
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and in vitro assessment of gold nanoparticles conjugated with extracts, sterols and pure compounds derived from marine sponges from the Indian and Pacific Oceans†
Avin Ramanjooloo ab,
Devesh Bekaha,
Samson A. Adeyemic,
Philemon Ubanakoc,
Lindokuhle Ngemac,
Yahya E. Choonarac,
David E. Williamsd,
Elena A. Polishchuke,
Raymond J. Andersend and
Archana Bhaw-Luximon*a
ab,
Devesh Bekaha,
Samson A. Adeyemic,
Philemon Ubanakoc,
Lindokuhle Ngemac,
Yahya E. Choonarac,
David E. Williamsd,
Elena A. Polishchuke,
Raymond J. Andersend and
Archana Bhaw-Luximon*a
aBiomaterials, Drug Delivery & Nanotechnology Unit, Centre for Biomedical & Biomaterials Research (CBBR), University of Mauritius, Réduit 80837, Mauritius. E-mail: a.luximon@uom.ac.mu
bMauritius Oceanography Institute, Avenue des Anchois, Morcellement de Chazal, Albion, Mauritius
cWits Advanced Drug Delivery Platform Research Unit, School of Therapeutic Science, Faculty of Health Sciences, Department of Pharmacy and Pharmacology, University of the Witwatersrand, Johannesburg, South Africa
dDepartments of Chemistry and Earth, Ocean and Atmospheric Sciences, University of British Columbia, 2036 Main Mall, Vancouver, B.C., V6T 1Z1, Canada
eDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, B.C., V6T 1Z1, Canada
First published on 11th November 2024
Abstract
Gold nanoparticles (AuNPs) exhibit different physical properties compared to small molecules, bulk materials and other nanoparticles. Their synthesis using plant extracts, particularly polyflavonoids as phytoreductants, for the conversion of Au(III) into Au(0) has been reported. In this study, AuNPs were synthesized with extracts, sterols and pure compounds derived from marine sponges using gold(III) chloride trihydrate. Extracts, hexane (JDH) and ethyl acetate (JDE), sterols (JC-2) and jaspamide were obtained from Jaspis diastra. Pure compounds, namely, contignasterol, ansellone A, motuporamines A and MN100 (a synthetic analog of pelorol), were also used. JC-2 was characterized using NMR and GC-MS, and the major constituent was determined to be β-sitosterol. β-Sitosterol has shown great promise as an anti-cancer molecule, but its poor aqueous solubility and bioavailability coupled with low targeting efficacy limit its therapeutic efficacy. Transmission electron microscopy (TEM) images revealed the formation of spherical AuNPs conjugated with JDH, JDE, JC-2, ansellone and contignasterol with average diameters of 21.1 ± 3.0 nm, 20.7 ± 2.1 nm, 26.2 ± 1.2 nm, 33.3 ± 5.1 nm and 30.8 ± 5.5 nm, respectively. No particle formation was seen with motuporamines A and MN100. Zeta potential values indicated that AuNPs-JC-2 was more stable than AuNPs-JDE, AuNPs-JDH and AuNPs-ansellone. Based on IC50 values, the cytotoxicity of AuNPs-JDH increased in A172, TERA, HeLa and HepG2 cells but showed similar activity in HaCaT cells compared to JDH. The cytotoxicity of AuNPs-JDE decreased in A172 and HaCaT cells but increased in TERA1, HeLa and HepG2 cells compared to JDE. AuNPs-JC-2 showed enhanced cytotoxicity with a decrease in IC50 values from 3.37 ± 0.19 μg mL−1 to 0.52 ± 0.09 μg mL−1 in A172 and from 2.28 ± 0.20 μg mL−1 to 0.78 ± 0.28 μg mL−1 in TERA1 compared to JC-2. The synergistic action of sterols in AuNPs-JC-2 seemed to favour enhanced anti-cancer activity. The presence of sterols increased the ability of transforming Au(III) into Au(0) to form AuNPs and further enhancing cellular uptake and, thus, anti-cancer activity. AuNPs-contignasterol displayed lower activity than contignasterol in the A172 cell line. No significant difference in activity was observed with AuNPs-ansellone A in the A172 and HaCaT cell lines compared to ansellone A.
Introduction
Among the treatment strategies for cancer, the design and development of nanoparticles (NPs) have emerged as a promising approach for cancer therapy.1–3 In 2005, the Food and Drug Administration (FDA) of the United States (US) approved the use of the nanoparticle nab-paclitaxel (albumin-bound paclitaxel) for the treatment of metastatic breast cancer. It is used in the US for the treatment of many solid tumors such as breast cancer, non-small-cell lung cancer and pancreatic cancer.4,5 Among NPs, AuNPs are FDA-approved metallic nanoparticles6 and have emerged as therapeutic agents with potential cancer-related applications in drug delivery, gene delivery, bioimaging, diagnostics, improved radiation treatment and light induction therapy.3,7,8 The forms (i.e. stars, nanospheres, nanorods, nanotriangles, nanocubes, nanobranches and nanobipyramids),3 charge and size of AuNPs are important to address cancer.1,9–12 Owing to their positive charge, they can readily enter the cell membrane—which contain negatively charged elements (i.e. lipids and phosphate group)—resulting in the production of reactive oxygen species (ROS), mitochondrial dysfunction, DNA damage and apoptosis, hence promoting cell death.13 AuNPs are naturally nonimmunogenic and have less toxicity.3 They can reduce issues associated with conventional drugs such as drug resistance, low drug distribution, biodegradability and early drug release. Moreover, they can reduce drug dosage and treatment frequency and are capable of transporting hydrophobic and insoluble drugs.6 Their unique physico-chemical and optical properties, low toxicity, high biocompatibility and great surface area allow loading of various drugs for delivery to target sites with controlled release.10,14 They are effective nano-carriers for small-molecule drugs as well as peptides, proteins, plasmid deoxynucleic acids (pDNAs), small interfering ribonucleic acids (siRNAs) and other chemotherapeutic agents.15 Ghafari et al., have provided comprehensive insights into AuNPs in biomedicine,16 whereas Zhang et al., (2023) have summarized clinical translation of AuNPs.17 Aurimune (CYT-6091) (PEGylated) (27 nm) has completed phase I for the treatment of patients with advanced solid tumors and phase II clinical trial agreement with the National Cancer Institute (NCI) has been signed.5,17 Auroshell (PEGylated 120 nm silica and 12 to 15 nm gold shell) efficacy for the treatment of various types of cancers including prostate cancer has been studied, which were very promising.17 Major limitations of AuNPs to clinical trials are the lack of quantity, small sizes of particles and limited understanding of the long-term consequences of gold accumulation.18Several methods have been developed for the synthesis of AuNPs. Panwar et al. (2024)3 have summarized various approaches used in the synthesis of AuNPs. In general, they can be classified as physical (ultrasonication, irradiation, laser ablation, microwave and electrochemical), chemical (chemical reduction, sol–gel and condensation) and biological or green synthesis methods (i.e. algae, enzymes, plant extracts and microbial sources).19 The Turkevich method is the most common method for producing spherical AuNPs with sizes in the range of 9–150 nm. For tumor therapy, the optimal particle size is 50 nm.20 In the biosynthesis and “green” syntheses of AuNPs, biological materials act as stabilizers (e.g., polysaccharides) and reducing agents (e.g., polyphenols).21,22 Biosynthesized AuNPs have been used to deliver anticancer drugs such as doxorubicin, paclitaxel and methotrexate.23 Marine natural products (bioactive compounds derived from marine organisms) are crucial in the field of pharmaceutical industry.24 Different marine bioresources such as seaweeds, micro algae, mangrove plants, invertebrates, bacteria, fungi and sponges have been used for the synthesis of metal and metal oxide nanoparticles.25 Recently, Rizki et al. (2023) have provided a comprehensive review of the synthesis of silver and AuNPs from marine organisms.26 Ameen et al., (2023) have reported the antioxidant, antibacterial and anticancer effects of AuNPs synthesized using the marine fungus Alternaria chlamydospore.27 Marine sponges and their associated microorganisms have contributed the majority of natural products displaying pharmacological activities.24 They are known to produce structurally novel and diverse secondary metabolites such as polyketides, peptides and alkaloids with anticancer, antimicrobial, antifungal and anti-infective properties.28 Marine sponges such as Acanthella elongate, Callyspongia diffusa, Haliclona sp., Haliclona exigua and Amphimedon sp. have been used for the synthesis of silver nanoparticles and silver nanocolloids.25,26 Spherical mono-dispersed AuNPs of size 15 nm were synthesized using the marine sponge Acanthella elongate.29
This study describes the synthesis of AuNPs using extracts, sterols and pure compounds from marine sponges. The hexane (JDH), ethyl acetate (JDE), sterols (JC-2) and jaspamide (1) (Fig. 1) were obtained from the marine sponge Jaspis diastra, whereas pure compounds contignasterol (2), ansellone A (3) and motuporamines A (4) (Fig. 1) were obtained from Petrosia contignata, Cadlina luteomarginata, Phorbas sp. and Xestospongia exigua respectively. MN100 (5) is a synthetic analog of pelorol from Dactylospongia elegans. Previous studies have reported the anticancer activity of JDH and JDE.30–33 However, to the best of our knowledge, there are no reported data on the synthesis of AuNPs with the extracts, sterols and pure compounds from marine sponges and their biological activities. The AuNPs were characterized for their physicochemical properties and the biological activity of conjugated AuNPs was evaluated on the human brain (A172), testicular (TERA1), normal immortalized human keratinocytes (HaCaT), cervical (HeLa) and liver (HepG2) cell lines. This study aims at demonstrating the importance of the presence of phytoreductants in extracts for the formation of AuNPs, the improved cellular uptake and the synergistic actions of molecules for anti-cancer action.
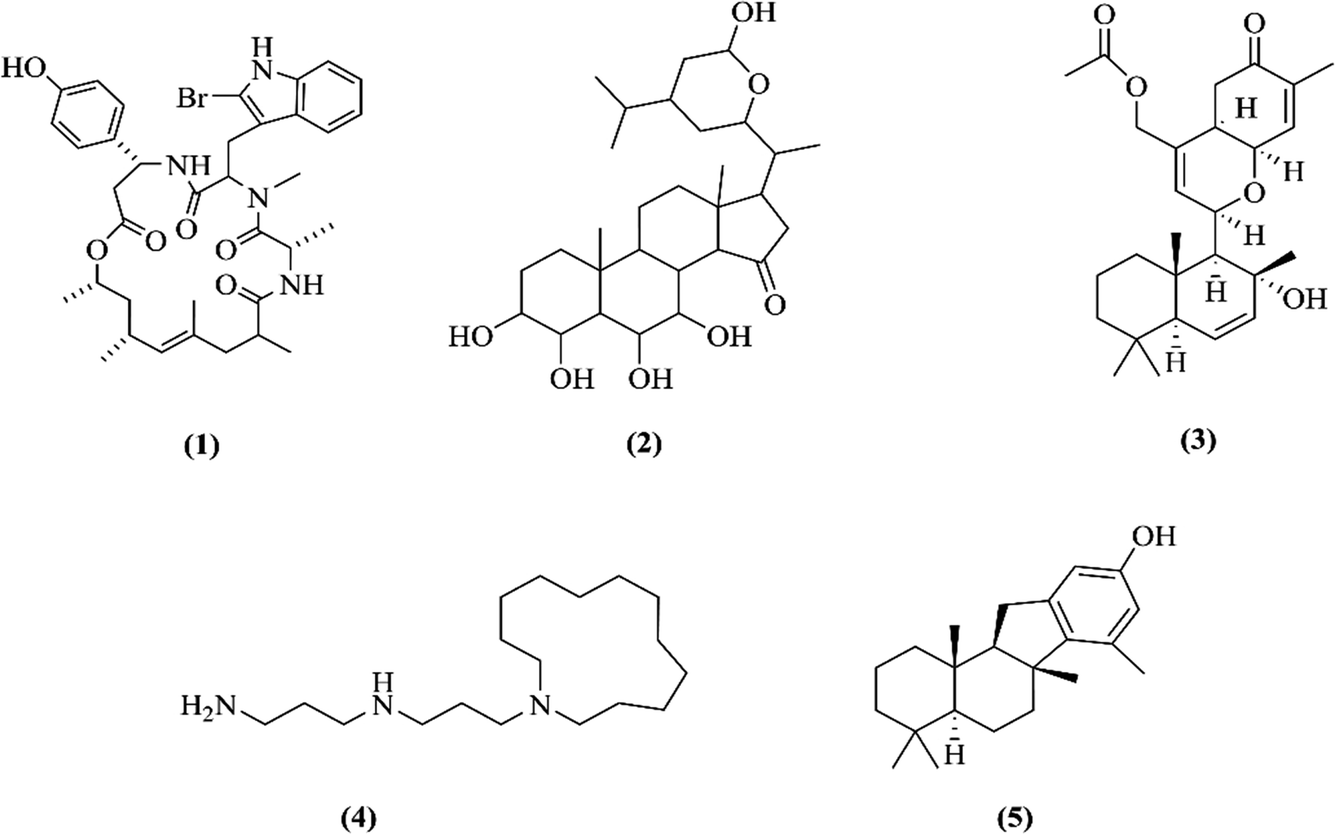 | ||
| Fig. 1 Chemical structures of jaspamide (1), contignasterol (2), ansellone A (3), motuporamine A (4) and MN100 (5). | ||
Results and discussion
Isolation and characterization of JC-2 and jaspamide
Our previous studies have showed significant cytotoxic activity of JDH, JDE and jaspamide—obtained from the sponge Jaspis diastra—on the TERA1, A172, DLD1 and HaCaT cell lines.33 As a continuation of our research work, further studies were performed on JDH, JDE and jaspamide. The purification of JDH by chromatography resulted in various fractions which were combined based on their TLCs. A precipitate (JC-2) was isolated from one of the combined fractions. The FTIR spectra of JC-2 (Fig. 1S†) showed absorption bands at 3433 cm−1 (O–H stretching); 2956–2865 cm−1 (C–H stretching) and 1642 cm−1 (absorption C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak). Other absorption frequencies were observed for cyclic methylene groups (CH2)n at 1462 cm−1, gem-dimethyl (–CH(CH3)2) group at 1380 cm−1 and C–OH (secondary alcohol) at 1052 cm−1.34,35
C peak). Other absorption frequencies were observed for cyclic methylene groups (CH2)n at 1462 cm−1, gem-dimethyl (–CH(CH3)2) group at 1380 cm−1 and C–OH (secondary alcohol) at 1052 cm−1.34,35
These IR signals corresponded to the presence of β-sitosterol and stigmasterol. 1H and 13C-NMR (Fig. 2S(A) and (B), Table 1S†) spectroscopy were performed to confirm the presence of these sterols. The spectra indicated a mixture of at least two compounds based on the intensities of peaks. The 1H-NMR spectrum of JC-2 showed characteristics of a sterol moiety due to the presence of a multiplet at δH 3.5, which was attributed to the proton at C-3. The presence of olefinic protons was observed at δH 5.02, δH 5.15 and δH 5.33. The presence of methyl groups was observed from δH 0.66 to δH 0.99. The 13C-NMR spectrum confirmed the presence of olefinic carbons at δC 140.78, δC 131.84, δC 129.30 and δC 121.74 and a secondary alcohol carbon δC at 71.83. With reference to the published 1H and 13C-NMR data of β-sitosterol and stigmasterol,36 it was concluded that JC-2 contained both β-sitosterol and stigmasterol. The main differences between the two compounds, as reported by Cayme and Ragasa (2004)36 are 13C-NMR signals for C-20, C-22, C-23 and C-24. The experimental 13C-NMR signals for JC-2 for C-20, C-22, C-23 and C-24 were δC 36.28, δC 33.92/138.34, δC 26.39/129.3 and δC 46.07/51.26, respectively. It can also be deduced from the intensities of the 13C-NMR signals that β-sitosterol was the major constituent, whereas stigmasterol was the minor constituent. Pierre and Moses (2015) have reported studies in which it was difficult to obtain β-sitosterol in pure state.37 Further characterization of JC-2 by GC-MS was performed after a derivatization procedure38 by silylation with N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) at 60 °C for 30 minutes (Fig. 2A). MSTFA as a silylating agent is the most volatile trimethylsilyl (TMS) donor available.39 In this derivatization reaction, the hydroxyl group is converted into TMS-ether derivatives. The sterols were identified as their trimethylsilyl ether (TMS ether) (Fig. 2B and C) and were β-sitosterol (31.97%), ergosta-5, 22-diene-3-ol/24-methylene cholesterol (14.83%), stigmasterol (16.16%), campesterol (10.38%) and cholesterol (4.08%).40 Analytical HPLC-DAD analysis of fraction JDH (15–21) showed the presence of various compounds with peak 7 as the major peak (Fig. 3S†). Peak 7 was isolated by preparative HPLC-DAD and was characterized by 1H-NMR spectroscopy (Fig. 4S†). By comparing its data with the reported values,33 it was concluded that peak 7 was attributed to jaspamide.
