
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of the VEGFR-2 inhibition activity of phthalazine derivatives: design, synthesis, cytotoxicity, ADMET, molecular docking and dynamic simulation†
Hatem Hussein Bayoumi a,
Mohamed-Kamal Ibrahima,
Mohammed A. Dahaba,
Fathalla Khedra and
Khaled El-Adl*ba
a,
Mohamed-Kamal Ibrahima,
Mohammed A. Dahaba,
Fathalla Khedra and
Khaled El-Adl*ba
aPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City 11884, Cairo, Egypt. E-mail: eladlkhaled74@yahoo.com; eladlkhaled74@azhar.edu.eg; khaled.eladl@hu.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
First published on 8th July 2024
Abstract
Novel phthalazine derivatives were designed, synthesized and evaluated against Hep G2 and MCF-7 as VEGFR-2 inhibitors. In particular, compounds 2g and 4a were found to be the most potent derivatives among all the tested compounds against MCF-7 and Hep G2 cancer cell lines with IC50 values of 0.15 and 0.12 and 0.18 and 0.09 μM respectively. Moreover, compounds 3a, 3c, 5a and 5b displayed excellent anticancer activities against MCF-7 and Hep G2 cancer cell lines. The highly active derivatives 2g, 3a, 3c, 4a, 5a and 5b were evaluated for their inhibitory activities against VEGFR-2. The tested compounds displayed high to low inhibitory activities with IC50 values ranging from 0.148 to 0.892 μM. Among them, compounds 2g and 4a were found to be the most potent derivatives that inhibited VEGFR-2 with IC50 values of 0.148 and 0.196 μM respectively. Compounds 3a, 3c, 5a and 5b exhibited good activity with IC50 values of 0.375, 0.892, 0.548 and 0.331 μM respectively. Sorafenib was used as a reference drug in this study. Molecular modeling studies were carried out for all compounds against the VEGFR-2 active site. The data obtained from biological testing highly correlated with those obtained from molecular modeling studies. Moreover, MD simulation results indicated the stability of ligand–target interaction. Furthermore, our derivatives 2g and 4a showed a good in silico calculated ADMET profile.
1. Introduction
Several phthalazine derivatives have been reported1–7 as promising anticancer agents as they are potent inhibitors of VEGFR-2.8–11 Phthalazine-1,4-diones have been reported as potent type II IMP dehydrogenase inhibitors and effective anti-proliferative agents against different human and murine tumor cells, particularly against hepatocellular carcinoma.3 Moreover, 1,4-disubstituted phthalazines have attracted considerable attention as promising and effective anticancer agents.3,5 In addition, many triazolo[3,4-a] phthalazine derivatives have been reported to have promising antitumor activities against MGC-803, EC-9706, HeLa and MCF-7 human cancer cell lines.12Increasing attention has been paid to the synthesis of numerous phthalazines during the past two decades as they are favorable drug candidates for cancer treatment. Several research efforts were made that led to the discovery of numerous phthalazine derivatives with diverse enzymatic and cellular targets. For example, AMG 900 I (Fig. 1) was synthesized by Amgen as an orally bioavailable, potent, and highly selective pan-aurora kinase inhibitor that is active in taxane resistant tumor cell lines.6 AMG 900 I was active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L).7 Thereafter, Cee et al. discovered two selective and orally bioavailable pyridinyl-pyrimidine phthalazine aurora kinase inhibitors.13 Vatalanib (PTK787) II14 (Fig. 1) inhibits both VEGFR-1 and VEGFR-2 with IC50 of 380 and 20 nM, respectively. Vatalanib showed good oral absorption and in vivo anticancer activities against a panel of human cancer xenograft models; however, vatalanib is presently in phase III clinical trials for colorectal cancer treatment.15,16 In addition, many anilino-phthalazines have been reported as potent inhibitors of VEGFR-2 as AAC789 III and IM-023911 IV with IC50 = 20 and 48 nM, respectively (Fig. 1).8–11,17,18 VEGFR-2 inhibitors can be classified into three categories: type I inhibitors inhibit the ATP binding region forming a hydrogen bond with the hinge region amino acid Cys919; type II inhibitors occupy the ATP binding site and extend over the gate area into the adjacent allosteric hydrophobic pocket; and type III inhibitors block the allosteric hydrophobic pocket forming hydrophobic interactions.19
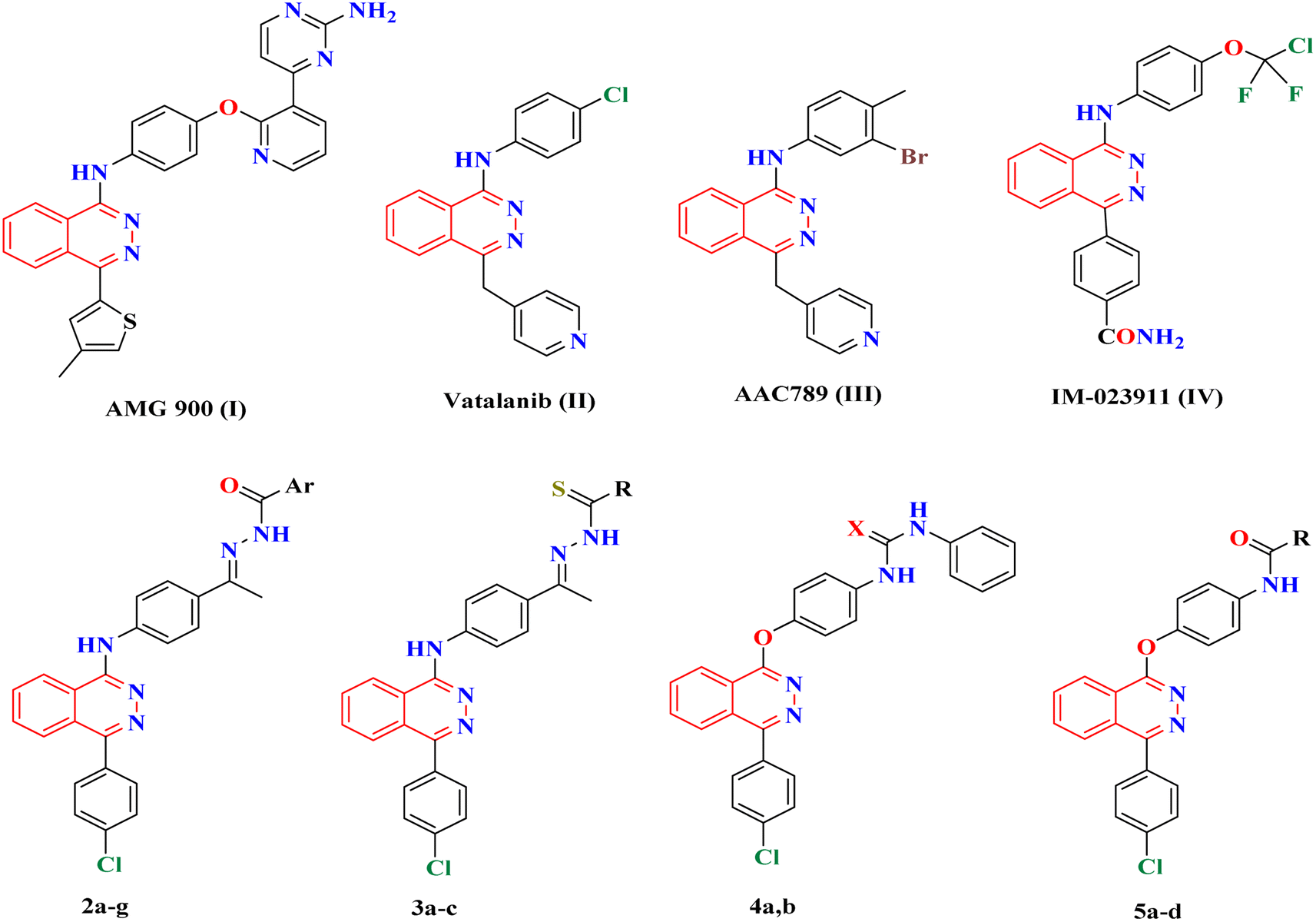 | ||
| Fig. 1 Structures of the lead anticancer phthalazine derivatives I–IV and our designed derivatives. | ||
In this context, a 1-substituted-4-phenylphthalazine scaffold, bearing different 4-substituted anilines and/or phenols, in particular, emerged as an interesting scaffold for designing VEGFR-2 inhibitors (Fig. 2). The abovementioned facts have aggravated us to design novel 4-phenylphthalazine derivatives in an attempt to obtain more potent anticancer agents. In continuation of our efforts to obtain new anticancer agents,20–34 the goal of the present work was the synthesis of new agents with the same essential pharmacophoric features of the reported and clinically used VEGFR-2 inhibitors (e.g. vatalanib and sorafenib).
