H. By analyzing the specific amount of nitrogen adsorption, the SSA, specific volume, and pore diameter of the material can be determined (Table 2). The SSA and pore diameter of the samples were measured using the BET and BJH methods, respectively.56 GO exhibited an SSA of 104.055 m2 g−1. After reduction, the values for rGO-D and rGO-H were considerably higher at 150.661 m2 g−1 and 249.572 m2 g−1, respectively. These values are similar to those reported in other studies (96–399 m2 g−1).57,58 However, these values are lower than the theoretical SSA of completely exfoliated and isolated graphene sheets (2630 m2 g−1).59,60 Nevertheless, the observed increase in the SSA during the reduction process is in good agreement with earlier reports.25,61 The difference between the observed values and the theoretical value is attributed to the accumulation or precipitation and partial overlapping of rGO sheets during the reduction process or analysis, which limits access to some surfaces.23,62 The pore diameters of GO (3.4 nm), rGO-D (3.8 nm), and rGO-H (3.8 nm) were very similar. GO has a relatively higher number of oxygen functional groups attached to carbon compared to rGO. While perfect single-layer graphene has a hexagonal honeycomb structure, GO generated through chemical exfoliation maintains a reduced number of carbon hexagonal structures due to oxygen functional group attachments. Similarly, rGO produced through chemical or thermal reduction removes oxygen functional groups from GO. This process leads to a partial restoration of hexagonal structures in some carbon atoms, thereby enhancing the structural stability. Therefore, due to the structural instability in GO caused by many carbon bonds, it is speculated that pore deformation may occur. This could be partially recovered in rGO, leading to a slight increase in the pore size. At a relative pressure of 0.02 in Fig. 4, both GO, rGO-H, and rGO-D exhibit a transition from monolayer to multilayer coverage. Similarly, they all show hysteresis, indicative of capillary condensation in the form of a type IV isotherm, indicating that they are all solid materials with micro- and mesopores, capable of interactions between gas molecules and the adsorbent mesopore surface.63,64
 |
| | Fig. 3 FT-IR spectra of GO, rGO-D, and rGO-H. | |
 |
| | Fig. 4 N2 adsorption–desorption isotherm of GO, rGO-D, and rGO-H. | |
Table 2 BET surface area, BJH pore volume, and BJH pore diameter. (a) GO; (b) rGO-D; (c) rGO-H
| |
Specific surface area (m2 g−1) |
Average pore volume (cc g−1) |
Average pore diameter (nm) |
| (a) GO |
104.055 |
0.175 |
3.412 |
| (b) rGO-D |
150.661 |
0.184 |
3.826 |
| (c) rGO-H |
249.572 |
0.400 |
3.819 |
The pore volume of rGO-H (0.400 cc g−1) was significantly higher than that of rGO-D (0.184 cc g−1). Compared to GO (0.175 cc g−1), the difference in the pore volume was not substantial in rGO-D, but it more than doubled in rGO-H. We propose that this increase in rGO-H can be attributed to the potential for interlayer adsorption. We will provide an explanation in conjunction with the structural analysis. The hysteresis loops caused by mesopore capillary condensation in rGO-D and rGO-H differed in appearance. The hysteresis loop of rGO-H resembled the H3 type, and the shape of the pores resembled aggregates of plate-like particles. The hysteresis loop of rGO-D more closely resembled the H4 type, and the material had slit-like pores. However, comparing the difference in the SSA based solely on the morphology of the pores is not sufficient. This can be explained further in connection with the structural forms mentioned later.