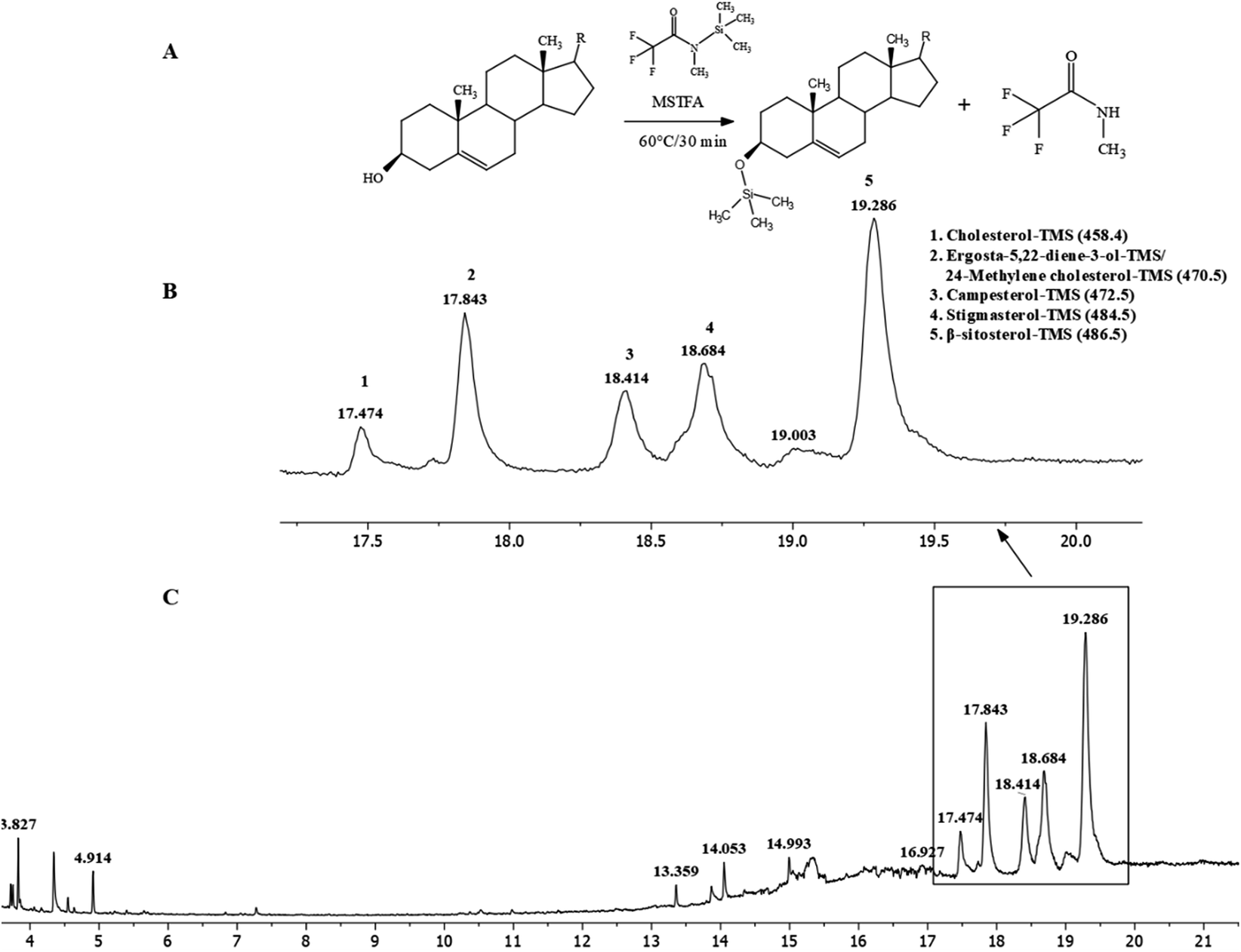 | ||
| Fig. 2 Derivatization reaction of JC-2 (A). Total ion chromatograms (TICs) of JC-2 (B) and (C). | ||
JDH-combined fractions and JC-2 were evaluated on the A172, TERA1, DLD1 and CCD18Co cell lines at different concentrations. Graphs of cell viability against the concentration of samples were plotted (Fig. 5S†), and fraction JDH (15–21) showed significant activity. Data were normalised to the control, and the IC50 values were calculated using GraphPad Prism software version 8.4.3. Fraction JDH (15–21) showed IC50 values of 1.09 ± 0.01 μg mL−1, 1.71 ± 0.21 μg mL−1, 0.78 ± 0.01 μg mL−1 and 0.67 ± 0.01 μg mL−1 on the A172, DLD1, TERA1 and CCD18Co cell lines, respectively. The presence of anticancer jaspamide (which was isolated and characterized as peak 7) in JDH (15–21) contributed to its activity.33 JC-2 displayed IC50 values of 3.37 ± 0.19 μg mL−1, 2.28 ± 0.20 μg mL−1, 4.94 ± 0.10 μg mL−1 and 2.47 ± 1.89 μg mL−1 on the A172, TERA1, DLD1 and CCD18Co cell lines, respectively. The presence of β-sitosterol (31.97%), stigmasterol (16.16%) and campesterol (10.38%)—which have reported anticancer activity—contributed to the activity of JC-2. β-Sitosterol displayed anticancer activities on various cancer cell lines and has been shown to act by enhancing apoptosis, inducing cell cycle arrest, bidirectionally regulating oxidative stress, improving metabolic reprogramming, inhibiting invasion and metastasis, modulating immunity and inflammation, and combating drug resistance. Preclinical evidence has shown that β-sitosterol exhibited multiple anticancer activities against different cancers such as liver, cervical, colon, stomach, breast, lung, pancreatic and prostate cancers, in addition to leukemia, multiple myeloma, melanoma and fibrosarcoma.41 However, its poor aqueous solubility and bioavailability coupled with low targeting efficacy limit its therapeutic efficacy and clinical application.41 Stigmasterol displayed anticancer activity on various types of cancers including breast, lung, liver, endometrial, skin, leukemia, gastric, gallbladder and ovarian by promoting apoptosis, inhibiting proliferation, metathesis and invasion.42 Campesterol showed apoptosis on U937 (human monocytic) cells and esophageal adenocarcinoma.43 It inhibited the growth of leukemia, hepatocarcinoma, prostate and ovarian cancer cells.44 It also increased the anticancer effects of anticancer agents on ovarian cancer cells and suppressed cell proliferation, cell cycle progression and cell aggregation.44
Synthesis of AuNPs using JDH, JDE, JC-2 and jaspamide
The synthesis of AuNPs normally takes place by mixing biological extracts such as plant, bacterial and fungal with a HAuCl4 solution in which Au3+ is reduced to gold atoms (Au0) by reducing agents present in the extracts. Growth and stabilization resulted in AuNP formation, which is indicated by a color change of the solution.19 Owing to the abundance of carboxyl, carbonyl, hydroxyl and phenol groups found in the natural source extracts, they reduced AuIII and stabilized the AuNPs with these groups.21 Marslin et al. (2018) have identified flavonoids, terpenoids, thiamine, phenolic compounds and/or carbohydrates, soluble proteins, polyphenols, terpenes, vitamins, hydroxyl group, carboxylic acid and polypeptides, among others, in plants for the reduction of AuIII.45The synthesis of AuNPs using the aqueous extract from the marine sponge Acanthella elongate has been reported by Inbakandan et al. (2010).29 The main challenge for the synthesis of AuNPs using JDH, JDE, JC-2 and jaspamide was their insolubility in water. Trial experiments performed with JDH, JDE and jaspamide at a concentration of 1 mg mL−1 in DMSO resulted in precipitation when added to water (10 mL) containing HAuCl4·3H2O (0.22 mM). JC-2 (1 mg mL−1) was not completely soluble in DMSO even when heated at 50 °C for 30 minutes. Jaspamide was soluble in DMSO but precipitated when added to a solution of HAuCl4·3H2O. Based on the modified Burst method by Masse et al. (2019) for the synthesis of ultrastable AuNPs as a new drug delivery system,46 and in a review by Ali et al., (2022) on the state-of-the-art silver and AuNP synthesis routes,47 new synthesis conditions were developed for the AuNPs using JDH, JDE and JC-2. Taking into consideration the solubility of JDH, JDE and JC-2 in ethyl acetate, the reaction was performed using mixtures of ethyl acetate, absolute ethanol and distilled water. JDH (3.83 mg), JDE (3.36) and JC-2 (3.24 mg) were dissolved in ethyl acetate (1 mL), and then absolute ethanol (3 mL) was added. The solutions were mixed vigorously using a vortex for complete solubility. In separate centrifuge tubes (50 mL), distilled water (27 mL) and solutions of JDH, JDE and JC-2 were added and mixed. Finally, HAuCl4·3H2O solution was added and mixed vigorously. The tubes were covered with aluminium foil and kept in dark for 24–48 h. The control consisted of distilled water (27 mL), ethanol (3 mL), ethyl acetate (1 mL) and HAuCl4·3H2O (0.22 mM). After 24–48 h, wine red solutions were obtained, which confirmed the synthesis of AuNPs. The formation of AuNPs in the control was slower in comparison to that of solutions containing JDE, JDH and JC-2, and ethanol acted as the reducing agent.48 The final concentration of HAuCl4·3H2O in JDH and JC-2 solutions was 0.22 mM, whereas for JDE, the concentration was reduced to 0.11 mM due to the aggregation of AuNPs after 24–48 h. The solutions were then subjected to dialysis (12–14 kDa dialysis) in distilled water for 2–3 days at room temperature to remove excess unreacted JDH/JDE/JC-2 and solvents. The percentage yield of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2—assuming full conversion of the initial mass of JDH, JDE and JC-2—with respect to HAuCl4·3H2O was 69.7%, 77.7% and 54.5%. The percentage yield of the control was 60.1%. The method was not suitable with jaspamide.