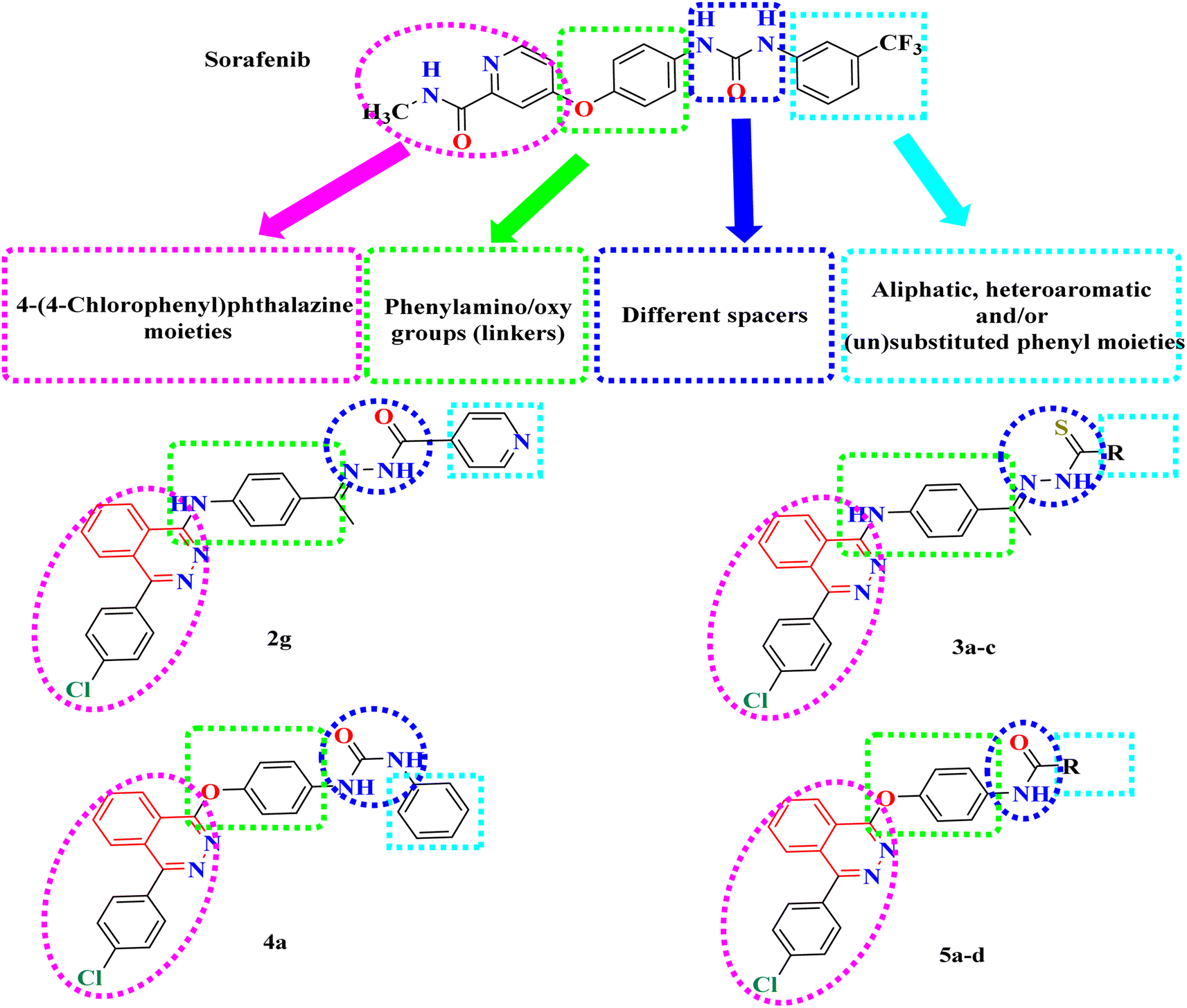 | ||
| Fig. 2 Basic structural features of VEGFR-2 inhibitors. | ||
2. Results and discussion
2.1. Rationale and structure-based design
Our derivatives were designed based on the essential pharmacophoric features of VEGFR-2 inhibitors20–23 (Fig. 2). We synthesized a series of new phthalazine derivatives encouraged by the bioisosteric modifications of VEGFR-2 inhibitor (sorafenib) at four different positions: (1) a flat heteroaromatic ring that occupies the ATP binding domain, (2) a central hydrophobic linker, (3) a spacer with a functional group that has both H-bond acceptor and donor properties to bind with key amino acids (Glu885 and Asp1046), and (4) a terminal hydrophobic moiety that fits into the allosteric hydrophobic pocket (Fig. 2).2.2. Chemistry
The synthetic strategy for the preparation of target compounds (2–5) is depicted in Schemes 1 and 2. The synthesis was started by cyclocondensation of 2-(4-chlorobenzoyl)benzoic acid with hydrazine hydrate to afford the corresponding 4-(4-chlorophenyl)phthalazin-1(2H)-one, which underwent chlorination by reaction with phosphorous oxychloride to afford 1-chloro-4-(4-chlorophenyl)phthalazine.3,4 The chloro derivative was refluxed with 4-aminoacetophenone to afford the corresponding acetyl derivative 1, which underwent condensation with the appropriate acid hydrazide, namely, benzohydrazide, 2-bromobenzohydrazide, 2-chlorobenzohydrazide, 2-hydroxybenzohydrazide, 4-aminobenzohydrazide, nicotinohydrazide and/or isonicotinohydrazide to afford the corresponding hydrazone derivatives 2a–g respectively. In addition, the acetyl derivative 1 was allowed to react with the appropriate thiosemicarbazide, 4-phenyl-3-thiosemicarbazide and/or methyl hydrazinecarbodithioate to give the corresponding derivatives 3a–c respectively (Scheme 1). Furthermore, the intermediate 1-chloro-4-(4-chlorophenyl)phthalazine was reacted with the appropriate urea and/or thiourea to give the corresponding derivatives 4a,b respectively. Moreover, it was reacted with the appropriate amide derivative to afford the corresponding acid amide derivatives 5a–d respectively (Scheme 2).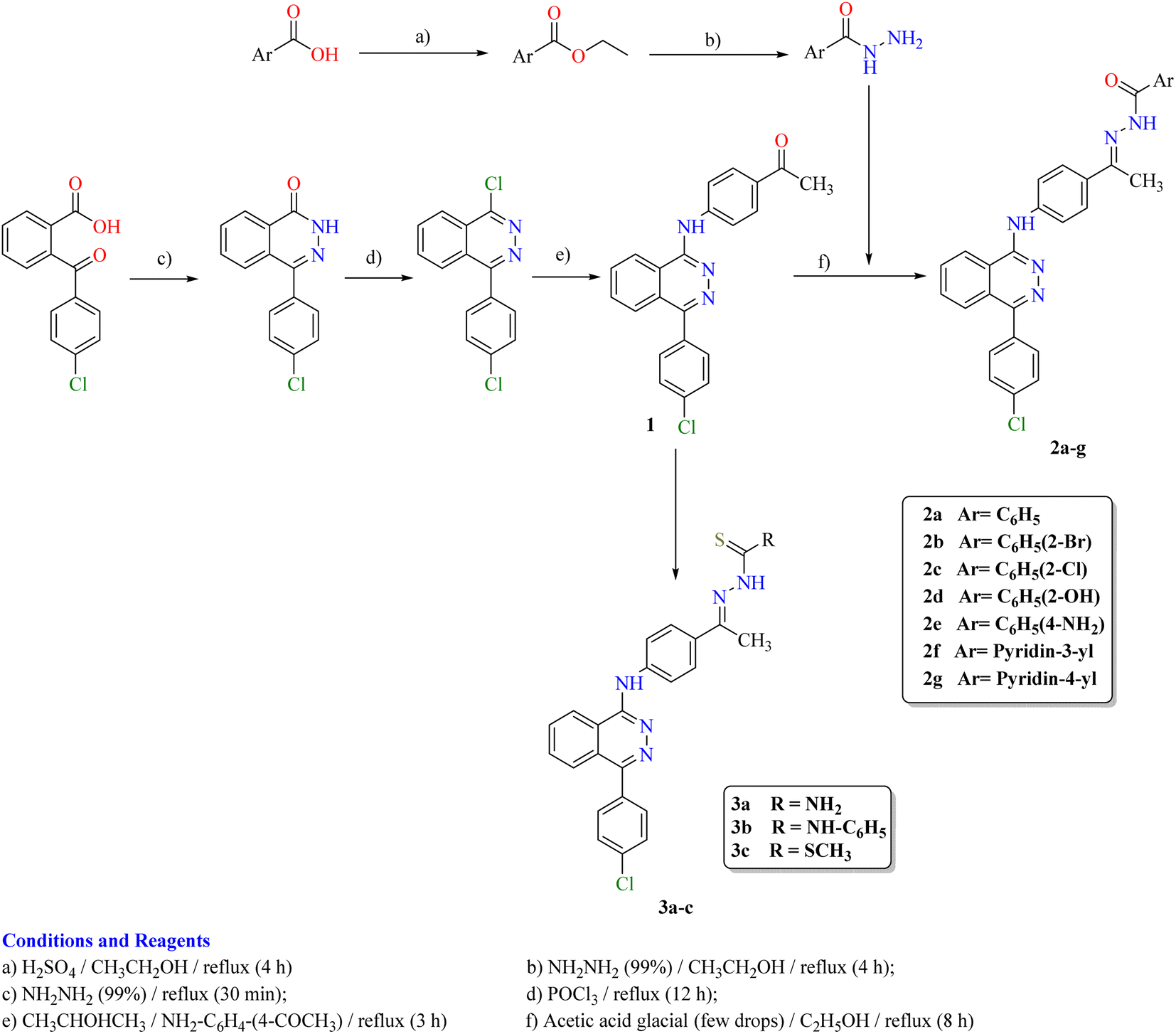 | ||
| Scheme 1 Synthetic route for the preparation of the target compounds 2a–g and 3a–c. | ||
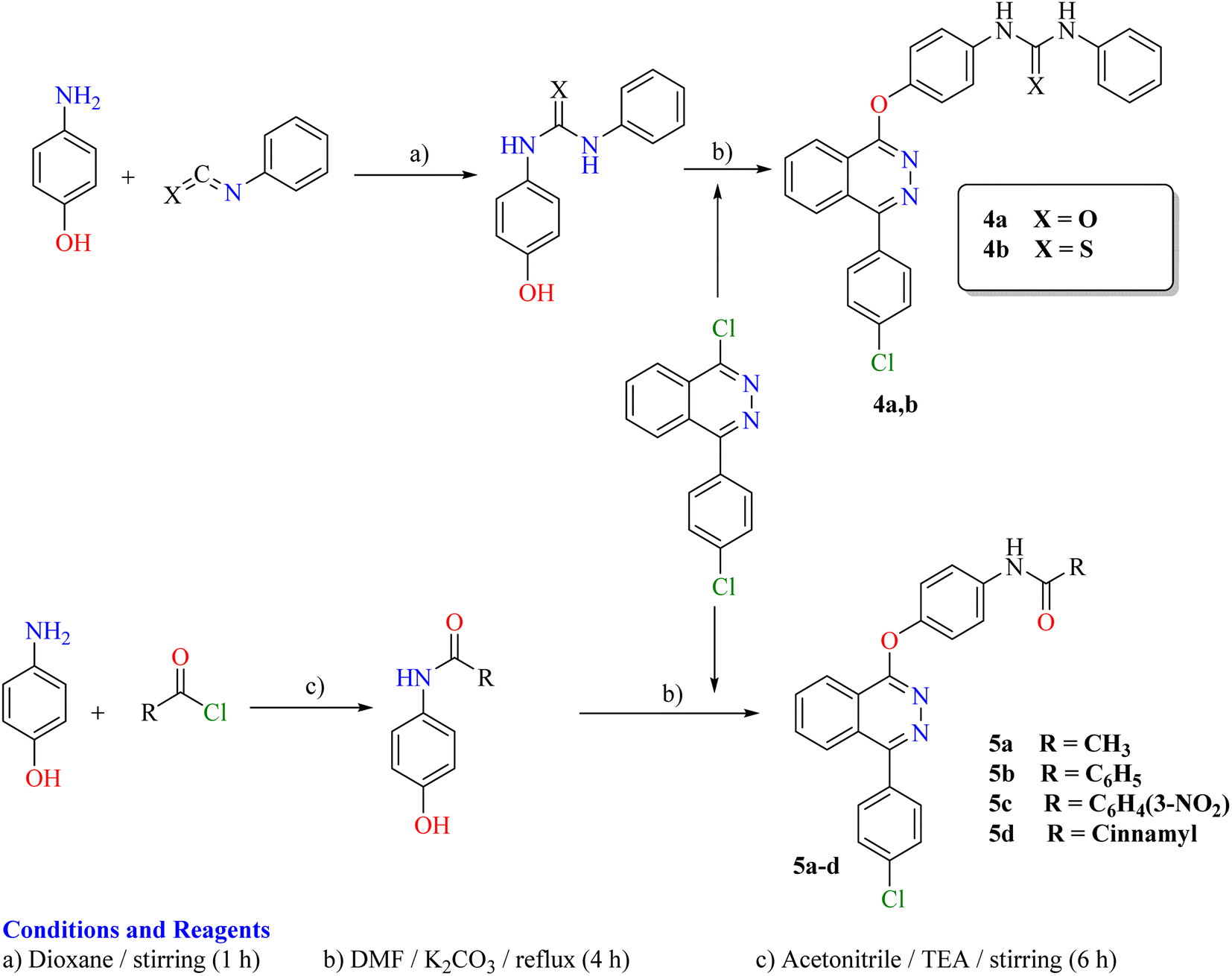 | ||
| Scheme 2 Synthetic route for the preparation of the target compounds 4a,b and 5a–d. | ||
2.3. In vitro anti-proliferative activity
The anti-proliferative activity of the newly synthesized phthalazine derivatives 2a–g–5a–d was examined against two human tumor cell lines, namely, hepatocellular carcinoma (Hep G2) and breast cancer (MCF-7) using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay, as described by Mosmann.35–37 Sorafenib was used as a reference cytotoxic drug (Table 1). From the obtained results, it was explicated that most of the prepared compounds displayed excellent to low growth inhibitory activity against the tested cancer cell lines. In particular, compounds 2g and 4a were found to be the most potent derivatives among all the tested compounds against the two MCF-7 and Hep G2 cancer cell lines with IC50 = 0.15, 0.12 and 0.18, 0.09 μM respectively.| Compound | IC50a (μM) | ||
|---|---|---|---|
| MCF-7 | Hep G2 | VEGFR-2 | |
| a IC50 values are the mean ± S.D. of three separate experiments.b NT = not tested. | |||
| 2a | 1.55 ± 0.01 | 0.77 ± 0.01 | NTb |
| 2b | 0.98 ± 0.01 | 0.63 ± 0.01 | NTb |
| 2c | 1.74 ± 0.01 | 0.59 ± 0.01 | NTb |
| 2d | 3.71 ± 0.07 | 2.18 ± 0.03 | NTb |
| 2e | 0.71 ± 0.02 | 1.19 ± 0.04 | NTb |
| 2f | 0.78 ± 0.01 | 0.75 ± 0.01 | NTb |
| 2g | 0.15 ± 01 | 0.12 ± 0.01 | 0.148 ± 0.01 |
| 3a | 0.18 ± 01 | 0.18 ± 01 | 0.375 ± 0.01 |
| 3b | 0.87 ± 0.02 | 1.53 ± 0.06 | NTb |
| 3c | 0.28 ± 01 | 0.20 ± 01 | 0.892 ± 0.13 |
| 4a | 0.18 ± 01 | 0.09 ± 0.01 | 0.196 ± 0.01 |
| 4b | 1.52 ± 0.01 | 1.63 ± 0.03 | NTb |
| 5a | 0.18 ± 01 | 0.26 ± 01 | 0.548 ± 0.02 |
| 5b | 0.15 ± 01 | 0.22 ± 01 | 0.331 ± 0.01 |
| 5c | 0.18 ± 01 | 1.37 ± 0.14 | NTb |
| 5d | 1.12 ± 0.02 | 1.26 ± 0.03 | NTb |
| Sorafenib | 0.05 ± 0.01 | 0.03 ± 0.01 | 0.059 ± 0.01 |
With respect to the MCF-7 cell line, compounds 3a, 3c, 5a, 5b and 5c displayed very good anticancer activities with IC50 ranging from 0.15 to 0.28 μM. Compounds 2b, 2e, 2f and 3b, with IC50 ranging from 0.71 to 0.98 μM, exhibited good cytotoxicity. Compounds 2a, 2c, 4b and 5d, with IC50 ranging from 1.12 to 1.74 μM displayed moderate cytotoxicity, while compound 2d with IC50 3.71 μM displayed the lowest cytotoxicity.
