
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical uncovering of the chalcogen element regulated ESDPT behaviors for 2,5-bis(2-benzoxazolyl)-hydroquinone derivatives†
Jiahe Chen and
Jinfeng Zhao *
*
College of Physical Science and Technology, Shenyang Normal University, Shenyang 110034, China. E-mail: jfzhao1990112@163.com
First published on 22nd August 2024
Abstract
Inspired by the captivating allure of exquisitely regulated characteristics exhibited by 2-(2-hydroxyphenyl)-benzoxazole and its derivatives in the realms of photochemistry and photophysics, our current endeavor primarily revolves around delving into the intricacies of photo-induced excited state reactions for derivatives of 2,5-bis(2-benzoxazolyl)-hydroquinone (BBHQ). Given the significant impact of chalcogen element doping, herein we predominantly focus on exploring the excited state behaviors of BBHQ-OO, BBHQ-SS, and BBHQ-SeSe fluorophores. Our simulations, resulting from variations in geometry and vertical excitation charge reorganization, reveal atomic-electronegativity-dependent hydrogen bonding interactions and charge recombination induced by photoexcitation that can significantly enhance the excited state intramolecular double proton transfer (ESDPT) reaction for BBHQ-OO, BBHQ-SS, and BBHQ-SeSe fluorophores. By constructing potential energy surfaces (PESs) and identifying transition states (TS), we unveil the ultrafast stepwise ESDPT mechanism due to the low potential barriers. Additionally, by employing heterosubstituted BBHQ-OS and BBHQ-OSe compounds, we rigorously validate the stepwise ESDPT mechanism regulated by chalcogen atomic electronegativity. We sincerely anticipate that the modulation of solvent polarity on excited state behaviors will pave the way for groundbreaking advancements in luminescent materials.
1. Introduction
In recent years, there has been growing interest in the academic research into organic compounds that exhibit excited state intramolecular proton transfer (ESIPT). This is primarily attributed to their remarkable fluorescence emission properties, characterized by a significant Stokes shift and the absence of reabsorption processes. Moreover, these compounds can readily undergo conversion into the keto form through proton transfer.1–6 The bathochromic-shifted emission observed during the photo-induced excited state process can mainly be ascribed to the “four-level cyclic” ESIPT mechanism. This mechanism culminates in a reverse proton transfer from the photoisomeric structure back to its original enol form in S0 state.7–9 The outstanding charge recombination before and after ESIPT reaction greatly amplifies the potential applications of these extraordinary organic molecular materials exhibiting ESIPT characteristics in diverse fields such as fluorescence probes, cell imaging, molecular switches, single molecule devices and more.10–14In recent years, there has been growing interest in the investigation of ESIPT reactions due to their potential applications in diverse fields such as optoelectronics and bioimaging. Consequently, researchers worldwide have extensively documented and reported numerous classical organic molecules that exhibit ESIPT characteristics. These molecules typically feature donor–acceptor systems incorporating hydrogen-bonding groups that facilitate efficient proton transfer upon excitation. The comprehension and advancement of ESIPT mechanisms, as well as the development of novel ESIPT-active materials, continue to thrive in research, showing promising outcomes for future technological advancements. Specifically, classical compounds such as 3-hydroxyflavone, methyl salicylate, oxadiazole, hydroxyl phenyl imidazole, imidazopyridine, and 2-(benzo[d]thiazol-2-yl)-phenol, along with their derivatives,15–23 have been extensively investigated for their ESIPT behavior. These investigations have led to the development of numerous fluorescent solid-state emitters, materials exhibiting aggregation-induced emission (AIE), and even emitters capable of generating high-quality white light.24–26 Furthermore, the successful utilization of inhibiting the ESIPT process subsequent to the introduction of a metal ion has been effectively employed for the detection and analysis of various metallic ions. Despite extensive literature on diverse ESIPT fluorophores and their applications, achieving an exceptionally large Stokes shift and high fluorescence quantum yield simultaneously is only feasible in a limited number of cases.
It is undeniable that, after nearly half a century of uninterrupted explorations and driven by the rapid advancement of theoretical and experimental techniques, significant progresses have been made in unraveling the reaction mechanism governing single proton transfer in excited states along a hydrogen bond chain. However, it must be acknowledged that our understanding of the dynamic behavior exhibited by novel molecular systems containing multiple hydrogen bond chains is still in its early stages of investigation. Among these systems, those encompassing excited state double proton transfer (ESDPT) compounds have attracted particular attention due to their status as the simplest and most fundamental class; they serve as an invaluable cornerstone for exploring scenarios involving multiple hydrogen bond sites.27–30 The research primarily focuses on investigating compounds with symmetrical or asymmetrical structures, which play a crucial role in facilitating proton transfer. Proton transfer refers to the movement of protons from one molecule to another and is an essential process in various chemical reactions and biological systems.
Understanding the reaction mechanism is pivotal for gaining profound insights into the photophysical and photochemical attributes exhibited by these excited-state compounds. For example, Chou and coworkers strategically synthesized and reported the typical 1,8-dihydroxy-2-naphthaldehyde (DHNA) fluorophore, which was confirmed to proceed the stepwise ESDPT relay reaction.31 Liao and colleagues designed and clarified the novel six-level ESDPT parent core that owns potential advantage as ideal gain materials for near-infrared organic lasers.32 Pan et al. designed the 2,5-bis(4,5-diphenyl-1H-imida-zol-2-yl)benzene-1,4-diol (BDIBD) derivatives featuring ESDPT characteristics that realizing the achievement of much broadened optical responses and full-color display.33 In summary, the theoretical and experimental investigations of ESDPT behavior associated with double hydrogen bonding chains have not only been recognized as the fundamental manifestation of multiple proton transfer phenomena but also made significant contributions to our understanding of molecular interactions. Moreover, these studies have inspired innovative approaches towards material design and technological advancements across various disciplines, shedding light on the intricate mechanisms underlying proton transfer processes in diverse chemical systems.