Structural defects of rGOs
The structural defects resulting from the reduction of GO were characterized using Raman spectra (Fig. 5a). The D-bands originated from the vibration of sp3 electronic configuration of defected carbon were seen at 1350–1360 cm−1. The G-bands associated with the vibration of sp2-bonded carbon appeared at 1579–1581 cm−1. The intensity ratio of the D-band to the G-band (ID/IG) increased as graphite (0.025) transformed into GO (1.014) (Table S1†). During oxidation and exfoliation processes, the oxygen functional groups synthesized on the carbon layer with sp3 electrons led to the formation of bonds, resulting in an increase in the intensity of the D-band. The reduction of GO is expected to simultaneously remove functional groups and restore sp2 bonding. Both rGO-D and rGO-H exhibited a decrease in the intensity of the D-band during the reduction process. However, rGO-D showed a significantly higher ID/IG compared to rGO-H (Table S1†). These results suggest that solvothermal reduction not only reduces oxygen functional groups but also simultaneously synthesizes nitrogen functional groups and methyl groups, indicating the presence of relatively more unrecovered sp2 (Fig. 2c and 3). Although the reduction process facilitated the transition of sp3 carbon in GO to sp2, the sp3 configuration was preserved to some extent. XRD provides further insight into the observed structural difference (Fig. 5b). The peak observed in the spectrum of rGO-H at 27.6° is similar to that in the spectrum of graphene obtained by chemical vapor deposition (CVD) at 26.3°, suggesting that carbon atoms may have severed their bonds with oxygen and re-established the sp2-hybridized structure by forming C–C or CC bonds.65 In contrast, the peaks in the spectrum of rGO-D (7.9° and 22.2°) are comparable to those in the spectra of GO (9.0°) and graphite (22.3°), respectively.66,67 The observation that the spectrum of rGO-D exhibits both GO and graphite peaks suggests that carbon atoms in rGO-D retain bonds with other atoms, indicating that its structure has a lower recovery of sp2 carbon than that of rGO-H. As a result, rGO-H is expected to have higher structural stability compared to rGO-D. This implies that it can potentially retain a greater extent of layered structure. However, it may also exacerbate the issue we mentioned earlier with graphene, namely, increased layer re-stacking phenomenon. By maintaining a layered structure through sp2 recovery, rGO-H could explain a higher SSA and pore volume due to the increased interlayer adsorption mentioned earlier. On the other hand, in rGO-D, where relatively less interlayer stacking occurs, it would explain a lower SSA and pore volume value due to relatively lower interlayer adsorption.
 |
| | Fig. 5 (a) Raman spectra of GO, rGO-D, rGO-H, and graphite; (b) XRD analysis of GO, rGO-D, rGO-H, and comparison with graphite, and CVD of graphene for reference. | |
Dispersibility
The degree of reduction in rGO-D and rGO-H is similar to that determined from the C/O ratio; however, the reduction process typically results in increased hydrophobicity and reduced dispersion owing to functional groups, which diminishes the polarity and dispersibility of rGO. Consequently, the advantages of rGO, such as its electrical conductivity and large surface area, are not optimally utilized. To address these issues, rGO-D via applied solvothermal reduction method was evaluated to identify the changes in dispersibility. In Table S2,† the zeta potentials of rGO-D and rGO-H were measured to be −32.22 mV and −26.35 mV, respectively. The zeta potential is a crucial factor in characterizing the stability of colloidal dispersions;68 particles with zeta potentials more positive than +30 mV or more negative than −30 mV typically form stable dispersions.69 Therefore, this indicates that rGO-D exhibits relatively higher particle repulsion compared to rGO-H, validating its higher dispersibility. The instability index of both rGO-D and rGO-H were analyzed using a LumiSizer dispersion analyzer, which measures the variation in the transmitted light over time and space due to centrifugal sedimentation70 and is commonly employed to assess the solvent dispersibility of a material. A higher instability index indicates greater solvent instability. In this study, rGOs were dispersed in D.I. water because electrode mixing was performed using D.I. water as a solvent. The instability index of rGO-D (0.96) was lower than that of rGO-H (1.34), indicating that rGO-D is more stable than rGO-H (Fig. 6a). Consequently, the analysis of the zeta potentials and instability indexes shows that rGO-D exhibits superior dispersibility relative to rGO-H. The dispersion stability of rGO-D and rGO-H in D.I. water over time was compared by analyzing the absorbance as a function of the elapsed time using a dispersion with a concentration of 15 μg of rGO in 1 mL of D.I. water (Fig. S8†). The absorbance was scanned in the range of 110–1900 nm for 72 h and continually decreased over this period (Fig. S6 and S7†). According to the Beer–Lambert law, the absorbance (A) is directly proportional to the concentration (C) of the dispersed particles.71,72| | |
A = ε × B × C (ε = molar absorptivity, B = path length)
| (1) |
 |
| | Fig. 6 (a) Instability index; (b) UV-vis absorbance in D.I. water versus retention time (where A and A0 are the absorbance of the dispersion at 400 nm). (c) Contact angle of rGO-D and rGO-H. | |
The absorbance of rGO-D is higher than that of rGO-H but decreases over time. To directly compare the dispersion stability of both rGOs, the initial absorbance (A0) was compared with the absorbance over time (At). A comparison of A/A0 at 400 nm as a function of time indicates that rGO-H experiences a greater reduction in absorbance than rGO-D over the 72 h period (Fig. 6b). Notably, rGO-H demonstrates a sharp decline in absorbance during the initial 9 h. Therefore, rGO-D shows higher dispersion stability than rGO-H in D.I. water, likely owing to the differences in the functional groups on the rGOs, with rGO-D containing methyl groups synthesized through the reaction between [NH2(CH3)2]+ derived from DMF and –COO− at the edges of GO, thereby enhancing the dispersibility.73 The contact angle of rGOs was measured using a 1 μL drop of D.I. water on the surfaces of pelletized rGO (Fig. 6c). The average contact angle of rGO-D (52.7 ± 2.97) was smaller than that of rGO-H (82.8 ± 3.86), indicating that rGO-D has a more hydrophilic surface, likely due to the affinity of the internal functional groups, especially methyl groups, toward water. Given these observations, the methylated rGO-D exhibits higher dispersibility than rGO-H, suggesting its potential to reduce the aggregation during electrode fabrication, thereby enhancing the performance by homogeneous dispersion.
