
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium(II) complexes bearing mesoionic carbene ligands: catalytic application in domino Sonogashira coupling/cyclization reactions for one-pot synthesis of benzofuran and indole derivatives†
Om Prakash Joshia,
Ramalingam Thirumoorthi a,
Ram T. Pardasania,
Sriparna Ray*b and
Chandrakanta Dash*a
a,
Ram T. Pardasania,
Sriparna Ray*b and
Chandrakanta Dash*a
aDepartment of Chemistry, School of Chemical Sciences and Pharmacy, Central University of Rajasthan, Bandarsindri, Ajmer 305817, Rajasthan, India. E-mail: ckdash@curaj.ac.in
bCatalytic Applications Laboratory, Department of Chemistry, School of Basic Sciences, Faculty of Science, Manipal University Jaipur, Dehmi Kalan, Jaipur 303007, Rajasthan, India. E-mail: sriparna.ray@gmail.com
First published on 27th August 2024
Abstract
Bioactive heterocycles such as benzofuran and indole derivatives were synthesized from commercially available 2-iodoarenes and alkynes via domino Sonogashira coupling followed by cyclization reaction using well-defined palladium PEPPSI (Pyridine Enhanced Precatalyst Preparation Stabilization and Initiation) complexes (2a and 2b). These reactions tolerate a variety of 2-iodoarenes and diversely substituted terminal alkynes, resulting in the corresponding product in moderate to good yields in an open-air atmosphere. In particular, two palladium(II) PEPPSI complexes 2a and 2b were synthesized in good yields from the reaction of corresponding 1,2,3-triazol-5-ylidene (MIC: mesoionic carbene), PdCl2, KI, and K2CO3 in pyridine at 110 °C and structurally characterized by various spectroscopic techniques including NMR spectroscopy, IR spectroscopy, HRMS and elemental analysis studies. Complex 2b is also characterized by X-ray crystallography.
Introduction
Benzofused five-membered heterocyclic scaffolds such as benzofuran and indole derivatives are extensively spread as a common structural skeleton in many biologically active compounds, natural products, and pharmaceuticals.1–6 Over the years, considerable effort has been devoted to synthesizing these heterocycles.6–10 Synthesis of benzofuran and indole derivatives via a domino approach has received more attention than the traditional multistep approach due to its high atom economy, environmentally benign nature, and operational simplicity.1,8,11–15 Along these lines, palladium-catalyzed domino Sonogashira coupling/cyclization reaction has emerged as an efficient method for the construction of the benzofuran and indole framework.16–19N-heterocyclic carbenes (NHCs) have been used as spectator ligands in organometallic chemistry for the past three decades.20–23 Their strong metal-NHC bond-forming tendency and high steric demands have made them much more popular and a good alternative to conventional phosphine ligands.20,24 An interesting counterpart of normal NHCs, 1,2,3-triazol-5-ylidene mesoionic carbene (1,2,3-triazolylidene) was first reported by Albrecht and co-workers in 2008.25 In these MICs, only one adjacent electronegative nitrogen atom at the donor site increased the sigma donor ability compared to NHCs.25 Triazolylidenes can be easily synthesized via copper-catalyzed Click reaction followed by an alkylation reaction.26–28 Several triazolylidene-based transition metal complexes are reported as catalysts in various organic transformations.27,29–38 PEPPSI (Pyridine Enhanced Precatalyst Preparation Stabilization and Initiation) themed39 palladium complexes bearing 1,2,3-triazolylidene were reported by Albrecht et al. in 2012.40
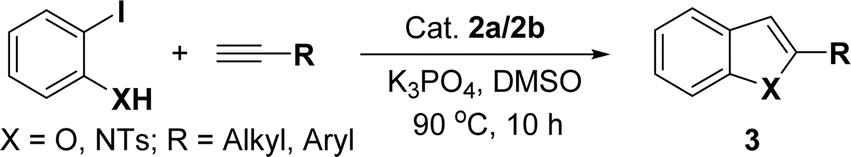
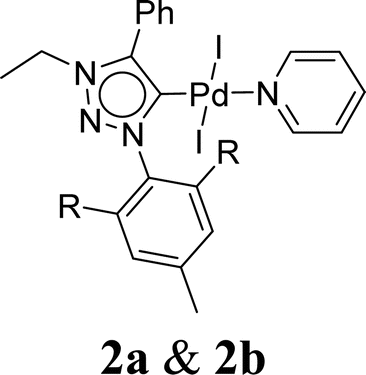