With respect to the Hep G2 hepatocellular carcinoma cell line, compounds 3a, 3c, 5a and 5b displayed very good anticancer activities with IC50 ranging from 0.18 to 0.26 μM. Compounds 2a, 2b, 2c and 2f with IC50 ranging from 0.59 to 0.77 μM exhibited good cytotoxicity. Compounds 2e, 3b, 4b, 5c and 5d with IC50 ranging from 1.19 to 1.63 μM displayed moderate cytotoxicity, while compound 2d with IC50 of 2.18 μM displayed the lowest cytotoxicity.
2.4. In vitro VEGFR-2 kinase assay
The highly active derivatives 2g, 3a, 3c, 4a, 5a and 5b were evaluated for their inhibitory activities against VEGFR-2 with an anti-phosphotyrosine antibody using an Alpha Screen system (PerkinElmer, USA).38 Sorafenib was used as positive control in this assay. The tested compounds displayed high to low inhibitory activity with IC50 values ranging from 0.148 to 0.892 μM (Table 1) (Fig. 3). Among them, compounds 2g and 4a were found to be the most potent derivatives that inhibited VEGFR-2 at IC50 values of 0.148 and 0.196 μM respectively. Compounds 3a and 5b exhibited good activity with IC50 values of 0.375 and 0.331 μM respectively. Moreover, compound 5a possessed moderate VEGFR-2 inhibition with an IC50 value of 0.548 μM. Finally, compound 3c displayed the lowest VEGFR-2 inhibition with an IC50 value of 0.892 μM.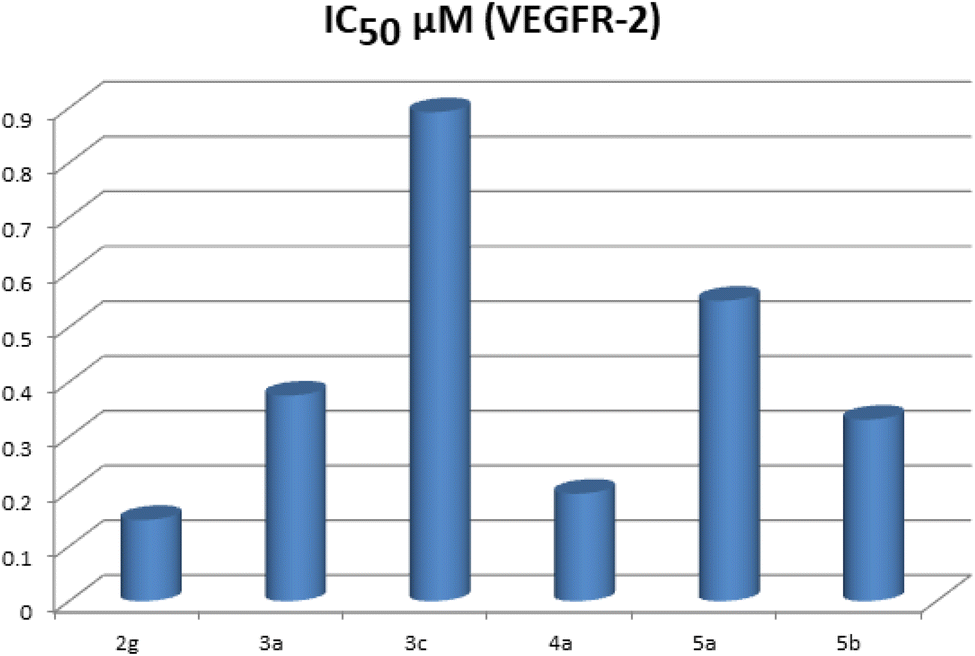 | ||
| Fig. 3 IC50 (μM) of the tested compounds as VEGFR-2 inhibitors. | ||
2.5. Structure–activity relationship (SAR)
The preliminary SAR study has focused on the effect of the hydrophobic and electronic nature of the substituents used in this study. Moreover, it focused on the effect of the type, length and number of spacers used and the distal moieties. The data obtained revealed that the tested compounds displayed different levels of anticancer activities. Generally, the 4-(4-chlorophenyl)phthalazine scaffold, bearing different 4-substituted anilines and/or phenols joined to the hydrophobic distal moieties through amide, hydrazide, thiosemicarbazide, urea and/or thiourea spacers containing (HBA–HBD). The lipophilicity and electronic nature of the distal moieties play an important role in VEGFR-2 inhibition and consequently the anticancer activities.In molecular docking studies, the presence of the hydrazide and urea spacers, e.g. 2g and 4a respectively, impart a higher VEGFR-2 binding affinity and consequently higher anticancer activities. This higher affinity of such derivatives may be attributed to the increased length of the molecular structures so as to enable the distal moieties to occupy the hydrophobic grooves formed by Asp1046, Cys1045, Hie1026, Ile888, Ile892 and Glu885. Moreover, our derivatives containing a phthalazine heterocyclic ring that inhibits the ATP binding domain, form an H-bond with Cys919, and then extend over the gate area into the adjacent allosteric hydrophobic moiety, which confirmed our compounds as type II inhibitors of VEGFR-2.
From the structure of the synthesized derivatives and the data shown in Table 1, we can divide these tested compounds into three groups. The first group contains acid hydrazide spacers as in compounds 2a–g. Generally, in this group, the derivative 2g with an isonicotinohydrazide spacer exhibited the highest anticancer activities through this group against both Hep G2 and MCF-7 tested cell lines. Compound 2e with the spacer benzohydrazide substituted at position-4 with the hydrophilic electron-donating NH2 group showed higher anticancer activities than those of compound 2f with isonicotinohydrazide and compound 2b with benzohydrazide substituted at position-2 with the hydrophobic electron-withdrawing Br one against MCF-7 cell lines, while against Hep G2, the order of activity is the reverse as follows: 2b > 2f > 2e. Moreover, compound 2b with the more hydrophobic electron-withdrawing Br group at position-2 displayed higher anticancer activities than those of 2a (unsubstituted benzohydrazide), 2c with the hydrophobic electron-withdrawing Cl and 2d with the hydrophilic electron-donating OH group against MCF-7 cell lines, while against Hep G2, the order of activity is 2c > 2b > 2a > 2d.
The second group 3a–c contains thiosemicarbazide and hydrazinecarbodithioate spacers. Compound 3a with a thiosemicarbazide spacer exhibited higher activities than those of compound 6 with a hydrazine carbodithioate spacer attached to the methyl group and 3c with thiosemicarbazide attached to the phenyl group against both the Hep G2 and MCF-7 cell lines.
The third group comprises compounds 4a,b and 5a–d that contain urea, thiourea and/or amide spacers. In this group, compound 5b with a benzamide spacer displayed higher activities than those of 4a with phenylurea, 5a with acetamide, 5c with 3-nitrobenzamide, and 5d with cinnamamide against MCF-7 cell lines, but against Hep G2, the order of activity is 4a > 5b > 5a > 5d > 5c > 4b.
2.6. Docking studies
All modeling experiments in the present work were performed using the Molsoft software. Each experiment used VEGFR-2 downloaded from the Brookhaven Protein Databank (PDB ID 4ASD).39Sorafenib suggested binding mechanism produced 5 H-bonds and an energy value of −99.50 kcal mol−1 (Table 2). It generated 2 hydrogen bonds with Cys919 (2.51 Å and 2.10 Å), two with Glu885 (1.77 Å and 2.75 Å), and one with Asp1046 (1.50 Å). The pocket created by Cys919, Phe918, Leu1035, Lys920, Glu917, Val848 and Leu840 was occupied by the N-methylpicolinamide group. Additionally, the hydrophobic groove formed by Cys1045, Leu1035, Val916, Lys868, and Val848 was occupied by the central phenyl linker. Moreover, Cys1045, Asp1046, Hie1026, Ile888, Ile892 and Glu885 formed a hydrophobic channel that was occupied by the terminal 3-trifluromethyl-4-chlorophenyl group (Fig. 4). The urea spacer had a crucial part in the binding of the VEGFR-2 enzyme, but it also played a crucial role in the high binding affinity of Sorafenib. These results encourage us to experiment with various spacers in order to obtain efficient VEGFR-2 inhibitors.