Consequently, rGO-H is likely to exhibit plate-like pores with numerous interconnected sp2 carbon structures, maintaining flat layers in a stacked configuration. In contrast, rGO-D is expected to feature slit-like pores between the sides of connected layers owing to the lower sp2-hybridized structures, which hinders the maintenance of a sheet-like formation. This structural difference is presumed to be a critical factor influencing the pore volume. rGO-H is anticipated to facilitate direct pore adsorption and enhance interlayer adsorption. Meanwhile, rGO-D is likely to exhibit a comparatively smaller pore volume, attributable solely to direct adsorption. rGO-H may be more adept at ion adsorption than rGO-D owing to its surface area and pore volume; however, a higher ion adsorption capacity does not unequivocally translate to proportional effectiveness in electrically-driven ion adsorption applications, such as CDI.
Electrochemical analysis and deionization efficiency
Fig. 7 shows the CVs at 10 cycles for the fabricated GO rGO-D, rGO-H, and the main material for the CDI electrode, AC, as single substances. Each single substance confirms a quasi-rectangular shape, indicative of pseudo capacitor-like materials. The electrochemical performance can be inferred from the area of the CV, with rGO-H showing the most promising results (Fig. 7a). In contrast, rGO-D exhibited slightly lower electrochemical performance compared to rGO-H at 10 cycles but slightly higher than that of AC. GO showed the lowest performance, which can be attributed to its lower proportion of sp2-hybridized carbon compared to rGO, as previously mentioned. The specific capacitance Cs was calculated from the area under the CV curve at 10 cycles (Table S3†), using eqn (2), which includes the CV curve area A, potential window ΔV(V), material mass m, and scan rate K (mV s−1).74,75 The calculation is based on the charge stored over time (eqn (3)), where Q is the charge (coulombs), I is the current (amperes), and t is the potential sweep time (seconds).76–78 eqn (2) is derived from eqn (3).| | |
Cs = A/(2 × ΔV × m × K)
| (2) |
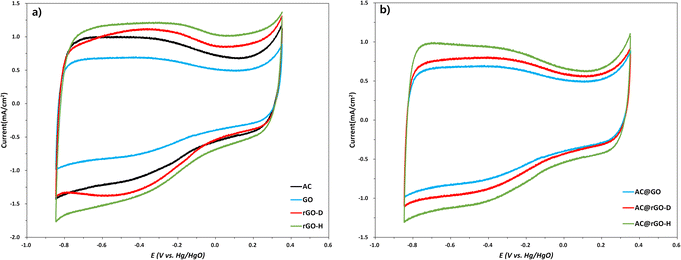 |
| | Fig. 7 (a) Cyclic voltammetry at 10 cycles of GO, rGO-D, rGO-H and AC, (b) cyclic voltammetry at 10 cycles of GO, rGO-D, rGO-H mixed with AC. | |
The rGO-H demonstrated the highest specific capacitance of 180.74 F g−1, while rGO-D showed about 177.23 F g−1. However, GO demonstrated the lowest specific capacitance of 89.80 F g−1. The relatively low proportion of sp2 hybridization present in GO mentioned earlier can be explained. We analyzed the electrochemical properties of materials mixed in the same ratio as the electrode material for CDI (Table 3a). Comparing the CV and specific capacitance accordingly, AC@rGO-H showed the highest performance (153.03 F g−1), followed by AC@rGO-D (128.50 F g−1) and AC@GO (112.05 F g−1). However, due to the different electrochemical mixing methods and electrode synthesis for CDI, it is difficult to conclude and compare their relative differences. Nevertheless, consistent characteristics were observed in the CV results of individual materials. Finally, Table 3b presents the synthesis of AC and rGO electrodes for CDI, showing their deionization efficiencies. Contrary to the expectations derived from CV and specific capacitance results, AC@rGO-D exhibited the highest deionization efficiency (25.43%). AC@rGO-H showed relatively lower deionization efficiency (22.05%), and AC@GO demonstrated the lowest efficiency (17.81%), which was attributed to the inferior electrical properties of GO79,80
Table 3 (a) Specific capacitance (F g−1) at 10 cycles of GO, rGO-D, rGO-H mixed with AC; (b) ion removal rate (%) of the CDI system based on the synthesized GO, rGO-D and rGO-H with AC
| |
(a) Specific capacitance (F g−1) at 10 cycles |
(b) Ion removal rate of the CDI system (%) |
| AC |
161.05 |
21.77 |
| AC@GO |
112.05 |
17.81 |
| AC@rGO-H |
153.03 |
22.05 |
| AC@rGO-D |
128.50 |
25.43 |