As widely recognized, doping with chalcogen elements significantly enhances the performance of organic molecular materials in terms of fluorescence and spin–orbit coupling, reduces efficiency roll-off, improves epitaxial quantum efficiency, narrows radiation spectrum width, and finds extensive applications across diverse fields.34–36 In contrast to the conventional dependence on solvent environment or regulation by push–pull electron groups for excited state processes, there has been relatively limited research on utilizing chalcogen element doping to regulate ESIPT/ESDPT behavior in recent years.37–39
As the dual fluorescent emissions with color-tunable luminophores, 2,5-bis(2-benzoxazolyl)-hydroquinone (BBHQ) and its derivatives not only present the potential advantages in the field of its dual-channel anionic sensing detection, but also own applications to optical properties in polymer.40 Further, the it was reported the forward and back PT could be strongly depended by the solvent polarities, which further affects the characteristics of luminescence.41 Although experimental and theoretical results have effectively elucidated the excited state dynamics of BBHQ and its derivatives, the impact of substituents containing oxygen group elements on the ESIPT reaction in systems with intramolecular hydrogen bonds remains unknown, despite potential insights from atomic electronegativity. Inspired by the excellent luminous properties of BBHQ with different chalcogen atoms, in this work, we mainly focus on exploring the chalcogen elements substituted BBHQ derivatives (i.e., abbreviated as BBHQ-OO, BBHQ-SS and BBHQ-SeSe) as shown in Fig. 1. As we elucidate chalcogen family elements dependent excited state dynamics, the new method of regulating the excited state ESDPT reaction of BBHQ derivatives by atomic electronegativity doping is also proposed.
 | ||
| Fig. 1 Structures of BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores. | ||
2. Computational details
In this work, Gaussian 16 software package was adopted to perform all the computational simulations.42 The DFT and TD-DFT methods were employed to explore the cases of S0 state and S1 states, respectively. Meanwhile, the B3LYP functional and TZVP basis set were employed, taking into account the D3 version of Grimme's dispersion for a more elegant expression,43,44 based on which the relative properties of S0 and S1 states for BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores could be determined. To ensure that the obtained structure is a local minimum, the corresponding infrared (IR) frequency was also calculated. In addition, we selected THF as solvent based on the IEFPCM solvation model,45–47 since the previous experimental results were reported in THF. Vertical excitation simulations were performed by TDDFT method with considering first six low-lying absorption transitions. To thoroughly probe into the reaction mechanism of ESDPT, we calculated and analyzed the IR vibrational behaviors, core-valence bifurcation (CVB) indexes, bond critical point (BCP) parameters, frontier molecular orbitals (MOs) and constructed the potential energy curves (PECs) by restrict optimization for BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores. Finally, using Berny optimization method,48 the S1-state transition state (TS) geometries were also searched along with propositional stepwise ESDPT paths.3. Results and discussion
3.1 Photo-induced geometrical variations
The optimized theoretical structures of BBHQ-OO, BBHQ-SS, and BBHQ-SeSe are depicted in Fig. 1. The corresponding single proton-transfer forms (BBHQ-OO-PT1, BBHQ-SS-PT1, and BBHQ-SeSe-PT1) as well as the dual proton-transfer forms (BBHQ-OO-PT2, BBHQ-SS-PT2, and BBHQ-SeSe-PT2) are provided in Fig. S1, ESI.† These configurations have been unequivocally established as highly stable based on all the IR spectral results obtained. Our preliminary investigation focuses on analyzing the molecular electrostatic potential (MEP) of the aforementioned compounds (BBHQ-OO, BBHQ-SS and BBHQ-SeSe) (seen in Fig. S2, ESI†). While investigating the potential correlation of hydrogen bonding interactions, we have made a noteworthy discovery that both oxygen (O) and nitrogen (N) atoms exhibit a negative electrostatic potential, whereas hydrogen (H) atoms display a positive electrostatic potential. Upon further investigation into this potential correlation, an intriguing observation has emerged. Our findings not only confirm the negative electrostatic potentials of both O and N atoms but also highlight their crucial roles in facilitating the formation of hydrogen bonds. These bonds are indispensable for stabilizing molecular structures and enabling various biological processes. Moreover, it is noteworthy that hydrogen (H) atoms exhibit a positive electrostatic potential. This characteristic renders them highly attractive to the negatively charged O or N atoms involved in hydrogen bonding interactions, thereby serving as bridges and contributing to the overall stability of chemical compounds. Consequently, spontaneous hydrogen bonding interactions can be inferred in the S0 state.Furthermore, this study investigates variations in the vibrational spectral behaviors of infrared (IR) radiation. The utilization of IR analyses has become an indispensable tool for probing excited state dynamics. Importantly, previous classical research convincingly demonstrated that changes in the observed vibrational modes can also serve as indicators of alterations in chemical bonding within these states.49–52 As displayed in Fig. 2, the IR stretching vibrations of O1–H2 and O4–H5 of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are provided. As revealed by the IR data, Fig. 2 directly reflects the strength of O1–H2⋯N3 and O4–H5⋯N6 interactions. The vibrational frequencies associated with dual O1–H2 and O4–H5 stretching are evidently 3331.69, 3237.76 and 3223.33 cm−1 in the S0 state for BBHQ-OO, BBHQ-SS and BBHQ-SeSe, respectively. These values undergo a transformation to become 2959.76, 2690.33 and 2666.82 cm−1 in the S1 state. Based on the given statement, it can be elaborated that excited state intramolecular dual hydrogen bonding interactions (O1–H2⋯N3 and O4–H5⋯N6) should be enhanced.49–52 For BBHQ-OO, BBHQ-SS and BBHQ-SeSe, the redshifts of dual O1–H2 and O4–H5 stretching IR between S0 and S1 states are 371.93, 547.43 and 566.51 cm−1, respectively. This larger redshift as atomic electronegativity decreases potentially reflects that low electronegativity may facilitate ESDPT behavior more easily.