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 2a | −83.68 | 3c | −79.21 |
| 2b | −87.16 | 4a | −95.96 |
| 2c | −84.73 | 4b | −90.40 |
| 2d | −84.35 | 5a | −74.05 |
| 2e | −89.05 | 5b | −75.66 |
| 2f | −86.66 | 5c | −77.44 |
| 2g | −98.84 | 5d | −88.90 |
| 3a | −81.79 | Sorafenib | −95.36 |
| 3b | −92.12 |
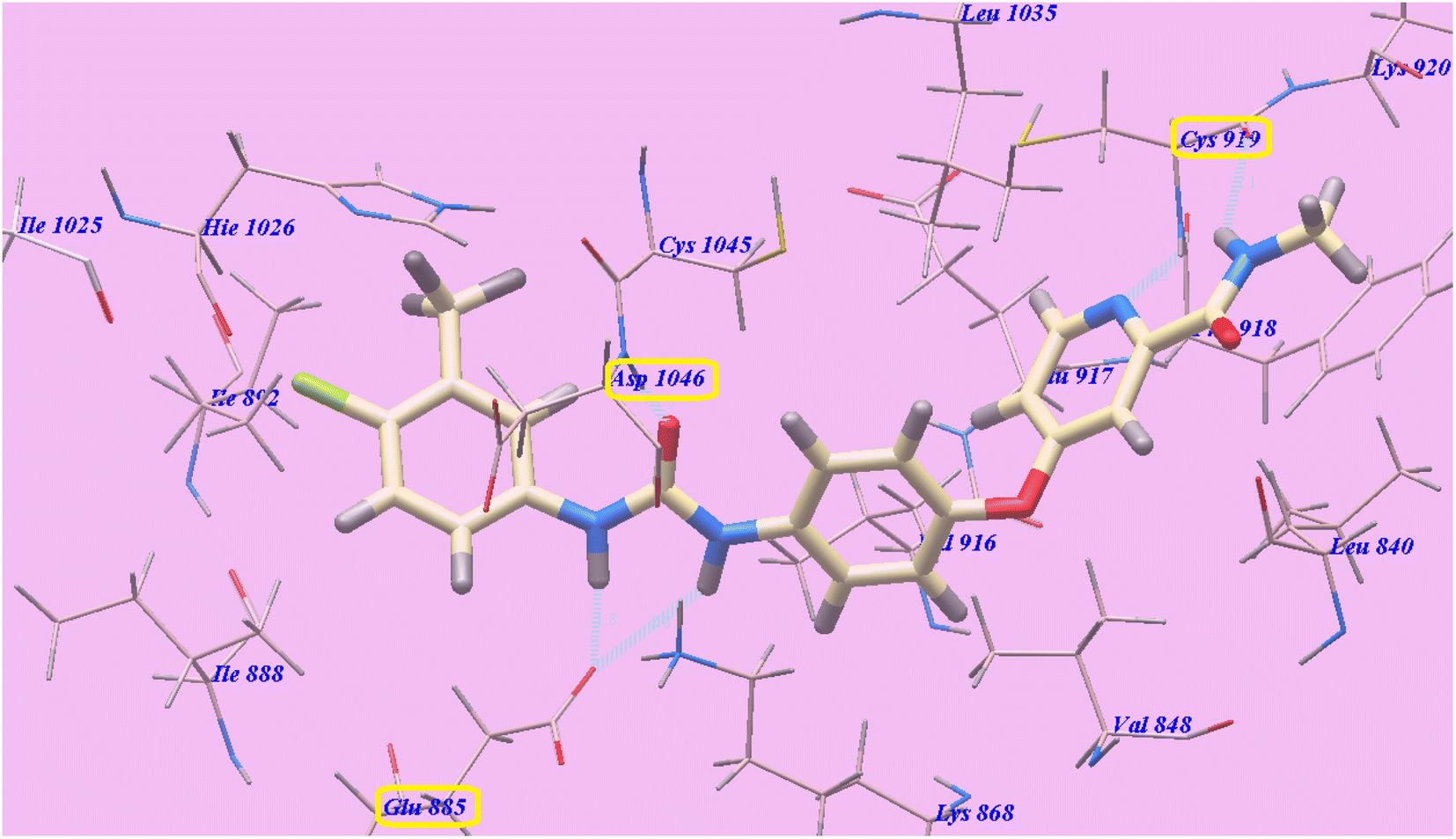 | ||
| Fig. 4 Sorafenib predicted mechanism of binding with 4ASD. Dotted lines represent atoms that are H-bonded. | ||
As planned, the proposed binding mode of compound 2g is virtually the same as that of sorafenib. It showed −98.84 kcal mol−1 and formed 6 H-bonds with Cys919 (2.28 Å), Val916 (2.96 Å), Glu883 (2.68 Å), Asp1044 (1.78 Å and 1.95 Å) and Ile892 (2.95 Å). The 4-(4-chlorophenyl)phthalazine moiety occupied the hydrophobic pocket created by Cys919, Phe918, Leu1035, Lys920, Glu917, Val848 and Leu840. Additionally, the central phenyl linker occupied the hydrophobic groove formed by Cys1045, Leu1035, Glu917, Val916, Lys868, and Val848. Furthermore, the terminal pyridine ring occupied a hydrophobic channel formed by Asp1046, Hie1026, Ile888, Ile892 and Glu885 (Fig. 5). These interactions may explain the highest anticancer activities of compound 2g.
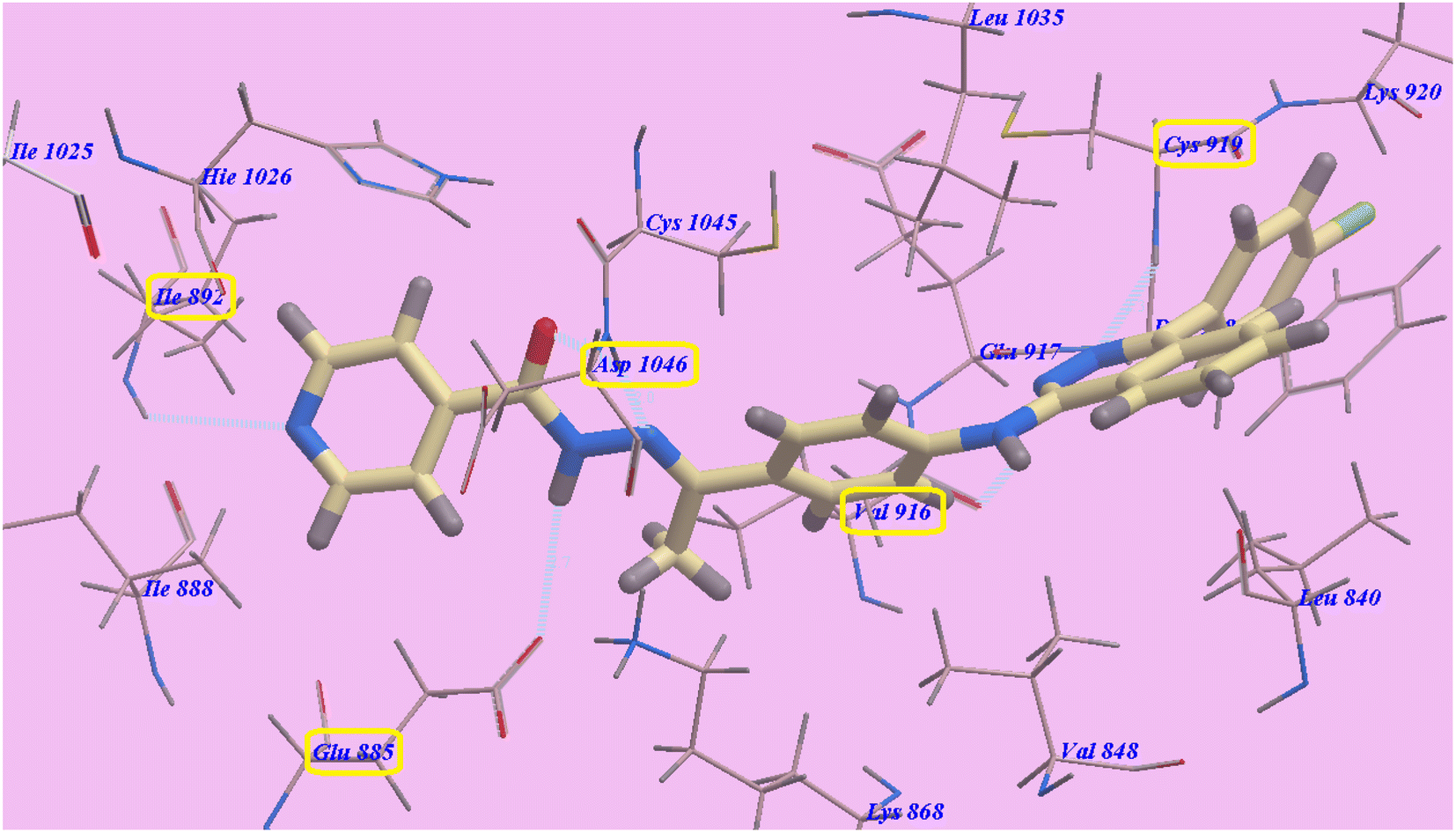 | ||
| Fig. 5 Predicted binding mode for 2g with 4ASD. | ||
The proposed binding mode of compound 4a is virtually the same as that of 2g. It showed −95.86 kcal mol−1 and formed 4 H-bonds with Cys919 (2.96 Å), Glu883 (2.05 Å and 2.95 Å), and Asp1044 (1.25 Å). The 4-(4-chlorophenyl)phthalazine moiety occupied the hydrophobic pocket created by Cys919, Phe918, Leu1035, Lys920, Glu917, Val848 and Leu840. Additionally, the central phenyl linker occupied the hydrophobic groove formed by Cys1045, Leu1035, Glu917, Val916, Lys868, and Val848. Furthermore, the terminal phenyl ring occupied a hydrophobic channel formed by Asp1046, Cys1045, Hie1026, Ile888, Ile892 and Glu885 (Fig. 6). These interactions of compound 4a may explain its high anticancer activity.