Based solely on CV and specific capacitance results, one might expect AC@rGO-H to achieve the highest deionization efficiency. Additionally, an increase in the SSA values could further strengthen this expectation. However, considering aspects such as contact angle measurement, dispersion stability, and other analyses, AC@rGO-D demonstrated the highest deionization efficiency, as demonstrated. These findings emphasize the critical role of rGO dispersibility in enhancing the performance of CDI systems. The mechanism of ion removal in CDI systems is predicated on electrostatic forces coupled with the adsorption of ionizable species within the porous electrode. Electrodes based on AC@rGO-H were expected to have a higher ion removal efficiency than those based on AC@rGO-D owing to the larger SSA and pore volume of rGO-H. Contrary to these expectations, the CDI system incorporating AC@rGO-D, which exhibited superior dispersibility, achieved higher ion removal efficiency. This outcome demonstrates that, despite the superior SSA, pore volume, and electrical properties of rGO-H, the CDI system utilizing AC@rGO-D outperformed the AC@rGO-H-based system with respect to the deionization efficiency. The enhanced dispersibility of rGO-D, achieved through thermal reduction, mitigates aggregation and promotes a more uniform distribution, thereby maximizing the effective SSA; thus, this study highlights the significance of the dispersibility of rGO in enhancing the electrical performance, especially EDLs, when synthesized with other significant materials. However, the low specific capacitance associated with the mixture of rGO-D than rGO-H is a critical aspect (Fig. 7b). Nitrogen functionalities synthesized during the reduction process, such as pyrrolic-N, pyridinic-N, and graphitic-N, may act as pseudocapacitive contributors, potentially causing this decrease.81,82 Conversely, rGO-H did not predominantly feature graphitic-N, and pyrrolic-N was not observed; thus, nitrogen contributes less to the current density of rGO-H. Moreover, pseudocapacitive materials like rGO undergo physical changes during several prolonged charge–discharge cycles, which remains a challenge.83
Conclusions
This study addressed the challenge associated with the low dispersibility in rGO—a material known for its superior electrical properties—and explored strategies to effectively overcome these properties in CDI systems. The study evaluated the impact of two reduction methods on the degree of reduction and, consequently, the electrical properties of rGO through the generation of π-conjugation within the electrode material. Both rGO-D and rGO-H displayed similar degrees of reduction, a critical factor for enhancing the electrical conductivity. Dispersion analysis indicated that rGO-D exhibited better dispersibility and reduced agglomeration when mixed with other electrode materials, which was attributed to the hydrophilic nature of the methyl groups introduced during its synthesis. The utilization of rGO is intended to increase the deionization efficiency by capitalizing on its electrical properties; however, a comparison of their deionization efficiencies revealed that electrodes based on rGO-D outperformed those based on rGO-H. This study highlights that although the electrical properties of rGO can be optimized by the reduction method, the low dispersibility of rGO mixed with various electrode components limits the full utilization of these properties. The dispersibility and electrical properties of the material can be improved applying the solvothermal reduction method and introducing hydrophilic functional groups; however, this did not significantly increase the observed specific capacitance, suggesting that further optimization of the rGO mixing ratio in the electrode composition is required.
This study highlights the potential improvements in the electrical properties that can be obtained through the optimization of the rGO mixing ratio during electrical application and will serve to guide future efforts to further enhance the performance.
Experimental section
Reagent
Graphite powder (natural, briquetting grade, 100 mesh) was acquired from Alfa Aesar. Activated carbon (P-60) was obtained from KURARAY. Potassium permanganate (KMnO4, reagent grade) powder, sulfuric acid (H2SO4, 95%) solution, hydrogen peroxide (H2O2, 30%) solution, N,N-dimethylformamide (HCON(CH3)2) solution, and ethyl alcohol (C2H6O, 99%) solution were procured from DUKSAN Pure Chemicals Inc., Republic of Korea. Hydrazine monohydrate (NH2NH2·H2O, 98%) was sourced from Kanto Chemical, Japan. All chemicals were of reagent grade. Distilled-deionized (D.I.) water was used for all experimental procedures.