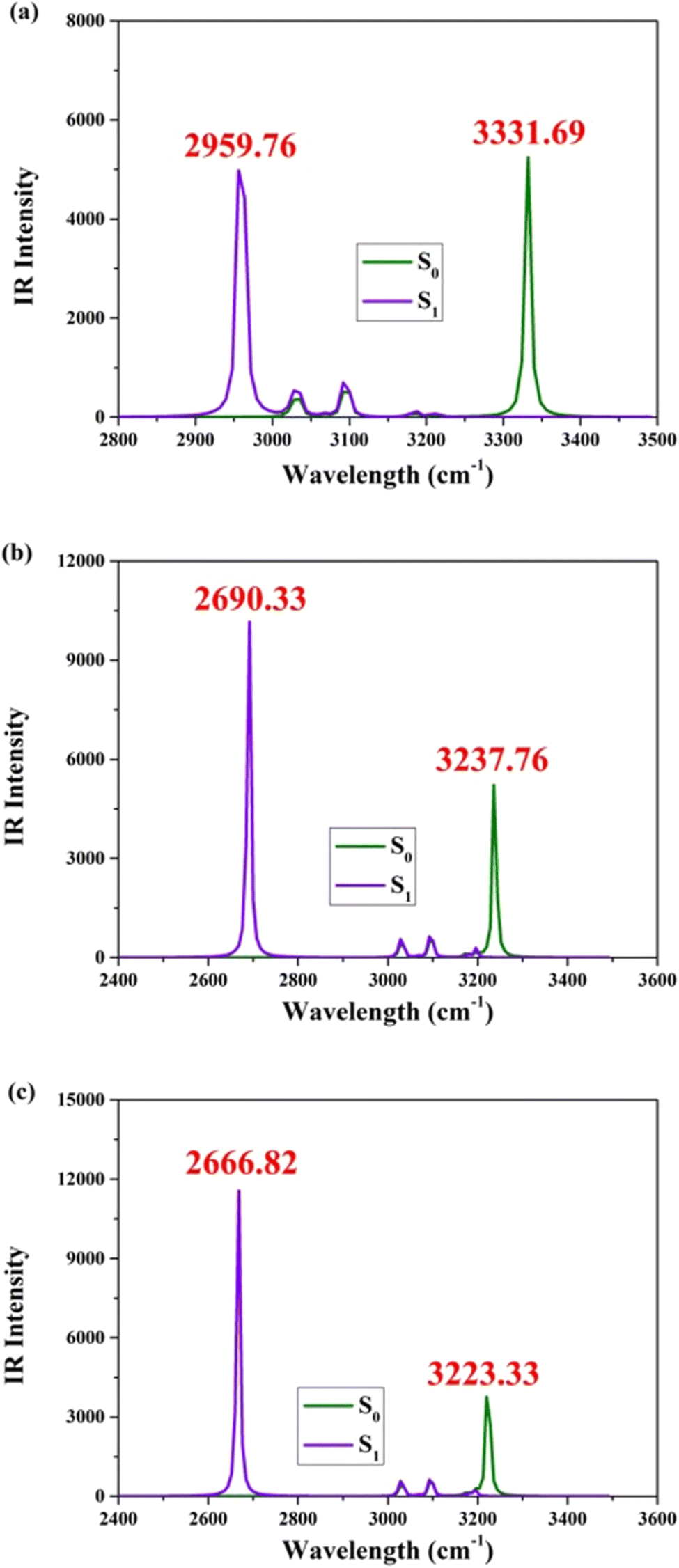 | ||
| Fig. 2 Computational IR vibrational behaviors of synergetic O1–H2 and O4–H5 stretching vibrational mode for (a) BBHQ-OO, (b) BBHQ-SS and (c) BBHQ-SeSe in S0 and S1 states. | ||
Herein, we begin the initial investigation into the changes in configurations of BBHQ-OO, BBHQ-SS and BBHQ-SeSe between its S0 and S1 states. As shown in Table 1, we present the optimized molecular structural parameters regarding dual intramolecular hydrogen bonds in a detailed manner for both the S0 and S1 states of BBHQ-OO, BBHQ-SS and BBHQ-SeSe dissolved in THF solvent. It is evident that due to the symmetrical nature of the structure, the parameters of the two hydrogen bonding chains also exhibit identical characteristics. Similarly, the optimized structural geometries of single proton-transfer and double proton-transfer tautomers are listed in Tables S1 and S2, ESI.† Compared to the S0 state in Table 1, it is clear that in the S1 state, dual hydroxyl lengths are elongated while distances of hydrogen bonds H2⋯N3 and H5⋯N6 are shortened, accompanied by an increase in bond angles (Δ(O1–H2⋯N3) and Δ(O4–H5⋯N6)). The difference between the two states is quite significant, as evidenced by the photo-induced excitation in the S1 state for BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores. When viewed from a lateral perspective, this excitation reveals an impressive increase in intramolecular hydrogen bonding interactions.49–52 Herein, to further check whether the solvent molecules could affect the excited state intramolecular hydrogen bonding interactions, we also consider the THF molecule as the display solvent. Taking the BBHQ-OO as an example, we show the MEP map of THF and BBHQ-OO in Fig. S3, ESI.† According to the positive and negative electrostatic potential regions by blue and red colors, we firstly construct the complex BBHQ-OO-THF form. After the geometrical optimization, the optimized configuration becomes the BBHQ-OO-THF-OPT form. It means solvent molecule plays insignificant roles in BBHQ-OO-THF complex. In addition, we also list the primary geometrical parameters including bond lengths and bond angles (seen in Table S3, ESI†). Comparing these results with Table 1 in the main text, it could be clearly found parameters of bond lengths (Å) and bong angles (Δ°) involved in O1–H2⋯N3 and O4–H5⋯N6 are consistent. Thus, we have reasons to believe solvent molecule THF have negligible influences on excited state hydrogen bonding interactions for BBHQ derivatives, which further reveals no effects on ESDPT reactions. This finding has important implications for understanding the behavior of chemical sensors and their ability to detect specific molecules or compounds. In particular, it suggests that changes in hydrogen bonding interactions can be used as a sensitive indicator of molecular recognition events. By monitoring these changes, researchers may be able to develop more effective and accurate chemical sensors for a wide range of applications.53–55 Furthermore, this discovery highlights the importance of considering multiple perspectives when studying complex systems like chemical sensors. By examining phenomena from different angles and using various techniques, we can gain a more complete understanding of how these systems work and how they can be optimized for specific tasks.