Very few well-defined palladium complexes bearing N-heterocyclic carbene ligands are used as catalysts in domino Sonogashira coupling/cyclization reactions for one-pot synthesis of benzofuran and indole derivatives.41–43 Mata and coworkers reported the triazole based N-heterocyclic carbene–palladium complex as catalyst for the synthesis of benzofuran derivatives.41 Ghosh and coworkers revealed the dipalladium complex stabilized by N-heterocyclic carbene ligand for benzofuran synthesis.42 Our group also reported various palladium complexes as catalysts for the synthesis of benzofurans via domino Sonogashira coupling and cyclization reactions.43,44 Bera and coworkers developed the annellated mesoionic carbene ligand stabilized palladium complexes and explored their catalytic activity for the synthesis of benzofuran, indole, isocoumarin, and isoquinolone derivatives using domino approach.7 Herein, we reported the synthesis of 1,2,3-triazol-5-ylidene mesoionic carbene-based palladium PEPPSI complexes, 2a and 2b (Fig. 1) and further utilized these catalysts for the synthesis of biologically active benzofuran and indole derivatives via domino Sonogashira coupling/cyclization reaction under copper and amine free condition.
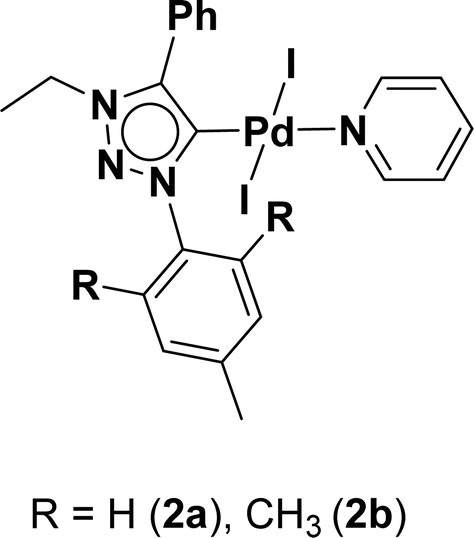 | ||
| Fig. 1 1,2,3-Triazol-5-ylidene based palladium PEPPSI complexes. | ||
Results and discussion
Synthesis and characterization of palladium PEPPSI complexes
The 1,2,3-triazolium triflate ligands 1a and 1b were synthesized by AgOTf-assisted N-ethylation of corresponding 1,2,3-triazole40,45 using ethyl iodide as ethylating reagent (Scheme 1). Formation of triazolium ligands was indicated by the appearance of a quartet peak of two protons which corresponds to triazolium methylene (trz–CH2–) peak in 1H NMR spectrum at 4.64 ppm (1a) and 4.76 ppm (1b), respectively. The 1,2,3-triazolylidene based palladium(II) complexes (2a and 2b) were synthesized with good yield (2a = 74%, 2b = 83%) from the reaction of corresponding triazolium triflate, PdCl2, K2CO3, and KI in pyridine at 110 °C in 5 days using a similar method reported earlier.35,46 The disappearance of a singlet of triazolium proton and the presence of a set of additional pyridine peaks in the aromatic region suggest the formation of palladium PEPPSI complexes. The 13C{1H} NMR spectrum of these complexes revealed Pd–Ccarbene peak at around 134.4 ppm for 2a and 135.6 ppm for 2b, respectively. After purification, both the complexes were obtained as pale-yellow solid.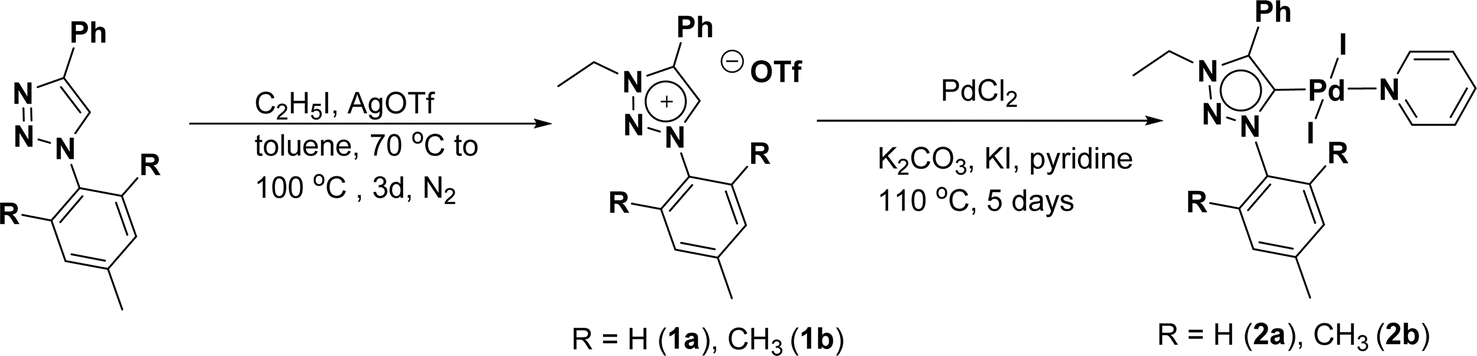 | ||
| Scheme 1 Synthesis of palladium-PEPPSI complex 2a and 2b. | ||
X-ray structure
A suitable single crystal for X-ray diffraction analysis was obtained for complex 2b by slow evaporation of solvent from a saturated solution of complex 2b in acetonitrile at room temperature. The formation of palladium PEPPSI complex 2b was confirmed by molecular structure analysis, and the geometry of the central palladium metal is found to be slightly distorted square planar in which 1,2,3-triazolylidene ligand located trans to pyridine and remaining coordination sites are occupied by iodido donors (Fig. 2). The average Pd–Ccarbene and Pd–Npyridine bond distances are 1.975(5) and 2.111(5), respectively, which are similar to analogous 1,2,3-trizolylidene ligand-based PEPPSI complexes previously reported in the literature.47–49 The average Pd–I bond lengths were also observed as close to the other reported PEPPSI type complexes containing iodide ligands.49 The average Ccarbene–Pd–Npyridine and I–Pd–I bond angles are 174.4 (2)° and 175.96(2)°, respectively, which are closer to the expected 180° for a square planar geometry.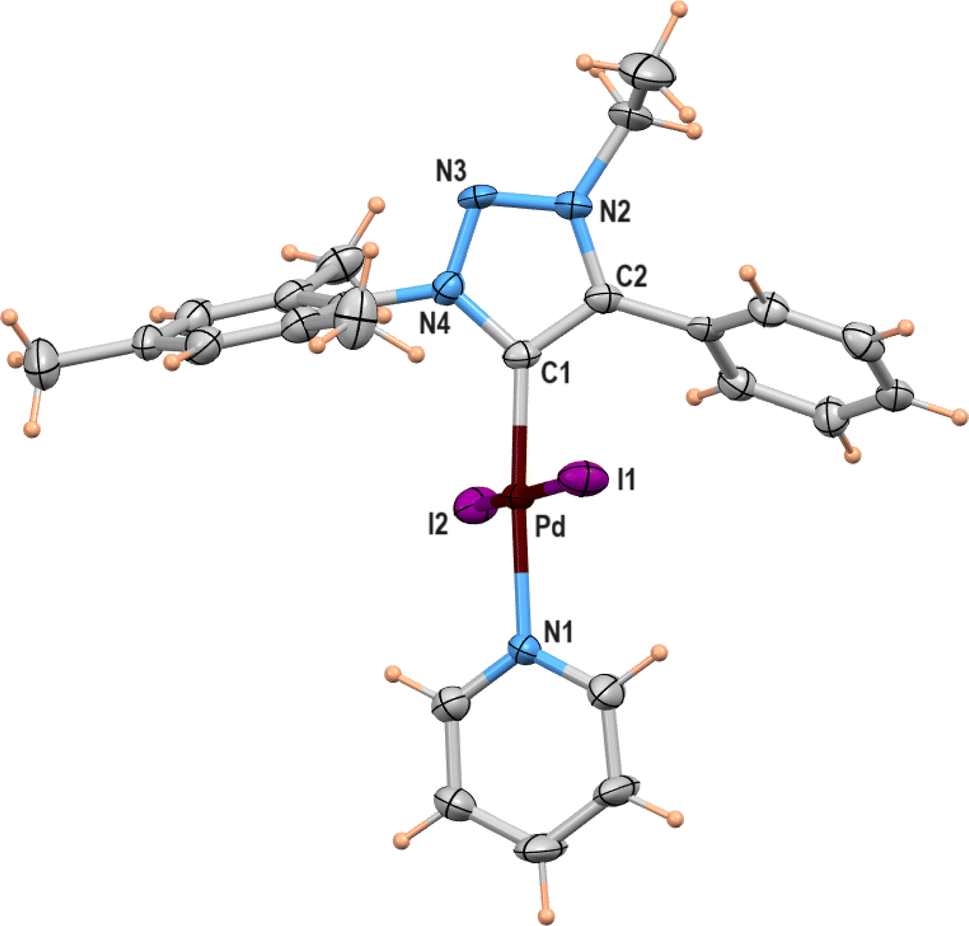 | ||