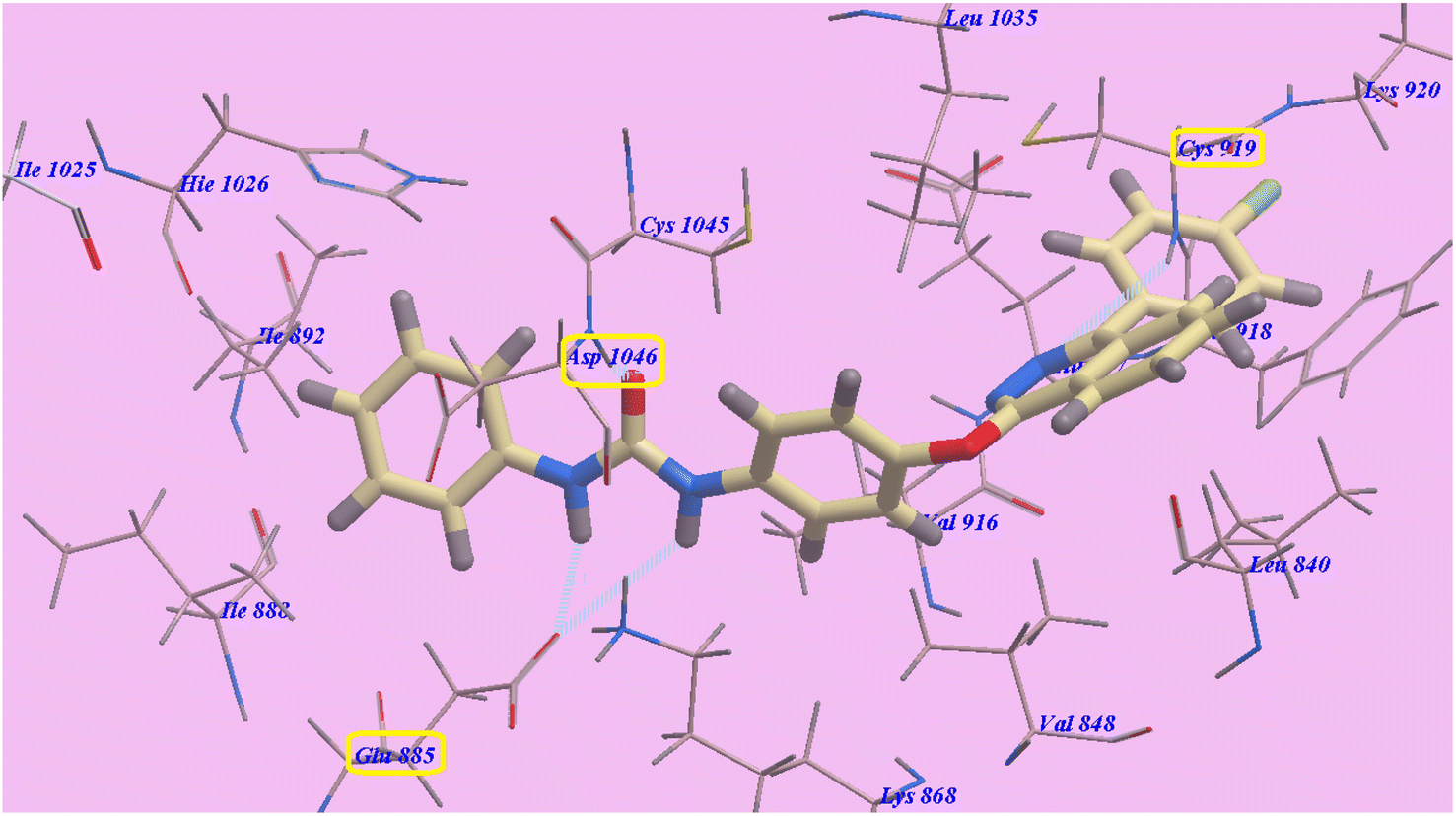 | ||
| Fig. 6 Predicted binding mode for 4a with 4ASD. | ||
The obtained results showed that our derivatives inhibited the ATP binding domain forming an H-bond with Cys919 and extended over the gate area into the adjacent allosteric hydrophobic moiety, which confirmed that our compounds were type II inhibitors of VEGFR-2.
2.7. Molecular dynamics simulation
The highly active derivatives 2g, 3a, 4a and 5b in the protein VEGFR-2 were simulated using molecular dynamics (MD). By utilizing Amber's MM/GBSA.py script and the trajectory, the receptor-ligand binding energy was calculated.40 Additionally, sorafenib was used as a positive control. With the help of GAFF241,42 and the force field AMBERff14SB for the protein, ligand force fields were produced.43 The monitored root-mean-square deviation (RMSD) validated the studied inhibitor compounds' considerable global stability inside the target's recognized active site throughout the 50 ns all-atom MD runs (Fig. 7). A given ligand's molecular divergence from a defined original/reference structure was estimated using RMSD. The selected MD simulation procedure was valid, indicating the stability of the ligand–target interaction.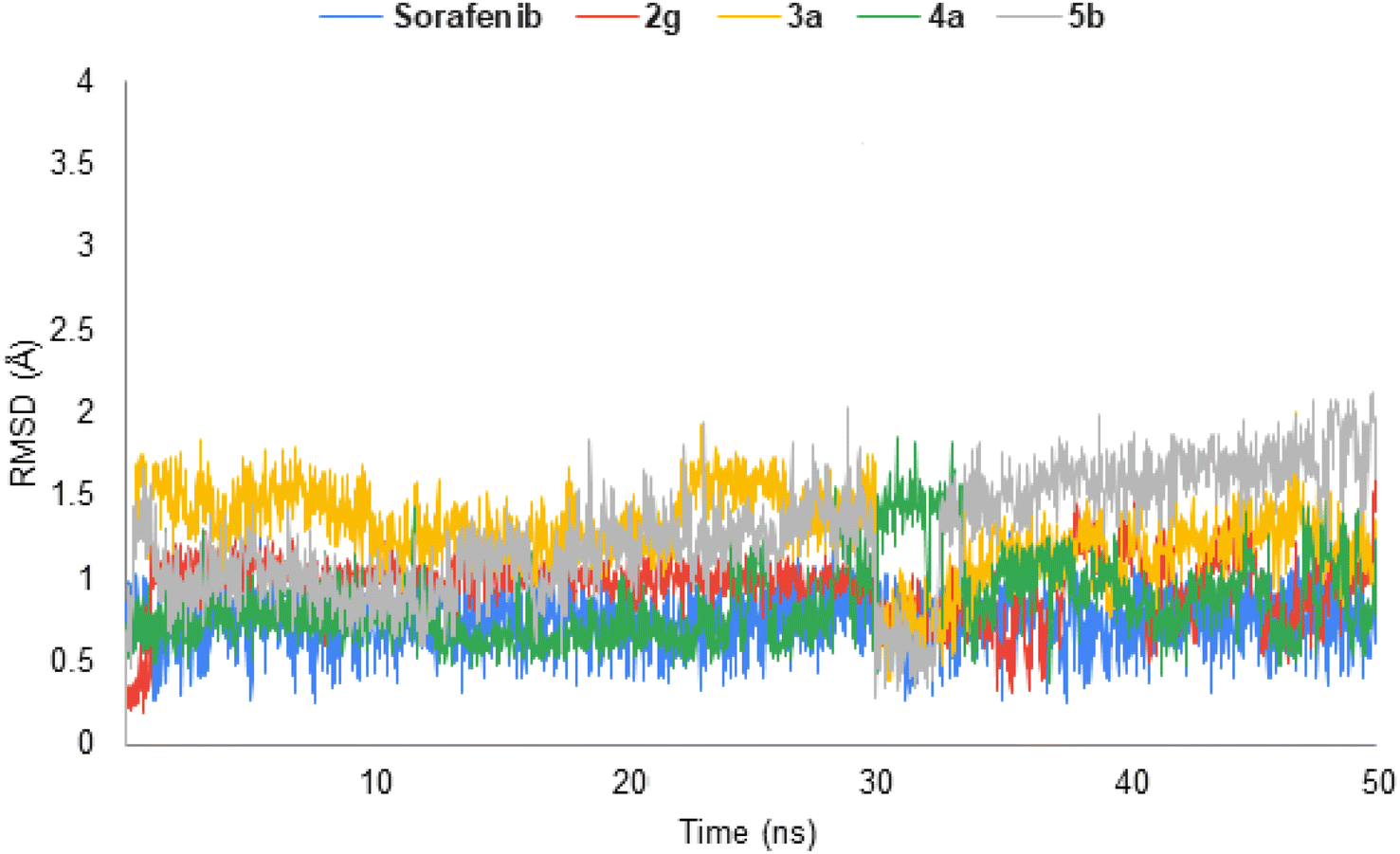 | ||
| Fig. 7 Analysis of VEGFR-2 protein RMSD throughout 50 ns for the ligand–protein complexes. | ||
2.8. ADMET; in silico studies profile
An in silico study of the highly active derivatives 2g and 4a was conducted for their physicochemical character evaluation and the proposed ADMET profile. It was predicted using the pkCSM descriptor algorithm procedures44 and matched to the rule of five described by Lipinski.45 Good absorption properties were expected for the molecules that accomplish at least four rules: (i) no more than five hydrogen bond donors, (ii) no more than 10 hydrogen bond acceptors, (iii) molecular weight less than 500, and (iv) not more than 5 for log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. In the current work, the standard anticancer agent sorafenib and our new compounds 2g and 4a violate the logP rule.
P. In the current work, the standard anticancer agent sorafenib and our new compounds 2g and 4a violate the logP rule.
As a result of obtaining data from Table 3, we can assume that compounds 2g and 4a have very good GIT absorption in humans (100 and 88.7 respectively), which indicates the different biological membranes to be easier to cross.46 Therefore, they may show a significantly high bioavailability through GIT. Concerning CNS penetrability, our prepared compounds can reach CNS (CNS permeability values −1.713 and −1.376 respectively), more than that of sorafenib (−2.007).
| Parameter | 2g | 4a | Sorafenib |
|---|---|---|---|
| Physicochemical properties | |||
| Molecular weight | 492.97 | 466.928 | 464.831 |
| logP |
6.2428 | 7.3865 | 5.5497 |
| Rotatable bonds | 6 | 5 | 5 |
| Acceptors | 6 | 4 | 4 |
| Donors | 2 | 2 | 3 |
| Surface area | 212.437 | 200.735 | 185.111 |
|
|||
| Absorption | |||
| Water solubility | −4.011 | −3.562 | −4.822 |
| Human intest. absorption | 100 | 88.707 | 89.043 |
| Substrate for P-glycoprotein | + | + | + |
| Inhibitor of P-glycoprotein I | + | + | + |
| Inhibitor of P-glycoprotein II | + | + | + |
|
|||
| Distribution | |||
| Permeability throughout BBB | −0.478 | −0.226 | −1.684 |
| Permeability to CNS | −1.713 | −1.376 | −2.007 |
|
|||
| Metabolism | |||
| CYP2D6 substrate | − | − | − |
| CYP3A4 substrate | + | + | + |
| Inhibition of CYP1A2 | + | + | + |
| Inhibition of CYP2C19 | + | + | + |
| Inhibition of CYP2C9 | + | + | + |
| Inhibition of CYP2D6 | − | − | − |
| Inhibition of CYP3A4 | + | − | + |
|
|||
| Excretion | |||
| Clearance | −0.167 | −0.23 | −0.219 |
|
|||
| Toxicity | |||
| Human max. tolerated dose | 0.367 | 0.515 | 0.549 |
| Acute toxic activity (LD50) | 3.12 | 3.665 | 2.538 |
| Chronic toxic activity (LOAEL) | 1.177 | 2.182 | 1.198 |
| Hepatotoxic effect | + | + | + |
It is well known that CYP3A4, the major drug-metabolizing enzyme, could be inhibited by sorafenib, and also our derivative 2g but our derivative 4a does not. Elimination was expected depending on the total clearance, which is a considerable factor in deciding dose intervals. The data showed that compound 2g exhibited a lower clearance rate than that of sorafenib and our compound 4a, which demonstrated high clearance values. Thus, it could be eliminated faster, and as a result, supposed to have shorter dosing intervals. Unlike sorafenib and 4a, the prepared compound 2g exhibited low clearance rate, which signifies a long duration of action and extended dosing intervals. Toxicity is the final ADMET profile studied factor. As presented in Table 3, sorafenib and the novel compounds 2g and 4a shared the drawback of unwanted hepatotoxic actions. Sorafenib and 4a demonstrated high maximum tolerated dose. These involve the advantage of the broad therapeutic index of sorafenib and our derivative 4a respectively. The oral acute toxic doses of the novel compound 2g and 4a are higher than that of sorafenib. Lastly, the oral chronic toxic dose of the novel compound 4a is higher than that of sorafenib.