Preparation of graphene materials
Graphite oxide (GtO) and GO. Graphite oxidation was achieved using the advanced Hummers' method and a Couette–Taylor flow reactor (LCTR – Lab II-HC, Laminar Co., Ltd, Republic of Korea). Initially, a mixture of graphite powder (7 g) and H2SO4 (245 mL) was cooled to below 10 °C using an ice bath and stirred for 30 min. Subsequently, KMnO4 (35 g) was gradually added and the mixture was further stirred for an additional 60 min at 10 °C. The mixture was subsequently processed in a Couette–Taylor flow reactor, with the inner cylinder rotating at 1500 rpm for 60 min, resulting in a brown-colored slurry, to which 5% H2SO4 solution (400 mL) was added to remove oxidized graphite, followed by the slow addition of 30% H2O2 solution (12 mL), inducing bubbling and stirring for 30 min to ensure complete oxidation. The mixture was washed three times with a washing solution (WS) prepared by mixing 30 mL of 95% H2SO4 (30 mL) and hydrogen peroxide (H2O2, 30%, 50 mL) in D.I. water to remove manganese and potassium, and further washed with D.I. water five times to remove sulfur. The purified product was freeze-dried to obtain GtO powder.84 To produce GO, GtO powder (1 g) was stirred in 1000 mL of D.I. water, sonicated for 60 min, and centrifuged at 6000 rpm for 30 min at room temperature (25 °C). The supernatant was freeze-dried to yield the GO powder.
Chemical reduced graphene oxide using hydrazine monohydrate (rGO-H). GO powder (1 g) was sonicated in D.I. water (1000 mL) and heated to 90 °C before hydrazine monohydrate solution (1 mL) was added, and the mixture was maintained at 90 °C for 120 min. After cooling to room temperature (25 °C), the product was washed three times with ethyl alcohol and D.I. water to remove nitrogen. The purified product was freeze-dried to obtain the rGO-H powder.
Solvothermally reduced graphene oxide using N,N-dimethylformamide (rGO-D). GO powder (0.5 mg) was sonicated in 1 mL of N,N-dimethylformamide (DMF, 1 mL) and heated to 153 °C for 60 min before being cooled to room temperature (25 °C) and subsequently purified three times with ethyl alcohol and D.I. water. The purified product was freeze-dried to obtain rGO-D powder.51
Characterization
The morphology and elemental composition of the rGOs were examined using field emission scanning electron microscopy and energy dispersive spectroscopy (FE-SEM-EDS, JEOL 7800F, Japan). X-ray photoelectron spectroscopy (XPS) was performed with a K-alpha (Thermo Fisher Scientific Inc., USA) to analyze the atomic bonds and elemental composition. Fourier-transform infrared spectroscopy (FT-IR) (Thermo Fisher Scientific Inc., USA) was used to identify the functional groups in GO and the two types of rGO. Nitrogen adsorption–desorption isotherms were measured at 77 K using an Autosorb IQ (Quantachrome Instruments, USA). X-ray diffraction (XRD) (Lab X XRD-6100, SHIMADZU, Japan) was conducted at 40 kV and 40 mA over a scan range of 5–40° to assess the carbon structure. Surface area was evaluated using the Brunauer–Emmett–Teller (BET) method, and the pore diameter was calculated using the Barrett, Joyner, and Halenda (BJH) model. Raman spectra were acquired using an XperRam35V confocal Raman imaging system (Nanobase, Seoul, South Korea) equipped with a 1800 g mm−1 grating, 532 nm neodymium–yttrium aluminium garnet laser, LTGL-532RL (Leading tech, Shanghai, China) and a MPLFLN 40× objective lens (Olympus, Tokyo, Japan). The laser power on a single cell was 2.0 mW, and the acquisition time of each spectrum was 20 s.
Dispersibility
The dispersibility of rGO was determined by measuring the zeta potential (ELSZ-1000, Otsuka Electronics Co., Ltd, Japan). The instability index was measured with a LUMiSizer® (LUM GmbH, Germany). Absorbance was assessed using a UV-vis spectrometer (DR-6000, Hach, USA) across a wavelength range of 190–1100 nm. The contact angle of rGOs was analyzed using a contact angle tester (FEMTOFAB SDS-TEZD, FEMTOFAB, Republic of Korea).