| Fig. 2 Molecular structure showing [LPdI2(pyridine)] (2b), ellipsoids are shown at 50% probability level. Only one of the molecules (out of two) present in the asymmetric unit is shown. Selected bond lengths (Å) and angles (deg): Pd(1)–N(1) 2.116(5), Pd(1)–C(1) 1.970(5), Pd(1)–I(1) 2.6208(6), Pd(1)–I(2) 2.5973(6), N(1)–Pd(1)–I(1) 92.12(14), N(1)–Pd(1)–I(2) 90.49(14), C(1)–Pd(1)–I(1) 87.90(17), C(1)–Pd(1)–I(2) 88.99(17); Pd(2)–N(5) 2.107(5), Pd(2)–C(25) 1.980(6), Pd(2)–I(3) 2.5801(6), Pd(2)–I(4) 2.6281(6), N(5)–Pd(2)–I(3) 90.93(14), N(5)–Pd(2)–I(4) 88.96(14), C(25)–Pd(2)–I(3) 89.24(16), C(25)–Pd(2)–I(4) 90.91(16). | ||
Domino Sonogashira coupling/cyclization reactions
Based on our previous study,43,44 2-iodophenol and phenylacetylene were chosen as model substrates for domino Sonogashira coupling/cyclization reactions. In the optimization experiments, the initial reaction condition included 2-iodophenol (0.50 mmol), phenylacetylene (0.60 mmol), K2CO3 (1 mmol), and catalyst 2a (2 mol%) in DMSO at 110 °C in air. Under this reaction condition, the desired benzofuran was obtained in 81% yield (entry 1, Table 1). Various other bases (entries 2–4, Table 1) and solvents (entries 5 and 6, Table 1) were screened for this reaction, with the result that K3PO4 base and DMSO solvent were found to be optimum for this reaction. Reducing the reaction time reduced the product yield to 65% (entry 7, Table 1). There was a negligible decrease in the product yield (83%) at 90 °C (entry 8, Table 1), but it decreased significantly to 41% at 60 °C (entry 9, Table 1). Reducing the catalyst loading to 1 mol% significantly decreases the product yield to 63% (entry 10, Table 1). The reaction with palladium precursor, i.e., Pd(CH3CN)2Cl2 gave a yield of 36% (entry 11, Table 1). In continuation, poor yield of the product was obtained under base-free conditions (entry 12, Table 1), while no reaction was observed in absence of a palladium catalyst (entry 13, Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base | Solvent | Time (h) | Temp. (°C) | % Yieldb |
| a Reaction conditions: 2-iodophenol (0.50 mmol), phenylacetylene (0.60 mmol), 2.0 mol% of catalyst, base (1.00 mmol), and solvent (2 mL).b Isolated yield.c Without base.d Without catalyst. nr = no reaction. | ||||||
| 1 | 2a (2.0) | K2CO3 | DMSO | 10 | 110 | 81 |
| 2 | 2a (2.0) | K3PO4 | DMSO | 10 | 110 | 84 |
| 3 | 2a (2.0) | KOtBu | DMSO | 10 | 110 | 51 |
| 4 | 2a (2.0) | KOH | DMSO | 10 | 110 | 42 |
| 5 | 2a (2.0) | K3PO4 | Dioxane | 10 | 110 | 71 |
| 6 | 2a (2.0) | K3PO4 | Toluene | 10 | 110 | 37 |
| 7 | 2a (2.0) | K3PO4 | DMSO | 4 | 110 | 65 |
| 8 | 2a (2.0) | K3PO4 | DMSO | 10 | 90 | 83 |
| 9 | 2a (2.0) | K3PO4 | DMSO | 10 | 60 | 41 |
| 10 | 2a (1.0) | K3PO4 | DMSO | 10 | 90 | 63 |
| 11 | Pd(CH3CN)2Cl2 (2.0) | K3PO4 | DMSO | 10 | 90 | 36 |
| 12 | 2a (2.0) | — | DMSO | 10 | 90 | 10c |
| 13 | — | K3PO4 | DMSO | 10 | 90 | nrd |
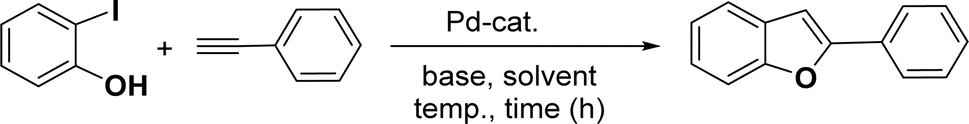
With the optimized reaction condition in hand (entry 8), various substituted benzofuran and indole derivatives were synthesized in moderate to good yields from commercially available terminal alkynes and 2-iodoarenes (Table 2). In comparison, benzofuran derivatives were obtained a higher yield (entries 1 to 7, Table 2) with respect to indole derivatives (entries 8 to 14, Table 2). Furthermore, there is not much difference in the catalytic activity of the catalysts 2a and 2b under the same reaction conditions was observed.
|
|||||
|---|---|---|---|---|---|
| Entry | 2-Iodoarenes | Alkynes | Product | Yieldb (%) | |
| 2a | 2b | ||||
| a Reaction conditions: 2-iodoarene derivative (0.50 mmol), alkyne (0.60 mmol), 2.0 mol% of 2a or 2b, K3PO4 (1.00 mmol), DMSO (2 mL), 90 °C, 10 h.b Isolated yield. | |||||