Characterization of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2
The UV spectra of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 showed a shift in the wavelength in comparison with the control (Fig. 3A). The intensity of the absorbance was higher for AuNPs-JDH in comparison with AuNPs-JC-2 and AuNPs-JDE. The formation of gold colloid is due to the surface plasmon resonance arising from the collective oscillation of free conducting electrons induced by an interacting electromagnetic field.49 A study by Woźniak et al. (2017) on the size- and shape-dependent cytotoxicity profile of AuNPs for biomedical applications showed that different shapes of AuNPs have different UV visible absorption spectra.50 Hence, it can be concluded that the JDH, JDE and JC-2 AuNPs were spherical in morphologies based on the reported absorption band of spherical AuNPs, which showed a wavelength (λ) of ∼530 nm.50 The size and morphology of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 were determined by TEM. The TEM images confirmed the spherical shape of the AuNPs (Fig. 3B). The average diameter of the AuNPs was calculated using the ImageJ software with measurement taken for over 100 nanoparticles. The average diameter of the AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 was 21.1 ± 3.0 nm, 20.7 ± 2.1 nm and 26.2 ± 1.2 nm, respectively (Fig. 3C), and was less than the average diameter of the control (28.3 ± 3.4 nm). EDX analysis showed the percentages of gold in AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 as 34.96 ± 4.39, 15.11 ± 4.91 and 27.23 ± 4.73, respectively. The percentage of gold in the AuNPs-control was 28.23 ± 4.48 (Fig. 3D).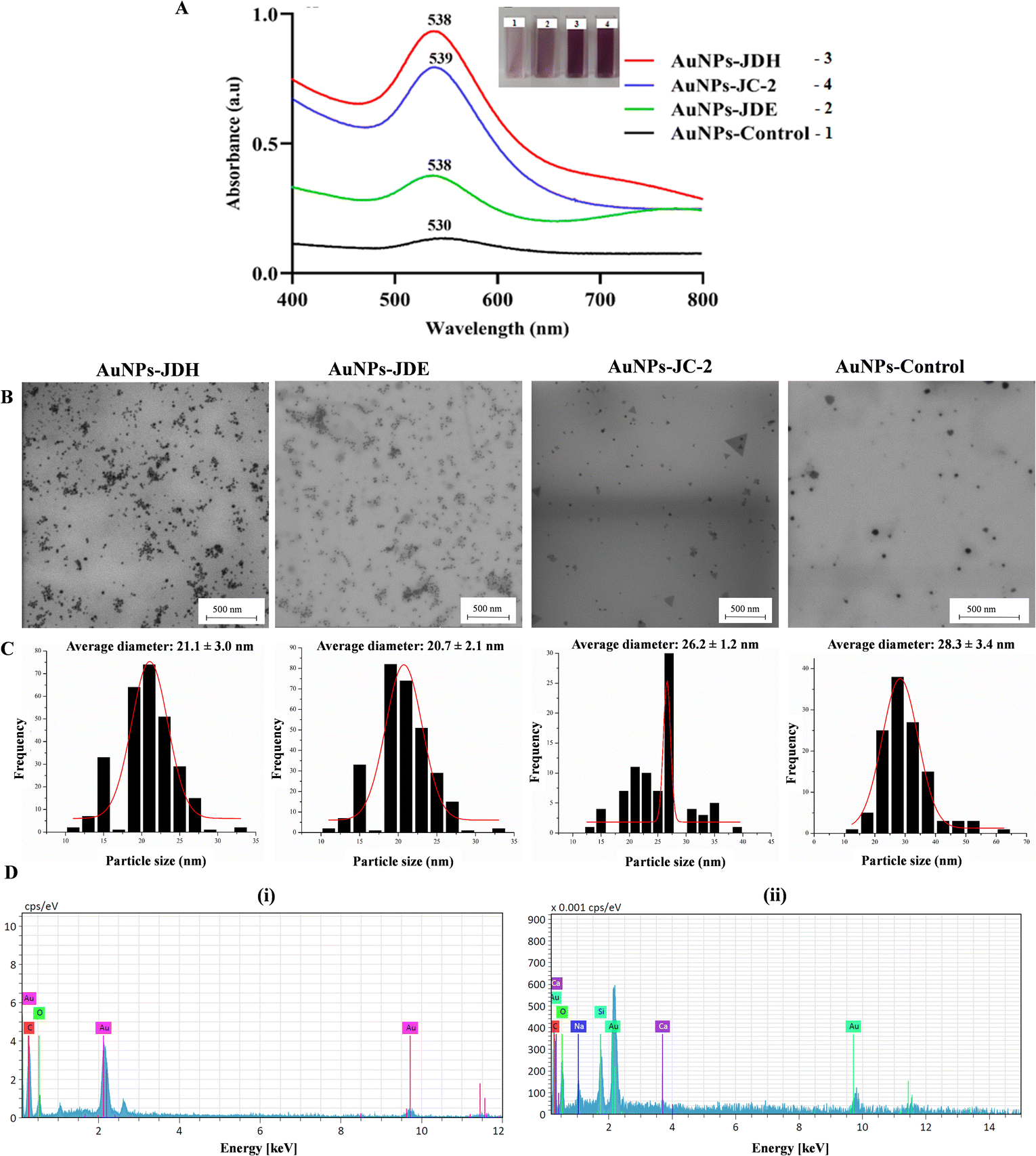 | ||
| Fig. 3 UV spectra of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 (A). TEM images (B) and average size distribution of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 in comparison with the control (AuNPs-control) (C). EDX spectra (D) of (i) AuNPs-JDH and (ii) AuNPs-control. | ||
The FTIR spectra of JDH, JDE and JC-2 together with their corresponding AuNPs, namely AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2, (Fig. 6S†) showed similar bands as follows: a band between 3421 and 3375 cm−1 assigned to the phenolic hydroxyl and aliphatic hydroxyl groups (–OH), between 2952 and 2849 cm−1 assigned to the symmetric stretching vibration of methyl (CH3) and methylene (CH2) groups, between 1710 and 1734 cm−1 assigned to the CO group and between 1376 and 1366 cm−1 attributed to the (C–H) bending and asymmetric stretching vibrations of the glycosidic bond (C–O–C).51
Biological activity of JDE, AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control
In order to assess the cytotoxicity of JDE, AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control, the A172, TERA1 and HaCaT cell lines were used. An MTT assay was carried out after 3 days of treatment of samples at concentrations of 25, 12.5, 6.3, 3.1, 1.6 and 0.8 μg mL−1 in triplicates with DMSO (0.25%) as the control (Fig. 4). Using percentage cell viability results, it can be deduced that AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control showed biocompatibility with healthy HaCaT cells at specific concentrations, except JDE extract which had a high cytotoxic effect on the healthy cells (Fig. 4A). AuNPs-control reduced cell viability on TERA1 and A172 cell lines. Data were normalised to the control, and the IC50 values were calculated using GraphPad Prism software version 8.4.3. AuNPs-JDE showed IC50 values of 2.82 ± 2.34 μg mL−1, 0.93 ± 0.50 μg mL−1 and 6.64 ± 2.62 μg mL−1 on A172, TERA1 and HaCaT cell lines, respectively. The reported IC50 values of native JDE were 1.60 ± 0.48 μg mL−1 on A172, 1.71 ± 0.42 μg mL−1 on TERA1 and 0.75 ± 0.04 μg mL−1 on HaCaT.33 AuNPs-JDH showed IC50 values of 1.19 ± 0.39 μg mL−1, 0.84 ± 0.20 μg mL−1 and 15.36 ± 2.16 μg mL−1 on A172, TERA and HaCaT cells, respectively. The reported IC50 values of native JDH were 2.98 ± 1.86 μg mL−1 on A172, 2.27 ± 1.29 μg mL−1 on TERA1 and 15.55 ± 3.82 μg mL−1 on HaCaT.33 The IC50 values of AuNPs-control on TERA1 and A172 were 0.93 ± 0.08 μg mL−1 and 2.11 ± 0.64 μg mL−1, respectively. Additional assays were performed with JDE, AuNPs-JDE, JDH, AuNPs-JDH and AuNPs-control on the HeLa and HepG2 cell lines. Pure jaspamide was also tested and nocodazole was used as the positive control. Using percentage cell viability results for samples tested at concentrations of 0.001, 0.01, 0.1 and 1 μg mL−1 (Fig. 5), it can be deduced that AuNPs-control was not toxic on HeLa and HepG2 cancer cells. On HeLa cells, JDE and JDH showed IC50 values of 2.41 ± 0.31 μg mL−1 and 8.79 ± 3.84 μg mL−1, respectively, whereas the IC50 values of AuNPs-JDE and AuNPs-JDH were 1.08 ± 0.09 μg mL−1 and 1.58 ± 0.11 μg mL−1. On HepG2 cells, the IC50 values of JDE and JDH were 0.96 ± 0.81 μg mL−1 and 3.76 ± 0.86 μg mL−1, respectively whereas the IC50 values of AuNPs-JDE and AuNPs-JDH were 0.40 ± 0.17 μg mL−1 and 1.88 ± 0.65 μg mL−1, respectively. The IC50 values of nocodazole on HeLa and HepG2 cells were 0.84 ± 0.04 μg mL−1 and 0.65 ± 0.26 μg mL−1, respectively, whereas the IC50 values of jaspamide on HeLa and HepG2 cells were 1.76 ± 0.02 μg mL−1 and 1.36 ± 0.21 μg mL−1, respectively. A summary of the IC50 values of JDH, AuNPs-JDH, JDE, and AuNPs-JDE on the A172, TERA1, HaCaT, HeLa and HepG2 cell lines is shown in Fig. 6. When JDH was coupled with AuNPs, the cytotoxicity was increased on A172, TERA, HeLa and HepG2 cells but showed similar activity on HaCaT cells. The cytotoxicity of JDE was reduced when coupled with AuNPs on A172 and HaCaT cells but increased on TERA1, HeLa and HepG2 cells. As shown by Ramanjooloo et al., (2023), anticancer jaspamide is the main constituent of JDE and JDH extracts and its content was 36.15 ± 0.73% and 0.60 ± 0.16%, respectively.33 The cytotoxicity of JDE and JDH was due to the contribution of jaspamide and its analogs, and the presence of other secondary metabolites reported from the genus Jaspis.33 Herein, the technique used to synthesize AuNPs was not successful with pure jaspamide. Therefore, less activity was expected with AuNPs-JDE and AuNPs-JDH, as jaspamide present in JDE and JDH could not be converted into AuNPs. However, the presence of other secondary metabolites in JDH and JDE such as sterols, jaspisamides, bengamides, sphingosine, bengazole alkaloids and nucleosides, reported from the genus Jaspis,52 could have been converted into their nanoparticle form and hence contributed to the enhanced activity of AuNPs-JDH on A172, TERA, HeLa and HepG2 and AuNPs-JDE on TERA1, HeLa and HepG2 cells. The enhanced anticancer activity of AuNPs-extracts could be attributed to the synergistic role of AuNPs and the extracts, the sustained released of bioactive compounds from the extracts, the size and surface of the AuNPs and their interactions with cancer cells.53,54 Ramanlingan et al., (2016) have shown that AuNPs caused apoptosis in A549 cells through oxidative stress, ROS generation, sensitization of mitochondrial membrane and cell cycle arrest.55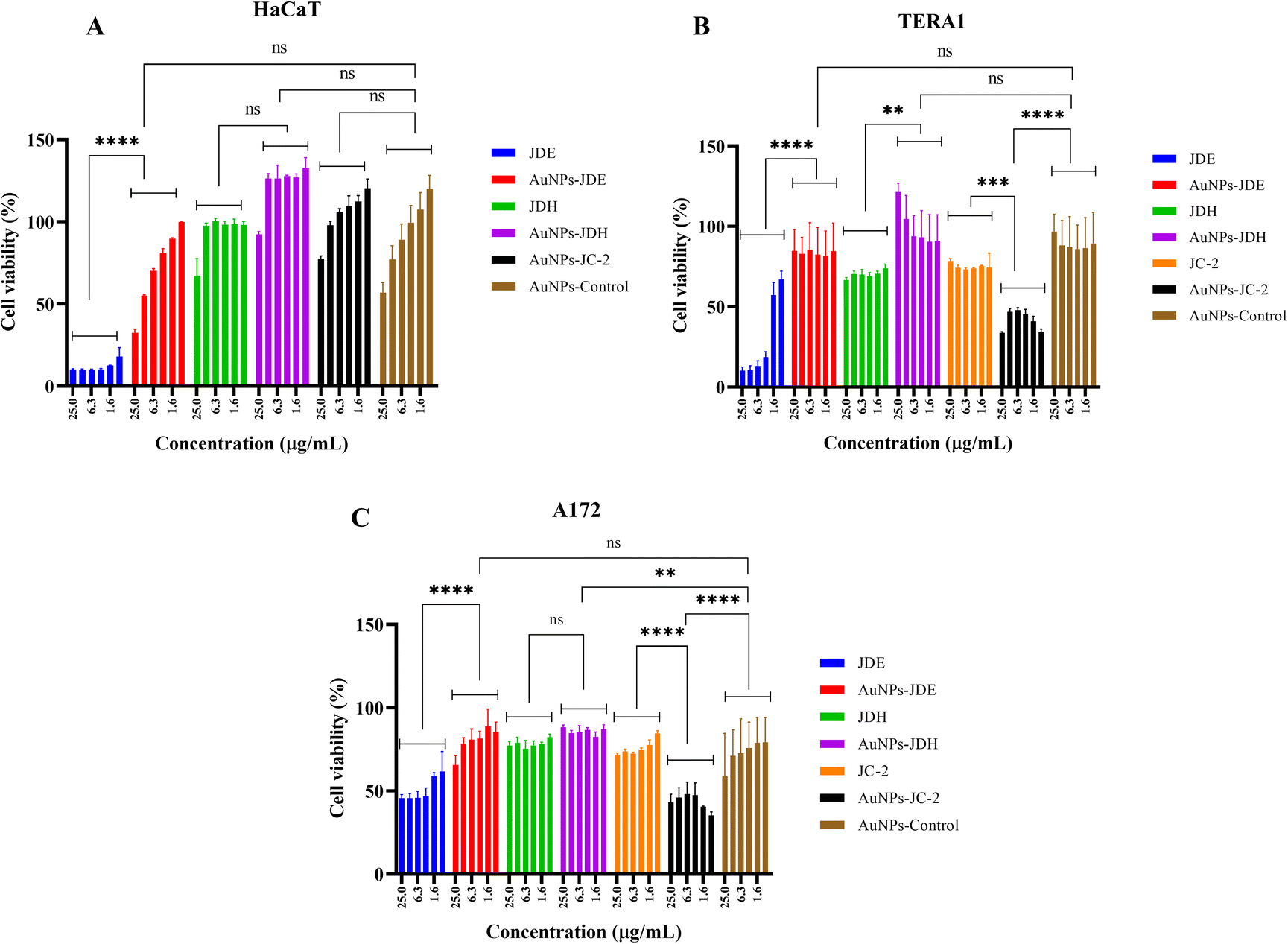 | ||