3. Conclusion
In summary, a new series of phthalazine derivatives have been designed, synthesized and evaluated for their anticancer activities against two human tumor cell lines, namely, hepatocellular carcinoma (Hep G2) and breast cancer (MCF-7) as VEGFR-2 inhibitors. All the tested compounds showed variable anticancer activities. The molecular modeling was performed to investigate the binding mode of the proposed compounds with the VEGFR-2 active site. The data obtained from biological testing highly correlated with those obtained from molecular modeling studies. In particular, compounds 2g and 4a were found to be the most potent derivatives among all the tested compounds against the two MCF-7 and Hep G2 cancer cell lines with IC50 = 0.15, 0.12 and 0.18, 0.09 μM respectively. Moreover, compounds 3a, 3c, 5a and 5b displayed very good anticancer activities against the two MCF-7 and Hep G2 cancer cell lines. The highly active derivatives 2g, 3a, 3c, 4a, 5a and 5b were evaluated for their inhibitory activities against VEGFR-2. The tested compounds displayed high to low inhibitory activities with IC50 values ranging from 0.148 to 0.892 μM. Among them, compounds 2g and 4a were found to be the most potent derivatives that inhibited VEGFR-2 at IC50 values of 0.148 and 0.196 μM respectively. Compounds 3a, 3c, 5a and 5b exhibited good activity with IC50 values of 0.375, 0.892, 0.548 and 0.331 μM respectively. Sorafenib was used as a reference drug in this study. Moreover, MD simulation procedure was valid, indicating the stability of the ligand–target interaction. Moreover, our derivatives 2g and 4a showed a good in silico calculated ADMET profile in comparison to sorafenib.4. Experimental
4.1. Chemistry
All melting point tests were carried out by an open capillary method using a Stuart melting point apparatus SMP30 and were uncorrected. The infrared spectra were recorded using a Bruker FT/IR Spectro-photometer at Microanalytical Unit, Faculty of Science, Cairo University, and expressed in wave number (cm−1). The 1H NMR spectra were recorded at 400 MHz, while 13C NMR spectra were run at 101 MHz, using a Bruker 400 MHz-NMR spectrophotometer at Nuclear Magnetic Unit, Faculty of Pharmacy, Mansoura University. Some of the 1H NMR spectra were recorded at 300 MHz at Micro Analytical Unit, Faculty of Science, Cairo University. Chemical shifts were expressed in δ (ppm) and recorded relative to the (DMSO-d6) solvent, using TMS as an internal reference standard. All coupling constant (J) values are given in Hertz. The mass spectra were recorded using a Varian MAT 311-A (70 e.v.) at the Regional Center for Mycology and Biotechnology, Faculty of Science, Al-Azhar University. Elemental analyses were performed at the Regional Center for Mycology and Biotechnology, Faculty of Science, Al-Azhar University. All compounds were within ±0.4 of the theoretical values.4-(4-Chlorophenyl)phthalazin-1(2H)-one and 1-chloro-4-(4-chlorophenyl)phthalazine were obtained according to the reported procedures.3,4
Yield, 80%; mp 176–8 °C; IRνmax (cm−1): 3426 (NH), 3049 (C–H aromatic), 2915 (C–H aliphatic), 1679 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1592 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.20 (d, J = 8.3 Hz, 1H), 8.16 (t, J = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 3H), 8.03 (d, J = 8.5 Hz, 2H), 7.97 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 197.26, 153.14, 152.61, 142.96, 136.17, 135.41, 133.62, 132.45, 130.51, 129.99, 129.50, 127.57, 125.45, 122.74, 121.61, 27.13; MS (m/z): 375.0982 (M+ +2, 2.72%), 373.0982 (M+, 8.13%), 79.04 (100%, base peak); Anal. calcd for C22H16ClN3O (373.84): C, 70.68; H, 4.31; N, 11.24. Found: C, 70.51; H, 4.43; N, 11.45%.
O), 1592 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.20 (d, J = 8.3 Hz, 1H), 8.16 (t, J = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 3H), 8.03 (d, J = 8.5 Hz, 2H), 7.97 (d, J = 8.2 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 197.26, 153.14, 152.61, 142.96, 136.17, 135.41, 133.62, 132.45, 130.51, 129.99, 129.50, 127.57, 125.45, 122.74, 121.61, 27.13; MS (m/z): 375.0982 (M+ +2, 2.72%), 373.0982 (M+, 8.13%), 79.04 (100%, base peak); Anal. calcd for C22H16ClN3O (373.84): C, 70.68; H, 4.31; N, 11.24. Found: C, 70.51; H, 4.43; N, 11.45%.
4.1.2.1. N′-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)benzohydrazide (2a). Light brown crystals; yield, 70%; mp 260–2 °C; IRνmax (cm−1): 3431 (NH), 2917 (CH aromatic), 2851 (CH aliphatic), 1617 (C
O), 1537 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.21 (s, 1H), 8.26–7.51 (m, 17H), 2.51 (d, J = 34.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.12, 152.82, 152.29, 135.75, 135.46, 134.59, 133.31, 132.31, 132.27, 129.92, 129.45, 129.35, 129.20, 128.82, 128.24, 127.98, 127.42, 125.44, 121.71, 121.38, 27.05; MS (m/z): 493.1513 (M+ + 2, 16.41%), 491.1513 (M+, 49.68%), 485.28 (100%, base peak); Anal. calcd For C29H22ClN5O (491.98): C, 70.80; H, 4.51; N, 14.24. Found: C, 71.06; H, 4.59; N, 14.43%.
4.1.2.2. 2-Bromo-N′-(1-[4-{(4-[4-chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)benzohydrazide (2b). Yellowish brown crystals; yield, 74%; mp 265–7 °C; IRνmax (cm−1): 3300, 3430 (NH), 3060 (CH aromatic), 2917 (CH aliphatic), 1659 (C
O), 1505 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H), 8.82 (s, 1H), 8.76–8.74 (m, 2H), 8.16–8.12 (m, 2H), 8.10–8.02 (m, J = 6.9, 3H), 7.95 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 8.4 Hz, 1H), 7.81–7.73 (m, 2H), 7.70–7.64 (m, 2H), 7.58–7.38 (m, 2H), 2.36 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 196.90, 170.74, 164.82, 139.51, 138.43, 134.67, 132.38, 132.11, 132.06, 131.70, 130.80, 130.03, 129.85, 129.19, 129.12, 127.63, 126.94, 126.57, 123.90, 120.05, 26.93, 15.00, 14.06; MS (m/z): 571.0618 (M+ + 2, 63.72%), 569.0618 (M+, 64.90%), 419.23 (100%, base peak); Anal. calcd for C29H21BrClN5O (570.88): C, 61.01; H, 3.71; N, 12.27. Found: C, 60.89; H, 3.84; N, 12.50%.
4.1.2.3. 2-Chloro-N′-(1-[4-{(4-[4-chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)benzohydrazide (2c). Dark yellow powder; yield, 73%; mp 255–7 °C; IRνmax (cm−1): 3422 (NH), 3209 (CH aromatic), 2918 (CH aliphatic), 1658 (C
O), 1594 (CN); 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 9.04 (s, 1H), 8.13–8.05 (m, 2H), 7.98–7.92 (m, 3H), 7.80–7.69 (m, 5H), 7.61–7.45 (m, 6H), 2.32 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 170.09, 163.46, 154.82, 148.48, 137.32, 136.27, 135.08, 133.83, 132.18, 131.61, 130.78, 130.20, 130.07, 129.32, 129.27, 129.02, 127.74, 127.06, 126.89, 122.62, 122.39, 120.54, 15.03; MS (m/z): 527.1123 (M+ + 2, 16.06%), 525.1123 (M+, 23.74%), 313.06 (100%, base peak); Anal. calcd For C29H21Cl2N5O (526.42): C, 66.17; H, 4.02; N, 13.30. Found: C, 65.98; H, 4.18; N, 13.42%.
4.1.2.4. N′-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)-2-hydroxybenzohydrazide (2d). Orange powder; yield, 75%; mp 262–4 °C; IRνmax (cm−1): 3771 (OH), 3432 (NH), 2917 (CH aromatic), 2850 (CH aliphatic), 1650 (C
O), 1609 (CN); 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 11.31 (s, 1H), 9.53 (s, 1H), 8.73 (d, J = 8.4 Hz, 1H), 8.71–8.12 (m, 8H), 7.74 (d, J = 8.1 Hz, 2H), 7.66 (d, J = 8.2 Hz, 2H), 7.44 (t, J = 7.7 Hz, 1H), 7.06–6.99 (m, 2H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.47, 157.07, 153.22, 152.98, 152.30, 142.43, 136.01, 134.04, 133.77, 132.95, 132.02, 131.87, 130.97, 129.02, 127.33, 126.16, 123.29, 120.46, 120.14, 119.07, 118.39, 117.36, 14.20; MS (m/z): 509.1462 (M+ + 2, 5.77%), 507.1462 (M+, 9.27%), 314.22 (100%, base peak); Anal. calcd for C29H22ClN5O2 (507.98): C, 68.57; H, 4.37; N, 13.79. Found: C, 68.73; H, 4.50; N, 14.06%.
4.1.2.5. 4-Amino-N′-(1-[4-{(4-[4-chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)benzohydrazide (2e). Reddish yellow powder; yield, 65%; mp 250–2 °C; IRνmax (cm−1): 3726, 3674, 3446 (NH, NH2), 3047 (CH aromatic), 2958 (CH aliphatic), 1628 (C
O), 1536 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.16 (s, 1H), 8.22 (d, J = 7.9 Hz, 2H), 8.16–8.09 (m, 4H), 8.04 (m, 2H), 7.99–7.74 (m, 8H), 6.76 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.04, 166.48, 153.12, 152.66, 144.50, 132.25, 132.23, 130.07, 129.84, 129.39, 129.26, 127.69, 127.27, 125.37, 124.64, 123.50, 121.22, 120.53, 114.51, 113.48, 25.42; MS (m/z): 508.1622 (M+ + 2, 4.00%), 506.1622 (M+, 11.31%), 263.63 (100%, base peak); Anal. calcd for C29H23ClN6O (506.99): C, 68.70; H, 4.57; N, 16.58. Found: C, 68.94; H, 4.65; N, 16.80%.
4.1.2.6. N′-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)nicotinohydrazide (2f). Brown powder; yield, 72%; mp 260–2 °C; IRνmax (cm−1): 3429, 3248 (NH), 3090 (CH aromatic), 2918 (CH aliphatic), 1641 (C
O), 1536 (CN); 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.99 (s, 1H), 8.77 (d, 1H), 8.31 (d, 1H), 8.10–7.92 (m, 7H), 7.72–7.60 (m, 7H), 2.42 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 196.90, 164.87, 162.80, 156.43, 152.69, 152.42, 148.99, 136.58, 134.94, 133.65, 132.14, 129.24, 129.14, 127.76, 127.21, 126.79, 124.46, 124.09, 122.20, 120.40, 120.33, 15.18; MS (m/z): 494.1465 (M+ + 2, 0.77%), 492.1465 (M+, 2.07%), 298.12 (100%, base peak); Anal. calcd for C28H21ClN6O (492.97): C, 68.22; H, 4.29; N, 17.05. Found: C, 68.43; H, 4.56; N, 17.24%.
4.1.2.7. N′-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)isonicotinohydrazide (2g). Light brown powder, yield, 79%; mp 261–3 °C; IRνmax (cm−1): 3429, 3248 (NH), 3090 (CH aromatic), 2918 (CH aliphatic), 1641 (C
O), 1536 (CN); 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.17 (s, 1H), 8.83 (m, 2H), 8.18–8.10 (m, 2H), 7.96 (m, 6H), 7.74–7.70 (m, 6H), 2.44 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.01, 162.61, 157.42, 152.51, 150.63, 149.70, 148.35, 142.56, 140.01, 135.42, 134.94, 132.23, 129.87, 128.01, 127.79, 125.01, 123.07, 122.85, 120.94, 15.36; MS (m/z): 492.1465 (M+, 3.13%), 76.34 (100%, base peak); Anal. calcd for C28H21ClN6O (492.97): C, 68.45; H, 4.29; N, 17.05. Found: C, 68.43; H, 4.47; N, 17.29%.