| BBHQ-OO | BBHQ-SS | BBHQ-SeSe | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| O1–H2 | 0.987 | 1.007 | 0.991 | 1.021 | 0.992 | 1.022 |
| H2⋯N3 | 1.793 | 1.699 | 1.739 | 1.621 | 1.735 | 1.615 |
| O4–H5 | 0.987 | 1.007 | 0.991 | 1.021 | 0.992 | 1.022 |
| H5⋯N6 | 1.793 | 1.699 | 1.739 | 1.621 | 1.735 | 1.615 |
| Δ(O1H2N3) | 145.7 | 148.4 | 146.5 | 149.8 | 146.6 | 149.9 |
| Δ(O4H5N6) | 145.7 | 148.4 | 146.5 | 149.8 | 146.6 | 149.9 |
3.2 Hydrogen bonding interactions
As is known, core-valence bifurcation (CVB) index offers researchers an alternative perspective to delve into electron density.56 Its employment enables comprehensive investigations into fundamental aspects of chemistry and related disciplines while providing valuable insights for practical applications ranging from drug design to material engineering. By Multiwfn,57 the CVB parameters could be obtained (listed in Table 2). It is widely acknowledged that a negative value indicates an exceptionally robust and covalent bond, while a near-zero value signifies a moderate level of strength. Weaker hydrogen bonds are generally characterized by positive values.56 From Table 2, it becomes evident that the CVB indexes exhibit negativity. Interestingly, the values of S1 state presents the more negative than the case of S0 state, unveiling the strengthening of hydrogen bonding effects in S1 state. Additionally, the Se-substituted BBHQ-SeSe molecule shows a much stronger negative CVB index in S1, which means that low atomic electronegativity of chalcogen family elements experiences a significantly increased hydrogen bonding when they are exposed to light.| States | BBHQ-OO | BBHQ-SS | BBHQ-SeSe | |||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| ELF(C-V,D) | 0.0997 | 0.1011 | 0.1014 | 0.1037 | 0.1016 | 0.1039 |
| ELF(DH-A) | 0.1669 | 0.2184 | 0.1982 | 0.2799 | 0.2030 | 0.2872 |
| CVB index | −0.0672 | −0.1173 | −0.0968 | −0.1762 | −0.1014 | −0.1833 |
Moreover, to quantitatively describe the energy of hydrogen bond interactions, we employed the atom in molecule method to analyze the distribution of electron density. The electron density ρ(r) at the bond critical point (BCP) parameters between acceptor and hydrogen atoms is presented in Table 3. As is known, the strength of chemical bonds is generally determined by electron density, thus, we could clearly find that S1-state ρ(r) values are more negative than those of S0 state unveiling excited state hydrogen bonding strengthening mentioned above. Following the formula (EHB (i.e., hydrogen bonding energy) ≈ −223.08 × ρ(r) + 0.7423 (ref. 58)), the predicted EHB values of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are listed in Table 3. Evidently, the hydrogen bonding in BBHQ-SeSe is expected to exhibit greater strength due to the presence of Se, which possesses the lowest atomic electronegativity among the elements involved. This phenomenon significantly facilitates the enhancement of the ESDPT reaction.