Electrochemical performance and deionization efficiency
The ink was prepared by the dispersion of 5 mg of graphene materials and 45 μL of Nafion solution in 1000 μL of ethanol, followed by ultrasonication. When mixed with AC, the ratio of AC with rGO is 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. 10 μL of the prepared ink was loaded on the working electrode and dried at room temperature. The electrochemical performances were measured using a three-electrode system (RDE 710, GAMRY Instruments, Inc., USA) with glassy carbon, Pt wire, and saturated Hg/HgO in 1 M KOH as the working, counter, and reference electrodes, respectively, with 0.1 M KOH (pH 10) solution as the electrolyte. Cyclic voltammetry (CV) was performed with a ZiVE sp1 potentiostat and Smart Manager Software (ZiVE LAB, Republic of Korea) at scan rates (ν) of 20 mV s−1, within a potential range between −0.85 V and 0.35 V (vs. V. Hg/HgO). Furthermore, the performance of rGO for the capacitive deionization (CDI) system was evaluated in a CDI cell with the synthesized AC (ESI†).
:1. 10 μL of the prepared ink was loaded on the working electrode and dried at room temperature. The electrochemical performances were measured using a three-electrode system (RDE 710, GAMRY Instruments, Inc., USA) with glassy carbon, Pt wire, and saturated Hg/HgO in 1 M KOH as the working, counter, and reference electrodes, respectively, with 0.1 M KOH (pH 10) solution as the electrolyte. Cyclic voltammetry (CV) was performed with a ZiVE sp1 potentiostat and Smart Manager Software (ZiVE LAB, Republic of Korea) at scan rates (ν) of 20 mV s−1, within a potential range between −0.85 V and 0.35 V (vs. V. Hg/HgO). Furthermore, the performance of rGO for the capacitive deionization (CDI) system was evaluated in a CDI cell with the synthesized AC (ESI†).
Data availability
The data generated or analyzed during the current study are available from the corresponding author upon reasonable request.
Author contributions
All authors contributed to the study. Conception and design were performed by [Junho Lee]. Material preparation and data collection were performed by [Junho Lee], [Seonghyeon Ju]. Analysis was performed by [Seonghyeon Ju], [Chaehwi Lim], [Jihoon Lee] and [Yeojoon Yoon]. The first draft of the manuscript was written by [Junho Lee] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research has been supported by the National Research Foundation of Korea (grant number: NRF-2019R1G1A1100560) funded by the Republic of Korea.
References
- W. Czepa, S. Witomska, A. Ciesielski and P. Samorì, Nanoscale, 2020, 12, 18733–18741 RSC
 .
. - F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC .
- P. Y. Liu, T. T. Yan, L. Y. Shi, H. S. Park, X. C. Chen, Z. G. Zhao and D. S. Zhang, J. Mater. Chem. A, 2017, 5, 13907–13943 RSC .
- P. S. Goh and A. F. Ismail, Desalination, 2015, 356, 115–128 CrossRef CAS .
- G. Wang, B. Q. Qian, Q. Dong, J. Y. Yang, Z. B. Zhao and J. S. Qiu, Sep. Purif. Technol., 2013, 103, 216–221 CrossRef CAS .
- L. M. Chang, Y. Y. Yu, X. Y. Duan and W. Liu, Sep. Sci. Technol., 2012, 48, 359–365 CrossRef CAS .
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS .
- J. B. Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC .
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS .
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. S. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS .
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed .
- M. Zhou, Y. M. Zhai and S. J. Dong, Anal. Chem., 2009, 81, 5603–5613 CrossRef CAS .
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S. E. Wu, S. F. Chen, C. P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS PubMed .
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS .
- C. H. Xu, B. H. Xu, Y. Gu, Z. G. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 RSC .
- J. T. Zhang, J. W. Jiang, H. L. Li and X. S. Zhao, Energy Environ. Sci., 2011, 4, 4009–4015 RSC .
- X. Huang, Z. Y. Zeng, Z. X. Fan, J. Q. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 CrossRef CAS PubMed .
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS .
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS .
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS .
- W. K. Park, H. Kim, T. Kim, Y. Kim, S. Yoo, S. Kim, D. H. Yoon and W. S. Yang, Carbon, 2015, 83, 217–223 CrossRef CAS .
- L. P. Lingamdinne, J. R. Koduru and R. R. Karri, J. Environ. Manage., 2019, 231, 622–634 CrossRef CAS PubMed .
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS .
- Z. Y. Lin, Y. G. Yao, Z. Li, Y. Liu, Z. Li and C. P. Wong, J. Phys. Chem. C, 2010, 114, 14819–14825 CrossRef CAS .
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC .
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed .
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS .
- A. J. Sellathurai, S. Mypati, M. Kontopoulou and D. P. J. Barz, Chem. Eng. J., 2023, 451, 138365 CrossRef CAS .
- S. Seo, Y. Yoon, J. Lee, Y. Park and H. Lee, ACS Nano, 2013, 7, 3607–3615 CrossRef CAS .