| Fig. 4 Cell viability (%) of JDE, AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control on HaCaT (A), TERA1 (B) and A172 (C) cell lines. Results are presented as mean ± standard deviation (n = 3). Asterisks represent significant differences between (i) JDE/JDH/JC-2 and their conjugated AuNPs and (ii) conjugated AuNPs and control (AuNPs-control); **p < 0.01, ***p < 0.001 and ****p < 0.001 were considered statistically significant, ns = non-significant. | ||
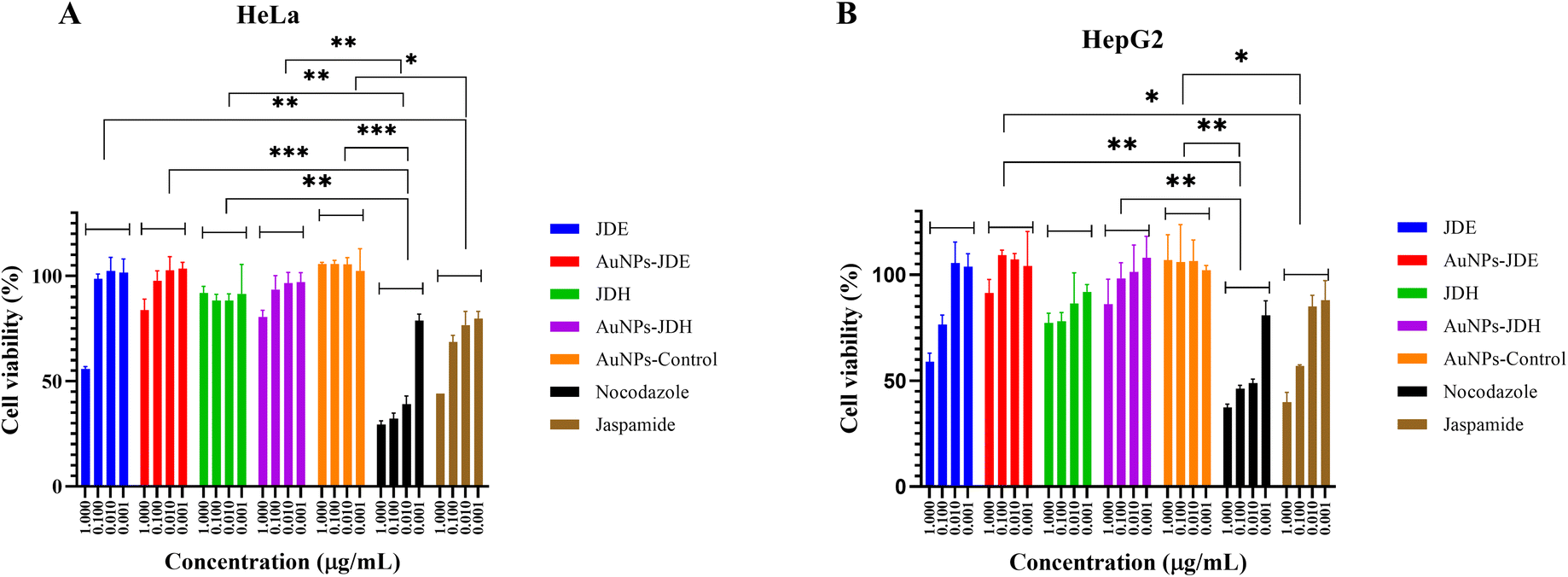 | ||
| Fig. 5 Cell viability (%) of JDE, AuNPs-JDE, JDH, AuNPs-JDH, AuNPs-control, nocodazole and jaspamide on the HeLa (A) and HepG2 (B) cell lines. Asterisks represent significant differences between significant comparisons only; *p < 0.05, **p < 0.01 and ***p < 0.001 were considered statistically significant. Other comparisons were non-significant. | ||
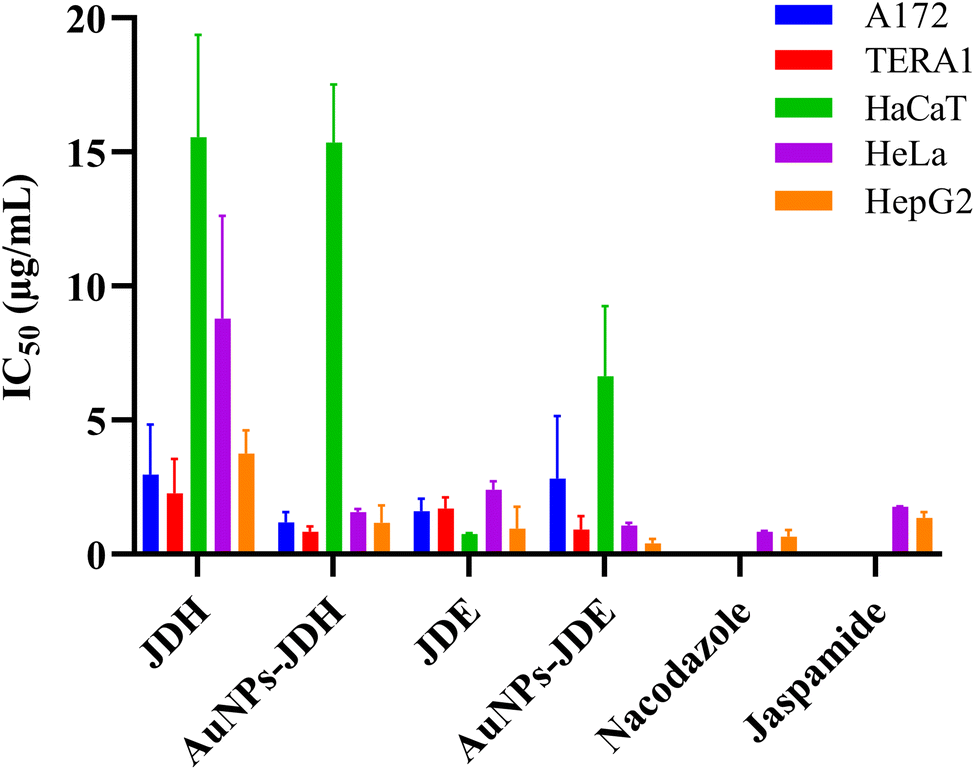 | ||
| Fig. 6 Comparison of IC50 values of JDH, AuNPs-JDH, JDE, AuNPs-JDE, nocodazole and jaspamide. | ||
To determine the toxicity of JDE, JDH, AuNPs-control, AuNPs-JDE, AuNPs-JDH and jaspamide, further investigation was performed by light microscopic imaging. HT-29 cells were treated with the samples at 0.1, 1, 10 and 100 μg mL−1, and light microscopic images were acquired (Fig. 7). A whitish color observed with HT-29 cells indicated toxicity.56 Thus, it was deduced that jaspamide was toxic at all concentrations. JDE was not toxic at 0.1 μg mL−1 but showed toxicity at 1, 10 and 100 μg mL−1. JDH was toxic at 100 μg mL−1 only. The nanoparticles, AuNPs-control, AuNPs-JDE and AuNPs-JDH were not toxic at 0.1, 1.0 and 10 μg mL−1. In a study of cell viability assay of AuNPs on HT-29, a significant reduction of viable cells was observed with no genotoxic effects.57,58
 | ||
| Fig. 7 Light microscopy images of HT-29 cells treated with jaspamide, JDE, JDH, AuNPs-control, AuNPs-JDE and AuNPs-JDH. The whitish colour indicates toxicity. | ||
AuNPs-JC-2 showed more activity than JC-2 on cancer cells A172 and TERA1 (Fig. 4B and C). Its IC50 values were 0.52 ± 0.09 μg mL−1 and 0.78 ± 0.28 μg mL−1 on A172 and TERA1 cells, respectively, whereas the IC50 values of JC-2 were 3.37 ± 0.19 μg mL−1 and 2.28 ± 0.20 μg mL−1, respectively on the same cell lines. AuNPs-JC-2 displayed an IC50 value of 6.81 ± 1.00 μg mL−1 on the HaCaT cell line. JC-2 was shown to be a mixture of sterols with β-sitosterol (31.97%), ergosta-5, 22-diene-3-ol/24-methylene cholesterol (14.83%), stigmasterol (16.16%) and campesterol (10.38%) as the main constituents. The enhanced activity of AuNPs-JC2 compared to JC-2 could be due to the more efficient uptake of the nanoparticles by the cancer cells. Similarly, Dhar et al. (2008) have reported enhanced activity of doxorubicin hydrochloride (DOX)-loaded AuNPs compared to free DOX due to improved internalization of drug-loaded nanoparticles by an endocytosis mechanism compared to the passive diffusion mechanism of free DOX into cells.59 Zhang et al. (2011) have shown that ultra-small AuNPs conjugated to doxorubicin were 20 times more cytotoxic in B16 melanoma cells than the equivalent concentration of doxorubicin alone. Doxorubicin remained highly active when conjugated to ultrasmall gold nanoparticles by entering into the cell nuclei.60
Synthesis of gold nanoparticles using contignasterol, ansellone, motuporamine A and MN100
Contignasterol, ansellone A, motuporamines A and MN100 were provided by Prof. Raymond Andersen.61 Contignasterol, a highly oxygenated steroid, was first isolated from the sponge Petrosia contignata by Burgoyne and Andersen in 1992.61,62 It displayed anti-allergic, anti-asthma and anti-thrombolytic activities and may be used for the prevention of inflammatory or allergic reactions or the treatment of cardiovascular or haemodynamic disorders.63 Ansellone A, a sesterpenoid, was isolated from the nudibranch Cadlina luteomarginata and the sponge Phorbas sp. It activates the cAMP signal transduction pathway without a ligand/receptor binding event64,65 and has gained interest due to its activity as an HIV latency-reversing agent (LRA).66 Williams et al., (1998) isolated a mixture of motuporamines A, B and C from the extracts of the sponge Xestospongia exigua. They showed a modest in vitro cytotoxicity with an IC50 value of 0.6 μg mL−1 against a panel of human cancer cell lines.67 The motuporamines inhibit the invasion of metastatic MDA-231 breast carcinoma cells and motuporamine A showed concentration-dependent activity with an IC50 value of 3 μM.68 MN100 synthesis was inspired from the structure of pelorol, isolated from the sponge Dactylospongia elegans.69 It showed three times greater SHIP1 (a new therapeutic target for cancer) activation than pelorol at the same concentration.70The synthesis of gold nanoparticles using contignasterol, ansellone A, motuporamine A and MN100 was performed using the new synthesis conditions. After 24 h, wine red solutions were obtained with contignasterol and ansellone whereas no colour change was observed with motuporamine A and MN100 as they precipitated in the solutions. With ansellone A, the concentration of HAuCl4·3H2O was reduced to 0.11 mM to avoid the aggregation of AuNPs. The red solutions were subjected to dialysis (12–14 kDa dialysis) in distilled water for 2–3 days at room temperature to remove excess of unreacted compounds and solvents. After dialysis, the conjugated AuNPs were characterized using UV-vis spectroscopy, TEM and EDX spectroscopy.