| Solvents | S0 | S1 | Δρ(S1 − S0) | ΔE(S1 − S0) | ||
|---|---|---|---|---|---|---|
| ρ | EHB | ρ | EHB | ρ | EHB | |
| BBHQ-OO | 0.04221 | −8.674 | 0.05255 | −10.913 | 0.01034 | −2.239 |
| BBHQ-SS | 0.04887 | −10.159 | 0.06457 | −13.662 | 0.01570 | −3.503 |
| BBHQ-SeSe | 0.04964 | −10.331 | 0.06575 | −13.926 | 0.01611 | −3.595 |
3.3 Photo-induced excitation
Charge reorganization, as the primary driving force, is of utmost importance in determining the behaviors and properties of excited states.1–3 It plays a pivotal role in various scientific fields such as chemistry, physics, and materials science. Furthermore, studying charge reorganization is crucial for developing new strategies to enhance energy storage technologies. Understanding charge reorganization is essential for unraveling biological processes involving excited states. For instance, it plays a vital role in photosynthesis by facilitating efficient energy transfer between pigment molecules during light absorption. Therefore, this study focuses on investigating the photo-induced absorption aspects of BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores. The vertical excitation results of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are presented in Table 4. Specifically, the absorption peaks of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are observed at 413.66, 440.10 and 447.82 nm in THF, respectively. This result shows a slight redshift in the absorption peak as electronegativity decreases. Furthermore, the results absorption peak of BBHQ-OO is consistent with experimental report (∼418 nm),41 which strongly support the validity of the current theoretical method used.| Transition | λ | f | Composition | CI (%) | |
|---|---|---|---|---|---|
| BBHQ-OO | S0 → S1 | 413.66 | 0.8776 | H → L | 98.54 |
| S0 → S2 | 342.73 | 0.8111 | H-1 → L | 97.26 | |
| S0 → S3 | 314.49 | 0.0001 | H-2 → L | 90.66 | |
| BBHQ-SS | S0 → S1 | 440.10 | 0.8054 | H → L | 98.71 |
| S0 → S2 | 361.73 | 0.5752 | H-1 → L | 97.24 | |
| S0 → S3 | 341.06 | 0.0000 | H-2 → L | 97.41 | |
| BBHQ-SeSe | S0 → S1 | 447.82 | 0.8116 | H → L | 98.80 |
| S0 → S2 | 375.46 | 0.2924 | H-1 → L | 97.98 | |
| S0 → S3 | 360.30 | 0.0000 | H-2 → L | 97.80 |
To better understand the charge/electron redistribution, we have also included a visualization of the frontier molecular orbitals (MOs) of BBHQ-OO, BBHQ-SS and BBHQ-SeSe in Fig. 3. It is important to note that the S0 → S1 transition observed for BBHQ-OO, BBHQ-SS and BBHQ-SeSe systems follow HOMO–LUMO transition, as indicated by CI (%) values exceeding 97% in Table 4. Thus, just these two orbitals of are provided in Fig. 3. Apparently, the S0 → S1 behavior pertains to the ππ*-type transition. During the HOMO → LUMO transition, the most intriguing aspect lies in the charge-altering behavior across O1–H2⋯N3 and O4–H5⋯N6 moieties. Our primary focus revolves around the reorganization of charges encompassing both hydrogen bonding donor and acceptor regions. Furthermore, in the analysis of charge density difference (CDD) maps, the violent signifies the increasing electron densities from S0 to S1, while wathet represents the decreasing electron densities. Evidently, we observe the shift in electron densities involved in hydrogen bonding moieties of BBHQ-OO, BBHQ-SS and BBHQ-SeSe towards N3 and N6 upon photoexcitation. Additionally, the phenomenon of ESIPT entails a significant modulation of electronic charge density distribution on heavy atoms induced by photoexcitation.
 | ||
| Fig. 3 HOMO and LUMO orbitals for BBHQ-OO, BBHQ-SS and BBHQ-SeSe. The simulated CDD maps between S1 and S0 are also provided. | ||
Herein, the computational atomic charge of O1, H2, N3, O4, H5, and N6 atoms of BBHQ-OO, BBHQ-SS and BBHQ-SeSe compound using Mulliken's charge and NPA charge across three solvents are presented in Table S4, ESI.† It becomes evident that the results obtained from these two distinct methodologies exhibit a harmonious pattern in their dynamic alterations. Herein lies a conspicuous observation: subsequent to photoexcitation, there is an augmentation in negative charge for hydrogen bonding donor atoms O1 and O4, while hydrogen bonding acceptor atoms N3 and N6 experience a reduction in negative charge. In essence, this redistribution of charges induced by excitation effectively facilitates the manifestation of ESDPT behavior demonstrated by BBHQ-OO, BBHQ-SS and BBHQ-SeSe fluorophores.
3.4 Chalcogen associated ESDPT behaviors