- F. T. Johra and W.-G. Jung, Appl. Surf. Sci., 2015, 357, 1911–1914 CrossRef CAS .
- J. H. Liu, J. W. An, Y. C. Zhou, Y. X. Ma, M. L. Li, M. Yu and S. M. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2870–2876 CrossRef CAS .
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed .
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS .
- Q. Bi, S. Jiang, C. Hu, L. Zhang, Y. Zhao, B. Yang, G. Li and J. Xue, J. Environ. Chem. Eng., 2023, 11, 109564 CrossRef CAS .
- A. Dolgov, D. Lopaev, C. J. Lee, E. Zoethout, V. Medvedev, O. Yakushev and F. Bijkerk, Appl. Surf. Sci., 2015, 353, 708–713 CrossRef CAS .
- B. Mandal, S. Saha, D. Das, J. Panda, S. Das, R. Sarkar and B. Tudu, Flatchem, 2022, 34, 100400 CrossRef CAS .
- Z. Li, W. He, X. Wang, X. Wang, M. Song and J. Zhao, Int. J. Hydrogen Energy, 2020, 45, 112–122 CrossRef CAS .
- Z. Chen, Y. Zhang, W. Gu, M. Yang, K. Yao, T. Cao and S. Li, Environ. Res., 2023, 220, 115198 CrossRef CAS PubMed .
- M.-S. Lee, M. Park, H. Y. Kim and S.-J. Park, Sci. Rep., 2016, 6, 23224 CrossRef CAS PubMed .
- H. He, Y. Hu, S. Chen, L. Zhuang, B. Ma and Q. Wu, Sci. Rep., 2017, 7, 3913 CrossRef PubMed .
- A. Alabadi, H. A. Abbood, Q. Li, N. Jing and B. Tan, Sci. Rep., 2016, 6, 38614 CrossRef CAS PubMed .
- K. H. An, C.-M. Yang, J. Y. Lee, S. C. Lim, C. Kang, J.-H. Son, M. S. Jeong and Y. H. Lee, J. Electron. Mater., 2006, 35, 235–242 CrossRef CAS .
- S. C. Rattana, N. Witit-anun, N. Nuntawong, P. Chindaudom, S. Oaew, C. Kedkeaw and P. Limsuwan, Procedia Eng., 2012, 32, 759–764 CrossRef .
- R. Al-Gaashani, A. Najjar, Y. Zakaria, S. Mansour and M. A. Atieh, Ceram. Int., 2019, 45, 14439–14448 CrossRef CAS .
- A. Konwar, S. Kalita, J. Kotoky and D. Chowdhury, ACS Appl. Mater. Interfaces, 2016, 8, 20625–20634 CrossRef CAS PubMed .
- N. Zhang, X. Liu, Y. Huang, M. Wang, S. Li, M. Zong and P. Liu, J. Colloid Interface Sci., 2019, 540, 218–227 CrossRef CAS PubMed .
- R. Wang, Y. Wang, C. Xu, J. Sun and L. Gao, RSC Adv., 2013, 3, 1194–1200 RSC .
- J. Porwal, N. Karanwal, S. Kaul and S. L. Jain, New J. Chem., 2016, 40, 1547–1553 RSC .
- Y. Miao, Y. Ma and Q. Wang, ACS Sustain. Chem. Eng., 2019, 7, 7597–7608 CrossRef CAS .
- S. Rani, M. Kumar, R. Kumar, D. Kumar, S. Sharma and G. Singh, Mater. Res. Bull., 2014, 60, 143–149 CrossRef CAS .
- K. L. Ai, Y. L. Liu, L. H. Lu, X. L. Cheng and L. H. Huo, J. Mater. Chem., 2011, 21, 3365–3370 RSC .
- X. Z. Tang, N. Srikanth, X. Q. Feng, C. K. Chua and K. Zhou, RSC Adv., 2016, 6, 102519–102527 RSC .
- J. Muzart, Tetrahedron, 2009, 65, 8313–8323 CrossRef CAS .
- H. N. Tien, V. H. Luan, T. K. Lee, B.-S. Kong, J. S. Chung, E. J. Kim and S. H. Hur, Chem. Eng. J., 2012, 211–212, 97–103 CrossRef CAS .
- P.-G. Ren, D.-X. Yan, X. Ji, T. Chen and Z.-M. Li, Nanotechnology, 2011, 22, 055705 CrossRef .
- M. Foschi, P. Capasso, M. A. Maggi, F. Ruggieri and G. Fioravanti, ACS Omega, 2021, 6, 16943–16954 CrossRef CAS PubMed .