4.1.3.1. 2-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)hydrazine-1-carbothioamide (3a). Light brown powder; yield, 70%; mp 203–5 °C; IRνmax (cm−1): 3858, 3739, 3440 (NH, NH2), 3055 (CH aromatic), 2918 (CH aliphatic), 1595 (C
N), 1402 (CS); 1H NMR (300 MHz, DMSO-d6) δ 11.11 (s, 1H), 9.04 (s, 1H), 8.20 (d, 2H), 8.07 (d, 2H), 8.00 (d, 2H), 7.82 (d, 2H), 7.74–7.69 (m, 4H), 6.55 (s, 2H), 2.23 (s, 3H); MS (m/z): 446.1080 (M+, 12.05%), 444.1080 (M+ - 2, 19.43%), 292.42 (100%, base peak); Anal. calcd for C23H19ClN6S (446.96): C, 61.81; H, 4.28; N, 18.80. Found: C, 61.97; H, 4.42; N, 18.95%.
4.1.3.2. 2-(1-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)-N-phenylhydrazine-1-carbothioamide (3b). Brown powder; yield, 66%; mp 243–5 °C; IRνmax (cm−1): 3424 (NH), 2918 (CH aromatic), 2850 (CH aliphatic), 1623 (C
N), 1406 (CS); 13C NMR (101 MHz, DMSO-d6) δ 166.02, 159.72, 152.87, 145.83, 142.18, 139.80, 138.80, 135.52, 134.50, 132.27, 131.69, 129.27, 128.32, 127.82, 126.88, 124.83, 122.59, 120.85, 115.26, 113.45, 12.24; MS (m/z): 522.1393 (M+, 28.63%), 422.47 (100%, base peak); Anal. calcd for C29H23ClN6S (523.06): C, 66.59; H, 4.43; N, 16.07. Found: C, 66.38; H, 4.51; N, 16.23%.
4.1.3.3. Methyl 2-(1-[4-{(4-[4-chlorophenyl]phthalazin-1-yl)amino}phenyl]ethylidene)hydrazine-1-carbodithioate (3c). Yellow powder; yield, 70%; mp 205–7 °C; IRνmax (cm−1): 3435 (NH), 3050 (CH aromatic), 2917 (CH aliphatic), 1591 (C
N), 1404 (CS); 1H NMR (300 MHz, DMSO-d6) δ 10.33 (s, 1H), 8.86 (s, 1H), 8.23–7.97 (m, 6H), 7.78–7.62 (m, 6H), 2.58 (s, 3H), 2.49 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.02, 157.74, 153.21, 152.62, 144.53, 135.12, 134.21, 133.90, 132.23, 129.88, 127.44, 126.84, 125.27, 124.38, 123.75, 121.14, 27.00, 13.69; MS (m/z): 477.0849 (M+, 12.81%), 368.38 (100%, base peak); Anal. calcd for C24H20ClN5S2 (478.03): C, 60.30; H, 4.22; N, 14.65. Found: C, 60.49; H, 4.37; N, 14.88%.
4.1.4.1. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)-3-phenylurea (4a). White solid; yield, 72%; mp 261–3 °C; IRνmax (cm−1): 3440, 3304 (NH), 3078 (CH aromatic), 2919 (CH aliphatic), 1642 (C
O), 1600 (CN), 1211 (C–O); 1H NMR (300 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.15 (s, 1H), 8.53–8.45 (m, 1H), 8.18–8.01 (m, 3H), 7.96 (d, J = 7.5 Hz, 2H), 7.78–7.68 (m, 4H), 7.64 (dd, J = 8.6, 3.6 Hz, 4H), 7.27 (q, J = 6.3 Hz, 3H); MS (m/z): 466.1197 (M+, 12.38), 301.62 (100%, base peak); Anal. calcd for C27H19ClN4O2 (466.93): C, 69.45; H, 4.10; N, 12.00. Found: C, 69.68; H, 4.21; N, 12.17%.
4.1.4.2. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)-3-phenylthiourea (4b). White solid; yield, 75%; mp 251–3 °C; IRνmax (cm−1): 3421 (NH), 3068 (CH aromatic), 2919 (CH aliphatic), 1631 (C
N), 1403 (CS), 1205 (C–O); MS (m/z): 482.0968 (M+, 8.65), 312.46 (100%, base peak); Anal. calcd for C27H19ClN4OS (482.99): C, 67.14; H, 3.97; N, 11.60. Found: C, 67.50; H, 4.13; N, 11.68%.
4.1.5.1. N-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)acetamide (5a). White solid; yield, 70%; mp 190–2 °C; IRνmax (cm−1): 3316 (NH), 3067 (CH aromatic), 2970, 2919 (CH aliphatic), 1664 (C
O), 1606 (CN), 1379 (CS), 1201 (C–O); 1H NMR (300 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.14–7.93 (m, 4H), 7.72–7.63 (m, 6H), 7.31–7.28 (d, J = 8.7 Hz, 2H), 2.07 (s, 3H); MS (m/z): 389.0931 (M+, 18.67), 264.11 (100%, base peak); Anal. calcd for C22H16ClN3O2 (389.84): C, 67.78; H, 4.14; N, 10.78. Found: C, 67.61; H, 4.25; N, 11.06%.
4.1.5.2. N-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)benzamide (5b). Grey powder; yield, 68%; mp 245–7 °C; IRνmax (cm−1): 3440 (NH), 3058 (CH aromatic), 2917 (CH aliphatic), 1650 (C
O), 1578 (CN), 1202 (C–O); MS (m/z): 451.1088 (M+, 5.43), 120.14 (100%, base peak); Anal. calcd for C27H18ClN3O2 (451.91): C, 71.76; H, 4.01; N, 9.30. Found: C, 71.53; H, 4.20; N, 9.47%.
4.1.5.3. N-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)-3-nitrobenzamide (5c). Pale white powder; yield, 70%; mp 242–4 °C; IRνmax (cm−1): 3427 (NH), 3073 (CH aromatic), 2917 (CH aliphatic), 1647 (C
O), 1527 (CN), 1380 (NO2), 1202 (C–O); 1H NMR (300 MHz, DMSO-d6) δ 8.84 (d, J = 2.5 Hz, 1H), 8.53–8.41 (m, 3H), 8.11 (dt, J = 17.3, 7.2 Hz, 2H), 7.97 (d, J = 7.9 Hz, 1H), 7.87 (dd, J = 12.3, 8.0 Hz, 3H), 7.72 (d, J = 8.1 Hz, 3H), 7.66 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H); MS (m/z): 496.0938 (M+, 3.14), 41.42 (100%, base peak); Anal. calcd for C27H17ClN4O4 (496.91): C, 65.26; H, 3.45; N, 11.28. Found: C, 65.50; H, 3.62; N, 11.44%.
4.1.5.4. N-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}oxy]phenyl)cinnamamide (5d). Light brown solid; yield, 78%; mp 248–9 °C; IRνmax (cm−1): 3423 (NH), 3060 (CH aromatic), 2965, 2918 (CH aliphatic), 1653 (C
O), 1618 (CC), 1572 (CN), 1202 (C–O); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.50 (d, J = 7.8 Hz, 1H), 8.15–8.05 (m, 2H), 7.96 (d, J = 8.0 Hz, 1H), 7.84–7.59 (m, 8H), 7.46–7.35 (m, 6H), 6.87 (d, J = 15.8 Hz, 1H). MS (m/z): 477.1244 (M+, 28.93), 468.93 (100%, base peak); Anal. calcd for C29H20ClN3O2 (477.95): C, 72.88; H, 4.22; N, 8.79. Found: C, 73.05; H, 4.39; N, 9.07%.
4.2. Biological testing
:1) and 0.1 nM of VEGFR-2 (Millipore, UK). Prior to catalytic initiation with ATP, the tested compounds at final concentrations ranging from 0 to 300 μg ml−1 and enzyme were incubated at room temperature for 5 min. The reactions were quenched by the addition of 25 μl of 100 mM EDTA, 10 μg ml−1 Alpha Screen streptavidin donor beads and 10 μg ml−1 acceptor beads in 62.5 mM HEPES pH 7.4, 250 mM NaCl, and 0.1% BSA. The plate was left to incubate in the darkness overnight and then analyzed using an ELISA reader (PerkinElmer, USA). Control reactions were conducted with wells containing the substrate and enzyme without compounds, while wells containing biotinylated poly-GluTyr (4:1) and the enzyme without ATP were used as basal controls. The percent inhibition was calculated by comparing the compounds treated to control incubations. The concentration of the test compound causing 50% inhibition (IC50) was determined from the concentration–inhibition response curve (triplicate determinations). Sorafenib (Sigma-Aldrich, USA) was used as a standard VEGFR-2 inhibitor for comparison.4.3. Docking studies
VEGFR-2 (PDB ID 4ASD)40 was used in the Molsoft program to carry out all docking studies. Each experiment used the VEGFR-2 receptor complexes with the Ligand downloaded from the Brookhaven Protein Databank. The compounds were drawn as a 3D structure and their energies were minimized. The ligand was extracted from the binding site and the compounds discussed herein were docked into the active site.4.4. Molecular dynamics simulation
The highly active derivatives 2g, 3a, 4a and 5b were simulated using molecular dynamics (MD) in VEGFR-2 with the help of GAFF2.40,41 The molecular dynamics utilizes Amber's MM/GBSA.py script and the trajectory, the receptor-ligand binding energy was calculated.40 The force field AMBERff14SB for the protein and ligand force fields were produced.434.5. ADMET profile
The in silico ADMET profile of the highly active derivatives 2g and 4a was predicted using pkCSM descriptor algorithm procedures.44Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no interest to declare.References
- A. A. El-Helby, R. R. A. Ayyad, H. Sakr, K. El-Adl, M. M. Ali and F. Khedr, Arch. Pharm., 2017, 350(12) DOI:10.1002/ardp.201700240.
- A. A. El-Helby, H. Sakr, R. R. A. Ayyad, K. El-Adl, M. M. Ali and F. Khedr, Anticancer Agents Med. Chem., 2018, 18(8), 1184–1196, DOI:10.2174/1871520618666180412123833.
- H. S. Abulkhair, A. Turky, A. Ghiaty, H. E. A. Ahmed and A. H. Bayoumi, Bioorg. Chem., 2020, 100, 103899, DOI:10.1016/j.bioorg.2020.103899.
- A. Turky, A. H. Bayoumi, A. Ghiaty, A. S. El-Azab, A. A.-M. Abdel-Aziz and H. S. Abulkhair, Bioorg. Chem., 2020, 104019, DOI:10.1016/j.bioorg.2020.104019.
- W. M. Eldehna, S. M. Abou-Seri, A. M. El Kerdawy, R. R. Ayyad, H. A. Ghabbour, M. M. Ali and D. A. Abou El Ella, Eur. J. Med. Chem., 2016, 113, 50–62, DOI:10.1016/j.ejmech.2016.02.029.
- T. L. Bush, M. Payton and S. Heller, et al., Mol. Cancer Ther., 2013, 12(11), 2356–2366, DOI:10.1158/1535-7163.MCT-12-1178.