To unveil the intricate process of reactions, we embark on a dedicated exploration and elucidation of the specific mechanisms occurring in excited states. By delving into these complex reactions, we aim to unravel the underlying principles governing chemical transformations at an atomic level. Through the construction of potential energy surfaces (PESs), we explore the domain of ESDPT reactions, providing deep quantitative understanding of reaction mechanisms and obstacles within the S1 state. This methodology has become widely accepted as a prominent pathway for elucidating dynamics in excited states.59,60 Considering the existence of intramolecular hydrogen bonds, we use O1–H2 and O4–H5 as two variables to construct the PESs of S0 and S1 states for BBHQ-OO, BBHQ-SS and BBHQ-SeSe, respectively. Via restricted optimization manner, we conduct simulations of PESs, while maintaining a fixed O1–H2 and O4–H5 bond lengths ranging from 0.90 to 2.10 Å with increments of 0.10 Å. The shape of PESs is symmetrical because of the symmetric molecular structures of BBHQ-OO, BBHQ-SS and BBHQ-SeSe. In fact, and the constructed PESs are basically similar for them. Herein, the S0-state PESs of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are provided in Fig. S4, ESI.† And the S1-state PESs of BBHQ-OO, BBHQ-SS and BBHQ-SeSe are provided in Fig. 4.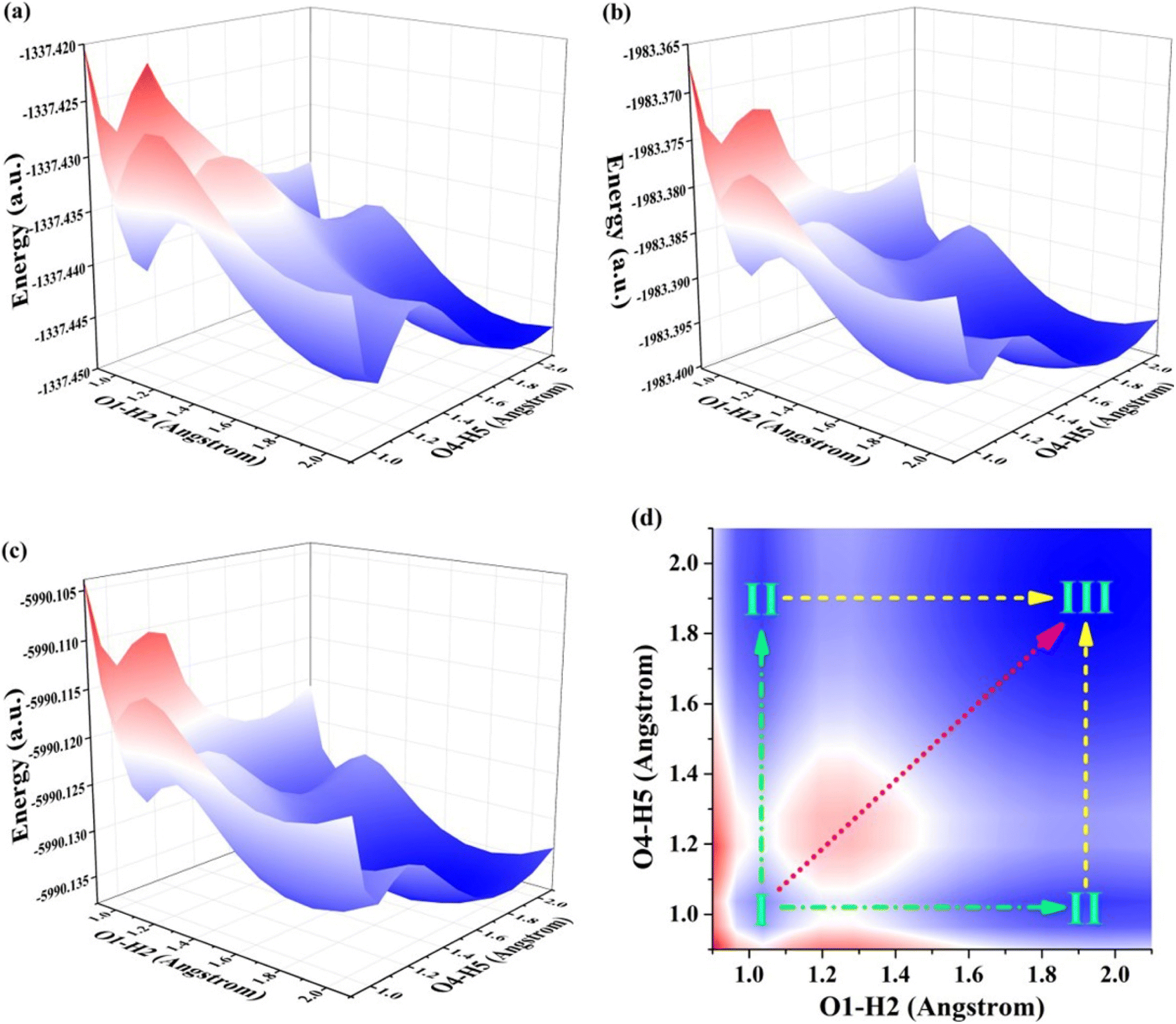 | ||
| Fig. 4 View of the constructed S1-state PES for (a) BBHQ-OO, (b) BBHQ-SS and (c) BBHQ-SeSe. Herein, for the convenience of description, (d) the projection diagram of S1-state PES is shown. | ||
It is worth noting that any increase in S0-state potential energy is accompanied by a corresponding increase in O1–H2 and O4–H5 bond length, which thermodynamically indicates the prohibited transfer of protons in S0 state for BBHQ-OO, BBHQ-SS and BBHQ-SeSe. For the S1 state, our findings indicate that the synergistic double proton transfer along with diagonal could be excluded due to the higher potential barriers. Herein, to check whether the first excited triplet (T1) state affects the excited state behavior for BBHQ derivatives, herein, we take the BBHQ-SS fluorophore as an example. Using the same manner of constructing PES, we show the T1-state PES of BBHQ-SS in Fig. S5, ESI.† Compared with the S1-state PES of BBHQ-SS in Fig. 4(b), it could be clearly found the energy gap between T1 and S1 states is more than 0.035 a.u. (i.e., 0.95 eV). As general, the 0.95 eV is too large to cause the interaction between T1 and S1 states. Thus, we have reasons to ignore the results of triplet states for BBHQ derivatives. In order to explain the specific behavior of the stepwise ESDPT process hereinafter, the projection diagram of S1-state PES is presented in Fig. 4(d). Obviously, in Fig. 4(d), I point stands for BBHQ-OO, BBHQ-SS and BBHQ-SeSe, II point presents the BBHQ-OO-PT1, BBHQ-SS-PT1 and BBHQ-SeSe-PT1, and III point means the BBHQ-OO-PT2, BBHQ-SS-PT2 and BBHQ-SeSe-PT2. To be more precise and to reveal the influence of chalcogen elements (O, S and Se), we further searched the TS configurations along with I → II → III path for BBHQ-OO, BBHQ-SS and BBHQ-SeSe compounds. All the TS forms are provided in ESI.† Further, the potential energy barriers along with stepwise I → II → III path could be calculated by the subtraction of the optimized structures and the TS forms, which have been listed in Table 5. Apparently, we can see that with the decrease of atomic electronegativity from O to S to Se, there is a decrease in the barrier size along the I → II → III path. Even though the potential barrier of synergetic I → III path is also reduced (6.493 kcal mol−1 for BBHQ-OO, 3.189 kcal mol−1 for BBHQ-SS and 2.801 kcal mol−1 for BBHQ-SeSe), the barrier of I → II path (3.398 kcal mol−1 for BBHQ-OO, 1.655 kcal mol−1 for BBHQ-SS and 1.478 kcal mol−1 for BBHQ-SeSe) is quite low. Thus, we have enough reasons to eliminate the synergistic ESDPT mechanism. Also, to check the difference of effects bringing from solvent and isolated BBHQ derivatives, we also consider the PES of ESDPT behaviors for the isolated BBHQ-OO, BBHQ-SS and BBHQ-SeSe in gas to compare the results with those in THF solvent. As shown in Fig. S6,† we present the gas-state PESs for BBHQ-OO (a), BBHQ-SS (b) and BBHQ-SeSe (c), respectively. To distinguish the differences with those in THF solvent, we also list the potential energy barriers in Table S5, ESI.† Clearly, it could be found along with the substitutions (O → S → Se), there is a decrease in the barrier size along the I → II → III path. Even though the potential barrier of synergetic I → III path is also reduced (i.e., 6.903 kcal mol−1 for BBHQ-OO, 3.676 kcal mol−1 for BBHQ-SS and 3.242 kcal mol−1 for BBHQ-SeSe) that is similar with the behaviors in THF solvent, the barrier of I → II path (3.550 kcal mol−1 for BBHQ-OO, 1.563 kcal mol−1 for BBHQ-SS and 1.502 kcal mol−1 for BBHQ-SeSe) is quite low. Obviously, the synergistic ESDPT is unsupported. As a whole, the results of potential energy barriers of the isolated BBHQ-OO, BBHQ-SS and BBHQ-SeSe are consistent with those in THF solvent environment. In brief, the atomic electronegativity of chalcogen elements could regulate the stepwise ESDPT behavior for BBHQ derivatives. That is, our calculation results show that the lower the electronegativity of an atom, the easier it is to promote the ESDPT reaction.