El-Seedi et al., (2019) has reported carbonyl, hydroxyl, amino and methoxide groups as the main functional groups involved in the reduction of metal ions.71 The formation of AuNPs was successful with contignasterol and ansellone A only. Although carbonyl and hydroxyl groups are present in jaspamide, AuNPs were not formed. The same observation was made with motuporamine A and MN100 although amines, tertiary amides and phenolic compounds have been reported for the reduction of Au(III).45
Characterization of AuNPs-contignasterol and AuNPs-ansellone A
The UV-visible spectra of AuNPs-contignasterol and AuNPs-ansellone A nanoparticles and the control showed maximum wavelengths of 539 nm, 536 nm and 540 nm, respectively (Fig. 8A) for the red colour of gold colloid and the formation of spherical gold nanoparticles.49 The intensity of the absorbance of AuNPs-contignasterol was higher than that of AuNPs-ansellone A due to the higher concentration of HAuCl4·3H2O used. The TEM images confirmed the spherical shape of AuNPs-ansellone A and AuNPs-contignasterol (Fig. 8B). The average diameters of AuNPs-control, AuNPs-ansellone A and AuNPs-contignasterol were calculated using the ImageJ software with measurements taken for over 100 nanoparticles and were 28.3 ± 4.8 nm, 33.3 ± 5.1 nm and 30.8 ± 5.5 nm, respectively (Fig. 8C). The EDX analysis of AuNPs-control, AuNPs-ansellone A and AuNPs-contignasterol showed percentages of gold as 44.99 ± 1.55, 34.63 ± 3.93 and 56.23 ± 2.42, respectively.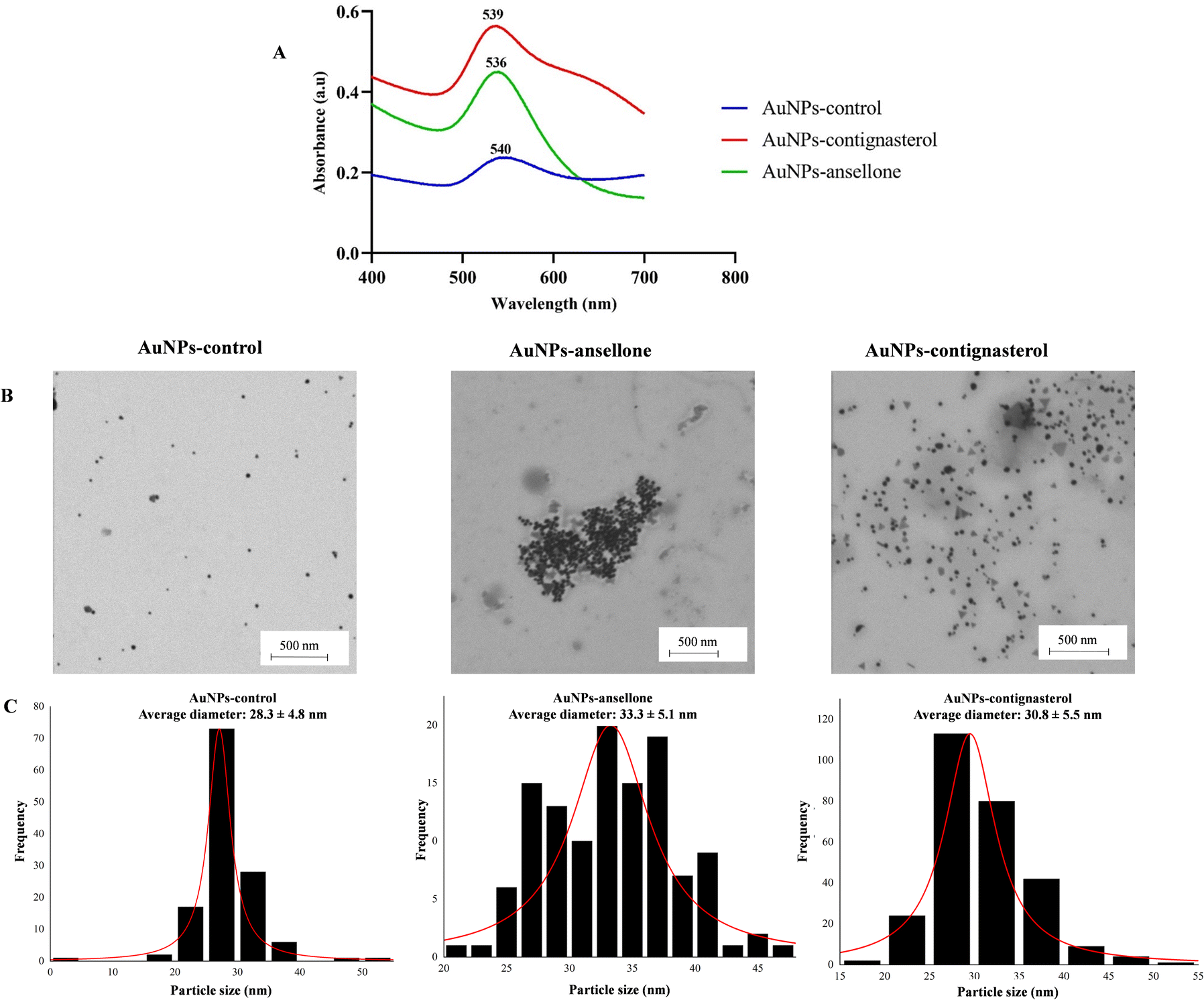 | ||
| Fig. 8 UV-visible spectra of AuNPs-contignasterol, AuNPs-ansellone A and AuNPs-control (A). TEM images (B) of AuNPs-control, AuNPs-ansellone A and AuNPs-contignasterol together with their particle size distribution (C). | ||
Biological activity of AuNPs-contignasterol and AuNPs-ansellone A
Contignasterol and ansellone A and their conjugated AuNPs were evaluated on the TERA1, A172 and HaCaT cell lines. The data were normalised to the control, and the IC50 values were calculated using GraphPad Prism software version 8.4.3. On the TERA1 cell line, no significant activity was observed with contignasterol and ansellone and their conjugated AuNPs. On the A172 cell line, ansellone A showed an IC50 value of 9.90 ± 2.50 μM, whereas the IC50 value of AuNPs-ansellone was 9.60 ± 5.44 μM. Contignasterol and AuNPs-contignasterol displayed IC50 values of 8.39 ± 1.74 μM and 18.22 ± 3.22 μM, respectively. On the HaCaT cell line, ansellone A and AuNPs-ansellone A showed IC50 values of 7.66 ± 1.46 μM and 8.80 ± 4.65 μM, respectively. Contignasterol and AuNPs-contignasterol displayed IC50 values of 7.42 ± 0.64 μM and 7.10 ± 2.20 μM, respectively. Thus, it can be deduced that the biological activity observed with contignasterol and ansellone A and their conjugated AuNPs were cell line specific. Based on the IC50 values, AuNPs-contignasterol displayed less activity than contignasterol on the A172 cell line and there was no significant difference of activity on the HaCaT cell lines. No significant difference in activity was also observed with AuNPs-ansellone A and ansellone A on A172 and HaCaT cell lines.Zeta potential analysis
The values of zeta potential of AuNPs-JDH, AuNPs-JC-2, AuNPs-JDE, AuNPs-ansellone, AuNPs-contignasterol and AuNPs-control were −32.03 ± 1.18 mV, −39.43 ± 0.90 mV, −34.73 ± 0.05 mV, −29.57 ± 0.91 mV, −14.57 ± 0.90 mV and −12.6 ± 1.56 mV, respectively. AuNPs with low zeta potentials favoured the enhanced permeability and retention (EPR) effect resulting in enhanced internalization of AuNPs by cancer cells, which have positively charged surfaces.72Internalization of AuNPs on A549 cells
The internalization of AuNPs on A549 cells73 was investigated by fluorescence microscopy74 and ICP-MS.75 A549 cells were exposed to AuNPs that were bound to fluorescein-5-isothiocyanate (FITC). FITC can effectively bind to AuNPs through electronically stable bonds with its isothiocyanante.76 Fig. 9 shows the images of green fluorescence, observed at the same intensity, of FITC-labeled AuNPs in comparison with controls (cells bound with and without FITC). It can be deduced that FITC-labelled AuNPs were more prominent than the controls, suggesting the internalization of AuNPs. | ||
| Fig. 9 Cellular uptake of gold nanoparticles (20× magnification). | ||
ICP-MS was performed to confirm the internalization of AuNPs. Fig. 10 shows the internalization of gold and number of AuNPs internalized per cell. The total amount of gold internalized ranged from 0.13 to 17.69 pg. AuNPs-JDE showed the highest uptake followed by AuNPs-JDH, AuNPs-ansellone, AuNPs-control, AuNPs-JC-2 and AuNPs-contignasterol. The number of AuNPs internalized per cell ranged from 2.62 to 9.65. AuNPs-ansellone showed the highest AuNPs internalized followed by AuNPs-contignasterol, AuNPs-control, AuNPs-JDH, AuNPs-JC-2 and AuNPs-JDE. AuNPs could be internalized in cells by either receptor-mediated endocytosis, or non-specific receptor independent endocytosis depending on the shape, size, surface charge and functionalization of the AuNPs.77 The common pathway for AuNPs with functionalized, charged or neutral charge is by clathrin-mediated endocytosis (i.e. binding of AuNPs to specific receptors on the surface of the cells).75,77
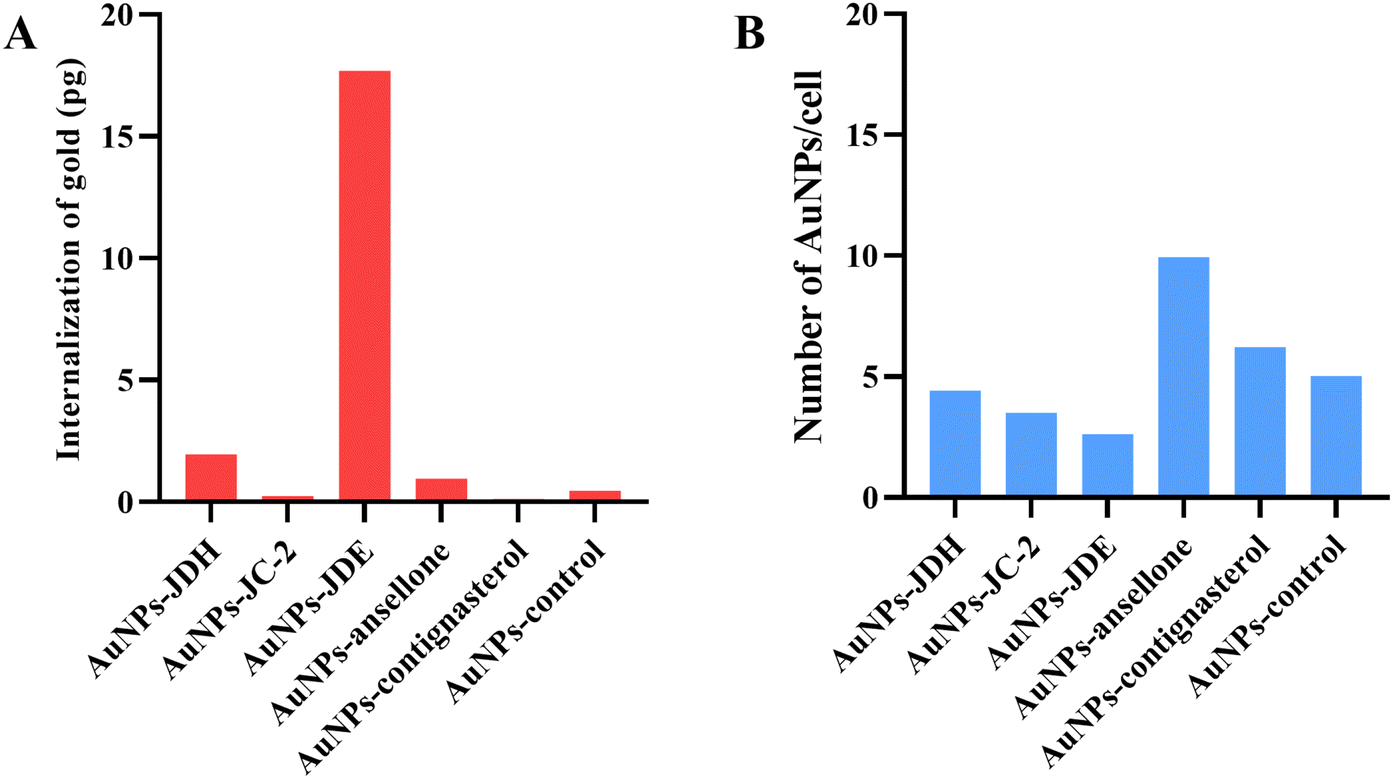 | ||
| Fig. 10 Internalization of gold nanoparticles (A) and number of AuNPs taken up per cell (B). | ||
Conclusion
AuNPs have been successfully synthesized using hexane (JDH), ethyl acetate (JDE) and sterols (JC-2) obtained from the marine sponge Jaspis diastra under new synthesis conditions. The method is suitable for poorly water-soluble extracts and sterols. The cytotoxicity of AuNPs-JDH increased on A172, TERA, HeLa and HepG2 cells but showed similar activity on HaCaT cells in comparison with JDH. For AuNPs-JDE, the cytotoxicity reduced on A172 and HaCaT cells but increased on TERA1, HeLa and HepG2 cells in comparison with JDE. AuNPs-JC-2 was more active than native JC-2. The synthesis of conjugated AuNPs was successful only with contignasterol and ansellone A. AuNPs-contignasterol displayed less activity than contignasterol on the A172 cell line, and there was no significant difference in the activity of AuNPs-ansellone A and ansellone A on the A172 and HaCaT cell lines. Future work would be evaluating further biological activity of AuNPs-JC-2 and elucidating its mechanism of action.Experimental
Materials and methods
Methanol, ethyl acetate, hexane and dichloromethane were acquired from LOBA CHIMIE PVT LTD. Silica gel, 60 (0.06–0.2 mm), used for column chromatography (70–230 mesh ASTM) was purchased from Scharlau, Turkey. Glass support (silica gel 60G F254) plates of dimensions 20 × 20 cm, suitable for thin-layer chromatography (TLC) were obtained from Merck, Germany. N-Methyl-N-trimethylsilyl-trifluoroacetamide and gold(III) chloride trihydrate (HAuCl4 3H2O) (≥99.9% trace metal basis) were purchased from Sigma-Aldrich. The cell lines TERA1, A172, CCD18Co and DLD1 were obtained from Cellonex, Randburg, South Africa. Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute (RPMI 1640) were purchased from Sigma-Aldrich (MO, USA). Additionally, gamma-irradiated and heat-inactivated fetal bovine serum (FBS), penicillin–streptomycin (PS), high glucose growth medium and phosphate-buffered saline (PBS) (1×) were purchased from PAN Biotech, Bavaria, Germany. The cell proliferation kit (MTT) was acquired from Sigma-Aldrich, USA.Sample collection and identification
The sponge used in this study was provided by the Mauritius Oceanography Institute (MOI). It was collected at Black River, Mauritius, by scuba diving at a depth of 5–32 m. Professor Rob Van Soest, from the Naturalis Biodiversity Center, in the Netherlands, taxonomically identified the sponge as Jaspis diastra. The sponge was given a voucher number of ZMAPOR21796 and was stored at the Zoological Museum of the Center. Vacelet and Vasseur first identified Jaspis diastra in 1965. It is orange and has a very soft texture with prominent oscules. Its family and order are Coppatiidae and Astrophorida, respectively.33,78Extraction procedures
The procedures described by Ramanjooloo et al. (2023) were used. Briefly, freeze-dried sponges (50.31 g) were macerated in methanol and dichloromethane (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 24–48 hours. This step extracts low-, medium- and high-polarity secondary metabolites. After maceration, the solution was filtered and concentrated using a rotatory evaporator, to give the crude extract (11.06 g). Distilled water was added to the crude extract to dissolve it and then selective extraction was performed with hexane and ethyl acetate. The hexane and ethyl acetate fractions were then concentrated to ¼ of their initial volume using a rotatory evaporator and any traces of water present in the fractions were removed using magnesium sulfate. The resulting fractions were filtered and concentrated to obtain hexane (JDH) (0.87 g) and ethyl acetate (JDE) (0.10 g) extracts.33,78
:1) for 24–48 hours. This step extracts low-, medium- and high-polarity secondary metabolites. After maceration, the solution was filtered and concentrated using a rotatory evaporator, to give the crude extract (11.06 g). Distilled water was added to the crude extract to dissolve it and then selective extraction was performed with hexane and ethyl acetate. The hexane and ethyl acetate fractions were then concentrated to ¼ of their initial volume using a rotatory evaporator and any traces of water present in the fractions were removed using magnesium sulfate. The resulting fractions were filtered and concentrated to obtain hexane (JDH) (0.87 g) and ethyl acetate (JDE) (0.10 g) extracts.33,78
Purification of JDH
The purification of JDH was performed by column chromatography. A glass column of diameter 3.5 cm and length 60 cm was used. It was packed with silica gel (30 g) that was first stirred in hexane (100 mL) before transferring to the column. The length of the packed silica in the column was about 15 cm. The sample was loaded on the column using the dry loading method. JDH (2.04 g) was transferred into a round-bottom flask and hexane was added to dissolve it. Silica (2.0 g) was then added and the mixture was dried using a rotary vacuum evaporator (temperature was set at 40 °C). The resulting dried silica, which contained JDH was added to the packed column. The gradual elution of solvents was done first with non-polar solvent (hexane), followed by mixtures of hexane and ethyl acetate (semi-polar solvent) in various ratios and then ethyl acetate only. Afterwards, a mixture of ethyl acetate and methanol (polar solvent) in various rations was used. The eluent (10–15 mL) was collected in test tubes. Similar fractions were combined together based on thin layer chromatography (TLC) observations. A total of twenty-five fractions were obtained and were combined as follows: JDH (2–3) – (469.2 mg), JDH (5–7) – (211.6 mg), JDH (10–14) – (64.3 mg), JDH (15–21) – (68.1 mg) and JDH (22–25) – (48.5 mg). White precipitates were observed in fractions JDH (8), (9) and (10–14). They were separated, dried and weighted. The mass of precipitates obtained from fractions JDH (8), JDH (9) and JDH (10–14) were 102 mg, 64 mg and 10 mg, respectively. TLC revealed that the precipitates were similar. Therefore, only precipitates from JDH (8), termed JC-2, were selected for biological evaluation. Cytotoxic assay was then conducted with these fractions and JC-2, which were stored at −20 °C until use.Biological assays of combined JDH fractions and JC-2