| 1 | 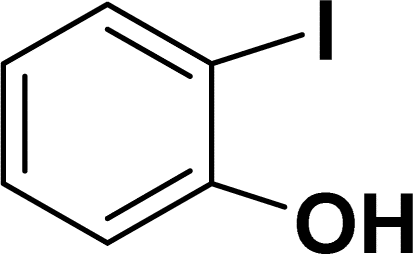 |
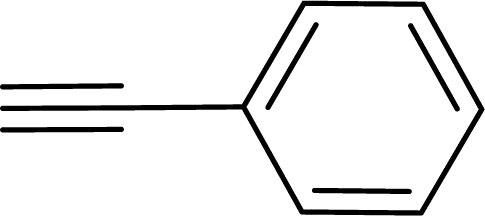 |
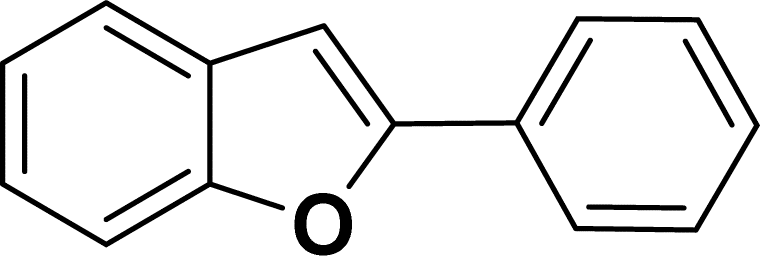 |
83 | 87 |
| 2 | 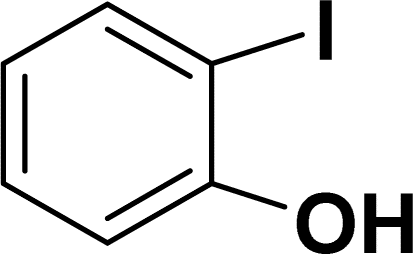 |
 |
 |
80 | 85 |
| 3 |  |
 |
 |
68 | 71 |
| 4 |  |
 |
 |
76 | 83 |
| 5 |  |
 |
 |
71 | 77 |
| 6 |  |
 |
 |
64 | 65 |
| 7 |  |
 |
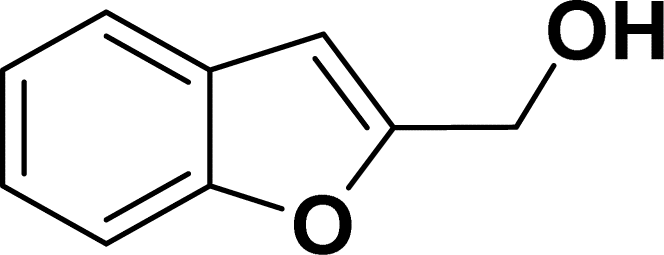 |
63 | 65 |
| 8 | 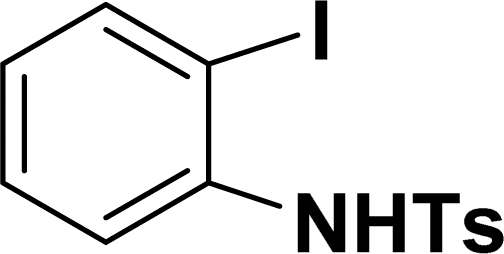 |
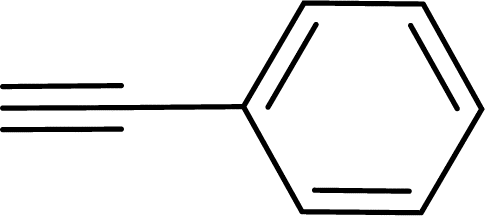 |
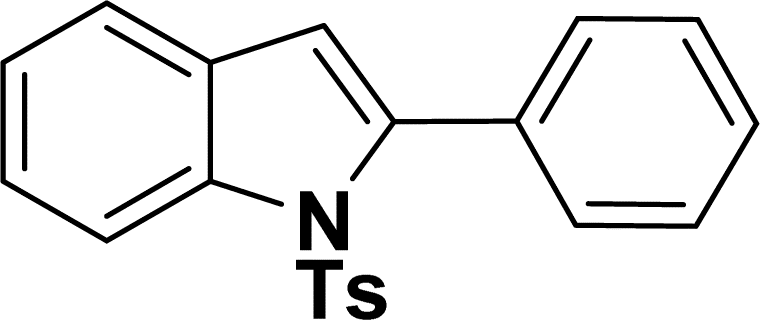 |
65 | 57 |
| 9 | 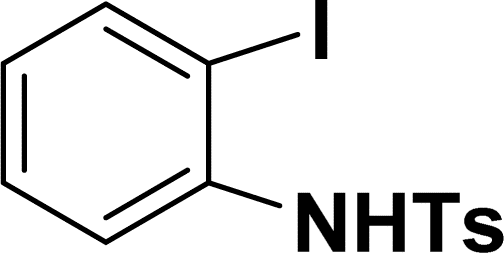 |
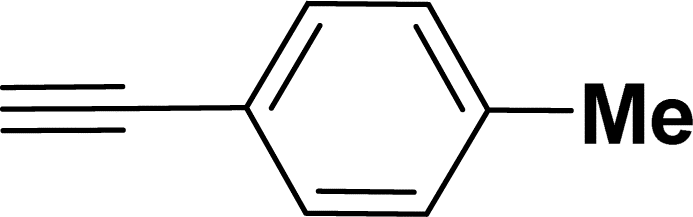 |
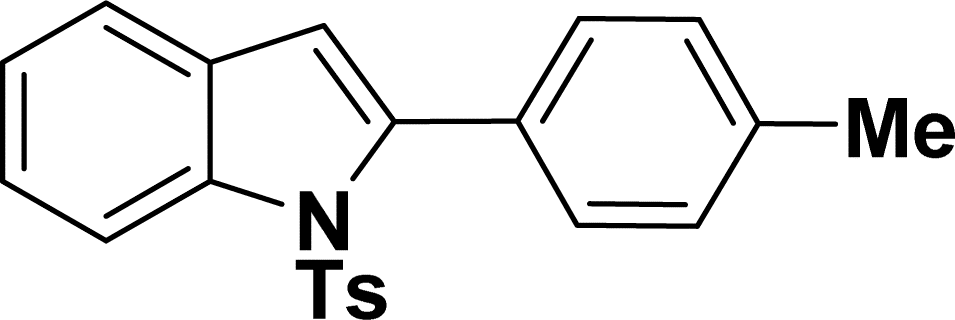 |
48 | 53 |
| 10 | 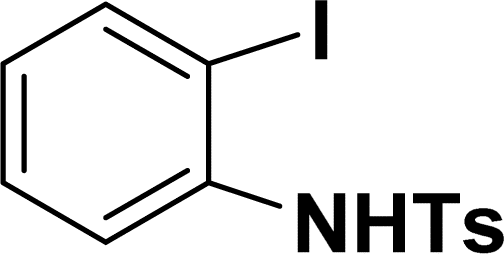 |
 |
 |
35 | 39 |
| 11 |  |
 |
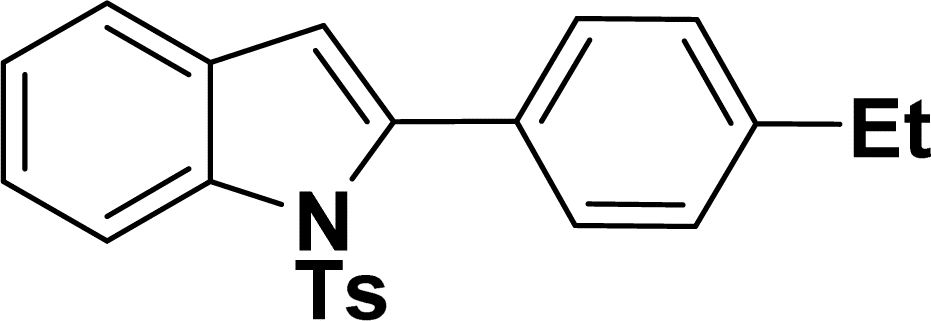 |
46 | 49 |
| 12 | 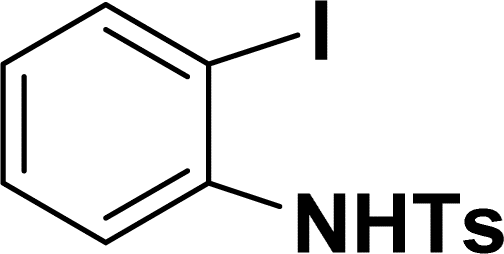 |
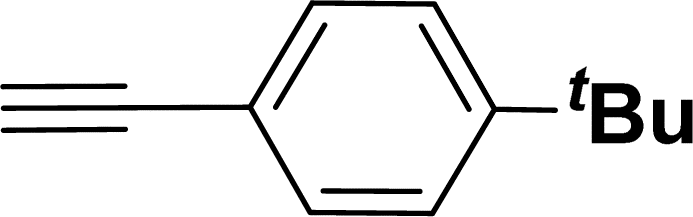 |
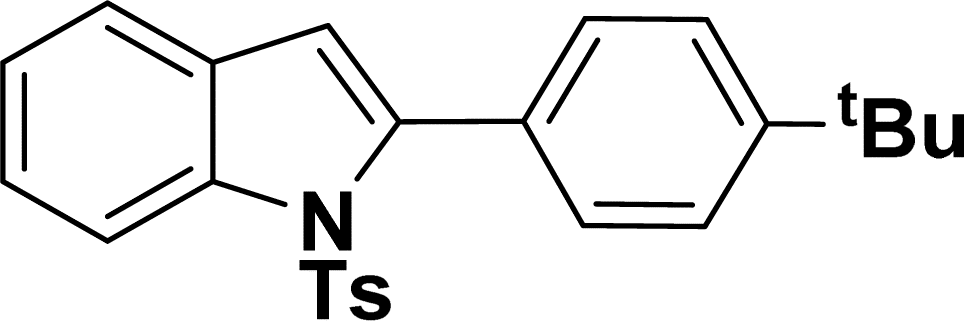 |
40 | 38 |
| 13 | 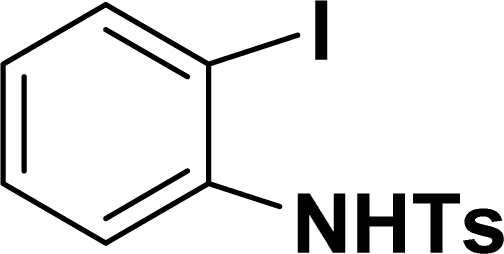 |
 |
 |
34 | 38 |
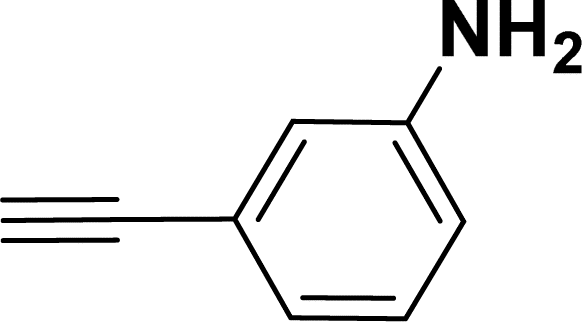
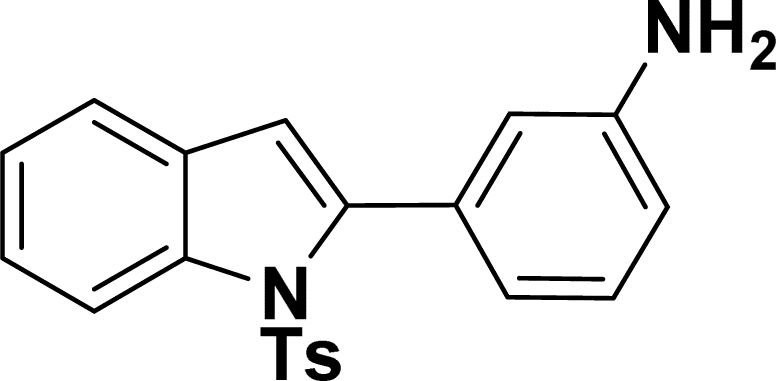
| 14 | 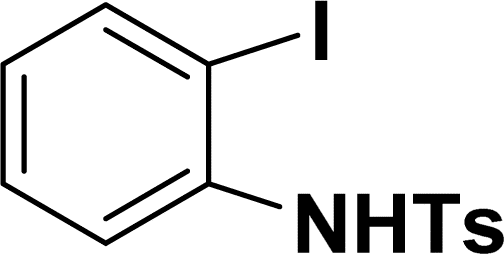 |
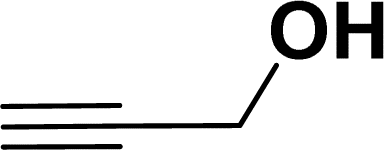 |
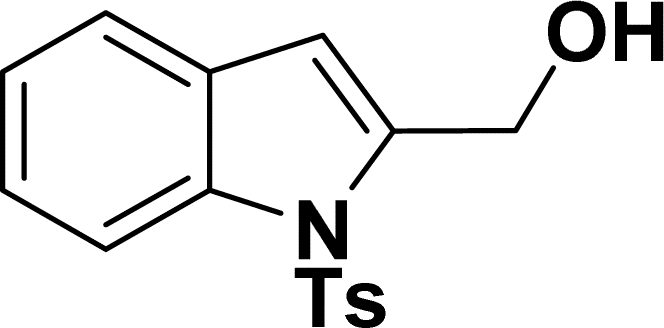 |
39 | 37 |
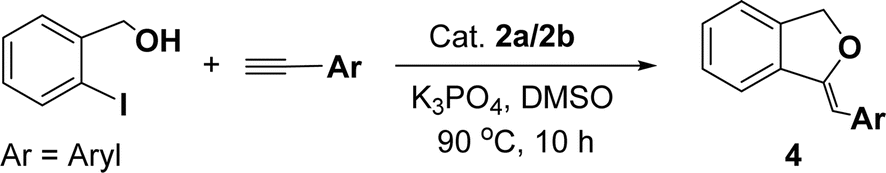
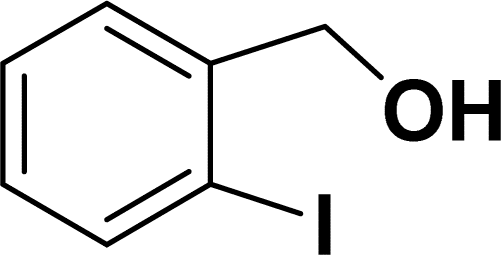
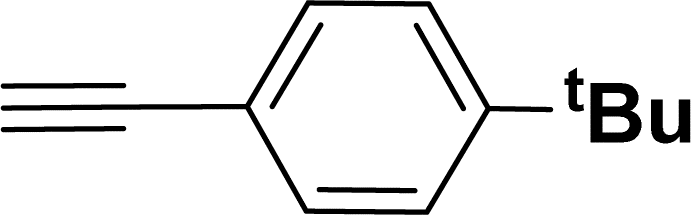
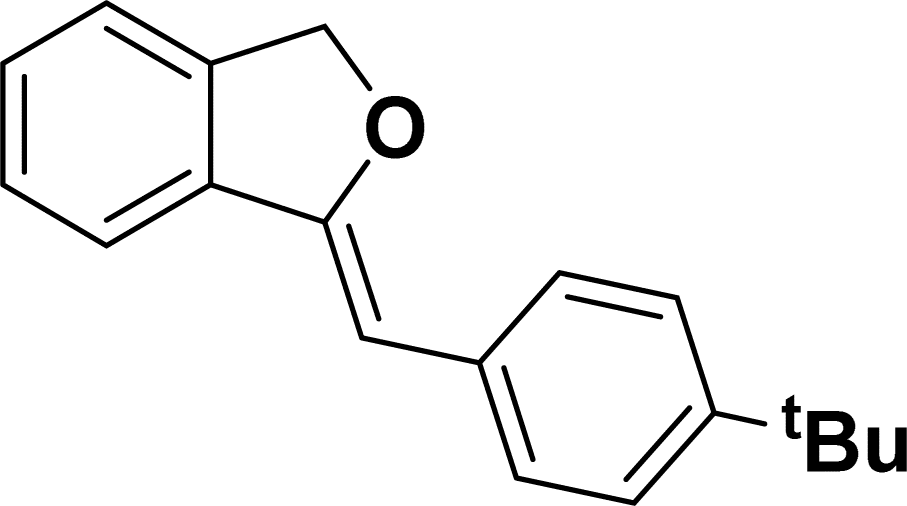
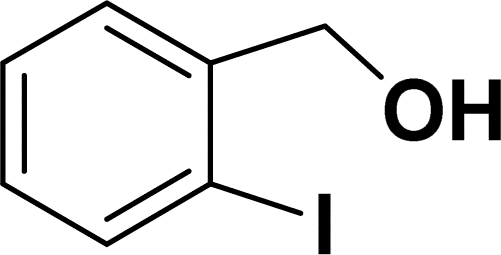
Under the same reaction condition, (Z)-1-benzylidene-1,3-dihydroisobenzofuran derivatives were also synthesized in good yields from 2-iodobenzyl alcohols and terminal alkynes (Table 3). Formation of (Z)-1-benzylidene-1,3-dihydroisobenzofuran was confirmed from the presence of a singlet of one proton at δ 5.98 ppm for methine proton and singlet of two protons at δ 5.54 ppm for methylene protons, which matches with the previously reported NMR data.50
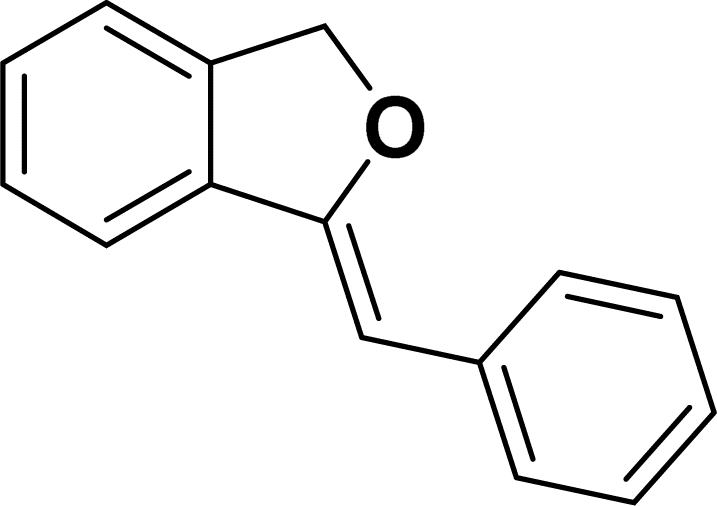
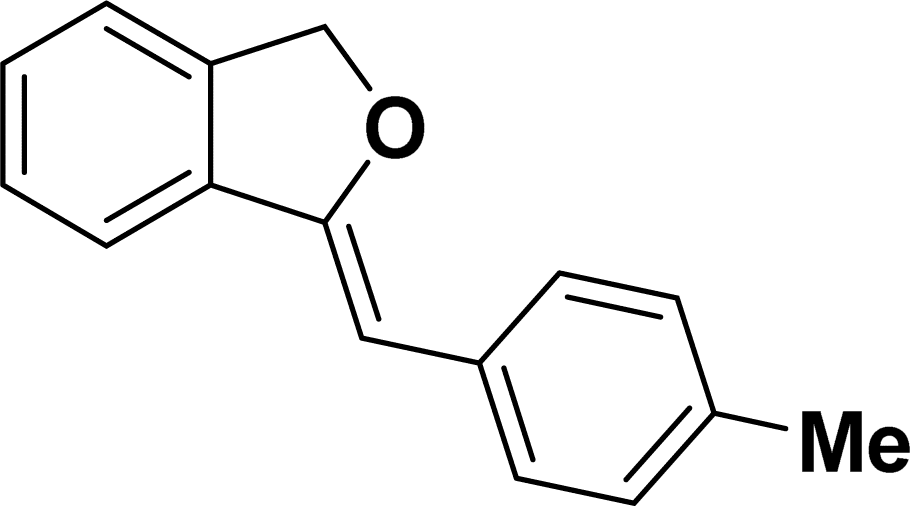
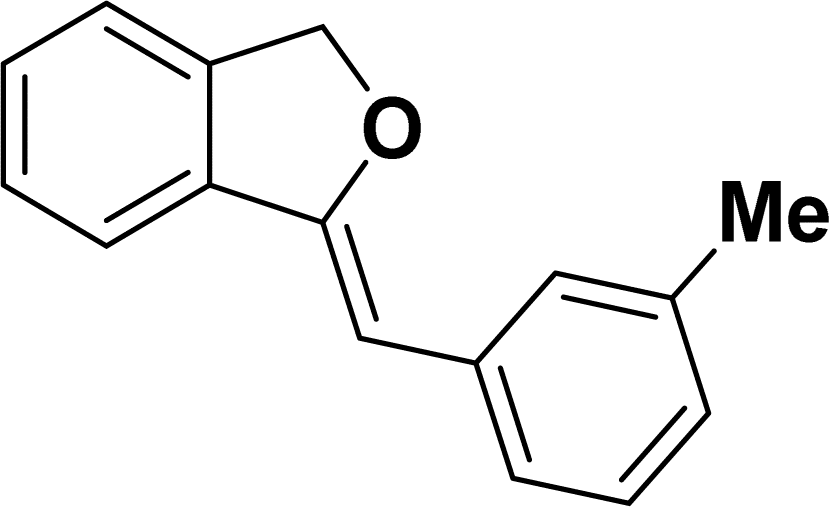
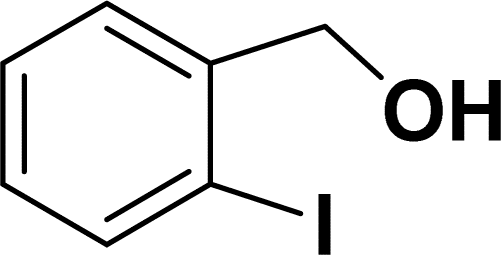
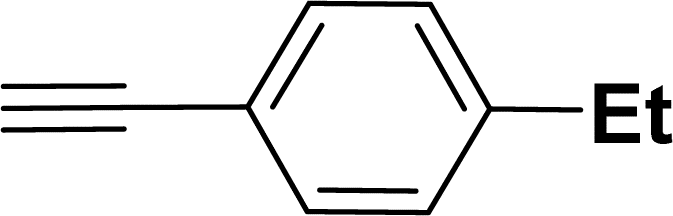
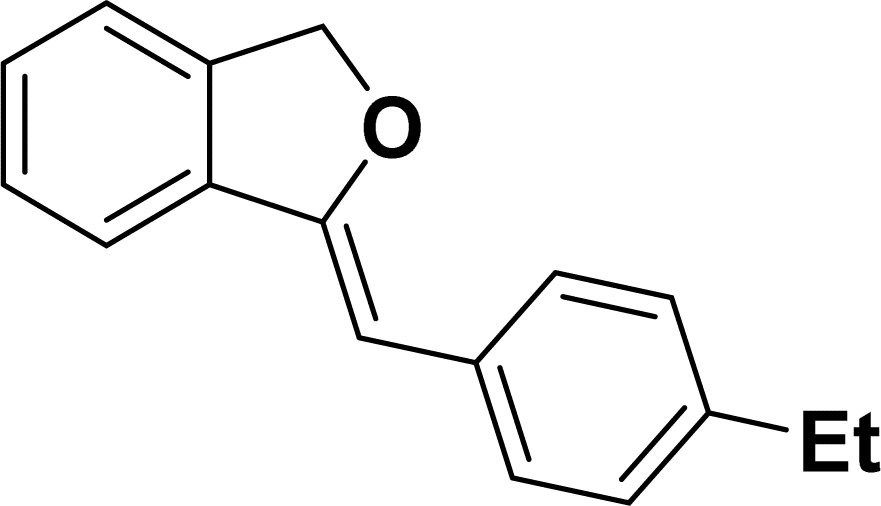
Based on previous literature reports and experimental observations,7,42,44 a plausible reaction mechanism pathway for the Sonogashira coupling/cyclization of 2-iodoarenes and terminal alkynes was proposed in Scheme 2.
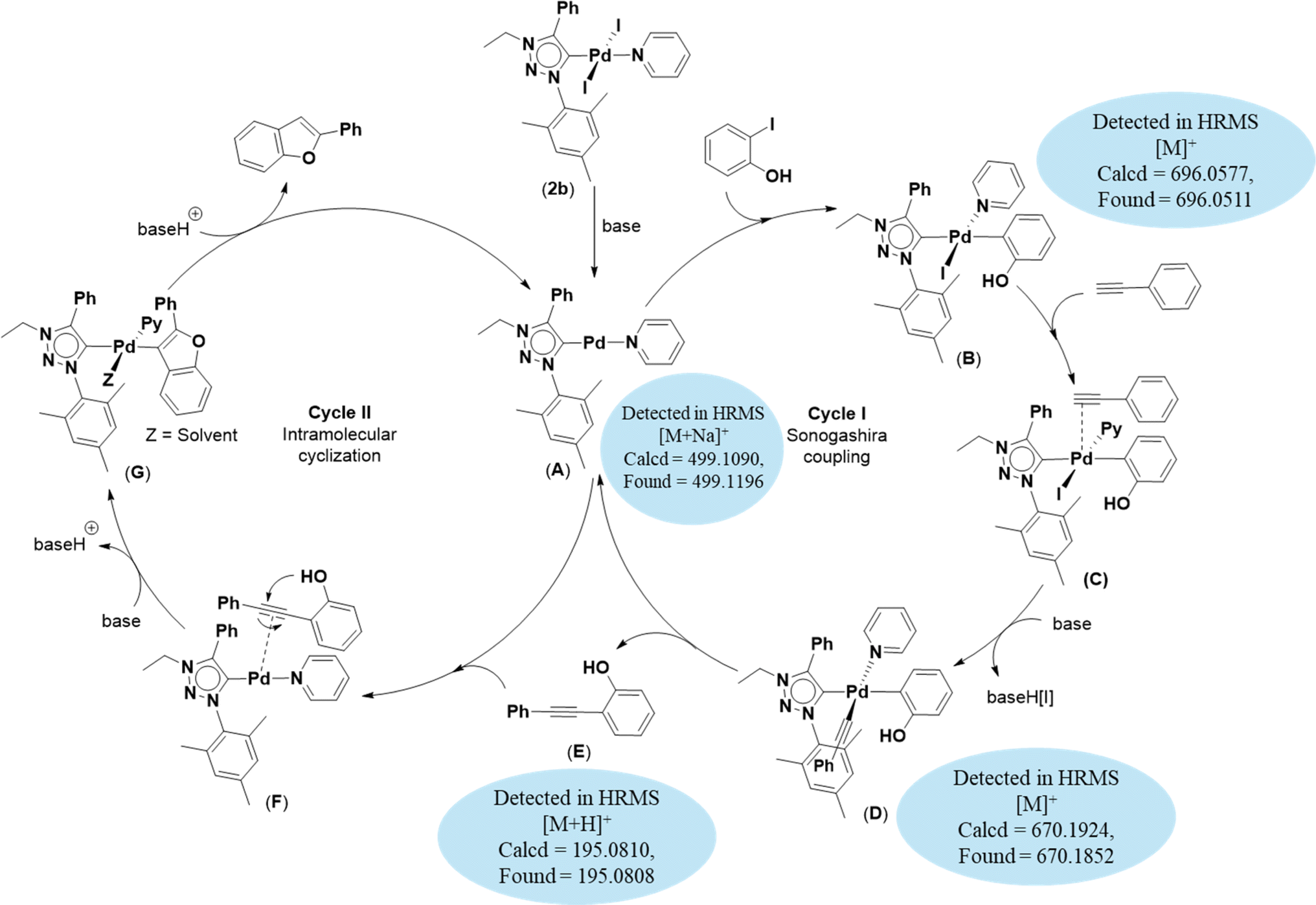 | ||
| Scheme 2 Proposed mechanism for the synthesis of benzofuran derivatives via domino Sonogashira coupling/cyclization reactions. | ||
Initially, the palladium(II) complex 2b was reduced to palladium(0) intermediate A as an active species. In cycle I, oxidative addition of 2-iodoarene led to the formation of intermediate B. This intermediate B coordinates with phenylacetylene to form intermediate C which is converted to intermediate D upon deprotonation of the coordinated phenylacetylene moiety. Next, through reductive elimination of intermediate D, 2-(phenylethynyl)arene and intermediate A are generated. Now, in cycle II, 2-(phenylethynyl)arene coordinated with intermediate A to give intermediate F, which is converted to intermediate G upon base-assisted deprotonation followed by palladium-assisted 5-endo-dig cyclization. Finally, protonolysis of intermediate G afforded the desired benzofuran along with the regeneration of intermediate A. This procedure yielded intermediates A (calcd = 499.1090; found = 499.1196), B (calcd = 696.0577; found = 696.0511), D (calcd = 670.1924; found = 670.1852), and E (calcd = 195.0810; found = 195.0808) detected by high-resolution mass spectrometry (HRMS).