- T. Chudziak, V. Montes-García, W. Czepa, D. Pakulski, A. Musiał, C. Valentini, M. Bielejewski, M. Carlin, A. Tubaro, M. Pelin, P. Samorì and A. Ciesielski, Nanoscale, 2023, 15, 17765–17775 RSC .
- J. Zhao, W. Ren and H.-M. Cheng, J. Mater. Chem., 2012, 22, 20197–20202 RSC .
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe, O. M. Yaghi, D. Materials and G. Discovery, Nature, 2004, 427, 523–527 CrossRef CAS .
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS .
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS .
- B. D. Ossonon and D. Bélanger, RSC Adv., 2017, 7, 27224–27234 RSC .
- R. Bardestani, G. S. Patience and S. Kaliaguine, Can. J. Chem. Eng., 2019, 97, 2781–2791 CrossRef CAS .
- T. K. Shruthi, M. S. Kumar, M. Arjunan, A. Pratap and N. Chandrasekaran, RSC Adv., 2015, 5, 93423–93432 RSC .
- M. Lockett, V. Sarmiento, M. Balingit, M. T. Oropeza-Guzmán and O. Vázquez-Mena, Carbon, 2020, 158, 202–209 CrossRef CAS .
- I. S. Sandhu, M. Chitkara, S. Rana, G. Dhillon, A. Taneja and S. Kumar, Opt. Quantum Electron., 2020, 52, 359 CrossRef CAS .
- A. M. Golsheikh, N. M. Huang, H. N. Lim and R. Zakaria, RSC Adv., 2014, 4, 36401–36411 RSC .
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS .
- R. J. Hunter, Foundations of Colloid Science, Oxford university press, 2001 Search PubMed .
- A. Zielinska, N. R. Ferreira, A. Durazzo, M. Lucarini, N. Cicero, S. E. Mamouni, A. M. Silva, I. Nowak, A. Santini and E. B. Souto, Molecules, 2019, 24, 2683 CrossRef CAS .
- W. Cheng, X. Zhang, N. Duan, L. Jiang, Y. Xu, Y. Chen, Y. Liu and P. Fan, Sci. Total Environ., 2022, 811, 152121 CrossRef CAS .
- B. Chen, H. Wu and S. F. Y. Li, Talanta, 2014, 120, 325–330 CrossRef CAS .
- Y. Zhang, H. Sun, W. Zhang, Z. Gao, P. Yang and J. Gu, Appl. Catal., A, 2015, 496, 9–16 CrossRef CAS .
- M. Isacfranklin, R. Yuvakkumar, G. Ravi, S. I. Hong, F. Shini, M. Thambidurai, C. Dang and D. Velauthapillai, Sci. Rep., 2020, 10, 19198 CrossRef CAS PubMed .
- V. Chaturvedi, M. Pawar, M. Thripuranthaka, R. Shivade and M. V. Shelke, Chem.–Asian J., 2023, 18, e202300121 CrossRef CAS PubMed .
- H. Jiang, L. Yang, C. Li, C. Yan, P. S. Lee and J. Ma, Energy Environ. Sci., 2011, 4, 1813–1819 RSC .
- V. Khomenko, E. Frackowiak and F. Béguin, Electrochim. Acta, 2005, 50, 2499–2506 CrossRef CAS .
- J. Yu, N. Fu, J. Zhao, R. Liu, F. Li, Y. Du and Z. Yang, ACS Omega, 2019, 4, 15904–15911 CrossRef CAS PubMed .
- A. Al-Hamry, H. Kang, E. Sowade, V. Dzhagan, R. D. Rodriguez, C. Müller, D. R. T. Zahn, R. R. Baumann and O. Kanoun, Carbon, 2016, 102, 236–244 CrossRef CAS .
- S. Some, Y. Kim, Y. Yoon, H. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 CrossRef .
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS .
- W. Yang, M. Ni, X. Ren, Y. Tian, N. Li, Y. Su and X. Zhang, Curr. Opin. Colloid Interface Sci., 2015, 20, 416–428 CrossRef CAS .
- K. K. Lee, S. Deng, H. M. Fan, S. Mhaisalkar, H. R. Tan, E. S. Tok, K. P. Loh, W. S. Chin and C. H. Sow, Nanoscale, 2012, 4, 2958–2961 RSC .
- W. K. Park, H. Kim, T. Kim, Y. Kim, S. Yoo, S. Kim, D. H. Yoon and W. S. Yang, Carbon, 2015, 83, 217–223 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: The experimental apparatus used to measure the deionization efficiency of CDI system, atomic scattering, and dispersion stability are included in the ESI. See DOI: https://doi.org/10.1039/d4ra03387f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Seonghyeon Ju
,
Seonghyeon Ju