The procedures described by Ramanjooloo et al. (2023) were used. Combined JDH fractions, namely JDH (2–3), JDH (5–7), JDH (10–14), JDH (15–21), JDH (22–25) and JC-2, were evaluated on three cancer cell lines, namely, human brain cancer—glioblastoma (A172), human colorectal carcinoma (DLD1), human testicular embryonal carcinoma (TERA1) and one normal cell line namely human colon cells (CCD18Co). Dulbecco's Modified Eagle's Medium (DMEM) was used to culture A172, CCD18Co and DLD1 cells, while Roswell Park Memorial Institute (RPMI)-1640 was used to culture TERA1 cells. Fetal Bovine Serum (FBS) 10% (v/v) and penicillin–streptomycin antibiotics 1% (v/v) were added to the growth media. A humidified incubator with a temperature set at 37 °C and 5% of carbon dioxide (CO2) was used to maintain the cells. A saturation of 90% was necessary for sub-culturing. Afterwards, the cells were detached and enumerated using a haemocytometer and microscope (inverted light). The cells were seeded in microplates (96 well) at the required densities.33Cell culture and treatment with JDH fractions and JC-2
Cells from A172, CCD18Co, DLD1 and TERA1 at a density of 6.0 × 104 cell per mL were transferred into 96-well plates. A cell suspension (90 μL) was added to each microplate well and incubated at 37 °C in a humidified atmosphere (5% CO2) for 24 h for cell attachment. Combined JDH fractions and JC-2 were dissolved in DMSO (100%). The adhered cells were then treated with diluted concentrations—prepared using PBS (1×)—of each fraction and JC-2 in triplicates to obtain final concentrations of 25, 12.5, 6.3, 3.1, 1.6 and 0.8 μg mL−1. After 72 h of incubation, an MTT cell viability assay was performed. The final concentration of DMSO in the well was 0.7% and the positive control was 10% of DMSO.Cell viability
The MTT colorimetric assay was used for cell viability. It uses 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to assess cellular metabolic activity in terms of viability, proliferation or cytotoxicity. Combined JDH fractions and JC-2 solutions (10 μL) and media (90 μL) were added to the cells and treated for 72 h. Afterwards, using a multichannel pipette, an MTT solution (10 μL, 5 mg mL−1 MTT) was transferred to each well and the plates were incubated in a humidified incubator (37 °C and 5% CO2) for 3 h. Subsequently, formazan crystals were formed and were dissolved by adding DMSO (100 μL). Complete solubilization of the crystals was achieved after overnight incubation. The blank was the microplate wells (in triplicate), which consisted of the growth medium and DMSO (0.7%). The absorbance was recorded at 570 nm using a microplate reader (Victor X3-multimode, PerkinElmer, Massachusetts, USA). The percentage cell viability was calculated using eqn (1).79
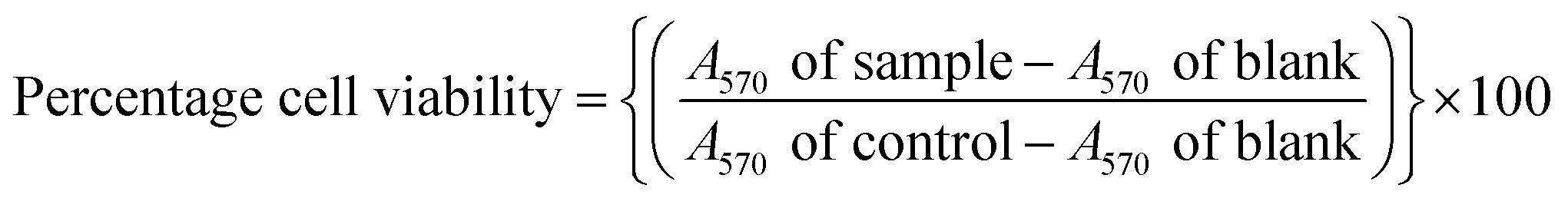 | (1) |
Isolation and characterization of pure compounds from fraction JDH (15–21)
Reverse-phase HPLC was performed to isolate pure compounds from fraction JDH (15–21). The HPLC used was Waters 1525 Binary HPLC pump attached to a Waters 2998 Photodiode Array Detector. All solvents were filtered through a 0.45 μm filter (Osmonics Inc.) prior to use. TLC was performed with Merck silica gel plates (Type 5554). JDH (15–21) (68.1 mg) was dissolved in acetonitrile (2 mL) before injection. The chromatographic conditions are as follows: column: (InertSustain column, C-18; 250 × 10 mm), mobile phase (ultra-pure water and acetonitrile); column temperature: room temperature; flow rate: 2.0 mL min−1; injection volume: 100 μL; wavelengths: 222 and 197 nm. Gradient elution was set up under the following conditions: from 0 to 65 min the mobile phase was water:acetonitrile (50:50)%, then the mobile phase was changed to acetonitrile (10:90)% from 65 to 80 min with 5 min of stabilization. Finally, at 90 min, the solvent system was water:acetonitrile (50:50)% with 15 min of stabilization. Each peak coming out of the column was collected individually. The procedures were repeated several times and the final volume collected was concentrated to dryness for NMR characterization. The mass obtained for peaks 1 to 8 was 0.6, 1.2, 1.7, 0.4, 0.4, 8.3 and 1.6 mg, respectively.
1H and 13C NMR spectra were recorded using a Bruker AV-600 spectrometer with a 5 mm CPTCI cryoprobe at the University of British Columbia (UBC). 1H chemical shifts were referenced to the DMSO-d6 signal (2.5 ppm) and 13C chemical shift was referenced to the DMSO-d6 solvent peak (39.5 ppm). Low-resolution ESI-QIT-MS were recorded using a Bruker-Hewlett Packard 1100 Esquire-LC system mass spectrometer.
Characterization of JC-2 from JDH (8)
JC-2 was characterized by FTIR spectroscopy and 1H and 13C NMR (250 MHz) available at the University of Mauritius. FTIR spectroscopy was performed using a Bruker Alpha ATR Infrared Spectrometer, where 32 scans were collected. 1H and 13C NMR (for JC-2) spectroscopies were performed using a 250 MHz Bruker Electrospin spectrometer in CDCl3.Derivatization of JC-2 and GC-MS (gas chromatography-mass spectrometry) analysis
The derivatization of the sterols in JC-2 was performed by adding N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) (40 μL) to JC-2 (∼2 mg) in a glass vial, which was kept at 60 °C for 30 minutes. After derivatization, the vial was dried under nitrogen at 60 °C and then reconstituted in n-heptane before GC-MS analysis.An Agilent GC/MSD (Gas chromatography/Mass selective detector) was used for the analysis of the derivatized sterols. A fused silica capillary GC column for the Rxi-5 ms (inner diameter, 0.25 mm; column length, 30 m; film thickness, 0.25 μm) was installed on a GC system with helium gas (purity: >99.9 vol%) flowing as the carrier gas. The spitless injection mode was employed. The injection room temperature was set to 60 °C and the injection volume was 1 μL. The setting for the oven is as follows: 60 °C (1.0 min), 120 °C (30°C min−1) for 3 minutes, 300 °C (15°C min−1) for 23 minutes. The mass ion source and quadrupole were programmed at 230 °C and 150 °C, respectively. MS data were recorded using the full scanning mode from 40 to 580 (amu). Ionization was achieved using the electron impact mode at 70 eV.
Synthesis of AuNPs
JDH (3.83 mg), JDE (3.36) and JC-2 (3.24 mg) were dissolved in ethyl acetate (1 mL), and then absolute ethanol (3 mL) was added and mixed vigorously using a vortex for complete solubility. In separate centrifuge tubes (50 mL), distilled water (27 mL) and solutions of JDH, JDE and JC-2 were added and mixed. A HAuCl4·3H2O (9.24 mM) solution was prepared by dissolving HAuCl4·3H2O (30.2 mg) in distilled water (8.3 mL). HAuCl4·3H2O solution (750 μL) was added to JDH and JC-2 solutions, whereas 375 μL was added to a JDE solution. The final concentration of HAuCl4·3H2O in JDH and JC-2 solutions was 0.22 mM, whereas for JDE, the concentration was 0.11 mM. The tubes were covered with aluminum foil and kept in the darkness for 24–48 h. The control was treated in the same way. After 24–48 h, solutions were then subjected to dialysis (12–14 kDa dialysis) in distilled water for 2–3 days at room temperature. After dialysis, the solutions were centrifuged at 4000 rpm in centrifugal filter tubes (15 mL) with a molecular weight cut off (MWCO) of 10 kDa using the Thermo Scientific SL 16R equipment. The procedure was repeated to obtain concentrated solutions of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2. They were then freeze-dried. The mass of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2 obtained were 4.61 mg, 3.69 mg and 3.28 mg, respectively. The mass obtained with the control was 1.67 mg. The theoretical yields of AuNPs-JDH, AuNPs-JDE and AuNPs-JC-2—assuming full conversion of the initial mass of JDH, JDE and JC-2—with respect to HAuCl4·3H2O used were 6.61 mg, 4.75 mg and 6.02 mg, respectively. The theoretical yield of the control was calculated as 2.78 mg.Characterization of AuNPs
UV-visible spectroscopy, Transmission Electron Microscopy (TEM), Energy-Dispersive X-ray (EDX) spectroscopy and Fourier Transform Infrared (FTIR) spectroscopy were performed to characterize the AuNPs. The UV-visible analysis was performed after dialysis of AuNPs-JDH, AuNPs-JDE, AuNPs-JC-2 and AuNPs-control with wavelengths spanning from 200 to 800 nm using the equipment Libra S2 UV/visible spectrophotometer (Biochrom Ltd). Samples (3–5 μL) were transferred onto TEM (carbon) grids and letting them to absorb for 24 h. The size and structure of the particles were investigated using TEM (TESCAN VEGA 3 LMU microscope). FTIR analysis of freeze-dried AuNPs was performed using a Bruker Alpha ATR Infrared Spectrometer (available at the University of Mauritius), where 32 scans were collected.Biological activity of JDE, AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control on A172, TERA and HaCaT cell lines
The procedures described above were used for the biological screening of JDE, AuNPs-JDE, JDH, AuNPs-JDH, JC-2, AuNPs-JC-2 and AuNPs-control on A172, HaCaT and TERA cell lines. The assays were performed in triplicates at final concentrations of 25, 12.5, 6.3, 3.1, 1.6 and 0.8 μg mL−1. The concentrations of DMSO used for negative and positive controls were 0.25% and 5%, respectively.Further biological activity
Further biological activity of JDE, AuNPs-JDE, JDH, AuNPs-JDH and AuNPs-control were conducted using HepG2 (human hepatocellular carcinoma) and HeLa (human cervical adenocarcinoma). Each cell line was incubated in Falcon T-25 vented tissue flasks (Corning Inc., NY) in DMEM supplemented with fetal bovine serum (10%) and penicillin/streptomycin (100 U mL−1) at 37 °C in air with 5% CO2. The medium was renewed two times weekly and the cells were sub-cultured when they reached confluence. Each cell line monolayer was detached from tissue culture flasks with trypsin 0.25% EDTA. All medium reagents were obtained from Life Technologies Inc. The cells were suspended in a fresh medium at a density of 1 × 105 cells per mL. The assays were performed in flat-bottom 96-well plates (Corning Inc., NY). Aliquots (50 μL) of this cell suspension were added to the plates. Sterile distilled water (100 μL per well) was added to perimeter wells to prevent evaporation from inner wells. The cells were incubated overnight at 37 °C in air with 5% CO2 before treatment with samples.Jaspamide, JDH and JDE were dissolved in dimethyl sulfoxide (DMSO, Fisher Sci.) to prepare a stock solution (6 mg mL−1), from which serial dilutions were prepared starting from 60 μg mL−1 to 20 μg mL−1, 2 μg mL−1, 0.2 μg mL−1, 0.02 μg mL−1 and 0.002 μg mL−1. AuNPs-JDE was diluted starting from 10 μg mL−1 and AuNPs-control and AuNPs-JDH were diluted starting from 20 μg mL−1 because of their limited amounts. Nocodazole was used as the positive control and its stock solution of 5 mg mL−1 in DMSO was diluted to concentrations of 50 μg mL−1, 20 μg mL−1, 2 μg mL−1, 0.2 μg mL−1, 0.02 μg mL−1 and 0.002 μg mL−1. Samples (50 μL) from each dilution point were added on the second day to the 96-well plates. The final sample concentrations were 1 μg mL−1, 0.1 μg mL−1, 0.01 μg mL−1 and 0.001 μg mL−1. DMSO (0.5%) was used as the negative control. For blank, the medium (50 μL) was added to the wells containing cells (50 μL). For positive control, DMSO (50 μL, 1%) was added to the wells containing cells (50 μL). The plate was incubated for 72 hours at 37 °C in air with 5% CO2. After 72 hours of incubation, MTT (50 μL, 2.5 mg mL−1) was added to each experimental well, including controls, and incubated as described above, for 3 hours. Then, the medium was carefully aspirated from each well. DMSO (100 μL) was added to each experimental well to lyse the cells and the plate was placed on the orbital shaker for 3 min to dissolve formazan crystals. The samples were read using a FilterMax F5 (Molecular Devices) plate reader at 570 nm. The percentage cell viability was calculated using eqn (1).