The comparison of catalytic activity of complexes 2a and 2b with the previously reported N-heterocyclic carbene-based palladium complexes for the synthesis of benzofuran from the 2-iodophenol and phenylacetylene via domino Sonogashira coupling/cyclization reactions was shown in Table 4. Initially, Peris and coworkers reported the domino synthesis of benzofuran via Sonogashira coupling/cyclization reactions using 1 mol% of 1,2,4-triazole derived PEPPSI complexes under copper- and amine-free conditions.41 Ghosh and coworkers group reported the same catalytic transformation employing 1 mol% of 1,2,4-triazole derived bis-NHC and phosphine ligand-based dipalladium complexes.42 Later, our group reported the same domino synthesis of benzofuran using 0.2 mol% of N-heterocyclic carbene ligated bimetallic palladium PEPPSI complexes.43 Recently, Bera group also developed an annellated mesoionic carbene ligand stabilized palladium complex and explored their catalytic activity in benzofuran synthesis via domino approach.7 Thus, a comparison of the performance with previously reported well-defined N-heterocyclic carbene-based palladium complexes revealed that these types of mesoionic palladium complexes were not as active as other normal N-heterocyclic carbene-based PEPPSI complexes in terms of catalyst loading.
Conclusions
We have reported the preparation and characterization of two 1,2,3-triazolylidene-based palladium PEPPSI complexes 2a and 2b and their application in domino Sonogashira coupling/cyclization reactions for the synthesis of benzofuran and indole derivatives. Both PEPPSI-themed palladium complexes were well characterized by spectroscopic techniques. The complex 2b was characterized by X-ray crystallography. The desired catalytic products were obtained in moderate to good yields with broad substrate scope at 2 mol% catalyst loading. This work further showed the catalytic potential of PEPPSI-themed complexes as effective catalysts for domino Sonogashira coupling/cyclization reactions.Experimental section
General methods and materials
All manipulations were carried out under an atmosphere of nitrogen or in air. NMR spectra were recorded at 298 K on Bruker 500 MHz and JEOL 400 MHz NMR spectrometer. The chemical shifts of proton and carbon are reported in ppm and referenced using residual proton (7.26 ppm) and carbon signals (77.16 ppm) of CDCl3.51 NMR annotations used: br. = broad, d = doublet, m = multiplet, s = singlet, t = triplet, sept = septet. Elemental analyses were performed using Thermo Quest FLASH 2000 SERIES (CHNS) Elemental Analyzer. Mass data are collected from Agilent 6545 LC/Q-Tof spectrometer. Solvents were purchased from commercial suppliers and used without further purification. CCDC-2336820 (2b) contains the supplementary crystallographic data for this paper. The data can be obtained free of charge at https://www.ccdc.cam.ac.uk/structures/.† Catalytic substrates 2-iodophenol, (2-iodophenyl)methanol, phenylacetylene, 4-ethynyltoluene, 3-ethynyltoluene, 4-ethylphenylacetylene, 4-tert-butylphenylacetylene, 4-ethynylanisole, 3-ethynylaniline, prop-2-yn-1-ol, AgOTf, C2H5I, and K3PO4 were purchased from Sigma-Aldrich and used as received without further purification. 1-(p-Tolyl)-4-phenyl-1H-1,2,3-triazole,45 phenyl-1H-1,2,3-triazole,40 and N-(2-iodophenyl)-4-methylbenzenesulfonamide52 were synthesised using literature procedures.Synthesis of ligand 1a
C2H5I (0.955 g, 6.12 mmol), AgOTf (1.57 g, 6.12 mmol) and anhydrous toluene (20 mL) were added in an aluminum foil covered 50 mL round bottom flask. The resulting mixture was heated at 60 °C for 5 hours under nitrogen atmosphere. In another 50 mL round bottom flask, 1-(p-tolyl)-4-phenyl-1H-1,2,3-triazole (1.20 g, 5.10 mmol) was mixed with anhydrous toluene (10 mL). After 5 hours, the mixture of C2H5I and AgOTf was transferred into the triazole containing round bottom flask by glass syringe, and the resulting reaction mixture was heated at 100 °C for 3 days under nitrogen atmosphere. The reaction mixture was cooled to room temperature and was decanted off the solvent. The residue was purified by column chromatography (MeOH/CHCl3 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :98), affording the product as a dark brownish solid (1.48 g, 70%). 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 3H), 7.33 (d, J = 8.0 Hz, 2H), 4.64 (q, J = 7.0 Hz, 2H), 2.39 (s, 3H), 1.58 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 143.7, 142.5, 132.6, 131.8, 130.8, 129.6, 129.6, 126.5, 121.7, 121.4, 47.9, 21.3, 14.1 ppm. IR data (KBr pellet) cm−1: 3094 (w), 2994 (w), 2947 (w), 2923 (w), 1613 (w), 1571 (w), 1518 (w), 1494 (w), 1457 (w), 1378 (w), 1265 (s), 1224 (m), 1153 (s), 1032 (m), 821 (w), 765 (w), 695 (w), 637 (w), 573 (w). HRMS (ESI) calcd for C18H18F3N3O3S+ [M + H]+ m/z 414.1093; found 414.1116.
:80, EtOAc:petroleum ether), affording desired product as an orange solid (0.366 g, 74%). 1H NMR (400 MHz, CDCl3): 8.78 (d, J = 8.0 Hz, 2H), 8.34 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 7.0 Hz, 2H), 7.60–7.55 (dd, J = 14.6, 7.2 Hz, 4H), 7.42 (d, J = 8.0 Hz, 2H), 7.22–7.15 (m, 2H), 4.34 (q, J = 7.3 Hz, 2H), 2.48 (s, 3H), 1.57–1.53 (m, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 153.8, 143.6, 140.1, 137.5, 137.3, 134.4 (MIC-Pd), 130.8, 130, 129.5, 128.8, 127.6, 125.2, 124.2, 46.2, 21.5, 14.7 ppm. IR data (KBr pellet) cm−1: 2984 (w), 1740 (w), 1599 (w), 1509 (w), 1444 (m), 1374 (w), 1291 (m), 1188 (m), 1071 (m), 1019 (m), 815 (m), 764 (s), 692 (s), 595 (w), 519 (w), 475 (w), 421 (w). HRMS (ESI) calcd for C22H22I2N4Pd+ [M + Na]+ m/z 724.8867; found 724.8820. Anal. calcd for C22H22I2N4Pd: C, 37.61; H, 3.16; N, 7.97; found: C, 37.59; H, 3.12; N, 7.42%.:MeOH/CHCl3 = 2:98), affording the product as a dark brownish solid (1.61 g, 80%). 1H NMR (500 MHz, CDCl3): 8.60 (s, 1H), 7.76 (d, J = 6.0 Hz, 2H), 7.55 (s, 3H), 7.02 (s, 2H), 4.76 q, J = 7.0 Hz, 2H), 2.34 (s, 3H), 2.08 (s, 6H), 1.58 (t, J = 7.0 Hz, 3H). 13C{1H} NMR(125 MHz, CDCl3): δ 143.8, 142.5, 134.4, 132.0, 131.4, 130.3, 129.89, 129.84, 129.77, 121.5, 48.3, 21.1, 17.2, 14.0 ppm. IR data (KBr pellet) cm−1: 3096 (m), 2924 (w), 2855 (w), 1615 (w), 1459 (w), 1267 (s), 1155 (m), 1030 (m), 637 (w). HRMS (ESI) calcd for C20H22F3N3O3S+ [M + H]+ m/z 442.1406; found 442.1396.:petroleum ether = 20:80) affording the desired product as an orange solid (0.308 g, 83%). 1H NMR (400 MHz, CDCl3): δ 8.67–8.65 (m, 2H), 8.00–7.98 (m, 2H), 7.63–7.53 (m, 4H), 7.14–7.11 (m, 2H), 7.05 (s, 2H), 4.42 (q, J = 7.0 Hz, 2H), 2.40 (s, 3H), 2.39 (s, 6H), 1.50 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 153.7, 144.7, 140.3, 137.5, 137.1, 135.7, 135.6 (MIC-Pd), 131.1, 130, 129.5, 128.6, 128.1, 124, 46.1, 21.4, 21.3, 14.8 ppm. IR data (KBr pellet) cm−1: 2921 (w), 1739 (w), 1600 (w), 1442 (m), 1371(m), 1284 (m), 1228 (w), 1174 (m), 1084 (m), 1025 (m), 853 (w), 760 (s), 693 (s), 600 (w), 548 (w), 489 (w). HRMS (ESI) calcd for C24H26I2N4Pd+ [M + K]+ m/z 768.8919; found 768.8943. Anal. calcd for C24H26I2N4Pd: C, 39.45; H, 3.59; N, 7.67; found: C, 38.90; H, 3.56; N, 7.20. Suitable single crystal for X-ray diffraction analysis of 2b was obtained by slow evaporation of acetonitrile solution at room temperature.
:98), affording the product as a dark brownish solid (1.48 g, 70%). 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 3H), 7.33 (d, J = 8.0 Hz, 2H), 4.64 (q, J = 7.0 Hz, 2H), 2.39 (s, 3H), 1.58 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 143.7, 142.5, 132.6, 131.8, 130.8, 129.6, 129.6, 126.5, 121.7, 121.4, 47.9, 21.3, 14.1 ppm. IR data (KBr pellet) cm−1: 3094 (w), 2994 (w), 2947 (w), 2923 (w), 1613 (w), 1571 (w), 1518 (w), 1494 (w), 1457 (w), 1378 (w), 1265 (s), 1224 (m), 1153 (s), 1032 (m), 821 (w), 765 (w), 695 (w), 637 (w), 573 (w). HRMS (ESI) calcd for C18H18F3N3O3S+ [M + H]+ m/z 414.1093; found 414.1116.