| BBHQ-OO | BBHQ-SS | BBHQ-SeSe | |
|---|---|---|---|
| I → II | 3.398 | 1.655 | 1.478 |
| II → III | 3.291 | 1.727 | 1.463 |
| I → III | 6.493 | 3.189 | 2.801 |
Hereby, to further verify our proposed atomic-electronegativity-regulated ESDPT mechanism, we further selected the heterosubstituted Oxygen–Sulphur (BBHQ-OS) and Oxygen–Selenium (BBHQ-OSe) compounds as the example. The structures of BBHQ-OS and BBHQ-OSe are provided in Fig. S7, ESI.† Clearly, different from BBHQ-OO, BBHQ-SS and BBHQ-SeSe, the BBHQ-OS and BBHQ-OSe are unsymmetrical. Thus, the related PESs should be also unsymmetrical. Based on the same method of constructing PESs as above, we present the S1-state PESs of BBHQ-OS and BBHQ-OSe in Fig. S8, ESI.† Interestingly, we can clearly find the stepwise ESDPT behavior should proceed following O1–H2⋯N3 → O4–H5⋯N6. Moreover, by comparing the potential barrier of the calculated potential curves, we also find that the potential barrier of BBHQ-OSe is lower than that of BBHQ-OS. All these confirm the atomic-electronegativity-regulated stepwise ESDPT mechanism mentioned above.
4. Conclusion
In summary, this study is dedicated to unraveling the ESDPT mechanism associated with hydrogen bonding interactions of three BBHQ derivatives (i.e., BBHQ-OO, BBHQ-SS and BBHQ-SeSe). The enhanced hydrogen bonding effects that facilitate the tendency for ESDPT are revealed by the critical chemical bond properties of BBHQ-OO, BBHQ-SS and BBHQ-SeSe. The CVB indexes and predicted EHB values reveal the atomic electronegativity of chalcogen elements has a great influence on the behavior of excited states. The pivotal ICT through the interaction between HOMO and LUMO, while the reorganization of charge densities facilitates the enhancement of ESDPT behavior. Simulated PESs profiles and simulated barriers suggest that ultrafast ESDPT reactions could occur with the stepwise manner. By the heterosubstituted BBHQ-OS and BBHQ-OSe compounds, we further verify the chalcogen atomic-electronegativity-regulated stepwise ESDPT mechanism. We sincerely wish our reports will pave the way for future design of novel luminescent materials.Data availability
Data are available on request from the corresponding author.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Liaoning Revitalization Talents Program (Grant No. XLYC2203155), the Support Program for Young and Middle-aged Scientific and Technological Innovation Talents of Shenyang City (Grant No. RC230960), the Major Incubation Program of Shenyang Normal University (Grant No. ZD202304), and Shenyang Normal University doctoral program (Grant No. BS202303). The main part of this work was carried out at Bianshui Riverside Supercomputing Center (BRSC) of China.References
- M. Huynh and T. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939 CrossRef CAS.
- A. Demchenko, K. Tang and P. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- P. Zhou and K. Han, Aggregate, 2022, 3, 160 CrossRef.
- L. Zhu, Y. Wan, Y. Wan, J. Gao, H. Yin and Y. Shi, Appl. Phys. Lett., 2023, 122, 061110 CrossRef.
- L. Zhu, Q. Zhou, Y. Wan, Q. Li, Y. Wan, H. Yin and Y. Shi, Phys. Chem. Chem. Phys., 2023, 25, 10661 RSC.
- G. Zhao, W. Shi, H. Zhuang, X. Xin, F. Ma and Y. Li, J. Lumin., 2023, 257, 119735 CrossRef CAS.
- Q. Zhou, D. Wang, P. Song and W. Wen, Chem. Phys. Lett., 2022, 805, 139933 CrossRef CAS.
- X. Xin, W. Shi, Y. Zhao, G. Zhao and Y. Li, Chem. Phys., 2023, 570, 111882 CrossRef CAS.
- C. Liu, J. Zhao, J. Chen, M. Wang, M. Hou and L. Yang, Phys. Chem. Chem. Phys., 2024, 26, 6335 RSC.
- Q. Li, L. Zhu, M. Guo, L. Yan, H. Yin and Y. Shi, J. Mol. Liq., 2023, 386, 122554 CrossRef CAS.
- J. Zhao and C. Liu, Molecules, 2023, 28, 5951 CrossRef CAS PubMed.
- P. Zhou, P. Li, Y. Zhao and K. Han, J. Phys. Chem. Lett., 2019, 10, 6929 CrossRef CAS PubMed.
- X. Xin, W. Shi, Y. Zhao, G. Zhao and Y. Li, J. Mol. Liq., 2023, 391, 123275 CrossRef CAS.
- X. Zhao, H. Yin, W. Zhang, J. Guo and Y. Shi, Phys. Chem. Chem. Phys., 2023, 25, 21604 RSC.