Light microscopy imaging
HT-29 (ATCC HTB-38) cells were plated at a density of 3.0 × 105 cells/25 cm2 flask and cultured in 35 mm 6-well plates for 48 hours (h) prior to treatment. Jaspamide, JDE, and JDH were dissolved in DMSO, whereas the gold nanoparticles AuNPs-control, AuNPs-JDH and AuNPs-JDE were suspended in sterile water prior to application to cells. Nocodazole (660 μM) was dissolved in DMSO and stored at −20 °C. For non-treated (NT) cells, DMSO was added at a final concentration of 0.4% (v/v) as a solvent vehicle control. Cells were cultivated for 18 hours before analysis by light microscopy imaging, which was performed using an Infinity 1 camera operated by the Infinity Capture imaging software (Lumenera Corporation, CA) on an Olympus CKX41 inverted microscope.56Biological activity of contignasterol, ansellone A and their conjugated AuNPs
The procedures described above (i.e. biological assays) were used for the biological screening of contignasterol, AuNPs-contignasterol, ansellone A and AuNPs-ansellone A on the A172, HaCaT and TERA cell lines. The assays were performed in triplicates at final concentrations of 25, 12.5, 6.3, 3.1, 1.6 and 0.8 μg mL−1. The concentrations of DMSO used for negative and positive controls were 0.25% and 10%, respectively. The data were normalised to the control, and the IC50 (half maximal inhibitory concentration) values were calculated using GraphPad Prism software version 8.4.3.Statistical analysis
GraphPad Prism™ version 8.4.3 was used for statistical analyses. One-way ANOVA for multiple groups, followed by Tukey's test for multiple comparisons, was used for determining statistical significance. The results were expressed as mean ± standard deviation (SD) with significance set at p values ≤0.05.80Zeta potential analysis
The zeta potential of the gold nanoparticles was determined using a modified method by Adeyemi et al., (2019).74 The analysis was conducted using a ZetaSizer instrument (NanoZS, Malvern Panalytical, Malvern, UK). Briefly, 10% v/v of each sample was prepared in distilled water, followed by sonication (Sonics Vibra Cell, Newtown, CT, USA) for 2 min. Samples were then filtered through 0.22 μm filters and transferred into disposable cuvettes for hydrodynamic size and polydispersity index (PDI) analysis, and thereafter into zeta potential cuvettes (DTS 1070) for surface charge analysis at 25 °C. The measurements were performed in triplicates (n = 3).Cellular internalization by fluorescence
Gold nanoparticle uptake in A549 cells was investigated by fluorescence using fluorescein-5-isothiocyanate (FITC) annexin V (Thermo Fisher Scientific) via a modified method developed by Adeyemi et al., (2019).74 FITC (20 μL) was added to annexin-binding buffer (5×) (80 μL) and mixed. The resulting solution (10 μL) was transferred to AuNPs (i.e. AuNPs-JDH, AuNPs-JDE, AuNPs-JC-2, AuNPs-contignasterol, AuNPs-ansellone and AuNPs control). The final concentration of the stock solution of AuNPs was 190 μg mL−1. The samples were incubated at 37 °C for 3 h, so that FITC can bind with the gold nanoparticles. After 24 h, samples were centrifuged and washed with distilled water to remove excess of FITC and re-centrifuged. A549 cells (1 × 105 cells per well) were cultured in a 12-well culture plate for 24 h in 1 mL of DMEM supplemented with FBS (10%) in a humidified atmosphere of 5% CO2 in air. After 24 h, the media was removed and replaced with fresh media containing AuNPs, from stock solutions (i.e. the final concentration of the AuNPs in the well was approx. 10 μg mL−1), and re-incubated for 24 h to allow sufficient time for AuNP internalization. The controls were cells treated with media with and without FITC. After 24 h, the media was removed and washed with PBS and the cells were observed using a Life Technologies EVOS FL Auto fluorescence microscope with a 20× objective. FITC is a fluorescent compound with an excitation peak at 491 nm and an emission peak at 516 nm.Internalization of AuNPs on A549 cells by ICP-MS
A modified method by Carnovale et al., (2019)75 was used for the internalization of AuNPs (i.e. AuNPs-JDH, AuNPs-JDE, AuNPs-JC-2, AuNPs-contignasterol, AuNPs-ansellone and AuNPs control) in A549 cells using ICPMS. A549 cells (1 × 105 cells per well) were cultured in 12-well culture plates for 24 h in 1 mL of DMEM supplemented with FBS (10%) in a humidified atmosphere of 5% CO2 in air. After 24 h, the media flasks were removed and replaced with fresh media containing AuNPs (i.e. the final concentration of the AuNPs in the well was 10 μg mL−1) and re-incubated for 24 h to allow sufficient time for AuNP internalization. The control consisted of only cells treated with media. Thereafter, cells were washed three times with DPBS to remove free AuNPs and treated with trypsin to detach the cells. Cells were then re-suspended in culture media and centrifuged (3500 rpm) for 10 min. The supernatant was discarded and the pellet (containing cells) was re-dispersed in a medium and a cell count was done. Centrifugation was performed and the samples were re-suspended in a solution of ethanol (i.e., 30%, 50%, 70%, 90% and 100%). The dehydrated cell pellets were then oven dried and weighed. For digestion, samples were transferred into digestion tubes and ultrapure water (20 mL) was added to the digestion tubes followed by concentrated nitric acid (5 mL). Samples were then transferred in a microwave digestion system (CEM MARS) (digestion modified AOAC 999.10, microwave digestion). After digestion, the solutions were transferred to tubes (50 mL) and centrifuged. The samples were filtered (0.45 μm) before ICP-MS analysis. The gold content was determined by ICP-MS (Thermo Scientific iCAP Q ICP-MS) and using a calibration curve obtained from a range of standard gold solutions (Gold ICP standard; ICP-22H-1, 1000 μg mL−1) (ISO 17294-1:2004 and ISO 17294-2:2016). Using the recorded values and corrections for dilution, the amount of gold in each sample was determined and hence the amount of gold internalized was calculated. The number of AuNPs taken up by each cell was calculated by dividing the total amount of gold detected by the total number of cells (obtained after post-treatment).Calculations for cellular internalization
The amount of gold (pg) internalized was calculated using the ICP-MS results and the weight of the cell pellets (after dehydration and oven drying). Additionally, the number of AuNPs internalized per cell was determined based on calculations and equations reported by Carnovale et al., (2019)75 as follows:(i) The total number of atoms per particle (Nparticle) for a spherical particle, is given by
| Nparticle = moles of Au per nanoparticle × NA |
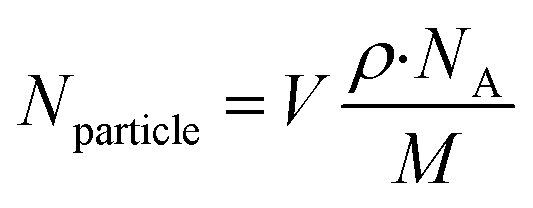 | (2) |
 ; density of gold (Au) ρ = 1.93 × 107 g m−3; molar mass (M) of Au = 196.97 g mol−1 and Avogadro's constant (NA) = 6.022 × 1023 mol−1.
; density of gold (Au) ρ = 1.93 × 107 g m−3; molar mass (M) of Au = 196.97 g mol−1 and Avogadro's constant (NA) = 6.022 × 1023 mol−1.
(ii) Eqn (2) can be simplified as follows:
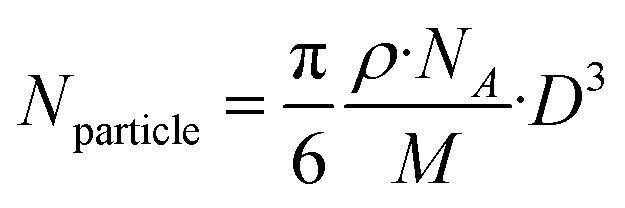 | (3) |
(iii) Taking into consideration that D is at the nano level, eqn (3) can be simplified as follows:
| Nparticle = 30.89·D3 | (4) |
(iv) The AuNPs internalized (Ninternalized) is calculated as follows:
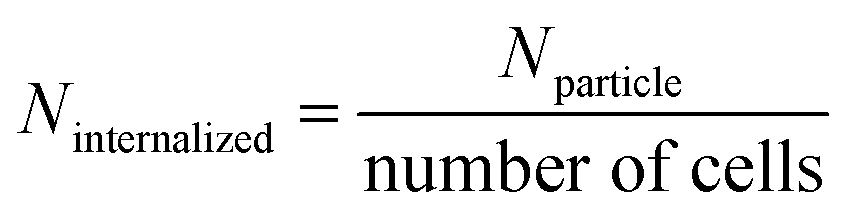 | (5) |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Mr Avin Ramanjooloo performed the investigation, methodology, data analysis, writing original draft, review and editing. Mr Devesh Bekah characterized the AuNPs. Dr Samson A. Adeyemi, Dr Philemon Ubanako, Prof. Yahya E. Choonara and Lindokuhle Ngema were involved with the in vitro studies on A172, TERA1, DLD1 and HaCaT cell lines. Dr David E. Williams helped in the identification and characterization of pure compounds. Dr Elena A. Polishchuk was involved in the in vitro studies on HeLa and HepG2 cell lines. Prof Raymond J. Andersen contributed to the scientific visit of Mr Avin Ramanjooloo at the University of British Columbia (UBC). Prof. Archana Bhaw-Luximon performed conceptualization of the study, writing original draft, review and editing, and resource and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mr Avin Ramanjooloo and Prof. Archana Bhaw-Luximon wish to acknowledge Mr Chettanand Samyan for sample sponge collection, Mr Mahmoud Kamel for GC-MS analysis, Jie Chen for biological assay at UBC, Prof. Golsteyn Roy for conducting light microscopy imaging and Prof. Rob Van Soest for sponge identification. Mr Avin Ramanjooloo wishes to thank the Higher Education Commission (Mauritius) for an MPhil/PhD fellowship and the Biomaterials, Drug Delivery and Nanotechnology Unit (CBBR) for funding this work through its project grant KB027. Mr Avin Ramanjooloo and Prof. Archana Bhaw-Luximon wish to thank Prof Raymond Andersen (University of British Columbia, Canada) for his support throughout the project. Mr Avin Ramanjooloo wishes to acknowledge Dr Itisha Chummun Phul for the internalization study.Notes and references
- K. K. Bharadwaj, B. Rabha, S. Pati, T. Sarkar, B. K. Choudhury, A. Barman, D. Bhattacharjya, A. Srivastava, D. Baishya and H. A. Edinur, Molecules, 2021, 26, 6389 CrossRef PubMed.
- F. Aflakian, F. Mirzavi, H. T. Aiyelabegan, A. Soleimani, J. G. Navashenaq, I. Karimi-Sani, A. R. Zomorodi and R. Vakili-Ghartavol, Eur. J. Pharm. Sci., 2023, 106515 CrossRef PubMed.
- C. Panwar, A. Phillips, A. Sangal and D. Gupta, Biointerface Res. Appl. Chem., 2024, 12, 11 Search PubMed.
- M. N. Kundranda and J. Niu, Drug Des., Dev. Ther., 2015, 9, 3767 CrossRef.
- Y. Yang, X. Zheng, L. Chen, X. Gong, H. Yang, X. Duan and Y. Zhu, Int. J. Nanomed., 2022, 2041–2067 CrossRef.
- N. R. S. Sibuyi, K. L. Moabelo, A. O. Fadaka, S. Meyer, M. O. Onani, A. M. Madiehe and M. Meyer, Nanoscale Res. Lett., 2021, 16, 1–27 CrossRef.
- F.-Y. Kong, J.-W. Zhang, R.-F. Li, Z.-X. Wang, W.-J. Wang and W. Wang, Molecules, 2017, 22, 1445 CrossRef.