:80, EtOAc:petroleum ether), affording desired product as an orange solid (0.366 g, 74%). 1H NMR (400 MHz, CDCl3): 8.78 (d, J = 8.0 Hz, 2H), 8.34 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 7.0 Hz, 2H), 7.60–7.55 (dd, J = 14.6, 7.2 Hz, 4H), 7.42 (d, J = 8.0 Hz, 2H), 7.22–7.15 (m, 2H), 4.34 (q, J = 7.3 Hz, 2H), 2.48 (s, 3H), 1.57–1.53 (m, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 153.8, 143.6, 140.1, 137.5, 137.3, 134.4 (MIC-Pd), 130.8, 130, 129.5, 128.8, 127.6, 125.2, 124.2, 46.2, 21.5, 14.7 ppm. IR data (KBr pellet) cm−1: 2984 (w), 1740 (w), 1599 (w), 1509 (w), 1444 (m), 1374 (w), 1291 (m), 1188 (m), 1071 (m), 1019 (m), 815 (m), 764 (s), 692 (s), 595 (w), 519 (w), 475 (w), 421 (w). HRMS (ESI) calcd for C22H22I2N4Pd+ [M + Na]+ m/z 724.8867; found 724.8820. Anal. calcd for C22H22I2N4Pd: C, 37.61; H, 3.16; N, 7.97; found: C, 37.59; H, 3.12; N, 7.42%.:MeOH/CHCl3 = 2:98), affording the product as a dark brownish solid (1.61 g, 80%). 1H NMR (500 MHz, CDCl3): 8.60 (s, 1H), 7.76 (d, J = 6.0 Hz, 2H), 7.55 (s, 3H), 7.02 (s, 2H), 4.76 q, J = 7.0 Hz, 2H), 2.34 (s, 3H), 2.08 (s, 6H), 1.58 (t, J = 7.0 Hz, 3H). 13C{1H} NMR(125 MHz, CDCl3): δ 143.8, 142.5, 134.4, 132.0, 131.4, 130.3, 129.89, 129.84, 129.77, 121.5, 48.3, 21.1, 17.2, 14.0 ppm. IR data (KBr pellet) cm−1: 3096 (m), 2924 (w), 2855 (w), 1615 (w), 1459 (w), 1267 (s), 1155 (m), 1030 (m), 637 (w). HRMS (ESI) calcd for C20H22F3N3O3S+ [M + H]+ m/z 442.1406; found 442.1396.:petroleum ether = 20:80) affording the desired product as an orange solid (0.308 g, 83%). 1H NMR (400 MHz, CDCl3): δ 8.67–8.65 (m, 2H), 8.00–7.98 (m, 2H), 7.63–7.53 (m, 4H), 7.14–7.11 (m, 2H), 7.05 (s, 2H), 4.42 (q, J = 7.0 Hz, 2H), 2.40 (s, 3H), 2.39 (s, 6H), 1.50 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 153.7, 144.7, 140.3, 137.5, 137.1, 135.7, 135.6 (MIC-Pd), 131.1, 130, 129.5, 128.6, 128.1, 124, 46.1, 21.4, 21.3, 14.8 ppm. IR data (KBr pellet) cm−1: 2921 (w), 1739 (w), 1600 (w), 1442 (m), 1371(m), 1284 (m), 1228 (w), 1174 (m), 1084 (m), 1025 (m), 853 (w), 760 (s), 693 (s), 600 (w), 548 (w), 489 (w). HRMS (ESI) calcd for C24H26I2N4Pd+ [M + K]+ m/z 768.8919; found 768.8943. Anal. calcd for C24H26I2N4Pd: C, 39.45; H, 3.59; N, 7.67; found: C, 38.90; H, 3.56; N, 7.20. Suitable single crystal for X-ray diffraction analysis of 2b was obtained by slow evaporation of acetonitrile solution at room temperature.General procedure for Sonogashira coupling/cyclization reactions of 2-iodoarenes and alkyne
In a reaction vial, 2-iodoarenes (0.50 mmol), phenylacetylene (0.60 mmol), and K3PO4 (1.00 mmol) were added. A palladium complex (2a or 2b, 2 mol%) and DMSO (ca. 2 mL) were added, and the resulting reaction mixture was heated at 90 °C for 10 hours. The reaction mixture was cooled to room temperature and quenched with water (ca. 20 mL). The resulting reaction mixture was extracted with EtOAc (3 × 20 mL) and dried on anhydrous Na2SO4. Solvent was evaporated under reduced pressure and further purification was done by column chromatography.1H NMR (500 MHz, CDCl3): δ 7.92–7.87 (m, 2H), 7.60 (d, J = 7.5 Hz, 1H), 7.56–7.54 (m, 1H), 7.48–7.45 (m, 2H), 7.40–7.35 (m, 1H), 7.33–7.28 (m, 1H), 7.28–7.23 (m, 1H), 7.04 (s, 1H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.79 (d, J = 8.0 Hz, 2H), 7.61–7.58 (m, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.31–7.23 (m, 2H), 7.16 (d, J = 8.0 Hz, 1H), 6.98 (s, 1H), 2.42 (s, 3H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.76 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.29–7.26 (m, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.06 (s, 1H), 2.48 (s, 3H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.88 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 7.5 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.34–7.30 (m, 1H), 7.05 (s, 1H), 2.78 (q, J = 7.0 Hz, 2H), 1.37 (t, J = 7.0 Hz, 3H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 7.5 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.32 (t, J = 7.5 Hz, 1H), 7.28–7.26 (m, 1H), 7.03 (s, 1H), 1.42 (s, 9H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 7.5 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.34–7.21 (m, 5H), 7.00 (s, 1H), 6.70–6.69 (m, 1H), 3.70 (br s, 2H) ppm.
1H NMR (500 MHz, CDCl3): δ 7.58 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.37–7.27 (m, 2H), 7.26–7.16 (m, 1H), 6.69 (s, 1H), 4.80 (s, 2H) ppm.
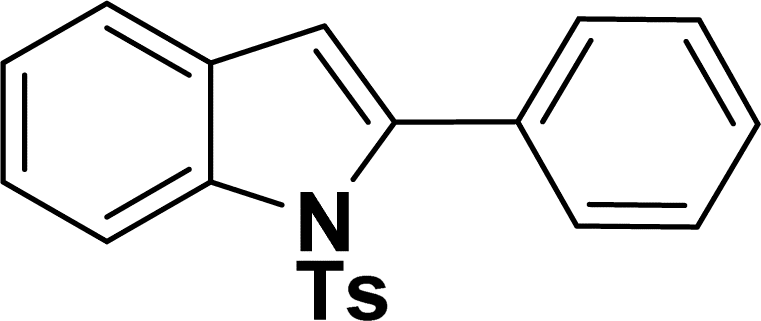
1H NMR (500 MHz, CDCl3): δ 8.23 (d, JHH = 7.0 Hz, 1H), 7.42–7.35 (m, 6H), 7.27–7.18 (m, 4H), 6.96–6.95 (m, 2H), 6.46 (s, 1H), 2.20 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 144.6, 142.2, 138.4, 134.9, 132.5, 130.6, 130.4, 129.3, 128.7, 127.6, 126.9, 124.8, 124.4, 120.8, 116.7, 113.6, 21.6 ppm.
1H NMR (500 MHz, CDCl3): δ 8.22 (d, JHH = 8.0 Hz, 2H), 7.35–7.31 (m, 3H), 7.27–7.24 (m, 1H), 7.21–7.14 (m, 5H), 6.96–6.94 (m, 2H), 6.42 (s, 1H), 2.36 (s, 3H), 2.19 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 144.5, 142.4, 138.7, 138.3, 134.8, 130.7, 130.3, 129.6, 129.2, 128.3, 126.9, 124.7, 124.3, 120.7, 116.8, 113.3, 29.8, 21.5 ppm.
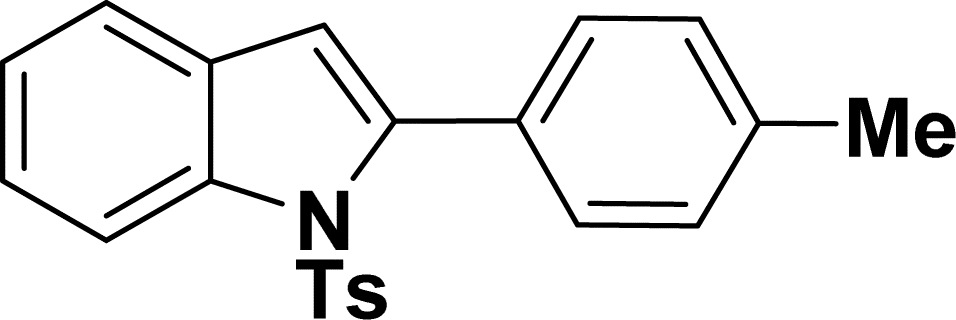
1H NMR (500 MHz, CDCl3): δ 8.22 (d, JHH = 8.0 Hz, 1H), 7.35–7.34 (m, 1H), 7.27–7.24 (m, 1H), 7.23–7.18 (m, 5H), 7.16–7.15 (m, 2H), 6.96–6.94 (m, 2H), 6.44 (s, 1H), 2.33 (s, 3H), 2.20 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 144.5, 142.4, 138.3, 137.0, 134.9, 132.4, 131.1, 130.6, 129.5, 129.2, 127.55, 127.5, 126.9, 124.7, 124.3, 120.7, 116.7, 113.4, 29.8, 21.6 ppm.
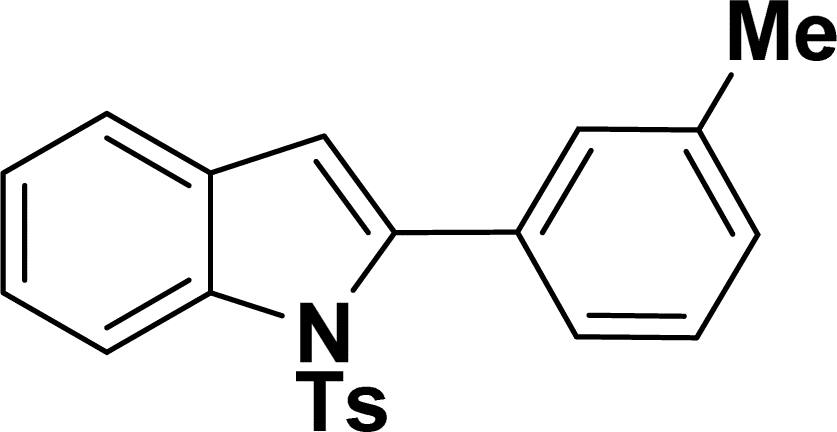
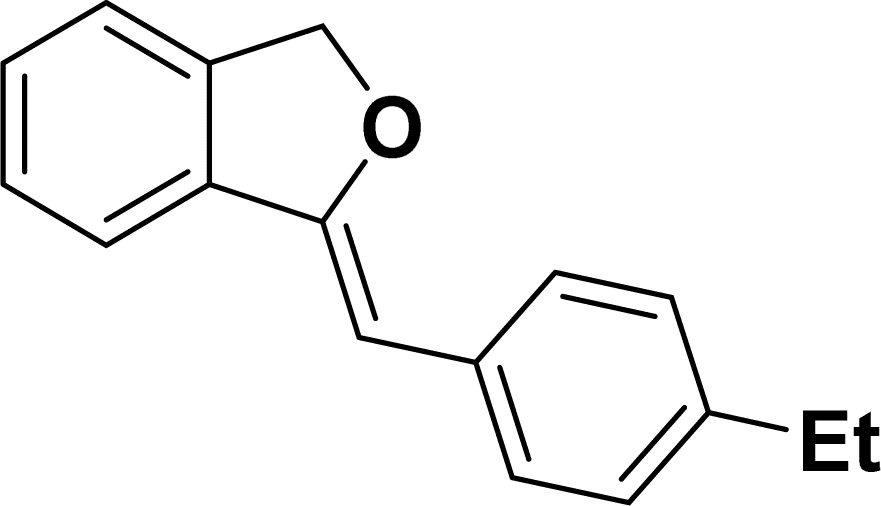
1H NMR (500 MHz, CDCl3): δ 8.21 (d, JHH = 8.0 Hz, 1H), 7.33–7.31 (m, 3H), 7.25–7.22 (m, 1H), 7.18–7.15 (m, 5H), 6.93–6.91 (m, 2H), 6.41 (s, 1H), 2.64 (q, JHH = 7.0 Hz, 2H), 2.17 (s, 3H), 1.21 (t, JHH = 7.0 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 144.9, 144.5, 142.4, 138.3, 134.8, 130.7, 130.3, 129.8, 129.2, 127.1, 126.9, 124.7, 124.3, 120.6, 116.7, 113.3, 28.8, 21.5, 15.4 ppm.