- J. Chen, J. Zhao and H. Dong, J. Mol. Model., 2024, 30, 225 CrossRef CAS.
- T. Qiao, W. Shi, H. Zhuang, X. Xin and Y. Li, J. Lumin., 2024, 269, 120413 CrossRef CAS.
- J. Guo, H. Yin, W. Zhang, X. Zhao, H. Zhao and Y. Shi, Dyes Pigm., 2023, 219, 111601 CrossRef CAS.
- Y. Yang, Y. Liu, Y. Liu and K. Jiang, J. Lumin., 2020, 227, 117587 CrossRef CAS.
- Y. Yang, Y. Liu, H. Zhai, X. Jia, Y. He, Q. Ma, R. Zhang, Y. Liu and K. Jiang, J. Lumin., 2020, 223, 117224 CrossRef CAS.
- K. Zhang, P. Song, F. Ma and Y. Li, Chin. Phys. B, 2024, 33, 068402 CrossRef.
- Z. Zang, F. Ma, P. Song and Y. Li, ACS Appl. Energy Mater., 2024, 7, 2362 CrossRef CAS.
- Q. Zhou, H. Wang and P. Song, New J. Chem., 2023, 47, 16059 RSC.
- K. Tang, M. Chang, T. Lin, H. Pan, T. Fang, K. Chen, W. Hung, Y. Hsu and P. Chou, J. Am. Chem. Soc., 2011, 133, 17738 CrossRef CAS PubMed.
- T. Lin, K. Tang, S. Yang, J. Shen, Y. Cheng, H. Pan, Y. Chi and P. Chou, J. Phys. Chem. A, 2012, 116, 4438 CrossRef CAS PubMed.
- W. Yang, R. Lai, J. Wu, Y. Yu, C. Yan, C. Sun, X. Wang and L. Liao, Adv. Funct. Mater., 2022, 32, 2204129 CrossRef CAS.
- W. Yang, C. Yan, X. Wang and L. Liao, Sci. China: Chem., 2022, 65, 1843 CrossRef CAS.
- J. Huang, K. Yu, H. Ma, S. Chai and B. Dong, Dyes Pigm., 2017, 141, 441 CrossRef CAS.
- J. Zhao, J. Chen, Y. Cui, J. Wang, L. Xia, Y. Dai, P. Song and F. Ma, Phys. Chem. Chem. Phys., 2015, 17, 1142 RSC.
- K. Tang, C. Chen, H. Chuang, J. Chen, Y. Chen, Y. Lin, J. Shen, W. Hu and P. Chou, J. Phys. Chem. Lett., 2011, 2, 3063 CrossRef CAS.
- C. Peng, J. Shen, Y. Chen, P. Wu, W. Hung, W. Hu and P. Chou, J. Am. Chem. Soc., 2015, 137, 14349 CrossRef CAS.
- C. Yan, Y. Liu, W. Yang, J. Wu, X. Wang and L. Liao, Angew. Chem., Int. Ed., 2022, 61, 202210422 CrossRef PubMed.
- S. Yi, B. Li, P. Fu, M. Pan and C. Su, ACS Appl. Mater. Interfaces, 2023, 15, 3172 CrossRef CAS.
- F. Chen, L. Zhao, X. Wang, Q. Yang, W. Li, H. Tian, S. Shao, L. Wang, X. Jing and F. Wang, Sci. China: Chem., 2021, 64, 547 CrossRef CAS.
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 17910 CrossRef CAS PubMed.
- I. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2022, 34, 2107951 CrossRef CAS.
- C. Sun, H. Zhao, X. Liu, H. Yin and Y. Shi, Org. Chem. Front., 2018, 5, 3435 RSC.
- C. Shang, Y. Cao, C. Sun and Y. Li, Spectrochim. Acta, Part A, 2022, 268, 120660 CrossRef CAS PubMed.
- D. Yang and Y. Yang, Spectrochim. Acta, Part A, 2024, 313, 123926 CrossRef CAS PubMed.
- Q. Chu, D. Medvetz and Y. Pang, Chem. Mater., 2007, 19, 6421 CrossRef CAS.
- P. Wnuk, G. Burdzinski, M. Sliwa, M. Kijak, A. Grabowska, J. Sepiol and J. kubicki, Phys. Chem. Chem. Phys., 2014, 16, 2542 RSC.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. Janesko, R. Gomperts, B. Mennucci, H. Hratchian, J. Ortiz, A. Izmaylov, J. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, J. Millam, M. Klene, C. Adamo, R. Cammi, J. Ochterski, R. Martin, K. Morokuma, O. Farkas, J. Foresman and D. Fox, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS.
- H. Schlegel, J. Comput. Chem., 1982, 3, 214 CrossRef CAS.
- G. Zhao and K. Han, J. Phys. Chem. A, 2007, 111, 2469 CrossRef CAS.
- P. Song and F. Ma, Int. Rev. Phys. Rev., 2023, 32, 589 Search PubMed.
- G. Zhao and K. Han, J. Phys. Chem. A, 2007, 111, 9218 CrossRef CAS PubMed.
- G. Zhao and K. Han, Acc. Chem. Res., 2012, 45, 404 CrossRef CAS PubMed.
- T. Chu and B. Liu, Int. Rev. Phys. Chem., 2016, 35, 187 Search PubMed.
- G. Li and K. Han, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1351 Search PubMed.
- T. Chu and J. Xu, J. Mol. Model., 2016, 22, 200 CrossRef PubMed.
- F. Fuster and B. Silvi, Theor. Chem. Acc., 2000, 104, 13 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868 CrossRef CAS PubMed.
- A. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 1999, 1, 3065 RSC.
- P. Zhou and K. Han, Acc. Chem. Res., 2018, 51, 1681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03443k |
| This journal is © The Royal Society of Chemistry 2024 |