- X. Li, Y. Zhang, G. Liu, L. Zhou, Y. Xue and M. Liu, RSC Adv., 2022, 12, 7635–7651 RSC.
- K. Bolaños, M. J. Kogan and E. Araya, Int. J. Nanomed., 2019, 14, 6387 CrossRef PubMed.
- K. Nejati, M. Dadashpour, T. Gharibi, H. Mellatyar and A. Akbarzadeh, J. Cluster Sci., 2022, 33, 1–16 CrossRef.
- M. Janát-Amsbury, A. Ray, C. Peterson and H. Ghandehari, Eur. J. Pharm. Biopharm., 2011, 77, 417–423 CrossRef.
- M. Souri and M. Soltani, in Gold Nanoparticles for Drug Delivery, Elsevier, 2024, pp. 495–509 Search PubMed.
- H. Khan, U. Shahab, A. Alshammari, A. R. Alyahyawi, R. Akasha, T. Alharazi, R. Ahmad, A. Khanam, S. Habib and K. Kaur, IUBMB Life, 2024, 1–17 Search PubMed.
- W. Wang, J. Wang and Y. Ding, J. Mater. Chem. B, 2020, 8, 4813–4830 RSC.
- S. Siddique and J. C. Chow, Appl. Sci., 2020, 10, 3824 CrossRef.
- Y. Ghafari, A. Asefnejad and D. O. Ogbemudia, Sci. Hypotheses, 2024, 1, 21–35 CrossRef.
- R. Zhang, F. Kiessling, T. Lammers and R. M. Pallares, Drug Delivery Transl. Res., 2023, 13, 378–385 CrossRef PubMed.
- L. Yao, D. Bojic and M. Liu, J. Pharm. Anal., 2023, 13, 960–967 CrossRef PubMed.
- P. B. Santhosh, J. Genova and H. Chamati, Chemistry, 2022, 4, 345–369 CrossRef CAS.
- E. Agunloye, A. Gavriilidis and L. Mazzei, Chem. Eng. Sci., 2017, 173, 275–286 CrossRef CAS.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- B. Mubaiwa, M. S. Lerata, N. R. Sibuyi, M. Meyer, T. Samaai, J. J. Bolton, E. M. Antunes and D. R. Beukes, Materials, 2023, 16, 1319 CrossRef CAS.
- M. Rai, A. P. Ingle, I. Gupta and A. Brandelli, Int. J. Pharm., 2015, 496, 159–172 CrossRef CAS PubMed.
- K. Kaliaperumal, L. Salendra, Y. Liu, Z. Ju, S. K. Sahu, S. Elumalai, K. Subramanian, N. M. Alotaibi, N. Alshammari and M. Saeed, Front. Microbiol., 2023, 14, 1216928 CrossRef PubMed.
- V. Ghosh, Encyclopedia of Mar. Biotechnol., 2020, vol. 3, pp. 1521–1534 Search PubMed.
- I. N. Rizki and W. Klaypradit, Sustainable Chem. Pharm., 2023, 31, 100888 CrossRef CAS.
- F. Ameen, K. S. Al-Maary, A. Almansob and S. AlNadhari, Appl. Nanosci., 2023, 13, 2233–2240 CrossRef CAS.
- J. Liang, J. She, J. Fu, J. Wang, Y. Ye, B. Yang, Y. Liu, X. Zhou and H. Tao, Mar. Drugs, 2023, 21, 236 CrossRef PubMed.
- D. Inbakandan, R. Venkatesan and S. A. Khan, Colloids Surf., B, 2010, 81, 634–639 CrossRef.
- G. Beedessee, A. Ramanjooloo, G. Aubert, L. Eloy, R. Surnam-Boodhun, R. W. Van Soest, T. Cresteil and D. E. Marie, Environ. Toxicol. Pharmacol., 2012, 34, 397–408 CrossRef.
- G. Beedessee, A. Ramanjooloo, G. Aubert, L. Eloy, D. Arya, R. W. Van Soest, T. Cresteil and D. E. Marie, Environ. Toxicol. Pharmacol., 2013, 36, 58–65 CrossRef.
- G. Beedessee, A. Ramanjooloo, I. Tiscornia, T. Cresteil, S. Raghothama, D. Arya, S. Rao, K. H. Gowd, M. Bollati-Fogolin and D. E. Marie, J. Pharm. Pharmacol., 2014, 66, 1317–1327 CrossRef PubMed.
- A. Ramanjooloo, M. Kamel, S. A. Adeyemi, P. Ubanako, B. Baudot, A. D. Thakoor, Y. E. Choonara and A. Bhaw-Luximon, New J. Chem., 2023, 47, 8854–8866 RSC.
- M. M. Ododo, M. K. Choudhury and A. H. Dekebo, Structure elucidation of β-sitosterol with antibacterial activity from the root bark of Malva parviflora, SpringerPlus, 2016, 5, 1–11 CrossRef.
- M. Amina, T. Amna, N. Al-Musayeib, S. A. Zabin, M. S. Hassan and M.-S. Khil, Appl. Biochem. Biotechnol., 2017, 182, 624–634 CrossRef PubMed.
- J.-M. C. Cayme and C. Y. Ragasa, Kimika, 2004, 20, 5–12 Search PubMed.
- L. L. Pierre, M. N. Moses and J. Innov, Pharm. Biol. Sci., 2015, 2, 88–95 CAS.
- J. A. Bowden, D. M. Colosi, D. C. Mora-Montero, T. J. Garrett and R. A. Yost, J. Chromatogr. B, 2009, 877, 3237–3242 CrossRef CAS.
- R. E. Kirk and D. F. Othmer, Encyclopedia of Chemical Technology, John Wiley & Sons, New Jersey, 5th edn, 2006 Search PubMed.
- P. Praveena, K. S. Singh, B. Naikc and P. Parameswaranb, J. Indian Chem. Soc., 2017, 94, 637–640 Search PubMed.
- H. Wang, Z. Wang, Z. Zhang, J. Liu and L. Hong, Adv. Nutr., 2023, 14, 1085–1110 CrossRef CAS PubMed.
- X. Zhang, J. Wang, L. Zhu, X. Wang, F. Meng, L. Xia and H. Zhang, Front. Oncol., 2022, 12, 1101289 CrossRef CAS.
- N. Shahzad, W. Khan, M. Shadab, A. Ali, S. S. Saluja, S. Sharma, F. A. Al-Allaf, Z. Abduljaleel, I. A. A. Ibrahim and A. F. Abdel-Wahab, Biomed. Pharmacother., 2017, 88, 786–794 CrossRef CAS.
- H. Bae, S. Park, C. Yang, G. Song and W. Lim, Antioxidants, 2021, 10, 379 CrossRef CAS PubMed.
- G. Marslin, K. Siram, Q. Maqbool, R. K. Selvakesavan, D. Kruszka, P. Kachlicki and G. Franklin, Materials, 2018, 11, 940 CrossRef PubMed.
- F. Masse, P. Desjardins, M. Ouellette, C. Couture, M. M. Omar, V. Pernet, S. Guérin and E. Boisselier, Molecules, 2019, 24, 2929 CrossRef CAS PubMed.
- F. Ali, M. Hamza, M. Iqbal, B. Basha, N. Alwadai and A. Nazir, J. Phys. Chem., 2022, 236, 291–326 CAS.
- J. Quinson, O. Aalling-Frederiksen, W. L. Dacayan, J. D. Bjerregaard, K. D. Jensen, M. R. Jørgensen, I. Kantor, D. R. Sørensen, L. Theil Kuhn and M. S. Johnson, Chem. Mater., 2023, 35, 2173–2190 CrossRef CAS.
- D. Philip, Spectrochim. Acta, Part A, 2010, 77, 807–810 CrossRef PubMed.
- A. Woźniak, A. Malankowska, G. Nowaczyk, B. F. Grześkowiak, K. Tuśnio, R. Słomski, A. Zaleska-Medynska and S. Jurga, J. Mater. Sci.: Mater. Med., 2017, 28, 1–11 CrossRef PubMed.
- K. M. Soto, I. Luzardo-Ocampo, J. M. López-Romero, S. Mendoza, G. Loarca-Piña, E. M. Rivera-Muñoz and A. Manzano-Ramírez, Pharmaceutics, 2022, 14, 2069 CrossRef CAS PubMed.
- D.-J. Jin, S.-A. Tang, G.-S. Xing, W.-J. Zhao, C. Zhao, H.-Q. Duan and W.-H. Lin, J. Asian Nat. Prod. Res., 2014, 16, 427–433 CrossRef CAS.
- R. Khandanlou, V. Murthy, D. Saranath and H. Damani, J. Mater. Sci., 2018, 53, 3106–3118 CrossRef CAS.
- S. Dutta, S. K. Mitra, A. Bir, T. Prabha and A. Ghosh, Cureus, 2023, 15, e43087 Search PubMed.
- V. Ramalingam, S. Revathidevi, T. Shanmuganayagam, L. Muthulakshmi and R. Rajaram, RSC Adv., 2016, 6, 20598–20608 RSC.
- L. Molina, D. E. Williams, R. J. Andersen and R. M. Golsteyn, Heliyon, 2021, 7, e07131 CrossRef CAS PubMed.
- T. Schneider, M. Westermann and M. Glei, Arch. Toxicol., 2017, 91, 3517–3527 CrossRef CAS.
- A. Sani, C. Cao and D. Cui, Biochem. Biophys. Rep., 2021, 26, 100991 Search PubMed.
- S. Dhar, E. M. Reddy, A. Shiras, V. Pokharkar and B. e. L. e. V. Prasad, Chem. - Eur. J., 2008, 14, 10244–10250 CrossRef PubMed.
- X. Zhang, H. Chibli, R. Mielke and J. Nadeau, Bioconjugate Chem., 2011, 22, 235–243 CrossRef PubMed.
- D. L. Burgoyne, R. J. Andersen and T. M. Allen, J. Org. Chem., 1992, 57, 525–528 CrossRef.
- M. A. Gammone, E. Gemello, G. Riccioni and N. D'Orazio, Mar. Drugs, 2014, 12, 2357–2382 CrossRef.
- R. J. Andersen, T. M. Allen and D. L. Burgoyne, U.S. Pat.5646138, 1997 Search PubMed.
- J. Daoust, A. Fontana, C. E. Merchant, N. J. de Voogd, B. O. Patrick, T. J. Kieffer and R. J. Andersen, Org. Lett., 2010, 12, 3208–3211 CrossRef PubMed.
- A. Caso, F. B. da Silva, G. Esposito, R. Teta, G. D. Sala, L. P. N. Cavalcanti, A. L. Valverde, R. C. C. Martins and V. Costantino, Mar. Drugs, 2021, 19, 667 CrossRef.
- M. Yanagihara, K. Murai, N. Kishimoto, T. Abe, S. Misumi and M. Arisawa, Org. Lett., 2021, 23, 1720–1725 CrossRef.
- D. E. Williams, P. Lassota and R. J. Andersen, J. Org. Chem., 1998, 63, 4838–4841 CrossRef.
- C. D. Roskelley, D. E. Williams, L. M. McHardy, K. G. Leong, A. Troussard, A. Karsan, R. J. Andersen, S. Dedhar and M. Roberge, Cancer Res., 2001, 61, 6788–6794 Search PubMed.
- L. Yang, D. E. Williams, A. Mui, C. Ong, G. Krystal, R. van Soest and R. J. Andersen, Org. Lett., 2005, 7, 1073–1076 CrossRef PubMed.
- R. J. Andersen, Biochem. Pharmacol., 2017, 139, 3–14 CrossRef.
- H. R. El-Seedi, R. M. El-Shabasy, S. A. Khalifa, A. Saeed, A. Shah, R. Shah, F. J. Iftikhar, M. M. Abdel-Daim, A. Omri and N. H. Hajrahand, RSC Adv., 2019, 9, 24539–24559 RSC.
- V. C. Thipe, A. Jatar, A. Raphael Karikachery, K. K. Katti and K. V. Katti, Nanotechnol., Sci. Appl., 2023, 19–40 CrossRef.
- K. Böse, M. Koch, C. Cavelius, A. K. Kiemer and A. Kraegeloh, Part. Syst. Charact., 2014, 31, 439–448 CrossRef.
- S. A. Adeyemi, Y. E. Choonara, P. Kumar, L. C. du Toit, T. Marimuthu, P. P. Kondiah and V. Pillay, Biomed. Pharmacother., 2019, 119, 109450 CrossRef.
- C. Carnovale, G. Bryant, R. Shukla and V. Bansal, ACS Omega, 2019, 4, 242–256 CrossRef.
- B. Giesen, A.-C. Nickel, A. G. Manjón, A. V. Toscano, C. Scheu, U. D. Kahlert and C. Janiak, J. Inorg. Biochem., 2020, 203, 110952 CrossRef PubMed.
- R. S. Darweesh, N. M. Ayoub and S. Nazzal, Int. J. Nanomed., 2019, 7643–7663 CrossRef PubMed.
- A. Ramanjooloo, I. C. Phul, N. Goonoo and A. Bhaw-Luximon, Int. J. Biol. Macromol., 2024, 259, 129218 CrossRef PubMed.
- F. Barzgar, S. Sadeghi-Mohammadi, Y. Aftabi, H. Zarredar, M. Shakerkhatibi, P. Sarbakhsh and A. Gholampour, Sci. Total Environ., 2023, 877, 162726 CrossRef PubMed.
- H. A. El-Naggar, M. A. Bashar, I. Rady, M. S. El-Wetidy, W. B. Suleiman, F. O. Al-Otibi, S. A. Al-Rashed, L. M. Abd El-Maoula, E.-S. S. Salem and E. M. Attia, Appl. Sci., 2022, 12, 1400 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. 1S–11S and Tables 1S–2S See DOI: https://doi.org/10.1039/d4ra04068f |
| This journal is © The Royal Society of Chemistry 2024 |