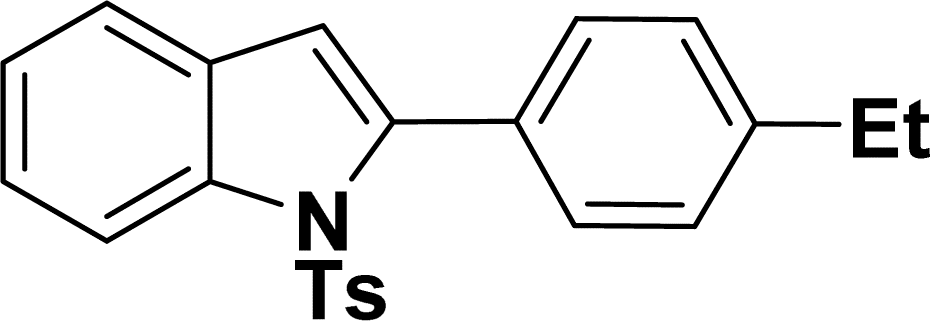
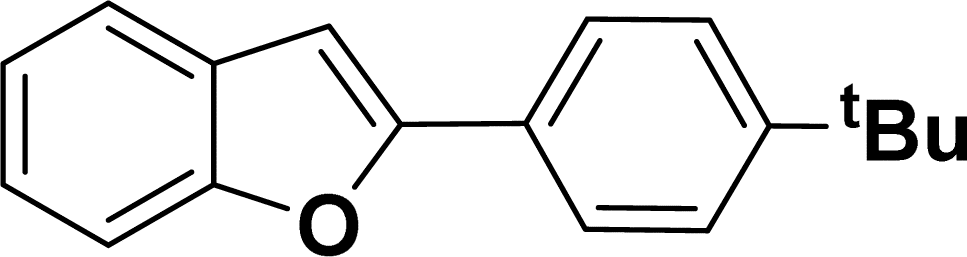
1H NMR (500 MHz, CDCl3): δ 8.22 (d, JHH = 8.0 Hz, 1H), 7.35–7.34 (m, 4H), 7.27–7.24 (m, 1H), 7.20–7.16 (m, 4H), 6.96–6.94 (m, 2H), 6.44 (s, 1H), 2.20 (s, 3H), 1.31 (s, 9H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 151.8, 144.5, 142.4, 138.4, 134.8, 130.7, 130.1, 129.5, 129.2, 127.0, 124.7, 124.5, 124.3, 120.7, 116.8, 113.4, 34.8, 31.5, 21.6 ppm.
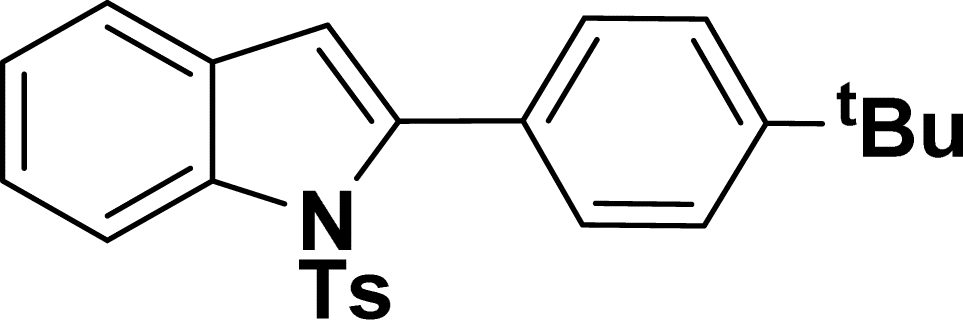
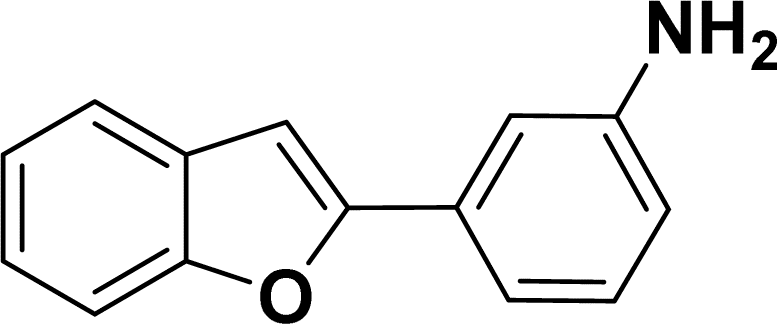
1H NMR (500 MHz, CDCl3): δ 8.21 (d, JHH = 8.0 Hz, 1H), 7.34–7.33 (m, 1H), 7.27–7.23 (m, 3H), 7.18–7.14 (m, 1H), 7.12–7.09 (m, 1H), 6.96–6.94 (m, 2H), 6.79–6.77 (m, 1H), 6.75 (s, 1H), 6.67–6.66 (m, 1H), 6.44 (s, 1H), 3.54 (br s, 2H), 2.19 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 145.6, 144.5, 142.5, 138.4, 134.8, 133.4, 130.6, 129.2, 128.5, 127.0, 124.7, 124.3, 120.77, 120.76, 117.4, 116.7, 115.6, 113.3, 21.6 ppm. HRMS (ESI) calcd for C21H18N2O2S+ [M + H]+ m/z 363.1169; found 363.1163.
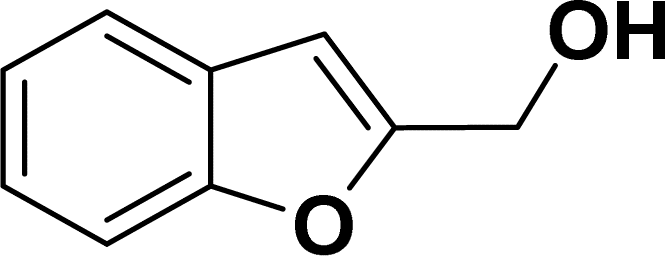
1H NMR (500 MHz, CDCl3): δ 7.97 (d, JHH = 8.0 Hz, 1H), 7.63 (d, JHH = 8.0 Hz, 2H), 7.39 (d, JHH = 8.0 Hz, 1H), 7.23–7.19 (m, 1H), 7.17–7.10 (m, 3H), 6.55 (s, 1H), 4.82 (s, 2H), 3.10 (br s, 1H), 2.24 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 145.2, 140.3, 137.1, 135.7, 130, 129.2, 126.5, 125, 123.8, 121.2, 114.4, 111.3. 58.6, 21.6 ppm.
1H NMR (500 MHz, CDCl3): δ 7.78–7.77 (m, 2H), 7.60–7.59 (m, 1H), 7.38–7.35 (m, 5H), 7.19–7.16 (m, 1H), 5.98 (s, 1H), 5.54 (s, 2H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 156.3, 139.3, 136.4, 134.9, 128.8, 128.4, 128.1, 127.8, 125.3, 121.2, 120.0, 96.3, 74.9, 29.7 ppm.
1H NMR (500 MHz, CDCl3): δ 7.56–7.54 (m, 2H), 7.47–7.46 (m, 1H), 7.26–7.23 (m, 3H), 7.07–7.06 (m, 2H), 5.84 (s, 1H), 5.42 (s, 2H), 2.26 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 155.7, 139.2, 135.1, 135.0, 133.6, 129.2, 128.6, 128.1, 127.8, 121.2, 119.9, 96.3, 74.9, 21.3 ppm.
1H NMR (500 MHz, CDCl3): δ 7.48–7.44 (m, 3H), 7.26–7.20 (m, 3H), 7.15–7.12 (m, 1H), 6.88–6.87 (m, 1H), 5.82 (s, 1H), 5.40 (s, 2H), 2.28 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 156.2, 139.3, 137.8, 136.3, 135.0, 128.7, 128.5, 128.3, 128.1, 126.2, 125.0, 121.2, 120.0, 96.4, 74.9, 21.6 ppm.
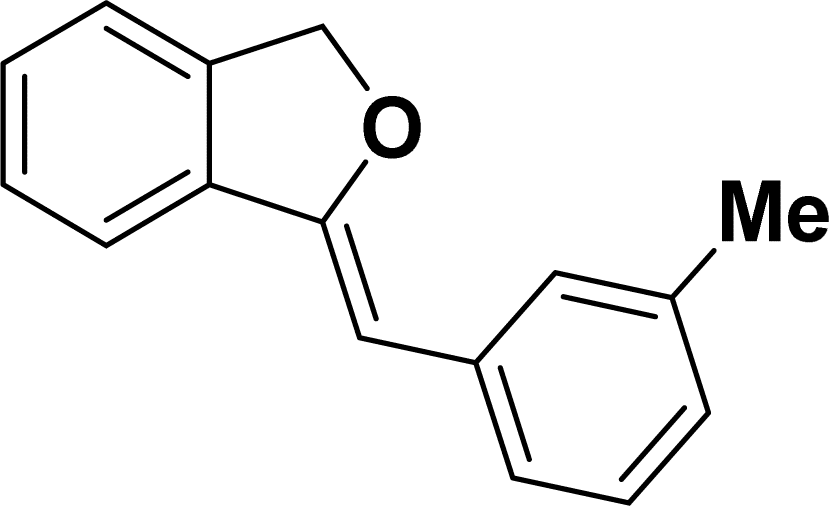
1H NMR (500 MHz, CDCl3): δ 7.59–7.57 (m, 2H), 7.48–7.47 (m, 1H), 7.28–7.24 (m, 3H), 7.10–7.08 (m, 2H), 5.85 (s, 1H), 5.42 (s, 2H), 2.56 (q, JHH = 7.5 Hz, 2H), 1.16 (t, JHH = 7.5 Hz, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 155.7, 141.5, 139.2, 135.1, 133.8, 128.6, 128.1, 128.0, 127.8, 121.2, 119.9, 96.3, 74.8, 28.7, 15.7 ppm.