- M. Payton, T. L. Bush, G. Chung, B. Ziegler, P. Eden, P. McElroy, S. Ross, V. J. Cee, H. L. Deak, B. L. Hodous, H. N. Nguyen, P. R. Olivieri, K. Romero, L. B. Schenkel, A. Bak, M. Stanton, I. Dussault, V. F. Patel, S. Geuns-Meyer, R. Radinsky and R. L. Kendall, Cancer Res., 2010, 70(23), 9846–9854 CrossRef CAS PubMed.
- M. A. J. Duncton, E. L. P. Chekler, R. Katoch-Rouse, D. Sherman, W. C. Wong, L. M. Smith, J. K. Kawakami, A. S. Kiselyov, D. L. Milligan, C. Balagtas, Y. R. Hadari, Y. Wang, S. N. Patel, R. L. Rolster, J. R. Tonra, D. Surguladze, S. Mitelman, P. Kussie, P. Bohlen and J. F. Doody, Bioorg. Med. Chem., 2009, 17(2), 731–740 CrossRef CAS PubMed.
- M. A. J. Duncton, E. L. Piatnitski, R. Katoch-Rouse, L. M. Smith, A. S. Kiselyov, D. L. Milligan, C. Balagtas, W. C. Wong, J. Kawakamia and J. F. Doody, Bioorg. Med. Chem. Lett., 2006, 16(6), 1579–1581 CrossRef CAS PubMed.
- A. S. Kiselyov, V. V. Semenov and D. Milligan, Chem. Biol. Drug Des., 2006, 68, 308–313 CrossRef CAS PubMed.
- E. L. Piatnitski, M. A. J. Duncton, A. S. Kiselyov, R. Katoch-Rouse, D. Sherman, D. L. Milligan, C. Balagtas, W. C. Wong, J. Kawakamia and J. F. Doody, Bioorg. Med. Chem. Lett., 2005, 15(21), 4696–4698 CrossRef CAS PubMed.
- D.-Q. Xue, X.-Y. Zhang, C.-J. Wang, L.-Y. Ma, N. Zhu, P. He, K.-P. Shao, P.-J. Chen, Y.-F. Gu, X.-S. Zhang, C.-F. Wang, C.-H. Ji, Q.-R. Zhang and H.-M. Liu, Eur. J. Med. Chem., 2014, 85, 235–244 CrossRef CAS PubMed.
- V. J. Cee, L. B. Schenkel, B. L. Hodous, H. L. Deak, H. N. Nguyen, P. R. Olivieri, K. Romero, A. Bak, X. Be, S. Bellon, T. L. Bush, A. C. Cheng, G. Chung, S. Coats, P. M. Eden, K. Hanestad, P. L. Gallant, Y. Gu, X. Huang, R. L. Kendall, M. J. Lin, M. J. Morrison, V. F. Patel, R. Radinsky, P. E. Rose, S. Ross, J. Sun, J. Tang, H. Zhao, M. Payton and S. D. Geuns-Meyer, J. Med. Chem., 2010, 53, 6368–6377 CrossRef CAS PubMed.
- G. Bold, J. Frei, P. Traxler, K.-H. Altmann, H. Mett, D. R. Stover and J. M. Wood, EP Pat., 98/00764, 1998 Search PubMed.
- G. Bold, K.-H. Altmann, J. Frei, M. Lang, P. W. Manley, P. Traxler, B. Wietfeld, J. Brüggen, E. Buchdunger, R. Cozens, S. Ferrari, P. Furet, F. Hofmann, G. Martiny-Baron, J. Mestan, J. Rosel, M. Sills, D. Stover, F. Acemoglu, E. Boss, € R. Emmenegger, L. Lasser, E. Masso, R. Roth, C. Schlachter, W. Vetterli, D. Wyss and € J. M. Wood, J. Med. Chem., 2000, 43, 2310e2323 Search PubMed.
- J. Dumas and J. A. Dixon, Expert Opin. Ther. Pat., 2005, 15, 647e658 Search PubMed.
- A. S. Kiselyov, M. Semenova, V. V. Semenov and E. L. Piatnitski, Chem. Biol. Drug Des., 2006, 68, 250–255 CrossRef CAS PubMed.
- J. C. Tille, J. Wood, S. J. Mandriota, C. Schnell, S. Ferrari, J. Mestan, Z. Zhu, L. Witte and M. S. Pepper, J. Pharmacol. Exp. Ther., 2001, 299, 1073e1085 Search PubMed.
- M. A. Abdullaziz, H. T. Abdel-Mohsen, A. M. El Kerdawy, F. A. F. Ragab, M. M. Ali, S. M. Abu-Bakr, A. S. Girgis and H. I. El Diwani, Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors, Eur. J. Med. Chem., 2017, 136, 315–329 CrossRef CAS PubMed.
- M. A. Abdelgawad, K. El-Adl, S. S. El-Hddad, M. M. Elhady, N. M. Saleh, M. M. Khalifa, F. Khedr, M. Alswah, A. A. Nayl and M. M. Ghoneim, Design, molecular docking, synthesis, anticancer and anti-hyperglycemic assessments of thiazolidine-2, 4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists, Pharmaceuticals, 2022, 15, 226 CrossRef CAS PubMed.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Phthalazine-based VEGFR-2 inhibitors: Rationale, design, synthesis, in silico, ADMET profile, docking, and anticancer evaluations, Arch. Pharm., 2021, e2100201, DOI:10.1002/ardp.202100201.
- A. K. B. Aljohani, K. El-Adl, B. Almohaywi, O. M. Alatawi, M. Alsulaimany, A. El-morsy, S. A. Almadani, H. Y. Alharbi, M. S. Aljohani, F. A. Abdulhaleem M, H. E. M. Osman and S. Mohamady, RSC Adv., 2024, 14, 7964–7980 RSC.
- M. M. Ghorab, A. M. Soliman, K. El-Adl and N. S. Hanafy, New quinazoline sulfonamide derivatives as potential anticancer agents: Identifying a promising hit with dual EGFR/VEGFR-2 inhibitory and radiosensitizing activity, Bioorg. Chem., 2023, 140, 106791, DOI:10.1016/j.bioorg.2023.106791.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218, DOI:10.1016/j.bioorg.2020.104218.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- A. E. Abdallah, I. H. Eissa, A. B. M. Mehany, H. Sakr, A. Atwa, K. El-Adl and M. A. El-Zahabi, Immunomodulatory quinazoline-based thalidomide analogs: Design, synthesis, apoptosis and anticancer evaluations, J. Mol. Struct., 2023, 1281, 135164, DOI:10.1016/j.molstruc.2023.135164.
- N. S. Hanafy, N. A. A. M. Aziz, S. S. A. El-Hddad, M. A. Abdelgawad, M. M. Ghoneim, A. F. Dawood, S. Mohamady, K. El-Adl and S. Ahmed, Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment, Arch. Pharm., 2023, e2300137, DOI:10.1002/ardp.202300137.
- D. Adel, K. El-Adl, T. Nasr, T. M. Sakr and W. Zaghary, Pyrazolo[3,4-d]pyrimidine derivatives as EGFRT790M and VEGFR-2 dual TK inhibitors: Design, synthesis, molecular docking, ADMET profile and anticancer evaluations, J. Mol. Struct., 2023, 1291, 136047, DOI:10.1016/j.molstruc.2023.136047.
- H. Elkady, K. El-Adl, H. Sakr, A. S. Abdelraheem, S. I. Eissa and M. A. El-Zahabi, Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: Design, synthesis, anticancer evaluation, and in silico ADMET analysis, Arch. Pharm., 2023, e2300097, DOI:10.1002/ardp.202300097.
- M. A. El-Zahabi, H. Elkady, H. Sakr, A. S. Abdelraheem, S. I. Eissa and K. El-Adl, Design, synthesis, anticancer evaluation, in silico docking and ADMET analysis of novel indole-based thalidomide analogs as promising immunomodulatory agents, J. Biomol. Struct. Dyn., 2023, 1–18, DOI:10.1080/07391102.2023.2187217.
- M. Alsulaimany, K. El-Adl, A. K. B. Aljohani, H. Y. Alharbi, O. M. Alatawi, M. S. Aljohani, A. El-Morsy, S. A. Almadani, A. A. Alsimaree, S. A. Salama, D. E. Keshek and A. A. Mohamed, Design, synthesis, docking, ADMET and anticancer evaluations of N-alkyl substituted iodoquinazoline derivatives as dual VEGFR-2 and EGFR inhibitors, RSC Adv., 2023, 13(51), 36301–36321, 10.1039/d3ra07700d.
- K. E. Anwer, S. S. A. El-Hddad, N. E. A. Abd El-Sattar, A. M. El-Morsy, F. Khedr, S. Mohamady, D. E. G. Keshek, S. A. Salama, K. El-Adl and N. S. Hanafy, Five and six membered heterocyclic rings endowed with azobenzene as dual EGFRT790M and VEGFR-2 inhibitors: design, synthesis, in silico ADMET profile, molecular docking, dynamic simulation and anticancer evaluations, RSC Adv., 2023, 13, 35321–35338 RSC , https://api.semanticscholar.org/CorpusID:265657522.
- M. S. A. El-Gaby, M. A. M. Abdel Reheim, Z. S. M. Akrim, B. H. Naguib, N. M. Saleh, A. A. A. M. El-Adasy, K. El-Adl and S. Mohamady, Drug Dev. Res., 2024, 85(1) DOI:10.1002/ddr.22143.
- A. A. Mohamed, S. S. A. El-Hddad, A. K. B. Aljohani, F. Khedr, O. M. Alatawi, D. E. Keshek, S. Ahmed, M. Alsulaimany, S. A. Almadani, K. El-Adl and N. S. Hanafy, Iodoquinazoline-derived VEGFR-2 and EGFRT790M dual inhibitors: Design, synthesis, molecular docking and anticancer evaluations, Bioorg. Chem., 2024, 143, 107062, DOI:10.1016/j.bioorg.2023.107062.
- T. Mosmann, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- D. A. Scudiero, R. H. Shoemaker, K. D. Paull, A. Monks, S. Tierney, T. H. Nofziger, M. J. Currens, D. Seniff and M. R. Boyd, Cancer Res., 1988, 48(17), 4827–4833 CAS.
- F. M. Freimoser, C. A. Jakob, M. Aebi and U. Tuor, Appl. Environ. Microbiol., 1999, 65(8), 3727–3729 CrossRef CAS PubMed.
- S. M. Abou-Seri, W. M. Eldehna, M. M. Ali and D. A. A. El Ella, Eur. J. Med. Chem., 2016, 107, 165–179 CrossRef CAS PubMed.
- S. A. H. El-Feky, H. A. Abd El-Fattah, N. A. Osman, M. Imran and M. N. Zedan, J. Chem. Pharm. Res., 2015, 7(7), 1154–1166 CAS.
- B. R. Miller III, T. D. J. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- S. El-Hddad, M. Sobhy, A. Ayoub and K. El-Adl, In silico molecular docking, dynamics simulation and repurposing of some VEGFR-2 inhibitors based on the SARS-CoV-2-main-protease inhibitor N3, J. Biomol. Struct. Dyn., 2022, 1–15, DOI:10.1080/07391102.2022.2148000.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58, 4066, DOI:10.1021/acs.jmedchem.5b00104.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- A. Beig, R. Agbaria and A. Dahan, Oral delivery of lipophilic drugs: the tradeoff between solubility increase and permeability decrease when using cyclodextrin-based formulations, PLoS One, 2013, 8, e68237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03459g |
| This journal is © The Royal Society of Chemistry 2024 |