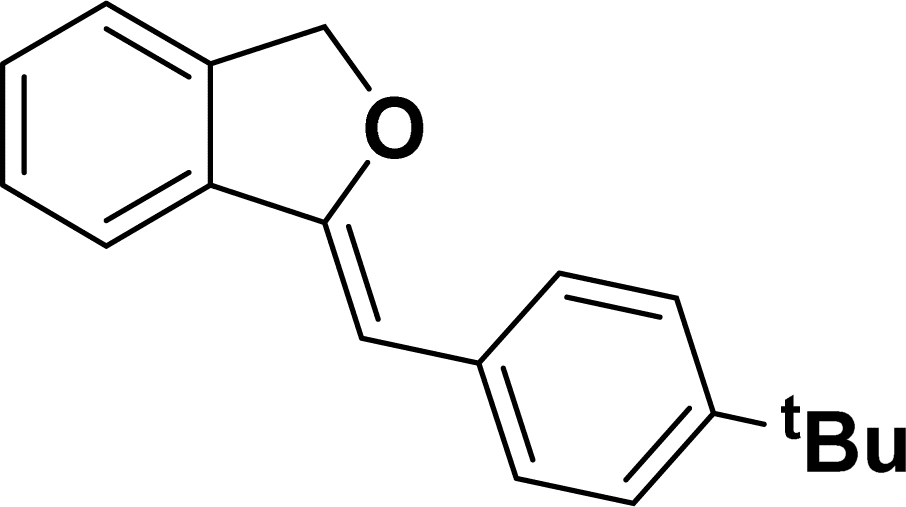
1H NMR (500 MHz, CDCl3): δ 7.60–7.58 (m, 2H), 7.47–7.45 (m, 1H), 7.29–7.27 (m, 2H), 7.26–7.21 (m, 3H), 5.85 (s, 1H), 5.40 (s, 2H), 1.24 (s, 9H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 155.9, 148.3, 139.3, 135.1, 133.6, 128.6, 128.1, 127.6, 125.3, 121.2, 120, 96.1, 74.8, 34.6, 31.4 ppm. HRMS (ESI) calcd for C19H20O+ [M − H]+ m/z 263.1434; Found 263.1432.
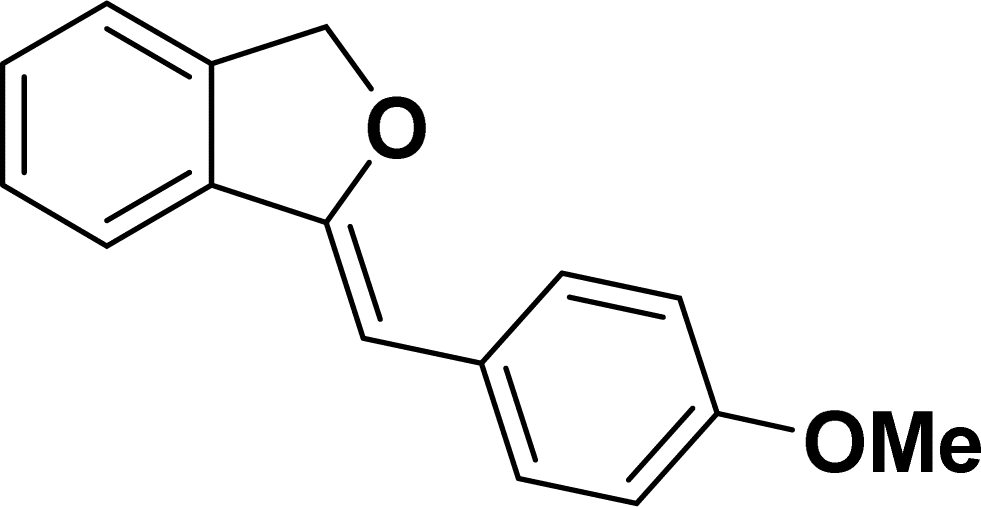
1H NMR (500 MHz, CDCl3): δ 7.60–7.59 (m, 2H), 7.45–7.44 (m, 1H), 7.25–7.22 (m, 3H), 6.81–6.80 (m, 2H), 5.82 (s, 1H), 5.41 (s, 2H), 3.73 (s, 3H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ 157.5, 154.8, 139, 135.1, 129.3, 129, 128.4, 128.1, 121.2, 119.7, 114, 95.9, 74.7, 55.3 ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DST-SERB, New Delhi, Grant No. CRG/2023/003598. Om Prakash Joshi thanks Central University of Rajasthan for Infrastructure facility and UGC for the Fellowship.References
- M. M. Heravi, V. Zadsirjan, H. Hamidi and P. H. Tabar Amiri, RSC Adv., 2017, 7, 24470–24521 RSC.
- H. Yuan, K.-J. Bi, B. Li, R.-C. Yue, J. Ye, Y.-H. Shen, L. Shan, H.-Z. Jin, Q.-Y. Sun and W.-D. Zhang, Org. Lett., 2013, 15, 4742–4745 CrossRef CAS PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- R. J. Reddy, J. J. Kumar, A. H. Kumari and G. R. Krishna, Adv. Synth. Catal., 2020, 362, 1317–1322 CrossRef CAS.
- N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS.
- Y. Shi, G. Wang, H. Wang, B. Deng, T. Gao, J. Wang, H. Guo, M. Wu and S. Sun, New J. Chem., 2020, 44, 14477–14480 RSC.
- A. Tyagi, N. U. D. Reshi, P. Daw and J. K. Bera, Dalton Trans., 2020, 49, 15238–15248 RSC.
- Y.-h. Miao, Y.-h. Hu, J. Yang, T. Liu, J. Sun and X.-j. Wang, RSC Adv., 2019, 9, 27510–27540 RSC.
- A. Savvidou, D. IoannisTzaras, G. S. Koutoulogenis, A. Theodorou and C. G. Kokotos, Eur. J. Org Chem., 2019, 2019, 3890–3897 CrossRef.
- S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544–6556 RSC.
- G. Battistuzzi, S. Cacchi and G. Fabrizi, Eur. J. Org Chem., 2002, 2002, 2671–2681 CrossRef.
- X.-Y. Liu, Y.-L. Liu and L. Chen, Adv. Synth. Catal., 2020, 362, 5170–5195 CrossRef.
- J. S. S. Neto and G. Zeni, Org. Chem. Front., 2020, 7, 155–210 RSC.
- L. Chiummiento, R. D'Orsi, M. Funicello and P. Lupattelli, Molecules, 2020, 25, 2327 CrossRef CAS.
- M. Alami and O. Provot, Org. Biomol. Chem., 2024, 22, 1323–1345 RSC.
- D. R. Kishore and G. Satyanarayana, J. Org. Chem., 2022, 87, 10158–10172 CrossRef CAS PubMed.
- R. Sanz, M. P. Castroviejo, Y. Fernández and F. J. Fañanás, J. Org. Chem., 2005, 70, 6548–6551 CrossRef CAS.
- W.-M. Dai and K. W. Lai, Tetrahedron Lett., 2002, 43, 9377–9380 CrossRef.
- R. Wang, S. Mo, Y. Lu and Z. Shen, Adv. Synth. Catal., 2011, 353, 713–718 CrossRef.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef PubMed.
- S. S. Bera and M. Szostak, ACS Catal., 2022, 12, 3111–3137 CrossRef CAS.
- Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- S. C. Sau, S. Santra, T. K. Sen, S. K. Mandal and D. Koley, Chem. Commun., 2012, 48, 555–557 RSC.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- R. Pretorius, M. R. Fructos, H. Müller-Bunz, R. A. Gossage, P. J. Pérez and M. Albrecht, Dalton Trans., 2016, 45, 14591–14602 RSC.
- M. Gazvoda, M. Virant, A. Pevec, D. Urankar, A. Bolje, M. Kočevar and J. Košmrlj, Chem. Commun., 2016, 52, 1571–1574 RSC.
- R. Maity and B. Sarkar, JACS Au, 2022, 2, 22–57 CrossRef CAS.
- D. Yuan and H. V. Huynh, Organometallics, 2012, 31, 405–412 CrossRef CAS.
- A. Bolje and J. Košmrlj, Org. Lett., 2013, 15, 5084–5087 CrossRef CAS PubMed.
- R. Maity, A. Mekic, M. van der Meer, A. Verma and B. Sarkar, Chem. Commun., 2015, 51, 15106–15109 RSC.
- R. Maity, A. Verma, M. van der Meer, S. Hohloch and B. Sarkar, Eur. J. Inorg. Chem., 2016, 2016, 111–117 CrossRef.
- R. Haraguchi, S. Hoshino, T. Yamazaki and S.-i. Fukuzawa, Chem. Commun., 2018, 54, 2110–2113 RSC.
- X. Marset, B. Saavedra, N. González-Gallardo, A. Beaton, M. M. León, R. Luna, D. J. Ramón and G. Guillena, Front. Chem., 2019, 7, 700 CrossRef PubMed.
- C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef PubMed.
- D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. M. Trzeciak and M. Albrecht, Chem.–Eur. J., 2012, 18, 6055–6062 CrossRef CAS PubMed.
- A. Zanardi, J. A. Mata and E. Peris, Organometallics, 2009, 28, 4335–4339 CrossRef CAS.
- A. Kumar, M. K. Gangwar, A. P. Prakasham, D. Mhatre, A. C. Kalita and P. Ghosh, Inorg. Chem., 2016, 55, 2882–2893 CrossRef CAS PubMed.
- S. Yadav, A. Singh, I. Mishra, S. Ray, S. M. Mobin and C. Dash, Appl. Organomet. Chem., 2019, 33, e4936 CrossRef.
- S. Yadav and C. Dash, Tetrahedron, 2020, 76, 131350 CrossRef CAS.
- Z.-X. Wang and H.-L. Qin, Chem. Commun., 2003, 2450–2451 RSC.
- R. Maity, M. van der Meer and B. Sarkar, Dalton Trans., 2015, 44, 46–49 RSC.
- D. Rendón-Nava, A. Álvarez-Hernández, A. L. Rheingold, O. R. Suárez-Castillo and D. Mendoza-Espinosa, Dalton Trans., 2019, 48, 3214–3222 RSC.
- M. K. Gangwar and R. J. Butcher, J. Organomet. Chem., 2020, 930, 121598 CrossRef.
- E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 6215–6221 CrossRef.
- C. Praveen, C. Iyyappan and P. T. Perumal, Tetrahedron Lett., 2010, 51, 4767–4771 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- H. A. Oskooie, M. M. Heravi and F. K. Behbahani, Molecules, 2007, 12, 1438–1446 CrossRef CAS PubMed.
- S. S. Palimkar, P. Harish Kumar, R. J. Lahoti and K. V. Srinivasan, Tetrahedron, 2006, 62, 5109–5115 CrossRef CAS.
- J. McNulty and K. Keskar, Eur. J. Org Chem., 2014, 2014, 1622–1629 CrossRef CAS.
- Z. Chen, X.-X. Shi, D.-Q. Ge, Z.-Z. Jiang, Q.-Q. Jin, H.-J. Jiang and J.-S. Wu, Chin. Chem. Lett., 2017, 28, 231–234 CrossRef CAS.
- Y.-L. Li, J. Li, A.-L. Ma, Y.-N. Huang and J. Deng, J. Org. Chem., 2015, 80, 3841–3851 CrossRef CAS.
- P. K. Mandali, A. K. Pati, A. K. Mishra and D. K. Chand, ChemistrySelect, 2017, 2, 5259–5265 CrossRef.
- K. Hiroya, R. Jouka, M. Kameda, A. Yasuhara and T. Sakamoto, Tetrahedron, 2001, 57, 9697–9710 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra, HRMS data, X-ray crystallographic data of the complexes, NMR data of catalytic products. CCDC 2336820. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03485f |
| This journal is © The Royal Society of Chemistry 2024 |